

Abstract
Mutations of the tumor suppressor TP53 are present in many forms of human cancer and are associated with increased tumor cell invasion and metastasis. Several mechanisms have been identified for promoting dissemination of cancer cells with TP53 mutations, including increased targeting of integrins to the plasma membrane. Here, we demonstrate a role for the filopodia-inducing motor protein Myosin-X (Myo10) in mutant p53–driven cancer invasion. Analysis of gene expression profiles from 2 breast cancer data sets revealed that MYO10 was highly expressed in aggressive cancer subtypes. Myo10 was required for breast cancer cell invasion and dissemination in multiple cancer cell lines and murine models of cancer metastasis. Evaluation of a Myo10 mutant without the integrin-binding domain revealed that the ability of Myo10 to transport β1 integrins to the filopodia tip is required for invasion. Introduction of mutant p53 promoted Myo10 expression in cancer cells and pancreatic ductal adenocarcinoma in mice, whereas suppression of endogenous mutant p53 attenuated Myo10 levels and cell invasion. In clinical breast carcinomas, Myo10 was predominantly expressed at the invasive edges and correlated with the presence of TP53 mutations and poor prognosis. These data indicate that Myo10 upregulation in mutant p53–driven cancers is necessary for invasion and that plasma-membrane protrusions, such as filopodia, may serve as specialized metastatic engines.
Introduction
Understanding the genesis of motile cancer cells capable of invading the surrounding stroma and disseminating into distant organs is of key importance, since metastases are the main cause of death of patients with cancer (1). Gene expression signatures of both primary cancers and metastatic cells have been defined for many cancer types (2–4), but how the genes linked with the metastatic genotypes drive metastasis remains largely unknown.
Filopodia are slender actin-containing plasma membrane protrusions. Their role in cell migration is well established in many important physiological processes, such as embryonic development, wound healing, and angiogenesis (5–7). Filopodia contain integrins and other cell-surface receptors as well as actin-nucleating proteins. Filopodia and filopodial proteins are required to form basement membrane–traversing actin protrusions called invadopodia (8). Fascin is actin-bundling and filopodia-promoting protein (9, 10) that has been linked to poor prognosis of patients with cancer (11, 12). Myo10 is a filopodia-inducing, unconventional, actin-based motor protein (13–15) with ubiquitous expression in various mammalian tissues (14, 16). It transports integrins to the filopodia tips and thus regulates filopodia stability (15). Although integrins are established mediators of cell motility and invasion, the role of Myo10 in cancer dissemination remains unknown.
Mutations of the TP53 tumor-suppressor gene are among the most frequent genetic alterations in human cancer. A majority of TP53 mutations not only impair the ability of p53 to bind WT p53–responsive elements on DNA, but the mutated protein may also gain new functions (17). A large body of evidence indicates that mutant p53 promotes cell migration, epithelial-to-mesenchymal transition, and metastasis (18–23). Interestingly, one of the mechanisms involved with metastasis downstream of mutant p53 is accelerated recycling of cell-adhesion receptors, β1 integrins, to the plasma membrane (19), suggesting that the integrins are necessary components for mutant p53–driven invasion.
Here we present evidence for a mutant p53–initiated, Myo10-dependent cell-invasion cascade. We show that Myo10 is frequently highly expressed in breast cancers and mediates adhesion, migration, invasion, and metastasis of breast cancer cells in vitro and in vivo. Our results indicate that targeting of integrins to the filopodia tips is critical for their proinvasive capability and that high Myo10 expression correlates with poor prognosis of patients with breast cancer. Expression of Myo10 is associated with expression of mutant p53 in clinical breast tumor samples and is required for mutant p53–driven cell invasion and metastasis. Overall, we describe a clinically important invasion cascade that might lead to new therapeutic interventions.
Results
Myosin-X is upregulated in basal-like and HER2-enriched breast carcinoma.
To investigate a possible link between filopodia and cancer invasiveness, we focused on breast cancer and searched for filopodia-related genes that are upregulated in carcinomas (24, 25). An in silico analysis of gene expression profiles of 2 independent breast cancer data sets (GEO GSE3494, 251 patients, ref. 25; and GEO GSE3985/GSE10783, 104 patients, ref. 4) revealed an intriguing candidate, MYO10. Expression of MYO10 was high in 2 aggressive and often metastatic breast cancer subtypes, basal-like and HER2-enriched cancer, as compared with the less aggressive subtypes (normal breast-like cancer and the luminal types A and B) (Figure 1A and Supplemental Figure 1; supplemental material available online with this article; doi: 10.1172/JCI67280DS1).
Figure 1. Myo10 is upregulated in breast cancer.

(A) The expression levels of MYO10 mRNA in 109 (GEO GSE3985/GSE19783) and 251 (GEO GSE3494) clinical breast tumor samples are shown as the log2 expression ratio. Clinical tumor classification, number of samples, and statistical significance are indicated below the box plots. (B) Western blot of Myo10 in the indicated breast cancer cell lines. Tubulin is shown for loading control. (C) Immunofluorescence staining of Myo10 in MDA-MB-231 cells. Right panel shows ROI (10 μm × 10 μm). Arrowheads point to filopodial, and arrows point to lamellipodial Myo10 localization. Scale bar: 10 μm. (D) Total internal reflection fluorescence microscope (TIRFM) image of MDA-MB-231 cell expressing EGFP-Myo10. Scale bar: 10 μm.
The MDA-MB-231 cell line is a widely used model for basal-like breast cancer (26, 27). In line with the in silico analysis, the Myo10 protein was highly expressed in these cells compared with the less aggressive and luminal-like MCF7 cell line (27) or to nontumorigenic breast epithelial MCF10A cells (ref. 28 and Figure 1B). The MDA-MB-231 cells display variable morphology in vitro, and endogenous Myo10 was found to be present in several types of protrusive structures including the tips of filopodia (Figure 1C, arrowheads) and in microspikes along the lamellipodia (Figure 1C, arrows). Importantly, the Myo10-positive protrusions were strongly associated with the matrix, since the Myo10-containing structures were readily visible when examined with total internal reflection fluorescence microscopy (Figure 1D).
Myo10 is required for breast cancer cell adhesion, migration, and invasion in vitro.
Since Myo10 is expressed in the protrusive structures of highly migratory breast cancer cells, we investigated the functional consequences of loss of Myo10. We observed that siRNA-mediated silencing of MYO10 in MDA-MB-231 cells induced slightly rounder cell morphology (Supplemental Figure 2, A and B) and significantly inhibited cell migration on the extracellular matrix (Matrigel; Supplemental Figure 2C). siRNA-mediated downregulation of Myo10 inhibited invasion of these cells through 3D Matrigel toward chemoattractants (an increasing serum gradient; Supplemental Figure 2D) without affecting proliferation or apoptosis (Supplemental Figure 3, A and B).
To validate these transient RNAi transfection–based data, we generated stable Myo10-silenced MDA-MB-231 clones using shRNA plasmids. Transfection of the Myo10-targeting shRNA reduced cell Myo10 levels, but did not downregulate important cell adhesion–related proteins β1 integrin and talin (Figure 2A) or influence proliferation or apoptosis (Supplemental Figure 3, C and D). Myo10 stable knockdown cell lines also displayed significantly rounder cell morphology (Figure 2B) and had reduced cell migration on Matrigel (Figure 2C) and attenuated invasion into Matrigel (Figure 2D). Conversely, overexpression of GFP-Myo10 in the Myo10-negative MCF7 cells significantly induced invasion of these poorly invasive cells into Matrigel (Figure 2E). Myo10 was also essential for adhesion and spreading on MDA-MB-231 cells. The Myo10–silenced cells adhered more slowly to fibronectin as compared with unmodified cells (Figure 2F), and most Myo10-silenced cells lacked endogenous filopodia (Figure 2G). These data demonstrate a previously unrecognized requirement of Myo10 for migration and invasion of breast cancer cells.
Figure 2. Myo10 regulates spreading, migration, and invasion.

(A) shMyo10–expressing MDA-MB-231 cell clones analyzed by Western blot for expression of Myo10, Talin-1, β1 integrin, and tubulin. (B) Morphology of shRNA-expressing cells on Matrigel. Shown are cell outlines, calculated cell area, and roundness (inverse of major axis/minor axis). Area units are pixels. n = 25 (shControl); n = 26 (shMyo10). Scale bar: 20 μm. (C) Random migration of shMyo10-expressing (pooled clones no. 1 and no. 2) MDA-MB-231 cells on Matrigel. Cumulative mean square displacement of tracked cells is shown. n = 75 (shControl) and n = 85 (shMyo10). (D) Invasion of shMyo10-expressing (pooled clones no. 1 and no. 2) MDA-MB-231 cells into Matrigel (4 days). Images show side views of invasion. Column graph shows mean invasion areas, and arrows indicate the direction of invasion. n = 10 (shControl); n = 14 (shMyo10) fields of view with ×20 objective. (E) Invasion of MCF-7 cells transfected with EGFP alone or EGFP-Myo10. Column graph shows the percentage of invaded GFP-positive cells from all GFP-positive cells. n = 10 (shControl); n = 8 (shMyo10) fields of view with ×20 objective. (F) Adhesion of shMyo10-expressing (pooled clones no. 1 and no. 2) MDA-MB-231 cells on fibronectin (5 μg/ml) was analyzed at 30- and 60-minute time points as 3 independent experiments. (G) Filopodial phenotype of shMyo10-expressing (pooled clones no. 1 and no. 2) MDA-MB-231 cells spreading actively on fibronectin (5 μg/ml). The number of cells expressing clear filopodial phenotype is shown. n = 21 (shControl); n = 25 (shMyo10). (H) Images show examples of the filopodia phenotype. Scale bar: 20 μm. Mean ± SEM and Mann-Whitney test P values are provided.
Myo10-mediated targeting of integrin to the tips of filopodia is required for cell invasion.
These findings linked the expression of Myo10 to adhesion-dependent functions that are regulated by integrins. Live-cell imaging of enhanced GFP–tagged (EGFP-tagged) Myo10-expressing cells labeled with a fluorescent β1 integrin antibody revealed cooperation of integrins and the motor protein Myo10 in filopodia maintenance. Integrins localize to the tip of the filopodia and remain stationary, presumably mediating adhesion of the tip (Figure 3A). In contrast, Myo10 moves back and forth along the filopodia and briefly overlaps with the integrins at the tip (Figure 3A and Supplemental Video 1).
Figure 3. Myo10-mediated targeting of integrin to the filopodia tip is critical for invasion.

(A) Cells expressing EGFP-Myo10 (green) stained with fluorescently labeled β1 integrin antibody (red). Time-lapse images of filopodia dynamics acquired at 10-second intervals. Still images of Myo10 moving back and forth along filopodia are shown. β1 Integrin is mostly seen at the filopodia tip. Scale bar: 5 μm. (B) MDA-MB-231 cells transfected with EGFP-Myo10 and EGFP-Myo10ΔFERM2 and analyzed for filopodia localization. Images show confocal slices at the bottom and the top of the cells. Graph shows the number of dorsal filopodia. Scale bar: 10 μm. (C) MDA-MB-231 cells transfected with EGFP-Myo10 (green) and treated with β1 integrin function–blocking (mAb13) and –activating (12G10) antibodies and stained for filamentous actin. Filopodia lengths were measured along filopodia shafts. Scale bar: 10 μm. (D) Myo10 shRNA–expressing MDA-MB-231 cells (pooled clones no. 1 and no. 2; shMyo10) transfected as indicated and imaged for localization of β1 integrin at the filopodia tips. Representative images are shown. Scale bar: 5 μm. (E, F) shMyo10 cells transfected with EGFP-Myo10 or EGFP-Myo10ΔFERM2 (green) to reconstitute Myo10 expression. Invasion into Matrigel (4 days) was analyzed by detecting all cells with Syto60 staining (white in the merged image) and the transfected cells by EGFP. Images show side view z-stacks. Arrows indicate the direction of invasion. The noninvaded cells at the bottom of the wells are below the red line. n = 6 (EGFP-Myo10) and n = 9 (EGFP-MYO10 ΔFERM) fields of view with ×20 objective. Mean ± SEM and Mann-Whitney test P values are provided.
We then employed a deletion mutant of Myo10 that is unable to bind integrins (Myo10ΔFERM2) but retains all other functional domains (15). Both WT and mutant Myo10 efficiently induced filopodia in cells. However, the filopodia induced by the WT Myo10 predominantly localized to the matrix-facing side of the cells, whereas Myo10ΔFERM2 induced more dorsal filopodia that did not adhere to the matrix (Figure 3B). Moreover, when cells expressing the WT EGFP-Myo10 were treated with an integrin function–blocking antibody (Mab13) (29), they displayed shortened and distorted filopodia as compared with the same cells treated with an integrin-activating antibody (12G10; Figure 3C and ref. 30). This underlines the requirement of integrin-mediated adhesion at the filopodia tip for stabilization of the filopodia. Accordingly, α5 integrin was transported efficiently to the tips of filopodia with WT Myo10 (Figure 3D, arrowheads), but failed to localize to the tips of the shorter and more distorted Myo10ΔFERM2-induced filopodia (ref. 15 and Figure 3D). Myo10 also colocalized more frequently with the active integrin close to the matrix compared with Myo10ΔFERM2 (Supplemental Figure 4, arrowheads). Finally, shMyo10 cells that reexpressed the WT Myo10 were more invasive than the same cells that reexpressed the Myo10ΔFERM2 construct (Figure 3E). Thus, the ability of Myo10 to mediate cell invasion is linked to its function as a transporter of integrin adhesion receptors to the filopodia tip.
Myo10 regulates dissemination of breast cancer cells in vivo.
Since the in vitro results suggested that Myo10 is an important regulator of cell adhesion and motility, we tested whether Myo10 is needed for dissemination of malignant cells in vivo using a tumor model in which zebrafish embryos (31) were implanted with human tumor cells. Control and Myo10-knockdown MDA-MB-231 cells were fluorescently labeled in vitro and injected into the perivitelline cavity of embryos 48 hours after fertilization. In agreement with previous data (32), the control cells disseminated widely and formed metastasis at 4 days after injection (Figure 4A), with multiple cells metastasizing through the vasculature into the tail region (Figure 4B). Remarkably, neither of the 2 independent Myo10-knockdown cell clones showed any significant invasion or metastasis (Figure 4, A and B). Metastases of the transiently siRNA-silenced MDA-MB-231 cells were also strongly suppressed in the same model (Supplemental Figure 5A).
Figure 4. Myo10 regulates invasion and metastasis in vivo.

(A and B) Labeled shControl and shMyo10 MDA-MB-231 cells were microinjected into zebrafish embryos, and dissemination (yellow arrowheads) was observed 4 days after implantation. Top panels show higher magnification regions of head-trunk and tail areas. Scale bars 500 μm (top panels); 100 μm (bottom panels). (C and D) Extravasation of shControl and shMyo10 MDA-MB-231 cells (C) or siMyo10 and siControl-transfected MDA-MB-468 and BT-474 breast cancer cells (D) into mouse lungs was studied in vivo. shMyo10/siMyo10 cells (green) and shControl/siControl cells (red) were coinjected (1:1) into the tail vein. After 48 hours, extravasated cells (arrowheads) were analyzed from lung sections visually (C) (12 sections/mouse) or by flow cytometry (D). Shown are representative lung sections stained with dapi. Scale bar: 300 μm (C and D). Area containing extravasated shControl cells is indicated with a dashed line. (E and F) Lung colonization of tail vein–injected unstained shControl and shMyo10 cells (4 weeks after injection). Shown are representative H&E stainings of lung tissue sections (E) (metastases indicated with asterisks) and FACS analysis of the human HLA or human vimentin–positive cells (%) of all the cells isolated from lungs (F). Scale bar: 100 μm. (G) Systemic spreading of shMyo10 or shControl cells injected orthotopically to mammary fat pads of nude mice was assessed after 6 weeks from frozen contralateral lymph nodes with vimentin staining, and primary tumors were stained for reference. Quantitation shows the number of mice with metastasis in the lymph nodes. Scale bar: 300 μm. Mean ± SEM and Mann Whitney test P values; n = 10 mice.
To further investigate the requirement of Myo10 for cancer invasiveness in vivo, we first investigated the effect of Myo10 in the extravasation step of metastasis. We injected control and shMyo10 cells i.v. into nude mice. The 2 cell populations were first stained red (control cells) or green (shMyo10 cells) with live cell dyes, following which they were coinjected in equal numbers into the same animal. This experiment suggested that Myo10 is required for extravasation of cancer cells into the lungs, as significantly reduced numbers of shMyo10 cells were detected in the lungs at 2 days after injection (Figure 4C). Similar results were obtained when Myo10 was transiently silenced in 2 other breast cancer cell lines harboring endogenous mutant p53 (MDA-MB-468 and BT-474, ref. 33; Figure 4D and Supplemental Figure 5B). We also observed that the Myo10-positive cells (shControl) were able to colonize in lungs and generate clearly visible metastases in mouse lungs when allowed to grow for 4 weeks after i.v. injection. However, the numbers of metastatic cells and visible metastatic lesions were significantly reduced in mice injected with shMyo10 cells analyzed by immunohistochemistry of lung sections (Figure 4E and Supplemental Figure 5C) and by flow cytometry of cells isolated from the lungs (Figure 4F) Finally, we investigated the requirement for Myo10 in breast cancer metastasis using orthotopic implantation of shControl and shMyo10 cells into the mammary fat pads of nude mice. In line with previous publications (22), we failed to detect lung metastasis of MDA-MB-231 cells. However, in 42% of the mice injected with shControl cells, metastasis was detected in the contralateral lymph nodes (indicative of systemic spread, ref. 22), while no metastasis were observed in the shMyo10 mice (Figure 4G and Supplemental Figure 5D). Taken together, these data demonstrate a functional requirement of Myo10 in dissemination of breast cancer cells in vivo.
High Myo10 expression correlates with poor prognosis in breast cancer.
The evidence for Myo10-dependent cell invasion and metastasis using the MDA-MB-231 cell model prompted us to investigate the clinical significance of Myo10. An analysis of MYO10 mRNA expression in the breast tumors of women included in the Oslo MicMa cohort (24) (109 patients) revealed that patients with high tumor MYO10 expression had less favorable survival compared with patients whose tumors expressed a low level of MYO10 (P = 0.011; Figure 5A).
Figure 5. High Myo10 expression in clinical breast cancer samples.

(A) MYO10 expression and survival (Kaplan-Meier curves) were analyzed in 104 patients with early breast cancer (GEO GSE3985/GSE19783 series; ref. 4). log-rank test P value, P = 0.011. (B) Myo10 protein immunostaining from the middle and at the invasive edges of the primary breast carcinomas. Scale bar: 200 μm. (C) Expression of Myo10 in a lymph node metastasis from the primary tumor shown in B. Scale bar: 100 μm (original magnification, ×10); 400 μm (original magnification, ×40). (D) Examples of immunostaining, and scoring of different Myo10 expression levels in TMA samples. Scale bar: 100 μm. (E) Survival of 1,326 Finnish patients with early breast cancer and high Myo10 expression (red) or low-intermediate/negative Myo10 expression (blue). log-rank test, P = 0.018. (F) Survival of the subset of 482 patients with regional lymph node–positive breast cancer (pN+) as in E. (G) Comparison of tumor Myo10 and p53 staining from adjacent TMA slices from a tumor with negative and positive Myo10 and p53 expression. Scale bar: 200 μm. (H) Analysis of MYO10 mRNA expression (log2 scale, P value, Student’s t test) and p53 mutations in breast cancer (n = 109; GEO GSE3985/GSE19783 series; ref. 4).
To investigate Myo10 at the protein level, we stained 31 human breast cancers for Myo10 using immunohistochemistry. Myo10 expression was higher at the invasive edge of the breast carcinomas as compared with the central parts of the tumors (P = 0.004, n = 31; Figure 5B), which is in line with the proinvasive function of Myo10. Overall, Myo10 expression was heterogeneous, and staining of the tumor edge regions ranged from strongly positive to negative. When metastatic lymph nodes from the same patients were immunostained, moderate Myo10 expression was detected in the metastases with the highest expression detected at the sites where cancer invaded into the surrounding axillary tissue (Figure 5C).
We next stained Myo10 with immunohistochemistry on a large human breast cancer tissue microarray (TMA) containing tumor tissue samples from 1,326 breast cancers diagnosed in Finland and categorized the tumors either as negative (–) or low to intermediate (+) or highly (++) positive with respect to Myo10 expression (Figure 5D). A Kaplan-Meier survival analysis showed a significant (P = 0.018) association between high tumor Myo10 expression and unfavorable breast cancer–specific survival (Figure 5E). Since the highest Myo10 expression occurred at the tumor edge, Myo10-expressing cells may have been underrepresented in the TMA. Despite this, for the subset of patients who had metastatic axillary lymph nodes at the time of the diagnosis (pN+, 580 or 43.7% out of the 1,326 patients), high tumor Myo10 expression was very significantly associated with unfavorable breast cancer–specific survival (P = 0.002; Figure 5F).
Myo10 and mutant p53 are coexpressed in breast cancer.
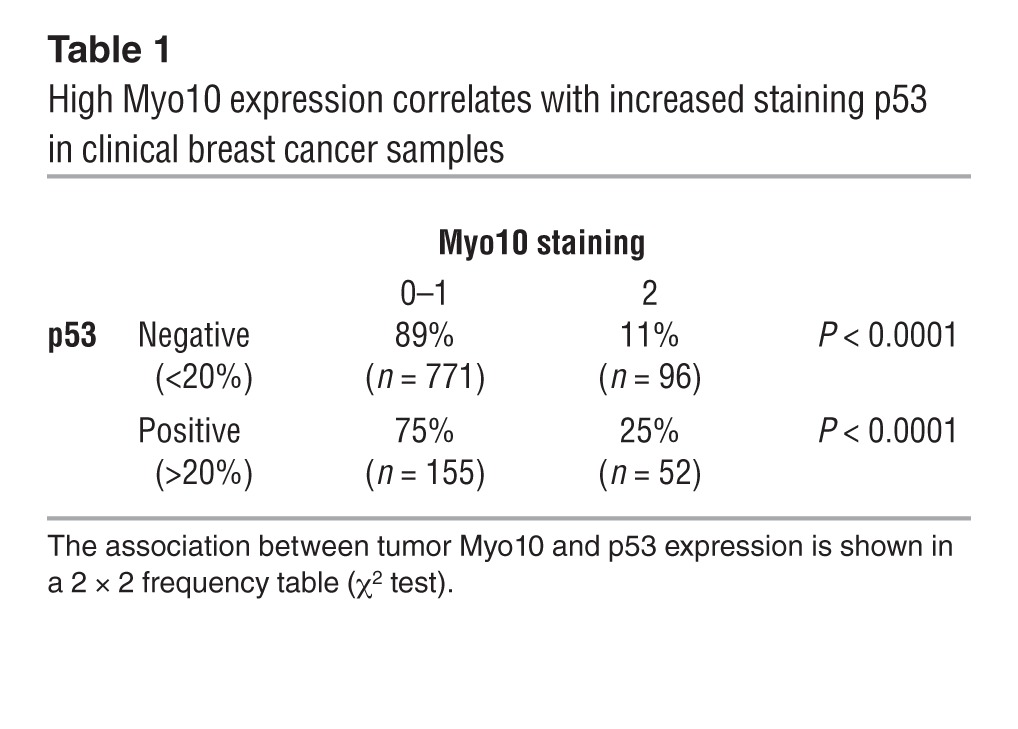
TP53 mutations, such as the R280K mutation present in the MDA-MB-231 cell line (34), result in a highly stable protein that accumulates in cancer cells and has gain-of-function properties (35). In general, presence of strong p53 immunostaining is associated with the presence of mutant TP53 (36, 37). The association between the TP53 gene mutation status and p53 immunostaining could not be studied in the current series, but in line with previous studies (36, 37), we found a strong positive correlation (P < 0.001) between the presence of mutated TP53 and positive p53 immunostaining in another series consisting of 649 primary breast cancers (38). Interestingly, we detected a strong association between positive p53 immunostaining and a high tumor expression of Myo10 in a microarray with tissue from 1,326 breast cancers (Figure 5G and Table 1). This finding was validated in the MicMa Oslo breast cancer cohort (4), where the presence of TP53 mutations was confirmed with DNA sequencing. MYO10 mRNA expression was significantly higher in breast tumors with mutant p53 (P < 0.0001; Figure 5H). Taken together, these data suggest that Myo10 could be functioning as a proinvasive protein that is often highly expressed in cancers that harbor mutant TP53 (20).
Table 1.
High Myo10 expression correlates with increased staining p53 in clinical breast cancer samples
Endogenous mutant p53 regulates Myo10 expression.
These clinical data suggested a previously unknown link between mutant TP53 and Myo10 expression in human breast cancer. In line with these data, Myo10 was highly expressed in endogenous mutant p53–expressing MDA-MB-231 cells compared with MCF7 and MCF10A cells that harbor WT p53 (Figure 6A). Several cell adhesion and matrix-linked genes are upregulated upon loss of WT p53 (39). However, loss of WT p53 did not strongly induce Myo10 expression, since Myo10 levels were low and nearly identical in HCT116 cells that expressed WT p53 and their p53-null counterparts (Supplemental Figure 6A). This suggests that the link between Myo10 and p53 is unrelated to loss of functional p53. In contrast, expression of mutant TP53 (R175H and R273H mutations frequently found in human cancer, IARC TP53 database [ http://p53.iarc.fr/] and Supplemental Figure 6B) in p53-null MCF7 breast cancer cells and HCT116 cells (R273H) increased Myo10 levels, suggesting that Myo10 upregulation may be linked with the mutant p53 gain-of-function properties (Figure 6B).
Figure 6. Mutant p53 regulates Myo10 expression.

(A) Western blot of Myo10, p53, and actin (loading control) in the indicated cell lines. (B) Western blot of mutant p53–transfected (R175H or R273H) MCF-7 and in HCT-116 p53–/– cells stably expressing mutant p53 (R273H). The number of experiments (n) and the quantification of Myo10 levels relative to the control are shown. (C) PDAC sections from mice expressing mutant p53 (p53R172H) and mice with deleted p53 (p53flox) were stained and quantified (Slidepath software, pixel count) for Myo10 expression. Scale bar: 100 μm. (P = 0.047, Mann Whitney test, n = 5 tumors). (D) Early PanIN (top panels), late PanIN (middle panels), and PDAC (bottom panels) sections were stained for Myo10 and mutant p53 expression. Scale bars: 100 μm. (E) Western blotting of Myo10, p53, and tubulin in human pancreatic cancer cell lines. (F, G) Myo10 expression in MDA-MB-231 cells upon shRNA-mediated mutant p53 silencing and reexpression of shRNA-insensitive R175H p53 or WT p53 (mean ± SEM, n = 4, Mann Whitney test).
To investigate whether mutant TP53 regulates Myo10 also in another cancer type in vivo, the expression of Myo10 was investigated within a series of different pancreatic ductal adenocarcinoma (PDAC) cancer mouse models. TP53 mutation occurs in 50%–75% of human PDACs. Furthermore, earlier work has shown that expression of mutant p53 (R172H), as compared with genetic loss of TP53, specifically promotes metastasis (40). Thus, investigation of the putative mutant p53–induced Myo10 expression is particularly relevant in this cancer type. First, expression of Myo10 was examined using the murine affymetrix HG_U133 Plus 2.0 chip and was found to be significantly upregulated only in mutant p53–expressing pancreatic cancers as compared with the other types of pancreatic tumors investigated, which had either lost p53 function or had other mutations that drive PDAC formation in the mouse (Supplemental Figure 6C and refs. 41, 42). Myo10 was upregulated in PDACs when compared with primary pancreatic ductal epithelial cells (PDEC) from WT mice (5.46, 1.57, and 1.32-fold for the 3 probes tested). To confirm these observations, quantitative RT-PCR (qRT-PCR) was performed on a subset of these murine PDACs. Myo10 mRNA was higher in the tumors carrying mutant p53 (R175H) as compared with p53 loss-of-function and PTEN-deleted PDACs (Supplemental Figure 6D). Strong Myo10 immunostaining was also detected in PDAC tissue sections from mice expressing mutant p53 (p53R172H) compared with p53-deleted mice (Figure 6C). Furthermore, increased Myo10 labeling correlated with p53 accumulation in late pancreatic intraepithelial neoplasia (late PanIN) and PDAC, whereas little Myo10 expression was detected in the p53-negative early PanIN tissue samples (Figure 6D). In line with these in vivo data, we observed a similar correlation between p53 gain-of-function mutations and Myo10 expression also in human PDAC cell lines (Figure 6E).
To study the role of mutant p53 in driving Myo10 expression in more detail, we silenced endogenous mutant p53 (R280K) in MDA-MB-231 cells. Efficient knockdown of p53 using shRNA resulted in significant downregulation of Myo10 as detected by Western blot (Figure 6F), and similar results were obtained with 3 independent p53-targeting siRNA oligonucleotides (Supplemental Figure 7A). Importantly, reintroduction of a different p53 mutant (RNAi-resistant R175H) to short hairpin p53 (shp53) expressing MDA-MB-231 cells fully restored Myo10 expression while reintroduction of RNAi resistant WT p53 had no effect on Myo10 levels (Figure 6F), further validating the functional link between mutant p53 status and Myo10 expression. Interestingly, the effect of silencing of mutant p53 on Myo10 expression was more prominent at the protein level, with a 42% to 55% reduction of the Myo10 protein, than on the mRNA level, with 10% to 45% reduction of the MYO10 mRNA (Supplemental Figure 7B), suggesting that Myo10 is regulated by mutant p53 on both protein and mRNA levels. In line with this, pulse-chase experiments using control and p53-silenced MDA-MB-231 cells revealed that in the absence of mutant p53, the Myo10 protein is less stable (Supplemental Figure 7C).
Next, we investigated the possible mechanism of mutant p53–dependent MYO10 mRNA regulation. Mutant p53 expression correlates with elevated MAPK/ERK pathway activity (ref. 43 and Supplemental Figure 7D) and increased levels of EGR1 transcription factor (44), which modifies expression of several genes involved in cytoskeletal regulation and filopodia formation (45). We observed that inhibition of the MAPK/ERK pathway efficiently reduced EGR1 and Myo10 in p53 mutant MDA-MB-231 cells but not p53 WT MCF10A cells (Figure 7A). Similar correlation between ERK activity and Myo10 expression was also detected in human PDAC cells with mutant p53 (Supplemental Figure 7E). In line with this observation, silencing of ERK1 and ERK2 kinases in MDA-MB-231 cells significantly inhibited invasion (Supplemental Figure 7F). We also observed that silencing of mutant p53 using 2 independent siRNAs significantly reduced both EGR1 and MYO10 mRNA levels (Figure 7B) and that EGR1 silencing alone was sufficient to significantly reduce MYO10 mRNA levels in MDA-MB-231 cells (Figure 7C and Supplemental Figure 8A). Finally, in line with the UCSC Genome Browser database prediction ( http://genome.ucsc.edu/), we observed direct binding of EGR1 to the MYO10 promoter in MDA-MB-231 cells using ChIP (Figure 7D). Taken together, these data suggest that mutant p53 regulates MYO10 mRNA levels via an MAPK/ERK- and EGR1-dependent pathway.
Figure 7. Myo10 is necessary for mutant p53–driven invasion.

(A) Western blot of MCF10A and MDA-MB-231 cells treated 16 hours with DMSO or MEK inhibitor (UO126, 10 μM). NT, non-treated. (B)TaqMan qRT-PCR of MYO10 and EGR1 mRNA levels in MDA-MB-231 cells silenced with 2 different p53 targeting oligos. n = 2 (sip53_13); n = 4 (sip53_3). (C) Western blot and TaqMan qRT-PCR showing Myo10 expression upon silencing of EGR1 for 48 hours. (D) ChIP analysis of mutant p53 or EGR1 binding to MYO10 promoter by PCR using Myo10 promoter–specific primers. IgG antibody was used as negative control. (E) Matrigel invasion of shMyo10 cells upon silencing of TP53 (areas of invasion, n = 10, ×20 objective). (F) Invasion of murine PDAC cells transfected with siMyo10 or siArpc2 (Arp2/3 component, positive control) into Matrigel-overlaid wounds. (G) MCF-7 cells were transfected as indicated, and invasion was analyzed as in E. (H) Invasion of p53-silenced MDA-MB-231 cells transfected as indicated (percentage of invaded/all GFP-positive cells; n = 9–14; ×20 objective. (I) Model of mutant p53–driven, integrin-dependent invasion. (a) Mutant p53 induces Myo10 that contributes to mutant p53–driven invasion by transporting integrin to filopodia tips. (b) Mutant p53 increases integrin and EGFR recycling to the plasma membrane (19) providing integrins to be transported to filopodia tips by Myo10. (c) Mutant p53 drives Myo10 by increasing EGR1 transcription directly and via MAPK/ERK signaling. (d) Increased integrin-EGFR recycling elevates PIP3 levels via PI3K/Akt pathway (19) and activates Myo10 (48). Mean ± SEM and Mann-Whitney test P values are shown.
Depletion of mutant p53 from MDA-MB-231 cells inhibits TGF-β–, serum-, and EGF-induced cell invasion into Matrigel (19, 22), and the present data link mutant p53 to expression of Myo10 and cancer invasion. This prompted us to investigate the link between Myo10 and mutant p53–driven cancer invasion. We observed a significant reduction in the invasion of shControl MDA-MB-231 cells upon p53 siRNA transfection. Interestingly, silencing of mutant p53 failed to inhibit the residual invasion of the Myo10 knockdown MDA-MB-231 cells (Figure 7E and Supplemental Figure 8B), and silencing of Myo10 in murine PDACs carrying mutant p53 (R175H) significantly inhibited invasion into Matrigel (Figure 7F and Supplemental Figure 8C), suggesting a role for Myo10 in mutant p53–driven invasion. These data were supported by the finding that mutant p53 expression not only induced Myo10 expression (Figure 6B), but also enhanced invasion of p53 WT MCF7 breast cancer cells (Figure 7G). Furthermore, overexpression of WT EGFP-Myo10 increased the invasive capacity of mutant p53–silenced MDA-MB-231 cells by 33%, whereas control transfection (EGFP alone) had no effect (Figure 7H and Supplemental Figure 8, D and E). Taken together, these results support the conclusion that Myo10 is a necessary component for mutant p53–driven cancer invasion.
Discussion
We describe what we believe is a previously unknown role for Myo10 in mutant p53–driven cancer invasion and metastasis. Myo10 was upregulated in breast carcinomas, mouse PDACs expressing mutant p53, and cancer cell lines with mutant p53. Myo10 promoted breast cancer invasion in vitro and metastasis in vivo. In addition, high Myo10 expression was associated with poor survival among patients with primary and lymph node–positive breast cancer. Mechanistically, Myo10-induced cell adhesion, migration, and invasion were dependent on stabilization of filopodia that protrude into the matrix using the cell adhesion receptor β1 integrin. Engaging in perturbation of the link between integrin and Myo10 by inducing a mutation (Myo10ΔFERM2) that makes Myo10 unable to bind the β1 integrin tail abolishes Myo10-driven cell invasion and filopodia stability. In line with the proinvasive activity of Myo10, high Myo10 staining was present at the invasive edges of breast carcinomas. Finally, Myo10 was found to be a necessary downstream component for mutant p53–driven invasion in breast cancer and PDAC cells.
Myo10 in cell migration and invasion.
Migratory cells polarize and form a defined front by activating Rac and PI3K at the leading edge following a directional cue (46). Formation and binding of PI(3,4,5)P3 activate Myo10, thereby allowing it to move along actin bundles that are parallel to the plasma membrane (47, 48). Activated Myo10 is targeted to fascin-containing microspikes that project outwards (49, 50) to induce formation of long filopodia in different cell types. Thus, the role of Myo10 in filopodia formation is well established, but little is known about the requirement of Myo10 for cell motility. In endothelial cells, Myo10 is required for bone morphogenetic protein–dependent migration (51), and it functions in the cranial neural crest cells during Xenopus embryogenesis (52, 53). However, evidence for a role of Myo10 in cancer cell motility has been lacking. We demonstrate a promigratory role for Myo10 in breast cancer cells. Silencing of Myo10 in MDA-MB-231 adenocarcinoma cells reduces cell migration and results in a rounder cell phenotype, probably indicating loss of front-rear polarity. We also demonstrate that silencing of Myo10 inhibits cell invasion in breast cancer and PDAC cells and that loss of Myo10 inhibits breast cancer cell extravasation from the vasculature to the lungs as well as lung colonization and systemic metastasis of orthotopically implanted breast cancer cells. Based on these findings, we conclude that Myo10-induced filopodia and plasma-membrane protrusions may be important in several distinct steps of the metastatic cascade, namely local invasion, intravasation, and extravasion. The identification of these cancer-relevant functions of Myo10 suggests that the induction of filopodia is a promigratory process that contributes to metastasis formation in human cancer.
Mutant p53–driven invasion and Myo10.
Mutant p53 drives metastasis in an integrin-dependent manner (19) and via mechanisms involving inhibition of p63-mediated transcriptional repression (22). However, p53 most likely plays many roles in cancer progression with several different molecular mechanisms (20, 54). For example, in some breast epithelial cells, expression of mutant p53 induces cell invasion, but loss of p63 induces cell detachment and death (19), indicating that mutant p53 also drives invasion via processes that are not dependent on blocking p63-mediated control of transcription. We found that Myo10 is upregulated in cells expressing mutant p53 (using 3 distinct TP53 mutations and exogenous and endogenous mutant p53 expression) and that in clinical breast cancer tissue samples, high Myo10 expression markedly correlates with the presence of mutant p53. This link between mutant p53–driven invasion and filopodia is new. Filopodia-inducing gene fascin associates with poor survival in breast and colorectal cancer and with the basal-like phenotype in breast cancer (12, 55, 56). Fascin expression does not, however, correlate with the presence of mutant p53 in breast cancer (56), which suggests different molecular mechanisms for upregulation of fascin and Myo10 in cancer invasion.
There appears to be at least 2 levels of regulation in mutant p53–induced Myo10 expression, since silencing of mutant p53 reduces Myo10 levels more on the protein level than on the mRNA level. We found that in mutant p53 cells, inhibition of ERK activity reduced EGR1 expression. Furthermore, EGR1 expression was necessary for Myo10 induction, and EGR1 directly bound to the Myo10 promoter. Thus, it is very likely that the mutant p53–induced transcriptional control of Myo10 involves this ERK/EGR1 pathway. However, this may be limited to mutant p53–expressing cells, since EGR1 overexpression is not sufficient to induce Myo10 expression in p53 WT MCF7 cells (not shown). Thus, EGR1 alone is not sufficient to drive Myo10 expression in breast cancer cells. This may be due to the fact that mutant p53 also induces the stability of the Myo10 protein, which could be related to diversion of Myo10 from a degradation route to the plasma membrane. Alternatively, mutant p53 may act together with EGR1 to induce Myo10 transcription by an unknown mechanism. Taken together, both transcriptional control and regulation at the protein level contribute to the cellular expression levels of Myo10.
PI3K-produced PI(3,4,5)P3 activates Myo10 and in the absence of PI(3,4,5)P3, Myo10 is monomeric, folded in an inactive conformation, and targeted to the Rab7-lysosomal pathway (48, 57). Mutant p53–dependent increased recycling of the EGFR–rab-coupling protein–integrin (EGFR-RCP-integrin) complex or the Met receptor (19, 43) to the plasma membrane provides the cell with increased numbers of cell-surface adhesion receptors and constitutively activates EGFR signaling to the PI3K/Akt pathway (19) and induces cellular ERK activity (43). Based on the data presented here and elsewhere (19), a model for the integrin-dependent component of mutant p53–driven invasion emerges (Figure 7I). Mutant p53 induces Myo10 expression via the ERK/EGR1 pathway. In addition, PI3K-produced PIP3 activates Myo10 motor protein, which transports the recycled and increasingly plasma membrane–targeted integrins to an invasive membrane protrusion to drive cell migration. In conclusion, mutant p53 has many activities that are critical for cancer progression (17), and the present study describes a new mutant p53–associated molecular mechanism that facilitates breast cancer invasion and dissemination. These data suggest that Myo10 may be an important new and potentially druggable therapeutic target, since myosin inhibitors are currently under development (58, 59).
Methods
Cell culture, reagents, DNA constructs, and siRNA transfections.
MDA-MB-231, MCF10A, MCF7, BT-474, MDA-MB-468, and HCT116-p53 (–/–) cells were cultured as described in Supplemental Experimental Procedures. To generate stable Myo10-silenced cell lines, MDA-MB-231 cells were transfected overnight with 2 μg of shMyo10 (sc-43241-SH; Santa Cruz Biotechnology Inc.) plasmid with Lipofectamine 2000 (Invitrogen), as described in Supplemental Experimental Procedures, and single cell clones were generated. Cloning of EGFP-Myo10 and EGFP-Myo10ΔFERM2 constructs have been described previously (15). The p53-R273H construct was cloned as described (60) and the cloning of EGR1 as described (61). The use of shRNAs and sRNAs is described in Supplemental Experimental Procedures.
qRT-PCR.
qRT-PCR using the Real-Time PCR HT7900 (Applied Biosystems) was performed as described in Supplemental Experimental Procedures.
Microscopy and image analysis.
Staining of the cells, image analysis, and microscopy specifications are described in ref. 62. “Roundness” is an ImageJ shape descriptor and is calculated as 4 × area(π × major axis2) or as the inverse of the aspect ratio (major axis/minor axis).
Cell migration, adhesion, and spreading assays.
Time-lapse microscopy was used to follow the migration of siRNA- and shRNA-mediated silencing of Myo10 as described in Supplemental Experimental Procedures. Cell adhesion assay is previously described in ref. 63 and in Supplemental Experimental Procedures. Cell spreading assay is described in Supplemental Experimental Procedures.
Matrigel invasion assay.
Fifty thousand cells were plated on a well of Ibidi 8-well microslides (Integrated Biodiagnostics). siRNA or plasmid transfections were done the next day. After 24 hours of transfection (8 hours in the case of plasmids), the assay was continued as described by Högnäs et al. (64). Invasion of EGFP and EGFP-Myo10 transfected cells was analyzed by measuring the ratio between invaded EGFP-transfected and noninvaded EGFP-transfected cells using ImageJ. Analysis was performed in a double-blind manner.
Proliferation and apoptosis assays.
The effect of Myo10 on cancer cell proliferation and apoptosis was studied in siMyo10 and shMyo10 MDA-MB-231 cells (2 individual shMyo10 clones and 2 different Myo10 targeting oligos). 5 × 103 cells were plated on 96-well plates. Proliferation was analyzed by cell proliferation reagent WST-1 reagent (Roche) according to the manufacturer’s instructions and with measuring the absorbance at 450 nm after 1 hour incubation at +37°C. Absorbance was measured every 24 hours for 4 days. Apoptosis was analyzed by measuring the caspase-3/7 activity by CellPlayer 96-Well Kinetic Caspase-3/7 Apoptosis Assay Kit (Essen BioScience). Cells were plated as in proliferation assay. DEVD-NucViewTM488 reagent was added to a final concentration of 5 nM, and the number of fluorescent cells was analyzed by Incuyte FLR.
Zebrafish invasion assay.
Zebrafish tumor models and the invasion assay are fully described in ref. 32 and in Supplemental Experimental Procedures.
Mouse lung extravasation and colonization assays.
Myo10 and control shRNA–transfected MDA-MB-231 (or siMyo10 transfected MDA-MB-468 and BT-474) cells were stained with live cell dyes (shControl/siControl, red CMTPX; shMyo10/siMyo10, green CMFDA) according to the manufacturer’s instructions, followed by a lung extravasation assay that was performed as previously described (26, 65). The number of CellTracker-stained cells was analyzed by counting the cells from frozen lung tissue sections (MDA-MB-231) or by flow cytometry (MDA-MB-468 and BT-474). Lung colonization assay was done as described (26, 59) with small modifications. Unstained shControl or shMyo10 MDA-MB-231 cells (3 × 105 cells to each mouse; 10 mice were used for both groups) were injected into tail veins and allowed to proliferate for 4 weeks. Left lungs were formalin fixed, and cells were isolated from the right lung and analyzed by flow cytometry as described in Supplemental Experimental Procedures.
Orthotopic breast cancer model.
In order to study breast cancer metastasis, shControl or shMyo10 MDA-MB-231 cells (8 × 105 cells/mouse) were injected into the mammary fat pads of nude female mice. After 3 weeks, primary tumors were removed. Assay was continued up to 6 weeks. Animals were sacrificed and examined for metastases to contralateral lymph nodes as described in Supplemental Experimental Procedures.
Mouse pancreatic cancer samples.
RNA was extracted using standard techniques from murine tumors that have arisen from the following genotypes: PDXCRE KRASG12D P53R172H/+ PDXCRE KRASG12D P53fl/+, PDXCRE KRASG12D PTENfl/+ PDXCRE KRASG12D LKB1fl/+ PDXCRE KRASG12D Apcfl/+ (40–42). We and others have previously shown that the WT allele of p53 is lost on both the P53fl/+ and P53R172H/+ tumors (40). Immunohistochemistry was performed on formalin-fixed mouse pancreatic samples. Antigen retrieval was carried out by boiling samples in sodium citrate buffer (10 mmol/l; pH 6.0) for 4 minutes using a pressure cooker. Anti-Myosin 10 (Novus Biologicals #22430002) was used at a 1 in 2,000 dilution and incubated at 4°C overnight. IHC slides were scanned using an Aperio slide scanner and positive pixel counts quantitated using Slidepath software.
Clinical studies.
Samples from a Finnish nationwide population-based breast cancer series were collected, and molecular breast cancer subtypes were defined as described (66, 67). A total of 1,326 patients were included in the series. Breast cancer TMA blocks were constructed as described (66). Immunohistochemical staining was performed as described in the Supplemental Experimental Procedures.
In silico data analysis.
The breast cancer raw data sets (GEO GSE3494, ref. 25; and GEO GSE3985/GSE19783, ref. 4) were downloaded for the public source GEO data repository (68). Annotation, normalization, and data analysis were done as described in Supplemental Experimental Procedures.
Western blotting.
Standard Western blotting techniques, the Odyssey LICOR imaging system, and Amersham ECL Plus Western blotting reagent were used. Cell lysates were collected using SDS sample buffer and scraping. Antibodies for Western blotting are described in Supplemental Experimental Procedures.
Statistics.
Frequency tables were analyzed using the χ2 test. Continuous distributions were compared with either the Mann-Whitney test or Student’s t test (2-tailed). Breast cancer–specific survival was calculated from the date of the diagnosis to the date of death from breast cancer. Patients who were alive or who died from causes other than breast cancer were censored on the last date when known to be alive or the date of death. Survival was analyzed using the Kaplan-Meier life table method, and survival between groups was compared with the log-rank test. All P values are 2-sided, and a P value of less than 0.05 was considered significant. In box-and-whisker plots, the box outline indicates the 95% CI, the line inside the box is the median, and the whiskers indicate the minimum and maximum values.
Study approval.
Permission to use breast tumor tissue for the study was provided by the Ministry of Health, Finland 15/12/1997 (as per legislation; permission 123/08/97). Animal experiments were performed under a valid animal permit (license number ESAVI/7522/04.10.03/2012). Permissions for animal experiments were received from the National Animal Experiment Board of Finland as required by the Finnish Act on Animal Experimentation.
Supplementary Material
Acknowledgments
We thank L. Lahtinen, J. Siivonen, M. Saari, J. Heiskanen, and S. Kollanus for excellent technical assistance. We are grateful to Heike Beck, B. Vogelstein, G. Selivanova, S. Strömblad, S. Piccolo, and R.E. Cheney for sharing their reagents, and we thank M. Salmi for critically reading the manuscript and S. Loi and C. Sotiriou for sharing their mutant p53 clinical data with us. This study was supported by the Academy of Finland, an ERC Starting Grant, the Sigrid Juselius Foundation, and the Finnish Cancer Organization. A. Arjonen has been supported by the V-S Cultural Foundation, the Ida Montin Foundation, the Orion-Farmos Foundation, the Finnish Cancer Organization, the K. Albin Johansson Foundation, and the Instrumentarium Foundation. A. Arjonen and R. Kaukonen are supported by the Turku Doctoral Program of Biomedical Sciences. R. Kaukonen has been supported by the V-S Cancer Organization.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Citation for this article: J Clin Invest. 2014;124(3):1069–1082. doi:10.1172/JCI67280.
Elmar Bucher’s present address is: Department of Biomedical Engineering, Oregon Health and Sciences University, Portland, Oregon, USA.
References
- 1.Weigelt B, Peterse JL, van ‘t Veer LJ. Breast cancer metastasis: markers and models. Nat Rev Cancer. 2005;5(8):591–602. doi: 10.1038/nrc1670. [DOI] [PubMed] [Google Scholar]
- 2.Sorlie T, et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci U S A. 2001;98(19):10869–10874. doi: 10.1073/pnas.191367098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Perou CM, et al. Molecular portraits of human breast tumours. Nature. 2000;406(6797):747–752. doi: 10.1038/35021093. [DOI] [PubMed] [Google Scholar]
- 4.Naume B, et al. Presence of bone marrow micrometastasis is associated with different recurrence risk within molecular subtypes of breast cancer. Mol Oncol. 2007;1(2):160–171. doi: 10.1016/j.molonc.2007.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Faix J, Rottner K. The making of filopodia. Curr Opin Cell Biol. 2006;18(1):18–25. doi: 10.1016/j.ceb.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 6.Mattila PK, Lappalainen P. Filopodia: molecular architecture and cellular functions. Nat Rev Mol Cell Biol. 2008;9(6):446–454. doi: 10.1038/nrm2406. [DOI] [PubMed] [Google Scholar]
- 7.Arjonen A, Kaukonen R, Ivaska J. Filopodia and adhesion in cancer cell motility. Cell Adh Migr. 2011;5(5):421–430. doi: 10.4161/cam.5.5.17723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schoumacher M, Goldman RD, Louvard D, Vignjevic DM. Actin, microtubules, and vimentin intermediate filaments cooperate for elongation of invadopodia. J Cell Biol. 2010;189(3):541–556. doi: 10.1083/jcb.200909113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Minn AJ, et al. Genes that mediate breast cancer metastasis to lung. Nature. 2005;436(7050):518–524. doi: 10.1038/nature03799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vignjevic D, Kojima S, Aratyn Y, Danciu O, Svitkina T, Borisy GG. Role of fascin in filopodial protrusion. J Cell Biol. 2006;174(6):863–875. doi: 10.1083/jcb.200603013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pelosi G, et al. Independent prognostic value of fascin immunoreactivity in stage I nonsmall cell lung cancer. Br J Cancer. 2003;88(4):537–547. doi: 10.1038/sj.bjc.6600731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yoder BJ, et al. The expression of fascin, an actin-bundling motility protein, correlates with hormone receptor-negative breast cancer and a more aggressive clinical course. Clin Cancer Res. 2005;11(1):186–192. [PubMed] [Google Scholar]
- 13.Kerber ML, Cheney RE. Myosin-X: a MyTH-FERM myosin at the tips of filopodia. J Cell Sci. 2011;124(pt 22):3733–3741. doi: 10.1242/jcs.023549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Berg JS, Derfler BH, Pennisi CM, Corey DP, Cheney RE. Myosin-X, a novel myosin with pleckstrin homology domains, associates with regions of dynamic actin. J Cell Sci. 2000;113(pt 19):3439–3451. doi: 10.1242/jcs.113.19.3439. [DOI] [PubMed] [Google Scholar]
- 15.Zhang H, et al. Myosin-X provides a motor-based link between integrins and the cytoskeleton. Nat Cell Biol. 2004;6(6):523–531. doi: 10.1038/ncb1136. [DOI] [PubMed] [Google Scholar]
- 16.Yonezawa S, et al. Mouse myosin X: molecular architecture and tissue expression as revealed by northern blot and in situ hybridization analyses. Biochem Biophys Res Commun. 2000;271(2):526–533. doi: 10.1006/bbrc.2000.2669. [DOI] [PubMed] [Google Scholar]
- 17.Oren M, Rotter V. Mutant p53 gain-of-function in cancer. Cold Spring Harb Perspect Biol. 2010;2(2):a001107. doi: 10.1101/cshperspect.a001107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Caulin C, et al. An inducible mouse model for skin cancer reveals distinct roles for gain- and loss-of-function p53 mutations. J Clin Invest. 2007;117(7):1893–1901. doi: 10.1172/JCI31721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Muller PA, et al. Mutant p53 drives invasion by promoting integrin recycling. Cell. 2009;139(7):1327–1341. doi: 10.1016/j.cell.2009.11.026. [DOI] [PubMed] [Google Scholar]
- 20.Muller PA, Vousden KH, Norman JC. p53 and its mutants in tumor cell migration and invasion. J Cell Biol. 2011;192(2):209–218. doi: 10.1083/jcb.201009059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Terzian T, et al. The inherent instability of mutant p53 is alleviated by Mdm2 or p16INK4a loss. Genes Dev. 2008;22(10):1337–1344. doi: 10.1101/gad.1662908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Adorno M, et al. A Mutant-p53/Smad complex opposes p63 to empower TGFbeta-induced metastasis. Cell. 2009;137(1):87–98. doi: 10.1016/j.cell.2009.01.039. [DOI] [PubMed] [Google Scholar]
- 23.Roger L, Jullien L, Gire V, Roux P. Gain of oncogenic function of p53 mutants regulates E-cadherin expression uncoupled from cell invasion in colon cancer cells. J Cell Sci. 2010;123(pt 8):1295–1305. doi: 10.1242/jcs.061002. [DOI] [PubMed] [Google Scholar]
- 24.Enerly E, et al. miRNA-mRNA integrated analysis reveals roles for miRNAs in primary breast tumors. PLoS One. 2011;6(2):e16915. doi: 10.1371/journal.pone.0016915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Miller LD, et al. An expression signature for p53 status in human breast cancer predicts mutation status, transcriptional effects, and patient survival. Proc Natl Acad Sci U S A. 2005;102(38):13550–13555. doi: 10.1073/pnas.0506230102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vuoriluoto K, et al. Vimentin regulates EMT induction by Slug and oncogenic H-Ras and migration by governing Axl expression in breast cancer. Oncogene. 2011;30(12):1436–1448. doi: 10.1038/onc.2010.509. [DOI] [PubMed] [Google Scholar]
- 27.Voss MJ, Moller MF, Powe DG, Niggemann B, Zanker KS, Entschladen F. Luminal and basal-like breast cancer cells show increased migration induced by hypoxia, mediated by an autocrine mechanism. BMC Cancer. 2011;11:158. doi: 10.1186/1471-2407-11-158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Soule HD, et al. Isolation and characterization of a spontaneously immortalized human breast epithelial cell line, MCF-10. Cancer Res. 1990;50(18):6075–6086. [PubMed] [Google Scholar]
- 29.Mould AP, Akiyama SK, Humphries MJ. The inhibitory anti-beta1 integrin monoclonal antibody 13 recognizes an epitope that is attenuated by ligand occupancy. Evidence for allosteric inhibition of integrin function. J Biol Chem. 1996;271(34):20365–20374. doi: 10.1074/jbc.271.34.20365. [DOI] [PubMed] [Google Scholar]
- 30.Mould AP, Garratt AN, Askari JA, Akiyama SK, Humphries MJ. Identification of a novel anti-integrin monoclonal antibody that recognises a ligand-induced binding site epitope on the beta 1 subunit. FEBS Lett. 1995;363(1–2):118–122. doi: 10.1016/0014-5793(95)00301-O. [DOI] [PubMed] [Google Scholar]
- 31.Lawson ND, Weinstein BM. In vivo imaging of embryonic vascular development using transgenic zebrafish. Dev Biol. 2002;248(2):307–318. doi: 10.1006/dbio.2002.0711. [DOI] [PubMed] [Google Scholar]
- 32.Lee SL, et al. Hypoxia-induced pathological angiogenesis mediates tumor cell dissemination, invasion, and metastasis in a zebrafish tumor model. Proc Natl Acad Sci U S A. 2009;106(46):19485–19490. doi: 10.1073/pnas.0909228106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lacroix M. Significance, detection and markers of disseminated breast cancer cells. Endocr Relat Cancer. 2006;13(4):1033–1067. doi: 10.1677/ERC-06-0001. [DOI] [PubMed] [Google Scholar]
- 34.Epstein CB, Attiyeh EF, Hobson DA, Silver AL, Broach JR, Levine AJ. p53 mutations isolated in yeast based on loss of transcription factor activity: similarities and differences from p53 mutations detected in human tumors. Oncogene. 1998;16(16):2115–2122. doi: 10.1038/sj.onc.1201734. [DOI] [PubMed] [Google Scholar]
- 35.Soussi T. The p53 tumor suppressor gene: from molecular biology to clinical investigation. Ann N Y Acad Sci. 2000;910:121–137. doi: 10.1111/j.1749-6632.2000.tb06705.x. [DOI] [PubMed] [Google Scholar]
- 36.Baas IO, Mulder JW, Offerhaus GJ, Vogelstein B, Hamilton SR. An evaluation of six antibodies for immunohistochemistry of mutant p53 gene product in archival colorectal neoplasms. J Pathol. 1994;172(1):5–12. doi: 10.1002/path.1711720104. [DOI] [PubMed] [Google Scholar]
- 37.Sihto H, Kukko H, Koljonen V, Sankila R, Bohling T, Joensuu H. Merkel cell polyomavirus infection, large T antigen, retinoblastoma protein and outcome in Merkel cell carcinoma. Clin Cancer Res. 2011;17(14):4806–4813. doi: 10.1158/1078-0432.CCR-10-3363. [DOI] [PubMed] [Google Scholar]
- 38.Loi S, et al. Somatic mutation profiling and associations with prognosis and trastuzumab benefit in early breast cancer. J Natl Cancer Inst. 2013;105(13):960–967. doi: 10.1093/jnci/djt121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Guo F, Zheng Y. Rho family GTPases cooperate with p53 deletion to promote primary mouse embryonic fibroblast cell invasion. Oncogene. 2004;23(33):5577–5585. doi: 10.1038/sj.onc.1207752. [DOI] [PubMed] [Google Scholar]
- 40.Morton JP, et al. Mutant p53 drives metastasis and overcomes growth arrest/senescence in pancreatic cancer. Proc Natl Acad Sci U S A. 2010;107(1):246–251. doi: 10.1073/pnas.0908428107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Morton JP, et al. LKB1 haploinsufficiency cooperates with Kras to promote pancreatic cancer through suppression of p21-dependent growth arrest. Gastroenterology. 2010;139(2):586–597. doi: 10.1053/j.gastro.2010.04.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kennedy AL, et al. Activation of the PIK3CA/AKT pathway suppresses senescence induced by an activated RAS oncogene to promote tumorigenesis. Mol Cell. 2011;42(1):36–49. doi: 10.1016/j.molcel.2011.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Muller PA, et al. Mutant p53 enhances MET trafficking and signalling to drive cell scattering and invasion. Oncogene. 2013;32(10):1252–1265. doi: 10.1038/onc.2012.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sauer L, Gitenay D, Vo C, Baron VT. Mutant p53 initiates a feedback loop that involves Egr-1/EGF receptor/ERK in prostate cancer cells. Oncogene. 2010;29(18):2628–2637. doi: 10.1038/onc.2010.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cermak V, Kosla J, Plachy J, Trejbalova K, Hejnar J, Dvorak M. The transcription factor EGR1 regulates metastatic potential of v-src transformed sarcoma cells. Cell Mol Life Sci. 2010;67(20):3557–3568. doi: 10.1007/s00018-010-0395-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sasaki AT, Chun C, Takeda K, Firtel RA. Localized Ras signaling at the leading edge regulates PI3K, cell polarity, and directional cell movement. J Cell Biol. 2004;167(3):505–518. doi: 10.1083/jcb.200406177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tokuo H, Mabuchi K, Ikebe M. The motor activity of myosin-X promotes actin fiber convergence at the cell periphery to initiate filopodia formation. J Cell Biol. 2007;179(2):229–238. doi: 10.1083/jcb.200703178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Plantard L, Arjonen A, Lock JG, Nurani G, Ivaska J, Stromblad S. PtdIns(3,4,5)P is a regulator of myosin-X localization and filopodia formation. J Cell Sci. 2010;123(pt 20):3525–3534. doi: 10.1242/jcs.069609. [DOI] [PubMed] [Google Scholar]
- 49.Svitkina TM, et al. Mechanism of filopodia initiation by reorganization of a dendritic network. J Cell Biol. 2003;160(3):409–421. doi: 10.1083/jcb.200210174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nagy S, et al. A myosin motor that selects bundled actin for motility. Proc Natl Acad Sci U S A. 2008;105(28):9616–9620. doi: 10.1073/pnas.0802592105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pi X, et al. Sequential roles for myosin-X in BMP6-dependent filopodial extension, migration, and activation of BMP receptors. J Cell Biol. 2007;179(7):1569–1582. doi: 10.1083/jcb.200704010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hwang YS, Luo T, Xu Y, Sargent TD. Myosin-X is required for cranial neural crest cell migration in Xenopus laevis. Dev Dyn. 2009;238(10):2522–2529. doi: 10.1002/dvdy.22077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nie S, Kee Y, Bronner-Fraser M. Myosin-X is critical for migratory ability of Xenopus cranial neural crest cells. Dev Biol. 2009;335(1):132–142. doi: 10.1016/j.ydbio.2009.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Girardini JE, et al. A Pin1/mutant p53 axis promotes aggressiveness in breast cancer. Cancer Cell. 2011;20(1):79–91. doi: 10.1016/j.ccr.2011.06.004. [DOI] [PubMed] [Google Scholar]
- 55.Hashimoto Y, Skacel M, Lavery IC, Mukherjee AL, Casey G, Adams JC. Prognostic significance of fascin expression in advanced colorectal cancer: an immunohistochemical study of colorectal adenomas and adenocarcinomas. BMC Cancer. 2006;6:241. doi: 10.1186/1471-2407-6-241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rodriguez-Pinilla SM, et al. Prognostic significance of basal-like phenotype and fascin expression in node-negative invasive breast carcinomas. Clin Cancer Res. 2006;12(5):1533–1539. doi: 10.1158/1078-0432.CCR-05-2281. [DOI] [PubMed] [Google Scholar]
- 57.Umeki N, Jung HS, Sakai T, Sato O, Ikebe R, Ikebe M. Phospholipid-dependent regulation of the motor activity of myosin X. Nat Struct Mol Biol. 2011;18(7):783–788. doi: 10.1038/nsmb.2065. [DOI] [PubMed] [Google Scholar]
- 58.Chinthalapudi K, et al. Mechanism and specificity of pentachloropseudilin-mediated inhibition of myosin motor activity. J Biol Chem. 2011;286(34):29700–29708. doi: 10.1074/jbc.M111.239210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fedorov R, et al. The mechanism of pentabromopseudilin inhibition of myosin motor activity. Nat Struct Mol Biol. 2009;16(1):80–88. doi: 10.1038/nsmb.1542. [DOI] [PubMed] [Google Scholar]
- 60.Bullock AN, et al. Thermodynamic stability of wild-type and mutant p53 core domain. Proc Natl Acad Sci U S A. 1997;94(26):14338–14342. doi: 10.1073/pnas.94.26.14338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Beck H, Semisch M, Culmsee C, Plesnila N, Hatzopoulos AK. Egr-1 regulates expression of the glial scar component phosphacan in astrocytes after experimental stroke. Am J Pathol. 2008;173(1):77–92. doi: 10.2353/ajpath.2008.070648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Arjonen A, Alanko J, Veltel S, Ivaska J. Distinct recycling of active and inactive β1 integrins. Traffic. 2012;13(4):610–625. doi: 10.1111/j.1600-0854.2012.01327.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pellinen T, Arjonen A, Vuoriluoto K, Kallio K, Fransen JA, Ivaska J. Small GTPase Rab21 regulates cell adhesion and controls endosomal traffic of β1-integrins. J Cell Biol. 2006;173(5):767–780. doi: 10.1083/jcb.200509019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Högnäs G, et al. Cytokinesis failure due to derailed integrin traffic induces aneuploidy and oncogenic transformation in vitro and in vivo. Oncogene. 2012;31(31):3597–3606. doi: 10.1038/onc.2011.527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pellinen T, Rantala JK, Arjonen A, Mpindi JP, Kallioniemi O, Ivaska J. A functional genetic screen reveals new regulators of beta1-integrin activity. J Cell Sci. 2012;125(pt 3):649–661. doi: 10.1242/jcs.090704. [DOI] [PubMed] [Google Scholar]
- 66.Joensuu H, et al. Amplification of erbB2 and erbB2 expression are superior to estrogen receptor status as risk factors for distant recurrence in pT1N0M0 breast cancer: a nationwide population-based study. Clin Cancer Res. 2003;9(3):923–930. [PubMed] [Google Scholar]
- 67.Sihto H, et al. Molecular subtypes of breast cancers detected in mammography screening and outside of screening. Clin Cancer Res. 2008;14(13):4103–4110. doi: 10.1158/1078-0432.CCR-07-5003. [DOI] [PubMed] [Google Scholar]
- 68.Edgar R, Domrachev M, Lash AE. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002;30(1):207–210. doi: 10.1093/nar/30.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.