

Abstract
Activating mutations of the PIK3CA gene occur frequently in breast cancer, and inhibitors that are specific for phosphatidylinositol 3-kinase (PI3K) p110α, such as BYL719, are being investigated in clinical trials. In a search for correlates of sensitivity to p110α inhibition among PIK3CA-mutant breast cancer cell lines, we observed that sensitivity to BYL719 (as assessed by cell proliferation) was associated with full inhibition of signaling through the TORC1 pathway. Conversely, cancer cells that were resistant to BYL719 had persistently active mTORC1 signaling, although Akt phosphorylation was inhibited. Similarly, in patients, pS6 (residues 240/4) expression (a marker of mTORC1 signaling) was associated with tumor response to BYL719, and mTORC1 was found to be reactivated in tumors from patients whose disease progressed after treatment. In PIK3CA-mutant cancer cell lines with persistent mTORC1 signaling despite PI3K p110α blockade (that is, resistance), the addition of the allosteric mTORC1 inhibitor RAD001 to the cells along with BYL719 resulted in reversal of resistance in vitro and in vivo. Finally, we found that growth factors such as insulin-like growth factor 1 and neuregulin 1 can activate mammalian target of rapamycin (mTOR) and mediate resistance to BYL719. Our findings suggest that simultaneous administration of mTORC1 inhibitors may enhance the clinical activity of p110α-targeted drugs and delay the appearance of resistance.
Introduction
The phosphatidylinositol 3-kinase (PI3K) pathway, which includes the PI3K holoenzyme and the downstream effectors Akt and mammalian target of rapamycin (mTOR), is essential for cell growth, proliferation, survival, and metabolism (1, 2). The PI3K family of enzymes is divided into three main classes (classes I to III), with class I being the most often implicated in human cancer (3, 4). Class IA PI3K is a heterodimer composed of a catalytic subunit (p110) and a regulatory subunit (p85) (5, 6). There are three p110 isoforms that can contribute to class IA PI3K signaling: p110α, p110β, and p110δ. PIK3CA, the gene encoding for p110α, is frequently mutated in human cancers (7, 8). In breast cancer, most activating mutations in this gene reside in the helical (E542K and E545K) or catalytic (H1047R) domains, and these mutations are found in ∼30% of estrogen receptor (ER)– and/or HER2-positive tumors (8–10). In addition, aberrant activation of the PI3K pathway may also occur as a result of HER2 amplification (11, 12) or loss of the PTEN or INPP4B tumor suppressors (13–15).
Direct pharmacologic inhibition of PI3K/Akt signaling is, therefore, an attractive therapeutic strategy for cancer. Recent clinical studies have demonstrated that pan–class I PI3K inhibitors (which inhibit p110α, β, and δ) can be therapeutic in patients with advanced cancer (16–19). In contrast to these broad-spectrum inhibitors, a new generation of PI3K p110α-specific inhibitors has been shown to be particularly effective in cancer cell lines harboring PIK3CA mutations in large cell line screens (20). Their specificity against cancer cells harboring PIK3CA mutations has led to the clinical development of one of these compounds, NVP BYL719, by Novartis exclusively for patients with tumors (including breast cancer) harboring PIK3CA mutations, and early clinical trials show promising clinical activity with a high response rate (20, 21). Nevertheless, despite the presence of activating PIK3CA mutations, not all patients benefit from BYL719, suggesting that their tumors may be intrinsically resistant to PI3K p110α inhibitors.
We sought to identify molecular determinants of sensitivity and resistance to BYL719 that could provide guidance for patient selection or for the choice of agents to be given in combination.
Results
Intrinsic resistance to BYL719 correlates with persistent mTORC1 activity
We determined the ability of BYL719 to inhibit proliferation and viability in a panel of 20 PIK3CA-mutant (PIK3CA-mut) breast cancer cell lines and in five control cell lines bearing wild-type PIK3CA (PIK3CA-WT) (Fig. 1A and Table 1). Overall, p110α-specific blockade with BYL719 decreased cell viability in PIK3CA-mut cell lines more than in PIK3CA-WT cells. Although all five tested PIK3CA-WT cell lines displayed a resistant phenotype [IC50 (median inhibitory concentration) >6 μM], the PIK3CA-mut cell lines showed variable response to BYL719 [IC50s ranging from 63 nM to 8 μM (Fig. 1A)]. At 1 μM, BYL719 blocks p110α without affecting the other p110 isoforms (20). Thus, we classified PIK3CA-mut cancer cells as BYL719-sensitive if the IC50 was lower than 1 μM and BYL719-resistant if it was higher than 1 μM. After treatment with 1 μM BYL719, sensitive cell lines showed, on average, a larger proportion of cells in cell cycle arrest (S-phase arrest) and more cells in apoptosis (as measured by annexin V staining) than did resistant lines (Fig. 1B).
Fig. 1. Correlation between mTORC1 activity and sensitivity to BYL719 in vitro.
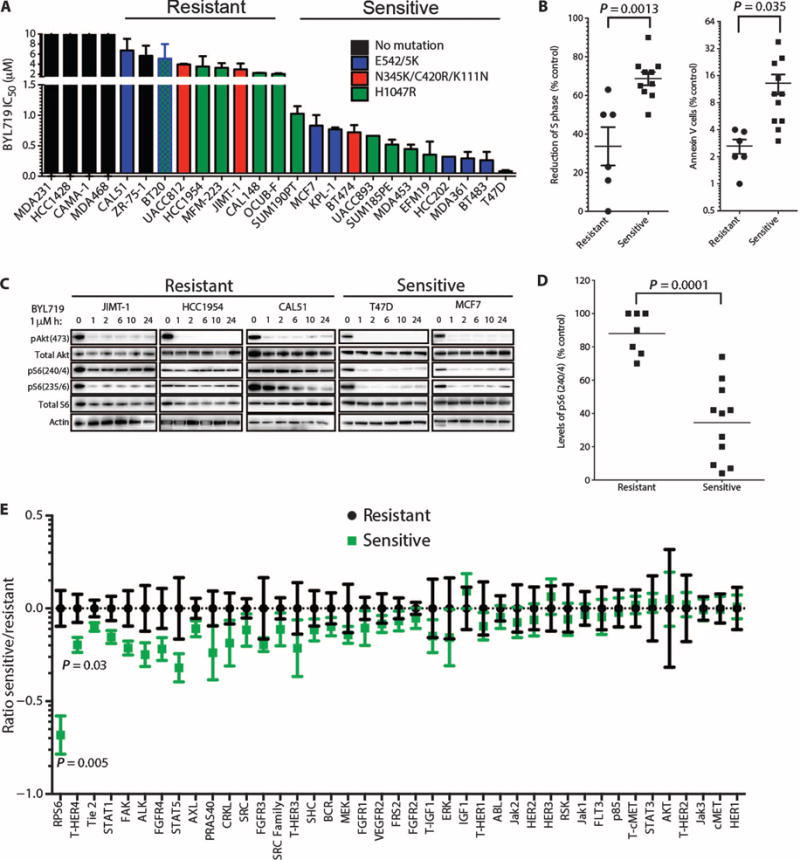
(A) IC50 for BYL719 effect on viability of 20 PIK3CA-mut and 5 PIK3CA-WT breast cancer cell lines after 3 days. (B) S-phase arrest and cell death (annexin V) in BYL719-sensitive and BYL719-resistant cells after 48 hours of treatment with 1 μM BYL719. Each dot represents the mean of two independent experiments performed in duplicate per cell line. (C) Western blot showing PI3K pathway signaling in BYL719-resistant and BYL719-sensitive cell lines. Protein lysates from cells treated with 1 μM BYL719 were extracted at different time points and analyzed by immunoblotting with the indicated antibodies. (D) pS6(240/4) determined by quantitative immunofluorescence (IF) after 2 hours of BYL719 treatment in resistant and sensitive cell lines (7 resistant and 11 sensitive PIK3CA-mut cell lines). Each dot represents the mean of three independent wells per cell line. (E) Changes in phospho or total (T) protein levels measured by cooperative, enhanced, enzyme immunoreactive (CEER) assay in sensitive cell lines relative to resistant cell lines treated with BYL719. The ratios between protein expression in treated versus control cells were log-transformed to achieve a normal distribution and to satisfy t test requirements. For visualization purposes, each protein was centered around the mean of the resistant samples. Experiments were run in triplicate per each cell line. Data are means ± SEM. P value was calculated using two-sided Student's t test.
Table 1. Breast cancer cell line information.
Twenty-five breast cancer cell lines are listed in increasing order of sensitivity to BYL719. HER2 and MYC amplification, as well as PIK3CA, PTEN, TP53, TSC2, and NRAS mutational status, is reported (TCGA and Cosmic database).
| Cell line | PIK3CA | PTEN | HER2 | TP53 | TSC2 | MYC | HRAS |
|---|---|---|---|---|---|---|---|
| MDA-MB-231 | WT | R280K | |||||
| HCC1428 | WT | ||||||
| CAMA-1 | WT | F278fs, D92H | R280T | Del | |||
| MDA-MB-468 | WT | V85_splice | |||||
| CAL51 | E542K | E288fs, T321fs | |||||
| ZR-75-1 | WT | L108R | E162K | ||||
| UACC812 | N345K | Amp | |||||
| JIMT-1 | C420R | Amp | R248W | ||||
| MFM223 | H1047R | c.395A>G, K132R | Amp | ||||
| CAL148 | D350N, H1047R | I253N, Q298* | E224K | ||||
| BT20 | P539R, H1047R | K132Q | |||||
| OCUB-F | H1047R | ||||||
| HCC1954 | H1047R | Amp | Y163C | Del | Amp | ||
| SUM190PT | H1047R | Amp | |||||
| MCF7 | E545K | ||||||
| BT474 | K111N | Amp | E285K | ||||
| KPL-1 | E545K | ||||||
| UACC893 | H1047R | Amp | R342* | ||||
| EFM19 | H1047L | Amp | H193R | ||||
| SUM185PE | H1047R | ||||||
| MDA453 | H1047R | E307K | Amp | ||||
| HCC202 | E545K, L866F | Amp | T284fs | ||||
| MDA-MB-361 | E545K, K567R | Amp | E56* | ||||
| BT483 | E542K | M246I | |||||
| T47D | H1047R | L194F |
Nonsense mutation.
These results were corroborated by long-term (8 to 11 days) proliferation assays and three-dimensional (3D) culture studies. In sensitive UACC893 cells, Ki67 staining (indicative of cell proliferation) was decreased, and cleaved caspase 3 staining (indicative of cell death) was increased after BYL719 treatment relative to those endpoints in resistant JIMT-1 cell (fig. S1). Together, these findings indicate that some PIK3CA-mut breast cancer cell lines are exquisitely sensitive to p110α inhibition, whereas others are relatively resistant. This set of observations is consistent with the results from our ongoing clinical trial in which BYL719 induces therapeutic responses in some, but not all, patients with tumors harboring PIK3CA mutations (21, 22).
Given our interest in understanding the determinants of sensitivity to p110α inhibition in PIK3CA mutant cells, we next assessed PI3K signaling in sensitive and resistant cell lines. To this end, we analyzed the phosphorylation status of Akt (pAkt), a proximal marker of PI3K inhibition, in PIK3CA-mut cell lines treated with 1 μM BYL719. A time course revealed that inhibition of pAkt was sustained for 24 hours in both sensitive and resistant cells (Fig. 1C). In contrast, the effects of BYL719 on mTORC1 inhibition, a more distal component of the PI3K signaling pathway, were varied. Only BYL719-sensitive cell lines displayed mTORC1 inhibition, as indicated by suppression of both phosphorylated S6 (pS6)(240/4) and pS6(235/6) (23). Furthermore, we measured by in-cell western assays the levels of pS6(240/4) in our panel of PIK3CA-mut cell lines after BYL719 treatment and found that sensitive and resistant cells clustered on the basis of the degree of inhibition of phosphorylation of residues 240/4 of S6 (Fig. 1D). The same result, although less pronounced, was obtained by measuring pS6(235/6), also modulated by the extracellular signal–regulated kinase (ERK) pathway (24) (fig. S2). Finally, these data were confirmed by an unbiased proteomic analysis that, among 42 total and phospho-proteins involved in cancer-related signaling pathways, identified expression of pS6(240/4) as the best predictor of BYL719 sensitivity in 12 (7 sensitive and 5 resistant) PIK3CA-mut cell lines treated with 1 μM BYL719 (Fig. 1E).
To determine whether the differential sensitivities observed in vitro would hold true in vivo, we evaluated the ability of BYL719 to inhibit growth of sensitive (MCF7) and resistant (JIMT-1) PIK3CA-mut xenograft tumors. With a daily dose (50 mg/kg) of BYL719, MCF7 xenografts did not show an increase in volume, whereas JIMT-1 xenografts grew almost as well as untreated xenograft tumors (Fig. 2A). Pharmacodynamic studies conducted in tumors collected after 3 days of treatment showed that this dose of BYL719 effectively inhibited pAkt in both MCF7 and JIMT-1 xenografts, but that pS6(240/4) and, to a lesser extent, pS6(235/6) levels were maintained at a higher level in the resistant xenografts (Fig. 2B). Consistently, phosphorylation of eukaryotic translation initiation factor 4E–binding protein 1 (4EBP1), another mTORC1 effector, was also less inhibited in JIMT-1 xenografts treated with BYL719 (fig. S3). These results point to a positive correlation between sustained mTORC1 activity and resistance to p110α inhibition.
Fig. 2. Sustained activation of mTORC1 in resistant cells after BYL719 treatment in vivo.
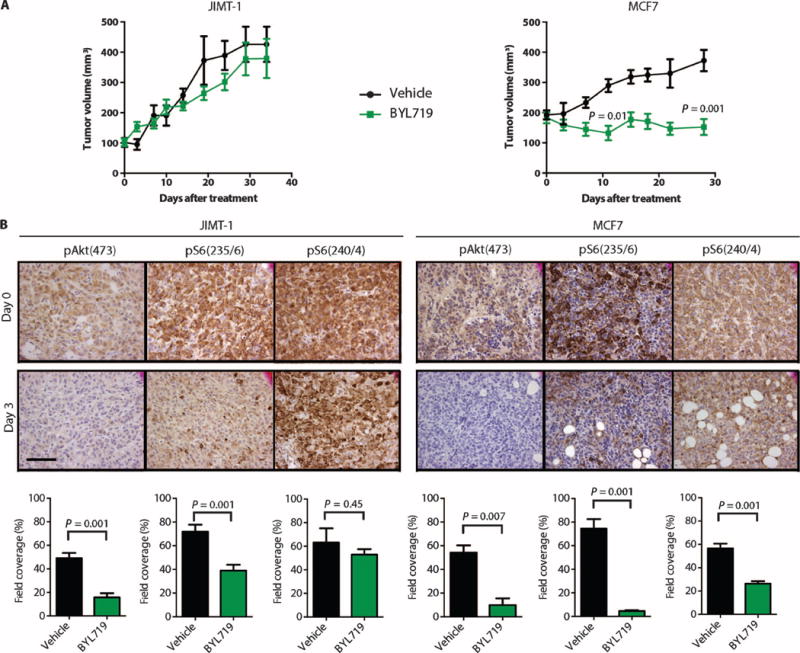
(A) Tumor growth curves of BYL719-resistant JIMT-1 (n = 10) and BYL719-sensitive MCF7 (n = 10) cell-derived xenografts upon daily treatment of mice with BYL719 (50 mg/kg). (B) Immunohistochemical (IHC) analysis of pAkt and pS6 before and after treatment with BYL719 (50 mg/kg) for 3 days. An average of six images of two independent tumors per condition was used for quantification. Quantification of IHC was performed by CellProfiler and is shown as bar graphs below each panel. Images were captured at ×40 magnification; scale bar, 100 μm. Data are means ± SEM. P value was calculated using two-sided Student's t test.
Persistent mTORC1 activation is sufficient to limit BYL719 sensitivity
We next investigated whether the mTORC1 activation status was altered in cells that acquired resistance to BYL719. We chose MDA-MB-453 (herein referred as MDA453) and T47D cell lines to generate these models of acquired resistance because they were among the most sensitive lines. Both cell lines were grown in increasing concentrations of BYL719 until their proliferation rate was undisturbed by constant inhibition of p110α with 1 μM BYL719 (∼6 months, Fig. 3A). At this concentration of BYL719, Akt phosphorylation was inhibited in both parental and resistant cells, suggesting that resistance was not due to lack of target inhibition. Although in the sensitive parental cells pS6 was almost undetectable after treatment with BYL719, S6 phosphorylation was present in both of the derived resistant cell lines (Fig. 3B). Similar results were observed for phosphorylated 4EBP1 (p4EBP1) expression. These results prompted us to explore whether mTORC1 was reactivated in cells with acquired resistance to GDC-0941, a molecule that inhibits all four isoforms of class I PI3K (25). We obtained MCF7 cells with acquired resistance to GDC-0941 (MCF7R) with the same strategy as that for MDA453R and T47DR cells (Fig. 3C). GDC-0941 suppressed Akt phosphorylation in both MCF7 and MCF7R cells, whereas pS6 levels were not fully suppressed in the resistant cells (Fig. 3D). These results suggest that failure to suppress mTORC1 signaling indicates a common resistance mechanism for different PI3K inhibitors. Indeed, BYL719-resistant MDA453R and T47DR cells were less sensitive to GDC-0941 treatment than were parental control cells (fig. S4A). Likewise, GDC-0941–resistant MCF7R cells were more resistant to BYL719 than were the parental counterparts (fig. S4B). Western blot analysis confirmed that neither BYL719 nor GDC-0941 prevented S6 phosphorylation in resistant cells (fig. S4).
Fig. 3. Resistance to PI3K inhibition induced by mTORC1 activation.
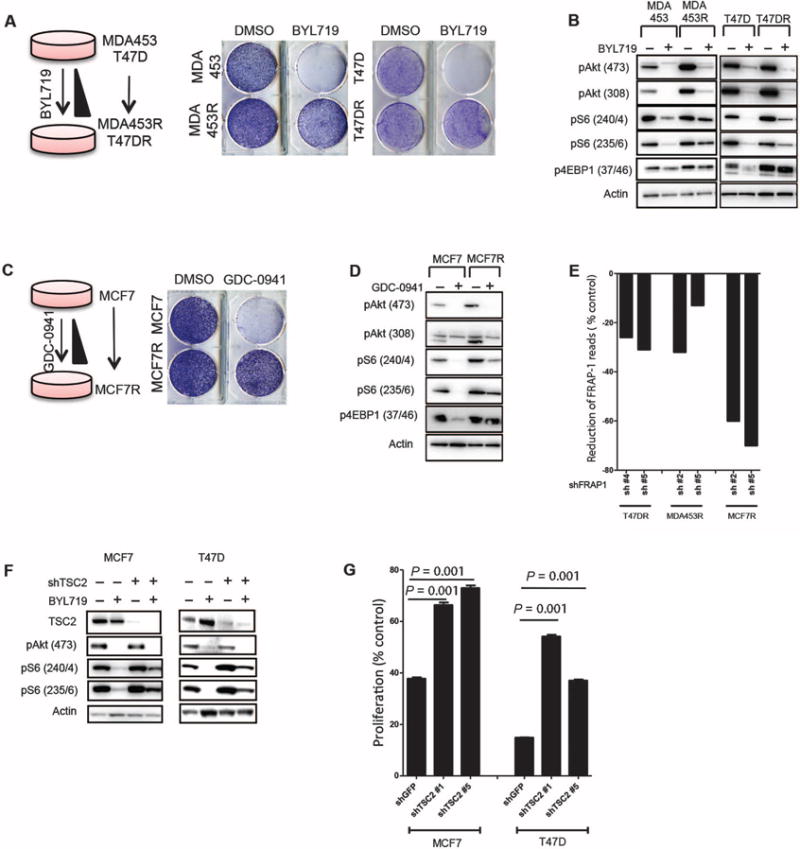
(A) Generation of MDA453 and T47D cell lines with acquired resistance to BYL719. (Right) Proliferation of parental and resistant (MDA453R and T47DR) cells in the presence of 1 μM BYL719. (B) Immunoblotting analysis of phosphorylated proteins in parental, MDA453R, and T47DR cell lines after 24 hours of treatment with 1 μM BYL719. (C) Generation of MCF7 cell line with acquired resistance to GDC-0941. (Right) Proliferation of parental and resistant (MCF7R) cells in the presence of 1 μM GDC-0941. (D) Immunoblotting analysis of phosphorylated proteins in MCF7 and MCF7R after 24 hours of treatment with 1 μM GDC-0941. (E) Reduction in the number of reads by individual FRAP1 shRNAs transfected into drug-resistant cells after 7 days of treatment with either BYL719 or GDC-0941 [normalized to dimethyl sulfoxide (DMSO)–treated controls]. (F) Immunoblotting analysis of phosphorylated proteins in MCF7 and T47D cells after knockdown of TSC2 with shTSC2 and treatment with 1 μM BYL719 for 2 hours. (G) Proliferation assay of MCF7 and T47D cells after TSC2 knockdown with shTSC2 in the presence of 1 μM BYL719 for 4 days. Graph shows one representative experiment of two performed. Data are means ± SEM. P value was calculated using two-sided Student's t test.
We then performed a short hairpin RNA (shRNA) screen to identify genes (fig. S5 and table S1) that could potentially be knocked down to resensitize MDA453R and T47DR cells to BYL719 or MCF7R to GDC-0941 (fig. S5). We found that the proliferation of cells transfected with shRNAs for FRAP1 (mTOR) and phosphoinositide-dependent kinase-1 (PDPK1) was strongly inhibited by BYL719 or GDC-0941 compared to DMSO-treated cells (as indicated by the reduction of shRNA reads, Fig. 3E and fig. S6). Notably, RICTOR or RAPTOR knockdown also resensitized both T47DR and MDA453R cells to BYL719 and MCF7R cells to GDC-0941 (fig. S6). These findings were corroborated by infecting the highly sensitive MCF7 and T47D cells with lentivirus encoding shRNAs targeting tuberous sclerosis protein 2 (TSC2), a direct mTOR suppressor (26, 27). This suppression of TSC2 expression abrogated BYL719-induced down-regulation of pS6 in both MCF7 and T47D cells (Fig. 3F) and was sufficient to significantly (P < 0.01) decrease their sensitivity to p110α inhibition (Fig. 3G).
Together, these results indicate that sustained activation of mTORC1 plays a pivotal role in limiting the antitumor activity of BYL719 and possibly other PI3K/Akt inhibitors.
mTORC1 inhibition correlates with clinical response in treated patients
To assess the clinical relevance of our findings, we studied the correlation between the magnitude of inhibition of S6 phosphorylation and clinical response to BYL719 in patients treated at our institutions as part of an ongoing first-in-human phase 1 clinical trial of BYL719 in patients with solid tumors harboring PIK3CA mutations (21). We used an Institutional Review Board–approved protocol to collect paired tumor biopsies before commencing BYL719 therapy and also on the 28th day of the second cycle of treatment between 4 and 6 hours after the daily drug administration. Twelve paired biopsies (before and on treatment) from six patients with advanced breast cancer were suitable for IHC analysis of pS6(240/4). Table 2 displays the breast tumor subtype, the BYL719 dose, the type of PIK3CA mutation, the amount of pS6 by H-score, the best tumor response to therapy by Response Evaluation Criteria in Solid Tumors (RECIST) criteria (28), and the tumor site from which the biopsies were taken in each patient (Table 2). A strong suppression of S6 phosphorylation was observed in three of the tumors (patients 4 to 6) that responded to therapy (>20% shrinkage of tumor size compared to baseline) (Fig. 4, A and B). In contrast, pS6 expression was only weakly inhibited in three tumors (patients 1 to 3) that did not respond to therapy (Fig. 4, A and B).
Table 2. Clinical information and mutational status in six patients with available paired tumor biopsies before and after therapy with BYL719.
Best tumor response was defined as maximum tumor volume change compared to pretreatment control and was determined per RECIST criteria. Pre, biopsy before the first dose of BYL719; post, biopsy collected at day 56 of treatment. pS6 was measured by H-score.
| Patient no. | Tumor subtype | Age | No. of previous therapies | BYL719 dose (mg) | PIK3CA-mut | pS6 pre | pS6 post | Best response | Lesion biopsied |
|---|---|---|---|---|---|---|---|---|---|
| 1 | ER+/PR+/HER2+ | 42 | 1 | 180 | H1047R | 300 | 285 | −1.50% | Breast |
| 2 | ER+/PR+/HER2+ | 49 | 2 | 400 | H1047R | 285 | 225 | −8.40% | Liver |
| 3 | ER+/PR+/HER2− | 55 | 6 | 450 | H1047R | 150 | 230 | +7.89% | Liver |
| 4 | ER+/PR+/HER2− | 53 | 2 | 400 | G1049Ar | 145 | 70 | −34.80% | Breast |
| 5 | ER+/PR+/HER2+ | 51 | 3 | 400 | H1047R | 300 | 75 | −26.30% | Breast |
| 6 | ER+/PR−/HER2− | 48 | 1 | 400 | H1047R | 285 | 140 | −24.90% | Breast |
Fig. 4. Correlation between pS6(240/4) inhibition and clinical response to BYL719 in breast cancer patients.
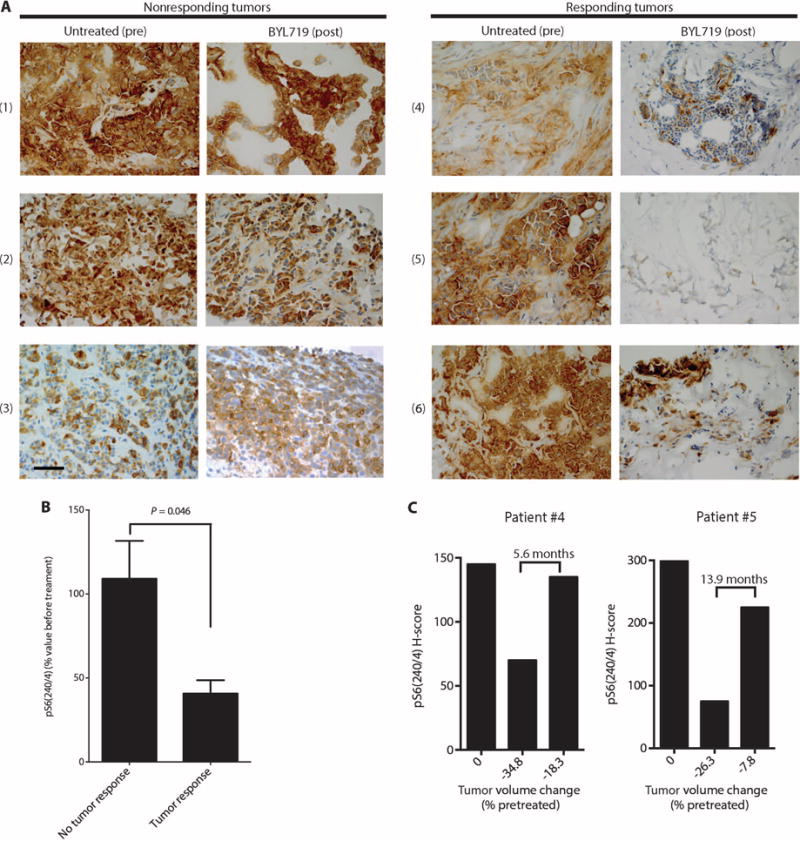
(A) Representative sections stained by IHC for pS6(240/4) in paired biopsies obtained from tumors of patients before (untreated) and at day 28 of BYL719 treatment. Patient numbers are indicated at left in parentheses. Images were captured at ×40 magnification; scale bar, 100 μm. (B) Differences in pS6(240/4) inhibition by BYL719 between the nonresponder (n = 3) and responder (n = 3) patients. pS6(240/4) was quantified in all the tumor cells present in the pretreatment biopsies and upon BYL719 treatment for each patient. (C) Levels of pS6(240/4) in tumor samples from patients 4 and 5 collected before initiation of BYL719 therapy (left bars), at 28 days after therapy initiation (middle bars), and at disease progression (right bars). Below the bars, the change in tumor volume (% of pretreatment size) by RECIST criteria is indicated at each time point.
Furthermore, we analyzed pS6(240/4) in tumor biopsies from two patients (patients 4 and 5) who initially responded to treatment but who later (after 5.6 and 13.9 months, respectively) showed tumor progression. Strikingly, pS6 in these regrowing lesions (also obtained 4 hours after the last drug administration) was present at levels comparable to those in the baseline pretreatment tumor samples (Fig. 4C). For the third responding patient (patient 6, who progressed on BYL719 after 3.6 months of treatment), pS6 could not be measured in the regrowing lesions because we were unable to perform a biopsy 4 hours after the last drug administration.
Together, these results from patients' biopsies are consistent with our observation that inhibition of mTORC1 activity in the tumor is necessary for PIK3CA mutant cancers to respond to BYL719. These results also corroborate our observations that pS6 is reactivated upon the development of acquired resistance.
mTORC1 inhibition enhances the efficacy of p110α inhibition and delays resistance
The effects of combined mTORC1 and p110α blockade were further examined in cells and tumors intrinsically resistant to BYL719. In proliferation/viability studies, the combination of BYL719 and RAD001 was more effective than either single agent in both JIMT-1 and HCC1954 cells (Fig. 5A and fig. S7). Similarly, cell cycle analysis showed that, unlike single-agent therapy, combined therapy induces both S-phase arrest and apoptosis (Fig. 5B and fig. S8A). As expected, the combination of BYL719 and RAD001 effectively inhibited both Akt and S6 phosphorylation (fig. S8B). Using 3D cell culture assays, we examined the effect of BYL719 and RAD001, either alone or in combination, in JIMT-1 and HCC1954 cell lines grown as spheroids. Whereas change in size and induction of cell death were minimal when BYL719 and RAD001 were used as single agents, concomitant p110α and mTORC1 suppression reduced spheroid size and induced apoptosis (the effect was more evident in HCC1954 cells) (Fig. 5C and fig. S9). We also tested the effect of targeting mTORC1 in our laboratory models of acquired resistance to PI3K inhibitors. RAD001 abolished pS6 expression in the MCF7 cell line with acquired resistance to BYL719 (MCF7R) and resensitized the cells to PI3K inhibition (fig. S10). We then tested the antitumor activity of this combination in BYL719-resistant PIK3CA-mut xenografts. We treated CAL51-derived xenografts with two different doses of BYL719, a fixed dose of RAD001, or the respective combinations. Although single agents had little or no effect in preventing tumor growth, the combinations of BYL719 and RAD001 had a pronounced antitumor effect (Fig. 5D). In a similar fashion, HCC1954-derived tumors were treated with a fixed dose of BYL719 (25 mg/kg), two different doses of RAD001, or the combinations. Consistently, single agents had minimal antitumor activity, whereas both combination arms showed markedly impaired tumor growth (Fig. 5E). The superiority of the BYL719/RAD001 combination in comparison to monotherapy was further confirmed in JIMT-1 xenografts (Fig. 5F).
Fig. 5. Antitumor effects of combined mTORC1 (RAD001) and p110α (BYL719) inhibition.
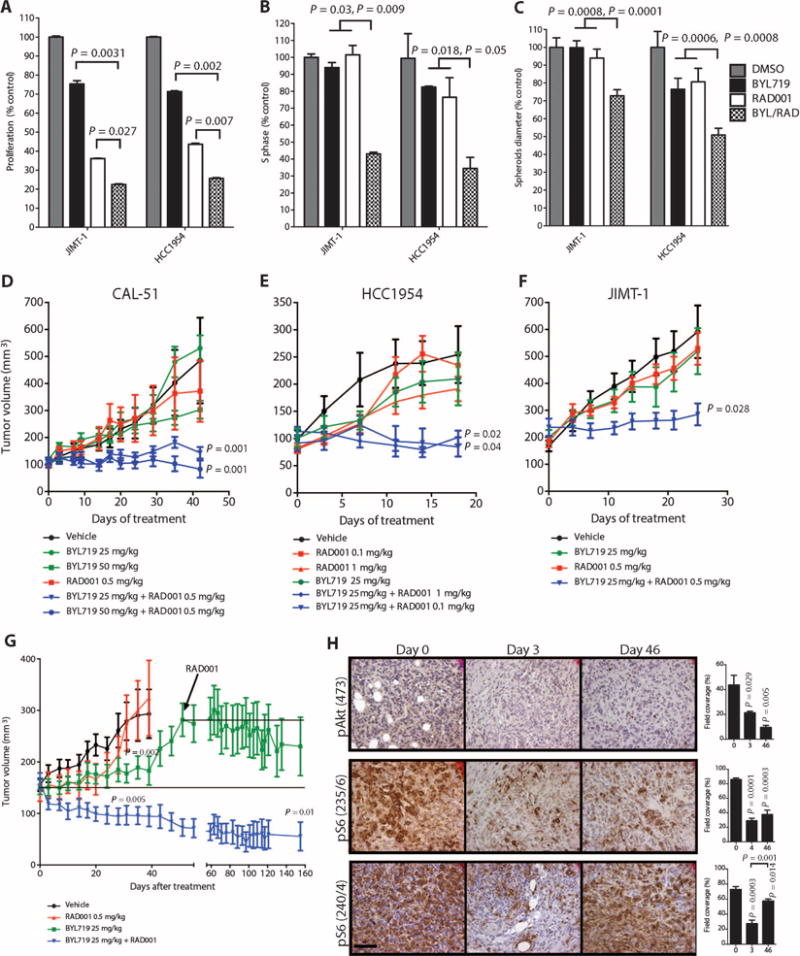
(A) Proliferation of the BYL719-resistant JIMT-1 and HCC1954 cell lines in the presence of BYL719 (0.5 μM), RAD001 (1 nM), or the combination for 72 hours. (B) Cell cycle analysis of JIMT-1 and HCC1954 cells with BYL719 (0.5 μM), RAD001 (1 nM), or the combination for 48 hours. (C) Spheroid size of JIMT-1 and HCC1954 cells treated with BYL719 (1 μM), RAD001 (10 nM), or the combination for 48 hours. In vitro experiments were in triplicates and repeated three times. (D) Tumor growth curves of BYL719-resistant CAL51-derived xenografts treated with RAD001 (0.5 mg/kg), BYL719 (50 or 25 mg/kg), or the described combinations (n = 10 for each treatment condition). (E) Tumor growth curves of BYL719-resistant HCC1954 cell–based xenografts treated with BYL719 (25 mg/kg), RAD001 (0.1 or 1 mg/kg), or the described combinations (n = 10 for each treatment condition). (F) Tumor growth curves of BYL719-resistant JIMT-1 cell–based xenografts treated with RAD001 (0.5 mg/kg), BYL719 (25 mg/kg), or the described combination (n = 10 for each treatment condition). (G) Tumor growth curve of BYL719-sensitive MCF7-derived xenografts treated as indicated with BYL719 (25 mg/kg), RAD001 (0.5 mg/kg), or the combination of both. RAD001 was added to the BYL719-treated cohort at day 46 (n = 10 for each treatment condition). (H) An average of 6 to 15 images of two to four independent tumors was used to quantify IHC for pAkt and pS6 on days 0, 3, and 46 of BYL719 treatment. Quantification of IHC was performed with CellProfiler and is shown in bar graphs to the right of each panel. Images were captured at ×40 magnification; scale bar, 100 μm. Data are means ± SEM. P value was calculated using two-sided Student's t test.
Next, we determined whether combined p110α and mTORC1 blockade would also be effective in tumors treated with BYL719 that initially responded to therapy but eventually progressed. MCF7-derived xenografts, sensitive to BYL719 (Fig. 2A), were treated with either BYL719 (25 mg/kg) or RAD001 (0.5 mg/kg). Both single-agent treatments delayed tumor growth compared to tumors in control vehicle– treated mice. The effect of RAD001 used as a single agent ceased after ∼20 days of treatment, whereas BYL719 suppressed tumor growth for ∼40 days. However, after 46 days, tumors started to grow in these animals despite BYL719 treatment, at a rate similar to that in vehicle-treated xenografts. At that time, RAD001 was added to the BYL719 given to this group of animals, and the treatment rapidly halted the tumor progression (Fig. 5G). This effect lasted until the end of the experiment (an additional 100 days). The cohort of mice treated with the combination of the two drugs from day 0 showed immediate and consistent reduction of tumor volume, and only 4 of 10 tumors were measurable after 120 days (Fig. 5G).
To further understand the link between mTORC1 activation and tumor escape from p110α inhibition, we measured S6 phosphorylation in MCF7 xenografts treated with BYL719 over time (Fig. 5H). At day 3 after beginning treatment, both pAkt and pS6 were suppressed. However, after 46 days of treatment, pS6(240/244) was again high despite continued inhibition of pAkt. Thus, these findings are consistent with data from our cell line models of acquired resistance and the patient biopsies of acquired resistance that showed restoration of pS6 in the resistant cancers. These results further suggest that the combination of BYL719 and RAD001 can overcome both intrinsic and acquired resistance to BYL719.
Insulin-like growth factor 1 and neuregulin 1 activate mTOR and limit the antitumor activity of BYL719
Recent reports have indicated that increased ligand secretion and ligandmediated activation of receptor tyrosine kinases are compensatory responses to therapy with kinase inhibitors that may in turn cause resistance to the kinase inhibitors (29–31). To identify possible ligands that mediate resistance to BYL719, we used a high-throughput platform (31) to screen for secreted proteins that, when added to BYL719-sensitive cell lines, would oppose the antiproliferative effects of the drug. The supernatants of human embryonic kidney (HEK) 293T cells that overexpressed a library of 2803 genes encoding secreted proteins were transferred to either MCF7 or T47D cells treated with 1 μM BYL719. Supernatants that induced resistance to BYL719 were identified by quantification of cell growth after an incubation period of 4 days (fig. S11). Although, as expected, most of the supernatants did not prevent BYL719-dependent inhibition of cell proliferation, insulin-like growth factor 1 (IGF1) and neuregulin 1 (NRG1) were able to reverse BYL719 inhibition of cell growth. We therefore identified these growth factors as candidates for mediating resistance to p110α inhibition in these cell lines (Fig. 6A).
Fig. 6. Effects of IGF1 and NRG1 in limiting tumor cell sensitivity to BYL719.
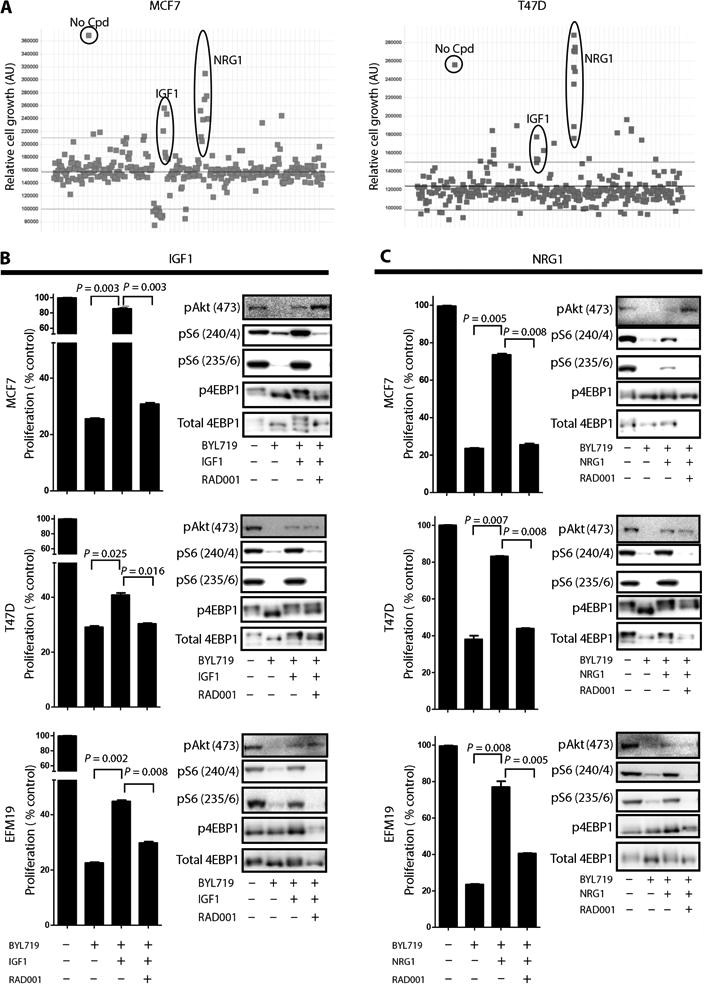
(A) A library of secreted proteins was screened for their ability to interfere with BYL719 inhibition of cell growth. MCF7 and T47D cells were treated for 4 days with 1 μM BYL719. Each dot represents the average of three replicates of cells conditioned with the same supernatant. No Cpd represents untreated cells (no compound and no supernatant). Y axes are arbitrary units (AU). (B) (Left) Proliferation of MCF7, T47D, and EFM19 cells treated for 6 days as indicated with or without recombinant IGF1 (50 ng/ml). (Right) Protein lysates from MCF7, T47D, and EFM19 cells treated as indicated for 4 hours were analyzed by immunoblotting against the indicated proteins. (C) (Left) Proliferation of MCF7, T47D, and EFM19 cells treated for 6 days as indicated with or without recombinant NRG1 (0.8 ng/ml). (Right) Protein lysates from MCF7, T47D, and EFM19 cells treated as indicated for 4 hours were analyzed by immunoblotting against the indicated proteins. Graphs show one representative experiment of two performed. Data are means ± SEM. P value was calculated using two-sided Student's t test.
To validate these findings, we examined the proliferation/viability of five BYL719-sensitive PIK3CA-mut cell lines treated with BYL719 in the presence of human recombinant IGF1 (Fig. 6B and fig. S12) or NRG1 (Fig. 6C and fig. S12). Exogenous supplementation with either IGF1 or NRG1 was sufficient to increase pS6 levels in the presence of BYL719 and limit the sensitivity to p110α inhibition in all the cell lines tested. Addition of RAD001 prevented IGF1/NRG1-dependent S6 phosphorylation and restored the sensitivity to BYL719. Hence, resistance to p110α may be caused by an abundance of certain ligands that have the capacity to activate mTORC1 in a p110α-independent fashion.
Discussion
We have shown that inhibition of mTORC1 is needed for optimal antitumor activity by BYL719, a specific PI3K p110α inhibitor currently under clinical development for tumors with PIK3CA mutations (21, 22). We found that high levels of residual pS6 in the tumor after BYL719 treatment positively correlated with either intrinsic or acquired resistance to this agent, in vitro, in vivo, and in patient biopsies. A causative role of sustained mTORC1 activation in limiting the effects of p110α inhibition by BYL719 was supported by the observation that concomitant inhibition of mTORC1 by RAD001 reversed resistance and by the finding that silencing of FRAP1 (mTOR), RICTOR, and RAPTOR with shRNA was effective in restoring sensitivity to BYL719. These results, however, do not rule out that mTORC2 may also contribute to the resistant phenotype. Strikingly, similar observations were independently made by Corcoran et al. in a paper published in this issue of Science Translational Medicine (32), which shows that mTORC1 suppression can predict responsiveness to RAF or MEK (mitogen-activated or extracellular signal–regulated protein kinase kinase) inhibitors in BRAF-mutant melanoma. This suggests that sustained activation of mTOR can function as a bottleneck for the efficacy of agents targeting both the PI3K/Akt and ERK pathways.
We were interested to find that exogenous supplementation of either IGF1 or NRG1 was able to reduce the activity of BYL719 by preventing suppression of mTORC1 in cells intrinsically sensitive to the drug. One possible explanation for this finding is that ligand-mediated maintenance of S6 phosphorylation in the presence of BYL719 may be a result of residual Akt phosphorylation through other p110 isoforms. On the basis of our findings, it is plausible that the persistent activation of mTOR despite BYL719 treatment in resistant cells could be the result of increased secretion of ligands such as NRG1 and IGF1.
Three recent reports converge in indicating that Myc amplification or overexpression may be a mechanism of resistance to PI3K inhibitors (33–35). However, among our panel of 25 cell lines, only HCC1954 and MFM223 cells (both BYL719-resistant) have a high copy number (more than seven) of Myc genes (36). Nevertheless, we cannot discount the possibility that MYC overexpression or deregulation, proposed to increase mTORC1 signaling (37), plays a role in sustaining mTORC1 activity independently of the gene copy number in these cells.
Our results suggest that combined inhibition of p110α and mTORC1 may be required for treatment of some PI3K mutant cancers. If this proves to be the case, what will be the preferred approach for simultaneously inhibiting both targets? Current choices include treatment with a dual PI3K and mTOR inhibitor such as BEZ235 (38) and GDC-0980 (39) or the use of a specific p110α inhibitor such as BYL719 plus a rapalog as we have done in the present study with RAD001. Although the first option would be convenient, it may also result in higher toxicities as a result of its effect on kinases other than PI3K p110α and mTORC1; this situation could necessitate the use of drug doses that do not effectively inhibit the two kinases. BYL719 appears to have a better therapeutic window (permitting more effective inhibition of p110α) than many of the dual PI3K/mTOR inhibitors or the pan-PI3K inhibitors currently under clinical development (16, 18, 21, 22, 40–42). In addition, our data show that only low doses of the rapalog RAD001 were required for efficacy, raising optimism that therapeutically effective doses will be tolerated. One limitation of this study is the impossibility to know a priori whether a patient would benefit from addition of an mTORC1 inhibitor to BYL719. This problem may be obviated by measuring the level of pS6 in tumor biopsies or fine-needle aspirations shortly after the initiation of therapy, which could then inform the decision to add an mTOR inhibitor to the therapy. However, the capability of detecting phospho-proteins by IHC may vary depending on the antibody used and the method of preservation of the sample. Moreover, the results obtained from a single biopsy may not accurately reflect the overall expression of the examined biomarker. Therefore, this method would need to be standardized to be used for patient stratification.
PI3K/mTOR inhibitors have been previously proposed to act synergistically with endocrine therapy in ER-expressing breast cancer patients (43, 44). Given that the vast majority of PIK3CA mutant tumors coexpress ER, our proposed therapeutic strategy may also benefit from concomitant antiestrogen agents.
In summary, our study provides evidence that mTORC1 mediates resistance to p110α and that combined inhibition of PI3K p110α and mTORC1 warrants further clinical investigation in patients bearing tumors with PIK3CA mutations.
Materials and Methods
Study design
The objective of our study was to identify molecular determinants of sensitivity to PI3K p110α inhibition in PIK3CA-mut breast tumors. We planned to apply BYL719 (p110α inhibitor) to breast cancer cell lines and tumors whose proliferation and growth were differently affected by BYL719, to identify the differences between responders and nonresponders, and then validate these findings in clinical samples. The number of animals in each group was calculated to measure a 25% difference between the means of placebo and treatment groups with a power of 80% and a P value of 0.01. Host mice carrying xenografts were randomly and equally assigned to either control or treatment groups. Animal experiments were conducted in a controlled and nonblinded manner. Quantification of pS6(240/4) in patient samples was performed in a blinded manner. In vitro experiments were performed at least two times and at least in triplicate for each replica. The original data for all experiments are available in table S3 (Excel file).
Cell lines and chemical compounds
OCUB-F cell line was obtained from Riken Gene Bank. SUM185PE and SUM190PT were provided by S. P. Ethier's laboratory. All other cell lines were purchased from commercial vendors. 293T cells were maintained in Dulbecco's modified Eagle's medium (DMEM) with 10% fetal calf serum (FCS). MDA-MB-361, MDA-MB-453, UACC893, KPL-1, MCF7, BT474, JIMT-1, BT20, CAL148, MFM223, UACC812, ZR-75-1, CAL51, MDA-MB-468, and MDA-MB-231 tumor cells were maintained in DMEM/F12 (1:1) with 10% FCS. T47D, CAMA-1, BT483, HCC1428, EFM19, HCC202, and HCC1954 tumor cells were grown in RPMI with 10% FCS. SUM185PE and SUM190PT were cultured in F12 with 5% FCS and Medium 199 without FCS, respectively. SUM185PE and SUM190PT were also supplemented with h-Insulin (5 μg/ml) (Sigma, I9278) and hydrocortisone (1 μg/ml) (Sigma, H-4001).
All cultured media contained 2 mM l-glutamine, penicillin (20 U/ml), and streptomycin (20 μg/ml). All cells were maintained at 37°C in a humidified atmosphere at 5% CO2.
MCF7R cells were obtained after chronic exposure to increasing concentrations of GDC-0941 for ∼6 months. MDA453R and T47DR were obtained after chronic exposure to increasing concentrations of BYL719 for ∼6 months. The p110α inhibitor BYL719 was provided by Novartis. The mTORC1 inhibitor RAD001 and the pan-p110 inhibitor GDC-0941 were provided by SU2C/PI3K Dream Team Mouse Pharmacy that obtains compounds from Shanghai Haoyuan Chemexpress Co. All compounds were dissolved in DMSO for in vitro experiments.
Determination of IC50
To calculate IC50, 5 × 105 cells were seeded in 96-well plates and treated with escalating concentration of BYL719 (from 20 nM to 12.5 μM). After 3 days of treatment, cell proliferation was analyzed with CellTiter-Glo Luminescent Cell Viability Assay (Promega) as described by the manufacturer. IC50 values were calculated with GraphPad Prism (GraphPad Software).
Determination of cell cycle, apoptosis, and proliferation
Cell cycle was quantified by flow cytometry as described (45). Briefly, cells were washed with phosphate-buffered saline (PBS), fixed in cold 70% ethanol, and then stained with propidium iodide (PI) while being treated with ribonuclease (Sigma-Aldrich).
Cell death was measured by annexin V–FITC (fluorescein isothiocyanate)/PI staining and evaluated by flow cytometry according to the manufacturer's protocol (BD Pharmingen). Cells (2 × 105) were washed twice with PBS and stained with 5 μl of annexin V–FITC and 10 μl of PI (5 μg/ml) in 1× binding buffer (BD) for 15 min at room temperature in the dark. The apoptotic cells were detected with an LSR flow cytometer. Both apoptotic (annexin V–positive, PI-negative) and necrotic (annexin V–positive and PI-positive) cells were included in cell death determinations. Quantitative analyses were carried out by FlowJo software.
For proliferation analysis, 5 × 105 cells were seeded in 24-well plates and treated with the appropriate drug(s) for 3 to 11 days. Cells were fixed and stained with crystal violet or Cyro60.
For IGF1/NRG1-induced proliferation experiments, 5 × 105 cells were seeded in 24-well plates, treated with BYL719 (1 μM) and/or RAD001 (1 nM) in the presence of IGF1 (50 ng/ml) or NRG1 (0.8 ng/ml) for 6 days, and then stained with crystal violet.
Western blotting
Cells were washed with ice-cold PBS and scraped into ice-cold radio-immunoprecipitation assay lysis buffer (Cell Signaling Technology) supplemented with phosphatase inhibitor cocktails (Complete Mini and PhosphoStop, Roche). Lysates were cleared by centrifugation at 13,000 rpm for 10 min at 4°C, and supernatants were removed and assayed for protein concentration with the Pierce BCA Protein Assay Kit (Thermo Scientific). Thirty-five micrograms of total lysate was resolved on NuPAGE 4 to 12% bis-tris gels (Life Technologies) and electrophoretically transferred to Immobilon transfer membranes (Millipore). Membranes were blocked for 1 hour in 5% nonfat dry milk in tris-buffered saline (TBS)–Tween and then hybridized with the primary antibodies in 5% bovine serum albumin (BSA)/TBS-Tween. List of primary antibodies and their working concentrations can be found in table S2. β-Actin was used as a loading control (1:5000, Sigma), also in 5% BSA/TBS-Tween. Mouse and rabbit horseradish peroxidase–conjugated secondary antibodies (1:50,000, Amersham Biosciences) were diluted in 2% nonfat dry milk in TBS-Tween. Protein-antibody complexes were detected by chemiluminescence with SuperSignal West Femto Chemiluminescent Substrate (Thermo Scientific), and images were captured with a G-BOX camera system.
Establishment of tumor xenografts and studies in nude mice
Six-week-old female athymic NU/NU nude mice purchased from Charles River were injected with 2 × 107 MCF7, 5 × 106 JIMT-1, and 1 × 107 CAL51 cells subcutaneously in a volume of 100 μl of culture medium/Matrigel (1:1) (BD Biosciences). For MCF7 xenografts, 1 μM 17β-estradiol was added to the drinking water as described (45).
For cell line–derived xenograft studies, animals were randomized at a tumor volume of 70 to 120 mm3, about 2 to 4 weeks after injection. Animals were randomized to four to six groups, with n = 8 to 10 tumors per group. Animals were treated daily with BYL719 [25 or 50 mg/kg in 0.5% carboxymethylcellulose sodium salt (CMC) (Sigma) in water] and/or RAD001 (0.5 or 0.1 mg/kg, freshly dissolved in water, and mixed in CMC or BYL719 in CMC solution). All agents were delivered by oral gavage. Tumor xenografts were measured with digital calipers, and tumor volumes were determined with the following formula: (length × width2) × (π/6). At the end of the experiment, animals were euthanized with CO2 inhalation. Tumor volumes are plotted as means ± SEM.
Mice were maintained and treated in accordance with Institutional Guidelines of Massachusetts General Hospital. Mice were housed in air-filtered laminar flow cabinets with a 12-hour light cycle and food and water ad libitum.
Patients
Tumor response was assessed according to the RECIST criteria. All potential sites of tumor lesions were assessed at screening/baseline with thoracic, abdominal, and pelvic computerized tomography or magnetic resonance imaging, complemented with brain scan in case of clinical evidence of brain metastatic disease. In addition, a bone scan was performed for all patients with clinical evidence of bone metastases. Subsequent scans were done at the end of cycle 2, and every 8 weeks thereafter (that is, at the end of cycles 4, 6, 8, etc.). A complete treatment cycle is defined as 4 weeks of daily continuous treatment with oral BYL719. All measurable lesions up to a maximum of 5 lesions per organ and 10 lesions in total, representative of all involved organs, were identified as target lesions and recorded and measured at baseline, and then followed throughout the study. A sum of the longest diameter for all target lesions at baseline was used as a reference by which the objective tumor response was characterized. Pretreatment biopsies were obtained within 2 weeks of starting the study agent. On-treatment tumor biopsies were collected at the end of cycle 2 (cycle 2 day 28), and progression tumor biopsy was performed at the time of disease progression. Both on-treatment and progression biopsies were collected 4 to 6 hours after dose.
Immunohistochemistry
Tumor xenografts
Tissue was fixed immediately after removal in a 10% buffered formalin solution for a maximum of 24 hours at room temperature before being dehydrated and paraffin-embedded under vacuum conditions for IHC or IF as described (46). List of primary antibodies and their working concentrations can be found in table S2. For pS6(235/6) staining, samples were blocked with normal goat serum (NGS) (vector S-1000), incubated in secondary biotinylated goat anti-rabbit (vector BA-1000), and stained with ABC (PK-6100) and DAB brown stain (vector SK-4100). For all other IHC staining, samples were blocked with NGS, incubated in secondary antibody (Cell Signaling Technology, #8114), and stained with DAB brown stain (Dako, #K401111). Analysis pipelines were designed for chromogen-based immunohistological images as previously described (47). For each IHC quantification, four random areas from four individual stained tumors were captured (magnification, ×40). The areas of the stained regions were quantified as percentages of the total tissue as identified by the CellProfiler software.
Patient samples
Biopsies were immediately snap-frozen and embedded in optimal cutting temperature (OCT). Samples were fixed in 10% neutral-buffered formalin, blocked with DEEB (Dako) blocking agent, and incubated with the primary antibody (1:200 dilution) overnight at 4°C. Incubation with secondary antibody and DAB staining was performed as described above. As positive and negative controls, we used snap-frozen tumor xenografts (untreated and treated with BYL719) with known content of pS6(240/4), embedded in OCT. Staining for pS6(240/4) was measured with a four-value intensity score (0, 1+, 2+, and 3+, H-score) and the percentage of the reactivity extent. Final scores were obtained by multiplying both intensity and extension values (range, 0 to 300). Only tumor cells were quantified.
Secreted protein screen
Secreted protein library complementary DNAs were reverse-transfected into HEK293T cells with FuGENE HD (4:1, transfection reagent to DNA ratio) and incubated for 4 days to allow accumulation of secreted proteins in supernatant. The supernatant was then transferred to MCF7 and T47D cells, followed by addition of BYL719 to a final concentration of 1 μM. After 96 hours, growth was measured with CellTiter-Glo.
Statistical analysis
Two-way t test was done with GraphPad Prism (GraphPad Software). Error bars represent the SEM. All the in vitro experiments were repeated at least three times. All the in vivo experiments were run in duplicate with at least n = 6 for each treatment arm.
Quantitative in-cell western
Cells were plated in 96-well black flat-bottom plates (BD Falcon) at a density of 3 × 105 cells per well. After treatment, the cells were immediately fixed with 3.7% paraformaldehyde in PBS for 20 min and then washed three times for 10 min each with 0.1% Triton X-100 in PBS with gentle shaking. Cells were blocked with blocking buffer (LI-COR) for 1 hour with gentle shaking. Cells were then hybridized with the following primary antibodies diluted in blocking buffer, with gentle shaking at 4°C overnight: pS6 (Ser240/4) (1:2000) and pS6 (Ser235/6) (1:100), all from Cell Signaling Technology. After incubation with primary antibody, cells were washed three times for 10 min each with PBS-Tween and incubated in goat anti-rabbit secondary IRDye (LI-COR, 1:1200), Sapphire 700 (LI-COR, 1:1000), and DRAQ5 (BioStatus, 1:2000) in blocking buffer for 1 hour with gentle shaking. Cells were washed again three times for 10 min each with PBS-Tween, and plates were dried and read with a LI-COR Odyssey Infrared Imager.
shRNA screen
Novartis Pharma generated the shRNA library. MCF7R, T47DR, and MDA453R cells were infected with shRNAs from the library, incubated for several days, selected with puromycin selection, and divided into two experiment arms (treatment and DMSO control, fig. S5). Because the total number of cells sometimes differed between the two arms, the number of shRNA reads for each arm was normalized to 5 × 106 and log-transformed. Quantile normalization was applied across all the screens to allow for comparison of results between cell lines. A fold change was determined for each shRNA in each screen by subtracting the log2 count in the control arm from the treatment arm.
A confidence value was calculated for each shRNA on the basis of percent knockdown of the gene target relative to other genes. For each gene K, we let {k1, k2, …, km} be the set of percent knockdowns for the shRNAs targeting K and let kmax denote the maximum percent knockdown in this set. The weights for the shRNAs against gene K were then given by . Thus, the weight of an shRNA corresponded to its percent knockdown of the target gene, normalized to the maximum percent knockdown for that target gene by any of its shRNAs.
For each gene, a score was calculated on the basis of the following formula:
where A = {a1, a2, …, am} are the indices of shRNAs targeting the gene and B = {b1, b2,…,bn} are the indices of all the shRNAs targeting other genes in the pool. FCx and Wx denote the fold change (compound versus DMSO) and confidence weight of the xth shRNA, as described above.
Thus, the score for a gene is the difference in the (weighted) log fold change of shRNAs targeting that gene versus all other genes. It therefore measures the degree to which targeting a specific gene differentially sensitizes or rescues the cell line from the effects of treatment compared to the other genes.
A permutation test was used to estimate the significance of the gene-level scores. The mapping of shRNAs to their gene targets was randomly permuted 10,000 times, and in each permutation, a score was calculated for each gene as described above. For any particular gene, the distribution of its 10,000 permuted scores approximated a normal distribution. Consequently, a P value could be determined from a two-sided comparison of the actual gene score against the fitted normal distribution derived from the permuted gene scores. The P values of all genes were also corrected for multiple hypotheses testing with the false discovery rate (FDR) method of Benjamini-Hochberg. Genes with an FDR <0.25 were considered to be significantly sensitizing or rescuing the cell line from the effects of drug treatment when compared to the average gene screened in the shRNA pool.
shRNA knockdown
shControl and shTSC [previously described in (48)] were purchased from Open Biosystems. 293T cells were seeded into 10-cm plates at a density of 0.5 × 105 cells per plate. Twenty-four hours later, cells were transfected with shRNA against TSC2 or Silencer negative control shRNA against green fluorescent protein with FuGENE (Roche) according to the manufacturer's instructions. Lentiviral shRNA was collected after 48 hours. Tumor cells were infected with lentiviral shRNA for 24 hours, and then cell lines were grown in medium supplemented with 10% fetal bovine serum in the presence of puromycin (2 μg/ml) for 5 to 8 days.
3D acinar morphogenesis assay and scoring of 3D structures
Cells were grown in DMEM/F12 or RPMI medium supplemented with 2% inactivated calf serum, which was replaced every 4 days for 2 to 3 weeks before treatment. Spheres were treated with DMSO, 1 μM BYL719, 10 nM RAD001, or combination for 48 hours and fixed. Phase-contrast microscopy was used to image 3D structures (10×) as described (49). The diameter of >50 spheroids in each condition was analyzed and measured by ImageJ.
IF and confocal microscopy
3D structures were fixed and stained as described (49). Secondary antibodies were as follows: Alexa Fluor 488 anti-rabbit immunoglobulin G (IgG) (1:200, Invitrogen) and Alexa Fluor 594 anti-mouse IgG (1:200, Invitrogen). For confocal imaging of 3D structures, we used Multispectral Multimode Laser Scanning Confocal Microscope: Nikon TE-200U inverted microscope with Nikon C1 point scanning confocal with spectral detection, wide range of Nikon 40×, 1.4 numerical aperture (NA) magnification/NA differential interference contrast (DIC) and phase optics, Nikon halogen trans-illuminator with 0.52 NA long working distance (LWD) and 0.85 NA Dry condenser, 405-, 488-, and 561-nm laser lines, Nikon EZ-C1 software, TMC vibration-isolation table. 3D images are shown as single sections from midstructure.
Cooperative, enhanced, enzyme immunoreactive assay
The extent of protein expression and activation in xenografts was determined by CEER (50). Briefly, targeted proteins were captured by antibodies printed on a nitrocellulose surface and successively detected by two different fluorescent-conjugated antibodies against different protein epitopes.
Supplementary Material
Acknowledgments
We thank M.-A. Bray for helping generating CellProfiler pipelines for IHC analysis, B. Jillian for the IHC staining, C. Benes for scientific discussion and providing cell lines, and E. Edelman and O. Litvin for statistical support.
Funding: This work was funded by a Stand Up To Cancer Dream Team Translational Cancer Research Grant, a program of the Entertainment Industry Foundation (SU2C-AACR-DT0209 to J.B.), the Breast Cancer Research Foundation (to J.B.), the European Research Council (AdG09250244 to J.B.), the Instituto de Salud Carlos III (Intrasalud PSO9/00623 to J.B.), and RO1CA137008 (to J.A.E.). M.E. is an International Sephardic Education Foundation postdoctoral fellow. A.B.C. holds a Translational Research Fellowship from the Spanish Society of Medical Oncology (SEOM). S.V. is supported by an ASCO YIA, American Association for Cancer Research–Genentech BioOncology Fellowship, and Terri Brodeur Breast Cancer Foundation research grant.
Footnotes
www.sciencetranslationalmedicine.org/cgi/content/full/5/196/196ra99/DC1
Fig. S1. Cell viability after BYL719 treatment.
Fig. S2. Inhibition of pS6(235/6) in sensitive and resistant cells.
Fig. S3. Immunostaining of p4EBP1 in JIMT-1 and MCF7 xenografts treated with BYL719.
Fig. S4. PI3K/Akt/mTOR pathway inhibition in drug-resistant cells treated with BYL719.
Fig. S5. Scheme of the shRNA screen to identify candidate genes for resistance to PI3K inhibition.
Fig. S6. Results of the shRNA resensitization screen.
Fig. S7. Proliferation of drug-resistant cells treated with BYL719, RAD001, and the combination.
Fig. S8. Apoptosis and PI3K/Akt/mTOR pathway inhibition in cells treated with BYL719, RAD001, or the combination.
Fig. S9. 3D culture of drug-resistant cells treated with BYL719, RAD001, or the combination.
Fig. S10. Biochemical and biological effects of BYL719, RAD001, or the combination on MCF7 parental and drug-resistant cells.
Fig. S11. Scheme of the secreted protein screen.
Fig. S12. Effects of IGF1 and NRG1 on BYL719-resistant cells.
Table S1. List of 132 genes tested in the shRNA screen.
Table S2. Antibody information.
Table S3. Original data (provided as a separate Excel file).
Author contributions: M.E., S.V., M.S., and J.B. designed the research. M.E., S.V., N.M., T.M., J.T., A.B.C., Y.H.I., and V.R.-O. performed the experiments. D.J., C.Q., V.S., J.R., and M.M.-K. collected and analyzed the human specimens. A.H. performed the secreted protein screening. M.L. performed the shRNA screen analysis. S.H., S.S., and P.K. performed the CEER array and its analysis. J.B., N.R., and J.A.E. assisted with research design. M.E. and M.S. prepared the figures. M.E., M.S., and J.B. wrote the manuscript.
Competing interests: J.B., J.A.E., and N.R. have consulted for Novartis Pharmaceuticals. J.A.E. has also consulted for Sanofi-Aventis, GlaxoSmithKline, Genentech, Abbott, Intellikine, Cell Signaling Technology, Pathway Therapeutics, and Chugai. J.A.E. receives research support from Novartis Pharmaceuticals. A.H. and M.L. are full-time employees of Novartis Pharmaceuticals. C.Q. holds equity in Novartis Pharmaceuticals. J.R. served on an advisory board for Novartis Pharmaceuticals. N.R. serves on advisory boards for AstraZeneca and Millennium. Novartis has filed a patent application for the combination of BYL719 and RAD001.
Data and materials availability: Requests for materials will be accommodated with material transfer agreements (MTAs). The shRNA and secretome screen technologies are owned or controlled by Novartis Pharmaceuticals. BYL719 and RAD001 were obtained with an MTA from Novartis Pharmaceuticals.
Contributor Information
Maurizio Scaltriti, Email: scaltrim@mskcc.org.
José Baselga, Email: baselgaj@mskcc.org.
References and Notes
- 1.Engelman JA, Luo J, Cantley LC. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat Rev Genet. 2006;7:606–619. doi: 10.1038/nrg1879. [DOI] [PubMed] [Google Scholar]
- 2.Cantley LC. The phosphoinositide 3-kinase pathway. Science. 2002;296:1655–1657. doi: 10.1126/science.296.5573.1655. [DOI] [PubMed] [Google Scholar]
- 3.Engelman JA. Targeting PI3K signalling in cancer: Opportunities, challenges and limitations. Nat Rev Cancer. 2009;9:550–562. doi: 10.1038/nrc2664. [DOI] [PubMed] [Google Scholar]
- 4.Hennessy BT, Smith DL, Ram PT, Lu Y, Mills GB. Exploiting the PI3K/AKT pathway for cancer drug discovery. Nat Rev Drug Discov. 2005;4:988–1004. doi: 10.1038/nrd1902. [DOI] [PubMed] [Google Scholar]
- 5.Brachmann SM, Ueki K, Engelman JA, Kahn RC, Cantley LC. Phosphoinositide 3-kinase catalytic subunit deletion and regulatory subunit deletion have opposite effects on insulin sensitivity in mice. Mol Cell Biol. 2005;25:1596–1607. doi: 10.1128/MCB.25.5.1596-1607.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhao L, Vogt PK. Helical domain and kinase domain mutations in p110α of phosphatidylinositol 3-kinase induce gain of function by different mechanisms. Proc Natl Acad Sci USA. 2008;105:2652–2657. doi: 10.1073/pnas.0712169105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kang S, Bader AG, Vogt PK. Phosphatidylinositol 3-kinase mutations identified in human cancer are oncogenic. Proc Natl Acad Sci USA. 2005;102:802–807. doi: 10.1073/pnas.0408864102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Samuels Y, Wang Z, Bardelli A, Silliman N, Ptak J, Szabo S, Yan H, Gazdar A, Powell SM, Riggins GJ, Willson JK, Markowitz S, Kinzler KW, Vogelstein B, Velculescu VE. High frequency of mutations of the PIK3CA gene in human cancers. Science. 2004;304:554. doi: 10.1126/science.1096502. [DOI] [PubMed] [Google Scholar]
- 9.Miller TW, Rexer BN, Garrett JT, Arteaga CL. Mutations in the phosphatidylinositol 3-kinase pathway: Role in tumor progression and therapeutic implications in breast cancer. Breast Cancer Res. 2011;13:224. doi: 10.1186/bcr3039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Isakoff SJ, Engelman JA, Irie HY, Luo J, Brachmann SM, Pearline RV, Cantley LC, Brugge JS. Breast cancer–associated PIK3CA mutations are oncogenic in mammary epithelial cells. Cancer Res. 2005;65:10992–11000. doi: 10.1158/0008-5472.CAN-05-2612. [DOI] [PubMed] [Google Scholar]
- 11.She QB, Chandarlapaty S, Ye Q, Lobo J, Haskell KM, Leander KR, DeFeo-Jones D, Huber HE, Rosen N. Breast tumor cells with PI3K mutation or HER2 amplification are selectively addicted to Akt signaling. PLoS One. 2008;3:e3065. doi: 10.1371/journal.pone.0003065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brachmann SM, Hofmann I, Schnell C, Fritsch C, Wee S, Lane H, Wang S, Garcia-Echeverria C, Maira SM. Specific apoptosis induction by the dual PI3K/mTor inhibitor NVP-BEZ235 in HER2 amplified and PIK3CA mutant breast cancer cells. Proc Natl Acad Sci USA. 2009;106:22299–22304. doi: 10.1073/pnas.0905152106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gewinner C, Wang ZC, Richardson A, Teruya-Feldstein J, Etemadmoghadam D, Bowtell D, Barretina J, Lin WM, Rameh L, Salmena L, Pandolfi PP, Cantley LC. Evidence that inositol polyphosphate 4-phosphatase type II is a tumor suppressor that inhibits PI3K signaling. Cancer Cell. 2009;16:115–125. doi: 10.1016/j.ccr.2009.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sun H, Lesche R, Li DM, Liliental J, Zhang H, Gao J, Gavrilova N, Mueller B, Liu X, Wu H. PTEN modulates cell cycle progression and cell survival by regulating phosphatidylinositol 3,4,5,-trisphosphate and Akt/protein kinase B signaling pathway. Proc Natl Acad Sci USA. 1999;96:6199–6204. doi: 10.1073/pnas.96.11.6199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wu X, Senechal K, Neshat MS, Whang YE, Sawyers CL. The PTEN/MMAC1 tumor suppressor phosphatase functions as a negative regulator of the phosphoinositide 3-kinase/Akt pathway. Proc Natl Acad Sci USA. 1998;95:15587–15591. doi: 10.1073/pnas.95.26.15587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bendell JC, Rodon J, Burris HA, de Jonge M, Verweij J, Birle D, Demanse D, De Buck SS, Ru QC, Peters M, Goldbrunner M, Baselga J. Phase I, dose-escalation study of BKM120, an oral pan-class I PI3K inhibitor, in patients with advanced solid tumors. J Clin Oncol. 2012;30:282–290. doi: 10.1200/JCO.2011.36.1360. [DOI] [PubMed] [Google Scholar]
- 17.Burris H, Rodon J, Sharma S, Herbst RS, Tabernero J, Infante JR, Silva A, Demanse D, Hackl W, Baselga J. First-in-human phase I study of the oral PI3K inhibitor BEZ235 in patients (pts) with advanced solid tumors. J Clin Oncol. 2010;28:3005. [Google Scholar]
- 18.Von Hoff DD, LoRusso P, Tibes R, Shapiro G, Weiss GJ, Ware JA, Fredrickson J, Mazina KE, Levy GG, Wagner AJ. A first-in-human phase I study to evaluate the pan-PI3K inhibitor GDC-0941 administered QD or BID in patients with advanced solid tumors. J Clin Oncol. 2010;28:2541. [Google Scholar]
- 19.Shapiro G, Kwak E, Baselga J, Rodon J, Scheffold C, Laird AD, Bedell C, Edelman G. Phase I dose-escalation study of XL147, a PI3K inhibitor administered orally to patients with solid tumors. J Clin Oncol. 2009;27:3500. [Google Scholar]
- 20.Huang A, Fritsch C, Wilson C, Reddy A, Liu M, Lehar J, Quadt C, Hofmann F, Schlegel R. Single agent activity of PIK3CA inhibitor BYL719 in a broad cancer cell line panel. Cancer Res. 2012;72 [Google Scholar]
- 21.Juric D, Argiles G, Burris HA, Gonzalez-Angulo AM, Saura C, Quadt C, Douglas M, Demanse D, De Buck S, Baselga J. Phase I study of BYL719, an alpha-specific PI3K inhibitor, in patients with PIK3CA mutant advanced solid tumors: Preliminary efficacy and safety in patients with PIK3CA mutant ER-positive (ER+) metastatic breast cancer (MBC) Cancer Res. 2012;72 [Google Scholar]
- 22.Juric D, Rodon J, Gonzalez-Angulo AM, Burris HA, Bendell J, Berlin JD, Middleton MR, Bootle D, Boehm M, Schmitt A, Rouyrre N, Quadt C, Baselga J. BYL719, a next generation PI3K alpha specific inhibitor: Preliminary safety, PK, and efficacy results from the first-in-human study. Cancer Res. 2012;72 [Google Scholar]
- 23.Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149:274–293. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Roux PP, Shahbazian D, Vu H, Holz MK, Cohen MS, Taunton J, Sonenberg N, Blenis J. RAS/ERK signaling promotes site-specific ribosomal protein S6 phosphorylation via RSK and stimulates cap-dependent translation. J Biol Chem. 2007;282:14056–14064. doi: 10.1074/jbc.M700906200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Folkes AJ, Ahmadi K, Alderton WK, Alix S, Baker SJ, Box G, Chuckowree IS, Clarke PA, Depledge P, Eccles SA, Friedman LS, Hayes A, Hancox TC, Kugendradas A, Lensun L, Moore P, Olivero AG, Pang J, Patel S, Pergl-Wilson GH, Raynaud FI, Robson A, Saghir N, Salphati L, Sohal S, Ultsch MH, Valenti M, Wallweber HJ, Wan NC, Wiesmann C, Workman P, Zhyvoloup A, Zvelebil MJ, Shuttleworth SJ. The identification of 2-(1H-indazol-4-yl)-6-(4-methanesulfonyl-piperazin-1-ylmethyl)-4-morpholin-4-yl-thieno[3,2-d]pyrimidine (GDC-0941) as a potent, selective, orally bioavailable inhibitor of class I PI3 kinase for the treatment of cancer. J Med Chem. 2008;51:5522–5532. doi: 10.1021/jm800295d. [DOI] [PubMed] [Google Scholar]
- 26.Zhang H, Cicchetti G, Onda H, Koon HB, Asrican K, Bajraszewski N, Vazquez F, Carpenter CL, Kwiatkowski DJ. Loss of Tsc1/Tsc2 activates mTOR and disrupts PI3K-Akt signaling through downregulation of PDGFR. J Clin Invest. 2003;112:1223–1233. doi: 10.1172/JCI17222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tee AR, Manning BD, Roux PP, Cantley LC, Blenis J. Tuberous sclerosis complex gene products, Tuberin and Hamartin, control mTOR signaling by acting as a GTPase-activating protein complex toward Rheb. Curr Biol. 2003;13:1259–1268. doi: 10.1016/s0960-9822(03)00506-2. [DOI] [PubMed] [Google Scholar]
- 28.Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, Dancey J, Arbuck S, Gwyther S, Mooney M, Rubinstein L, Shankar L, Dodd L, Kaplan R, Lacombe D, Verweij J. New response evaluation criteria in solid tumours: Revised RECIST guideline (version 1.1) Eur J Cancer. 2009;45:228–247. doi: 10.1016/j.ejca.2008.10.026. [DOI] [PubMed] [Google Scholar]
- 29.Straussman R, Morikawa T, Shee K, Barzily-Rokni M, Qian ZR, Du J, Davis A, Mongare MM, Gould J, Frederick DT, Cooper ZA, Chapman PB, Solit DB, Ribas A, Lo RS, Flaherty KT, Ogino S, Wargo JA, Golub TR. Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature. 2012;487:500–504. doi: 10.1038/nature11183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wilson TR, Fridlyand J, Yan Y, Penuel E, Burton L, Chan E, Peng J, Lin E, Wang Y, Sosman J, Ribas A, Li J, Moffat J, Sutherlin DP, Koeppen H, Merchant M, Neve R, Settleman J. Widespread potential for growth-factor-driven resistance to anticancer kinase inhibitors. Nature. 2012;487:505–509. doi: 10.1038/nature11249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Harbinski F, Craig VJ, Sanghavi S, Jeffery D, Liu L, Sheppard KA, Wagner S, Stamm C, Buness A, Chatenay-Rivauday C, Yao Y, He F, Lu CX, Guagnano V, Metz T, Finan PM, Hofmann F, Sellers WR, Porter JA, Myer VE, Graus-Porta D, Wilson CJ, Buckler A, Tiedt R. Rescue screens with secreted proteins reveal compensatory potential of receptor tyrosine kinases in driving cancer growth. Cancer Discov. 2012;2:948–959. doi: 10.1158/2159-8290.CD-12-0237. [DOI] [PubMed] [Google Scholar]
- 32.Corcoran RB, Rothenberg SM, Hata AN, Faber AC, Piris A, Nazarian RM, Brown RD, Godfrey JT, Winokur D, Walsh J, Mino-Kenudson M, Maheswaran S, Settleman J, Wargo JA, Flaherty KT, Haber DA, Engelman JA. TORC1 suppression predicts responsiveness to RAF and MEK inhibition in BRAF-mutant melanoma. Sci Transl Med. 2013;5:196ra98. doi: 10.1126/scitranslmed.3005753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu P, Cheng H, Santiago S, Raeder M, Zhang F, Isabella A, Yang J, Semaan DJ, Chen C, Fox EA, Gray NS, Monahan J, Schlegel R, Beroukhim R, Mills GB, Zhao JJ. Oncogenic PIK3CA-driven mammary tumors frequently recur via PI3K pathway–dependent and PI3K pathway–independent mechanisms. Nat Med. 2011;17:1116–1120. doi: 10.1038/nm.2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Muellner MK, Uras IZ, Gapp BV, Kerzendorfer C, Smida M, Lechtermann H, Craig-Mueller N, Colinge J, Duernberger G, Nijman SM. A chemical-genetic screen reveals a mechanism of resistance to PI3K inhibitors in cancer. Nat Chem Biol. 2011;7:787–793. doi: 10.1038/nchembio.695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ilic N, Utermark T, Widlund HR, Roberts TM. PI3K-targeted therapy can be evaded by gene amplification along the MYC-eukaryotic translation initiation factor 4E (eIF4E) axis. Proc Natl Acad Sci USA. 2011;108:E699–E708. doi: 10.1073/pnas.1108237108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.S. Institute. CONAN CGP copy number analysis. http://www.sanger.ac.uk/research/areas/humangenetics/cnv/
- 37.Wall M, Poortinga G, Stanley KL, Lindemann RK, Bots M, Chan CJ, Bywater MJ, Kinross KM, Astle MV, Waldeck K, Hannan KM, Shortt J, Smyth MJ, Lowe SW, Hannan RD, Pearson RB, Johnstone RW, McArthur GA. The mTORC1 inhibitor everolimus prevents and treats Eμ-Myc lymphoma by restoring oncogene-induced senescence. Cancer Discov. 2013;3:82–95. doi: 10.1158/2159-8290.CD-12-0404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Maira SM, Stauffer F, Brueggen J, Furet P, Schnell C, Fritsch C, Brachmann S, Chène P, De Pover A, Schoemaker K, Fabbro D, Gabriel D, Simonen M, Murphy L, Finan P, Sellers W, García-Echeverría C. Identification and characterization of NVP-BEZ235, a new orally available dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor with potent in vivo antitumor activity. Mol Cancer Ther. 2008;7:1851–1863. doi: 10.1158/1535-7163.MCT-08-0017. [DOI] [PubMed] [Google Scholar]
- 39.Wallin JJ, Edgar KA, Guan J, Berry M, Prior WW, Lee L, Lesnick JD, Lewis C, Nonomiya J, Pang J, Salphati L, Olivero AG, Sutherlin DP, O'Brien C, Spoerke JM, Patel S, Lensun L, Kassees R, Ross L, Lackner MR, Sampath D, Belvin M, Friedman LS. GDC-0980 is a novel class I PI3K/mTOR kinase inhibitor with robust activity in cancer models driven by the PI3K pathway. Mol Cancer Ther. 2011;10:2426–2436. doi: 10.1158/1535-7163.MCT-11-0446. [DOI] [PubMed] [Google Scholar]
- 40.Markman B, Tabernero J, Krop I, Shapiro GI, Siu L, Chen LC, Mita M, Melendez Cuero M, Stutvoet S, Birle D, Anak O, Hackl W, Baselga J. Phase I safety, pharmacokinetic, and pharmacodynamic study of the oral phosphatidylinositol-3-kinase and mTOR inhibitor BGT226 in patients with advanced solid tumors. Ann Oncol. 2012;23:2399–2408. doi: 10.1093/annonc/mds011. [DOI] [PubMed] [Google Scholar]
- 41.Wagner AJ, Bendell JC, Dolly S, Morgan JA, Ware JA, Fredrickson J, Mazina KE, Lauchle JO, Burris HA, De Bono JS. A first-in-human phase I study to evaluate GDC-0980, an oral PI3K/mTOR inhibitor, administered QD in patients with advanced solid tumors. J Clin Oncol. 2011;29:3020. [Google Scholar]
- 42.Tanaka H, Yoshida M, Tanimura H, Fujii T, Sakata K, Tachibana Y, Ohwada J, Ebiike H, Kuramoto S, Morita K, Yoshimura Y, Yamazaki T, Ishii N, Kondoh O, Aoki Y. The selective class I PI3K inhibitor CH5132799 targets human cancers harboring oncogenic PIK3CA mutations. Clin Cancer Res. 2011;17:3272–3281. doi: 10.1158/1078-0432.CCR-10-2882. [DOI] [PubMed] [Google Scholar]
- 43.Bachelot T, Bourgier C, Cropet C, Ray-Coquard I, Ferrero JM, Freyer G, Abadie-Lacourtoisie S, Eymard JC, Debled M, Spaëth D, Legouffe E, Allouache D, El Kouri C, Pujade-Lauraine E. Randomized phase II trial of everolimus in combination with tamoxifen in patients with hormone receptor–positive, human epidermal growth factor receptor 2–negative metastatic breast cancer with prior exposure to aromatase inhibitors: A GINECO study. J Clin Oncol. 2012;30:2718–2724. doi: 10.1200/JCO.2011.39.0708. [DOI] [PubMed] [Google Scholar]
- 44.Baselga J, Campone M, Piccart M, Burris HA, III, Rugo HS, Sahmoud T, Noguchi S, Gnant M, Pritchard KI, Lebrun F, Beck JT, Ito Y, Yardley D, Deleu I, Perez A, Bachelot T, Vittori L, Xu Z, Mukhopadhyay P, Lebwohl D, Hortobagyi GN. Everolimus in postmenopausal hormone-receptor–positive advanced breast cancer. N Engl J Med. 2012;366:520–529. doi: 10.1056/NEJMoa1109653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.García-García C, Ibrahim YH, Serra V, Calvo MT, Guzmán M, Grueso J, Aura C, Pérez J, Jessen K, Liu Y, Rommel C, Tabernero J, Baselga J, Scaltriti M. Dual mTORC1/2 and HER2 blockade results in antitumor activity in preclinical models of breast cancer resistant to anti-HER2 therapy. Clin Cancer Res. 2012;18:2603–2612. doi: 10.1158/1078-0432.CCR-11-2750. [DOI] [PubMed] [Google Scholar]
- 46.Ibrahim YH, García-García C, Serra V, He L, Torres-Lockhart K, Prat A, Anton P, Cozar P, Guzmán M, Grueso J, Rodríguez O, Calvo MT, Aura C, Díez O, Rubio IT, Pérez J, Rodón J, Cortés J, Ellisen LW, Scaltriti M, Baselga J. PI3K inhibition impairs BRCA1/2 expression and sensitizes BRCA-proficient triple-negative breast cancer to PARP inhibition. Cancer Discov. 2012;2:1036–1047. doi: 10.1158/2159-8290.CD-11-0348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Elkabets M, Gifford AM, Scheel C, Nilsson B, Reinhardt F, Bray MA, Carpenter AE, Jirström K, Magnusson K, Ebert BL, Pontén F, Weinberg RA, McAllister SS. Human tumors instigate granulin-expressing hematopoietic cells that promote malignancy by activating stromal fibroblasts in mice. J Clin Invest. 2011;121:784–799. doi: 10.1172/JCI43757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Choi YJ, Di Nardo A, Kramvis I, Meikle L, Kwiatkowski DJ, Sahin M, He X. Tuberous sclerosis complex proteins control axon formation. Genes Dev. 2008;22:2485–2495. doi: 10.1101/gad.1685008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Debnath J, Muthuswamy SK, Brugge JS. Morphogenesis and oncogenesis of MCF-10A mammary epithelial acini grown in three-dimensional basement membrane cultures. Methods. 2003;30:256–268. doi: 10.1016/s1046-2023(03)00032-x. [DOI] [PubMed] [Google Scholar]
- 50.Kim PS, Von Ahsen O, Schmitz A, Schatz C, Magonova K, Lee T, Harvie G, Barham R, Leesman G, Kuller A, Lin F, Gong H, Krahn T, Singh S. Pathway profiling of signal transduction proteins in paired tumor and adjacent normal tissues obtained from breast cancer patients. Cancer Res. 2010;70 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.