

Abstract
Many pathogens infiltrate the body and initiate infection via mucosal surfaces. Hence, eliciting cellular immune responses at mucosal portals of entry is of great interest for vaccine development against mucosal pathogens. We describe a pulmonary vaccination strategy combining Toll-like receptor (TLR) agonists with antigen-carrying lipid nanocapsules [interbilayer-crosslinked multilamellar vesicles (ICMVs)], which elicit high-frequency, long-lived, antigen-specific effector memory T cell responses at multiple mucosal sites. Pulmonary immunization using protein- or peptide-loaded ICMVs combined with two TLR agonists, polyinosinic-polycytidylic acid (polyI:C) and monophosphoryl lipid A, was safe and well tolerated in mice, and led to increased antigen transport to draining lymph nodes compared to equivalent subcutaneous vaccination. This response was mediated by the vast number of antigen-presenting cells (APCs) in the lungs. Nanocapsules primed 13-fold more T cells than did equivalent soluble vaccines, elicited increased expression of mucosal homing integrin α4β7+, and generated long-lived T cells in both the lungs and distal (for example, vaginal) mucosa strongly biased toward an effector memory (TEM) phenotype. These TEM responses were highly protective in both therapeutic tumor and prophylactic viral vaccine settings. Together, these data suggest that targeting cross-presentation–promoting particulate vaccines to the APC-rich pulmonary mucosa can promote robust T cell responses for protection of mucosal surfaces.
Introduction
Mucosal immunity is critical for optimal protection against pathogens invading through the respiratory, gastrointestinal, or reproductive tracts. Infections at mucosal sites elicit immune responses concentrated at the site of exposure; for example, respiratory or gastrointestinal infections elicit high levels of specific immunoglobulin G (IgG) and secretory IgA at the airway and gut mucosal surfaces, respectively (1). In a similar manner, vaccines administered to mucosal surfaces elicit strong immune responses concentrated at the application site (2, 3). However, preclinical and clinical studies have also demonstrated crosstalk between mucosal compartments, permitting immunization at one mucosal surface to establish immunity at distal mucosal sites (2). Together, these findings suggest that needle-free vaccination strategies such as aerosol or intranasal delivery might be capable of protecting against not only airway or systemic infections but also gastrointestinal or reproductive tract pathogens.
Neutralizing antibody responses are clinical correlates of protection for existing mucosal vaccines such as FluMist and the oral polio vaccine (4). However, cellular immunity may also have an important role to play in mucosal protection through memory T cell populations that reside in mucosal tissues and respond rapidly to infection directly at sites of pathogen entry (5, 6). Evidence suggests that effector memory CD8+ T cells (TEM) are composed of both noncirculating memory cells that reside permanently (or with low turnover rates) in peripheral tissues and migratory cells that traffic between the periphery and the blood (5). Although effector memory cells show a lower proliferative capacity than do central memory T cells (TCM) that recirculate through the blood and lymphoid organs, TEM can immediately recognize and kill infected target cells. These cells therefore can facilitate early containment of nascent infections, and tissue-resident T cells have been shown to protect against respiratory (7), intravaginal (8), and skin infections (9). Given these findings, the design of vaccines capable of eliciting robust effector memory populations in target mucosal tissues is of great interest.
Safety concerns with attenuated live replicating vaccine vectors motivate the question of whether subunit vaccines based on purified pathogen components may be capable of achieving similar TEM-biased mucosal immune responses. Pulmonary delivery of plasmid DNA formulated with polyethyleneimine (10), lipid complexes (11), or liposomes (12) has been shown to elicit mucosal immune responses characterized by mucosal IgA and CTL (cytotoxic T lymphocyte) responses in genital, rectal, and gut-associated tissues in mice. In addition, it has been shown that subunit vaccines composed of HIV peptides and experimental adjuvants administered via intrarectal (13) or intranasal (14) routes can elicit HIV-specific cytotoxic T cells resident in mucosal tissues. However, clinical trials to date have failed to replicate the immunogenicity of DNA vaccines seen with small-animal models (15); this is thought to be due in part to poorer transfection and differences in DNA-sensing Toll-like receptor 9 (TLR9) expression in humans and mice. Polypeptide vaccines, on the other hand, generally remain substantially less immunogenic than live vectors. Hence, vaccine systems that can elicit more potent immune responses with whole proteins or peptides as antigens are being investigated.
Most of the studies of mucosal protein vaccines have focused exclusively on humoral immune responses, in part because protein vaccines have traditionally elicited strong humoral immunity but weak to nonexistent cytotoxic T cell responses. However, strategies based on the use of synthetic nanoparticles to promote cross-presentation of antigen have begun to show promise in eliciting robust CTL responses. We recently reported the synthesis of interbilayer-crosslinked multilamellar vesicles (ICMVs), nanoscale capsules with walls composed of stacked lipid bilayers “stapled” to one another via bilayer-to-bilayer chemical cross-links, which exhibited greatly enhanced stability in serum compared to traditional liposomes (16, 17). Parenteral immunization with ICMVs coloaded with antigen and a TLR agonist used in a Food and Drug Administration (FDA)–approved HPV vaccine, monophosphoryl lipid A (MPLA), promoted both strong humoral and CTL responses. This was thought to be mediated by enhanced antigen delivery and persistence in draining lymph nodes (dLNs) and efficient cross-presentation of ICMV-associated antigen by LN-resident dendritic cells (DCs), resulting in elicitation of a TCM-biased response (16, 17). However, antigen delivery to LNs could still be enhanced because the vast majority of antigen-presenting cells (APCs) in LNs still do not acquire antigen after parenteral ICMV immunizations (16). In addition, recent studies suggest that high levels of antigen presentation and prolonged early antigen exposure can skew cellular immunity toward an effector memory response (18). Therefore, we sought to further improve antigen accumulation and target antigen delivery to mucosal tissue–draining LNs to drive potent TEM-biased responses directed against mucosal tissues.
Efficient draining of nanoparticle carriers is typically engineered by optimizing particle properties (19), for example, by choosing particle size for efficient trafficking to lymphatics, introducing PEGylation to reduce matrix binding, or functionalizing particles with targeting moieties to promote uptake by DCs. Here, we tested a complementary strategy, optimizing the site of vaccine administration instead of the vaccine carrier. Exploiting the immunomonitoring mechanisms of the alveolar spaces, we vaccinated mice by pulmonary administration of ICMVs to the lung mucosa, where high densities of APCs constantly sample antigen across the airway epithelium (20). We found that nanocapsules were efficiently phagocytosed by lung APCs, a subset of which trafficked antigen to dLNs, leading to large increases in the amount of antigen delivered to LN DCs, compared to the same vaccines given via parenteral vaccination. Antigen delivered in ICMVs was detected in dLNs for at least 7 days, whereas pulmonary immunization with soluble vaccines led to rapid antigen clearance. These changes in antigen trafficking correlated with robust generation of TEM cells, which homed to the local lung tissue, systemic lymphoid compartments, and distant mucosal sites. T cell responses elicited by pulmonary ICMV vaccination enhanced protection in therapeutic tumor and prophylactic viral challenge models. These results demonstrate a strategy for eliciting potent mucosal TEM responses with protein- or peptide-based vaccines and provide a complementary approach for promoting protective T cell responses at mucosal surfaces as a stand-alone vaccine or component of prime-boost regimens with potent viral vectors.
Results
Pulmonary vaccination leads to nanocapsule transport to dLNs and enhances T cell priming
We first identified the optimal dose and configuration of molecular adjuvants to combine with ICMV capsules in pulmonary vaccination. We focused on the TLR3 agonist polyI:C (polyinosinic-polycytidylic acid) and the TLR4 agonist MPLA based on our previous results with MPLA-loaded ICMVs (16, 17) and studies of polyI:C demonstrating stimulation of airway epithelial cells (21) and promotion of DC cross-presentation (22). Pilot immunizations of C57BL/6 mice by intratracheal instillation showed that the strongest T cell responses were primed by the combination of soluble polyI:C mixed with ICMVs encapsulating the model antigen ovalbumin (OVA), with MPLA embedded in the capsule walls (fig. S1). ICMV vaccination with this combination of TLR agonists elicited large frequencies of antigen-specific CD8+ T cells [identified by staining with SIINFEKL peptide–MHC (major histocompatibility complex) tetramers (OVA-tetramers)] and cytokine-producing CD8+ T cells in the blood, spleen, and lungs. We thus focused our studies on this configuration of antigen and adjuvant for nanocapsule vaccination (hereinafter “ICMV vaccines”) with 10 μg of OVA, 0.3 of μg MPLA, and 10 μg of polyI:C.
To evaluate ICMV transport to dLNs, we administered nanocapsules intratracheally and analyzed fluorescent Alexa647-OVA+ cells in the lungs and in the draining mediastinal LNs (mdLNs). For comparison, we immunized mice subcutaneously at the tail base, a site where ICMVs elicit potent systemic immune responses and that drains to the inguinal LNs (ingLNs). Lung OVA+ macrophages, identified as highly autofluorescent CD11c+F4/80+MHC-II− cells, internalized more OVA than did lung DCs (characterized as non-autofluorescent CD11c+F4/80−CD11b+MHC-II+ cells), but 40 ± 6% (SEM) of lung DCs acquired antigen (fig. S2, A and B). Comparative analysis of antigen delivery to the dLNs showed that pulmonary immunization led to more OVA+ DCs in the mdLNs compared to OVA+ DCs in ingLNs after subcutaneous immunization (Fig. 1A). To obtain an aggregate measure of the total amount of OVA taken up by LN DCs that would account for both the number of antigen-acquiring cells and the total protein internalized, we defined relative OVA uptake as the product of the number of OVA+ DCs multiplied by their mean OVA median fluorescence intensity (MFI). Pulmonary ICMV administration led to a 58-fold increase in the total amount of antigen delivered to dLN DCs compared to parenteral vaccination (Fig. 1A), reflecting the enormous population of OVA+ DCs in the lung tissue itself. OVA+ DCs in the lungs and mdLNs after ICMV vaccination were MHC-II+ and heterogeneous for CD11b expression, suggesting that no single DC population acquired the particles (fig. S2C).
Fig. 1. Pulmonary vaccination increases DC uptake of nanocapsules, enhances T cell priming, and promotes imprinting of mucosal homing receptors.
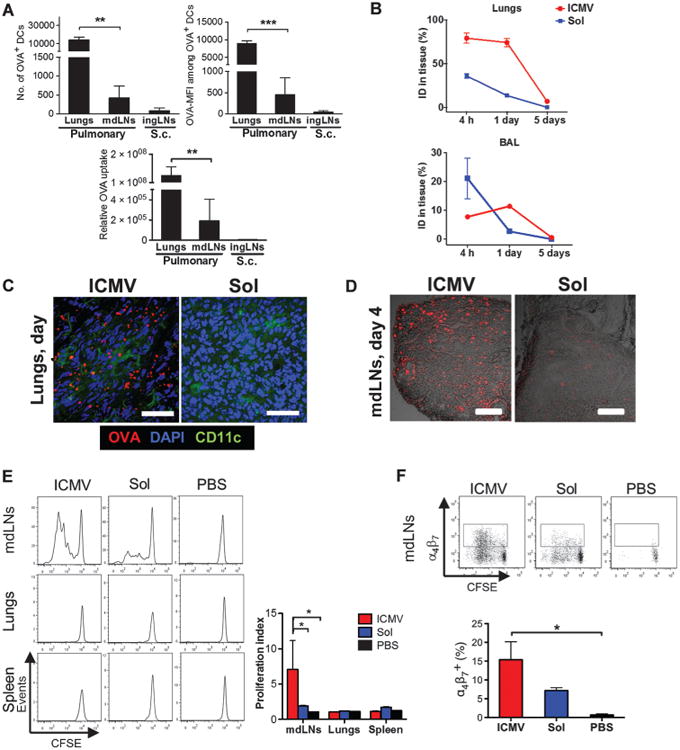
C57BL/6 mice were immunized subcutaneously or intratracheally with OVA in either soluble or ICMV formulations with MPLA and polyI:C adjuvants. (A) Numbers of DCs containing OVA and the MFI of OVA in individual cells were measured in the lungs and mdLNs on day 2 after intratracheal administration. Numbers were compared to those in ingLNs after subcutaneous (s.c.) administration. Relative OVA uptake was calculated by multiplying OVA+ DC counts by mean OVA-MFI. (B) The amount of antigen remaining in the lungs and BAL was measured over time by fluorescence spectroscopy. ID, injected dose. (C and D) Representative cryosections after intratracheal immunization with fluorescent OVA (red) from lungs on day 1 (C; scale bars, 50 μm) and mdLNs on day 4 (D; scale bars, 200 μm). Lung sections were costained with anti-CD11c and 4′,6-diamidino-2-phenylindole (DAPI). (E and F) Tissues harvested 3 days after intratracheal immunization were homogenized, and recovered cells were cocultured with CFSE-labeled OT-I CD8+ T cells. (E) The proliferation index was calculated as the total number of cell divisions divided by the number of cells that went into division. (F) Expression of α4β7 on OT-I CD8+ T cells determined by flow cytometry after 3 days of coculture. Data are means ± SEM of two to three independent experiments conducted with n = 3 to 4 animals per group. *P < 0.05, **P < 0.01, ***P < 0.001, by one-way analysis of variance (ANOVA) (A and F) or two-way ANOVA (E).
To assess whether particulate formulation is important for LN delivery through the lung mucosa, we administered ICMV vaccines or the same dose of antigen and TLR agonists in soluble form and characterized antigen accumulation in the lung tissue, bronchoalveolar lavage (BAL) fluid, and mdLNs. Soluble antigen was rapidly cleared from both the BAL and lungs, whereas ∼75% of the antigen dose delivered by ICMVs was still present in the lungs at 24 hours (Fig. 1, B and C). Histological analysis showed that soluble antigen was absent from mdLNs 4 days after vaccine administration but still prominent in the mdLNs of ICMV-treated groups (Fig. 1D). ICMV-delivered antigen was detectable in mdLNs until day 7.
To determine whether the persistence of ICMV-associated antigen in mdLNs also led to prolonged antigen presentation, we analyzed cross-priming of T cells by lung and spleen APCs after vaccination. Groups of mice were immunized as before, and after 3 days, cells from the mdLNs, lungs, or spleens were cocultured with carboxyfluorescein diacetate succinimidyl ester (CFSE)–labeled naïve syngeneic OVA-specific mouse OT-I CD8+ T cells. Only mdLN cells stimulated OT-I cell proliferation, and APCs from nanocapsule-immunized mice elicited 3.7-fold greater proliferation than did those from soluble vaccine–immunized animals (Fig. 1E). In addition, APCs from ICMV-immunized mice induced a nearly twofold higher frequency of OT-I cells expressing the mucosal homing integrin α4&β7+ (Fig. 1F). Thus, mucosal vaccination with nanocapsules provided greater antigen targeting to LNs than did parenteral vaccination and enhanced both cross-presentation by APCs and imprinting of mucosal homing receptors on CD8+ T cells relative to equivalent soluble vaccines.
Mucosal nanocapsule vaccination enhances T cell expansion and functionality and establishes memory
To analyze the expansion, persistence, and homing patterns of antigen-specific T cells after pulmonary immunization, we immunized animals with ICMV or soluble vaccines on days 0 and 28, and evaluated the frequency of OVA-specific CD8+ T cells over time. ICMVs triggered much greater T cell responses than did soluble vaccines, with 13-fold more OVA-tetramer+ CD8+ T cells in the mdLNs and 5.3-fold more in the lungs and spleen at the post-boost peak on day 35 (Fig. 2A). ICMV-immunized animals also had a greater frequency of T cells producing multiple cytokines (Fig. 2B) and a greater frequency of granzyme B+ cells (Fig. 2C). Eleven weeks after priming, nanocapsule vaccination established a substantially greater memory population than did soluble immunization, with higher frequencies of OVA-specific CD8+ cells in both lungs and spleen (Fig. 2A).
Fig. 2. ICMV vaccination elicits durable CD8+ T cell responses with enhanced functionality.
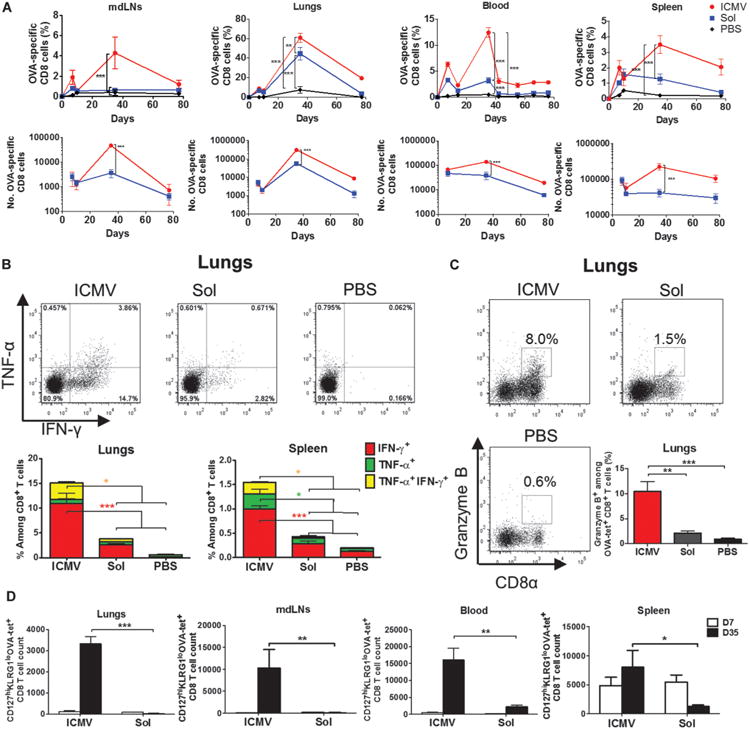
C57BL/6 mice were immunized intratracheally with ICMV or soluble OVA vaccines on day 0 and boosted on day 28. (A) Frequencies and absolute numbers of OVA-specific CD8+ T cells in mdLNs, lungs, blood, and spleen were tracked over time by SIINFEKL-MHC tetramer staining. (B and C) On day 35, cells isolated from lungs and spleen were restimulated ex vivo with SIINFEKL peptide, and expression of intracellular interferon-γ (IFN-γ) and/or tumor necrosis factor–α (TNF-α) (B) and granzyme B (C) among CD8+ T cells was determined by flow cytometry. (D) Frequency of long-lived effector memory (CD127hiKLRG1lo) CD8+ T cell precursors in multiple compartments was determined on day 7 after prime and boost by flow cytometry analysis of tetramer+ CD8+ T cells. Data are means ± SEM of two to three independent experiments conducted with n = 3 to 4 animals per group. *P < 0.05, **P < 0.01, ***P < 0.001, by two-way ANOVA (A, B, and D) or one-way ANOVA (C).
Consistent with these data, by analyzing the expression of markers associated with long-lived memory cell precursors (23) on day 7 after boost, we found that ICMV immunization increased the absolute number of OVA-specific CD127hiKLRG1lo T cells by ∼110-fold in the lungs and mdLNs compared with soluble vaccines (Fig. 2D), indicating a much greater population of T cells entering the memory pool after vaccination with ICMVs. Moreover, we confirmed that the enhanced T cell responses observed were not achieved at the expense of humoral immunity because antibody responses trended toward higher titers after ICMV compared to soluble pulmonary vaccination (fig. S3).
Mucosal immunization with ICMVs induces disseminated effector memory–biased immunity
To assess the capacity of nanocapsule vaccination to induce distal mucosal immunity, we first used an adoptive transfer model and traced the proliferation and trafficking of luciferase-expressing OT-I CD8+ T cells (OT-I–luc). Twenty-four hours after transfer of naïve OT-I–luc T cells, animals were immunized via intratracheal or subcutaneous routes. By day 3 after pulmonary vaccination, ICMVs or soluble vaccines primed OT-I–luc expansion only in the mdLNs, whereas subcutaneous vaccination induced T cell proliferation confined to the draining ingLNs (Fig. 3A). By day 5, the pulmonary ICMV group showed the greatest OT-I expansion, with OT-I–luc cells detected not only in LNs and spleen but also across the gut and in the reproductive tract (Fig. 3, A and B). ICMV vaccination uniquely promoted strong homing of OT-I–luc cells into the cecum and Peyer's patches along the small intestine as assessed by whole-tissue imaging (Fig. 3, A and C) and tetramer staining (Fig. 3D). Further, only pulmonary nanocapsule vaccination induced significant expression of the mucosal homing integrin α4β7 in tetramer+ peripheral blood OT-I cells by day 5 (Fig. 3E).
Fig. 3. Pulmonary ICMV immunization induces dissemination of CD8 T cells from mdLNs to distant mucosal tissues.
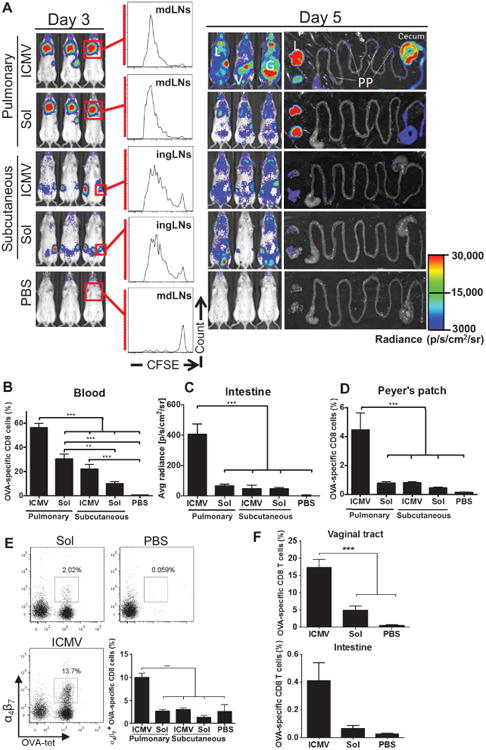
(A to E) OT-I–luc CD8+ T cells were adoptively transferred into C57BL/6 mice (n = 5) 1 day before ICMV or soluble OVA vaccine immunization via intratracheal or subcutaneous routes. (A) Trafficking and proliferation of OT-I–luc T cells were monitored by flow cytometry and bioluminescence imaging on days 3 and 5 after immunization. Flow cytometry histograms show representative CFSE dilutions in transferred T cells on day 3 after immunization at dLNs (mdLNs for intratracheal vaccines and ingLNs for subcutaneous vaccinations). Lungs (L) and gastrointestinal tracts (G) were dissected on day 5 and imaged to identify T cell localization. V, vaginal tract; PP, Peyer's patches. (B to E) Flow cytometry and imaging analyses of OT-I T cells on day 5. Shown are frequencies of OVA-specific CD8+ T cells in blood (B) and Peyer's patches (D), quantification of bioluminescence signal from small intestines (C), and integrin α4β7+ cells (E) in blood. (F) C57BL/6 mice (no adoptive transfers, n = 3 per group) were immunized with ICMV or soluble OVA vaccines on days 0 and 28. The frequency of OVA-specific CD8+ T cells in the vaginal tract and in the small intestine on day 7 after boost was assessed by flow cytometry. Data are means ± SEM of two independent experiments. **P < 0.01, ***P < 0.001, by one-way ANOVA.
To confirm that similar enhanced mucosal homing was elicited for endogenous T cells after mucosal ICMV vaccination, we analyzed antigen-specific memory CD8+ T cells in the reproductive tract and gut post-boost after pulmonary immunization of C57BL/6 mice (no adoptive transfers). Nanocapsule immunization elicited greater frequencies of OVA-specific CD8+ T cells in the reproductive tract and gut relative to soluble vaccines (Fig. 3F and fig. S4). To characterize the memory phenotype of this disseminated T cell response, we analyzed memory markers of OVA-specific T cells at 11 weeks after vaccination. Both soluble and ICMV vaccines adjuvanted with MPLA and polyI:C elicited T cells biased toward an effector memory (CD44hiCD62Llo) phenotype, but mucosal nanocapsule vaccination elicited more TEM cells in both systemic and mucosal tissues compared to pulmonary soluble vaccines or parenteral nanocapsule vaccines (fig. S5). Thus, pulmonary ICMV vaccination drives a T cell response strongly biased toward effector memory populations, with lymphocytes expressing mucosal homing receptors to populate both local and distal mucosal tissues.
Mucosal nanocapsule vaccination enhances protective immune responses in tumor and viral challenge models
To determine whether the strong TEM responses elicited by pulmonary vaccination with ICMVs enhanced the protection elicited by subunit vaccines, we tested ICMV efficacy in both therapeutic tumor and prophylactic viral challenge models. First, we inoculated C57BL/6 mice subcutaneously with OVA-expressing B16F10 mouse melanoma tumor cells and administered either soluble or ICMV OVA pulmonary vaccines on days 3 and 10. Soluble OVA vaccination delayed tumor growth but did not improve the ultimate survival of animals compared to phosphate-buffered saline (PBS)–treated controls (Fig. 4A). In contrast, ICMV vaccination led to 100% rejection of tumors and long-term survival of all mice.
Fig. 4. Mucosal ICMV vaccination enhances protection against tumors and pulmonary viral infection.
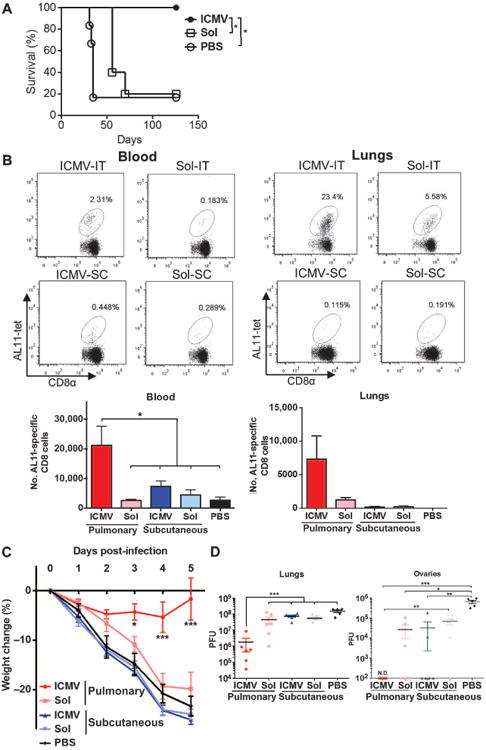
(A) C57BL/6 mice (n = 5 to 6 per group) inoculated with 5 × 105 B16F10-OVA cells subcutaneously in the flank on day 0 were treated on days 3 and 10 with intratracheal administration of ICMVs or soluble OVA vaccines. Survival of tumor-bearing mice was tracked for 18 weeks. *P < 0.05, log-rank (Mantel-Cox) test. (B) C57BL/6 mice were immunized subcutaneously (SC) or intratracheally (IT) on days 0 and 28 with AL11 and PADRE peptide ICMV or soluble vaccines. Frequencies of AL11-specific CD8+ T cells in blood and lungs were determined on day 77 by flow cytometry. (C and D) Groups of mice were immunized as in (B) and then challenged by intratracheal administration of SIV gag–expressing vaccinia virus (1 × 106 PFU) on day 42. (C) Body weight changes over time. (D) Viral titer counts in lungs and ovaries harvested 5 days after infection; dots show titers from individual animals. Data are means ± SEM with n = 3 to 7 animals per group. *P < 0.5, **P < 0.01, ***P< 0.001, by two-way ANOVA (C) or one-way ANOVA (B and D). N.D., not detectable.
We next assessed the efficacy of ICMVs as a mucosal peptide vaccine in a mucosal viral challenge model, using a viral target antigen instead of OVA. Groups of mice were immunized intratracheally with vaccines composed of an immunodominant CTL epitope from simian immunodeficiency virus (SIV) gag, the antigen AL11 (AAVKNWMTQTL) (24), and the universal CD4+ T cell helper epitope PADRE (25), each encapsulated in separate ICMVs or in soluble form together with MPLA and polyI:C on days 0 and 28. Control mice received equivalent vaccines administered subcutaneously. Pulmonary ICMV vaccination significantly increased the numbers of antigen (AL11)–specific cells in both blood and lung tissue by 11 weeks after prime compared to soluble vaccines or subcutaneous administration of ICMVs (Fig. 4B). To test the protective efficacy of this response, we challenged animals intratracheally with SIV gag–expressing vaccinia virus on day 43. Mice receiving subcutaneous ICMV vaccines or pulmonary soluble peptide vaccines showed steady weight loss similar to unvaccinated control mice, reaching the criterion for euthanasia (weight loss >20%) by day 5 after challenge (Fig. 4C).
Plaque assays on the lungs and ovaries at day 5 showed that subcutaneous or soluble pulmonary vaccines had a minor impact on viral titers in the lungs compared to PBS-treated mice, and most of the animals showed viral dissemination to the ovaries at high levels (Fig. 4D). In contrast, mice that received the mucosal ICMV vaccines exhibited only minor weight loss that was fully recovered by day 5 with no mortality, achieved a 2-log reduction in vaccinia plaque-forming units (PFU) in the lungs, and completely blocked dissemination of virus to the ovaries (Fig. 4, C and D). Thus, pulmonary nanocapsule vaccination provided substantially enhanced protection against a mucosal viral challenge compared to soluble vaccines administered at the same site or parenteral nanocapsule vaccination.
Memory CD8+ cells elicited by pulmonary vaccination were ∼95% effector memory cells; this was true for AL11 vaccines as well as OVA (figs. S5 and S6). However, LN-homing TCM are capable of massive proliferation on antigen challenge (6), making it possible that a minor population of TCM could play an important role in the enhanced protection of ICMV vaccines. Thus, we characterized the cells mediating the antiviral protection elicited by ICMV vaccination. Antibody depletion of CD8+ or CD4+ T cells from vaccinated mice before challenge (fig. S7A) revealed that CD8+, but not CD4+, T cells were required for protection from vaccinia challenge (as revealed by protection from weight loss) (Fig. 5A). To determine whether protection was mediated by TCM or TEM cells, we treated vaccinated mice before challenge with FTY720, a sphingosine-1-phosphate analog that traps CCR7+ naïve and TCM cells in LNs by blocking LN egress, thereby rendering TCM incapable of participating in antiviral responses in tissues (26). As expected, FTY720-treated mice showed a strong depletion of circulating (mostly naïve) CD8+ T cells from the blood (Fig. 5B) and reduced the number of AL11-specific TCM in the blood by ∼10-fold (fig. S7B). However, tissue-resident AL11-specific TEM remained unaffected after treatment with FTY720 (Fig. 5B). Subsequent challenge of vaccinated mice showed that survival (Fig. 5C) and viral replication in the lungs and ovaries (Fig. 5D) was identical for control and FTY720-treated mice, confirming that protection was not dependent on TCM.
Fig. 5. ICMV vaccination confers protection via CD8+ TEM cells.
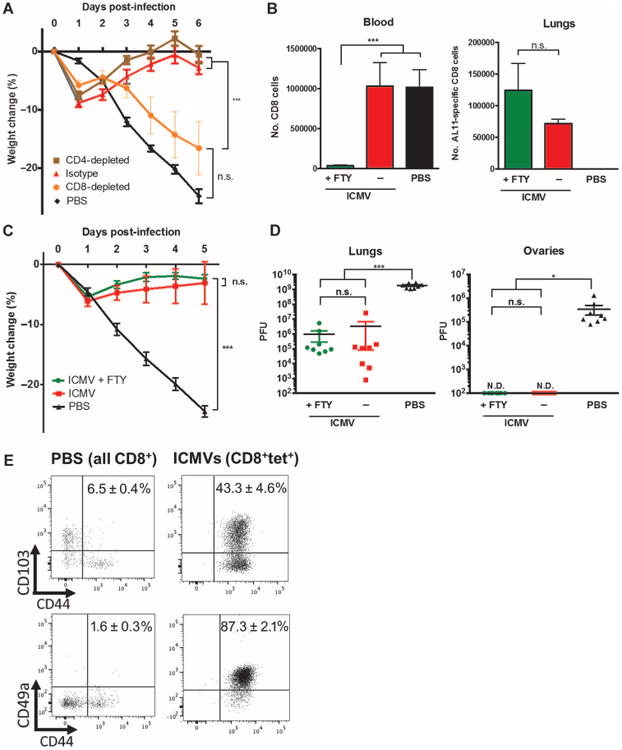
(A) C57BL/6 mice were immunized intratracheally with AL11/PADRE-loaded ICMVs as in Fig. 4B. Immunized animals were treated with depleting antibodies against CD4, CD8, or isotype controls on days 40, 42, 44, and 46 and challenged with vaccinia virus (1 × 106 to 2 ×106 PFU) on day 42. Animal weights were tracked for 6 days after challenge. (B to D) Groups of mice were immunized as in Fig. 4B; controls received only PBS administration. Some animals were treated with FTY720 on days 38 to 47 via oral gavage. All animals were challenged with vaccinia virus (1 × 106 to 2 × 106 PFU) on day 42. (B) Total CD8+ T cells in blood and antigen-specific T cells in the lungs were quantified on day 41. (C) Animal weights over time after challenge. (D) Viral plaques in the lungs and ovaries on day 5 after viral challenge. (E) Mice were immunized with ICMV AL11/PADRE vaccines intratracheally as in Fig. 4B. Expression of CD103/CD49a and CD44 by antigen-specific CD8+ T cells in immunized animals and total CD8+ T cells in PBS control animals were assessed by flow cytometry on day 41. Representative plots are shown, with mean percentage ± SEM of CD44+CD103+or CD44+CD49a+ among analyzed populations. Data in (A) to (D) are means ± SEM with n= 4 to 8. *P <0.5, ***P< 0.001, by one-way ANOVA (B and D) or two-way ANOVA (A and C). n.s., not significant; N.D., not detectable.
To determine whether tissue-resident TEM cells could play a role in the pulmonary protection from vaccinia, we assessed the expression of CD49a and CD103—adhesion receptors that promote entry into the lung (27) and retention in the lung tissue (28), respectively. Most of the AL11-specific T cells taken from the lungs after ICMV vaccination expressed CD49a, and 40 to 50% of these cells expressed CD103 (Fig. 5E), consistent with a tissue-resident phenotype. Thus, ICMV vaccines mediated effective protection after pulmonary vaccination via the generation of circulating and tissue-resident TEM cell populations that could protect the local tissue without the need for restimulation in dLNs.
Pulmonary nanocapsule vaccination is safe and well tolerated
Although several recent studies have reported the safe use of TLR agonists in intratracheal (29) or intranasal administration (30, 31), potential toxicity in the respiratory tract is still a chief concern for mucosal vaccine development. Intratracheal immunization with ICMV vaccines elicited no significant weight loss after prime or boost (fig. S8). There was a transient presence of interleukin-6 (IL-6), TNF-α, and IFN-γ that resolved within 24 hours, which was similar for soluble and nanocapsule vaccines (Fig. 6A). Chemokines associated with immune cell recruitment were also transiently elevated in lung tissues of vaccinated mice (fig. S9). IL-1β, a potent innate inflammatory cytokine, was detected only transiently after boost and at fourfold lower levels with ICMVs compared to soluble vaccines (fig. S9). Cytokines were mainly confined to the site of administration and were not detected in serum. IL-2, IL-4, IL-10, and IL-17 were only detected at background levels in the lungs, BAL fluid, and serum (fig. S9).
Fig. 6. Pulmonary nanocapsule vaccination is nontoxic and does not induce lung pathology.
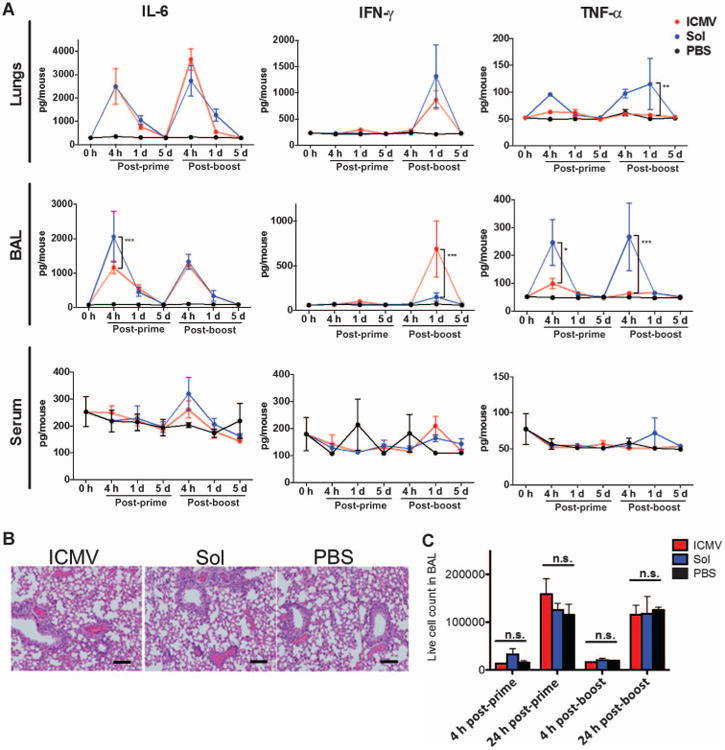
Groups of C57BL/6 mice (n = 3 to 4 per group) were immunized with ICMV or soluble OVA vaccines as in Fig. 1. (A) Supernatants from lung homogenates, BAL, and sera were collected over time and analyzed for cytokine levels by bead-based enzyme-linked immunosorbent assay (ELISA). (B) Representative hematoxylin and eosin (H&E) staining on lung sections on day 1 after boost (day 29 after prime). Control mice received PBS. Scale bars, 100 mm. (C) Enumeration of live cells infiltrating the BAL after prime and boost. Data are means ± SEM of two independent experiments. **P < 0.01, ***P < 0.001, n.s. not significant, by two-way ANOVA.
Histologically, lung tissues from mice vaccinated with ICMVs or soluble antigen were indistinguishable from saline-treated controls (Fig. 6B). Scoring of tissue sections from groups of mice by a blinded pathologist showed no signatures of toxicity (eosinophils, alveolar damage, and necrosis) but did show evidence of vaccine responses after boost [perivascular leukocyte infiltrates and expansion of bronchial-associated lymphoid tissue (BALT)] (Table 1). Interstitial pneumonitis and granulomas were absent in all mice, as well as pneumonia, bronchitis, and pleuritis (only present in rare mice in all groups). In line with the histology, quantification of total infiltrating leukocytes in BAL at 4 or 24 hours after prime and after boost showed no significant difference from saline-treated control animals for both vaccine groups (Fig. 6C). Thus, intratracheal immunization with either soluble or ICMV vaccine induced only transient inflammatory responses in the lungs and no tissue toxicity.
Table 1. Histopathological analysis of pulmonary immunization responses.
C57BL/6 mice (n = 4 to 5 per group) were immunized with OVA-ICMVs, soluble OVA vaccines, or PBS on days 0 and 28. Lung tissues (one from each mouse) collected on days 1, 7, 29, and 35 were stained with H&E. A blinded pathologist scored the toxicity (eosinophils, alveolar damage, and necrosis) and vaccine responses (perivascular leukocyte infiltrates and expansion of BALT). +, present; −, absent; +/−, indeterminate. The number of samples with each score is shown.
| Toxicity/hypersensitivity | Immune response | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
|
|
|||||||||||||||
| Eosinophil infiltrates | Diffuse alveolar damage | Necrosis | BALT expansion | Perivascular leukocyte infiltration | ||||||||||||
| Sample | Day | + | − | +/− | + | − | +/− | + | − | +/− | + | − | +/− | + | − | +/− |
| OVA-ICMV | 1 | 4 | 4 | 4 | 4 | 1 | 3 | |||||||||
| 7 | 4 | 4 | 4 | 4 | 1 | 3 | ||||||||||
| 29 | 4 | 4 | 4 | 4 | 4 | |||||||||||
| 35 | 5 | 5 | 5 | 3 | 2 | 5 | ||||||||||
| Soluble OVA vaccine | 1 | 4 | 4 | 4 | 4 | 2 | 1 | 1 | ||||||||
| 7 | 4 | 4 | 4 | 1 | 3 | 3 | 1 | |||||||||
| 29 | 4 | 4 | 4 | 4 | 4 | |||||||||||
| 35 | 5 | 5 | 5 | 2 | 3 | 5 | ||||||||||
| PBS | 1 | 4 | 4 | 4 | 4 | 4 | ||||||||||
| 7 | 4 | 4 | 4 | 4 | 4 | |||||||||||
| 29 | 4 | 4 | 4 | 4 | 4 | |||||||||||
| 35 | 4 | 4 | 4 | 4 | 4 | |||||||||||
Discussion
Nanoparticles are of great interest as carriers of subunit vaccines owing to their ability to codeliver antigen and molecular adjuvants, promote cross-presentation of antigen to CD8+ T cells, and augment humoral immunity (19). However, as with any vaccine formulation, the efficiency of particle-associated antigen delivery to DC populations in the LNs plays a major role in determining the strength of the immune response. To date, strategies to enhance responses elicited by synthetic nanoparticle vaccines have largely focused on engineering the vaccine carrier itself, for example, to obtain optimal surface chemistry or particle size (19). However, the site of vaccine administration may also play a critical role in the response to particulate vaccines. Pulmonary vaccination with small (30 nm) antigen-conjugated poly(propylene sulfide) nanoparticles was shown to promote strong local mucosal T cell responses in the lungs of mice that had greater protection against tuberculosis challenge compared to the same vaccine given intradermally (32). Antigen-encoding DNA/poly(ethyleneimine) polyplex vaccines protected mice from lethal flu challenge when vaccinated in the lungs, but not when given intramuscularly (10). However, whether these outcomes after pulmonary versus parenteral immunization reflect differences in antigen trafficking, overall magnitude of T cell priming, differences in T cell expression of homing receptors, or other factors has remained unclear. In addition, the absolute magnitude of antigen-specific T cell responses elicited by these vaccines remains modest.
A recent clinical study with human papillomavirus (HPV) viruslike particles demonstrated that mucosal vaccination by bronchial aerosolization targeting the lower respiratory tract elicited strong humoral immune responses with high antigen-specific IgG and IgA titers in sera and the genital tract, whereas true intranasal vaccination with nasal nebulization was poorly immunogenic (33). Because intranasal vaccination is an efficient route to elicit humoral immune responses to soluble antigens, this finding was attributed to the substantially larger population and superior ability of DCs in the trachea and lungs to acquire nanoparticles compared to DCs in the nasal-associated lymphoid tissues. The lung mucosa is a site densely lined with APCs, which have been shown to constitutively and actively collect antigen from across the epithelial barrier for delivery to the dLNs (20).
Motivated by such data, here we evaluated the impact of targeting lipid nanocapsule vaccines directly to the lung mucosa via intratracheal instillation, an experimentally facile model of human aerosol vaccination. We found that pulmonary vaccination increased antigen transport to APCs by ∼60-fold relative to parenteral injection. Increased antigen delivery coupled with enhanced cross-presentation induced by the lipid nanocapsule vaccine led to potent CD8+ T cell expansion and induction of mucosal homing receptors after pulmonary ICMV vaccination. Mucosal nanocapsule vaccination primed CD8+ T cells with greater functionality than did equivalent soluble pulmonary vaccines and generated a highly effector memory–biased T cell response with antigen-specific CD8+ T cells persisting at both local and distal mucosal effector sites at about fivefold greater levels than did soluble vaccines. Antigen-specific TEM cells taken from the lungs after ICMV vaccination expressed CD49a, and 40 to 50% expressed CD103, similar to previous reports of lung tissue–resident memory cells (27, 28).
Signals controlling TCM/TEM fate decisions of memory cells are still being defined but are believed to be determined at the early stages of vaccination (34). Initial signal strength, concentration of antigen, stimulation duration, and costimulatory molecule expression can determine effector/central memory differentiation (18). Strong antigen stimulation promotes cell survival and responsiveness to IL-7 and IL-15, which is closely associated with expansion of TEM (18). In addition, studies artificially altering the relative abundance of antigen-specific T cells versus antigen-bearing DCs by T cell adoptive transfer or Fms-like tyrosine kinase 3 ligand (Flt3L) treatment (35, 36) have shown that low DC/T cell ratios preferentially generate TCM, whereas high ratios tend to generate TEM. Consistent with the above evidence, we found that administration of antigen encapsulated in ICMVs to increase and prolong antigen delivery, combined with administration to a site with high APC density (lungs), successfully stimulated a TEM-biased CD8 immune response.
Equally important with efficacy is vaccine safety, especially for targeting mucosal sites. Several preclinical studies as well as clinical trials have demonstrated that aerosolized vaccines had comparable safety profiles to parenteral vaccines (37). Here, we have shown that two TLR agonists, MPLA and polyI:C, could be combined in pulmonary delivery into the lungs without evidence for destructive pathology, systemic inflammation, or clinical signs of distress. These findings are consistent with the established low toxicity of MPLA, which is an adjuvant in an FDA-approved HPV vaccine and a hepatitis B vaccine licensed in Europe. On the other hand, a clinical formulation of polyI:C known as polyICLC has been shown to elicit both local reactions and systemic flu-like symptoms in a small fraction of recipients (38). Derivatives of polyI:C engineered for further safety are in various clinical trials that could further enable this TLR combination to be used safely in the clinic (39). Unexpectedly, our initial studies (fig. S1) revealed that ICMVs mixed with soluble polyI:C elicited stronger CTL responses than did particles encapsulating the TLR3 agonist. Formulation of the vaccine with soluble TLR agonist is advantageous for translation to humans because the dose of polyI:C could be titrated with a single good manufacturing practice formulation of nanocapsules to identify the optimal human TLR agonist dose in clinical trials.
This study used mice as a relevant animal model for detailed mechanistic characterization of the immune response to pulmonary ICMV vaccination. However, it will be important to verify in future research whether these findings fully translate to large-animal models (for example, macaques) and humans. The trafficking and functional properties of tissue-resident versus circulating memory cells, in particular, are still being defined in both small animals and humans, and it remains to be determined if there are any important species-dependent differences in response to vaccines that promote distinct memory phenotypes.
In conclusion, we have demonstrated a strategy to elicit strong effector memory–biased CD8+ T cell responses to protein and peptide vaccines by combining a potent cross-presentation–promoting lipid nanocapsule delivery system with needle-free administration to the lung mucosal surfaces, which are highly enriched in antigen-sampling, migratory APC populations. This vaccine strategy primes TEM that disseminate to local mucosal tissues, systemic compartments, and distal mucosal sites and provides enhanced protection to both local and systemic challenges compared to nonparticulate pulmonary vaccination. This approach is likely generalizable to other nanoparticle vaccine platforms in preclinical and clinical development, and may provide a strategy for protection of distal sites such as the rectal and reproductive tract mucosa without the danger of directly inducing inflammation at these sites by local vaccination. A key next step is evaluation of the safety and efficacy of ICMV vaccination in a large-animal model closer to humans using relevant disease antigens (for example, HIV or mucosal tumor antigens), which could provide critical guidance for clinical translation of this vaccination approach.
Materials and Methods
Study design
Rationale and design of study
Immunization studies were designed to assess the immunogenicity (strength of T cell and antibody responses) and/or protection against challenge with vaccinia virus.
Sample size justification and endpoint selection
Immunogenicity studies were carried out in C57BL/6 mice in groups of three or more mice per treatment, with group sizes chosen based on pilot experiments to provide 80% power to detect a 20% difference in tetramer-or cytokine-positive T cell populations. Vaccinia challenge studies were performed with five to seven animals per group to provide the same power to detect a 20% difference in weight loss.
Treatment of outliers
All data points were included in the analyses, and no outliers were excluded in calculations of means/statistical significance.
Replication
Immunogenicity and vaccinia challenge studies were repeated in at least three independent experiments, with the number of mice per group indicated above.
Blinding
Histopathology analyses were carried out by a blinded, licensed pathologist (M.A.S.).
Synthesis of ICMVs
Lipid nanocapsules were prepared as described previously (16, 17) with slight modifications. Dried lipid films consisting of DOPC (1,2-dioleoyl-sn-glycero-3-phosphocholine) and MPB {1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-[4-(p-maleimidophenyl)butyramide]} at 1:1 molar ratio and MPLA were rehydrated with cargo protein/peptide solutions, followed by sonication and addition of dithiothreitol and CaCl2 at final concentrations of 3 and 40 mM, respectively, to induce fusion and cross-linking of lipid bilayers, followed by washing and PEGylation, as described in Supplementary Materials and Methods.
Immunizations
Animals were cared for following federal, state, and local guidelines. Groups of 6- to 10-week-old female C57BL/6 mice (The Jackson Laboratory) were immunized via intratracheal or subcutaneous injection (at the tail base) with antigens (OVA, AL11 peptide, or PADRE), each encapsulated in separate ICMVs, in optimized doses (10 μg of OVA or 10 μg of AL11 + 3.3 μg of PADRE), with or without 0.3 μg of MPLA and 10 μg of polyI:C on days 0 and 28. Control groups included immunization with the equivalent doses of soluble antigen and TLR agonists or intratracheal administration of PBS. Intratracheal instillations were performed as described previously (40) and in Supplementary Materials and Methods.
In some experiments, C57BL/6 albino mice were adoptively transferred with purified 7.5 × 105 CD8+ T cells from luciferase-expressing OT-I mice (OT-I–luc) 1 day before immunization, and bioluminescence signals from OT-I–luc CD8+ T cells in these mice were acquired with a Xenogen IVIS Spectrum Imaging System before and after necropsies on days 3 and 5 after vaccination. Immune responses were characterized by flow cytometric analysis of antigen-specific T cell frequency (peptide-MHC tetramer staining), phenotypic marker expression, and intracellular cytokine staining, and ELISA analysis was carried out to determine antibody responses. These assays are described in Supplementary Materials and Methods.
Challenge studies
Tumor challenge
Mice were inoculated with 500,000 B16F10-OVA cells subcutaneously in the flank. On days 3 and 10, the mice were immunized with ICMV or soluble OVA vaccines intratracheally.
Vaccinia challenge
Six weeks after priming, mice were infected with vaccinia expressing SIV gag via a single intratracheal administration (1 × 106 PFU in 75 μl). Weights of mice were recorded daily after challenge. Animals were euthanized when their weight decreased by 20%. Tissue homogenates from lungs and ovaries were used to infect CV-1 cells in culture, and the resulting plaques were used to calculate viral titers (41). To determine the subset of T cells responsible for protection, we treated immunized mice with either intraperitoneal injection of 400 μg of depleting antibodies against CD8, CD4, or isotype control antibodies on days 40, 42, 44, and 46, or daily oral gavage treatment with FTY720 (3 mg/kg on day 38 and then 1 mg/kg until day 47). On day 42, the treated animals were infected with virus (1 × 106 to 2 × 106 PFU), and animal weights were tracked over 5 to 6 days.
Statistical analyses
Statistical analyses were performed with GraphPad Prism version 5. Analysis of groups was performed by confirming that the data conformed to a normal distribution, and was compared with one- or two-way ANOVA with Bonferroni correction. Comparison of survival curves after tumor challenge was performed with the log-rank test.
Supplementary Material
Fig. S1. ICMV nanoparticles adjuvanted with TLR agonists elicit CD8+ T cell responses after pulmonary vaccination.
Fig. S2. Phenotype of OVA+ APCs in lungs and mdLNs.
Fig. S3. Pulmonary vaccination elicits humoral immune responses in the blood and vaginal tract.
Fig. S4. OVA-tetramer+ T cells from reproductive tract and gut after pulmonary immunization. Fig. S5. Pulmonary nanocapsule vaccination promotes TEM cell response in systemic and mucosal compartments.
Fig. S6. Immunization with AL11/PADRE peptide ICMVs induces effector memory–biased T cell responses.
Fig. S7. The role of TEM and TCM cells in ICMV immunization.
Fig. S8. Pulmonary nanocapsule vaccination does not induce weight loss in healthy animals after vaccination.
Fig. S9. Chemokine and cytokine levels in lungs, serum, and BAL after pulmonary nanocapsule vaccination.
Acknowledgments
Funding: This work was supported in part by the Koch Institute Support (core) Grant P30-CA14051 from the National Cancer Institute; the Ragon Institute of MGH, MIT, and Harvard; the Gates Foundation; the Department of Defense (contract W911NF-07-D-0004); and the NIH (AI095109). AL11 MHC-I tetramer was obtained through the NIH Tetramer Facility. D.J.I. is an investigator of the Howard Hughes Medical Institute. We wish to dedicate this paper to the memory of Officer Sean Collier, for his caring service to the MIT community.
Footnotes
Supplementary Materials: www.sciencetranslationalmedicine.org/cgi/content/full/5/204/204ra130/DC1
Materials and Methods
Author contributions: A.V.L., J.J.M., and D.J.I. designed the experiments, analyzed the data, carried out statistical analyses, and wrote the paper. A.V.L. and J.J.M. carried out all experiments. W.A., J.E., H.S., and M.Y. provided technical support for in vivo studies. E.-J.I. and D.H.B. provided SIV gag– expressing vaccinia and guided vaccinia challenge studies. M.H.F. expanded and titered SIV gag–expressing vaccinia. M.A.S. designed histopathological analyses on lung tissues and scored blinded histology slides.
Competing interests: D.J.I. and J.J.M. are inventors on patents related to the ICMV technology. These patents have been licensed to Vedantra Pharmaceuticals Inc., in which D.J.I. holds equity and is a cofounder.
References
- 1.Cerutti A, Chen K, Chorny A. Immunoglobulin responses at the mucosal interface. Annu Rev Immunol. 2011;29:273–293. doi: 10.1146/annurev-immunol-031210-101317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kozlowski PA, Williams SB, Lynch RM, Flanigan TP, Patterson RR, Cu-Uvin S, Neutra MR. Differential induction of mucosal and systemic antibody responses in women after nasal, rectal, or vaginal immunization: Influence of the menstrual cycle. J Immunol. 2002;169:566–574. doi: 10.4049/jimmunol.169.1.566. [DOI] [PubMed] [Google Scholar]
- 3.Czerkinsky C, Holmgren J. Mucosal delivery routes for optimal immunization: Targeting immunity to the right tissues. Curr Top Microbiol Immunol. 2012;354:1–18. doi: 10.1007/82_2010_112. [DOI] [PubMed] [Google Scholar]
- 4.Plotkin SA. Immunologic correlates of protection induced by vaccination. Pediatr Infect Dis J. 2001;20:63–75. doi: 10.1097/00006454-200101000-00013. [DOI] [PubMed] [Google Scholar]
- 5.Woodland DL, Kohlmeier JE. Migration, maintenance and recall of memory T cells in peripheral tissues. Nat Rev Immunol. 2009;9:153–161. doi: 10.1038/nri2496. [DOI] [PubMed] [Google Scholar]
- 6.Sheridan BS, Lefrançois L. Regional and mucosal memory T cells. Nat Immunol. 2011;12:485–491. doi: 10.1038/ni.2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Teijaro JR, Turner D, Pham Q, Wherry EJ, Lefrançois L, Farber DL. Cutting edge: Tissue-retentive lung memory CD4 T cells mediate optimal protection to respiratory virus infection. J Immunol. 2011;187:5510–5514. doi: 10.4049/jimmunol.1102243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shin H, Iwasaki A. A vaccine strategy that protects against genital herpes by establishing local memory T cells. Nature. 2012;491:463–467. doi: 10.1038/nature11522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mackay LK, Stock AT, Ma JZ, Jones CM, Kent SJ, Mueller SN, Heath WR, Carbone FR, Gebhardt T. Long-lived epithelial immunity by tissue-resident memory T (TRM) cells in the absence of persisting local antigen presentation. Proc Natl Acad Sci U S A. 2012;109:7037–7042. doi: 10.1073/pnas.1202288109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bivas-Benita M, Bar L, Gillard GO, Kaufman DR, Simmons NL, Hovav AH, Letvin NL. Efficient generation of mucosal and systemic antigen-specific CD8+ T-cell responses following pulmonary DNA immunization. J Virol. 2010;84:5764–5774. doi: 10.1128/JVI.02202-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Klavinskis LS, Barnfield C, Gao L, Parker S. Intranasal immunization with plasmid DNA-lipid complexes elicits mucosal immunity in the female genital and rectal tracts. J Immunol. 1999;162:254–262. [PubMed] [Google Scholar]
- 12.Wang D, Christopher ME, Nagata LP, Zabielski MA, Li H, Wong JP, Samuel J. Intranasal immunization with liposome-encapsulated plasmid DNA encoding influenza virus hemagglutinin elicits mucosal, cellular and humoral immune responses. J Clin Virol. 2004;31(Suppl. 1):99–106. doi: 10.1016/j.jcv.2004.09.013. [DOI] [PubMed] [Google Scholar]
- 13.Belyakov IM, Derby MA, Ahlers JD, Kelsall BL, Earl P, Moss B, Strober W, Berzofsky JA. Mucosal immunization with HIV-1 peptide vaccine induces mucosal and systemic cytotoxic T lymphocytes and protective immunity in mice against intrarectal recombinant HIV-vaccinia challenge. Proc Natl Acad Sci U S A. 1998;95:1709–1714. doi: 10.1073/pnas.95.4.1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tai W, Roberts L, Seryshev A, Gubatan JM, Bland CS, Zabriskie R, Kulkarni S, Soong L, Mbawuike I, Gilbert B, Kheradmand F, Corry DB. Multistrain influenza protection induced by a nanoparticulate mucosal immunotherapeutic. Mucosal Immunol. 2011;4:197–207. doi: 10.1038/mi.2010.50. [DOI] [PubMed] [Google Scholar]
- 15.Liu MA, Ulmer JB. Human clinical trials of plasmid DNA vaccines. Adv Genet. 2005;55:25–40. doi: 10.1016/S0065-2660(05)55002-8. [DOI] [PubMed] [Google Scholar]
- 16.Moon JJ, Suh H, Bershteyn A, Stephan MT, Liu H, Huang B, Sohail M, Luo S, Um SH, Khant H, Goodwin JT, Ramos J, Chiu W, Irvine DJ. Interbilayer-crosslinked multilamellar vesicles as synthetic vaccines for potent humoral and cellular immune responses. Nat Mater. 2011;10:243–251. doi: 10.1038/nmat2960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Moon JJ, Suh H, Li AV, Ockenhouse CF, Yadava A, Irvine DJ. Enhancing humoral responses to a malaria antigen with nanoparticle vaccines that expand Tfh cells and promote germinal center induction. Proc Natl Acad Sci U S A. 2012;109:1080–1085. doi: 10.1073/pnas.1112648109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Obar JJ, Lefrançois L. Memory CD8+ T cell differentiation. Ann N Y Acad Sci. 2010;1183:251–266. [Google Scholar]
- 19.Swartz MA, Hirosue S, Hubbell JA. Engineering approaches to immunotherapy. Sci Transl Med. 2012;4:148rv9. doi: 10.1126/scitranslmed.3003763. [DOI] [PubMed] [Google Scholar]
- 20.Thornton EE, Looney MR, Bose O, Sen D, Sheppard D, Locksley R, Huang X, Krummel MF. Spatiotemporally separated antigen uptake by alveolar dendritic cells and airway presentation to T cells in the lung. J Exp Med. 2012;209:1183–1199. doi: 10.1084/jem.20112667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ritter M, Mennerich D, Weith A, Seither P. Characterization of Toll-like receptors in primary lung epithelial cells: Strong impact of the TLR3 ligand poly(I:C) on the regulation of Toll-like receptors, adaptor proteins and inflammatory response. J Inflamm. 2005;2:16. doi: 10.1186/1476-9255-2-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhu Q, Egelston C, Gagnon S, Sui Y, Belyakov IM, Klinman DM, Berzofsky JA. Using 3 TLR ligands as a combination adjuvant induces qualitative changes in T cell responses needed for antiviral protection in mice. J Clin Invest. 2010;120:607–616. doi: 10.1172/JCI39293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kaech SM, Tan JT, Wherry EJ, Konieczny BT, Surh CD, Ahmed R. Selective expression of the interleukin 7 receptor identifies effector CD8 T cells that give rise to long-lived memory cells. Nat Immunol. 2003;4:1191–1198. doi: 10.1038/ni1009. [DOI] [PubMed] [Google Scholar]
- 24.Barouch DH, Pau MG, Custers JHHV, Koudstaal W, Kostense S, Havenga MJE, Truitt DM, Sumida SM, Kishko MG, Arthur JC, Korioth-Schmitz B, Newberg MH, Gorgone DA, Lifton MA, Panicali DL, Nabel GJ, Letvin NL, Goudsmit J. Immunogenicity of recombinant adenovirus serotype 35 vaccine in the presence of pre-existing anti-Ad5 immunity. J Immunol. 2004;172:6290–6297. doi: 10.4049/jimmunol.172.10.6290. [DOI] [PubMed] [Google Scholar]
- 25.Alexander J, Sidney J, Southwood S, Ruppert J, Oseroff C, Maewal A, Snoke K, Serra HM, Kubo RT, Sette A, Grey HM. Development of high potency universal DR-restricted helper epitopes by modification of high affinity DR-blocking peptides. Immunity. 1994;1:751–761. doi: 10.1016/s1074-7613(94)80017-0. [DOI] [PubMed] [Google Scholar]
- 26.Cyster JG, Schwab SR. Sphingosine-1-phosphate and lymphocyte egress from lymphoid organs. Annu Rev Immunol. 2012;30:69–94. doi: 10.1146/annurev-immunol-020711-075011. [DOI] [PubMed] [Google Scholar]
- 27.Sandoval F, Terme M, Nizard M, Badoual C, Bureau MF, Freyburger L, Clement O, Marcheteau E, Gey A, Fraisse G, Bouguin C, Merillon N, Dransart E, Tran T, Quintin-Colonna F, Autret G, Thiebaud M, Suleman M, Riffault S, Wu TC, Launay O, Danel C, Taieb J, Richardson J, Zitvogel L, Fridman WH, Johannes L, Tartour E. Mucosal imprinting of vaccine-induced CD8+ T cells is crucial to inhibit the growth of mucosal tumors. Sci Transl Med. 2013;5:172ra20. doi: 10.1126/scitranslmed.3004888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wakim LM, Gupta N, Mintern JD, Villadangos JA. Enhanced survival of lung tissue-resident memory CD8+ T cells during infection with influenza virus due to selective expression of IFITM3. Nat Immunol. 2013;14:238–245. doi: 10.1038/ni.2525. [DOI] [PubMed] [Google Scholar]
- 29.Honko AN, Mizel SB. Mucosal administration of flagellin induces innate immunity in the mouse lung. Infect Immun. 2004;72:6676–6679. doi: 10.1128/IAI.72.11.6676-6679.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.El-Kamary SS, Pasetti MF, Mendelman PM, Frey SE, Bernstein DI, Treanor JJ, Ferreira J, Chen WH, Sublett R, Richardson C, Bargatze RF, Sztein MB, Tacket CO. Adjuvanted intranasal Norwalk virus-like particle vaccine elicits antibodies and antibody-secreting cells that express homing receptors for mucosal and peripheral lymphoid tissues. J Infect Dis. 2010;202:1649–1658. doi: 10.1086/657087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nembrini C, Stano A, Dane KY, Ballester M, van der Vlies AJ, Marsland BJ, Swartz MA, Hubbell JA. Nanoparticle conjugation of antigen enhances cytotoxic T-cell responses in pulmonary vaccination. Proc Natl Acad Sci U S A. 2011;108:E989–E997. doi: 10.1073/pnas.1104264108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ballester M, Nembrini C, Dhar N, de Titta A, de Piano C, Pasquier M, Simeoni E, van der Vlies AJ, McKinney JD, Hubbell JA, Swartz MA. Nanoparticle conjugation and pulmonary delivery enhance the protective efficacy of Ag85B and CpG against tuberculosis. Vaccine. 2011;29:6959–6966. doi: 10.1016/j.vaccine.2011.07.039. [DOI] [PubMed] [Google Scholar]
- 33.Nardelli-Haefliger D, Lurati F, Wirthner D, Spertini F, Schiller JT, Lowy DR, Ponci F, De Grandi P. Immune responses induced by lower airway mucosal immunisation with a human papillomavirus type 16 virus-like particle vaccine. Vaccine. 2005;23:3634–3641. doi: 10.1016/j.vaccine.2005.02.019. [DOI] [PubMed] [Google Scholar]
- 34.Sallusto F, Geginat J, Lanzavecchia A. Central memory and effector memory T cell subsets: Function, generation, and maintenance. Annu Rev Immunol. 2004;22:745–763. doi: 10.1146/annurev.immunol.22.012703.104702. [DOI] [PubMed] [Google Scholar]
- 35.van Faassen H, Saldanha M, Gilbertson D, Dudani R, Krishnan L, Sad S. Reducing the stimulation of CD8+ T cells during infection with intracellular bacteria promotes differentiation primarily into a central (CD62LhighCD44high) subset. J Immunol. 2005;174:5341–5350. doi: 10.4049/jimmunol.174.9.5341. [DOI] [PubMed] [Google Scholar]
- 36.Marzo AL, Klonowski KD, Bon AL, Borrow P, Tough DF, Lefrançois L. Initial T cell frequency dictates memory CD8+ T cell lineage commitment. Nat Immunol. 2005;6:793–799. doi: 10.1038/ni1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Blank F, Stumbles P, von Garnier C. Opportunities and challenges of the pulmonary route for vaccination. Expert Opin Drug Deliv. 2011;8:547–563. doi: 10.1517/17425247.2011.565326. [DOI] [PubMed] [Google Scholar]
- 38.Caskey M, Lefebvre F, Filali-Mouhim A, Cameron MJ, Goulet JP, Haddad EK, Breton G, Trumpfheller C, Pollak S, Shimeliovich I, Duque-Alarcon A, Pan L, Nelkenbaum A, Salazar AM, Schlesinger SJ, Steinman RM, Sékaly RP. Synthetic double-stranded RNA induces innate immune responses similar to a live viral vaccine in humans. J Exp Med. 2011;208:2357–2366. doi: 10.1084/jem.20111171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Adams M, Navabi H, Jasani B, Man S, Fiander A, Evans AS, Donninger C, Mason M. Dendritic cell (DC) based therapy for cervical cancer: Use of DC pulsed with tumour lysate and matured with a novel synthetic clinically non-toxic double stranded RNA analogue poly [I]:poly [C12U] (Ampligen) Vaccine. 2003;21:787–790. doi: 10.1016/s0264-410x(02)00599-6. [DOI] [PubMed] [Google Scholar]
- 40.DuPage M, Dooley AL, Jacks T. Conditional mouse lung cancer models using adenoviral or lentiviral delivery of Cre recombinase. Nat Protoc. 2009;4:1064–1072. doi: 10.1038/nprot.2009.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nakaya Y, Zheng H, García-Sastre A. Enhanced cellular immune responses to SIV Gag by immunization with influenza and vaccinia virus recombinants. Vaccine. 2003;21:2097–2106. doi: 10.1016/s0264-410x(02)00781-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. ICMV nanoparticles adjuvanted with TLR agonists elicit CD8+ T cell responses after pulmonary vaccination.
Fig. S2. Phenotype of OVA+ APCs in lungs and mdLNs.
Fig. S3. Pulmonary vaccination elicits humoral immune responses in the blood and vaginal tract.
Fig. S4. OVA-tetramer+ T cells from reproductive tract and gut after pulmonary immunization. Fig. S5. Pulmonary nanocapsule vaccination promotes TEM cell response in systemic and mucosal compartments.
Fig. S6. Immunization with AL11/PADRE peptide ICMVs induces effector memory–biased T cell responses.
Fig. S7. The role of TEM and TCM cells in ICMV immunization.
Fig. S8. Pulmonary nanocapsule vaccination does not induce weight loss in healthy animals after vaccination.
Fig. S9. Chemokine and cytokine levels in lungs, serum, and BAL after pulmonary nanocapsule vaccination.