

Abstract
Pathogenic T helper cells and myeloid cells are involved in the pathogenesis of Multiple Sclerosis (MS) and Experimental Autoimmune Encephalomyelitis (EAE), an animal model of MS. The JAK/STAT pathway is utilized by numerous cytokines for signaling, and is critical for development, regulation and termination of immune responses. Dysregulation of the JAK/STAT pathway has pathological implications in autoimmune and neuroinflammatory diseases. Many of the cytokines involved in MS/EAE, including IL-6, IL-12, IL-23, IFN-γ and GM-CSF, use the JAK/STAT pathway to induce biological responses. Thus, targeting JAKs has implications for treating autoimmune inflammation of the brain. We have utilized AZD1480, a JAK1/2 inhibitor, to investigate the therapeutic potential of inhibiting the JAK/STAT pathway in models of EAE. AZD1480 treatment inhibits disease severity in MOG-induced classical and atypical EAE models by preventing entry of immune cells into the brain, suppressing differentiation of Th1 and Th17 cells, deactivating myeloid cells, inhibiting STAT activation in the brain, and reducing expression of pro-inflammatory cytokines and chemokines. Treatment of SJL/J mice with AZD1480 delays disease onset of PLP-induced relapsing-remitting disease, reduces relapses and diminishes clinical severity. AZD1480 treatment was also effective in reducing ongoing paralysis induced by adoptive transfer of either pathogenic Th1 or Th17 cells. In vivo AZD1480 treatment impairs both the priming and expansion of T-cells, and attenuates antigen-presentation functions of myeloid cells. Inhibition of the JAK/STAT pathway has clinical efficacy in multiple pre-clinical models of MS, suggesting the feasibility of the JAK/STAT pathway as a target for neuroinflammatory diseases.
Introduction
Multiple Sclerosis (MS) is an autoimmune disease of the central nervous system (CNS) characterized by demyelination, inflammatory lesions, axonal damage, activation of IFN-γ-producing Th1 cells and IL-17-producing Th17 cells, inappropriate activation of innate immune cells (macrophages, dendritic cells (DCs), neutrophils, microglia), and aberrant production of cytokines/chemokines (1, 2). Th1 cells, Th17 cells and innate immune cells are also implicated in Experimental Autoimmune Encephalomyelitis (EAE), an animal model of MS (3, 4). The pathogenesis of MS and EAE is associated with the overexpression of cytokines including IL-12, IFN-γ IL-6, IL-21 and IL-23, which function in part to promote differentiation of effector Th1 and Th17 cells (1, 3, 5, 6).
The JAK/STAT signaling pathway is utilized by numerous cytokines, and is critical for initiating innate immunity, orchestrating adaptive immunity, and ultimately constraining immune responses (7). Cytokines are of paramount importance in regulating the development, differentiation and function of myeloid cells and T-cells (8, 9), thus, unrestrained activation of the JAK/STAT pathway has pathological implications for autoimmune diseases (7, 10, 11). In MS and EAE, there is evidence for aberrant functionality of the JAK/STAT pathway. T-cells and monocytes from MS patients during relapse have elevated levels of activated STAT3 compared to cells from patients in remission (12), and high levels of activated STAT3 in T-cells from patients with clinically isolated syndrome predict conversion to clinically defined MS (13). In EAE, IL-6 has a deleterious role by activation of STAT3, which is pivotal for induction of pathogenic Th17 cells (14-16). Loss of STAT3 in T-cells renders mice resistant to EAE disease (17, 18). STAT target genes, including IL-23R, IL-6, IL-17A and IL-17F, are implicated in contributing to MS and EAE. The JAK/STAT pathway has received attention as a therapeutic target in autoimmune diseases and cancers (7, 11). JAK inhibitors have demonstrated clinical efficacy in rheumatoid arthritis and other inflammatory diseases (19-21). Indeed, Bright et al., previously demonstrated that tyrphostin B42, a JAK2 inhibitor, reduced severity of EAE (22). JAK inhibitors interrupt signaling downstream of multiple cytokines, a useful approach for EAE and MS, which are characterized by a “cytokine storm” in the periphery and CNS. Simultaneous inhibition of cytokine signaling by JAK inhibitors may break the cycle of inflammation characteristic of neuroinflammatory diseases.
AZD1480, an ATP competitive inhibitor of JAK1 and JAK2, has beneficial effects in cancer models by suppressing downstream activation of STATs, particularly STAT3 (23, 24). We demonstrate that AZD1480 is effective in suppressing clinical symptoms in five pre-clinical models of MS. AZD1480 treatment was associated with diminished STAT activation in the CNS, reduced pathogenic Th1 and Th17 cell responses, alterations in DC and macrophage functionality, decreased infiltration of immune cells into the CNS, reduced demyelination and suppression of pro-inflammatory cytokine/chemokine expression in vivo. Using AZD1480 for proof-of-principle, we demonstrate that inhibiting the JAK/STAT pathway has striking clinical efficacy in multiple models of EAE.
Materials and Methods
Mice
C57BL/6 and MOG35-55-T-cell receptor transgenic 2D2 mice (25) were bred in the animal facility at the University of Alabama at Birmingham (UAB). SJL/J mice were purchased from NCI Frederick National Lab (Frederick, MD). SOCS3 floxed transgenic (SOCS3fl/fl) mice (26) were the generous gift of Dr. Warren Alexander (Walter and Eliza Hall Institute of Medical Research, Victoria, Australia), and were bred at UAB. SOCS3 conditional knockout (LysMCre-SOCS3fl/fl) mice were generated by serial breeding of SOCS3fl/fl mice with mice expressing Cre-recombinase under the control of the LysM promoter (27). All experiments were reviewed and approved by the institutional animal care and use committee of UAB.
JAK Inhibitor
AZD1480 (Supplementary Fig. 1), a JAK1/JAK2 inhibitor, was synthesized and provided by AstraZeneca R&D (Waltham, Massachusetts), and re-suspended in DMSO, as previously described (23, 24).
Peptides, Antibodies, and Cytokines
MOG35-55 peptide was synthesized by New England Peptide, and PLP139-151 peptide was from CPC Scientific. LPS was from Sigma-Aldrich. Neutralizing antibodies (Abs) to IL-4 and IFN-γ, and Abs against mouse CD3, B220, Gr-1, CD4, IFN-γ and IL-17A, and human CD3, CD4, CD14 and HLA-DR used for flow cytometry are from eBioscience. Mouse IL-6, IFN-γ, IL-12, IL-23, and GM-CSF, and human IFN-γ and TGF-β1 are from R&D Systems. Abs against mouse and human CD3 and CD28 are from BioLegend. Abs against phospho-STAT1 (Tyr701) and phospho-STAT3 (Tyr705) used for flow cytometry are from Cell Signaling Technology, and phospho-STAT4 (Tyr693) Ab is from BD Biosciences. Abs against phospho-STAT1 (Tyr701), phospho-STAT3 (Tyr705), phospho-STAT4 (Tyr693), phospho-STAT5 (Tyr694), phospho-STAT6 (Tyr641), phospho-p65 (Ser536), phospho-JAK2 (Tyr221), STAT1, STAT3, STAT4, STAT5, STAT6, p65 and JAK2 used for immunoblotting are from Cell Signaling Technology. Ab against GAPDH is from Abcam, and Abs against Ly-6C, CD11c, MHC class II, CD40, CD80, CD86, CD11b and CD45 are from BD Pharmingen.
EAE Induction, Assessment and Treatment with AZD1480
Active and adoptive transfer EAE was induced as previously described (27, 28). Eight-12 week old C57BL/6 or LysMCre-SOCS3fl/fl mice were immunized s.c. with 200 μg of MOG35-55 emulsified in CFA (supplemented with 2 mg/ml of Mycobacterium tuberculosis) and injected i.p. on days 0 and 2 with 500 ng pertussis toxin. For adoptive transfer EAE, C57BL/6 mice were immunized with MOG35-55 for 10 days, then splenic and axillary lymph node cells were isolated and re-stimulated with MOG35–55 (10 μg/ml) and 10 ng/ml of IL-12 for Th1 cell differentiation, or with 20 ng/ml of IL-6, 20 ng/ml of IL-23, and 3 ng/ml of TGF-β1 for Th17 cell differentiation for 3 days. Thirty × 106 cells were transferred into healthy recipients. The frequency of donor CD4+ T-cells producing IL-17A or IFN-γ was assessed by fluorescence activated cell sorting (FACS) prior to transfer (data not shown). Relapsing-remitting (RR) EAE was induced as follows: female SJL/J mice (8-12 week old) were immunized with a s.c. injection of 150 μg of PLP139–151 peptide emulsified in CFA (29). Assessment of classical EAE was as follows: 0, no disease; 1, decreased tail tone; 2, hind limb weakness or partial paralysis; 3, complete hind limb paralysis; 4, front and hind limb paralysis; and 5, moribund state. Assessment of atypical EAE was done using the scoring system as described previously (27): 0, no disease; 1, hunched appearance, slight head tilt; 2, ataxia, scruffy coat; 3, severe head tilt, slight axial rotation, staggered walking; 4, severe axial rotation, spinning; and 5, moribund. For treatment of mice with active classical EAE, AZD1480 was administered at 25 mg/kg or DMSO (Sigma-Aldrich) as vehicle control i.p. daily from day 9 post-immunization for 10 days, or when disease score reached 1.5 for 12 days, and for atypical active EAE at day 5 post-immunization. For treatment of adoptive transfer EAE, AZD1480 was administered at 25 mg/kg or DMSO as vehicle control i.p. daily from day 7 (Th1) or day 8 (Th17) post-immunization for 10 days. For treatment of RR EAE, mice were administered AZD1480 (25 mg/kg) or vehicle control by oral gavage daily from day 7 post-immunization for 10 days, and again at day 60 for 10 days. Mice were weighed and examined daily for disease symptoms. Mice were killed, perfused, and mononuclear cells were isolated from the CNS (cerebellum and spinal cord) by a 30/70% Percoll gradient, or from draining cervical lymph nodes and spleen as previously described (27), and cell phenotype determined by surface and intracellular staining by flow cytometry.
Cell Preparation
Bone marrow cells were cultured with RPMI-1640 medium containing 10% (vol/vol) FBS and 20 ng/ml of mouse M-CSF for 7 days to obtain BMDMs (30). Naïve CD4+ T-cells were obtained from spleen or lymph nodes of 8- to 12-week-old mice and isolated by Dynabeads® CD4 Positive Isolation Kit (Invitrogen). T-cells were cultured in RPMI-1640 medium supplemented with 10% (vol/vol) FBS, 2 mM glutamine, 100 IU/ml penicillin, 0.1 mg/ml streptomycin, 10 mM HEPES, 1 mM sodium pyruvate and nonessential amino acids, and 2 μM β-mercaptoethanol.
Human Subjects
Cryopreserved peripheral blood mononuclear cells (PBMC) obtained by standard Histopaque density centrifugation from healthy donors were used in this study. These subjects were recruited from the Alabama Vaccine Research Clinic at the University of Alabama at Birmingham. To obtain monocytes, PBMC (3 × 106 cells/ml) were incubated in serum-free medium (RPMI-1640) supplemented with 2 mM glutamine, 100 IU/ml penicillin, 0.1 mg/ml streptomycin and 10 mM HEPES for 3 h at 37°C. Adherent cells were obtained by washing four times with pre-warmed serum-free medium. For T-cells, non-adherent PBMC were collected after the 3 h period of plastic adherence, and washed three times with serum-free medium. Written informed consent was obtained from all donors who participated in this study. The Institutional Review Board of the University of Alabama at Birmingham approved the study (IRB#X090708004).
Stimulation of Human PBMC
For T cell stimulation, non-adherent PBMC were stimulated in vitro with anti-CD3 (5 μg/ml) and anti-CD28 (2 μg/ml) Abs in the absence or presence of AZD1480 (0.25 and 0.5 μM) for 4 days, and tyrosine phosphorylation of STAT1, STAT3, and STAT4 was examined in gated CD3+CD4+ T-cells. For human monocytes, adherent PBMCs were pretreated in the absence or presence of AZD1480 (0.25 and 0.5 μM) for 2 h, then stimulated with IFN-γ (100 U/ml) for 1 h. The extent of STAT1 and STAT3 tyrosine phosphorylation within the CD3-CD14+ monocyte population was assessed by intracellular flow cytometry.
In Vitro T Helper Cell Differentiation
Purified murine CD4+CD25- T-cells were stimulated with antibodies to CD3 (1 μg/ml) and CD28 (1 μg/ml) under Th1 cell differentiation conditions (10 ng/ml IL-12 and 10 μg/ml anti-IL-4), and Th17 cell differentiation conditions (20 ng/ml IL-6, 10 ng/ml IL-23, 5 ng/ml TGF-β1, 10 μg/ml anti-IL-4, and 10 μg/ml anti-IFN-γ) in the presence of vehicle control (DMSO) or AZD1480. For antigen specific differentiation, spleen and lymph nodes cells from MOG35–55 immunized mice were restimulated with MOG35–55 (10 μg/ml) for 3 days in Th1 or Th17 cell differentiation conditio ns in the absence or presence of AZD1480.
Intracellular Staining
Cells were stimulated for 4 h with PMA/Ionomycin (25 ng/ml and 1 μg/ml) plus GolgiStop (BD Pharmingen), and analyzed for intracellular production of cytokines by staining with anti-cytokine Abs and subsequent flow cytometry, as described (27). All flow data shown are representative of three individual experiments.
Immunohistology Analysis
Mice were killed and cerebellum and spinal cord immersion fixed in Bouin's fixative and paraffin embedded. Sections were stained with H&E and Luxol Fast Blue (LFB). Digital images were captured with a Zeiss Axiovision software. Images were collected at the same time using identical settings with respect to image exposure time and image compensation settings as described (27).
RNA Isolation, RT-PCR, and TaqMan Gene Expression Assays
Total RNA was isolated from the cerebellum and spinal cord of mice, and RT reactions performed as described (27). Five hundred nanograms of RNA was used to reverse transcribe into cDNA and subjected to qRT-PCR. The data were analyzed using the comparative Ct method to obtain relative quantitation values.
Immunoblotting
Thirty micrograms of cell lysate or brain homogenate was separated by electrophoresis and probed with antibodies as described previously (27). All immunoblots are representative of three individual experiments.
Cell Proliferation Assay In Vitro and In Vivo
For in vitro proliferation, CFSE labeled CD4+CD25- T-cells were stimulated with anti-CD3 (5 μg/ml) and anti-CD28 (2 μg/ml) Abs, and cell proliferation was assessed by CFSE dilution detected by flow-cytometric analysis 72 h post-stimulation. For in vivo proliferation, C57BL/6 mice were immunized with MOG35-55 (200 μg), and then administrated vehicle or AZD1480 (25 mg/kg) at day 7 post-immunization. Mice were then injected with 5-ethynyl-2′-deoxyuridine (EdU) on day 13. CNS-infiltrating mononuclear cells were isolated from the cerebellum and spinal cord 12 h later, and EdU incorporation was detected for proliferation.
Statistical Analysis
Levels of significance for comparison between two groups were determined by one-sided two-sample Mann-Whitney rank sum test and the Student's t-test distribution. A value of p<0.05 was considered statistically significant.
Results
AZD1480 Inhibits Th1 and Th17 Cell Differentiation In Vitro
AZD1480 potently inhibits the JAK/STAT pathway in tumor cells (23, 24), but has not been evaluated in primary immune cells. AZD1480 at 0.25 μM partially inhibits STAT1 and STAT4 tyrosine phosphorylation in T-cells cultured in Th1 cell polarizing conditions (Fig. 1A). The differentiation of naïve T-cells to Th1 cells was strongly inhibited by AZD1480 treatment, as assessed by decreased IFN-γ production, and decreased mRNA levels of IFN-γ and T-bet (Fig. 1B). As some of the key cytokines for Th17 differentiation, including IL-6 and IL-23, signal through JAK1/2, we investigated the influence of inhibiting the JAK/STAT pathway on Th17 cell polarization. Using the Th17 cell differentiation cocktail of IL-6, IL-23 and TGF-β, we observed that treatment with AZD1480 partially inhibited STAT3 tyrosine phosphorylation (Fig. 1C), which led to reduced differentiation of naïve T-cells to Th17 cells (Fig. 1D). AZD1480 treatment also inhibited mRNA levels of the STAT3 target genes IL-17A, RORγt, IL-22 and IL-23R (Fig. 1D). Th17 cells generated in the absence of TGF-β (with IL-6 + IL-23 or IL-6 + IL-23 + IL-1β), so called Th17(23) cells, are more pathogenic in vivo (31). The differentiation of naïve T-cells to Th17 cells in the presence of IL-6 + IL-23(Supplementary Fig. 2A) or IL-6 + IL-23 + IL-1β (Supplementary Fig. 2B) was also substantially inhibited by AZD1480 treatment. Furthermore, the expression of STAT3 target genes under those differentiation conditions was inhibited by AZD1480 (Supplementary Fig. 2). These results clearly indicate that inhibition of the JAK/STAT pathway in T-cells suppresses differentiation of Th1 and Th17 cells.
Figure 1. Inhibition of the JAK/STAT Pathway Suppresses Th1 and Th17 Cell Differentiation In Vitro.
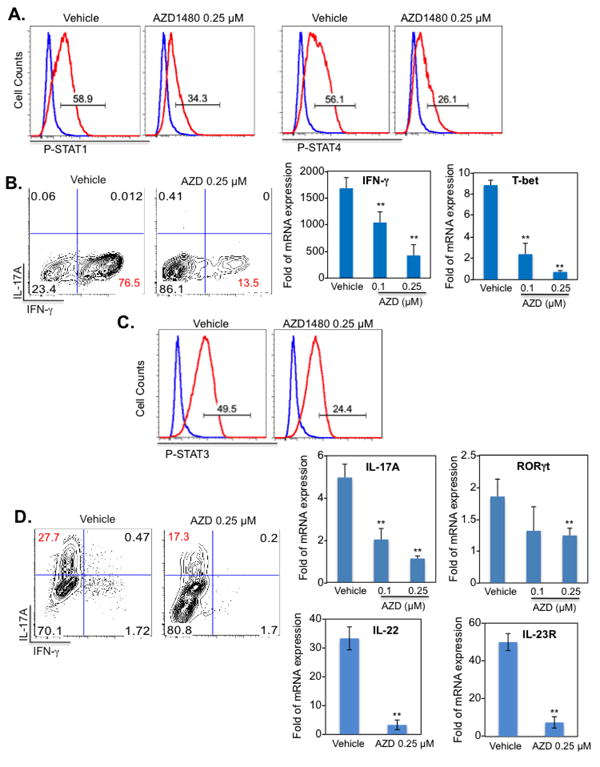
Naïve CD4+ T-cells were isolated from the spleen of C57BL/6 mice. Dendritic cells (DCs) were used as antigen-presenting cells at a 1:5 ratio to CD4+ T-cells in co-culture. Vehicle control or AZD1480 (0.25 μM) was added simultaneously to the Th1 or Th17 differentiation cocktail. (A). Th1 cells were differentiated with anti-CD3 (1 μg/ml), anti-CD28 (1 μg/ml), IL-12 (10 ng/ml) plus anti-IL-4 Ab (10 μg/ml) for 4 days. STAT1 and STAT4 tyrosine phosphorylation was examined by flow cytometry (red line) with isotype control shown in blue. (B). At day 4, cells were stimulated with PMA/Ionomycin plus GolgiStop for 4 h, stained for the surface marker CD4 and by intracellular flow to detect IFN-γ expression. For mRNA expression, cells were collected at day 4, and mRNA was analyzed by qRT-PCR for IFN-γ and T-bet. (C). Th17 cells were differentiated with anti-CD3 (1 μg/ml), anti-CD28 (1 μg/ml), TGF-β (5 ng/ml), IL-6 (20 ng/ml), IL-23 (10 ng/ml) plus anti-IFN-γ Ab (10 μg/ml) and anti-IL-4 Ab (10 μg/ml) for 4 days. STAT3 tyrosine phosphorylation (red line) was examined by flow cytometry, with isotype control shown in blue. (D). At day 4, cells were stimulated with PMA/Ionomycin plus GolgiStop for 4 h, stained for the surface marker CD4 and by intracellular flow to detect IL-17A expression. For mRNA expression, cells were collected at day 4, and mRNA was analyzed by qRT-PCR for IL-17A, RORγt, IL-22 and IL-23R. *p<0.05 and **p<0.001.
To assess whether the inhibitory effect of AZD1480 was a direct or indirect effect, short-term experiments were performed. As shown in Supplementary Fig. 3A and 3B, AZD1480 inhibited the phosphorylation of STAT1, JAK2, STAT3 and STAT5 in naïve T-cells, suggesting a direct inhibitory effect on the JAK/STAT pathway.
AZD1480 Blocks JAK/STAT Activation and Gene Expression in Macrophages and Dendritic Cells
Many cytokines important in activating macrophages and DCs, including IFN-γ and GM-CSF, utilize the JAK/STAT pathway for signaling (7, 30). Bone marrow-derived macrophages (BMDM) were stimulated with IFN-γ, a key cytokine in polarizing macrophages to the pro-inflammatory M1 phenotype (30, 32). IFN-γ stimulation led to strong STAT1 and STAT3 tyrosine phosphorylation, which was inhibited by AZD1480 treatment (Fig. 2A). GM-CSF, another important cytokine in polarization of M1 macrophages and inflammatory DCs (32, 33), induced STAT5 tyrosine phosphorylation, and AZD1480 inhibited this response (Fig. 2B). To examine the effect of AZD1480 on upstream JAK activation, BMDM were stimulated with IL-6/sIL-6R to activate JAK2 and STAT3. AZD1480 pretreatment inhibited phosphorylation of both JAK2 and STAT3 (Fig. 2C). We examined the influence of AZD1480 on IL-4 signaling, which through activation of STAT6, induces polarization of macrophages to the M2 anti-inflammatory phenotype (34). IL-4 activation of STAT6 was largely unaffected by AZD1480 (Supplementary Fig. 3C), indicating that there are selective effects of AZD1480 on different STATs, dependent upon cytokine stimulation. Interestingly, when BMDM were stimulated with LPS, rapid activation of the NF-κB pathway was not affected, while delayed activation of STAT1 and STAT3 at 2-4 h (via IFN induction) was inhibited (Fig. 2D), indicating the specificity of AZD1480 treatment.
Figure 2. AZD1480 Blocks JAK/STAT Activation and Gene Expression in Macrophages and DCs.
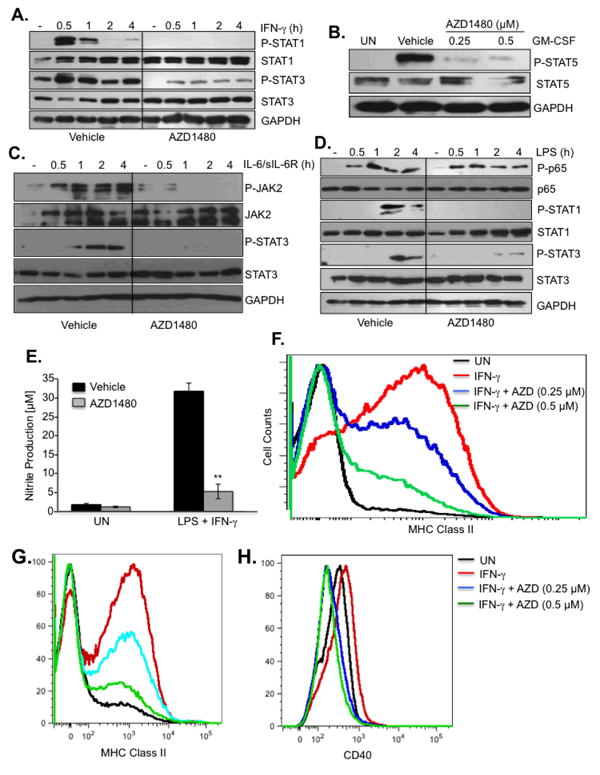
(A). BMDMs were treated with Vehicle Control or AZD1480 (0.25 μM) for 2 h, stimulated with medium (-) or IFN-γ (10 ng/ml) for up to 4 h, then cell lysates were subjected to immunoblotting with the indicated antibodies. (B). BMDMs were treated with Vehicle Control or AZD1480 (0.25 and 0.5 μM) for 2 h, stimulated with medium (-) or GM-CSF (10 ng/ml) for 30 min, then cell lysates were subjected to immunoblotting with the indicated antibodies. (C). BMDMs were treated with Vehicle or AZD1480 (0.25 μM) for 2 h, and stimulated with medium (-) or IL-6 (10 ng/ml) plus sIL-6R (25 ng/ml) for up to 4 h. Cell lysates were subjected to immunoblotting with the indicated antibodies. (D). BMDMs were treated with Vehicle Control or AZD1480 (0.25 μM) for 2 h, stimulated with medium (-) or LPS (10 ng/ml) for up to 4 h, then cell lysates were subjected to immunoblotting with the indicated antibodies. (E). BMDMs were treated with Vehicle Control or AZD1480 (0.25 μM) for 2 h, stimulated with medium (UN) or LPS (10 ng/ml) plus IFN-γ (10 ng/ml) for 24 h, and nitrite production was measured by Griess. (F). BMDMs were treated with Vehicle Control or AZD1480 (0.25 and 0.5 μM) for 2 h, stimulated with medium (UN) or IFN-γ (10 ng/ml) for 24 h, then MHC Class II expression analyzed by flow cytometry. DCs were treated with Vehicle Control or AZD1480 (0.25 and 0.5 μM) for 2 h, stimulated with medium (UN) or IFN-γ (10 ng/ml) for 24 h, then MHC Class II (G) and CD40 (H) expression was analyzed by flow cytometry. **p<0.001.
Nitric oxide (NO) is typically produced by M1 macrophages upon triggering of IFN-γ and TLR pathways (30). The expression of NO and its reactive derivative peroxynitrite has been implicated in the pathogenesis of MS and EAE (35, 36). Pretreatment of BMDM with AZD1480 significantly inhibited LPS/IFN-γ-induced production of nitrite (Fig. 2E). IFN-γ inducible MHC class II expression is mediated by activation of STAT1 (37). AZD1480, in a dose-dependent manner, inhibited MHC class II expression in BMDM (Fig. 2F) and DCs (Fig. 2G). IFN-γ-inducible CD40 co-stimulatory molecule expression on DCs was also inhibited by AZD1480 (Fig. 2H). This suggests that AZD1480 may regulate the antigen presentation capability of these cells. Taken together, these findings indicate that AZD1480 inhibits JAK/STAT signaling in macrophages and DCs.
Inhibition of the JAK/STAT Pathway Suppresses Classical, Atypical, and Relapsing-Remitting EAE
Given the striking effect of AZD1480 in inhibiting Th1 and Th17 cell differentiation, as well as blocking IFN-γ-inducible gene expression in myeloid cells, we assessed the therapeutic potential of AZD1480 in EAE. First, we evaluated the effect of AZD1480 in naïve mice by analyzing proliferation of naïve CD4+ T-cells and hematological parameters. Dosage was determined based on previous studies (23, 24), and on preliminary dose response experiments (data not shown). No significant differences were noted in the proliferation of CD4+ T-cells stimulated with anti-CD3 and anti-CD28, as well as in any hematological parameters in mice administrated AZD1480 (Figs. 3A and 3B). These results demonstrate that in vivo AZD1480 treatment does not induce toxicity when assessing circulating populations of the major cell types, consistent with studies using other JAK inhibitors, which are well-tolerated in vivo (21, 38).
Figure 3. Effect of AZD1480 on CD4+ T-cell Proliferation and Hematologic Parameters.
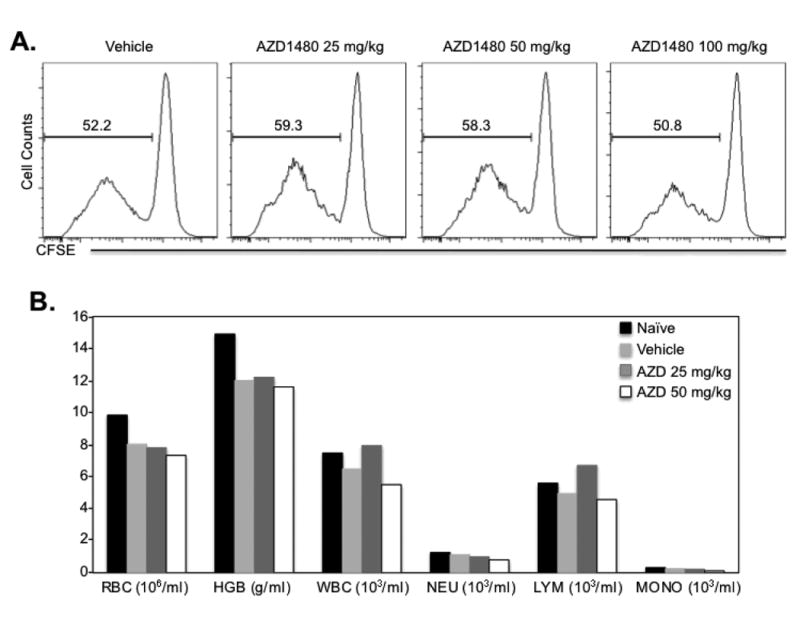
(A). C57BL/6 mice were injected i.p. with Vehicle Control (DMSO) or AZD1480 (25 mg/kg, 50 mg/kg or 100 mg/kg) for 4 days. Näive CD4+ T-cells isolated from the spleen of Vehicle Control or AZD1480 treated mice were labeled with carboxyfluorescein succinimidyl ester (CFSE), and stimulated with anti-CD3 (5 μg/ml) and anti-CD28 (2 μg/ml) Abs for 4 days. T-cell proliferation was analyzed by flow cytometry based on the dilution of CFSE intensity. (B). Peripheral blood from Vehicle Control or AZD1480 (25 mg/kg or 50 mg/kg) treated mice (n=3) was analyzed with HEMAVET®950. The number of red blood cells, hemoglobin, white blood cells, neutrophils, lymphocytes and monocytes were calculated.
C57BL/6 mice were immunized with MOG35-55 peptide to induce classical EAE, and AZD1480 (25 mg/kg) or vehicle control was administrated i.p. daily for 10 days starting at the onset of disease on day 9. Animals were scored daily for signs of ascending motor paralysis graded on a scale of 0-5. Most strikingly, AZD1480 treatment resulted in a significant reduction in disease severity compared to vehicle treatment (Fig. 4A). Evaluation of the spinal cord revealed strong inhibition of STAT3 and STAT4 tyrosine phosphorylation, and a modest reduction in expression of STAT3 and STAT4 protein was also observed with AZD1480 treatment (Fig. 4B). Concordant with disease attenuation, the absolute number of mononuclear cells in the spinal cord was markedly decreased in AZD1480 treated mice, and the absolute numbers of neutrophils, monocytes, macrophages, microglia, DCs, CD4+ T-cells and B-cells were significantly reduced in the spinal cord (Fig. 4C). There was also a marked reduction of IFN-γ, T-bet and IL-17A mRNA levels (Fig. 4D), indicating an inhibitory effect on Th1 and Th17 cell entry into the CNS. AZD1480 treatment also inhibited mRNA levels of numerous pro-inflammatory cytokines and chemokines in the spinal cord (Fig. 4D). Histological characterization of the spinal cord revealed less inflammatory infiltrates and less demyelination in the AZD1480 treated group (Fig. 4E). To investigate whether AZD1480 treatment influenced mice with established active EAE, AZD1480 (25 mg/kg) or vehicle control was administrated to C57BL/6 mice immunized with MOG35-55 after they developed a clinical score of 1.5 (Fig. 4F). This therapeutic administration of AZD1480 resulted in significant reduction of ongoing disease severity (Fig. 4F).
Figure 4. The JAK1/2 Inhibitor AZD1480 Suppresses Classical EAE.
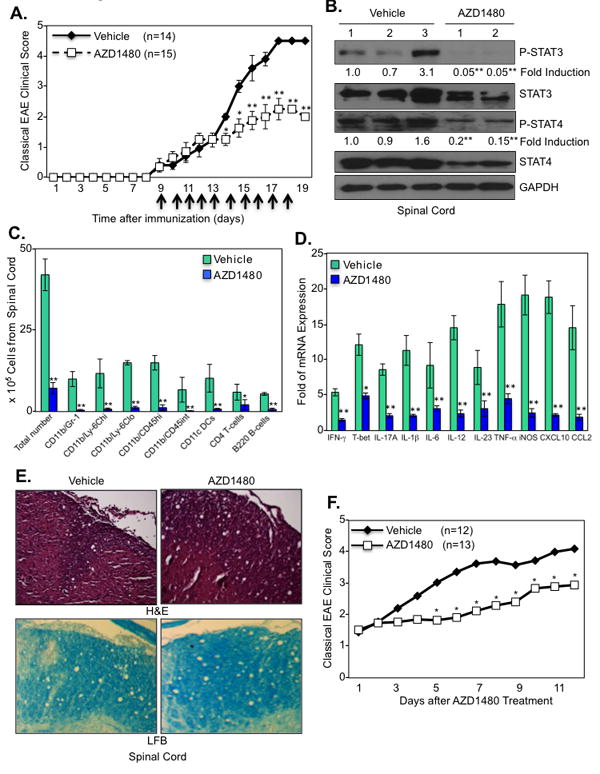
(A). C57BL/6 mice were immunized with MOG35-55 peptide (200 μg) emulsified in CFA containing M. tuberculosis. The mice received i.p. injections of 250 ng of pertussis toxin on days 0 and 2. Vehicle Control (0.1% DMSO) (n=14) or AZD1480 (25 mg/kg) (n=15) was administrated i.p. daily for 10 days starting at the onset of EAE (day 9). Mean ± S.D. of classical EAE clinical scores. (B). Protein extracts from the spinal cord of Vehicle Control or AZD1480 treated mice at day 16 were immunoblotted with the indicated antibodies. (C). CNS-infiltrating mononuclear cells were isolated from the spinal cord of Vehicle Control or AZD1480 treated mice at day 16. Cells were stained with trypan blue and counted. Cells were stained with Abs to CD4, CD11b, Gr-1, CD45, CD11c, Ly-6C and B220, and the percentage of CD11b+/Gr-1+ neutrophils, CD11b+/Ly-6Chi and CD11b+/Ly-6Clo monocytes, CD11b+/CD45hi macrophages, CD11b+/CD45int microglia, CD11c+ dendritic cells, CD4+ T-cells and B220+ B-cells were gated. The absolute number of cells is shown. (D). Mice were perfused, spinal cord removed, and mRNA from spinal cord of Vehicle Control or AZD1480 treated mice at day 16 was analyzed by qRT-PCR. (E). Sections from the spinal cord of Vehicle Control or AZD1480 treated mice at day 16 were stained with H&E for inflammatory infiltrates and luxol fast blue (LFB) for demyelination. (F). C57BL/6 mice were immunized with MOG35-55 peptide (200 μg) emulsified in CFA containing M. tuberculosis. The mice received i.p. injections of 250 ng of pertussis toxin on days 0 and 2. Vehicle Control (0.1% DMSO) (n=12) or AZD1480 (25 mg/kg) (n=13) was administrated i.p. after mice reached a clinical score of ∼1.5 for up to 12 days. *p<0.05 and **p<0.001.
We previously demonstrated that mice with conditional knockout of Suppressor Of Cytokine Signaling 3 (SOCS3) in cells of the myeloid lineage (LysMCre-SOCS3fl/fl mice) develop early onset of a severe and non-resolving disease with features of atypical EAE, which is associated with hyperactivation of the JAK/STAT signaling pathway in the CNS (27). Thus, we evaluated the effect of inhibiting the JAK/STAT pathway in this EAE model. AZD1480 treated mice had delayed onset of disease and significantly reduced severity of atypical EAE disease (Fig. 5A). In a separate experiment, after treatment was stopped at day 14, the protective effect of AZD1480 was observed until day 22, at which point clinical scores started to increase (Supplementary Fig. 4). AZD1480 treatment prevented activation of STAT1 and STAT3 in the cerebellum, and decreased total levels of STAT1 (Fig. 5B). Decreased infiltration of neutrophils, monocytes, microglia, DCs and B-cells in the cerebellum was observed (Fig. 5C), with reduced expression of Th1 markers (IFN-γ and T-bet) and Th17 markers (IL-17A), and reduced expression of cytokines/chemokines in the cerebellum comparable to unimmunized mice (UN) (Fig. 5D). Decreased inflammation and demyelination was observed in the cerebellum of AZD1480 treated mice compared to control mice (Fig. 5E). Oral gavage of AZD1480 produced a comparable protective effect in both classical and atypical EAE as i.p. treatment (data not shown). Collectively, these results indicate that AZD1480 has the desired, selective effect on its target (STATs) in the tissue region of interest (the brain), and that suppression of STAT activation by AZD1480 has a protective effect in classical and atypical EAE models.
Figure 5. The JAK1/2 Inhibitor AZD1480 Suppresses Development of Atypical EAE.
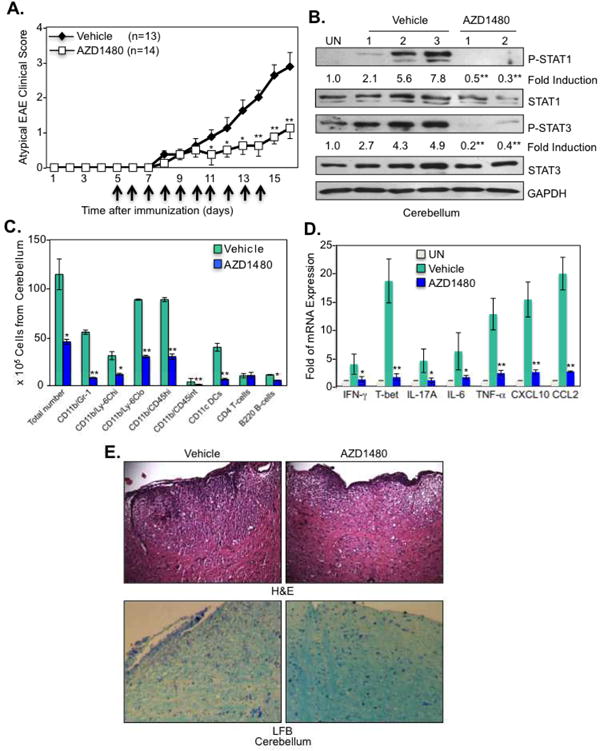
(A). LysMCre-SOCS3fl/fl mice were immunized with MOG35-55 peptide (200 μg) emulsified in CFA containing M. tuberculosis. The mice received i.p. injections of 250 ng of pertussis toxin at days 0 and 2. Vehicle Control (n=13) or AZD1480 (25 mg/kg) (n=14) was administrated i.p. daily for 10 days starting at day 5 post-immunization. Mean ± S.D. of atypical EAE clinical scores. (B). Mice were perfused, cerebellum removed, and protein extracts from unimmunized (UN) or MOG35-55-immunized Vehicle Control or AZD1480 treated mice at day 14 were immunoblotted with the indicated antibodies. (C). CNS-infiltrating mononuclear cells were isolated from the cerebellum of Vehicle Control or AZD1480 treated mice at day 14 after MOG immunization. Cells were stained with Abs to CD4, CD11b, Gr-1, CD45, CD11c, Ly-6C and B220, and the percentage of CD11b+/Gr-1+ neutrophils, CD11b+/Ly-6Chi and CD11b+/Ly-6Clo monocytes, CD11b+/CD45hi macrophages, CD11b+/CD45int microglia, CD11c+ dendritic cells, CD4+ T-cells and B220+ B-cells were gated. The absolute number of cells is shown. (D). Mice were perfused, cerebellum removed, and mRNA was analyzed by qRT-PCR. (E). Sections from the cerebellum of Vehicle Control or AZD1480 treated mice at day 14 were stained with H&E for inflammatory infiltrates and LFB for demyelination. *p<0.05 and **p<0.001.
To determine whether the protective effect of AZD1480 was specific to the MOG-C57BL/6 EAE model, we examined the RR model of EAE induced in SJL/J mice by PLP139-151. We observed that disease onset was significantly delayed with the first AZD1480 treatment at day 7, with no clinical symptoms until ∼day 23 (Fig. 6A). A second AZD1480 treatment at day 60 inhibited further relapse (Fig. 6A). Examination of spinal cord revealed inhibition of STAT1 and STAT3 tyrosine phosphorylation, and a reduction in expression levels of STAT3 protein (Fig. 6B). AZD1480 treatment also inhibited IL-6, IL-12p40 and IL-23 p19 mRNA expression in the spinal cord (Fig. 6C).
Figure 6. Inhibition of the JAK/STAT Pathway Suppresses PLP-induced Relapsing-Remitting EAE Disease.
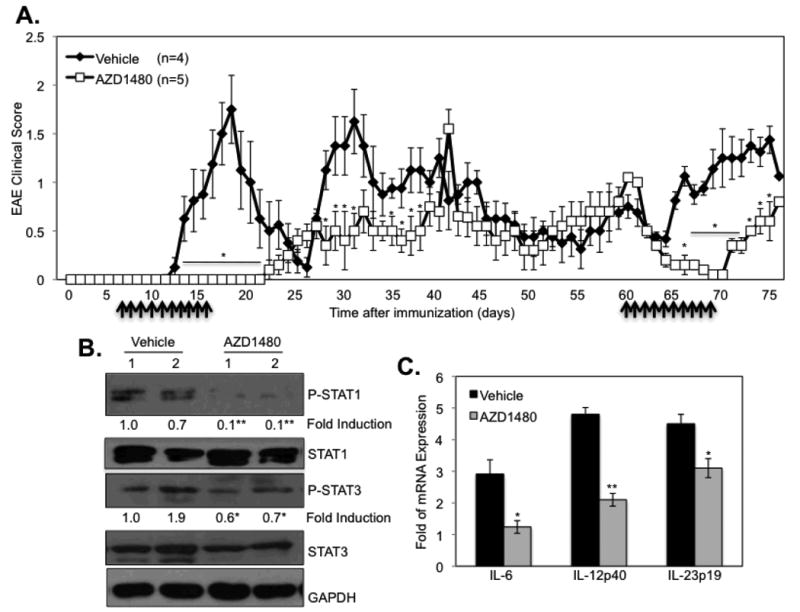
(A). SJL/J mice were immunized with PLP139-151 peptide (200 μg) emulsified in CFA containing M. tuberculosis. Vehicle Control (0.1% DMSO) (n=4) or AZD1480 (25 mg/kg) (n=5) was administrated by oral gavage daily starting at day 7 for 10 days, and then a second treatment at day 60 for 10 days. Mean ± S.D. of EAE clinical scores. (B). Mice were perfused, spinal cord removed, and protein extracts from PLP139-151-immunized Vehicle Control or AZD1480 treated mice at day 75 were immunoblotted with the indicated antibodies, and (C) mRNA was analyzed by qRT-PCR. *p<0.05 and **p<0.001.
Inhibition of the JAK/STAT Pathway Influences the Priming and Expansion Phases of EAE
Next, we examined the underlying cellular mechanisms leading to clinical improvement of actively-induced EAE by AZD1480 treatment. As MOG35-55-specific T-cells are first primed to differentiate into specialized effector subsets in the periphery (3), the effect of AZD1480 on the priming phase of EAE was investigated. C57BL/6 mice were immunized with MOG35-55, and vehicle or AZD1480 (25 mg/kg) was administered at day 2 post-immunization for 3 days (Fig. 7A). Purified CD4+ T-cells were isolated from the draining lymph nodes and spleen of these mice on day 10 post-immunization, re-stimulated with MOG35-55 peptide under Th1 and Th17 polarizing conditions, and the percentage of MOG35-55-specific T-cells measured by flow cytometry. Treatment with AZD1480 led to a significant decrease of MOG35-55-specific Th1 and Th17 cells in the draining lymph nodes and, to a lesser degree, in the spleen (Figs. 7B and 7C). Next, the effect of AZD1480 on expansion of MOG-specific T-cells was examined. C57BL/6 mice were immunized with MOG35-55, and administrated vehicle or AZD1480 (25 mg/kg) at day 7 post-immunization for 3 days (Fig. 7D). Purified CD4+ T-cells were isolated and stimulated as above. Decreased MOG35-55-specific Th1 cells in the draining lymph nodes were observed (Fig. 7E), and diminished percentages of MOG-specific Th17 cells were detected in draining lymph nodes and spleen (Fig. 7F). These results indicate that inhibition of the JAK/STAT pathway influences both the priming and expansion phases of EAE by suppressing MOG35-55-specific T-cell responses in secondary lymphoid tissues.
Figure 7. Inhibition of the JAK/STAT Pathway Regulates the Priming and Expansion Phases of EAE, and Proliferation of CD4+ T-cells and CD11b+ Cells.
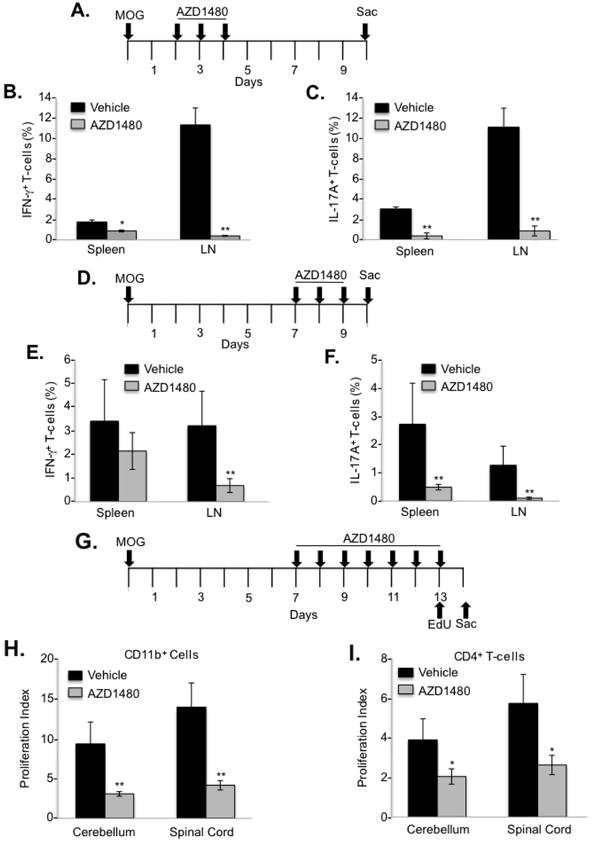
C57BL/6 mice were immunized with MOG35-55 peptide (200 μg) emulsified in CFA containing M. tuberculosis. Vehicle Control or AZD1480 (25 mg/kg) was administrated i.p. daily for 3 days starting at day 2 post-immunization (A) or at day 7 post-immunization (D), and mice were sacrificed at day 10. Purified CD4+ T-cells isolated from the lymph nodes (LN) and spleen of C57BL/6 mice at day 10 were re-stimulated with MOG35-55 peptide (10 μg/ml) under Th1 and Th17 differentiation conditions for 4 days. At day 4, cells were stimulated with PMA/Ionomycin plus GolgiStop for 4 h, stained for the surface marker CD4 and by intracellular flow for IFN-γ (B and E) or IL-17A (C and F). C57BL/6 mice were immunized with MOG35-55 peptide (200 μg) emulsified in CFA containing M. tuberculosis. The mice received i.p. injections of 250 ng of pertussis toxin at days 0 and 2. Vehicle Control or AZD1480 (25 mg/kg) was administrated i.p. daily starting at day 7 post-immunization for 7 days. Mice were injected i.p. with 100 μg 5-ethynyl-2′-deoxyuridine (EdU) in PBS at day 13. (G). CNS-infiltrating mononuclear cells were isolated from the cerebellum or spinal cord 12 h later, stained with Abs to CD11b (H) or CD4 (I), and EdU was detected by the Click reaction and flow cytometry. *p<0.05 and **p<0.001.
JAK/STAT Pathway Inhibition Suppresses Proliferation of CD4+ T-cells and CD11b+ Cells
The decreased number of CNS infiltrating cells in AZD1480 treated mice may be due to reduced cell proliferation, thus, the effect of AZD1480 on T-cell and myeloid cell proliferation during EAE development was assessed. C57BL/6 mice were immunized with MOG35-55, and then administrated vehicle or AZD1480 (25 mg/kg) at day 7 post-immunization. Mice were then injected with 5-ethynyl-2′-deoxyuridine (EdU) on day 13 and sacrificed on day 14 (Fig. 7G). CNS-infiltrating mononuclear cells were isolated from the cerebellum and spinal cord 12 h later, stained for CD4 or CD11b, and EdU incorporation measured. Importantly, proliferation of CD11b+ myeloid cells was significantly inhibited in AZD1480 treated mice (Fig. 7H). Similarly, significant inhibition of CD4+ T-cell proliferation was observed in AZD1480 treated mice (Fig. 7I). Thus, in vivo AZD1480 treatment suppresses proliferation of both CD11b+ myeloid cells and CD4+ T-cells in the CNS compartment, which may contribute to the clinical improvement of EAE.
JAK/STAT Pathway Inhibition Exerts a Protective Effect in Th1 and Th17-induced EAE
Transfer of myelin-specific CD4+ T-cells induces EAE in naïve recipients, thus we tested whether AZD1480 treatment would be effective in treating EAE induced by adoptive transfer of pathogenic CD4+ Th1 or Th17 cells. C57BL/6 mice were adoptively transferred with MOG35-55 specific Th1 or Th17 cells to induce EAE, and AZD1480 was administrated i.p. daily for 10 days starting at day 7 for Th1-induced EAE (Fig. 8A) or on day 8 for Th17-induced EAE (Fig. 8D). Notably, we observed a delay in onset of disease from day 10 to day 14, and diminished disease severity in Th1-induced EAE (Fig. 8A). When AZD1480 treatment was discontinued, a protective effect was still noted at days 17 and 18, but was lost at days 19 and 20. A substantial reduction of STAT1 and STAT3 tyrosine phosphorylation in the spinal cord of AZD1480 treated mice was observed (Fig. 8B), and mRNA levels of pro-inflammatory cytokines were markedly decreased (Fig. 8C). AZD1480 treatment suppressed mRNA levels of IFN-γ and T-bet in Th1-induced EAE at the site of disease in the spinal cords of treated mice (Fig. 8C). It should be noted that IL-17A and RORγt mRNA expression was not detected in mice with Th1-induced EAE (data not shown). In Th17-induced EAE, disease onset and severity were significantly inhibited by AZD1480 treatment (Fig. 8D), although the mice developed clinical signs of EAE after AZD1480 withdrawal. STAT1 and STAT3 tyrosine phosphorylation was inhibited in Th17-induced EAE by AZD1480, as were total levels of STAT1 and STAT3 protein (Fig. 8E). In addition, markers of Th1 and Th17 cells and pro-inflammatory cytokine mRNA levels were inhibited by AZD1480 treatment (Fig. 8F). These data indicate that AZD1480, by inhibiting the JAK/STAT pathway, can diminish peripheral T-cell mediated damage against the CNS in vivo.
Figure 8. Effect of AZD1480 on Th1- and Th17-induced Adoptive Transfer EAE.
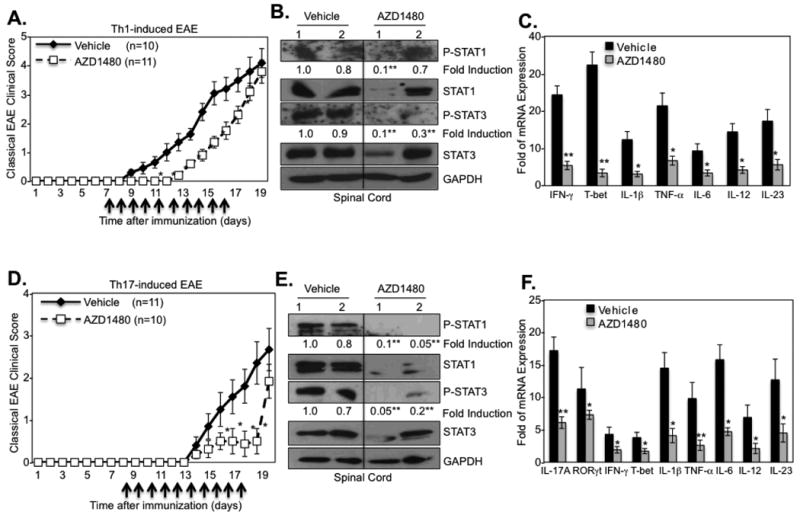
(A). C57BL/6 mice were immunized with MOG35-55 peptide (200 μg) for 10 days, splenocytes and lymph node cells were isolated, then MOG-specific T-cells were cultured with MOG35-55 peptide (10 μg/ml) under Th1 differentiation conditions for 3 days. Thirty × 106 cells were injected i.v.into C57BL/6 mice. Vehicle Control (n=10) or AZD1480 (25 mg/kg) (n=11) was administrated daily by i.p. starting at day 7 for 10 days. Mean ± S.D. of classical EAE clinical scores. (B). Mice were perfused, spinal cord removed, and protein extracts from spinal cord of Vehicle Control or AZD1480 treated mice at day 17 were immunoblotted with the indicated antibodies. (C). Mice were perfused, spinal cord removed, and mRNA from Vehicle Control or AZD1480 treated mice at day 17 was analyzed by qRT-PCR. (D). C57BL/6 mice were immunized with MOG35-55 peptide (200 μg) for 10 days, splenocytes and lymph node cells were isolated, then MOG-specific T-cells were cultured with MOG35-55 peptide (10 μg/ml) under Th17 differentiation conditions for 3 days. Thirty × 106 cells were injected i.v. into C57BL/6 mice. Vehicle Control (n=11) or AZD1480 (25 mg/kg) (n=10) was administrated daily by i.p. starting at day 8 for 10 days. Mean ± S.D. of classical EAE clinical scores. (E). Mice were perfused, spinal cord removed, and protein extracts from spinal cord of Vehicle Control or AZD1480 treated mice at day 18 were immunoblotted with the indicated antibodies. (F). Mice were perfused, spinal cord removed, and mRNA from spinal cord of Vehicle Control or AZD1480 treated mice at day 18 was analyzed by qRT-PCR. *p<0.05 and **p<0.001.
AZD1480 Treatment In Vivo Affects the Function of Myeloid Cells and Th1 Cells
Myeloid cells are pivotal for the development of EAE by modulating T-cell responses due to the production of proinflammatory cytokines and presentation of antigen. To assess the functional influence of JAK/STAT inhibition in myeloid cells, C57BL/6 mice were injected with vehicle or AZD1480 (25 mg/kg) for 4 days, and BMDM isolated from these mice were examined for JAK/STAT pathway activation. IL-6-induced STAT3 tyrosine phosphorylation was strongly inhibited in BMDM obtained from AZD1480 treated mice (Fig. 9A), as was IFN-γ-induced STAT1 tyrosine phosphorylation (Fig. 9B). In addition, IFN-γ-inducible MHC class II expression was suppressed by in vivo AZD1480 treatment (Fig. 9C). Consistent with the in vitro data, LPS activation of the NF-κB pathway was not affected (Fig. 9D).
Figure 9. The JAK1/2 Inhibitor AZD1480 Has Direct Effects on Myeloid APCs and Th1 Cells.
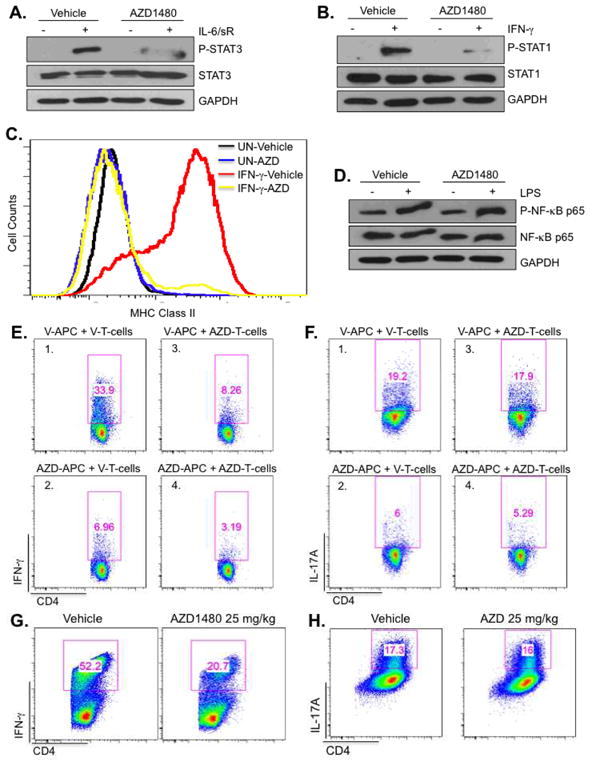
C57BL/6 mice were administrated Vehicle Control or AZD1480 (25 mg/kg) i.p. daily for 4 days. At that time, BMDM were isolated from mice and stimulated with medium (-) or IL-6 (10 ng/ml) plus sIL-6R (25 ng/ml) for 30 min (A), or IFN-γ (10 ng/ml) for 30 min (B), then cell lysates were subjected to immunoblotting with the indicated antibodies. (C). BMDM isolated from Vehicle Control or AZD1480 treated mice after 4 days of treatment were stimulated with medium (UN) or IFN-γ (10 ng/ml) for 24 h, then MHC Class II expression was analyzed by flow cytometry. (D). BMDM obtained as described above were stimulated with medium or LPS (10 ng/ml) for 30 min, then cell lysates subjected to immunoblotting with the indicated antibodies. (E and F). C57BL/6 or MOG35-55 TCR transgenic mice (2D2) were administrated Vehicle Control or AZD1480 (25 mg/kg) i.p. daily for 5 days. CD11b+/11c+ cells sorted from the spleen of Vehicle Control (V) or AZD1480 (AZD) treated C57BL/6 mice were used as APCs in co-culture with naïve CD4+ T-cells from Vehicle- or AZD1480-treated 2D2 mice. Polarization of MOG-specific Th1 cells was determined by intracellular cytokine staining for IFN-γ after 3 days of culture with MOG peptide (10 μg/ml), IL-12 and anti-IL-4 Ab (E). Polarization of MOG-specific Th17 cells was determined by intracellular cytokine staining for IL-17A after 3 days of culture with MOG peptide (10 μg/ml), IL-23, IL-6, TGF-β, anti-IFN-γ and anti-IL-4 Abs (F). (G and H). C57BL/6 mice were administrated Vehicle Control or AZD1480 (25 mg/kg) i.p. daily for 3 days. Naïve CD4+ T-cells were isolated from the spleen. Vehicle control DCs were used as APCs at a 1:5 ratio to CD4+ T-cells in co-culture under Th1 or Th17 differentiation cocktail conditions with anti-CD3 and anti-CD28 Abs for 4 days, then stained for the surface marker CD4 and by intracellular flow for IFN-γ (G) or IL-17A (H).
To address whether inhibiting the JAK/STAT pathway modulates the APC functions of myeloid cells, purified CD11b+/CD11c+ cells from vehicle- or AZD1480-treated C57BL/6 mice were used as APCs for stimulation of naïve MOG35-55-specific 2D2 CD4+ T-cells under Th1- or Th17-polarizing conditions. Notably, we found that APCs from AZD1480 treated mice (AZD-APC) were less efficient than those from vehicle-treated mice (V-APC) in promoting MOG35-55-specific Th1 cell differentiation (Fig. 9E, compare panels 1 and 2) and Th17 cell differentiation (Fig. 9F, compare panels 1 and 2). These results indicate that CD11b+/CD11c+ APCs from AZD1480 treated mice are defective in their ability to induce MOG35-55-specific Th1 and Th17 cell differentiation.
Next, the response of purified naïve MOG35-55-specific CD4+ T-cells from vehicle- or AZD1480 treated 2D2 mice to CD11b+/CD11c+ APCs from vehicle-treated C57BL/6 mice was examined. CD4+ MOG-specific T-cells from AZD1480 treated 2D2 mice (AZD-T-cells) were unable to differentiate into Th1 cells when provided with vehicle-treated APCs (Fig. 9E, compare panels 1 and 3), and there was a further decrease when MOG-specific T-cells from AZD1480-treated 2D2 mice were cultured with AZD1480-treated APCs (Fig. 9E, panel 4). However, CD4+ T-cells from AZD1480 treated 2D2 mice were capable of differentiating into Th17 cells in the presence of APCs (Fig. 9F, compare panels 1 and 3). We next investigated whether the inhibitory effect of AZD1480 on Th1 cell differentiation was antigen specific. Purified naïve CD4+ T-cells from vehicle- or AZD1480-treated C57BL/6 mice were stimulated with anti-CD3 and anti-CD28 Abs under Th1- or Th17-differentiation conditions. Consistent with the results from MOG35-55-specific T-cells, in vivo treatment with AZD1480 resulted in partial inhibition of Th1 cell polarization (Fig. 9G), while Th17 cell polarization was not affected (Fig. 9H). These findings indicate that in vivo inhibition of the JAK/STAT pathway has an inhibitory effect on the ability of CD4+ T-cells to become polarized to the Th1 phenotype, but does not appear to affect the Th17 polarization program.
AZD1480 Inhibits the JAK/STAT Pathway in Human T-cells and Monocytes
The findings thus far demonstrate that AZD1480 suppresses the function of murine T-cells and macrophages. To extend these findings to humans, PBMCs were stimulated in vitro with anti-CD3 and anti-CD28 Abs in the absence or presence of AZD1480, and phosphorylation of STAT1, STAT3, and STAT4 was examined in gated CD3+CD4+ T-cells. Treatment with AZD1480 led to substantially reduced levels of STAT1 and STAT3 tyrosine phosphorylation, and partial inhibition of STAT4 tyrosine phosphorylation (Fig. 10A). In addition, mRNA levels of IFN-γ and IL-17A were significantly reduced by AZD1480 (Fig. 10B). We also assessed the effect of AZD1480 on the JAK/STAT signaling pathway in human monocytes. CD3-CD14+ monocytes were stimulated with IFN-γ in the absence or presence of AZD1480, and phosphorylation of STAT1 and STAT3 examined. IFN-γ induced strong phosphorylation of STAT1 and moderate phosphorylation of STAT3, both of which were inhibited by AZD1480 (Fig. 10C). Furthermore, consistent with the inhibition of IFN-γ-induced STAT1 phosphorylation, the expression of IFN-γ inducible HLA-DR expression in AZD1480 treated monocytes was strongly suppressed (Fig. 10D). These in vitro experiments demonstrate that activated human T-cells and monocytes are direct targets of AZD1480.
Figure 10. AZD1480 Inhibits Activation of the JAK/STAT Pathway and Gene Expression in Human T-cells and Monocytes.
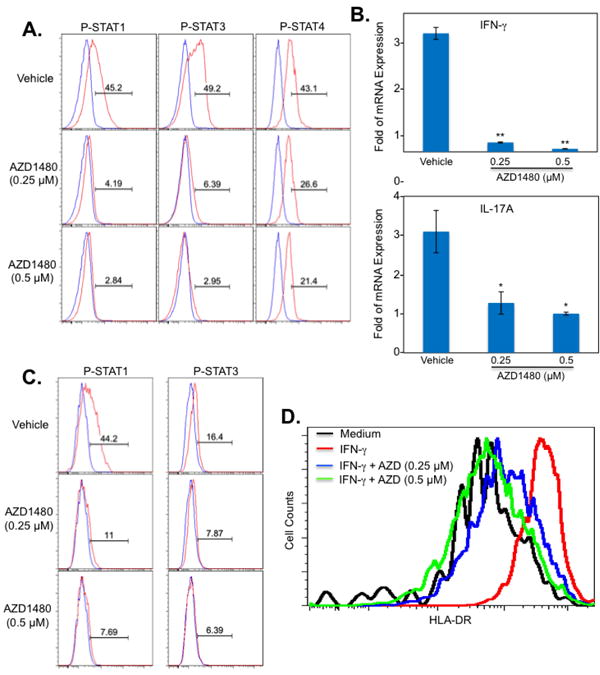
(A). PBMCs were stimulated with anti-CD3 (5 μg/ml) and anti-CD28 (2 μg/ml) Abs in the absence or presence of AZD1480 (0.25 and 0.5 μM) for 4 days. The extent of STAT phosphorylation within the CD4+ T-cell population was assessed by intracellular flow cytometry using anti-phospho-STAT1Tyr701, anti-phospho-STAT3Tyr705, and phospho-STAT4Tyr693 (red), with isotype control shown in blue. (B). For mRNA expression, cells were collected at day 4, and mRNA was analyzed by qRT-PCR for IFN-γ and IL-17A. (C). Adherent PBMCs were pretreated with Vehicle Control or AZD1480 (0.25 and 0.5 μM) for 2 h, then stimulated with IFN-γ (100 U/ml) for 1 h. The extent of STAT phosphorylation within the CD3-/CD14+ monocyte population was assessed by intracellular flow cytometry using anti-phospho-STAT1Tyr701 and anti-phospho-STAT3Tyr705 (red), with isotype control shown in blue. (D). Adherent PBMCs were pretreated with Vehicle Control or AZD1480 (0.25 μM or 0.5 μM) for 2 h, stimulated with medium or IFN-γ (100 U/ml) for 24 h, then HLA-DR expression was analyzed by flow cytometry. *p< 0.05 and**p< 0.001.
Discussion
Genome-wide association studies have shown that cytokines, their receptors, JAKs, STATs and SOCS proteins are associated with human autoimmune diseases, especially pathways leading to STAT3 and STAT4 activation (39, 40). STAT3 has been identified as an MS susceptibility gene (41, 42), and independent replication supports the association between STAT3 and increase in MS risk (43). We demonstrate that inhibition of the JAK/STAT pathway, specifically inhibition of STAT1, STAT3 and STAT4 activation, has broad actions on cells of the innate and adaptive immune systems, leading to amelioration of clinical disease in EAE models. Treatment with AZD1480, an inhibitor of JAK1 and JAK2, effectively prevents infiltration of immune cells into the CNS, inhibits STAT activation in the CNS, suppresses differentiation and proliferation of CD4+ cells, inhibits proliferation and deactivates myeloid cells, and reduces expression of pro-inflammatory cytokines and chemokines.
In MS and EAE, the pivotal role of pathogenic Th1 and Th17 cells, as well as myeloid cells, has been well documented (44-46). Inhibition of the JAK/STAT pathway by AZD1480 suppressed the in vitro differentiation of naïve T-cells to Th1 cells and also suppressed in vitro Th17 cell differentiation induced by three different polarization conditions: IL-6 + IL-23 + TGF-β; IL-6 + IL-23; and IL-6 + IL-23 + IL-1β. As STAT3 signaling is a central component of Th17 cell differentiation and Th17-dependent autoimmune processes (17, 47), using a JAK inhibitor that suppresses STAT3 activation is a promising strategy for therapeutic intervention. Disease supporting functions of macrophages, DCs and microglia have been described for CNS autoimmune diseases (45, 48, 49). In vitro results indicate that AZD1480 suppresses IFN-γ induction of genes that mediate some of the detrimental effects of macrophages and DCs, including MHC class II, CD40 and NO. IFN-γ promotes the polarization of macrophages to the classically activated, M1 phenotype, characterized by high levels of IL-6, IL-1, IL-12 and IL-23, increased levels of reactive oxygen species, MHC Class II and CD40, and low levels of IL-10 (50). M1 macrophages have detrimental effects in a number of CNS diseases, including EAE/MS, Alzheimer's Disease and spinal cord injury (51-55). We demonstrated that AZD1480 inhibited IFN-γ-induced NO release, which contributes in a significant manner to EAE pathogenesis and axonal damage (35). AZD1480 suppressed IFN-γ induction of MHC class II and CD40, which are critical for antigen presentation functions of macrophages/DCs. IFN-γ induction of MHC class II and CD40 expression is STAT1-dependent (56, 57), indicating that inhibition of the JAK/STAT pathway is an effective means of suppressing these genes. In macrophages, inhibited GM-CSF activation of STAT5, and IL-6 activation of STAT3, while not influencing IL-4 activation of STAT6. IFN-γ, GM-CSF and IL-6 signaling utilize JAK1 and/or JAK2, which explains the inhibitory effect of AZD1480. IL-4 signaling involves both JAK1 and JAK3, thus, inhibition of JAK1 by AZD1480 may not suffice to inhibit this pathway due to the kinase activity of JAK3.
Inhibition of the JAK/STAT pathway by AZD1480 has beneficial effects in mitigating clinical symptoms in five EAE models: chronic progressive disease in C57BL/6 mice; atypical disease with involvement of the cerebellum in LysMCre-SOCS3fl/fl mice; RR disease in SJL/J mice; adoptive Th1 cell EAE; and adoptive Th17 cell EAE. Improvement in clinical scores was associated with suppression of inflammatory responses in lymph node and spleen, indicating that inhibition of the JAK/STAT pathway affects the pathogenic potential of T-cells outside the CNS. The most reliable parameter of JAK inhibition is that of downstream inhibition of STAT activation (23, 24). AZD1480 treatment inhibited STAT1, STAT3 and/or STAT4 activation within the CNS. These STATs are activated downstream of JAK1 and/or JAK2, and are essential for development and differentiation of Th1 and Th17 cells, and maturation of myeloid cells. Thus, inhibition of STAT activation may be responsible for the reduction of inflammatory lesions and diminished expression of proinflammatory mediators in the CNS. Total levels of STAT proteins were also inhibited by AZD1480 treatment, most notably that of STAT1. STAT1 is itself a STAT1-inducible gene (37), therefore, inhibiting STAT activity will function in a negative feedback loop to inhibit STAT expression. Mice deficient in STAT3 in CD4+ T-cells are resistant to EAE (17, 18), as are mice deficient in STAT4 (58), while mice lacking STAT1 are highly susceptible to EAE (25). It is possible that in the absence of STAT1, STAT3 and/or STAT4 signaling may compensate to drive Th1 cell responses, as these mice were characterized by IFN-γ producing Th1 cells (25). Nonetheless, our results demonstrate that pharmacologic inhibition of STAT1, STAT3 and STAT4 signaling contributes to significant lessening of EAE disease severity.
The beneficial effects of inhibiting the JAK/STAT pathway on EAE pathogenesis appear to involve a number of mechanisms, including effects on both APCs and T-cells. First, both the primary and expansion phases of EAE were inhibited by AZD1480 treatment, as reflected by suppressed antigen-specific Th1 and Th17 responses in secondary lymphoid tissues, indicating that inhibition of the JAK/STAT pathway affects the early stages of the immunological cascade leading to EAE. Second, this, in turn, leads to diminished infiltration of immune cells into the CNS. Third, of the T-cells and myeloid cells that did extravasate into the CNS, AZD1480 treatment diminished their proliferative capacity, which we believe contributes to the therapeutic effectiveness of AZD1480 treatment. Fourth, in vivo AZD1480 treatment inhibits both the innate and adaptive arms of the immune system that are associated with EAE. AZD1480 treatment in vivo was effective in inhibiting the ability of T-cells to differentiate into Th1 cells, in both antigen-specific and non-antigen specific manners. This intriguing finding needs to be examined in more detail, but may relate to the effectiveness of AZD1480 in inhibiting IFN-γ and IL-12 signaling, which are critical for Th1 cell differentiation. This suggests a direct effect of AZD1480 on Th1 differentiation. In contrast, although our in vitro data documented an inhibitory effect of AZD1480 on Th17 cell differentiation, we did not observe a direct inhibitory effect on Th17 cell polarization in vivo. This may reflect the complexities of promoting Th17 cell differentiation by diverse cytokines including TGF-β1, TGF-β3, IL-1, IL-6, IL-23, and inhibition by IL-2 and IL-27 (59, 60). We do know that AZD1480 is a strong inhibitor of IL-6 and IL-23 signaling, and suppresses expression of the IL-23R on T-cells, a critical determinant of Th17 cell polarization. However, the influence of inhibiting the JAK/STAT pathway on TGF-β1, TGF-β3 and IL-27 signaling is unknown, and IL-1β signaling is unaffected (data not shown). Inhibition of the JAK/STAT pathway has potent suppressive effects on myeloid cell functions in vivo. AZD1480 treatment renders myeloid cells non-responsive to IFN-γ and IL-6 signal transduction, resulting in inhibition of STAT1 and STAT3 activation, and downstream inhibition of gene expression such as MHC class II. Further, AZD1480 treatment has a direct effect on APC functions of myeloid cells as demonstrated by the findings that CD11b+/CD11c+ APCs from AZD1480 treated mice inhibited the development of MOG35-55 specific Th1 and Th17 cells. This is in line with observations on the in vivo treatment of mice with Glatiramer Acetate (GA), an approved therapy for RRMS (61), fumarates (62), which were approved by the FDA on March 27, 2013 for treatment of RRMS, and laquinimod, which has been successfully evaluated in Phase II/III studies of RRMS (63, 64). All of these agents had direct effects on cells of the innate immune system, and importantly, skewed macrophages and DCs towards a regulatory phenotype. Characteristics of the regulatory, anti-inflammatory phenotype included reduced STAT1 phosphorylation, decreased production of IL-6, IL-12, and IL-23, and elevated secretion of IL-10 (61-64). At this point, we do not know if in vivo treatment with JAK inhibitors skews monocytes/macrophages and DCs to an anti-inflammatory phenotype, but we do demonstrate a suppression of the M1 pro-inflammatory phenotype by AZD1480. Thus, collectively, AZD1480 functions to promote immune modulation by a direct effect on myeloid APCs, which impacts the differentiation of Th1 and Th17 cells in vivo, as well as a direct effect of AZD1480 in vivo on Th1 cell differentiation.
Studies have implicated the JAK/STAT axis in regulating clinical manifestations of EAE. Peroxisome proliferator activated receptor-γ (PPARγ) and COX2 inhibitors suppress EAE severity, in part, by inhibiting IL-12-induced activation of the JAK/STAT pathway, and subsequent suppression of Th1 cell differentiation (65, 66). The protective effect of GA in EAE is in part due to inhibition of STAT4 and STAT3 phosphorylation in T-cells, altering Th1 and Th17 cell differentiation, respectively (67). Two herbal compounds, plumbagin (PL) and berberine, exert protective effects in EAE models by inhibiting STAT activation and Th1 and Th17 cell differentiation (68, 69). In this study, we have utilized a specific inhibitor of the JAK/STAT pathway, AZD1480, and documented a striking beneficial immunomodulatory effect in five different models of EAE. Importantly, AZD1480 treatment was administered at the onset of disease and in a therapeutic manner after the appearance of clinical symptoms, with potent clinical efficacy. These findings collectively suggest the JAK/STAT axis may serve as a therapeutic target for intervention in MS, as well as other neuroinflammatory conditions such as Parkinson's Disease, Spinal Cord Injury and Alzheimer's Disease.
Supplementary Material
Acknowledgments
We thank the UAB Neuroscience Molecular Detection Core [NS47466], and the Rheumatic Diseases Core Center (P30 Flow Core) [AR48311] for advice and technical assistance. Dr. Dennis Huszar, Cancer Bioscience, AstraZeneca R & D, Waltham, MA generously provided AZD1480 and helpful discussions. We thank Dr. Kui Zhang, Section on Statistical Genetics, Department of Biostatistics, School of Public Health at UAB, for assistance with statistical analysis of the EAE data. The authors thank Dr. Warren Alexander for the generous gift of the SOCS3fl/fl mice, and members of the Benveniste laboratory for helpful discussions.
Footnotes
This work was supported in part by National Institutes of Health Grants [NS45290 and NS57563 to E.N.B.], [NS64261 to P.D.], [DK84082 to L.E.H.], [AI27767 to S.S.], and [AI76562 to C.R.]; National Multiple Sclerosis Society Grants [RG-3891 to C.R.], [CA-1059-A-13 to E.N.B.] and [RG-4885-A-14 to E.N.B.], and a grant from the American Brain Tumor Foundation in honor of Paul Fabbri [B.C.M.].
References
- 1.Bhat R, Steinman L. Innate and adaptive autoimmunity directed to the central nervous system. Neuron. 2009;64:123–132. doi: 10.1016/j.neuron.2009.09.015. [DOI] [PubMed] [Google Scholar]
- 2.Ransohoff RM. Chemokines and chemokine receptors: standing at the crossroads of immunobiology and neurobiology. Immunity. 2009;31:711–721. doi: 10.1016/j.immuni.2009.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Goverman J. Autoimmune T cell responses in the central nervous system. Nat Rev Immunol. 2009;9:393–407. doi: 10.1038/nri2550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ransohoff RM. Animal models of multiple sclerosis: the good, the bad and the bottom line. Nat Neurosci. 2012;15:1074–1077. doi: 10.1038/nn.3168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Domingues HS, Mues M, Lassmann H, Wekerle H, Krishnamoorthy G. Functional and pathogenic differences of Th1 and Th17 cells in experimental autoimmune encephalomyelitis. PloS One. 2010;5:e15531. doi: 10.1371/journal.pone.0015531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Segal BM. Th17 cells in autoimmune demyelinating disease. Semin Immunopathol. 2010;32:71–77. doi: 10.1007/s00281-009-0186-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.O'Shea JJ, Plenge R. JAK and STAT signaling molecules in immunoregulation and immune-mediated disease. Immunity. 2012;36:542–550. doi: 10.1016/j.immuni.2012.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Weaver CT, Hatton RD, Mangan PR, Harrington LE. IL-17 family cytokines and the expanding diversity of effector T-cell lineages. Ann Rev Immunol. 2007;25:821–852. doi: 10.1146/annurev.immunol.25.022106.141557. [DOI] [PubMed] [Google Scholar]
- 9.Geissmann F, Manz MG, Jung S, Sieweke MH, Merad M, Ley K. Development of monocytes, macrophages, and dendritic cells. Science. 2010;327:656–661. doi: 10.1126/science.1178331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Baker BJ, Akhtar LN, Benveniste EN. SOCS1 and SOCS3 in the control of CNS immunity. Trends Immunol. 2009;30:392–400. doi: 10.1016/j.it.2009.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Seavey MM, Dobrzanski P. The many faces of Janus kinase. Biochem Pharmacol. 2012;83:1136–1145. doi: 10.1016/j.bcp.2011.12.024. [DOI] [PubMed] [Google Scholar]
- 12.Frisullo G, Angelucci F, Caggiula M, Nociti V, Iorio R, Patanella AK, Sancricca C, Mirabella M, Tonali PA, Batocchi AP. pSTAT1, pSTAT3, and T-bet expression in peripheral blood mononuclear cells from relapsing-remitting multiple sclerosis patients correlates with disease activity. J Neurosci Res. 2006;84:1027–1036. doi: 10.1002/jnr.20995. [DOI] [PubMed] [Google Scholar]
- 13.Frisullo G, Nociti V, Iorio R, Patanella AK, Marti A, Mirabella M, Tonali PA, Batocchi AP. The persistency of high levels of pSTAT3 expression in circulating CD4+ T cells from CIS patients favors the early conversion to clinically defined multiple sclerosis. J Neuroimmunol. 2008;205:126–134. doi: 10.1016/j.jneuroim.2008.09.003. [DOI] [PubMed] [Google Scholar]
- 14.Quintana A, Muller M, Frausto RF, Ramos R, Getts DR, Sanz E, Hofer MJ, Krauthausen M, King NJ, Hidalgo J, Campbell IL. Site-specific production of IL-6 in the central nervous system retargets and enhances the inflammatory response in experimental autoimmune encephalomyelitis. J Immunol. 2009;183:2079–2088. doi: 10.4049/jimmunol.0900242. [DOI] [PubMed] [Google Scholar]
- 15.Chen Z, Laurence A, Kanno Y, Pacher-Zavisin M, Zhu BM, Tato C, Yoshimura A, Hennighausen L, O'Shea JJ. Selective regulatory function of SOCS3 in the formation of IL-17-secreting T cells. Proc Natl Acad Sci USA. 2006;103:8137–8142. doi: 10.1073/pnas.0600666103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang XO, Panopoulos AD, Nurieva R, Chang SH, Wang D, Watowich SS, Dong C. STAT3 regulates cytokine-mediated generation of inflammatory helper T cells. J Biol Chem. 2007;282:9358–9363. doi: 10.1074/jbc.C600321200. [DOI] [PubMed] [Google Scholar]
- 17.Harris TJ, Grosso JF, Yen HR, Xin H, Kortylewski M, Albesiano E, Hipkiss EL, Getnet D, Goldberg MV, Maris CH, Housseau F, Yu H, Pardoll DM, Drake CG. Cutting edge: an in vivo requirement for STAT3 signaling in TH17 development and TH17-dependent autoimmunity. J Immunol. 2007;179:4313–4317. doi: 10.4049/jimmunol.179.7.4313. [DOI] [PubMed] [Google Scholar]
- 18.Liu X, Lee YS, Yu CR, Egwuagu CE. Loss of STAT3 in CD4+ T cells prevents development of experimental autoimmune diseases. J Immunol. 2008;180:6070–6076. doi: 10.4049/jimmunol.180.9.6070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ghoreschi K, Jesson MI, Li X, Lee JL, Ghosh S, Alsup JW, Warner JD, Tanaka M, Steward-Tharp SM, Gadina M, Thomas CJ, Minnerly JC, Storer CE, LaBranche TP, Radi ZA, Dowty ME, Head RD, Meyer DM, Kishore N, O'Shea JJ. Modulation of innate and adaptive immune responses by tofacitinib (CP-690,550) J Immunol. 2011;186:4234–4243. doi: 10.4049/jimmunol.1003668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stump KL, Lu LD, Dobrzanski P, Serdikoff C, Gingrich DE, Dugan BJ, Angeles TS, Albom MS, Ator MA, Dorsey BD, Ruggeri BA, Seavey MM. A highly selective, orally active inhibitor of Janus kinase 2, CEP-33779, ablates disease in two mouse models of rheumatoid arthritis. Arthr Res Ther. 2011;13:R68. doi: 10.1186/ar3329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.van Vollenhoven RF, Fleischmann R, Cohen S, Lee EB, Garcia Meijide JA, Wagner S, Forejtova S, Zwillich SH, Gruben D, Koncz T, Wallenstein GV, Krishnaswami S, Bradley JD, Wilkinson B. Tofacitinib or adalimumab versus placebo in rheumatoid arthritis. N Engl J Med. 2012;367:508–519. doi: 10.1056/NEJMoa1112072. [DOI] [PubMed] [Google Scholar]
- 22.Bright JJ, Du C, Sriram S. Tyrphostin B42 inhibits IL12-induced tyrosine phosphorylation and activation of Janus kinase-2 and prevents experimental allergic encephalomyelitis. J Immunol. 1999;162:6255–6262. [PubMed] [Google Scholar]
- 23.Hedvat M, Huszar D, Herrmann A, Gozgit JM, Schroeder A, Sheehy A, Buettner R, Proia D, Kowolik CM, Xin H, Armstrong B, Bebernitz G, Weng S, Wang L, Ye M, McEachern K, Chen H, Morosini D, Bell K, Alimzhanov M, Ioannidis S, McCoon P, Cao ZA, Yu H, Jove R, Zinda M. The JAK2 inhibitor AZD1480 potently blocks Stat3 signaling and oncogenesis in solid tumors. Cancer Cell. 2009;16:487–497. doi: 10.1016/j.ccr.2009.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McFarland BC, Ma JY, Langford CP, Gillespie GY, Yu H, Zheng Y, Nozell SE, Huszar D, Benveniste EN. Therapeutic potential of AZD1480 for the treatment of human glioblastoma. Mol Cancer Ther. 2011;10:2384–2393. doi: 10.1158/1535-7163.MCT-11-0480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bettelli E, Sullivan B, Szabo SJ, Sobel RA, Glimcher LH, Kuchroo VK. Loss of T-bet, but not STAT1, prevents the development of experimental autoimmune encephalomyelitis. J Exp Med. 2004;200:79–87. doi: 10.1084/jem.20031819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Croker BA, Krebs DL, Zhang JG, Wormald S, Willson TA, Stanley EG, Robb L, Greenhalgh CJ, Förster I, Clausen BE, Nicola NA, Metcalf D, Hilton DJ, Roberts AW, Alexander WS. SOCS3 negatively regulates IL-6 signaling in vivo. Nat Immunol. 2003;4:540–545. doi: 10.1038/ni931. [DOI] [PubMed] [Google Scholar]
- 27.Qin H, Yeh WI, De Sarno P, Holdbrooks AT, Liu Y, Muldowney MT, Reynolds SL, Yanagisawa LL, Fox THI, Park K, Harrington LE, Raman C, Benveniste EN. Signal transducer and activator of transcription-3/suppressor of cytokine signaling-3 (STAT3/SOCS3) axis in myeloid cells regulates neuroinflammation. Proc Natl Acad Sci USA. 2012;109:5004–5009. doi: 10.1073/pnas.1117218109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Axtell RC, de Jong BA, Boniface K, van der Voort LF, Bhat R, De Sarno P, Naves R, Han M, Zhong F, Castellanos JG, Mair R, Christakos A, Kolkowitz I, Katz L, Killestein J, Polman CH, de Waal Malefyt R, Steinman L, Raman C. T helper type 1 and 17 cells determine efficacy of interferon-beta in multiple sclerosis and experimental encephalomyelitis. Nat Med. 2010;16:406–412. doi: 10.1038/nm.2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.De Sarno P, Axtell RC, Raman C, Roth KA, Alessi DR, Jope RS. Lithium prevents and ameliorates experimental autoimmune encephalomyelitis. J Immunol. 2008;181:338–345. doi: 10.4049/jimmunol.181.1.338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Qin H, Holdbrooks AT, Liu Y, Reynolds SL, Yanagisawa LL, Benveniste EN. SOCS3 deficiency promotes M1 macrophage polarization and inflammation. J Immunol. 2012;189:3439–3448. doi: 10.4049/jimmunol.1201168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ghoreschi K, Laurence A, Yang XP, Tato CM, McGeachy MJ, Konkel JE, Ramos HL, Wei L, Davidson TS, Bouladoux N, Grainger JR, Chen Q, Kanno Y, Watford WT, Sun HW, Eberl G, Shevach EM, Belkaid Y, Cua DJ, Chen W, O'Shea JJ. Generation of pathogenic T(H)17 cells in the absence of TGF-beta signalling. Nature. 2011;467:967–971. doi: 10.1038/nature09447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fleetwood AJ, Lawrence T, Hamilton JA, Cook AD. Granulocyte-macrophage colony-stimulating factor (CSF) and macrophage CSF-dependent macrophage phenotypes display differences in cytokine profiles and transcription factor activities: implications for CSF blockade in inflammation. J Immunol. 2007;178:5245–5252. doi: 10.4049/jimmunol.178.8.5245. [DOI] [PubMed] [Google Scholar]
- 33.Sonderegger I, Iezzi G, Maier R, Schmitz N, Kurrer M, Kopf M. GM-CSF mediates autoimmunity by enhancing IL-6-dependent Th17 cell development and survival. J Exp Med. 2008;205:2281–2294. doi: 10.1084/jem.20071119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gordon S, Martinez FO. Alternative activation of macrophages: mechanism and functions. Immunity. 2010;32:593–604. doi: 10.1016/j.immuni.2010.05.007. [DOI] [PubMed] [Google Scholar]
- 35.Kroenke MA, Carlson TJ, Andjelkovic AV, Segal BM. IL-12- and IL-23-modulated T cells induce distinct types of EAE based on histology, CNS chemokine profile, and response to cytokine inhibition. J Exp Med. 2008;205:1535–1541. doi: 10.1084/jem.20080159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hooper DC, Bagasra O, Marini JC, Zborek A, Ohnishi ST, Kean R, Champion JM, Sarker AB, Bobroski L, Farber JL, Akaike T, Maeda H, Koprowski H. Prevention of experimental allergic encephalomyelitis by targeting nitric oxide and peroxynitrite: implications for the treatment of multiple sclerosis. Proc Natl Acad Sci USA. 1997;94:2528–2533. doi: 10.1073/pnas.94.6.2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lee YJ, Benveniste EN. STAT-1α expression is involved in IFN-γ induction of the class II transactivator and class II MHC genes. J Immunol. 1996;157:1559–1568. [PubMed] [Google Scholar]
- 38.Fridman JS, Scherle PA, Collins R, Burn TC, Li Y, Li J, Covington MB, Thomas B, Collier P, Favata MF, Wen X, Shi J, McGee R, Haley PJ, Shepard S, Rodgers JD, Yeleswaram S, Hollis G, Newton RC, Metcalf B, Friedman SM, Vaddi K. Selective inhibition of JAK1 and JAK2 is efficacious in rodent models of arthritis: preclinical characterization of INCB028050. J Immunol. 2010;184:5298–5307. doi: 10.4049/jimmunol.0902819. [DOI] [PubMed] [Google Scholar]
- 39.Oksenberg JR, Baranzini SE. Multiple sclerosis genetics—is the glass half full, or half empty? Nat Rev Neurol. 2010;6:429–437. doi: 10.1038/nrneurol.2010.91. [DOI] [PubMed] [Google Scholar]
- 40.Sawcer S, Hellenthal G, Pirinen M, et al. Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature. 2011;476:214–219. doi: 10.1038/nature10251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jakkula E, Leppa V, Sulonen AM, Varilo T, Kallio S, Kemppinen A, Purcell S, Koivisto K, Tienari P, Sumelahti ML, Elovaara I, Pirttila T, Reunanen M, Aromaa A, Oturai AB, Sondergaard HB, Harbo HF, Mero IL, Gabriel SB, Mirel DB, Hauser SL, Kappos L, Polman C, De Jager PL, Hafler DA, Daly MJ, Palotie A, Saarela J, Peltonen L. Genome-wide association study in a high-risk isolate for multiple sclerosis reveals associated variants in STAT3 gene. Am J Hum Gen. 2010;86:285–291. doi: 10.1016/j.ajhg.2010.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Baranzini SE, Galwey NW, Wang J, Khankhanian P, Lindberg R, Pelletier D, Wu W, Uitdehaag BM, Kappos L, Polman CH, Matthews PM, Hauser SL, Gibson RA, Oksenberg JR, Barnes MR. Pathway and network-based analysis of genome-wide association studies in multiple sclerosis. Hum Mol Gen. 2009;18:2078–2090. doi: 10.1093/hmg/ddp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lill CM, Schjeide BM, Akkad DA, Blaschke P, Winkelmann A, Gerdes LA, Hoffjan S, Luessi F, Dorner T, Li SC, Steinhagen-Thiessen E, Lindenberger U, Chan A, Hartung HP, Aktas O, Lohse P, Kumpfel T, Kubisch C, Epplen JT, Zettl UK, Bertram L, Zipp F. Independent replication of STAT3 association with multiple sclerosis risk in a large German case-control sample. Neurogenetics. 2012;13:83–86. doi: 10.1007/s10048-011-0305-6. [DOI] [PubMed] [Google Scholar]
- 44.Nikic I, Merkler D, Sorbara C, Brinkoetter M, Kreutzfeldt M, Bareyre FM, Bruck W, Bishop D, Misgeld T, Kerschensteiner M. A reversible form of axon damage in experimental autoimmune encephalomyelitis and multiple sclerosis. Nat Med. 2011;17:495–499. doi: 10.1038/nm.2324. [DOI] [PubMed] [Google Scholar]
- 45.Davalos D, Ryu JK, Merlini M, Baeten KM, Le Moan N, Petersen MA, Deerinck TJ, Smirnoff DS, Bedard C, Hakozaki H, Gonias Murray S, Ling JB, Lassmann H, Degen JL, Ellisman MH, Akassoglou K. Fibrinogen-induced perivascular microglial clustering is required for the development of axonal damage in neuroinflammation. Nature Commun. 2012;3:1227–1241. doi: 10.1038/ncomms2230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mildner A, Mack M, Schmidt H, Bruck W, Djukic M, Zabel MD, Hille A, Priller J, Prinz M. CCR2+Ly-6Chi monocytes are crucial for the effector phase of autoimmunity in the central nervous system. Brain. 2009;132:2487–2500. doi: 10.1093/brain/awp144. [DOI] [PubMed] [Google Scholar]
- 47.Durant L, Watford WT, Ramos HL, Laurence A, Vahedi G, Wei L, Takahashi H, Sun HW, Kanno Y, Powrie F, O'Shea JJ. Diverse targets of the transcription factor STAT3 contribute to T cell pathogenicity and homeostasis. Immunity. 2010;32:605–615. doi: 10.1016/j.immuni.2010.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Heppner FL, Greter M, Marino D, Falsig J, Raivich G, Hovelmeyer N, Waisman A, Rulicke T, Prinz M, Priller J, Becher B, Aguzzi A. Experimental autoimmune encephalomyelitis repressed by microglial paralysis. Nat Med. 2005;11:146–152. doi: 10.1038/nm1177. [DOI] [PubMed] [Google Scholar]
- 49.Greter M, Heppner FL, Lemos MP, Odermatt BM, Goebels N, Laufer T, Noelle RJ, Becher B. Dendritic cells permit immune invasion of the CNS in an animal model of multiple sclerosis. Nat Med. 2005;11:328–334. doi: 10.1038/nm1197. [DOI] [PubMed] [Google Scholar]
- 50.Mantovani A, Locati M. Orchestration of macrophage polarization. Blood. 2009;114:3135–3136. doi: 10.1182/blood-2009-07-231795. [DOI] [PubMed] [Google Scholar]
- 51.Mikita J, Dubourdieu-Cassagno N, Deloire MS, Vekris A, Biran M, Raffard G, Brochet B, Canron MH, Franconi JM, Boiziau C, Petry KG. Altered M1/M2 activation patterns of monocytes in severe relapsing experimental rat model of Multiple Sclerosis. Amelioration of clinical status by M2 activated monocyte administration. Mult Scler. 2011;17:2–15. doi: 10.1177/1352458510379243. [DOI] [PubMed] [Google Scholar]
- 52.Reed-Geaghan EG, Reed QW, Cramer PE, Landreth GE. Deletion of CD14 attenuates Alzheimer's Disease pathology by influencing the brain's inflammatory milieu. J Neurosci. 2010;30:15369–15373. doi: 10.1523/JNEUROSCI.2637-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kigerl KA, Gensel JC, Ankeny DP, Alexander JK, Donnelly DJ, Popovich PG. Identification of two distinct macrophage subsets with divergent effects causing either neurotoxicity or regeneration in the injured mouse spinal cord. J Neurosci. 2009;29:13435–13444. doi: 10.1523/JNEUROSCI.3257-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jimenez S, Baglietto-Vargas D, Caballero C, Moreno-Gonzalez I, Torres M, Sanchez-Varo R, Ruano D, Vizuete M, Gutierrez A, Vitorica J. Inflammatory response in the hippocampus of PS1M146L/APP751SL mouse model of Alzheimer's disease: age-dependent switch in the microglial phenotype from alternative to classic. J Neurosci. 2008;28:11650–11661. doi: 10.1523/JNEUROSCI.3024-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kuo HS, Tsai MJ, Huang MC, Chiu CW, Tsai CY, Lee MJ, Huang WC, Lin YL, Kuo WC, Cheng H. Acid fibroblast growth factor and peripheral nerve grafts regulate th2 cytokine expression, macrophage activation, polyamine synthesis, and neurotrophin expression in transected rat spinal cords. J Neurosci. 2011;31:4137–4147. doi: 10.1523/JNEUROSCI.2592-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nguyen VT, Benveniste EN. Involvement of STAT-1α and ets family members in interferon-γ induction of CD40 transcription in macrophages/microglia. J Biol Chem. 2000;271:23674–23684. doi: 10.1074/jbc.M002482200. [DOI] [PubMed] [Google Scholar]
- 57.O'Keefe GM, Nguyen VT, Tang LP, Benveniste EN. IFN-γ regulation of class II transactivator promoter IV in macrophages and microglia: involvement of the suppressors of cytokine signaling-1 protein. J Immunol. 2001;166:2260–2269. doi: 10.4049/jimmunol.166.4.2260. [DOI] [PubMed] [Google Scholar]
- 58.Chitnis T, Najafian N, Benou C, Salama AD, Grusby MJ, Sayegh MH, Khoury SJ. Effect of targeted disruption of STAT4 and STAT6 on the induction of experimental autoimmune encephalomyelitis. J Clin Invest. 2001;108:739–747. doi: 10.1172/JCI12563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lee Y, Awasthi A, Yosef N, Quintana FJ, Xiao S, Peters A, Wu C, Kleinewietfeld M, Kunder S, Hafler DA, Sobel RA, Regev A, Kuchroo VK. Induction and molecular signature of pathogenic T(H)17 cells. Nat Immunol. 2012;13:991–999. doi: 10.1038/ni.2416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Quintana FJ, Jin H, Burns EJ, Nadeau M, Yeste A, Kumar D, Rangachari M, Zhu C, Xiao S, Seavitt J, Georgopoulos K, Kuchroo VK. Aiolos promotes TH17 differentiation by directly silencing Il2 expression. Nat Immunol. 2012;13:770–777. doi: 10.1038/ni.2363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Weber MS, Prod'homme T, Youssef S, Dunn SE, Rundle CD, Lee L, Patarroyo JC, Stuve O, Sobel RA, Steinman L, Zamvil SS. Type II monocytes modulate T cell-mediated central nervous system autoimmune disease. Nat Med. 2007;13:935–943. doi: 10.1038/nm1620. [DOI] [PubMed] [Google Scholar]
- 62.Ghoreschi K, Bruck J, Kellerer C, Deng C, Peng H, Rothfuss O, Hussain RZ, Gocke AR, Respa A, Glocova I, Valtcheva N, Alexander E, Feil S, Feil R, Schulze-Osthoff K, Rupec RA, Lovett-Racke AE, Dringen R, Racke MK, Rocken M. Fumarates improve psoriasis and multiple sclerosis by inducing type II dendritic cells. J Exp Med. 2011;208:2291–2303. doi: 10.1084/jem.20100977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Thone J, Ellrichmann G, Seubert S, Peruga I, Lee DH, Conrad R, Hayardeny L, Comi G, Wiese S, Linker RA, Gold R. Modulation of autoimmune demyelination by laquinimod via induction of brain-derived neurotrophic factor. Amer J Pathol. 2012;180:267–274. doi: 10.1016/j.ajpath.2011.09.037. [DOI] [PubMed] [Google Scholar]
- 64.Schulze-Topphoff U, Shetty A, Varrin-Doyer M, Molnarfi N, Sagan SA, Sobel RA, Nelson PA, Zamvil SS. Laquinimod, a quinoline-3-carboxamide, induces type II myeloid cells that modulate central nervous system autoimmunity. PloS One. 2012;7:e33797. doi: 10.1371/journal.pone.0033797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Natarajan C, Bright JJ. Peroxisome proliferator-activated receptor-gamma agonists inhibit experimental allergic encephalomyelitis by blocking IL-12 production, IL-12 signaling and Th1 differentiation. Genes Immun. 2002;3:59–70. doi: 10.1038/sj.gene.6363832. [DOI] [PubMed] [Google Scholar]
- 66.Muthian G, Raikwar HP, Rajasingh J, Bright JJ. 1,25 Dihydroxyvitamin-D3 modulates JAK-STAT pathway in IL-12/IFNgamma axis leading to Th1 response in experimental allergic encephalomyelitis. J Neurosci Res. 2006;83:1299–1309. doi: 10.1002/jnr.20826. [DOI] [PubMed] [Google Scholar]
- 67.Chen C, Liu X, Wan B, Zhang JZ. Regulatory properties of copolymer I in Th17 differentiation by altering STAT3 phosphorylation. J Immunol. 2009;183:246–253. doi: 10.4049/jimmunol.0900193. [DOI] [PubMed] [Google Scholar]
- 68.Jia Y, Jing J, Bai Y, Li Z, Liu L, Luo J, Liu M, Chen H. Amelioration of experimental autoimmune encephalomyelitis by plumbagin through down-regulation of JAK-STAT and NF-kappaB signaling pathways. PloS One. 2011;6:e27006. doi: 10.1371/journal.pone.0027006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Qin X, Guo BT, Wan B, Fang L, Lu L, Wu L, Zang YQ, Zhang JZ. Regulation of Th1 and Th17 cell differentiation and amelioration of experimental autoimmune encephalomyelitis by natural product compound berberine. J Immunol. 2010;185:1855–1863. doi: 10.4049/jimmunol.0903853. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.