

Abstract
The use of B-cell targeted therapies for the treatment of systemic lupus erythematosus (SLE) has generated great interest owing to the multiple pathogenic roles carried out by B cells in this disease. Strong support for targeting B cells is provided by genetic, immunological and clinical observations that place these cells at the center of SLE pathogenesis, as initiating, amplifying and effector cells. Interest in targeting B cells has also been fostered by the successful use of similar interventions to treat other autoimmune diseases such as rheumatoid arthritis, and by the initial promise shown by B-cell depletion to treat SLE in early studies. Although the initial high enthusiasm has been tempered by negative results from phase III trials of the B-cell-depleting agent rituximab in SLE, renewed vigor should be instilled in the field by the convergence of the latest results using agents that inhibit B-cell-activating factor (BAFF, also known as BLyS and tumor necrosis factor ligand superfamily, member 13b), further analysis of data from trials using rituximab and greatly improved understanding of B-cell biology. Combined, the available information identifies several new avenues for the therapeutic targeting of B cells in SLE.
Introduction
B cells carry out central roles in the pathogenesis of the autoimmune disease systemic lupus erythematosus (SLE) through a combination of antibody-mediated and antibody-independent actions. These actions include the presentation of autoantigens, induction of CD4+ helper T cells (type 1 T-helper [TH1] cells, TH2, TH17) and CD8+ effector T cells, maintenance of T-cell memory, inhibition of regulatory T (TREG) cells, secretion of proinflammatory cytokines and chemokines, and organization of tertiary lymphoid tissue, all of which might promote the generation and/or amplification of autoimmune responses in target organs (Figure 1; reviewed elsewhere1–3).
Figure 1.
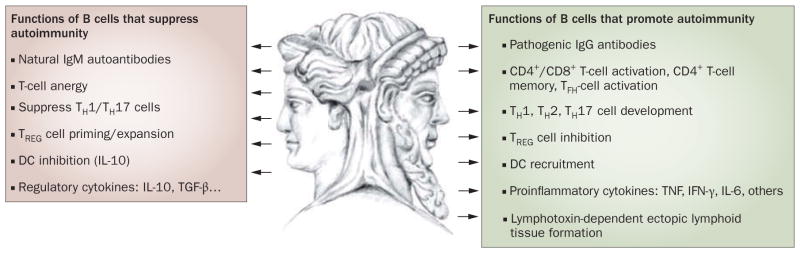
The Janus nature of B cells. B cells carry out multiple functions, through the production of antibodies (either protective natural autoantibodies or pathogenic IgG autoantibodies), and in an antibody-independent fashion. Pathogenic antibody-independent functions include the formation of ectopic lymphoid tissue (through lymphotoxin β receptor signaling) and lymphotoxin-independent functions, including the promotion of multiple effector CD4+ and CD8+ T-cell subsets, T-cell memory, DC recruitment and inhibition of TREG cells. Many of these functions are mediated by B-cell production of proinflammatory cytokines and chemokines. However, B cells also carry out essential protective functions that might prevent or suppress autoimmunity, including induction of T-cell anergy, suppression of effector TH cells, inhibition of DCs and expansion of TREG cells. Whether these functions are carried out by specialized, irreversibly committed B-cell populations (or by more plastic cells), their role in disease manifestations and progression, and their impact on treatment outcome, remain to be fully understood. Abbreviations: DC, dendritic cell; IFN, interferon; IL, interleukin; TFH, T follicular helper cell; TH, helper T cell; TREG cell, regulatory T cell; TGF-β, transforming growth factor-β; TNF, tumor necrosis factor. Reprinted by permission from Macmillan Publishers Ltd: Journal of Investigative Dermatology 129, 278–288 © 2008.
Such a strong rationale, combined with the success of B-cell depletion for the treatment of rheumatoid arthritis (RA), provided the impetus for the investigation of this approach in SLE almost 10 years ago, and the results from early studies created tremendous expectations for SLE. However, randomized, placebo-controlled trials of the B-cell-depleting agent rituximab, which targets CD20, failed to meet their primary or secondary clinical endpoints for renal and nonrenal SLE. These unexpected findings have caused confusion and resulted in considerable skepticism regarding the future of B-cell therapies for SLE, despite the existence of numerous observational studies of efficacy.4,5 However, the efficacy of other anti-B-cell agents provides renewed support for the concept of B-cell targeting for the treatment of SLE and has generated considerable impetus to pursue this approach.6 In this Review, we discuss the rationale, limitations and challenges for this approach and provide an overview of current and future agents for global or selective targeting of B cells in SLE.
B-cell targeting in SLE: status quo
SLE has long been considered a disease associated with the presence of autoreactive immune complexes that induce a type III hypersensitivity reaction and also, more recently, with the ability of some of these complexes to induce the release of type I interferons (IFNs) by Toll-like receptor (TLR)-mediated signaling in plasma cytoid dendritic cells.7 Accordingly, the rationale for, and early goal of, B-cell targeted therapies was the elimination of autoantibodies. Such design, however, has been tempered by the realization that B-cell depletion as currently achieved using anti-CD20 agents such as rituximab fails to achieve substantial depletion of some autoantibody species and that clinical benefit might be dissociated from the observed changes in autoantibody levels in terms of timing and magnitude.5,8,9 These clinical observations are consistent with the existence of separate compartments of short-lived and long-lived plasma cells,10 which have been considered to have life spans of a few weeks and several months or even years, respectively. A current model, yet to be formally tested, posits that the levels of auto-antibodies produced by short-lived plasma cells (such as anti-double-stranded [ds]DNA autoantibodies) would decline considerably over a period of a few months after therapy, whereas long-lived plasma cells would generate autoantibodies that are more ‘stable’, such as anti-RNA-binding protein (RBP) autoantibodies (Ro, La, Smith, RNP), and protective anti microbial anti bodies.9,11 The long-term stability of anti-RBP autoantibodies and antimicrobial antibodies would be explained by the prolonged survival of long-lived plasma cells in the absence of precursor B cells and by the lack of expression of CD20, which renders them impervious to direct attack by anti-CD20 agents. By contrast, a relatively small fraction of short-lived plasma cells expressing some level of surface CD20 could be depleted by the direct action of anti-CD20 agents, with the rest of these cells being depleted over the course of time by attrition in the absence of replenishment by precursor B cells. The discordant levels of different autoantibodies could also be the result of their generation by distinct populations of B cells with differential sensitivity to B-cell-depletion therapy owing to differences in their anatomical location or activation pathways.
The considerable clinical benefit that B-cell depletion imparts before the observation of substantial changes in autoantibody levels supports the evidence that, as previously discussed, B cells also have powerful pathogenic antibody-independent functions. As further discussed in the following section, interruption of pathogenic functions is likely to account for a considerable portion of the benefit provided by B-cell-depletion therapy and, therefore, a precise understanding of the nature of pathogenic B-cell subsets and their function should identify targets for more-selective B-cell interventions that would spare the undesirable elimination of protective B cells likely to occur with global depletion.8,12
Strategies for B-cell targeting
Several strategies that target B cells are currently available (summarized in Table 1 and Figure 2). The approaches include: direct killing using depleting antibodies; inhibition of factors involved in the survival or differentiation (or both) of B cells; induction of B-cell inhibitory receptors; interruption of signaling through the B-cell receptor (BCR) or co-stimulatory receptors (or both); and deletion or functional inactivation of antigen-specific autoreactive B cells. Pathogenic B-cell functions could also be interrupted by altering the migration patterns of these cells or eliminating the secondary and tertiary lymphoid microarchitecture that facilitates the selection and expansion of autoimmune B cells.
Table 1.
Strategies for therapeutic targeting of B cells and plasma cells in SLE*
| Molecular target | Effect on cellular target | Drug | |
|---|---|---|---|
| B cells | Plasma cells | ||
| Depletion | |||
| CD205 | Killing | Depletes precursors; kills some PBs | Rituximab, others |
| CD1986 | Killing | Depletes precursors; kills PBs and some PCs | Antibodies |
| Proteasome72 | Kills activated B cells or GC cells | Kill PCs | Bortezomib, car3lzomib |
| Depletion and inhibition | |||
| CD22,49 CD79α/β58 | Depletes and inhibits proliferation | Depletes precursors, inhibits differentiation | Epratuzumab, other antibodies |
| B-cell receptor inhibition | |||
| Syk, PI3K53–55 | ↓ Activation and survival | ↓ Generation | Inhibitors |
| Survival and/or differentiation | |||
| BAFF37 | ↓ Naive-cell and transitional-cell survival; promotes tolerance; ↓ GC survival43 | Depletes some precursors | Belimumab |
| BAFF + APRIL37 | ↓ Naive-cell and transitional-cell survival; promotes tolerance; ↓ GC survival | Depletes SLPC, spleen LLPC; ↓ BM PCs | Atacicept |
| Type I IFN66 | ↓ Naive-cell activation; ↓ memory-cell differentiation | ↓ Generation | Antibodies to IFN, IFN receptor |
| IL-2187–89 | ↓ Naive-cell differentiation; inhibits CC and memory-cell differentiation, survival | ↓ Generation | Antibodies‡ |
| IL-690 | ↓ Memory-cell differentiation | ↓ Generation | Tocilizumab |
| TNF91 | ↓ Memory-cell formation; ↓ GC reactions | ↓ Survival | Anti-TNF agents |
| IL-1792,93 | Prolongs GC reactions | ↑ Generation | Antibodies‡ |
| IFN-γ69 | ↓ TH1 cell-dependent autoantibodies | ↓ Generation? | Antibodies§ |
| Co-stimulation | |||
| CD40 ligand94,95 | Inhibits early-B-cell activation, GC formation | ↓ Generation? | Antibodies|| |
| TLR65 | Inhibits B-cell activation | ↓ Differentiation | Inhibitors |
| ICOS96 | ↓ CC differentiation, B-cell activation | ↓ Generation? | Antibodies¶ |
| BAFF or APRIL or IFN | As above | As above | As above |
| CD30–CD30 ligand97 | Inhibits B-cell activation, GC reactions | Unknown | Antibodies‡ |
| Homing and positioning | |||
| CXCR4–CXCL1210,98,99 | Inhibits GC reactions | ↓ BM homing, survival | Antagonists, antibodies |
| CXCR5–CXCL13100,101 | Inhibits GC, ectopic lymphogenesis | ↓ Generation | Antagonists,‡ antibodies§ |
| CXCR3–CXCL9–11102 | Inhibits/kills CXCR3+ effector B cells | Inhibits migration to target organs | Antagonists,‡ antibodies‡ |
| LTβR103 | Disrupts GC, ectopic lymphogenesis | ↓ Generation | Baminercept |
| Induction of BREG cells# | |||
| CD40 | Expands IL-10-producing T2 B cells | Unknown | Agonistic CD40 antibody13 |
| IL-15 receptor | Expands IL-10-producing B cells | Unknown | GM-CSF–IL-15 fusokine14 |
Mechanisms range from preclinical studies to phase III clinical studies, and the main known biologic effects are derived on the basis of primarily animal experiments but also human data when available. In many instances, the impact on PCs was deduced from the effect of the intervention on antibody levels and not necessarily from direct cellular evidence.
Preclinical studies.
Antibodies or small molecules under development.
Study interrupted owing to thromboembolic complications.
In phase I trial.
Induction of BREG cells could also be accomplished through B-cell depletion104 or infusion of BREG cells expanded in vitro.
Abbreviations: APRIL, a proliferation-inducing agent; BAFF, B-cell activating factor; BM, bone marrow; BREG cell, regulatory B cell; CC, centrocyte; CXCL, CXC-chemokine ligand; CXCR, CXC-chemokine receptor; ICOS, inducible T-cell co-stimulator; IFN, interferon; IL, interleukin; GC, germinal center; GM-CSF, granulocyte-macrophage colony stimulating factor; LLPC, long-lived plasma cell; LTβR, lymphotoxin β receptor; PB, plasmablast; PC, plasma cell; PI3K, phosphoinositide 3-kinase; SLPC, short-lived plasma cell; TH1 cell, type 1 T-helper cell; TLR, Toll-like receptor; TNF, tumor necosis factor.
Figure 2.

Strategies for B-cell targeting in SLE. B cells can be targeted at various stages of their development. Targeting surface antigens with depleting antibodies will provide differential breadth of depletion. Inhibition of B-cell co-stimulation and/or survival can also be therapeutically desirable and could be achieved by targeting surface receptors (both in the early phases of activation [1] and GC development and in later GC phases [2]). GC formation is dependent on the LTβR, and its structure and survival can also be influenced by targeting organizing chemokines and BAFF, respectively (3).105,106 Chemokine blockade could also influence plasma cell homing or survival (or both) in different locales. The retention of GC B cells and autoantibody output can be powerfully influenced by IL-17 (4).92 Blockade of other critical co-stimulatory and survival/differentiation factors could be therapeutically beneficial (indicated in Table 1). The bars at the top indicate the phases and types of B-cell response preferentially affected by blockade of the corresponding factor. A more-comprehensive listing of the molecules involved is provided in Supplementary Figure 1 online. Abbreviations: APRIL, a proliferation-inducing ligand; BAFF, B-cell activating factor; CB, centroblast; CC, centrocyte; CXCR, CXC-chemokine receptor; FDC, follicular dendritic cell; GC, germinal center; IL-17, interleukin-17; LTβR, lymphotoxin β receptor; TFH, T follicular helper cell; TH, helper T cell.
An alternative strategy that could be developed for future human use involves the expansion of regulatory B cells (BREG cells).13,14 BREG cells are currently defined as cells that produce interleukin (IL)-10 and possibly other immunoregulatory cytokines such as transforming growth factor-β (TGF-β) and that are capable of inhibiting the proinflammatory functions of either TH cells or dendritic cells.15 Mouse BREG cells have been observed within the transitional, B1 and marginal zone compartments.15 Of notable interest, human transitional B cells also have an IL-10-mediated regulatory function, which, in preliminary studies, seems to be defective in patients with SLE.16 In addition, autoantibody production could be minimized using agents capable of depleting plasma cells.
Strategies that target B cells will be discussed in the following sections, with special emphasis on agents that are either currently available or under development, and with the purpose of outlining concepts that illustrate proof-of-principle. Notably, all B-cell-targeting interventions will have to contend with one of the main obstacles faced in the treatment of SLE—namely, the high degree of disease diversity, which may be mediated at least in part by genetic heterogeneity.17 This issue is of consider able importance for both the design of therapies targeted to specific disease subsets and the need to identify patients most likely to benefit from such therapies.
B-cell-depleting antibodies
Anti-CD20 antibodies
As exemplified by rituximab, antibodies directed against CD20 target a large proportion of human B cells, from pre-B cells to a small fraction of peripheral, immature plasmablasts. Several mechanisms have been invoked to explain the killing action of anti-CD20 antibodies, such as complement-induced lysis and direct apoptosis, but antibody-dependent cell-mediated cytotoxicity is likely to represent the main mechanism of action of rituximab.18
Two large, randomized trials of rituximab in moderate- to-severe active nonrenal lupus and in lupus nephritis (the EXPLORER and LUNAR trials, respectively) failed to meet their superiority endpoints—that is, they were unable to show that rituximab was clinically superior to placebo when added to standard care.19 In these studies, rituximab was compared with placebo following the addition of either intervention to conventional therapy (corticosteroids plus stable background immunosuppressive therapy in the case of EXPLORER, and mycophenolate mofetil for LUNAR).19 However, despite failing to meet all other primary and secondary endpoints, a benefit in major clinical response and partial clinical response at 1 year in African-American and Hispanic patients with SLE and improved platelet counts in patients with baseline thrombocytopenia were detected in EXPLORER.19 Rituximab-treated patients showed an increase in complement levels and a decrease in levels of anti-dsDNA autoantibodies (in patients without anti-RBP autoantibodies). Considerable immunological effect was also inferred by reductions in levels of CD8+ memory T cells and anticardiolipin antibodies.
Despite glimpses of benefit, the disappointing results of the EXPLORER and LUNAR trials contrast with the perceived benefit of rituximab in clinical practice, mostly when used as rescue therapy in combination with cyclophosphamide for refractory disease.4 Reasons for the conflicting results are likely to include disease heterogeneity, aggressive background therapy that shows good efficacy, and limitations of available outcome ‘instruments’ with very sensitive cutoffs for adjudicating lack of response to treatment (in the case of EXPLORER).19,20 In addition, follow-up periods of longer than the 1 year used in these studies might be required to demonstrate the superiority of rituximab over conventional treatment in SLE studies.
Consideration of the immunological variables that might influence this therapy and help inform clinical practice and future studies is, however, important. Thus, both LUNAR and EXPLORER included a second cycle of B-cell depletion at 6 months, which would have eliminated the transitional and naive B cells that typically emerge after profound depletion in SLE. We and other researchers have shown that a prolonged dominance of early cells at the expense of memory cells is a feature that persists for several years in patients who respond well to rituximab, whereas rapid re-accumulation of memory cells (which are more likely to be responsible for the pathogenic effects) characterizes poor responders.8,21 It is plausible, therefore, that repeated B-cell depletion could have blunted or delayed a late phase of improvement (Figure 3), which might be revealed on longer follow-up. This possibility is supported by the observation that both human transitional and naive B cells have been shown to produce IL-10, and human transitional cells have IL-10-mediated regulatory properties that might be defective in SLE patients.16,22 A similar defect has been reported for naive B cells in patients with multiple sclerosis, which seems to be repaired on repopulating naive cells after B-cell-depletion therapy.22
Figure 3.

Phases of B-cell depletion: a working model. This model assumes that disease is balanced by protective and pathogenic B-cell functions with active disease characterized by a pathogenic B-cell environment with dominance of effector B cells and/or autoantibodies (owing to expansion of pathogenic cells or deficit of regulatory cells). The benefit of early B-cell depletion would depend on: the extent of initial B-cell killing and early re-expansion; the relative impact of depletion on protective versus pathogenic B cells; the relative importance of effector versus regulatory B-cell functions at the time of treatment; and the pathogenic contribution of short-lived autoantibodies. Long-term outcome would depend on: the relative numerical and functional balance between pathogenic and regulatory B cells; and the pathogenic contribution of long-lived autoantibodies, which would continue to be produced by long-lived plasma cells unless they are directly targeted by other agents or eventually decreased by chronic B-cell depletion. Long-term remission could be achieved if immunological tolerance is restored during the reconstitution period owing to the confluence of factors discussed elsewhere.1,21 This model provides a template for understanding the phases and underpinnings of B-cell depletion, how to design combination therapies either for induction or maintenance, and how to evaluate the success or failure of B-cell targeted therapies. Abbreviation: TREG cell, regulatory T cell. Permission obtained from Drug Discovery Today: Therapeutic Strategies 6, Sanz, I. Indications of rituximab in autoimmune diseases, 13–19 © (2009) Elsevier Ltd, with permission from Elsevier.
Another issue is the failure of the trials to carry out high-sensitivity analysis to properly evaluate complete depletion (defined as less than 5 cells per μl); the extent of depletion in SLE patients might vary in the absence of concomitant cyclophosphamide treatment, and depletion to levels as low as 0.01 cells per μl has been shown to correlate with greater benefit in RA and correlated with good responses in our original phase I–II study in SLE.23–25 In this scenario, excess circulating B-cell-activating factor (BAFF, also known as BLyS and tumor necrosis factor [TNF] ligand superfamily, member 13b) consistently observed after rituximab treatment could expand any residual B cells and impair the censoring of autoreactive B cells.26,27 All these considerations emphasize the need for high- sensitivity B-cell quantification methods and multiparameter flow cytometry protocols to reproducibly discriminate between transitional, naive, memory and effector B-cell subsets (a feat that cannot be accomplished using current methods [IgD, CD38 and CD27 staining]).28 Validated standard operating procedures for these protocols have been developed (Figure 4) and are currently being validated for multicenter studies through the NIH-sponsored Autoimmunity Centers of Excellence network (I. Sanz, C. Wei, J. Anolik and F. E.-H. Lee, unpublished data). A proper understanding of the immunological basis for the success or failure of B-cell depletion (or other B-cell-directed interventions) will also require measurements of B-cell autoreactivity, the extent of B-cell depletion in lymphoid tissue and bone marrow, B-cell cytokine secretion and the impact on T-cell immunity.
Figure 4.

Variable B-cell homeostasis in human SLE. The challenge of tailoring specific B-cell therapies for a heterogeneous B-cell disease is illustrated by the high variability of B-cell profiles displayed by individual SLE patients. This variability applies to both populations with regulatory potential (transitional and marginal zone-like cells) and pathogenic potential (memory, effector and plasma cells). This figure represents a heat map showing B-cell profiles for normal individuals and patients with SLE. Each column represents an individual and each row corresponds to a B-cell subset (specified on the right according to previously published classifications). Both individuals and cell populations are hierarchically clustered and reordered accordingly. Color maps correspond to the log of the percentage of the cell subset with respect to the CD19+ subset (except for the 9G4+ group, which are expressed relative to their immediate parent population). The healthy and SLE groups were clustered separately, each using correlation as the distance measure and complete linkage. There were 25 healthy individuals and 49 patients with SLE. This type of data visualization clearly demonstrates both high interindividual variability as well as clustering of patient subsets, and should help design and evaluate B-cell therapies in SLE. Abbreviations: CXCR, CXC-chemokine receptor; SLE, systemic lupus erythematosus.
Other B-cell depleting antibodies
Other B-cell depleting antibodies include anti-CD19 antibodies, which, in human CD19 transgenic mice, deplete a wider spectrum of B cells than does rituximab, including pro-B cells and a fraction of plasma cells, a result that is consistent with the decreased levels of total serum immunoglobulins seen in this model.29 Whether targeting a wider range of B cells translates into higher clinical efficacy in SLE remains to be determined.
A different approach to B-cell depletion is provided by antibodies, such as anti-CD22 (epratuzumab), which target inhibitory receptors. Theoretically, at least, the effect of these antibodies would depend on their activity—agonistic or antagonistic—towards the receptor in question, as well as on their killing ability. Thus, epratuzumab could inhibit the recruitment into the mature compartment of autoimmune cells by preferentially depleting transitional and naive B cells while, at least in vitro, also inhibiting B-cell activation and proliferation.30 This drug has shown safety in short-term studies and potential for clinical efficacy, and warrants further exploration for the treatment of SLE.31 Fc receptor γ2b (FcRγ2b) is a potent inhibitory receptor for B-cell activation; its crosslinking might induce feedback suppression and apoptosis of B cells and plasma cells (thereby providing a mechanistic explanation for the therapeutic benefit of intravenous immunoglobulin).32 Given the strong immunological and genetic rationale that implicates defective FcRγ2b activity in SLE, agonistic antibodies could show strong therapeutic potential, although their efficacy could be compromised by the decreased expression of this receptor in SLE B cells and plasma cells.33–35 Moreover, interventions designed to target this pathway in SLE patients will need to contend with the possibility that this intervention could potentiate a type I IFN response.36
BAFF and APRIL blockade
Inhibiting B-cell stimulation induced by BAFF or APRIL (a proliferation-inducing ligand, also known as TNF ligand superfamily member 13), or both, represents a different paradigm for B-cell targeting in SLE (reviewed elsewhere37). Human BAFF signals through three different receptors—BAFF receptor (BAFF-R), TACI (also known as TNF receptor superfamily member 13b) and BCMA (also known as TNF receptor superfamily member 17; binds with low avidity). By contrast, APRIL only signals through TACI and BCMA. Accordingly, anti-BAFF antibodies (belimumab) or BAFF-R–Ig provide selective BAFF blockade, whereas a TACI decoy receptor (TACI–Ig fusion protein; atacicept) that is capable of binding circulating BAFF and APRIL inhibits both signaling pathways.
While the relative expression of the three BAFF-family receptors and the relative roles of BAFF and APRIL in the homeostasis of different B-cell subsets are well established in mice, these important aspects remain to be fully elucidated in humans. However, emerging human data outlined below indicate considerable similarities between the two species.38–40 BAFF is essential for the survival of murine late transitional, follicular naive and marginal zone B cells, but does not influence the survival of memory cells. Of note, low levels of BAFF induce the negative selection of early autoreactive B cells and, under conditions of excess BAFF, their selection into the mature compartment is instead facilitated.41 In contrast to B cells, murine short-lived and long-lived plasma cells preferentially express TACI and BCMA, respectively, and their survival is compromised by the combined blockade of BAFF and APRIL with TACI–Ig, but not by selective BAFF blockade.37
In most lupus models, TACI–Ig preferentially depletes short-lived IgM plasma cells while sparing long-lived IgG bone marrow plasma cells. The same agent, however, was able to deplete IgM and IgG plasma cells from the spleens of lupus-prone MRLlpr mice.42 Of note, BAFF blockade induces clinical improvement in murine lupus despite the persistence of autoantibodies, which again indicates a pathogenic role for antibody-independent B-cell functions. This interpretation, however, is complicated by the fact that blockade of BAFF or APRIL (or both) can also considerably influence several other immune, inflammatory and stromal cells.37,43
Results from belimumab trials
The initial impetus for the use of anti-BAFF and/or anti-APRIL agents to treat SLE came from data from mouse studies that supported a role for increased levels of BAFF in the pathogenesis of lupus, the amelioration of disease in response to BAFF blockade, and the elevated values of serum BAFF that characterize human SLE.44 Consistent with this evidence, promising results have been released from two phase III clinical trials of belimumab for the treatment of SLE, BLISS-52 and BLISS-76, which treated 865 and 819 patients, respectively. In both trials, the primary efficacy endpoint was the SLE Responder Index (SRI) at 52 weeks based on intention-to-treat. In BLISS-52, belimumab considerably reduced SLE disease activity (as measured using the SRI), SLE flare rates and the need for prednisone therapy, and increased the time to first SLE flare in patients with active SLE.6,45 Belimumab was well tolerated with rates of serious and infection adverse events comparable to placebo. Similar results for BLISS-76 data analyzed at 52 weeks have also been announced.46
These results provide a much needed boost to the concept of B-cell targeting in SLE, although many important questions remain to be addressed through the analysis of available data and additional studies. Although it was statistically significant, the additional clinical improvement observed with belimumab as compared to the control arm was modest. Therefore, it will be particularly important to learn how quickly significant clinical benefit ensues after starting treatment (only the results at 52 weeks have been released), what the immunological correlates of disease improvements are, and whether specific patient subsets or disease manifestations are more responsive to therapy than others. Such information will be essential to determine whether BAFF blockade should be used mainly as maintenance therapy after induction therapy with other modalities, as background therapy to prevent flares and disease progress ion, or possibly as part of combination induction therapies.
The study of a small subset of patients treated with belimumab for up to 2.5 years in an extension of a phase II trial has also provided helpful information regarding the effect of chronic BAFF blockade on human B-cell homeostasis.40 Chronic BAFF inhibition induced slow but sustained decreases in the numbers of transitional cells and naive cells, with statistical significance reached after 3 months of treatment. These kinetics contrast with those seen in response to rituximab, which induced major depletion within 2 weeks and maximal depletion typically within 1–2 months.47 The numbers of memory cells and plasma cells did not decrease appreciably indicating that, similar to their mouse counterparts, these human B-cell subsets are independent of BAFF for survival. By contrast, a subset of isotype-switched CD27− B cells, the expansion of which correlates with active SLE, also underwent significant and sustained depletion.48,49 More-extensive and detailed immunological analysis will be required to establish the value of specific B-cell changes as biomarkers of a clinical response to belimumab. Additional studies will also be required to resolve the counterintuitive observation that, in contrast to studies of universal B-cell depletion, clinical improvement was associated with a relative expansion of memory and plasma cells and a decrease in the numbers of populations with a potential regulatory role.21,50
Results from atacicept trials
Preclinical models showing profound depletion of plasma cells and results from early phase I studies using atacicept are highly appealing on theoretical grounds, but the clinical benefit and risk:benefit ratio of combined BAFF–APRIL blockade with this drug remain to be determined.51 Of some concern, a phase II trial using this drug in combination with mycophenolate mofetil for lupus nephritis was discontinued owing to increased infection rates.52 However, a phase II–III trial of this drug for nonrenal lupus is ongoing.
Targeting B-cell signaling pathways
SLE is characterized by B-cell hyperactivity that, at least in part, might result from polymorphisms in genes encoding B-cell signaling components that increase disease susceptibility.34 Hence, dampening BCR stimulation represents an attractive therapeutic approach.
The concept of targeting the BCR signaling pathways is illustrated by the promising results shown by inhibition of spleen tyrosine kinase (Syk) in B-cell lymphomas and RA.53 Syk inhibition also ameliorates lupus in NZB/NZW mice, although such improvement was not associated with changes in the levels of autoantibodies and might arise, at least in part, from T-cell modulation.53,54 Of note, Syk is a key mediator of Fc receptor signaling and its inhibition can also affect other inflammatory cells, including dendritic cells, macrophages and neutrophils.
The aforementioned studies also provide proof-of- concept for the inhibition of molecules that signal downstream of Syk, including components of the phosphatidylinositol 3-kinase–Akt–mammalian target of rapamycin (PI3K–Akt–mTOR) pathway, the hyper activity of which has been implicated in the pathogenesis of murine SLE.55 Inhibition of the PI3Kδ isoform has shown clinical benefit in lupus-prone MRLlpr mice, bringing about a considerable decrease in the numbers of B cells and autoantibodies, at least in part by CD4 T-cell inactivation.56 These results highlight the therapeutic potential of specific inhibitors of PI3Kδ, which has an essential and nonredundant role in B-cell development.57
Antibodies to CD79, a transmembrane protein that associates with the BCR, represent another approach to targeting B cells through a combination of depletion and interruption of BCR signaling. These antibodies can efficiently delete a large fraction of mouse B cells, including follicular, marginal zone and IgD+ B cells and possibly IgG2a+ B cells, while sparing newly formed and IgG1+ B cells, which express low levels of CD79. Anti-CD79 antibodies have been shown to decrease tissue damage and the levels of antichromatin antibodies and to improve survival in MRLlpr mice.58
Targeting B cells through TLR and IFN inhibition
TLRs provide essential B-cell co-stimulation and represent appealing targets for pharmacological inhibition in SLE. In mice, TLR9 is critical for the generation of anti-DNA antibodies and TLR7 controls the production of anti-RBP antibodies and determines clinical severity.59 Moreover, TLR signaling amplifies a pathogenetic loop that integrates two critical SLE cytokines, IFN-α and BAFF. In B cells, TLR stimulation upregulates the expression of BAFF receptors to create synergy between BAFF and BCR stimulation. Similarly, type I IFN produced by TLR-activated plasmacytoid dendritic cells enhances TLR7-induced and TLR9-induced B-cell activation and enhances BAFF production by myeloid dendritic cells.60–63
These observations strongly suggest that blockade of either TLR or type I IFN stimulation could be beneficial in SLE, at least in part through B-cell inhibition. This notion is also supported by the considerable benefit imparted by antimalarial drugs, which represent a corner stone of SLE therapy and can inhibit TLR9 or TLR7. Currently, TLR inhibitors that are more powerful and more specific than those currently available are under development for SLE,64 with initial reports indicating clinical efficacy in murine lupus.65 Similarly, anti-IFN agents are currently in the early phases of clinical development and a phase Ia trial of a fully human monoclonal antibody that inhibits most IFN-α subtypes has shown promising biological activity and safety in patients with mild-to-moderate SLE when added to standard therapy.66 In another ongoing phase I study, a humanized anti-IFN-α monoclonal antibody (rontalizumab) is being evaluated for safety and efficacy in patients with moderate-to-severe active SLE.66
However, as with all biologic agents, it will be important to check for unanticipated adverse effects that could be triggered by inhibition of either TLR or type I IFN, given the multifaceted nature of the signaling events that could be triggered by these molecules. Thus, murine lupus can be exacerbated by a deficiency in TLR9,59 and TLR activation of B cells might control autoimmune T-cell responses.67,68 Similarly, type I IFN has been shown to protect against murine lupus and might protect against inflammation through IL-10-producing B cells.69,70
Targeting plasma cells
As discussed above, most B-cell-targeted therapies fail to eliminate plasma cells and long-lived autoantibodies (defined above as ‘stable’ autoantibodies represented by those that recognize RBP). Therefore, there is appreciable interest in developing agents that can carry out this function. In addition to the aforementioned combined BAFF–APRIL blockade, several other strategies might achieve this goal (Table 1 and Figure 2), including the blockade of cytokines known to promote B-cell differentiation into plasma cells, such as IL-21, IL-6 and IL-17. Also of particular interest is the role of proteasome inhibitors, such as bortezomib. A selective inhibitor of the 26S proteasome, bortezomib induces the terminal unfolded protein response (UPR), which eventually leads to cell-cycle arrest and apoptosis. Owing to their extremely high rate of immunoglobulin synthesis, which initially triggers the UPR, plasma cells are particularly sensitive to the effect of proteasome inhibitors.71 Proteasome inhibitors also block nuclear factor κB (NFκB) and can inhibit activated B cells, germinal center B cells and dendritic cells, and the release of NFκB-induced proinflammatory cytokines (including TNF, IL-1 and IL-6) from activated T cells, effects which could also confer therapeutic benefit.72,73
Bortezomib has been approved for the initial treatment of multiple myeloma and has shown impressive biologic and clinical activity in murine lupus.72,74 However, its use is limited by the occurrence of painful peripheral neuropathy.74 Other agents such as carfilzomib seem to show improved safety compared with bortezomib, and a phase I dose escalation study of this drug is under development by the National Institute of Allergy and Infectious Diseases (NIAID)-sponsored Autoimmunity Centers of Excellence Network. An important safety concern related to all proteasome inhibitors is that by inducing universal depletion of plasma cells, including long-lived plasma cells, these agents could be expected to decrease not only pathogenic autoantibodies but also the main source of protective antimicrobial antibodies.
Future directions and challenges
Global and sustained B-cell depletion is predicated on the flawed premise that B cells have only pathogenic roles in SLE; yet, experimental evidence for protective roles for B cells in this disease continues to grow, both in mice and humans.3,14,15,68,75–82 Thus, in addition to their ability to induce T-cell anergy, B cells produce the anti-inflammatory cytokines IL-10 and TGF-β, induce TREG cells and directly suppress effector TH cells. It is therefore obvious that, as summarized in Figure 1, B cells often carry out opposing functions and accordingly, the ultimate outcome of B-cell depletion will be determined by the preferential elimination or reconstitution of specialized B-cell subsets (Figure 3). Alternatively, favorable long-term outcomes could be achieved by reversing pathogenic functions or inducing functions that confer protection in plastic B-cell subsets, perhaps through modification of the cytokine milieu responsible for the initiation or amplification of SLE (type I IFNs, BAFF, IFN-γ, TNF, IL-17, IL-21, and so on). Accordingly, B-cell therapies could induce early benefit by eliminating or inhibiting pathogenic effector B cells, whereas delayed and more sustained improvement could depend on the preferential expansion of B cells with regulatory functions.
This working model (Figure 3) would favor aggressive B-cell depletion for induction therapy in active SLE, a view that is also consistent with available data that correlate the depth of initial depletion with the extent of clinical improvement in RA and SLE.9,25 However, the model also suggests that chronic B-cell depletion might not be a suitable maintenance therapy for diseases or disease subsets that might critically depend on the dominance of BREG cells for remission. Extending this argument, it could be suggested that—in at least some cases—chronic B-cell depletion might prevent the re-establishment of immunologic tolerance, if this process is dependent on the expansion of BREG cells (whether these cells are envisioned as producers of IL-10 or TGF-β or simply as immature or naive B cells that induce T-cell anergy). It is also important to bear in mind that, as shown in animal studies,83 there might be situations in which a deleterious immune response could be kept in check by BREG cells and, therefore, even acute B-cell depletion could exacerbate disease. Such a risk has been highlighted by a report of hyperacute cellular renal allograft rejection in patients treated with rituximab as pretransplantation desensitizing therapy,84 and by isolated reports of secondary autoimmune diseases occurring after B-cell depletion.85
Another possibility for the disappointing results of rituximab therapy in SLE might be its inability to eliminate long-lived autoantibodies, including those implicated in the induction of type I IFN production. This limitation of B-cell depletion could be overcome by therapies that are capable of effectively eliminating long-lived plasma cells.
Conclusions
A rather complicated picture emerges from all the above considerations. It seems obvious that a much more detailed understanding of human B-cell biology is sorely needed in order to design therapies that are more specific and more effective than those currently available. In particular, the heterogeneity of human B-cell populations, their division of labor and their homeostatic regulation need to be elucidated, as a better understanding of the functional balance between pathogenic and protective B-cell populations will be of the essence. SLE is a disease characterized by multiple B-cell abnormalities and extended B-cell profiles vary considerably from patient to patient (Figure 4). Understanding the consequence of this variability, whether it is related to disease subtype, disease duration or degree of activity will be central to our ability to design B-cell-directed therapies, time their administration appropriately and monitor their outcome. A deep understanding of the biologic and clinical relevance of B-cell profiles in SLE will also provide a much needed rational basis for the formulation of combination biologic therapies, an approach that, although difficult to evaluate, might well represent the best way to treat SLE. Finally, B-cell profiling could also provide information that might affect the choice of therapy for induction versus maintenance therapy (Figures 3 and 4).
Supplementary Material
Key points.
B cells have antibody-dependent and antibody-independent functions that can either promote or inhibit autoimmunity
B-cell-targeted therapies would ideally eliminate pathogenic B cells or promote the expansion and function of protective B cells, or both
Despite clinical observations of benefit and results from early trials, two large phase III, randomized placebo-controlled trials of rituximab have failed to meet their primary or secondary clinical endpoints
More-selective B-cell depletion achieved by blocking B-cell-activating factor (using belimumab) has demonstrated considerable clinical benefit
Improved knowledge of the biology of B cells and plasma cells opens the door to several additional strategies for the therapeutic targeting of B cells
Review criteria.
Publications listed in this Review and other papers used to obtain the information presented here were obtained through MEDLINE searches using the Boolean terms “SLE” and “B-cell lymphocytes” in combination. Selected papers and reviews on similar topics were also used a source of references. Papers, reviews and abstracts published between 1995 and 2010 were used. Only English-language papers were included. In addition, relevant publications were selected from other reviews on similar topics and from the authors’ own bibliographic files.
Acknowledgments
Supported in part by NIH grants U19 AI56390 (Rochester Autoimmunity Center of Excellence) and R01 AI049660 to I. Sanz, and K23 AI67501 to F. E.-H. Lee.
Footnotes
Désirée Lie, Univesity of California, Orange, CA, is the author of and is solely responsible for the content of the learning objectives, questions and answers of the MedscapeCME-accredited continuing medical education activity associated with this article.
Competing interests
I. Sanz declares associations with the following companies: Genentech, GlaxoSmithKline and Biogen. See the article online for full details of the relationships. F. E.-H. Lee and the Journal Editor J. Buckland declare no competing interests. The CME questions author D. Lie has served as a nonproduct speaker for “Topics in Health” for Merck Speaker Services.
Supplementary information is linked to the online version of the paper at www.nature.com/nrrheum
References
- 1.Manjarrez-Orduno N, Quach TD, Sanz I. B cells and immunological tolerance. J Invest Dermatol. 2009;129:278–288. doi: 10.1038/jid.2008.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yanaba K, et al. B-lymphocyte contributions to human autoimmune disease. Immunol Rev. 2008;223:284–299. doi: 10.1111/j.1600-065X.2008.00646.x. [DOI] [PubMed] [Google Scholar]
- 3.Lund FE. Cytokine-producing B lymphocytes—key regulators of immunity. Curr Opin Immunol. 2008;20:332–338. doi: 10.1016/j.coi.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lu T, et al. A retrospective seven-year analysis of the use of B cell depletion therapy in systemic lupus erythematosus at University College London Hospital: the first fifty patients. Arthritis Rheum. 2009;61:482–487. doi: 10.1002/art.24341. [DOI] [PubMed] [Google Scholar]
- 5.Leandro MJ, de la Torre I. Translational Mini-Review Series on B Cell-Directed Therapies: The pathogenic role of B cells in autoantibody-associated autoimmune diseases; lessons from B cell-depletion therapy. Clin Exp Immunol. 2009;157:191–197. doi: 10.1111/j.1365-2249.2009.03978.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Navarra S, et al. Belimumab, a BLyS-specific inhibitor, reduced disease activity, flares and prednisone use in patients with active SLE: efficacy and safety results from the phase 3 BLISS-52 study. Presented at the American College of Rheumatology 2009 Annual Scientific Meeting; 2009. [Google Scholar]
- 7.Lovgren T, et al. Induction of interferon-alpha by immune complexes or liposomes containing systemic lupus erythematosus autoantigen- and Sjogren’s syndrome autoantigen-associated RNA. Arthritis Rheum. 2006;54:1917–1927. doi: 10.1002/art.21893. [DOI] [PubMed] [Google Scholar]
- 8.Sanz I. The conundrum of B cell depletion in SLE. Nat Rev Rheumatol. 2009;5:304–305. doi: 10.1038/nrrheum.2009.100. [DOI] [PubMed] [Google Scholar]
- 9.Anolik JH, et al. Rituximab improves peripheral B cell abnormalities in human systemic lupus erythematosus. Arthritis Rheum. 2004;50:3580–3590. doi: 10.1002/art.20592. [DOI] [PubMed] [Google Scholar]
- 10.Radbruch A, et al. Competence and competition: the challenge of becoming a long-lived plasma cell. Nat Rev Immunol. 2006;6:741–750. doi: 10.1038/nri1886. [DOI] [PubMed] [Google Scholar]
- 11.Cambridge G, et al. B cell depletion therapy in systemic lupus erythematosus: relationships among serum B lymphocyte stimulator levels, autoantibody profile and clinical response. Ann Rheum Dis. 2008;67:1011–1016. doi: 10.1136/ard.2007.079418. [DOI] [PubMed] [Google Scholar]
- 12.Matsushita T, Yanaba K, Bouaziz JD, Fujimoto M, Tedder TF. Regulatory B cells inhibit EAE initiation in mice while other B cells promote disease progression. J Clin Invest. 2008;118:3420–3430. doi: 10.1172/JCI36030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Blair PA, et al. Selective targeting of B cells with agonistic anti-CD40 is an efficacious strategy for the generation of induced regulatory T2-like B cells and for the suppression of lupus in MRL/lpr mice. J Immunol. 2009;182:3492–3502. doi: 10.4049/jimmunol.0803052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rafei M, et al. A granulocyte-macrophage colony-stimulating factor and interleukin-15 fusokine induces a regulatory B cell population with immune suppressive properties. Nat Med. 2009;15:1038–1045. doi: 10.1038/nm.2003. [DOI] [PubMed] [Google Scholar]
- 15.Fillatreau S, Gray D, Anderton SM. Not always the bad guys: B cells as regulators of autoimmune pathology. Nat Rev Immunol. 2008;8:391–397. doi: 10.1038/nri2315. [DOI] [PubMed] [Google Scholar]
- 16.Blair PA, et al. CD19(+)CD24(hi)CD38(hi) B cells exhibit regulatory capacity in healthy individuals but are functionally impaired in systemic lupus erythematosus patients. Immunity. 2010;32:129–140. doi: 10.1016/j.immuni.2009.11.009. [DOI] [PubMed] [Google Scholar]
- 17.Barcellos LF, et al. High-density SNP screening of the major histocompatibility complex in systemic lupus erythematosus demonstrates strong evidence for independent susceptibility regions. PLoS Genet. 2009;5:e1000696. doi: 10.1371/journal.pgen.1000696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Clark EA, Ledbetter JA. How does B cell depletion therapy work, and how can it be improved? Ann Rheum Dis. 2005;64(Suppl):iv77–iv80. doi: 10.1136/ard.2005.042507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Merrill JT, et al. Efficacy and safety of rituximabin moderately-to-severely active systemic lupus erythematosus: The randomized, double-blind, phase ii/iii systemic lupus erythematosus evaluation of rituximab trial. Arthritis Rheum. 2010;62:222–233. doi: 10.1002/art.27233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wallace D. New therapies in systemic lupus erythematosus—trials, troubles and tribulations—working towards a solution: part 2—the politically incorrect version. Lupus. 2009;18:101–103. doi: 10.1177/0961203308100843. [DOI] [PubMed] [Google Scholar]
- 21.Anolik J, et al. Delayed memory B cell recovery in peripheral blood and lymphoid tissue in systemic lupus erythematosus after B cell depletion therapy. Arthritis Rheum. 2007;56:3044–3056. doi: 10.1002/art.22810. [DOI] [PubMed] [Google Scholar]
- 22.Duddy M, et al. Distinct effector cytokine profiles of memory and naive human B cell subsets and implication in multiple sclerosis. J Immunol. 2007;178:6092–6099. doi: 10.4049/jimmunol.178.10.6092. [DOI] [PubMed] [Google Scholar]
- 23.Looney RJ, et al. B cell depletion as a novel treatment for systemic lupus erythematosus: A phase I/II dose-escalation trial of rituximab. Arthritis Rheum. 2004;50:2580–2589. doi: 10.1002/art.20430. [DOI] [PubMed] [Google Scholar]
- 24.Albert D, et al. Variability in the biological response to anti-CD20 B cell depletion in systemic lupus erythematosus. Ann Rheum Dis. 2008;67:1724–1731. doi: 10.1136/ard.2007.083162. [DOI] [PubMed] [Google Scholar]
- 25.Dass S, et al. Highly sensitive B cell analysis predicts response to rituximab therapy in rheumatoid arthritis. Arthritis Rheum. 2008;58:2993–2999. doi: 10.1002/art.23902. [DOI] [PubMed] [Google Scholar]
- 26.Cambridge G, et al. B cell depletion therapy in systemic lupus erythematosus: relationships among serum B lymphocyte stimulator levels, autoantibody profile and clinical response. Ann Rheum Dis. 2008;67:1011–1016. doi: 10.1136/ard.2007.079418. [DOI] [PubMed] [Google Scholar]
- 27.Cancro MP. The BLyS/BAFF family of ligands and receptors: key targets in the therapy and understanding of autoimmunity. Ann Rheum Dis. 2006;65 (Suppl 3):34–36. doi: 10.1136/ard.2006.058412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sanz I, Wei C, Lee FEH, Anolik J. Phenotypic and functional heterogeneity of human memory B cells. Semin Immunol. 2008;20:67–82. doi: 10.1016/j.smim.2007.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yazawa N, Hamaguchi Y, Poe JC, Tedder TF. Immunotherapy using unconjugated CD19 monoclonal antibodies in animal models for B lymphocyte malignancies and autoimmune disease. Proc Natl Acad Sci USA. 2005;102:15178–15183. doi: 10.1073/pnas.0505539102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jacobi AM, et al. Differential effects of epratuzumab on peripheral blood B cells of patients with systemic lupus erythematosus versus normal controls. Ann Rheum Dis. 2008;67:450–457. doi: 10.1136/ard.2007.075762. [DOI] [PubMed] [Google Scholar]
- 31.Petri M, et al. Clinically meaningful improvements with epratuzumab (anti-CD22 mAb targeting B-cells) in patients with moderate/severe systemic lupus erythematosus (SLE) flares: results from 2 randomized controlled trials [abstract 1087] Arthritis Rheum. 2008;58 (Suppl):S571. [Google Scholar]
- 32.Tarasenko T, Dean JA, Bolland S. FcγRIIB as a modulator of autoimmune disease susceptibility. Autoimmunity. 2007;40:409–417. doi: 10.1080/08916930701464665. [DOI] [PubMed] [Google Scholar]
- 33.Xiang Z, et al. FcγRIIb controls bone marrow plasma cell persistence and apoptosis. Nat Immunol. 2007;8:419–429. doi: 10.1038/ni1440. [DOI] [PubMed] [Google Scholar]
- 34.Moser KL, Kelly JA, Lessard CJ, Harley JB. Recent insights into the genetic basis of systemic lupus erythematosus. Genes Immun. 2009;10:373–379. doi: 10.1038/gene.2009.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mackay M, et al. Selective dysregulation of the FcγIIB receptor on memory B cells in SLE. J Exp Med. 2006;203:2157–2164. doi: 10.1084/jem.20051503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dhodapkar KM, et al. Selective blockade of the inhibitory Fcγ receptor (FcγRIIB) in human dendritic cells and monocytes induces a type I interferon response program. J Exp Med. 2007;204:1359–1369. doi: 10.1084/jem.20062545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Moisini I, Davidson A. BAFF: a local and systemic target in autoimmune diseases. Clin Exp Immunol. 2009;158:155–163. doi: 10.1111/j.1365-2249.2009.04007.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Moir S, et al. Decreased survival of B cells of HIV-viremic patients mediated by altered expression of receptors of the TNF superfamily. J Exp Med. 2004;200:587–600. [PubMed] [Google Scholar]
- 39.Avery DT, et al. BAFF selectively enhances the survival of plasmablasts generated from human memory B cells. J Clin Invest. 2003;112:286–297. doi: 10.1172/JCI18025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jacobi AM, et al. Effect of long-term belimumab treatment on B cells in systemic lupus erythematosus: extension of a phase II, double-blind, placebo-controlled, dose-ranging study. Arthritis Rheum. 2010;62:201–210. doi: 10.1002/art.27189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lesley R, et al. Reduced competitiveness of autoantigen-engaged B cells due to increased dependence on BAFF. Immunity. 2004;20:441–453. doi: 10.1016/s1074-7613(04)00079-2. [DOI] [PubMed] [Google Scholar]
- 42.Liu W, et al. Control of spontaneous B lymphocyte autoimmunity with adenovirus-encoded soluble TACI. Arthritis Rheum. 2004;50:1884–1896. doi: 10.1002/art.20290. [DOI] [PubMed] [Google Scholar]
- 43.Kalled SL. Impact of the BAFF/BR3 axis on B cell survival, germinal center maintenance and antibody production. Semin Immunol. 2006;18:290–296. doi: 10.1016/j.smim.2006.06.002. [DOI] [PubMed] [Google Scholar]
- 44.Petri M, et al. Association of plasma B lymphocyte stimulator levels and disease activity in systemic lupus erythematosus. Arthritis Rheum. 2008;58:2453–2459. doi: 10.1002/art.23678. [DOI] [PubMed] [Google Scholar]
- 45.Wallace DJ, et al. A phase II, randomized, double-blind, placebo-controlled, dose-ranging study of belimumab in patients with active systemic lupus erythematosus. Arthritis Rheum. 2009;61:1168–1178. doi: 10.1002/art.24699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.GlaxoSmithKline. GlaxoSmithKline and Human Genome Sciences announce positive results in second of two phase 3 trials of Benlysta in systemic lupus erythematosus. 2009 [online], http://www.gsk.com/media/pressreleases/2009/2009_pressrelease_10121.htm.
- 47.Anolik JH, et al. Rituximab improves peripheral B cell abnormalities in human systemic lupus erythematosus. Arthritis Rheum. 2004;50:3580–3590. doi: 10.1002/art.20592. [DOI] [PubMed] [Google Scholar]
- 48.Wei C, et al. A new population of cells lacking expression of CD27 represents a notable component of the B cell memory compartment in systemic lupus erythematosus. J Immunol. 2007;178:6624–6633. doi: 10.4049/jimmunol.178.10.6624. [DOI] [PubMed] [Google Scholar]
- 49.Jacobi AM, et al. Activated memory B cell subsets correlate with disease activity in systemic lupus erythematosus: Delineation by expression of CD27, IgD, and CD95. Arthritis Rheum. 2008;58:1762–1773. doi: 10.1002/art.23498. [DOI] [PubMed] [Google Scholar]
- 50.Odendahl M, et al. Disturbed peripheral B lymphocyte homeostasis in systemic lupus erythematosus. J Immunol. 2000;165:5970–5979. doi: 10.4049/jimmunol.165.10.5970. [DOI] [PubMed] [Google Scholar]
- 51.Dall’Era M, et al. Reduced B lymphocyte and immunoglobulin levels after atacicept treatment in patients with systemic lupus erythematosus: results of a multicenter, phase Ib, double-blind, placebo-controlled, dose-escalating trial. Arthritis Rheum. 2007;56:4142–4150. doi: 10.1002/art.23047. [DOI] [PubMed] [Google Scholar]
- 52.Dall’Era M, Wofsy D. Systemic lupus erythematosus clinical trials—an interim analysis. Nat Rev Rheumatol. 2009;5:348–351. doi: 10.1038/nrrheum.2009.79. [DOI] [PubMed] [Google Scholar]
- 53.Ghosh D, Tsokos GC. Spleen tyrosine kinase: An Src family of non-receptor kinase has multiple functions and represents a valuable therapeutic target in the treatment of autoimmune and inflammatory diseases. Autoimmunity. 2010;43:48–55. doi: 10.3109/08916930903374717. [DOI] [PubMed] [Google Scholar]
- 54.Bahjat FR, et al. An orally bioavailable spleen tyrosine kinase inhibitor delays disease progression and prolongs survival in murine lupus. Arthritis Rheum. 2008;58:1433–1444. doi: 10.1002/art.23428. [DOI] [PubMed] [Google Scholar]
- 55.Xie C, et al. PI3K/AKT/mTOR hypersignaling in autoimmune lymphoproliferative disease engendered by the epistatic interplay of Sle1b and FASlpr. Int Immunol. 2007;19:509–522. doi: 10.1093/intimm/dxm017. [DOI] [PubMed] [Google Scholar]
- 56.Barber DF, et al. PI3Kγ inhibition blocks glomerulonephritis and extends lifespan in a mouse model of systemic lupus. Nat Med. 2005;11:933–935. doi: 10.1038/nm1291. [DOI] [PubMed] [Google Scholar]
- 57.Rommel C, Camps M, Ji H. PI3Kδ and PI3Kγ: partners in crime in inflammation in rheumatoid arthritis and beyond? Nat Rev Immunol. 2007;7:191–201. doi: 10.1038/nri2036. [DOI] [PubMed] [Google Scholar]
- 58.Li Y, et al. B cell depletion with anti-CD79 mAbs ameliorates autoimmune disease in MRL/lpr mice. J Immunol. 2008;181:2961–2972. doi: 10.4049/jimmunol.181.5.2961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Christensen SR, et al. Toll-like receptor 7 and TLR9 dictate autoantibody specificity and have opposing inflammatory and regulatory roles in a murine model of lupus. Immunity. 2006;25:417–428. doi: 10.1016/j.immuni.2006.07.013. [DOI] [PubMed] [Google Scholar]
- 60.Braun D, Caramalho I, Demengeot J. IFN-α/β enhances BCR-dependent B cell responses. Int Immunol. 2002;14:411–419. doi: 10.1093/intimm/14.4.411. [DOI] [PubMed] [Google Scholar]
- 61.Bekeredjian-Ding IB, et al. Plasmacytoid dendritic cells control TLR7 sensitivity of naive B cells via type I IFN. J Immunol. 2005;174:4043–4050. doi: 10.4049/jimmunol.174.7.4043. [DOI] [PubMed] [Google Scholar]
- 62.Giordani L, et al. IFN-α amplifies human naive B cell TLR-9-mediated activation and Ig production. J Leukoc Biol. 2009;86:261–271. doi: 10.1189/jlb.0908560. [DOI] [PubMed] [Google Scholar]
- 63.Thibault D, et al. Type I IFN receptor controls B cell expression of nucleic acid sensing toll-like receptors and autoantibody production in a murine model of lupus. Arthritis Res Ther. 2009;11:R112. doi: 10.1186/ar2771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Barrat FJ, Coffman RL. Development of TLR inhibitors for the treatment of autoimmune diseases. Immunol Rev. 2008;223:271–283. doi: 10.1111/j.1600-065X.2008.00630.x. [DOI] [PubMed] [Google Scholar]
- 65.Barrat FJ, Meeker T, Chan JH, Guiducci C, Coffman RL. Treatment of lupus-prone mice with a dual inhibitor of TLR7 and TLR9 leads to reduction of autoantibody production and amelioration of disease symptoms. Eur J Immunol. 2007;37:3582–3586. doi: 10.1002/eji.200737815. [DOI] [PubMed] [Google Scholar]
- 66.Yao Y, et al. Neutralization of interferon-α/β-inducible genes and downstream effect in a phase I trial of an anti-interferon-alpha monoclonal antibody in systemic lupus erythematosus. Arthritis Rheum. 2009;60:1785–1796. doi: 10.1002/art.24557. [DOI] [PubMed] [Google Scholar]
- 67.Lenert P, Brummel R, Field EH, Ashman RF. TLR-9 activation of marginal zone B cells in lupus mice regulates immunity through increased IL-10 production. J Clin Immunol. 2005;25:29–40. doi: 10.1007/s10875-005-0355-6. [DOI] [PubMed] [Google Scholar]
- 68.Lampropoulou V, et al. TLR-activated B cells suppress T cell-mediated autoimmunity. J Immunol. 2008;180:4763–4773. doi: 10.4049/jimmunol.180.7.4763. [DOI] [PubMed] [Google Scholar]
- 69.Hron JD, Peng SL. Type I IFN protects against murine lupus. J Immunol. 2004;173:2134–2142. doi: 10.4049/jimmunol.173.3.2134. [DOI] [PubMed] [Google Scholar]
- 70.Li J, et al. Deficiency of type I interferon contributes to SLE2-associated component lupus phenotypes. Arthritis Rheum. 2005;52:3063–3072. doi: 10.1002/art.21307. [DOI] [PubMed] [Google Scholar]
- 71.Obeng EA, et al. Proteasome inhibitors induce a terminal unfolded protein response in multiple myeloma cells. Blood. 2006;107:4907–4916. doi: 10.1182/blood-2005-08-3531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Neubert K, et al. The proteasome inhibitor bortezomib depletes plasma cells and protects mice with lupus-like disease from nephritis. Nat Med. 2008;14:748–755. doi: 10.1038/nm1763. [DOI] [PubMed] [Google Scholar]
- 73.van der Heijden JW, et al. The proteasome inhibitor bortezomib inhibits the release of NFκB-inducible cytokines and induces apoptosis of activated T cells from rheumatoid arthritis patients. Clin Exp Rheumatol. 2009;27:92–98. [PubMed] [Google Scholar]
- 74.San Miguel JF, et al. Bortezomib plus melphalan and prednisone for initial treatment of multiple myeloma. N Engl J Med. 2008;359:906–917. doi: 10.1056/NEJMoa0801479. [DOI] [PubMed] [Google Scholar]
- 75.Tian J, et al. Lipopolysaccharide-activated B cells down-regulate Th1 immunity and prevent autoimmune diabetes in nonobese diabetic mice. J Immunol. 2001;167:1081–1089. doi: 10.4049/jimmunol.167.2.1081. [DOI] [PubMed] [Google Scholar]
- 76.Fillatreau S, Sweenie CH, McGeachy MJ, Gray D, Anderton SM. B cells regulate autoimmunity by provision of IL-10. Nat Immunol. 2002;3:944–950. doi: 10.1038/ni833. [DOI] [PubMed] [Google Scholar]
- 77.Wei B, et al. Mesenteric B cells centrally inhibit CD4+ T cell colitis through interaction with regulatory T cell subsets. Proc Natl Acad Sci USA. 2005;102:2010–2015. doi: 10.1073/pnas.0409449102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zhong X, et al. Reciprocal generation of Th1/Th17 and Treg by B1 and B2 B cells. Eur J Immunol. 2007;37:2400–2404. doi: 10.1002/eji.200737296. [DOI] [PubMed] [Google Scholar]
- 79.Hu CY, et al. Treatment with CD20-specific antibody prevents and reverses autoimmune diabetes in mice. J Clin Invest. 2007;117:3857–3867. doi: 10.1172/JCI32405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Chen X, Jensen PE. Cutting Edge: primary B lymphocytes preferentially expand allogeneic FoxP3+ CD4 T cells. J Immunol. 2007;179:2046–2050. doi: 10.4049/jimmunol.179.4.2046. [DOI] [PubMed] [Google Scholar]
- 81.Evans JG, et al. Novel suppressive function of transitional 2 B cells in experimental arthritis. J Immunol. 2007;178:7868–7878. doi: 10.4049/jimmunol.178.12.7868. [DOI] [PubMed] [Google Scholar]
- 82.Yanaba K, et al. A regulatory B cell subset with a unique CD1dhiCD5+ phenotype controls T cell-dependent inflammatory responses. Immunity. 2008;28:639–650. doi: 10.1016/j.immuni.2008.03.017. [DOI] [PubMed] [Google Scholar]
- 83.Matsushita T, Yanaba K, Bouaziz JD, Fujimoto M, Tedder TF. Regulatory B cells inhibit EAE initiation in mice while other B cells promote disease progression. J Clin Invest. 2008;118:3420–3430. doi: 10.1172/JCI36030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Clatworthy MR, et al. B-cell-depleting induction therapy and acute cellular rejection. N Engl J Med. 2009;360:2683–2685. doi: 10.1056/NEJMc0808481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Dass S, Vital EM, Emery P. Development of psoriasis after B cell depletion with rituximab. Arthritis Rheum. 2007;56:2715–2718. doi: 10.1002/art.22811. [DOI] [PubMed] [Google Scholar]
- 86.Yazawa N, Hamaguchi Y, Poe JC, Tedder TF. Immunotherapy using unconjugated CD19 monoclonal antibodies in animal models for B lymphocyte malignancies and autoimmune disease. Proc Natl Acad Sci USA. 2005;102:15178–15183. doi: 10.1073/pnas.0505539102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Sawalha AH, et al. Genetic association of IL-21 polymorphisms with systemic lupus erythematosus. Ann Rheum Dis. 2008;67:458–461. doi: 10.1136/ard.2007.075424. [DOI] [PubMed] [Google Scholar]
- 88.Ettinger R, et al. IL-21 and BAFF/BLyS synergize in stimulating plasma cell differentiation from a unique population of human splenic memory B cells. J Immunol. 2007;178:2872–2882. doi: 10.4049/jimmunol.178.5.2872. [DOI] [PubMed] [Google Scholar]
- 89.Avery DT, et al. B cell-intrinsic signaling through IL-21 receptor and STAT3 is required for establishing long-lived antibody responses in humans. J Exp Med. 2010;207:155–171. doi: 10.1084/jem.20091706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Illei GG, et al. Tocilizumab in systemic lupus erythematosus: Data on safety, preliminary efficacy, and impact on circulating plasma cells from an open-label phase I dosage-escalation study. Arthritis Rheum. 2010;62:542–552. doi: 10.1002/art.27221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Anolik JH, et al. Cutting Edge: anti-tumor necrosis factor therapy in rheumatoid arthritis inhibits memory B lymphocytes via effects on lymphoid germinal centers and follicular dendritic cell networks. J Immunol. 2008;180:688–692. doi: 10.4049/jimmunol.180.2.688. [DOI] [PubMed] [Google Scholar]
- 92.Hsu HC, et al. Interleukin 17-producing T helper cells and interleukin 17 orchestrate autoreactive germinal center development in autoimmune BXD2 mice. Nat Immunol. 2008;9:166–175. doi: 10.1038/ni1552. [DOI] [PubMed] [Google Scholar]
- 93.Doreau A, et al. Interleukin 17 acts in synergy with B cell-activating factor to influence B cell biology and the pathophysiology of systemic lupus erythematosus. Nat Immunol. 2009;10:778–785. doi: 10.1038/ni.1741. [DOI] [PubMed] [Google Scholar]
- 94.Grammer AC, Lipsky PE. CD154–CD40 interactions mediate differentiation to plasma cells in healthy individuals and persons with systemic lupus erythematosus. Arthritis Rheum. 2002;46:1417–1429. doi: 10.1002/art.10287. [DOI] [PubMed] [Google Scholar]
- 95.Huang W, et al. The effect of anti-CD40 ligand antibody on B cells in human systemic lupus erythematosus. Arthritis Rheum. 2002;46:1554–1562. doi: 10.1002/art.10273. [DOI] [PubMed] [Google Scholar]
- 96.Vinuesa CG, Sanz I, Cook MC. Dysregulation of germinal centres in autoimmune disease. Nat Rev Immunol. 2009;9:845–857. doi: 10.1038/nri2637. [DOI] [PubMed] [Google Scholar]
- 97.Gaspal FMC, et al. Mice deficient in OX40 and CD30 signals lack memory antibody responses because of deficient CD4 T cell memory. J Immunol. 2005;174:3891–3896. doi: 10.4049/jimmunol.174.7.3891. [DOI] [PubMed] [Google Scholar]
- 98.Murdoch C. CXCR4: chemokine receptor extraordinaire. Immunol Rev. 2000;177:175–184. doi: 10.1034/j.1600-065x.2000.17715.x. [DOI] [PubMed] [Google Scholar]
- 99.Nanki T, et al. Chemokine receptor expression and functional effects of chemokines on B cells: implication in the pathogenesis of rheumatoid arthritis. Arthritis Res Ther. 2009;11:R149. doi: 10.1186/ar2823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Linterman MA, et al. Follicular helper T cells are required for systemic autoimmunity. J Exp Med. 2009;206:561–576. doi: 10.1084/jem.20081886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Zheng B, et al. CXCL13 neutralization reduces the severity of collagen-induced arthritis. Arthritis Rheum. 2005;52:620–626. doi: 10.1002/art.20768. [DOI] [PubMed] [Google Scholar]
- 102.Yellin M, et al. A phase II, randomized, double-blind, placebo-controlled study to evaluate the efficacy and safety of MDX-1100, a fully human anti-CXCL10 monoclonal antibody, in combination with methotrexate (MTX) in patients with rheumatoid arthritis (RA) [abstract 414] Arthritis Rheum. 2009;60 (Suppl):S153. doi: 10.1002/art.34330. [DOI] [PubMed] [Google Scholar]
- 103.Genovese MC, et al. Efficacy and safety of baminercept in the treatment of rheumatoid arthritis (RA)—results of the phase 2B study in the TNF-IR population [abstract 417] Arthritis Rheum. 2009;60 (Suppl):S154. [Google Scholar]
- 104.Palanichamy A, et al. Novel human transitional B cell populations revealed by B cell depletion therapy. J Immunol. 2009;182:5982–5993. doi: 10.4049/jimmunol.0801859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Ngo VN, et al. Lymphotoxin α/β and tumor necrosis factor are required for stromal cell expression of homing chemokines in B and T cell areas of the spleen. J Exp Med. 1999;189:403–412. doi: 10.1084/jem.189.2.403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Allen CDC, Okada T, Cyster JG. Germinal-center organization and cellular dynamics. Immunity. 2007;27:190–202. doi: 10.1016/j.immuni.2007.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.