

Abstract
The MYC oncoprotein is an essential transcription factor that regulates the expression of many genes involved in cell growth, proliferation, and metabolic pathways. Thus, it is important to keep MYC activity in check in normal cells in order to avoid unwanted oncogenic changes. Normal cells have adapted several ways to control MYC levels, and these mechanisms can be disrupted in cancer cells. One of the major ways in which MYC levels are controlled in cells is through targeted degradation by the ubiquitin–proteasome system (UPS). Here, we discuss the role of the UPS in the regulation of MYC protein levels and review some of the many proteins that have been shown to regulate MYC protein stability. In addition, we discuss how this relates to MYC transcriptional activity, human cancers, and therapeutic targeting.
MYC has strong growth-promoting activity, so its abundance must be controlled to avoid oncogenesis. One of the most prominent control mechanisms involves degradation of MYC by the ubiquitin–proteasome system.
MYC is a multifunctional transcription factor that regulates many genes involved in multiple biological processes, including cell growth, proliferation, and apoptosis (Cole 1986; Prendergast 1999; Dang 2012). In fact, MYC is thought to regulate most, if not all, actively transcribed genes within a given cell (Lin et al. 2012). MYC functions as a transcription factor through heterodimerization with MAX. Together, MYC/MAX heterodimers bind to E-box motifs (CACGTG) within the promoters of target genes and recruit transcriptional coactivators to activate transcription (Dang 1999; Eisenman 2001).
The MYC protein contains several domains that play important roles in MYC function, and a variety of proteins that mediate posttranslational modifications that regulate MYC activity and stability interact with these domains (Fig. 1). Within the amino-terminal domain are several conserved regions, known as MYC boxes (MBI, II, III, and IV). MBI and MBII are located within the transactivation domain (TAD), a 143-amino-acid acidic domain that is required for MYC transcriptional and cell-transforming activity (Kato et al. 1990). MBIII has been shown to be important for transcriptional repression (Kurland and Tansey 2008) and for MYC’s pro-apoptotic activity (Herbst et al. 2005). MBIV is also important for MYC transcriptional activity and MYC-induced apoptosis (Cowling et al. 2006). In addition to these conserved regions, there is a canonical nuclear localization signal (NLS) at amino acids 320–328 (Dang and Lee 1988). The carboxy-terminal region of MYC includes the basic, helix–loop–helix, and leucine zipper domains (B-HLH-LZ), which mediate dimerization with other HLH LZ proteins and DNA binding (Blackwood and Eisenman 1991).
Figure 1.
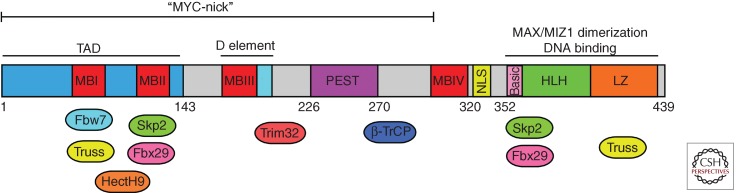
Structure of MYC. Elements known to regulate MYC protein localization, function, and stability are shown. The substrate recognition subunit of known E3 ubiquitin ligases whose interaction sites have been defined are shown.
Given the strong growth-promoting activity of MYC, it is not surprising that MYC abundance is controlled at multiple steps in normal cells. MYC gene transcription is stimulated by mitogens and controlled at the level of initiation and elongation (Spencer and Groudine 1990; Liu and Levens 2006). In addition, MYC mRNA is inherently unstable, with a half-life of ∼30 min (Dani et al. 1984), and MYC mRNA translation is tightly regulated and responsive to cell growth-signaling pathways (Wall et al. 2008). Finally, MYC protein is rapidly degraded following its synthesis (half-life of ∼20 min in non-transformed cells) (Hann and Eisenman 1984). One of the most prominent mechanisms to ensure proper regulation of MYC levels involves degradation by the ubiquitin–proteasome system (UPS) (Thomas and Tansey 2011). In this review, we discuss the role of the UPS in the regulation of MYC protein levels and how this impacts MYC transcriptional activity. We also discuss the multiple proteins that have been shown to regulate MYC protein stability. Finally, we discuss connections between the UPS-mediated control of MYC and human cancers, with an eye toward therapeutics.
DEGRADATION OF MYC
Calpain-Dependent Cleavage
Although the UPS, which we discuss below, mediates the bulk turnover of MYC in cells, it is not the only way in which MYC can be processed, because cleavage of MYC by calpains has been reported (Small et al. 2002). Calpain-dependent cleavage is calcium dependent and occurs in the cytosol (proteasomal degradation appears to occur mostly in the nucleus). It has been shown that cleavage by calpains functions to inactivate MYC transcriptional activity by removing the carboxyl terminus. Like calpain-mediated cleavage of other proteins, calpains cause partial cleavage of MYC rather than complete degradation. Calpain cleavage of MYC generates “MYC-nick,” a 298-amino-acid amino-terminal segment that has been shown to regulate microtubules to promote muscle cell differentiation (Conacci-Sorrell et al. 2010; Conacci-Sorrell and Eisenman 2011). More research is needed to determine if the generation of MYC-nick by calpains is important in other cell types or processes.
Proteasomal Degradation
The most prominent route for MYC degradation in cells is through the UPS. Ubiquitin-mediated degradation is a highly specific, ATP-dependent process. Proteins are targeted for degradation by the proteasome in a two-step process in which (1) ubiquitin molecules are covalently added to the target protein, and (2) poly-ubiquitinated proteins are degraded by the 26S proteasome. Conjugation of ubiquitin to target proteins occurs through a three-step process involving three different enzymes. First, ubiquitin is activated by an ubiquitin-activating enzyme (E1), a process that involves adenylation of the ubiquitin molecule in an ATP-dependent manner. Second, the activated ubiquitin is transferred to an E2 ubiquitin-conjugating enzyme. Third, in conjunction with an E3 ubiquitin ligase bound to the target protein, the E2 enzyme catalyzes transfer of the activated ubiquitin molecule to a lysine (K) residue in the target substrate. Successive reactions lead to the attachment of additional ubiquitin molecules to lysine 48 (K48) in the previously added ubiquitin to form poly-ubiquitin chains. It is this K48 poly-ubiquitin chain that is recognized by the 26S proteasome. The proteasome will bind to proteins containing the correct number of ubiquitin moieties (four or more) and type of linkages (K48), and subsequently deubiquitinate, unfold, and degrade them into small peptide fragments (Sorokin et al. 2009).
Cells usually contain only a few E1 enzymes, approximately 50 E2s, and approximately 500 E3s. Although the E2s help determine the type of ubiquitin chain assembled, it is the E3 ubiquitin ligases that generally confer substrate specificity to the UPS (Nandi et al. 2006). There are several different families of E3 ligases, which differ in domain homology and mechanism of action. The majority of E3 ligases belong to the RING-FINGER/U-box family. RING-FINGER domain E3s do not form a catalytic intermediate with ubiquitin but, instead, serve as scaffold proteins that bring together the E2 and ubiquitination substrate. In this case, it is the E2 that transfers the ubiquitin to the substrate. RING-FINGER E3s function as multi-subunit complexes. These complexes usually contain a RING-FINGER domain E3 ligase (such as Rbx), a Cullin scaffold protein, an adaptor (such as Skp1), and a substrate-specific binding protein (such as F-box proteins), which usually give the E3 ligase its name. For example, the SCFFbw7 complex contains the Rbx1 RING-FINGER domain E3 ligase, the Skp1 adaptor, Cul1 scaffold, and the F box and WD-repeat-domain-containing seven (Fbw7) substrate-binding subunit, which is often referred to as the Fbw7 E3 ligase. E3 ligases may also contain RING-FINGER-related domains, such as U box and PHD domains. Fewer E3 ligases belong to the HECT (“homologous to E6AP carboxyl terminus”) family, which, in contrast to the RING-FINGER/U-box family, form a catalytic intermediate with ubiquitin and directly transfer ubiquitin to the substrate (Dikic and Robertson 2012; Metzger et al. 2012).
The selectivity of the UPS means that proteins must contain elements to control their own degradation. As shown in Figure 1, degron elements known to interact with E3s and regulate MYC protein stability overlap with the TAD and include MBI with its conserved serine 62 and threonine 58 residues and a degron sequence overlapping MBII (amino acid residues 127–158) (Flinn et al. 1998; Salghetti et al. 1999; Sears et al. 1999). In some cases, E3s have been mapped to both the TAD and the carboxyl terminus of MYC. In addition, the D element has been shown to be important for proteolysis but not ubiquitination (Herbst et al. 2004), and deletion of the PEST sequence stabilizes MYC without reducing overall ubiquitination of MYC (Gregory and Hann 2000).
PROTEINS THAT REGULATE MYC UBIQUITINATION AND PROTEIN STABILITY
Several E3 ubiquitin ligases for MYC have been described, which we summarize below. In addition, several other proteins that have been implicated in the regulation of MYC protein stability are discussed. Table 1 summarizes these proteins.
Table 1.
Proteins involved in the regulation of MYC protein stability
| Protein | Effect on MYC stability | Effect on MYC activity | Phase of cell cycle |
|---|---|---|---|
| Fbw7 | Decrease | Decrease | G1–S |
| Pinl | Decrease | Increase | — |
| Usp28 | Increase | Increase | G1–S |
| β-TrCP | Increase | Increase | S–G2 |
| Skp2 | Decrease | Increase | G1–S |
| HectH9 | — | Increase | G1–S |
| Truss | Decrease | Decrease | — |
| Trim32 | Decrease | — | — |
| Fbx29 | Decrease | Decrease | — |
| CHIP | Decrease | Decrease | — |
| SIRT2 | Increase | — | — |
| NEDD4 | Decrease | — | — |
| NEMO | Increase | Increase | — |
The effect of each protein on MYC stability and MYC transcriptional activity is given, if known. In addition, the phase of the cell cycle where this regulation occurs is shown, if known.
Fbw7
The best-studied E3 ubiquitin ligase for MYC is SCFFbw7. Fbw7 is the F-box substrate-specificity component of this SCF-type (Skp–Cullin–F box) RING-FINGER domain ubiquitin ligase complex (Deshaies 1999). Human Fbw7 encodes three isoforms—Fbw7α, Fbw7β, and Fbw7γ—which differ in their subcellular localizations (Kimura et al. 2003). Both the Fbw7α (nucleoplasmic) and Fbw7γ (nucleolar) isoforms have been implicated in the regulation of MYC protein turnover (Grim et al. 2008). Fbw7 uses the E2 cdc34 to add K48-linked ubiquitin chains to MYC. Studies have shown that MYC is a direct target for Fbw7-mediated ubiquitination and that SCFFbw7 triggers proteasomal degradation of MYC (Welcker et al. 2004b; Yada et al. 2004).
Regulation of c-MYC stability by Fbw7 is dependent on MYC phosphorylation. Two conserved phosphorylation sites within MBI, threonine 58 (T58) and serine 62 (S62), are part of a phospho-degron sequence recognized by Fbw7, and they control Fbw7-mediated turnover of MYC (Welcker et al. 2004a,b; Yada et al. 2004). Work in several laboratories has elucidated a signaling pathway that regulates these phosphorylation events (Lutterbach and Hann 1994; Pulverer et al. 1994; Sears et al. 1999, 2000). As shown in Figure 2, following cell growth stimulation, MYC is stabilized upon phosphorylation of serine 62 (pS62) by ERK and/or CDKs (Sears 2004; Bachireddy et al. 2005). In conjunction with Pin1-mediated proline isomerization, S62 phosphorylation increases MYC transcriptional activity at pro-proliferative target genes (Hydbring et al. 2010; Farrell et al. 2013; Sanchez-Arevalo Lobo et al. 2013). S62 phosphorylation also primes subsequent phosphorylation at threonine 58 (pT58) by GSK-3β (Gregory et al. 2003), which allows a second Pin1-mediated isomerization step to facilitate Protein Phosphatase 2A (PP2A)-B56α-mediated dephosphorylation of the stabilizing phosphate at S62 (Yeh et al. 2004; Arnold and Sears 2006). pT58-MYC is recognized by the E3 ubiquitin ligase SCFFbw7 and degraded by the 26S proteasome (Welcker et al. 2004b; Yada et al. 2004).
Figure 2.

pS62/pT58 MYC degradation pathway. Proteins in red stabilize and/or activate MYC. Proteins in green facilitate MYC degradation.
The scaffold protein Axin1 helps coordinate these events by facilitating the formation of a MYC degradation complex containing GSK-3β, Pin1, and PP2A-B56α (Arnold et al. 2009). Interestingly, Axin1 can be detected at MYC target gene promoters by chromatin immunoprecipitation (Arnold et al. 2009) along with Fbw7, GSK-3β, Pin1, PP2A, and components of the 26S proteasome (Farrell et al. 2013), suggesting that this mode of MYC degradation involves transcriptionally active chromatin-bound MYC.
Pin1
The Pin1 peptidyl-prolyl isomerase is a phosphorylation-directed proline isomerase that adds an additional posttranslational modification to phosphorylated substrates through catalyzing trans–cis or cis–trans isomerization at proline residues followed by a phosphorylated serine or threonine (Joseph et al. 2003; Lu 2003; Lippens et al. 2007; Lu and Zhou 2007). Recent data suggest that Pin1 functions at two points in the above pS62/pT58 MYC degradation pathway, where it first catalyzes proline 63 in pS62-MYC from trans to cis to enhance its DNA binding and transcriptional activity, and subsequently catalyzes proline 63 in pS62/pT58-MYC from cis to trans to facilitate PP2A-mediated dephosphorylation of S62, and in this way contributes to pT58-MYC degradation via the Fbw7 E3 ligase (Farrell et al. 2013; Sanchez-Arevalo Lobo et al. 2013). These studies support a coupled relationship between MYC’s transcriptional activity and its degradation (see discussion below).
Usp28
Opposing Fbw7α-mediated MYC ubiquitination, the deubiquitinating enzyme Usp28 was first identified as a MYC regulator using a short hairpin RNA (shRNA) screen to identify genes required for MYC function (Popov et al. 2007b). Usp28 is a ubiquitin-specific protease (USP) that cleaves ubiquitin chains to antagonize the activity of ubiquitin ligases (Nijman et al. 2005). Popov and colleagues found that Usp28 binds MYC via interaction with Fbw7α and stabilizes MYC. In addition, they found that Usp28-mediated stabilization of MYC was required for tumor cell proliferation (Popov et al. 2007b). Subsequently they showed that, in response to UV irradiation, Usp28 dissociates from Fbw7α, allowing for enhanced Fbw7-mediated MYC ubiquitination and degradation upon DNA damage (Popov et al. 2007a).
Other USPs have recently been discovered for MYC. For example, a USP called Puf was identified in Drosophila as an enhancer of dMyc growth (D Ling and RN Eisenman, pers. comm.). Puf binds dMYC and the Fbw7 ortholog Ago (Moberg et al. 2004) and regulates cyclin E turnover and MYC-dependent cell growth. In addition, Usp36, a novel deubiqutinating enzyme for MYC, is localized in the nucleolus and interacts directly with Fbw7γ, but not Fbw7α, thus complementing the activity of Usp28 (M-S Dai and RC Sears, unpubl.). Usp36 associates with MYC and deubiquitinates MYC in cells and in vitro, increasing MYC stability. Usp36-mediataed stabilization of MYC enhances MYC’s transcriptional activity and promotes cell proliferation. Furthermore, Usp36 itself is a MYC target gene, suggesting that Usp36 and MYC form a positive-feedback regulatory loop (M-S Dai and RC Sears, unpubl.).
β-TrCP
Ubiquitination of MYC mediated by Fbw7 is thought to be important for controlling MYC levels in the G1 and early S phases of the cell cycle. However, during subsequent phases of the cell cycle, MYC can be ubiquitinated by another RING-FINGER E3 ligase, SCFβ-TrCP. Popov et al. (2010) showed that, in contrast to Fbw7 action on MYC, the F-box protein β-TrCP stabilizes MYC. MYC contains a phospho-recognition sequence for β-TrCP binding at amino acids 278–283 (Fig. 1), and mutation of these residues abolished MYC binding to β-TrCP and decreased MYC protein stability. Furthermore, they showed that SCFβ-TrCP is a bona fide E3 ligase for MYC and that it recruits the UbcH5 ubiquitin-conjugating enzyme to directly ubiquitinate MYC. Interestingly, both Fbw7 and β-TrCP mediate direct ubiquitination of the amino terminus of MYC; however, SCFβ-TrCP forms heterotypic poly-ubiquitin chains composed of K63 and K48 linkages, but SCFFbw7 forms only K48-linked chains on MYC. Finally, they showed that ubiquitination of MYC by β-TrCP is required for cell cycle reentry after S-phase arrest, suggesting that β-TrCP functions to stabilize MYC protein by antagonizing Fbw7-mediated ubiquitination upon recovery from S-phase arrest (Popov et al. 2010).
Skp2
A third RING-FINGER SCF ubiquitin ligase F-box protein identified for MYC is Skp2. Skp2, a known oncogene, has been implicated in the turnover of many cell cycle regulatory proteins, including p27Kip1 (von der Lehr et al. 2003). Skp2 recognizes MYC through both MBII and HLH-LZ motifs (amino acids 367–439) (Fig. 1) and promotes MYC poly-ubiquitination and degradation (Kim et al. 2003; von der Lehr et al. 2003). To our knowledge, specific lysine linkages have not been reported, although K48 is likely. In addition, Skp2-mediated regulation of MYC degradation does not appear to be phosphorylation dependent. von der Lehr et al. (2003) showed that SCFSkp2 regulates MYC protein turnover at the G1-to-S phase transition in lymphocytes.
Intriguingly, Skp2 expression stimulated MYC-induced S-phase entry (von der Lehr et al. 2003). Thus, unlike Fbw7, which stimulates MYC degradation and inhibits MYC activity, Skp2 promotes MYC transcriptional activity, acting as a transcriptional coactivator (Kim et al. 2003; von der Lehr et al. 2003). This function for Skp2 was shown to require Skp2’s F-box domain, involved in SCF complex binding, suggesting that E3 ubiquitin ligase activity is important for Skp2’s ability to stimulate MYC transcriptional activity (von der Lehr et al. 2003). In addition, Skp2 was found to be associated with MYC target gene promoters, along with proteasome subunits, suggesting a link between SCFSkp2-mediated ubiquitination, MYC transcriptional activation, and degradation (see below for further discussion).
An additional layer of complexity exists here because Skp2 is a direct MYC target gene (Bretones et al. 2011). Thus, MYC can augment expression of Skp2, possibly contributing to oncogenesis by both increasing MYC transcriptional activity, while controlling its level, and inducing the degradation of p27.
HectH9
Another ubiquitin ligase for MYC is HectH9. HectH9 belongs to the HECT-domain family of ubiquitin ligases, which are characterized by a conserved carboxy-terminal catalytic domain (Huibregtse et al. 1995). HectH9 was originally identified in a yeast two-hybrid screen to find novel interacting proteins of Miz1, a transcription factor inhibited by its interaction with MYC (Adhikary et al. 2005). Additionally, they found that HectH9 also interacted with MYC via its TAD and catalyzed K63-linked ubiquitination of a cluster of lysines overlapping the NLS. This ubiquitination, which did not trigger proteasomal degradation of MYC, was inhibited by Miz1. Moreover, mutation of lysine residues in MYC targeted by HectH9, which did not interfere with its nuclear localization despite their location within MYC’s NLS, reduced recruitment of p300 and suppressed transactivation of a subset of MYC target genes involved in cellular metabolism and protein synthesis. Consequently, this MYC mutant had a reduced ability to promote proliferation after serum starvation (Adhikary et al. 2005). These data suggest that HectH9-mediated ubiquitination does not trigger MYC degradation but, instead, increases MYC transcriptional activity. Thus, as is the case with Skp2 (von der Lehr et al. 2003), these studies suggest a strong link between MYC ubiquitination and its transcriptional activity (see below for further discussion).
TRUSS
TRUSS (tumor necrosis factor receptor-associated ubiquitous scaffolding and signaling protein) is an adaptor for the DDB1–CUL4 ubiquitin ligase complex, which belongs to the cullin–RING-FINGER ubiquitin ligase superfamily (Petroski and Deshaies 2005). TRUSS was identified using a proteomic screen for proteins that interact with N-MYC (Choi et al. 2010). TRUSS was subsequently shown to bind both c-MYC and N-MYC, and to mediate the interaction between MYC and the DDB1–CUL4 E3 ligase, thereby stimulating MYC ubiquitination and degradation. Domain mapping indicated that TRUSS interacts with the carboxyl terminus of MYC, which contains the HLH-LZ motif, but that elements near the amino terminus are additionally required for TRUSS-mediated degradation. MYC transactivation of target genes was also reduced in response to TRUSS, as was MYC-induced cell transformation (Choi et al. 2010). Thus, like Fbw7, TRUSS negatively regulates MYC function by reducing MYC protein levels.
TRIM32
One of the least-well-characterized E3 ligases for MYC is TRIM32, a RING-FINGER ubiquitin ligase. TRIM32 has been shown to regulate stability of several proteins and activity of specific microRNAs, including Let-7a, to control the balance between differentiating and progenitor daughter cell types produced from neural progenitor cells in the mouse neocortex. This work identified c-MYC as a ubiquitination target of TRIM32 and showed that TRIM32 promotes degradation of MYC (Schwamborn et al. 2009). At this time, little is known about TRIM32-mediated regulation of MYC protein stability. More work is needed to determine how TRIM32 interacts with MYC and whether the effect on MYC is direct or indirect.
Fbx29
Fbx29 (also known as FBXW8), a substrate recognition component for the Skp1-Cul7-ROC1-containing E3 ubiquitin ligase complex (Dias et al. 2002), was identified as a MYC-interacting protein in a proteomic screen. Mapping experiments indicated that MBII and the carboxy-terminal HLH-LZ domains were important for MYC’s interaction with Fbx29. Although these studies did not directly measure MYC ubiquitination, they found that overexpression of Fbx29 decreased MYC protein levels and transactivation activity (Koch et al. 2007). Thus, it remains to be seen whether MYC is a direct target of Fbx29. Because the domains that are required for this interaction are the same as those identified for Skp2 binding, it is possible that Skp2 and Fbx29 might compete for binding to MYC. It will be interesting to determine whether this occurs and what the biological consequences might be.
CHIP
The most recent ubiquitin ligase to be identified for MYC is CHIP (carboxyl terminus of Hsc70-interacting protein) (Paul et al. 2013). CHIP is a chaperone-associated U-box-containing E3 ligase that links a chaperone to the 26S proteasome machinery by ubiquitinating chaperone substrates and directing them toward the proteasome (Ballinger et al. 1999). Ballinger et al. (1999) showed that CHIP interacts with and ubiquitinates MYC, targeting MYC for degradation by the 26S proteasome. They showed that this involved interaction with the chaperone protein Hsp70 and to a lesser extent, Hsp90. The increase in MYC degradation mediated by CHIP correlated with decreased MYC transcriptional activity and reduced expression of MYC target genes (Paul et al. 2013). More studies are required to determine whether the MYC–CHIP interaction is direct, and if it is, to map the regions of MYC important for the interaction, as well as determine the physiological relevance of this interaction.
SIRT2 and NEDD4
It was recently shown that SIRT2 indirectly stabilizes MYC protein and promotes cancer cell proliferation (Liu et al. 2013). SIRT2 is a class III histone deacetylase (HDAC) that shows a strong preference for acetylated lysine 16 of histone H4 (H4K16) (Vaquero et al. 2006), an acetylation mark commonly lost in cancer cells (Fraga et al. 2005). Liu et al. (2013) showed that MYC up-regulates SIRT2 protein expression in neuroblastoma and pancreatic cancer cells and that SIRT2 then represses transcription of the HECT-domain E3 ubiquitin ligase NEDD4 by directly binding to the NEDD4 promoter and deacetylating H4K16. Although NEDD4 has not been previously described as an E3 ligase for MYC, they additionally showed that NEDD4 directly binds MYC to target it for ubiquitination and degradation. Therefore, repression of NEDD4 expression by SIRT2 leads to reduced MYC ubiquitination and subsequent stabilization (Liu et al. 2013). This study suggests a possible new E3 ligase for MYC and reveals a novel pathway for the stabilization of MYC in cancer cells.
NEMO
Another indirect regulator of Myc stability is NEMO (NF-κB essential modulator), the regulatory subunit of the IKK complex. NEMO was recently shown to suppress MYC turnover (Kim et al. 2010). NEMO plays a critical role in the activation of the NF-κB pathway, likely by acting as a scaffold protein in the IKK complex (Yamaoka et al. 1998). Kim et al. (2010) found that NEMO induced MYC up-regulation through protein stabilization and that this involved direct interaction between MYC and NEMO in the nucleus. Additionally, they showed that NEMO reduced ubiquitination of MYC by inhibiting the ubiquitinating activity of SCFFbw7, and that this resulted in enhanced expression of select MYC target genes (Kim et al. 2010). They subsequently showed that stabilization of MYC by NEMO resulted in resistance to ionizing radiation through the specific up-regulation of γ-GCS (γ-glutamyl-cysteine synthetase), a MYC target gene. Up-regulation of γ-GCS upon NEMO-mediated MYC stabilization led to an increase in the intracellular glutathione levels, which rendered cells more resistant to ionizing radiation (Kim et al. 2011). These studies suggest that the NEMO/MYC interaction might be a good target in the development of strategies to overcome radiotherapy resistance (Kim et al. 2011).
It is clear from the discussion above that many proteins have been identified that regulate MYC stability through directly or indirectly affecting its ubiquitination, and many of these have been mapped to overlapping domains in MYC (see Fig. 1). Although a few studies have defined relationships between these players, in most cases, they have been studied in isolation, and thus it is difficult to make comprehensive conclusions about the regulation of MYC ubiquitination and stability. Hopefully, future research will begin to probe the inter-relationships between these proteins and how they coordinately regulate MYC expression level as well as activity.
THE INTERPLAY BETWEEN MYC UBIQUITINATION AND ACETYLATION
MYC is known to interact with several cofactors that have histone acetyltransferase (HAT) activity, including CBP/p300, TIP60, and GCN5 (Vervoorts et al. 2003). Although these HATs are known to be important for MYC-dependent transcriptional activation through the acetylation of histones (Adhikary et al. 2005), it has been shown that MYC is also an acetylation target, and because both ubiquitination and acetylation occur on lysine residues, acetylation could potentially interfere with MYC ubiquitination. Indeed, it has been shown that acetylation competes with ubiquitination of lysine residues in several other proteins, including p53 (Li et al. 2002), Runx3 (Jin et al. 2004), SMAD7 (Gronroos et al. 2002), and RelA (Li et al. 2012).
Vervoorts et al. (2003) found that MYC was an acetylation target of CBP/p300 and that CBP-mediated MYC acetylation had no effect on MYC DNA binding. Instead, acetylation reduced MYC ubiquitination resulting in increased protein stability. Zhang et al. (2005) subsequently identified six lysine residues in human MYC that were acetylated by p300: K143, K157, K275, K317, K323, and K371. Additionally, Patel et al. (2004) showed that MYC is similarly acetylated by GCN5 and TIP60, resulting in increased MYC protein stability. Despite the location of some of these acetylation sites, MYC nuclear localization and dimerization with Max were not affected by GCN5-mediated acetylation. More recent work has indicated that MYC can also be targeted directly by deacetylases. Yuan et al. (2009) found that the protein deacetylase SIRT1, which is a transactivated MYC target gene, interacts with and deacetylates MYC, and this results in decreased MYC protein stability. They proposed that MYC and SIRT1 form a negative-feedback loop that inhibits MYC-induced transformation, suggesting that SIRT1 functions as a tumor suppressor (Yuan et al. 2009). Further studies are needed to determine if the proposed feedback loop is relevant to human tumors and whether other deacetylases are important in controlling MYC protein stability and activity. However, together, these studies show that MYC ubiquitination and acetylation are likely connected. Further studies are required to better understand this interplay and determine its functional significance.
A LINK BETWEEN MYC UBIQUITINATION AND TRANSCRIPTIONAL ACTIVATION
The work described above for Skp2, HectH9, and Pin1/Fbw7 supports the idea adopted by the Tansey laboratory, termed “transcription factor licensing.” This model suggests that activation of some transcription factors is coupled to their ubiquitination and degradation (Salghetti et al. 2000, 2001). Indeed, Zhang et al. (2013) have recently shown that ubiquitination of six lysine residues in the TAD of murine MYC (K51, K52, K127, K144, K149, and K158) is required for induction of canonical E-box-containing target genes and that this is important for transformation. Furthermore, they showed that loss of TAD ubiquitination leads to the induction of the non-canonical MYC target gene Egr1, resulting in apoptosis. This loss of TAD ubiquitination and subsequent switch to apoptotic activity was mediated by ARF, which they showed inhibits the interaction between MYC and Skp2, and Skp2-mediated ubiquitination of MYC, resulting in MYC stabilization. Overexpression of Skp2, which occurs in many tumors, prevents ARF recruitment and inhibits apoptosis. Thus, these studies suggest that ubiquitination not only controls MYC protein levels, but also controls MYC transcriptional and biological activity. As discussed above, this might involve competition between overlapping acetylation and ubiquitination sites within the TAD of MYC.
The idea that MYC activation is coupled to its degradation is reminiscent of a negative-feedback loop in signaling, where MYC activity contributes to its own down-regulation. It has been shown that proteasome subunits can be detected at MYC target gene promoters (Salghetti et al. 2000; von der Lehr et al. 2003; Farrell et al. 2013). By linking transcriptional activity to degradation, MYC function can be more finely tuned and responsive to the cellular environment and fluctuations in MYC expression levels. This would allow for more precise control of MYC-mediated cell fate decisions. In addition, the data with Pin1 suggest that dynamic MYC DNA binding appears to contribute to optimal MYC transcriptional activity (Farrell et al. 2013). Pin1 regulates MYC at two points in normal cells: (1) target gene promoter binding and cofactor recruitment, leading to transcriptional activation; and (2) subsequent release from the promoter associated with Fbw7-mediated degradation. In cancer cells with increased Myc stability due to defects in the pS62/pT58 MYC degradation pathway downstream from Pin1, Pin1 no longer facilitates MYC degradation. However, Pin1 is still able to mediate MYC transcriptional activation. Interestingly, rapid dissociation of MYC from target gene promoters was still observed in cancer cells with more stable MYC. However, unlike non-transformed cells, a new peak of MYC binding at target gene promoters was observed in the absence of new protein synthesis, and this was dependent on Pin1 and presumably coming from remaining pools of pS62-MYC present in cancer cells. This results in cyclic, or biphasic MYC DNA binding, which appears to be important for optimal MYC transcriptional activity. It is possible that the binding and release of MYC is in some way tied to the release of paused RNA polymerases and in this way contributes to continued firing of gene transcription (Rahl et al. 2010; Giraud et al. 2012).
MYC STABILITY AND CANCER
MYC E3 Ubiquitin Ligases and Cancer
Deregulated expression of MYC plays a significant role in tumorigenesis. MYC protein is overexpressed in ∼70% of human cancers, but on average only 20% of these tumors have a MYC gene amplification or translocation that could help explain the high expression of MYC protein (Nesbit et al. 1999). Deregulation of E3 ubiquitin ligases can contribute to the increased MYC levels and protein stability seen in human cancers. Indeed, aberrant expression and/or mutation/inactivation have been shown for some MYC E3 ligases. Specifically, Fbw7 is a known tumor suppressor (Minella and Clurman 2005) that can be inactivated by point mutations or whose expression can be lost in human cancers (O’Neil et al. 2007; Tan et al. 2008). Genetic deletion of FBW7 was reported in ∼30% of human cancers (Knuutila et al. 1999), and analysis of Fbw7 mutational status in primary human tumors showed an overall mutation rate of 6% (although this varies significantly depending on the tumor type) (Akhoondi et al. 2007). Usp28, which antagonizes Fbw7α activity on MYC, has been shown to be overexpressed in cancer (Popov et al. 2007b). Studies have also found levels of TRUSS, another E3 ligase that negatively regulates MYC protein, to be low in many human cancer cell lines (Choi et al. 2010). In addition, studies suggest that CHIP might be a tumor suppressor, because CHIP has been shown to negatively correlate with malignancy of human breast cancer tissues (Kajiro et al. 2009). Likewise, Paul et al. (2013) found that knockdown of CHIP in rat glioma cell lines enhanced their metastatic properties, and that CHIP was down-regulated in glioblastoma compared with normal brain tissue.
In contrast to the above, E3 ligases that positively regulate MYC transcriptional activity, such as Skp2 and HectH9, might be expected to be overexpressed in human cancers. Indeed, Skp2 is considered to be an oncogene (Gstaiger et al. 2001) and is overexpressed in many human tumors (Chan et al. 2010). In addition, using tissue microarrays, Adhikary et al. (2005) found overexpression of HectH9 in many primary human tumors, including 43% of breast cancers, 46% of lung tumors, 52% of colon tumors, 18% of liver tumors, 20% of pancreatic carcinomas, and 9% of thyroid tumors examined.
Alterations in Cell Signaling Pathways that Impact MYC Protein Stability in Cancer
Given that many of the signaling proteins involved in the pS62/pT58 MYC degradation pathway controlling Fbw7-mediated MYC turnover (Fig. 2) are often misregulated in human cancers, altered S62 and T58 phosphorylation levels and increased MYC stability could help explain MYC’s frequent overexpression without gene amplification in tumors. Highlighting the importance of this degradation pathway in cancer, three of the four original MYC-containing retroviruses and many Burkitt lymphomas have mutations in MYC at or around T58 that impair phosphorylation at this site, increase phosphorylation at S62, and inhibit Fbw7-mediated degradation of MYC (Bhatia et al. 1993; Bahram et al. 2000; Gregory and Hann 2000). Studies using hematopoietic stem cells transduced with MYC T58A or ROSA26-MYC T58A or S62A phosphorylation mutant knock-in mice with conditional expression in the mammary gland, have shown that MYC T58A, which is resistant to PP2A and has increased S62 phosphorylation, has increased tumorigenic potential (Hemann et al. 2005; Wang et al. 2011). Furthermore, knock-in of MYC T58A into the endogenous MYC locus in mice results in aberrant self-renewal of hematopoietic progenitors and the late appearance of lymphoid and myeloid neoplasia (B Freie and RN Eisenman, pers. comm.). Although MYC is not mutated in most human cancers aside from Burkitt lymphoma, analysis of MYC phosphorylation and stability in human leukemia and breast cancer cell lines, as well as primary human tumors, showed that wild-type MYC has high S62 phosphorylation and low T58 phosphorylation and is aberrantly stabilized in many of these cancer cell lines and patient samples relative to normal controls (Malempati et al. 2006; Zhang et al. 2012). An example of high pS62-MYC in breast cancer is shown in Figure 3. Similar changes in MYC phosphorylation and MYC protein stability are seen in pancreatic cancer (AS Farrell et al., in prep.). Importantly, in conjunction with the high Pin1 observed in many cancers (Ayala et al. 2003; Lu 2003; Miyashita et al. 2003; Ryo et al. 2003; Wulf et al. 2003; Lam et al. 2008), this pS62-MYC present in cancer cells is expected to be highly transcriptionally active (Farrell et al. 2013; Sanchez-Arevalo Lobo et al. 2013). Studies exploring signaling mechanisms that could contribute to this altered MYC phosphorylation and stabilization have observed, in addition to the common activation of MEK/ERK signaling, decreased expression of PP2A-B56α and altered Axin1 splicing in some cancer cell lines that express S62-phosphorylated and stabilized MYC (Mannava et al. 2012; Zhang et al. 2012; RC Sears, unpubl.). Taken together, these studies provide evidence that impairment of the pathway that regulates MYC T58/S62 phosphorylation and Fbw7-mediated degradation could represent a novel mechanism for oncogenic activation of MYC in human cancers, and a focus for therapeutic targeting.
Figure 3.
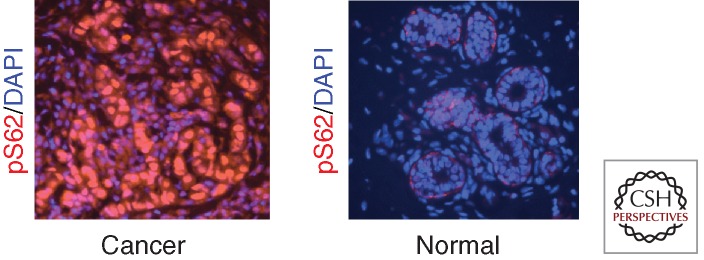
Patient-matched normal and breast tumor tissue were analyzed for pS62-MYC expression by immunofluorescence.
Targeting Myc through PP2A Inhibitors CIP2A and SET
PP2A, the major serine/threonine-specific phosphatase in mammalian cells, can dephosphorylate S62 and decrease MYC stability. PP2A refers to a large family of heterotrimeric protein phosphatases containing a common catalytic C subunit whose activity is regulated by a diverse set of regulatory B subunits (Sablina and Hahn 2008). PP2A is a critical tumor-suppressor gene that negatively regulates multiple important signal transduction pathways in addition to MYC (Eichhorn et al. 2009). Inhibition of PP2A has been shown to be essential for cell transformation and can occur through inactivation by viral oncogenes, mutation of specific subunits, or by overexpression of endogenous inhibitors (Sablina and Hahn 2008; Westermarck and Hahn 2008). Several naturally occurring inhibitors of PP2A have been identified, including SET (also known as I2PP2A) and Cellular Inhibitor of PP2A (CIP2A).
CIP2A has been described as an important PP2A inhibitor in multiple cancer types (Junttila et al. 2007). CIP2A overexpression cooperates with Ras and MYC to transform mouse primary embryo fibroblasts, whereas its suppression inhibits tumor growth (Sablina and Hahn 2008). CIP2A interacts with MYC and PP2A and interferes with PP2A-mediated S62 dephosphorylation of MYC leading to stabilization of MYC. CIP2A is up-regulated in head and neck squamous cell carcinoma, colon cancer, and many gastric cancers, and this is associated with reduced overall survival (Sablina and Hahn 2008; Khanna et al. 2009). In addition, ∼33% of breast cancers overexpress CIP2A, where it is associated with clinical aggressiveness (Come et al. 2009). Furthermore, CIP2A is frequently overexpressed in human pancreatic cancer (AS Farrell et al., in prep.).
The phosphoprotein SET, a PP2A inhibitor, was originally identified as the SET–CAN fusion gene in acute myeloid leukemia (AML) (von Lindern et al. 1992) and is also up-regulated in multiple cancer types, including chronic myelogenous leukemia, Wilm’s tumors, malignant brain tumors, tumors of the head and neck, and testicular cancers (Westermarck and Hahn 2008). Furthermore, SET expression levels have been correlated with more aggressive disease in ovarian cancer (Ouellet et al. 2006), AML (Cristobal et al. 2011), and chronic lymphocytic leukemia (Christensen et al. 2011). In addition, it is frequently overexpressed in human breast (M Janghorban et al., in prep.) and pancreatic (AS Farrell et al., in prep.) cancers.
Thus, because SET and CIP2A overexpression occurs in multiple human cancers, antagonizing these PP2A inhibitors to restore PP2A activity in cancer cells could be an approach for targeting posttranslational activation of MYC in human cancers. Indeed, recent experiments show that knockdown of SET or CIP2A increases PP2A activity and MYC degradation and decreases the tumorigenic potential of breast and pancreatic cancer cell lines both in vitro and in vivo (AS Farrell et al., in prep.; M Janghorban et al., in prep.). Although pharmacological antagonists of CIP2A have not been developed, treatment with the SET inhibitor OP449 (Christensen et al. 2011) shows activation of PP2A, increased degradation of MYC, significant reduction in proliferation, and attenuation of proliferative and survival signaling in breast and pancreatic cancer cell lines (AS Farrell et al., in prep.; M Janghorban et al., in prep.).
CONCLUDING REMARKS
Because MYC is a driver of cell growth and metabolism, multiple cellular controls act to regulate its levels. One of the most important mechanisms to control MYC levels is regulated degradation via the ubiquitin–proteasome system. Many E3 ubiquitin ligases have been shown to act on MYC; however, not all of these are equivalent in their capacity to control MYC abundance through degradation (see Table 1). Some E3 ligases clearly stimulate MYC degradation, whereas others stabilize MYC. Furthermore, E3 ligases that destabilize MYC can either inhibit MYC activity or increase MYC activity, involving a complex relationship between MYC ubiquitination and its transcriptional function. In addition, there is a potentially important interplay between MYC ubiquitination and acetylation. All of these points are critical in understanding the regulation of MYC in normal cells and how MYC deregulation occurs in cancer cells. Ultimately, more knowledge of the different pathways that posttranslationally regulate MYC protein stability and activity will be beneficial in designing new cancer therapeutics targeting MYC.
Footnotes
Editors: Chi V. Dang and Robert N. Eisenman
Additional Perspectives on MYC and the Pathway to Cancer available at www.perspectivesinmedicine.org
REFERENCES
- Adhikary S, Marinoni F, Hock A, Hulleman E, Popov N, Beier R, Bernard S, Quarto M, Capra M, Goettig S, et al. 2005. The ubiquitin ligase HectH9 regulates transcriptional activation by Myc and is essential for tumor cell proliferation. Cell 123: 409–421 [DOI] [PubMed] [Google Scholar]
- Akhoondi S, Sun D, von der Lehr N, Apostolidou S, Klotz K, Maljukova A, Cepeda D, Fiegl H, Dafou D, Marth C, et al. 2007. FBXW7/hCDC4 is a general tumor suppressor in human cancer. Cancer Res 67: 9006–9012 [DOI] [PubMed] [Google Scholar]
- Arnold HK, Sears RC 2006. Protein phosphatase 2A regulatory subunit B56α associates with c-Myc and negatively regulates c-Myc accumulation. Mol Cell Biol 26: 2832–2844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold HK, Zhang X, Daniel CJ, Tibbitts D, Escamilla-Powers J, Farrell A, Tokarz S, Morgan C, Sears RC 2009. The Axin1 scaffold protein promotes formation of a degradation complex for c-Myc. EMBO J 28: 500–512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayala G, Wang D, Wulf G, Frolov A, Li R, Sowadski J, Wheeler TM, Lu KP, Bao L 2003. The prolyl isomerase Pin1 is a novel prognostic marker in human prostate cancer. Cancer Res 63: 6244–6251 [PubMed] [Google Scholar]
- Bachireddy P, Bendapudi PK, Felsher DW 2005. Getting at MYC through RAS. Clin Cancer Res 11: 4278–4281 [DOI] [PubMed] [Google Scholar]
- Bahram F, von der Lehr N, Cetinkaya C, Larsson LG 2000. c-Myc hot spot mutations in lymphomas result in inefficient ubiquitination and decreased proteasome-mediated turnover. Blood 95: 2104–2110 [PubMed] [Google Scholar]
- Ballinger CA, Connell P, Wu Y, Hu Z, Thompson LJ, Yin LY, Patterson C 1999. Identification of CHIP, a novel tetratricopeptide repeat-containing protein that interacts with heat shock proteins and negatively regulates chaperone functions. Mol Cell Biol 19: 4535–4545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatia K, Huppi K, Spangler G, Siwarski D, Iyer R, Magrath I 1993. Point mutations in the c-Myc transactivation domain are common in Burkitt’s lymphoma and mouse plasmacytomas. Nat Genet 5: 56–61 [DOI] [PubMed] [Google Scholar]
- Blackwood EM, Eisenman RN 1991. Max: A helix–loop–helix zipper protein that forms a sequence-specific DNA-binding complex with Myc. Science 251: 1211–1217 [DOI] [PubMed] [Google Scholar]
- Bretones G, Acosta JC, Caraballo JM, Ferrandiz N, Gomez-Casares MT, Albajar M, Blanco R, Ruiz P, Hung WC, Albero MP, et al. 2011. SKP2 oncogene is a direct MYC target gene and MYC down-regulates p27KIP1 through SKP2 in human leukemia cells. J Biol Chem 286: 9815–9825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan CH, Lee SW, Wang J, Lin HK 2010. Regulation of Skp2 expression and activity and its role in cancer progression. ScientificWorldJournal 10: 1001–1015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi SH, Wright JB, Gerber SA, Cole MD 2010. Myc protein is stabilized by suppression of a novel E3 ligase complex in cancer cells. Genes Dev 24: 1236–1241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen DJ, Chen Y, Oddo J, Matta KM, Neil J, Davis ED, Volkheimer AD, Lanasa MC, Friedman DR, Goodman BK, et al. 2011. SET oncoprotein overexpression in B-cell chronic lymphocytic leukemia and non-Hodgkin lymphoma: A predictor of aggressive disease and a new treatment target. Blood 118: 4150–4158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole MD 1986. The myc oncogene: Its role in transformation and differentiation. Annu Rev Genet 20: 361–384 [DOI] [PubMed] [Google Scholar]
- Come C, Laine A, Chanrion M, Edgren H, Mattila E, Liu X, Jonkers J, Ivaska J, Isola J, Darbon JM, et al. 2009. CIP2A is associated with human breast cancer aggressivity. Clin Cancer Res 15: 5092–5100 [DOI] [PubMed] [Google Scholar]
- Conacci-Sorrell M, Eisenman RN 2011. Post-translational control of Myc function during differentiation. Cell Cycle 10: 604–610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conacci-Sorrell M, Ngouenet C, Eisenman RN 2010. Myc-nick: A cytoplasmic cleavage product of Myc that promotes α-tubulin acetylation and cell differentiation. Cell 142: 480–493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowling VH, Chandriani S, Whitfield ML, Cole MD 2006. A conserved Myc protein domain, MBIV, regulates DNA binding, apoptosis, transformation, and G2 arrest. Mol Cell Biol 26: 4226–4239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cristobal I, Garcia-Orti L, Cirauqui C, Cortes-Lavaud X, Garcia-Sanchez MA, Calasanz MJ, Odero MD 2011. Overexpression of SET is a recurrent event associated with poor outcome that contributes to protein phosphatase 2A inhibition in acute myeloid leukemia. Haematologica 97: 543–550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang CV 1999. c-Myc target genes involved in cell growth, apoptosis, and metabolism. Mol Cell Biol 19: 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang CV 2012. MYC on the path to cancer. Cell 149: 22–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang CV, Lee WM 1988. Identification of the human c-Myc protein nuclear translocation signal. Mol Cell Biol 8: 4048–4054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dani C, Blanchard JM, Piechaczyk M, El Sabouty S, Marty L, Jeanteur P 1984. Extreme instability of myc mRNA in normal and transformed human cells. Proc Natl Acad Sci 81: 7046–7050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshaies RJ 1999. SCF and Cullin/Ring H2-based ubiquitin ligases. Annu Rev Cell Dev Biol 15: 435–467 [DOI] [PubMed] [Google Scholar]
- Dias DC, Dolios G, Wang R, Pan ZQ 2002. CUL7: A DOC domain-containing cullin selectively binds Skp1•Fbx29 to form an SCF-like complex. Proc Natl Acad Sci 99: 16601–16606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dikic I, Robertson M 2012. Ubiquitin ligases and beyond. BMC Biol 10: 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eichhorn PJ, Creyghton MP, Bernards R 2009. Protein phosphatase 2A regulatory subunits and cancer. Biochim Biophys Acta 1795: 1–15 [DOI] [PubMed] [Google Scholar]
- Eisenman RN 2001. Deconstructing myc. Genes Dev 15: 2023–2030 [DOI] [PubMed] [Google Scholar]
- Farrell AS, Pelz C, Wang X, Daniel CJ, Wang Z, Su Y, Janghorban M, Zhang X, Morgan C, Impey S, et al. 2013. Pin1 regulates the dynamics of c-Myc DNA binding to facilitate target gene regulation and oncogenesis. Mol Cell Biol 33: 2930–2949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flinn EM, Busch CM, Wright AP 1998. Myc boxes, which are conserved in Myc family proteins, are signals for protein degradation via the proteasome. Mol Cell Biol 18: 5961–5969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraga MF, Ballestar E, Villar-Garea A, Boix-Chornet M, Espada J, Schotta G, Bonaldi T, Haydon C, Ropero S, Petrie K, et al. 2005. Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nat Genet 37: 391–400 [DOI] [PubMed] [Google Scholar]
- Giraud M, Yoshida H, Abramson J, Rahl PB, Young RA, Mathis D, Benoist C 2012. Aire unleashes stalled RNA polymerase to induce ectopic gene expression in thymic epithelial cells. Proc Natl Acad Sci 109: 535–540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory MA, Hann SR 2000. c-Myc proteolysis by the ubiquitin-proteasome pathway: Stabilization of c-Myc in Burkitt’s lymphoma cells. Mol Cell Biol 20: 2423–2435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory MA, Qi Y, Hann SR 2003. Phosphorylation by glycogen synthase kinase-3 controls c-Myc proteolysis and subnuclear localization. J Biol Chem 278: 51606–51612 [DOI] [PubMed] [Google Scholar]
- Grim JE, Gustafson MP, Hirata RK, Hagar AC, Swanger J, Welcker M, Hwang HC, Ericsson J, Russell DW, Clurman BE 2008. Isoform- and cell cycle–dependent substrate degradation by the Fbw7 ubiquitin ligase. J Cell Biol 181: 913–920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gronroos E, Hellman U, Heldin CH, Ericsson J 2002. Control of Smad7 stability by competition between acetylation and ubiquitination. Mol Cell 10: 483–493 [DOI] [PubMed] [Google Scholar]
- Gstaiger M, Jordan R, Lim M, Catzavelos C, Mestan J, Slingerland J, Krek W 2001. Skp2 is oncogenic and overexpressed in human cancers. Proc Natl Acad Sci 98: 5043–5048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hann SR, Eisenman RN 1984. Proteins encoded by the human c-myc oncogene: Differential expression in neoplastic cells. Mol Cell Biol 4: 2486–2497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemann MT, Bric A, Teruya-Feldstein J, Herbst A, Nilsson JA, Cordon-Cardo C, Cleveland JL, Tansey WP, Lowe SW 2005. Evasion of the p53 tumour surveillance network by tumour-derived MYC mutants. Nature 436: 807–811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbst A, Salghetti SE, Kim SY, Tansey WP 2004. Multiple cell-type-specific elements regulate Myc protein stability. Oncogene 23: 3863–3871 [DOI] [PubMed] [Google Scholar]
- Herbst A, Hemann MT, Tworkowski KA, Salghetti SE, Lowe SW, Tansey WP 2005. A conserved element in Myc that negatively regulates its proapoptotic activity. EMBO Rep 6: 177–183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huibregtse JM, Scheffner M, Beaudenon S, Howley PM 1995. A family of proteins structurally and functionally related to the E6-AP ubiquitin-protein ligase. Proc Natl Acad Sci 92: 5249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hydbring P, Bahram F, Su Y, Tronnersjo S, Hogstrand K, von der Lehr N, Sharifi HR, Lilischkis R, Hein N, Wu S, et al. 2010. Phosphorylation by Cdk2 is required for Myc to repress Ras-induced senescence in cotransformation. Proc Natl Acad Sci 107: 58–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin YH, Jeon EJ, Li QL, Lee YH, Choi JK, Kim WJ, Lee KY, Bae SC 2004. Transforming growth factor-β stimulates p300-dependent RUNX3 acetylation, which inhibits ubiquitination-mediated degradation. J Biol Chem 279: 29409–29417 [DOI] [PubMed] [Google Scholar]
- Joseph JD, Yeh ES, Swenson KI, Means AR, Winkler 2003. The peptidyl-prolyl isomerase Pin1. Prog Cell Cycle Res 5: 477–487 [PubMed] [Google Scholar]
- Junttila MR, Puustinen P, Niemela M, Ahola R, Arnold H, Bottzauw T, Ala-aho R, Nielsen C, Ivaska J, Taya Y, et al. 2007. CIP2A inhibits PP2A in human malignancies. Cell 130: 51–62 [DOI] [PubMed] [Google Scholar]
- Kajiro M, Hirota R, Nakajima Y, Kawanowa K, So-ma K, Ito I, Yamaguchi Y, Ohie SH, Kobayashi Y, Seino Y, et al. 2009. The ubiquitin ligase CHIP acts as an upstream regulator of oncogenic pathways. Nat Cell Biol 11: 312–319 [DOI] [PubMed] [Google Scholar]
- Kato GJ, Barrett J, Villa-Garcia M, Dang CV 1990. An amino-terminal c-Myc domain required for neoplastic transformation activates transcription. Mol Cell Biol 10: 5914–5920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khanna A, Bockelman C, Hemmes A, Junttila MR, Wiksten JP, Lundin M, Junnila S, Murphy DJ, Evan GI, Haglund C, et al. 2009. MYC-dependent regulation and prognostic role of CIP2A in gastric cancer. J Natl Cancer Inst 101: 793–805 [DOI] [PubMed] [Google Scholar]
- Kim SY, Herbst A, Tworkowski KA, Salghetti SE, Tansey WP 2003. Skp2 regulates Myc protein stability and activity. Mol Cell 11: 1177–1188 [DOI] [PubMed] [Google Scholar]
- Kim BY, Yang JS, Kwak SY, Zhang XK, Han YH 2010. NEMO stabilizes c-Myc through direct interaction in the nucleus. FEBS Lett 584: 4524–4530 [DOI] [PubMed] [Google Scholar]
- Kim BY, Kwak SY, Yang JS, Han YH 2011. Phosphorylation and stabilization of c-Myc by NEMO renders cells resistant to ionizing radiation through up-regulation of γ-GCS. Oncol Rep 26: 1587–1593 [DOI] [PubMed] [Google Scholar]
- Kimura T, Ishizuka H, Yoshida A, Morii M, Takeguchi N, Asano S 2003. Quantity and quality control of gastric proton pump in the endoplasmic reticulum by ubiquitin/proteasome system. Biochemistry 42: 4771–4779 [DOI] [PubMed] [Google Scholar]
- Knuutila S, Aalto Y, Autio K, Bjorkqvist AM, El-Rifai W, Hemmer S, Huhta T, Kettunen E, Kiuru-Kuhlefelt S, Larramendy ML, et al. 1999. DNA copy number losses in human neoplasms. Am J Pathol 155: 683–694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch HB, Zhang R, Verdoodt B, Bailey A, Zhang CD, Yates JR 3rd, Menssen A, Hermeking H 2007. Large-scale identification of c-MYC-associated proteins using a combined TAP/MudPIT approach. Cell Cycle 6: 205–217 [DOI] [PubMed] [Google Scholar]
- Kurland JF, Tansey WP 2008. Myc-mediated transcriptional repression by recruitment of histone deacetylase. Cancer Res 68: 3624–3629 [DOI] [PubMed] [Google Scholar]
- Lam PB, Burga LN, Wu BP, Hofstatter EW, Lu KP, Wulf GM 2008. Prolyl isomerase Pin1 is highly expressed in Her2-positive breast cancer and regulates erbB2 protein stability. Mol Cancer 7: 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Luo J, Brooks CL, Gu W 2002. Acetylation of p53 inhibits its ubiquitination by Mdm2. J Biol Chem 277: 50607–50611 [DOI] [PubMed] [Google Scholar]
- Li H, Wittwer T, Weber A, Schneider H, Moreno R, Maine GN, Kracht M, Schmitz ML, Burstein E 2012. Regulation of NF-κB activity by competition between RelA acetylation and ubiquitination. Oncogene 31: 611–623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CY, Loven J, Rahl PB, Paranal RM, Burge CB, Bradner JE, Lee TI, Young RA 2012. Transcriptional amplification in tumor cells with elevated c-Myc. Cell 151: 56–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippens G, Landrieu I, Smet C 2007. Molecular mechanisms of the phospho-dependent prolyl cis/trans isomerase Pin1. FEBS J 274: 5211–5222 [DOI] [PubMed] [Google Scholar]
- Liu J, Levens D 2006. Making Myc. Curr Top Microbiol Immunol 302: 1–32 [DOI] [PubMed] [Google Scholar]
- Liu PY, Xu N, Malyukova A, Scarlett CJ, Sun YT, Zhang XD, Ling D, Su SP, Nelson C, Chang DK, et al. 2013. The histone deacetylase SIRT2 stabilizes Myc oncoproteins. Cell Death Differ 20: 503–514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu KP 2003. Prolyl isomerase Pin1 as a molecular target for cancer diagnostics and therapeutics. Cancer Cell 4: 175–180 [DOI] [PubMed] [Google Scholar]
- Lu KP, Zhou XZ 2007. The prolyl isomerase PIN1: A pivotal new twist in phosphorylation signalling and disease. Nat Rev Mol Cell Biol 8: 904–916 [DOI] [PubMed] [Google Scholar]
- Lutterbach B, Hann SR 1994. Hierarchical phosphorylation at N-terminal transformation-sensitive sites in c-Myc protein is regulated by mitogens and in mitosis. Mol Cell Biol 14: 5510–5522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malempati S, Tibbitts D, Cunningham M, Akkari Y, Olson S, Fan G, Sears RC 2006. Aberrant stabilization of c-Myc protein in some lymphoblastic leukemias. Leukemia 20: 1572–1581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mannava S, Omilian AR, Wawrzyniak JA, Fink EE, Zhuang D, Miecznikowski JC, Marshall JR, Soengas MS, Sears RC, Morrison CD, et al. 2012. PP2A-B56α controls oncogene-induced senescence in normal and tumor human melanocytic cells. Oncogene 31: 1484–1492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzger MB, Hristova VA, Weissman AM 2012. HECT and RING finger families of E3 ubiquitin ligases at a glance. J Cell Sci 125: 531–537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minella AC, Clurman BE 2005. Mechanisms of tumor suppression by the SCFFbw7. Cell Cycle 4: 1356–1359 [DOI] [PubMed] [Google Scholar]
- Miyashita H, Mori S, Motegi K, Fukumoto M, Uchida T 2003. Pin1 is overexpressed in oral squamous cell carcinoma and its levels correlate with cyclin D1 overexpression. Oncol Rep 10: 455–461 [PubMed] [Google Scholar]
- Moberg KH, Mukherjee A, Veraksa A, Artavanis-Tsakonas S, Hariharan IK 2004. The Drosophila F box protein archipelago regulates dMyc protein levels in vivo. Curr Biol 14: 965–974 [DOI] [PubMed] [Google Scholar]
- Nandi D, Tahiliani P, Kumar A, Chandu D 2006. The ubiquitin–proteasome system. J Biosci 31: 137–155 [DOI] [PubMed] [Google Scholar]
- Nesbit CE, Tersak JM, Prochownik EV 1999. MYC oncogenes and human neoplastic disease. Oncogene 18: 3004–3016 [DOI] [PubMed] [Google Scholar]
- Nijman SM, Luna-Vargas MP, Velds A, Brummelkamp TR, Dirac AM, Sixma TK, Bernards R 2005. A genomic and functional inventory of deubiquitinating enzymes. Cell 123: 773–786 [DOI] [PubMed] [Google Scholar]
- O’Neil J, Grim J, Strack P, Rao S, Tibbitts D, Winter C, Hardwick J, Welcker M, Meijerink JP, Pieters R, et al. 2007. FBW7 mutations in leukemic cells mediate NOTCH pathway activation and resistance to γ-secretase inhibitors. J Exp Med 204: 1813–1824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouellet V, Le Page C, Guyot MC, Lussier C, Tonin PN, Provencher DM, Mes-Masson AM 2006. SET complex in serous epithelial ovarian cancer. Int J Cancer 119: 2119–2126 [DOI] [PubMed] [Google Scholar]
- Patel JH, Du Y, Ard PG, Phillips C, Carella B, Chen CJ, Rakowski C, Chatterjee C, Lieberman PM, Lane WS, et al. 2004. The c-MYC oncoprotein is a substrate of the acetyltransferases hGCN5/PCAF and TIP60. Mol Cell Biol 24: 10826–10834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul I, Ahmed SF, Bhowmik A, Deb S, Ghosh MK 2013. The ubiquitin ligase CHIP regulates c-Myc stability and transcriptional activity. Oncogene 32: 1284–1295 [DOI] [PubMed] [Google Scholar]
- Petroski MD, Deshaies RJ 2005. Function and regulation of cullin-RING ubiquitin ligases. Nat Rev Mol Cell Biol 6: 9–20 [DOI] [PubMed] [Google Scholar]
- Popov N, Herold S, Llamazares M, Schulein C, Eilers M 2007a. Fbw7 and Usp28 regulate myc protein stability in response to DNA damage. Cell Cycle 6: 2327–2331 [DOI] [PubMed] [Google Scholar]
- Popov N, Wanzel M, Madiredjo M, Zhang D, Beijersbergen R, Bernards R, Moll R, Elledge SJ, Eilers M 2007b. The ubiquitin-specific protease USP28 is required for MYC stability. Nat Cell Biol 9: 765–774 [DOI] [PubMed] [Google Scholar]
- Popov N, Schulein C, Jaenicke LA, Eilers M 2010. Ubiquitylation of the amino terminus of Myc by SCFβ-TrCP antagonizes SCFFbw7-mediated turnover. Nat Cell Biol 12: 973–981 [DOI] [PubMed] [Google Scholar]
- Prendergast GC 1999. Mechanisms of apoptosis by c-Myc. Oncogene 18: 2967–2987 [DOI] [PubMed] [Google Scholar]
- Pulverer BJ, Fisher C, Vousden K, Littlewood T, Evan G, Woodgett JR 1994. Site-specific modulation of c-Myc cotransformation by residues phosphorylated in vivo. Oncogene 9: 59–70 [PubMed] [Google Scholar]
- Rahl PB, Lin CY, Seila AC, Flynn RA, McCuine S, Burge CB, Sharp PA, Young RA 2010. c-Myc regulates transcriptional pause release. Cell 141: 432–445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryo A, Liou YC, Lu KP, Wulf G 2003. Prolyl isomerase Pin1: A catalyst for oncogenesis and a potential therapeutic target in cancer. J Cell Sci 116: 773–783 [DOI] [PubMed] [Google Scholar]
- Sablina AA, Hahn WC 2008. SV40 small T antigen and PP2A phosphatase in cell transformation. Cancer Metastasis Rev 27: 137–146 [DOI] [PubMed] [Google Scholar]
- Salghetti SE, Kim SY, Tansey WP 1999. Destruction of Myc by ubiquitin-mediated proteolysis: Cancer-associated and transforming mutations stabilize Myc. EMBO J 18: 717–726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salghetti SE, Muratani M, Wijnen H, Futcher B, Tansey WP 2000. Functional overlap of sequences that activate transcription and signal ubiquitin-mediated proteolysis. Proc Natl Acad Sci 97: 3118–3123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salghetti SE, Caudy AA, Chenoweth JG, Tansey WP 2001. Regulation of transcriptional activation domain function by ubiquitin. Science 293: 1651–1653 [DOI] [PubMed] [Google Scholar]
- Sanchez-Arevalo Lobo VJ, Doni M, Verrecchia A, Sanulli S, Faga G, Piontini A, Bianchi M, Conacci-Sorrell M, Mazzarol G, Peg V, et al. 2013. Dual regulation of Myc by Abl. Oncogene 32: 5261–5271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwamborn JC, Berezikov E, Knoblich JA 2009. The TRIM-NHL protein TRIM32 activates microRNAs and prevents self-renewal in mouse neural progenitors. Cell 136: 913–925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sears RC 2004. The life cycle of c-Myc: From synthesis to degradation. Cell Cycle 3: 1133–1137 [PubMed] [Google Scholar]
- Sears R, Leone G, DeGregori J, Nevins JR 1999. Ras enhances Myc protein stability. Mol Cell 3: 169–179 [DOI] [PubMed] [Google Scholar]
- Sears R, Nuckolls F, Haura E, Taya Y, Tamai K, Nevins JR 2000. Multiple Ras-dependent phosphorylation pathways regulate Myc protein stability. Genes Dev 14: 2501–2514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Small GW, Chou TY, Dang CV, Orlowski RZ 2002. Evidence for involvement of calpain in c-Myc proteolysis in vivo. Arch Biochem Biophys 400: 151–161 [DOI] [PubMed] [Google Scholar]
- Sorokin AV, Kim ER, Ovchinnikov LP 2009. Proteasome system of protein degradation and processing. Biochemistry (Mosc) 74: 1411–1442 [DOI] [PubMed] [Google Scholar]
- Spencer CA, Groudine M 1990. Molecular analysis of the c-Myc transcription elongation block. Implications for the generation of Burkitt’s lymphoma. Ann NY Acad Sci 599: 12–28 [DOI] [PubMed] [Google Scholar]
- Tan Y, Sangfelt O, Spruck C 2008. The Fbxw7/hCdc4 tumor suppressor in human cancer. Cancer Lett 271: 1–12 [DOI] [PubMed] [Google Scholar]
- Thomas LR, Tansey WP 2011. Proteolytic control of the oncoprotein transcription factor Myc. Adv Cancer Res 110: 77–106 [DOI] [PubMed] [Google Scholar]
- Vaquero A, Scher MB, Lee DH, Sutton A, Cheng HL, Alt FW, Serrano L, Sternglanz R, Reinberg D 2006. SirT2 is a histone deacetylase with preference for histone H4 Lys 16 during mitosis. Genes Dev 20: 1256–1261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vervoorts J, Luscher-Firzlaff JM, Rottmann S, Lilischkis R, Walsemann G, Dohmann K, Austen M, Luscher B 2003. Stimulation of c-MYC transcriptional activity and acetylation by recruitment of the cofactor CBP. EMBO Rep 4: 484–490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- von der Lehr N, Johansson S, Wu S, Bahram F, Castell A, Cetinkaya C, Hydbring P, Weidung I, Nakayama K, Nakayama KI, et al. 2003. The F-box protein Skp2 participates in c-Myc proteosomal degradation and acts as a cofactor for c-Myc-regulated transcription. Mol Cell 11: 1189–1200 [DOI] [PubMed] [Google Scholar]
- von Lindern M, van Baal S, Wiegant J, Raap A, Hagemeijer A, Grosveld G 1992. Can, a putative oncogene associated with myeloid leukemogenesis, may be activated by fusion of its 3′ half to different genes: Characterization of the set gene. Mol Cell Biol 12: 3346–3355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wall M, Poortinga G, Hannan KM, Pearson RB, Hannan RD, McArthur GA 2008. Translational control of c-MYC by rapamycin promotes terminal myeloid differentiation. Blood 112: 2305–2317 [DOI] [PubMed] [Google Scholar]
- Wang X, Cunningham M, Zhang X, Tokarz S, Laraway B, Troxell M, Sears RC 2011. Phosphorylation regulates c-Myc’s oncogenic activity in the mammary gland. Cancer Res 71: 925–936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welcker M, Orian A, Grim JE, Eisenman RN, Clurman BE 2004a. A nucleolar isoform of the Fbw7 ubiquitin ligase regulates c-Myc and cell size. Curr Biol 14: 1852–1857 [DOI] [PubMed] [Google Scholar]
- Welcker M, Orian A, Jin J, Grim JE, Harper JW, Eisenman RN, Clurman BE 2004b. The Fbw7 tumor suppressor regulates glycogen synthase kinase 3 phosphorylation-dependent c-Myc protein degradation. Proc Natl Acad Sci 101: 9085–9090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westermarck J, Hahn WC 2008. Multiple pathways regulated by the tumor suppressor PP2A in transformation. Trends Mol Med 14: 152–160 [DOI] [PubMed] [Google Scholar]
- Wulf G, Ryo A, Liou YC, Lu KP 2003. The prolyl isomerase Pin1 in breast development and cancer. Breast Cancer Res 5: 76–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yada M, Hatakeyama S, Kamura T, Nishiyama M, Tsunematsu R, Imaki H, Ishida N, Okumura F, Nakayama K, Nakayama KI 2004. Phosphorylation-dependent degradation of c-Myc is mediated by the F-box protein Fbw7. EMBO J 23: 2116–2125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaoka S, Courtois G, Bessia C, Whiteside ST, Weil R, Agou F, Kirk HE, Kay RJ, Israel A 1998. Complementation cloning of NEMO, a component of the IκB kinase complex essential for NF-κB activation. Cell 93: 1231–1240 [DOI] [PubMed] [Google Scholar]
- Yeh E, Cunningham M, Arnold H, Chasse D, Monteith T, Ivaldi G, Hahn WC, Stukenberg PT, Shenolikar S, Uchida T, et al. 2004. A signalling pathway controlling c-Myc degradation that impacts oncogenic transformation of human cells. Nat Cell Biol 6: 308–318 [DOI] [PubMed] [Google Scholar]
- Yuan J, Minter-Dykhouse K, Lou Z 2009. A c-Myc–SIRT1 feedback loop regulates cell growth and transformation. J Cell Biol 185: 203–211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K, Faiola F, Martinez E 2005. Six lysine residues on c-Myc are direct substrates for acetylation by p300. Biochem Biophys Res Commun 336: 274–280 [DOI] [PubMed] [Google Scholar]
- Zhang X, Farrell AS, Daniel CJ, Arnold H, Scanlan C, Laraway BJ, Janghorban M, Lum L, Chen D, Troxell M, et al. 2012. Mechanistic insight into Myc stabilization in breast cancer involving aberrant Axin1 expression. Proc Natl Acad Sci 109: 2790–2795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Spears E, Boone DN, Li Z, Gregory MA, Hann SR 2013. Domain-specific c-Myc ubiquitylation controls c-Myc transcriptional and apoptotic activity. Proc Natl Acad Sci 110: 978–983 [DOI] [PMC free article] [PubMed] [Google Scholar]