

Abstract
Tau-tubulin kinase-1 (TTBK1) is a central nervous system (CNS)–specific protein kinase implicated in the pathological phosphorylation of tau. TTBK1-transgenic mice show enhanced neuroinflammation in the CNS. Double-transgenic mice expressing TTBK1 and frontotemporal dementia with parkinsonism-17–linked P301L (JNPL3) tau mutant (TTBK1/JNPL3) show increased accumulation of oligomeric tau protein in the CNS and enhanced loss of motor neurons in the ventral horn of the lumbar spinal cord. To determine the role of TTBK1-induced neuroinflammation in tauopathy-related neuropathogenesis, age-matched TTBK1/JNPL3, JNPL3, TTBK1, and non-transgenic littermates were systematically characterized. There was a striking switch in the activation phenotype and population of mononuclear phagocytes (resident microglia and infiltrating macrophages) in the affected spinal cord region: JNPL3 mice showed accumulation of alternatively activated microglia, whereas TTBK1 and TTBK1/JNPL3 mice showed accumulation of classically activated infiltrating peripheral monocytes. In addition, expression of chemokine ligand 2, a chemokine important for the recruitment of peripheral monocytes, was enhanced in TTBK1 and TTBK1/JNPL3 but not in other groups in the spinal cord. Furthermore, primary cultured mouse motor neurons showed axonal degeneration after transient expression of the TTBK1 gene or treatment with conditioned media derived from lipopolysaccharide-stimulated microglia; this was partially blocked by silencing of the endogenous TTBK1 gene in neurons. These data suggest that TTBK1 accelerates motor neuron neurodegeneration by recruiting proinflammatory monocytes and enhancing sensitivity to neurotoxicity in inflammatory conditions.
Tau tubulin kinase 1 (TTBK1), located on chromosome 6p21.1, belongs to the casein kinase 1 superfamily. This kinase, highly conserved from Caenorhabditis elegans to human, is a serine/threonine/tyrosine kinase and is specifically expressed in the brain, spinal cord, and testis in mammals.1,2 TTBK1 directly phosphorylates the tau protein at Ser198, Ser199, Ser202, and Ser422, which are known phosphorylation sites of paired helical filament-tau.3–8 TTBK1 levels are up-regulated in brains of human patients with Alzheimer disease (AD) compared with age-matched non-AD controls,1 and genetic variations of the TTBK1 gene are associated with late-onset AD in two large cohorts of Chinese and Spanish populations,9,10 further validating the potential significance of the TTBK1 gene in the neuropathogenesis of tauopathy-related neurodegenerative disorders, including AD. We have generated a transgenic (Tg) mouse model harboring an entire bacterial artificial chromosome fragment of the human TTBK1 genomic region,1,11 which expresses full-length human TTBK1 under the control of the endogenous promoter. We have previously demonstrated that TTBK1-Tg mice show significant age-dependent memory impairment, as determined by the radial arm water maze test.2 This impairment is associated with enhancement of tau and neurofilament phosphorylation, increased levels of p25 and p35, both activators of cyclin dependent kinase 5 (CDK5), another tau-kinase, and enhanced calpain I activity. Enhanced mononuclear phagocytosis (by brain resident microglia and infiltrating peripheral monocytes) in cortical and hippocampal regions is an additional major phenotype of TTBK1 mice.2,11 Double-transgenic mice expressing TTBK1 and P301L tau mutant (JNPL3) show increased accumulation of oligomeric tau protein in the forebrain and spinal cord, and enhanced reduction of motor neurons in the ventral horn of the lumbar spinal cord with severe muscle weakness, pathological features that are associated with neuroinflammation.11 However, precise neuroinflammatory pathological features of TTBK1 and TTBK1/JNPL3 mice are poorly understood.
Microglial activation parallels to neurofibrillary tangle (NFT) formation in AD brains, and induction of systemic inflammation with lipopolysaccharide (LPS) significantly induces tau hyperphosphorylation12,13 in AD mouse models. These findings suggest that neuroinflammation could contribute to disease progression in tauopathies. However, the mechanism(s) through which this might occur is unknown. Transgenic mice expressing P301S tau mutant show age-dependent neurodegeneration in the entorhinal cortex and hippocampus, which is preceded by extensive microglial activation that can be suppressed by an immunosuppressive agent, FK506.14 This is indicative of nonautonomous neuronal cell loss in the P301S tau mouse brain. Similarly, other P301S tau mouse models show extensive neurodegeneration in both the brainstem and spinal cord, which is accompanied by neuroinflammation and inflammatory mononuclear phagocyte accumulation.15,16 Furthermore, disruption of the CX3CR1 gene in hTau mice enhances microglial activation and tauopathy-related neuropathological features.17 These studies reveal that neuroinflammation is significantly involved in tauopathy-related neuropathogenesis.
Neuroinflammation is triggered by the innate immune response, in which mononuclear phagocytes play a major role. When these phagocytes recognize pathogens or damaged cell molecules, they become activated, and this activation can be classified into two phenotypes: classic (M1) and alternative (M2) activation.18 M1-skewed activation of mononuclear phagocytes causes the release of proinflammatory cytokines, such as interferon-γ, tumor necrosis factor-α, IL-6, IL-12, IL-1β, IL-23, and reactive oxygen/nitrogen intermediates induced by the expression of nitric oxide synthase (NOS) and NADPH oxidases.19–21 In contrast, M2-skewed activation is characterized by abundant levels of nonopsonic receptors (eg, the mannose receptor) and production of high levels of anti-inflammatory cytokines.19 However, there is no comprehensive characterization of central nervous system mononuclear phagocytes and their activation status (M1 or M2) in tauopathy-related neurodegenerative disorders.
In this study, we characterized tauopathy-related neurodegeneration and the profile of mononuclear phagocytes in the spinal cord of TTBK1 mice crossed with JNPL3 in vivo, and in primary tissue culture of neurons and microglia in vitro. Our data show that TTBK1 is involved in the induction of motor neuron degeneration by M1-skewed mononuclear phagocytes, thereby shedding light on how up-regulation of AD-associated molecules can induce cell nonautonomous neurodegeneration in the context of tauopathy pathogenesis.
Materials and Methods
Transgenic Animal Models
Generation of TTBK1, JNPL3, and TTBK1/JNPL3 transgenic mouse lines has been described previously.1,11 TTBK1 transgenic (TTBK1-Tg) mice (line 141) harboring human TTBK1 genomic DNA (57 kb) were used in this study. Briefly, the founders in the B6/SJL F1 background were backcrossed to the B6/129 F1 strain (Jackson Laboratories, Bar Harbor, ME) for five or fewer generations before the study. Transgenic JNPL3 mice expressing the P301L mutant of 4-repeat tau without amino-terminal inserts (4R0N for tau isoform nomenclature22,23) were backcrossed to the B6/129 F1 strain for five or more generations and crossed with TTBK1 mice to generate TTBK1, JNPL3, TTBK1/JNPL3, and non-Tg mice. All animal use procedures were strictly reviewed by the Laboratory Animal Safety Committee at Boston University School of Medicine (Boston, MA).
Tissue Preparation
Age-matched (10 to 11 months) mice were euthanized with a ketamine/xylazine mixture and perfused transcardially with PBS. L4-L5 spinal cords were rapidly removed, fixed in 4% paraformaldehyde for 48 hours, and cryoprotected by successive 24-hour immersions in 15% and 30% sucrose. Fixed, cryoprotected spinal cords were embedded in optimal cutting temperature compound (Fisher Scientific, Pittsburgh, PA), frozen, and divided into sections (10 μm thick) in the coronal plane using a cryostat (Leica, Bannockburn, IL).
Immunohistochemistry
Serially prepared slides (10 μm thick) of spinal cord sections were dehydrated, followed by antigen retrieval using 10% formic acid. Endogenous peroxidase activity was inhibited with 3% hydrogen peroxide incubation. Samples were permeabilized with 1% Triton X-100 and incubated in 1% bovine serum albumin/5% normal goat serum blocking buffer (Sigma-Aldrich, St. Louis, MI), followed by incubation with the following primary antibodies: ionized calcium-binding adapter molecule 1 (IBA1; 1:1000; WAKO Chemical, Tokyo, Japan), Ym-1 (1:1000; Stemcell Technologies, Vancouver, BC, Canada), and CD206 (1:100; Santa Cruz Biotechnology, Santa Cruz, CA). Twenty-four hours later, sections were incubated in secondary antibody for 1 hour (Envision HRP; Dako, Carpinteria, CA). 3,3′-Diaminobenzidine (DAB; Vector Laboratories, Burlingame, CA) staining was used as a chromogen, and cresyl violet was used for counterstaining. Motor neuron number was analyzed by counting cell bodies in the ventral horn of the spinal cord using a Nikon Eclipse E600 microscope and a color charge-coupled device camera (Nikon Instruments, Melville, NY).
Immunofluorescence
The fixed cells or tissue sections were subjected to immunofluorescence with the following antibodies: CD169 mouse monoclonal (1:200; AbD Serotec, Raleigh, NC), NOS2 rabbit polyclonal (1:500; Santa Cruz Biotechnology), CD11c hamster polyclonal (1:50; eBioscience, San Diego, CA), β-3 tubulin mouse monoclonal (1:1000, for neurite density assay; Promega, Fitchburg, WI), IBA1 rabbit polyclonal (1:1000; Wako, Tokyo, Japan), and TTBK1 mouse monoclonal (clone F287-1.1-1E91), followed by incubation with species-specific Alexa Fluor secondary antibodies (1:1000; Molecular Probes/Invitrogen, Grand Island, NY). Immunostained images were captured using an inverted fluorescence microscope attached to a monochromatic charge-coupled device camera (model TE-2000U; Nikon Instruments), and the cell number or fluorescence intensity was quantified with ImageJ software version 1.43 (NIH, Bethesda, MD). The axon length was quantified by NeuronJ plug-in (http://www.imagescience.org/meijering/software/neuronj, last accessed January 22, 2014). The number of apoptotic bodies (determined by condensed/fragmented nuclei) was also quantified with Hoechst 33342 staining (10 μg/mL; Invitrogen).
mRNA Analysis
RNA was extracted from 100 mg of 4% paraformaldehyde fixed spinal cord, as described previously,24 by using the RecoverAll Total Nucleic Acid Isolation Kit (Invitrogen), according to the manufacturer's instructions. Quantitative PCR was performed with a QuantiFast SYBR Green PCR Kit (Qiagen, Valencia, CA) with the Eppendorf realplex system. Melting curves were determined to ensure the amplification of a single product. The primers used were as follows: mouse chemokine ligand (CCL) 2, 5′-TTAAAAACCTGGATCGGAACCAA-3′ (forward); 5′-GCATTAGCTTCAGATTTACGGGT-3′ (reverse); and mouse glyceraldehyde-3-phosphate dehydrogenase, 5′-CATGTTCCAGTATGACTCCACTC-3′ (forward); and 5′-GGCCTCACCCCATTTGATGT-3′ (reverse).25
siRNA Vectors
Four putative siRNA sequences (clone 19, 5′-GCTCTTAAGGACGAAACCAACATGAGTGG-3′; clone 304, 5′-TTTAACTATGTGGTGATGCAGCTCCAGGG-3′; clone 1624, 5′-AAGGAGTGGGTCATTATTGACAAGGAGAC-3′; and clone 3152, 5′-TCTTGTTGTTCTGAAGAGGATACAGGCTCA-3′) were selected and subcloned into pVL-EGFP-Puro vectors (Capital Biosciences, Rockville, MD).
Motor Neuron Culture
Mouse primary motor neurons were obtained and cultured according to published protocol.26 E13.5-15 mice were removed from the pregnant mother, and 10 to 12 embryonic spinal cords were incubated with fresh trypsin (0.05% in HBSS) for 15 to 20 minutes at 37°C. After incubation, tissues were triturated. The dissociated tissue was undisturbed for 1 to 2 minutes to allow tissue debris to settle. Supernatants from the digestions were pooled and filtered through a 70-μm pore nylon mesh. Cell-containing supernatants were then centrifuged for 5 minutes at 500 × g at 4°C. Cells were resuspended in Dulbecco's minimum essential media containing 10% heat-inactivated fetal calf serum and 5% heat-inactivated horse serum (all from Invitrogen/Gibco, Carlsbad, CA), seeded into a 100-mm uncoated dish, and incubated in a 5% CO2 incubator for 30 minutes to allow tissue debris and nonneuronal cells to adhere to the bottom of the dish. Supernatants were collected and cells were plated into 24-well plates precoated with poly-d-lysine at a concentration of 1 × 105 cells per well. The following day, the medium was replaced with serum-free Neurobasal media supplemented with 2% B27 supplement and 2 mmol/L glutamine (all from Invitrogen). Cells were transfected with plasmid DNAs using Lipofectamine 2000 (Invitrogen) or were treated with microglial conditioned media. After 24 hours, cells were fixed for immunofluorescence.
Microglial Culture
Microglial cultures were maintained in Dulbecco's minimum essential media containing 10% fetal bovine serum during the first 24 hours after plating, then washed twice with serum-free medium to remove any traces of fetal bovine serum and stimulated with or without LPS (from Escherichia coli, 100 ng/mL; Sigma-Aldrich) or IL-4 (20 ng/mL; R&D Systems, Minneapolis, MN). After 1 hour of stimulation, cultures were washed twice and maintained in new medium for an additional 24 hours. Microglia-conditioned media (MCM) were then collected, centrifuged at 15,000 × g, filtered through a 0.22-μm pore membrane (EMD Millipore, Billerica, MA), and stored at −80°C until use.
Results
TTBK1 Accelerates Motor Neuron Degeneration in JNPL3 Mice
We have previously demonstrated that TTBK1 expression induced motor impairment by comparison between JNPL3 and TTBK1/JNPL3 mice. TTBK1/JNPL3 mice showed a 30% decrease in time on accelerating rotarod and a 20% decrease in forelimb grip strength compared with JNPL3 mice.11 Because JNPL3 mice are also known to show motor deficits and motor neuron degeneration in the ventral horn,23 we examined the pathological characteristics of neurons in the ventral horn of the L4-L5 spinal cord in non-Tg, TTBK1, JNPL3, and TTBK1/JNPL3 mice. Quantification of motor neurons by a cresyl violet Nissl stain on serially prepared sections (10 μm thick) in non-Tg, TTBK1, JNPL3, and TTBK1/JPNPL3 murine spinal cords revealed a significant loss of spinal cord motor neurons in all transgenic mice, compared with non-Tg mice (P < 0.001) (Figure 1). Furthermore, motor neuron loss was most prominent in TTBK1/JNPL3 mice, followed by TTBK1 and JNPL3 mice.
Figure 1.

Reduction of motor neurons in ventral horn in TTBK1, JNPL3, and TTBK1/JNPL3 spinal cord. Ventral horn images of coronal spinal cord frozen sections (L4-L5) with Nissl staining. Original magnification, ×40. Quantification of motor neuron counts per mm2 in the ventral horn (N = 4 per group). ∗∗P < 0.01, ∗∗∗P < 0.001, as determined by one-way analysis of variance and Tukey post hoc test, respectively. Scale bar = 50 μm.
TTBK1 Alters Mononuclear Phagocyte Population in the Spinal Cord
Our previous study demonstrated accelerated ionized calcium binding adaptor molecule 1–positive (IBA1+) mononuclear phagocytosis in the spinal cord ventral horn of TTBK1/JNPL3 mice, compared with age-matched JNPL3 littermates.11 However, whether this mononuclear phagocytosis causes motor neuron degeneration is unknown. Several lines of evidence show enhanced microgliosis in the JNPL3 mouse brain, but not in the spinal cord.27,28 To assess this point, we first investigated the morphological characteristics of IBA1+ mononuclear phagocytes in the spinal cord ventral horn in JNLP3, TTBK1, and TTBK1/JNPL3 mice. This revealed that ramified IBA1+ cells were increased in the ventral horn in JNPL3 mice, but there was no difference in ramified IBA1+ cells between non-Tg, TTBK1, and TTBK1/JNPL3 mice. On the contrary, IBA1+ cells were predominantly amoeboid in TTBK1 and TTBK1/JNPL3 mice (Figure 2, A and B). Thus, microglial activation with ramified morphological characteristics was exclusively observed in JNPL3 mice, and infiltrated macrophages or amoeboid microglia were predominantly observed in TTBK1 and TTBK1/JNPL3 mice.
Figure 2.

Morphological characteristics and distribution of IBA1+ cells in the spinal cord. A: Ventral horn images of the coronal spinal cord frozen sections (L4-L5) DAB immunostained for IBA1 (brown) and counterstained with cresyl violet (purple). G, gray matter; W, white matter. Original magnifications: ×10; ×40 (insets). Scale bar = 50 μm. B: Quantification of ramified (white bars) and amoeboid (black bars) IBA1+ cells per mm2 in the ventral horn (N = 4 per group). ∗∗∗P < 0.001, as determined by Student's t-test.
TTBK1 Induces a Switch of Cell Population from CD11c+ Microglia to Infiltrating Activated Monocytes in the Spinal Cord of JNPL3 Mice
Next, to classify the mononuclear phagocytes and activation phenotype, spinal cord sections were immunostained for CD169, a peripheral macrophage marker28,29) (Figure 3A) and NOS2, an M1 marker.18,21 There were no NOS2+ cells and few CD169+ cells in the ventral horn of JNPL3 mice. In contrast, there were many CD169+ cells in both the white and gray matter of TTBK1/JNPL3 mice (Figure 3A) and, to a lesser degree, in TTBK1 mice. In TTBK1/JNPL3 mice, 80% and 10% of CD169+ cells were NOS2+ in the gray and white matter, respectively (Figure 3B). CD169+NOS2+ cells were also observed in TTBK1 spinal cord. Taken together, these data demonstrate that IBA1+ cells are mostly CD169-NOS2− ramified microglia in JNPL3 mice, and CD169+ infiltrated macrophages are mostly CD169+NOS2+ M1-activated monocytes in the gray matter of TTBK1 and TTBK1/JNPL3 mice. This suggests a gray matter–specific proinflammatory activation mechanism in TTBK1/JNPL3 mice.
Figure 3.

CD169+ infiltrated macrophages are mostly M1 skewed in the spinal cord gray matter of TTBK1 and TTBK1/JNPL3 mice. A: Immunofluorescence of CD169 (peripheral macrophage marker, green) and NOS2 (M1 marker, red) in the L4-L5 spinal cord. Original magnifications: ×10; ×40 (insets). Scale bar = 50 μm. B: Quantification of NOS2+CD169+ cells in gray (white bars) and white (black bars) matter. ∗∗∗P < 0.001, as determined by Student's t-test (N = 4 per group).
To verify further the phenotypes of CD169−NOS2−IBA1+ cells, spinal cord sections were immunostained for YM1 (an M2 marker18), CD206 (a mannose receptor and an M2 marker18), CD11c (a dendritic cell marker, commonly seen in M2-skewed microglia30), and IBA1. YM1+ cells and CD206+ cells were detected in JNPL3 mice (Figure 4A), whereas no YM1+CD206+ cells were detected in non-Tg, TTBK1, or TTBK1/JNPL3 mice. CD11c+ cells were exclusively in the gray matter of JNPL3 mice and mostly colocalized with IBA1+ cells in this region (Figure 4B). There were no CD11c+ cells in the gray or white matter of non-Tg, TTBK1, or TTBK1/JNPL3 mice (Figure 4B). Collectively, these data suggest that most IBA1+ cells in JNPL3 mice are M2-skewed CD11c+ dendritic cell–like microglia, which are absent in TTBK1/JNPL3 mice.
Figure 4.

M2 phenotype of microglia in JNPL3, but not in TTBK1 or TTBK1/JNPL3, spinal cord. A: Ventral horn images of coronal spinal cord frozen sections (L4-L5) DAB immunostained for M2 markers [brown, YM-1 (top panels); CD206 (bottom panels)], and counterstained with cresyl violet (purple). B: Immunofluorescence of IBA1 (green) and CD11c (red) of ventral horn. Original magnifications: ×10; ×40 (inset). Scale bar = 50 μm (A and B).
A subset of infiltrating peripheral monocytes is known to be recruited to spinal cord by CCL2/C-C chemokine receptor 2 signaling in experimental autoimmune encephalomyelitis and amyotrophic lateral sclerosis (ALS) mouse models.29,31 This suggests that CCL2 levels are enhanced in the spinal cord of TTBK1 and TTBK1/JNPL3 mice. To verify this point, total RNA was isolated from spinal cord from all of the mouse groups and subjected to quantification of CCL2 mRNA by real-time PCR. As shown in Figure 5, CCL2 mRNA levels were significantly enhanced in TTBK1 and TTBK1/JNPL3 mice, compared with the JNPL3 or non-Tg group. These data suggest that CCL2 induction is responsible for the infiltration of CD169+NOS2+ cells in TTBK1 and TTBK1/JNPL3 mice.
Figure 5.

CCL2 mRNA expression in non-Tg, JNPL3, TTBK1, and TTBK1/JNPL3 spinal cord. Semiquantification of CCL2 mRNA levels by real-time RT-PCR normalized by glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA levels in spinal cords (N = 4 to 6 per group). ∗P < 0.05 versus either non-Tg or JNPL3 group, as determined by one-way analysis of variance and Tukey post hoc test.
M1-Skewed Microglia Suppress Neurite Extension of Primary Cultured Motor Neurons
Based on our results suggesting peripheral monocyte infiltration in TTBK1-overexpressing mice, we hypothesized that TTBK1 induced danger-associated molecular pattern molecules (DAMPs; eg, ATP, DNA, S100, and chromatin-associated molecules released from injured neurons), which activate an M1-like innate immunity response of mononuclear phagocytes and production of chemotactic factors (CCL2), leading to the recruitment of peripheral macrophages into the affected brain region.32–34 To test this hypothesis, we first showed that overexpression of TTBK1 in primary mouse motor neurons at day 7 differentiation in vitro (div) was associated with axonal degeneration, compared with untreated control (Figure 6). Degenerating axons are known to release DAMPs, which trigger the Toll-like receptor–mediated innate immunity response.34 Our next concern was to determine whether phenotype-skewed mononuclear phagocytes affected motor neuron degeneration. To investigate microglial polarization, we cultured mouse primary microglia and harvested the conditioned media after treating cells with LPS (to induce M1), IL-4 (to induce M2), or PBS, and examined their effect on mouse primary cultured motor neurons at day 7 div (Figure 7). LPS was used to induce M1 activation because it specifically stimulates Toll-like receptor 4, a known receptor stimulated by several DAMPs, such as Mrp8 and Mrp14.35 The conditioned media from PBS-treated microglia demonstrated significant enhancement of neurite density, compared with control (no treatment with microglia-conditioned media) (Figure 7, A, B, and E). This is because, under noninflammatory conditions, microglia are known to release neurotrophic factors that can support the growth of neurons.36,37 On the other hand, M1-skewed microglia conditioned media significantly reduced neurite density, compared with the PBS-treated microglia group (P < 0.01), whereas M2-skewed microglia showed no difference from the PBS-treated microglia group. These data suggest the following: i) un-stimulated microglia secrete molecules enhancing neurite extension, ii) M1 skewing of microglia diminishes the neurotrophic effect, and iii) M2 skewing has no significant effect on neurite extension.
Figure 6.
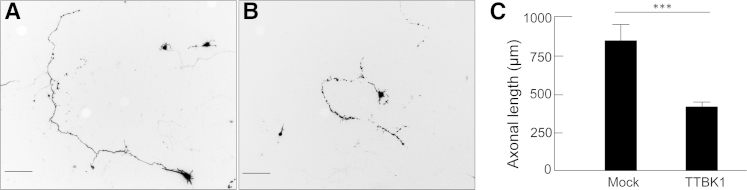
Reduction of axonal length by the expression of TTBK1 in primary motor neurons. Primary cultured motor neurons were transfected with DNA plasmids encoding GFP and pcDNA3.1 (mock, A) or TTBK1 (B) at 5 days div and fixed at 8 days div. The axonal lengths of transfected cells (as determined by GFP expression) were determined by NeuronJ plug-in and subjected to statistical analysis (C). ∗∗∗P < 0.001, as determined by Student's t-test (N = 100 per group). Scale bar = 100 μm.
Figure 7.

Microglia-induced neurite extension of primary motor neurons is attenuated by LPS. Mouse primary motor neurons were harvested from embryonic spinal cord and div for 7 days. A: No MCM control. MCM was collected from primary cultured mouse microglia after stimulation with PBS (B), LPS (C), or IL-4 (D), and applied to motor neurons. Cells were fixed 24 hours after the incubation for analysis of neurite density per neuron. E: Quantification. ∗P < 0.05, as determined by one-way analysis of variance and Tukey post hoc test (N = 50 per group). Scale bar = 10 μm.
TTBK1 Is Involved in the Induction of Motor Neuron Degeneration by M1-Skewed Microglia in Vitro
To deduce whether TTBK1 induces motor neuron degeneration in a dose-dependent manner, we used a mix of shRNA expression vectors against murine TTBK1 gene (clones 19, 304, and 1624), which significantly suppressed TTBK1 protein expression (Figure 8A). After the silencing of TTBK1 gene expression, motor neurons were treated with M1-skewed microglia conditioned media (Figure 8B). Silencing of TTBK1 (siTTBK1) alone or direct treatment of motor neurons with LPS had no effect on motor neuron degeneration, as determined by the formation of apoptotic bodies (No MCM groups ± LPS) (Figure 8B). The conditioned media from PBS-treated microglia rather suppressed apoptotic body formation (PBS MCM group), which is consistent with its effect on neurite growth. Conditioned media from LPS-treated microglia significantly enhanced motor neuron degeneration, which was neutralized by siTTBK1 pretreatment (LPS MCM ± siTTBK1 groups). This suggests that endogenous TTBK1 plays a significant role in M1-skewed mononuclear phagocyte neurotoxicity.
Figure 8.

Silencing of endogenous TTBK1 reduces M1-skewed microglia-induced neuronal cell death. A: The silencing effect of lentiviral vectors expressing shRNA targeting different mRNA sequences of murine TTBK1 was quantified by immunofluorescence and presented as percentage TTBK1 intensity versus control groups. B: Primary cultured mouse motor neurons were transduced with siTTBK1 vectors co-expressing GFP at 5 days div, then treated with MCM from PBS- or LPS-treated microglia (PBS or LPS MCM) at 8 days div, and fixed at 9 days div. The number of apoptotic bodies out of siTTBK1 or control viral vector transduced (GFP+) cells was counted and presented as percentage cell death over background. ∗P < 0.05, ∗∗∗P < 0.001 versus control (A) or LPS MCM (B), as determined by one-way analysis of variance and Tukey post hoc test.
Discussion
This study shows the following: i) TTBK1 accelerates motor neuron loss in the spinal cord of JNPL3 mice, ii) JNPL3 mice show mostly M2-skewed microglial accumulation in the spinal cord ventral horn, whereas TTBK1/JNPL3 mice show M1-skewed infiltrating monocytes in the same region, iii) TTBK1 up-regulation reduces axonal length, whereas resting microglia enhance neurite extension, and iv) motor neurons are sensitive to neurotoxicity induced by M1-activated microglia, and this is dependent on endogenous TTBK1 gene expression. Figure 9 depicts our proposed mechanism: In JNPL3 mice, motor neuron loss is evident in the ventral horn of the lumbar spinal cord, which is accompanied by accumulation of CD11c+ neuroprotective microglia, thereby limiting neurodegeneration. TTBK1/JNPL3 mice, on the other hand, show a dramatic conversion of the cell population from CD11c+ microglia to M1-skewed infiltrating monocytes. This could be because of the accelerated neurodegeneration by TTBK1 up-regulation and enhanced generation of danger-associated molecular pattern molecules (M1-skewing ligands) from degenerating motor neurons, which leads to proinflammatory activation, generation of CCL2 chemokine, and infiltration of peripheral monocytes. This may lead to the acceleration of neurodegeneration via bystander killing of neurons.
Figure 9.

Scheme of neuroinflammation and neurodegeneration in JNPL3 and TTBK1/JNPL3 mice. In JNPL3 mice, CD11c+ neuroprotective microglia accumulate in the gray matter of the ventral horn, thereby protecting from accelerated neurodegeneration. Conversely, in TTBK1/JNPL3 mice, the cell population is shifted from CD11c+ microglia to M1-skewed infiltrating NOS2+CD169+ monocytes, resulting in enhanced neuroinflammation and accelerated motor neuron loss.
A possible explanation of how TTBK1 influences motor neuron degeneration is that TTBK1 may enhance the sensitivity of motor neurons to proinflammatory cytokine or reactive oxygen/nitrogen intermediate-mediated neurotoxicity. Because TTBK1 phosphorylates both tau and tubulin, which are enriched in axons, TTBK1 may mediate proinflammatory cytokine-induced destabilization of microtubules through phosphorylation of the two molecules, and up-regulation of TTBK1 may accelerate the destabilization.
In the human AD brain, increased expression of proinflammatory cytokines or chemokines is accompanied by M1-skewed microglial activation.38–40 In addition, CCL2 levels are known to be associated with cognitive decline during the early stage of AD in patients.41–43 These findings are reproduced in several AD mouse models, such as the APP+PS1 mouse,44 which shows a distinctive age-dependent shift from M2 to M1 mononuclear cell activation in the hippocampus.45 We could not detect differences of inflammatory changes in hippocampus between TTBK1 and TTBK1/JNPL3 murine models (data not shown), because in the JNPL3 mice, NFTs are primarily located in the spinal cord and hindbrain, with fewer NFTs in the midbrain, amygdala, and hypothalamus.23 However, the inflammation mechanism of TTBK1 mouse spinal cord could be a useful model for future investigation of tauopathy-mediated neuroinflammatory changes in AD brains.
The phenomenon of nonautonomous neuron death induced by infiltrating monocytes in TTBK1 or TTBK1/JNPL3 mice is reminiscent of established neuropathogenesis in mutant Cu/Zn superoxide dismutase (SOD1) mice or patients with ALS, although some aspects of the mechanism of monocyte infiltration may be different. In spinal cords of SOD1G93A rats46 and patients with ALS,47 a significant decrease in expression of tight junction molecules was observed, suggesting the effect of SOD1 mutants in endothelium. In contrast, TTBK1 expression was absent in endothelial cells, and the expression levels of tight junction molecules (occludin and claudin-5) and aquaporin-4 (an astrocyte endfeet-specific marker) were unchanged (data not shown), suggesting a different molecular mechanism. Another potential mechanism is that low expression of TTBK1 may enhance monocyte recruitment or activation. However, TTBK1 expression was negative in any type of myeloid cells, as determined by various markers (IBA1, CD169, NOS2, YM1, and CD206) both in spinal cord and spleen (data not shown). In addition, transient expression of TTBK1 gene in murine microglia cell line BV-2 had no effect on cell migration or phagocytosis of apoptotic cells in vitro (data not shown). These results show that it is unlikely that TTBK1 gene expression in mononuclear phagocytes or myeloid cells contributes to their phenotypic changes. Thus, the neuroinflammatory mechanism in TTBK1 and TTBK1/JNPL3 mouse models appears to be distinct from that of ALS models.
Populations of the Kii Peninsula in Japan and the US territory Guam have historically shown a high incidence of ALS.48–50 Symptoms of the unique ALS complex in patients in those areas are not restricted to motor neuron signs but include parkinsonism and dementia. Pathologically, patients display characteristic appearance of NFT-tau, especially in the temporal cortex, hippocampus, amygdala, brainstem, and spinal cord.49 These clinical symptoms, pathological tau phosphorylation, and affected areas are similar to those of TTBK1/JNPL3 mice. Many patients with ALS complex from certain areas in Kii Peninsula and Guam Island have familial traits, but those from other areas in Kii Peninsula are almost always sporadic cases. Genetic studies show that SOD1, tau, apolipoprotein E, and neurofilament heavy chain are not genetically linked to patients with ALS from these areas.50 TTBK1 gene is a noteworthy candidate for further investigation of these motor neuron disease subtypes.
In conclusion, this study demonstrates that TTBK1 expression plays a unique role in accelerating motor neuron degeneration in JNPL3 mice, and may be a pivotal neuronal molecule for the accumulation and activation of infiltrating monocytes in the central nervous system. Thus, TTBK1 is a potential therapeutic target of neuroinflammation-associated neurodegeneration, such as that seen in AD and tauopathy-related ALS complex. In addition, TTBK1/JNPL3 mice are symptomatically and pathologically similar to a certain subset of patients with motor neuron disease and, therefore, can be used as a valuable research tool for delineating therapeutic treatments.
Footnotes
This work is supported in part by BrightFocus Foundation for Alzheimer Disease Research (H.A.), NIH grant 5T32GM008541 (M.W.), Boston University Undergraduate Research Opportunities Program Award (G.M.S.Y.), Alzheimer's Art Quilt Initiative (T.I.), and Boston University Faculty Recruit Fund (T.I.).
Disclosures: None declared.
References
- 1.Sato S., Xu J., Okuyama S., Martinez L.B., Walsh S.M., Jacobsen M.T., Swan R.J., Schlautman J.D., Ciborowski P., Ikezu T. Spatial learning impairment, enhanced CDK5/p35 activity, and downregulation of NMDA receptor expression in transgenic mice expressing tau-tubulin kinase 1. J Neurosci. 2008;28:14511–14521. doi: 10.1523/JNEUROSCI.3417-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sato S., Cerny R.L., Buescher J.L., Ikezu T. Tau-tubulin kinase 1 (TTBK1), a neuron-specific tau kinase candidate, is involved in tau phosphorylation and aggregation. J Neurochem. 2006;98:1573–1584. doi: 10.1111/j.1471-4159.2006.04059.x. [DOI] [PubMed] [Google Scholar]
- 3.Iqbal K., Grundke-Iqbal I., Smith A.J., George L., Tung Y.C., Zaidi T. Identification and localization of a tau peptide to paired helical filaments of Alzheimer disease. Proc Natl Acad Sci U S A. 1989;86:5646–5650. doi: 10.1073/pnas.86.14.5646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Morishima-Kawashima M., Hasegawa M., Takio K., Suzuki M., Yoshida H., Titani K., Ihara Y. Proline-directed and non-proline-directed phosphorylation of PHF-tau. J Biol Chem. 1995;270:823–829. doi: 10.1074/jbc.270.2.823. [DOI] [PubMed] [Google Scholar]
- 5.Morishima-Kawashima M., Hasegawa M., Takio K., Suzuki M., Yoshida H., Watanabe A., Titani K., Ihara Y. Hyperphosphorylation of tau in PHF. Neurobiol Aging. 1995;16:365–371. doi: 10.1016/0197-4580(95)00027-c. [DOI] [PubMed] [Google Scholar]
- 6.Hanger D.P., Betts J.C., Loviny T.L., Blackstock W.P., Anderton B.H. New phosphorylation sites identified in hyperphosphorylated tau (paired helical filament-tau) from Alzheimer's disease brain using nanoelectrospray mass spectrometry. J Neurochem. 1998;71:2465–2476. doi: 10.1046/j.1471-4159.1998.71062465.x. [DOI] [PubMed] [Google Scholar]
- 7.Vega I.E., Cui L., Propst J.A., Hutton M.L., Lee G., Yen S.H. Increase in tau tyrosine phosphorylation correlates with the formation of tau aggregates. Brain Res Mol Brain Res. 2005;138:135–144. doi: 10.1016/j.molbrainres.2005.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lund H., Cowburn R.F., Gustafsson E., Strömberg K., Svensson A., Dahllund L., Malinowsky D., Sunnemark D. Tau-tubulin kinase 1 expression, phosphorylation and co-localization with phospho-Ser422 tau in the Alzheimer's disease brain. Brain Pathol. 2013;23:378–389. doi: 10.1111/bpa.12001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vázquez-Higuera J.L., Martínez-Garcia A., Sánchez-Juan P., Rodriguez-Rodriguez E., Mateo I., Pozueta A., Frank A., Valdivieso F., Berciano J., Bullido M.J., Combarros O. Genetic variations in tau-tubulin kinase-1 are linked to Alzheimer's disease in a Spanish case-control cohort. Neurobiol Aging. 2011;32 doi: 10.1016/j.neurobiolaging.2009.12.021. 550.e5–e9. [DOI] [PubMed] [Google Scholar]
- 10.Yu N.N., Yu J.T., Xiao J.T., Zhang H.W., Lu R.C., Jiang H., Xing Z.H., Tan L. Tau-tubulin kinase-1 gene variants are associated with Alzheimer's disease in Han Chinese. Neurosci Lett. 2011;491:83–86. doi: 10.1016/j.neulet.2011.01.011. [DOI] [PubMed] [Google Scholar]
- 11.Xu J., Sato S., Okuyama S., Swan R.J., Jacobsen M.T., Strunk E., Ikezu T. Tau-tubulin kinase 1 enhances prefibrillar tau aggregation and motor neuron degeneration in P301L FTDP-17 tau-mutant mice. FASEB J. 2010;24:2904–2915. doi: 10.1096/fj.09-150144. [DOI] [PubMed] [Google Scholar]
- 12.Lee D.C., Rizer J., Selenica M.L., Reid P., Kraft C., Johnson A., Blair L., Gordon M.N., Dickey C.A., Morgan D. LPS-induced inflammation exacerbates phospho-tau pathology in rTg4510 mice. J Neuroinflammation. 2010;7:56. doi: 10.1186/1742-2094-7-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kitazawa M., Oddo S., Yamasaki T.R., Green K.N., LaFerla F.M. Lipopolysaccharide-induced inflammation exacerbates tau pathology by a cyclin-dependent kinase 5-mediated pathway in a transgenic model of Alzheimer's disease. J Neurosci. 2005;25:8843–8853. doi: 10.1523/JNEUROSCI.2868-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yoshiyama Y., Higuchi M., Zhang B., Huang S.M., Iwata N., Saido T.C., Maeda J., Suhara T., Trojanowski J.Q., Lee V.M. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron. 2007;53:337–351. doi: 10.1016/j.neuron.2007.01.010. [DOI] [PubMed] [Google Scholar]
- 15.Allen B., Ingram E., Takao M., Smith M.J., Jakes R., Virdee K., Yoshida H., Holzer M., Craxton M., Emson P.C., Atzori C., Migheli A., Crowther R.A., Ghetti B., Spillantini M.G., Goedert M. Abundant tau filaments and nonapoptotic neurodegeneration in transgenic mice expressing human P301S tau protein. J Neurosci. 2002;22:9340–9351. doi: 10.1523/JNEUROSCI.22-21-09340.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bellucci A., Westwood A.J., Ingram E., Casamenti F., Goedert M., Spillantini M.G. Induction of inflammatory mediators and microglial activation in mice transgenic for mutant human P301S tau protein. Am J Pathol. 2004;165:1643–1652. doi: 10.1016/S0002-9440(10)63421-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bhaskar K., Konerth M., Kokiko-Cochran O.N., Cardona A., Ransohoff R.M., Lamb B.T. Regulation of tau pathology by the microglial fractalkine receptor. Neuron. 2010;68:19–31. doi: 10.1016/j.neuron.2010.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gordon S. Alternative activation of macrophages. Nat Rev Immunol. 2003;3:23–35. doi: 10.1038/nri978. [DOI] [PubMed] [Google Scholar]
- 19.Mantovani A., Sica A., Sozzani S., Allavena P., Vecchi A., Locati M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004;25:677–686. doi: 10.1016/j.it.2004.09.015. [DOI] [PubMed] [Google Scholar]
- 20.Benoit M., Desnues B., Mege J.-L. Macrophage polarization in bacterial infections. J Immunol. 2008;181:3733–3739. doi: 10.4049/jimmunol.181.6.3733. [DOI] [PubMed] [Google Scholar]
- 21.Brown G.C. Mechanisms of inflammatory neurodegeneration: iNOS and NADPH oxidase. Biochem Soc Trans. 2007;35:1119–1121. doi: 10.1042/BST0351119. [DOI] [PubMed] [Google Scholar]
- 22.Tatebayashi Y., Miyasaka T., Chui D.H., Akagi T., Mishima K., Iwasaki K., Fujiwara M., Tanemura K., Murayama M., Ishiguro K., Planel E., Sato S., Hashikawa T., Takashima A. Tau filament formation and associative memory deficit in aged mice expressing mutant (R406W) human tau. Proc Natl Acad Sci U S A. 2002;99:13896–13901. doi: 10.1073/pnas.202205599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lewis J., McGowan E., Rockwood J., Melrose H., Nacharaju P., Van Slegtenhorst M., Gwinn-Hardy K., Paul Murphy M., Baker M., Yu X., Duff K., Hardy J., Corral A., Lin W.L., Yen S.H., Dickson D.W., Davies P., Hutton M. Neurofibrillary tangles, amyotrophy and progressive motor disturbance in mice expressing mutant (P301L) tau protein. Nat Genet. 2000;25:402–405. doi: 10.1038/78078. [DOI] [PubMed] [Google Scholar]
- 24.Cronin M., Pho M., Dutta D., Stephans J.C., Shak S., Kiefer M.C., Esteban J.M., Baker J.B. Measurement of gene expression in archival paraffin-embedded tissues: development and performance of a 92-gene reverse transcriptase-polymerase chain reaction assay. Am J Pathol. 2004;164:35–42. doi: 10.1016/S0002-9440(10)63093-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Izhak L., Wildbaum G., Weinberg U., Shaked Y., Alami J., Dumont D., Friedman B., Stein A., Karin N. Predominant expression of CCL2 at the tumor site of prostate cancer patients directs a selective loss of immunological tolerance to CCL2 that could be amplified in a beneficial manner. J Immunol. 2010;184:1092–1101. doi: 10.4049/jimmunol.0902725. [DOI] [PubMed] [Google Scholar]
- 26.Jiang X.Y., Fu S.L., Nie B.M., Li Y., Lin L., Yin L., Wang Y.X., Lu P.H., Xu X.M. Methods for isolating highly-enriched embryonic spinal cord neurons: a comparison between enzymatic and mechanical dissociations. J Neurosci Methods. 2006;158:13–18. doi: 10.1016/j.jneumeth.2006.05.014. [DOI] [PubMed] [Google Scholar]
- 27.Sasaki A., Kawarabayashi T., Murakami T., Matsubara E., Ikeda M., Hagiwara H., Westaway D., George-Hyslop P.S., Shoji M., Nakazato Y. Microglial activation in brain lesions with tau deposits: comparison of human tauopathies and tau transgenic mice TgTauP301L. Brain Res. 2008;1214:159–168. doi: 10.1016/j.brainres.2008.02.084. [DOI] [PubMed] [Google Scholar]
- 28.Chiu I.M., Phatnani H., Kuligowski M., Tapia J.C., Carrasco M.A., Zhang M., Maniatis T., Carroll M.C. Activation of innate and humoral immunity in the peripheral nervous system of ALS transgenic mice. Proc Natl Acad Sci U S A. 2009;106:20960–20965. doi: 10.1073/pnas.0911405106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Butovsky O., Siddiqui S., Gabriely G., Lanser A.J., Dake B., Murugaiyan G., Doykan C.E., Wu P.M., Gali R.R., Iyer L.K., Lawson R., Berry J., Krichevsky A.M., Cudkowicz M.E., Weiner H.L. Modulating inflammatory monocytes with a unique microRNA gene signature ameliorates murine ALS. J Clin Invest. 2012;122:3063–3087. doi: 10.1172/JCI62636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Butovsky O., Koronyo-Hamaoui M., Kunis G., Ophir E., Landa G., Cohen H., Schwartz M. Glatiramer acetate fights against Alzheimer's disease by inducing dendritic-like microglia expressing insulin-like growth factor 1. Proc Natl Acad Sci U S A. 2006;103:11784–11789. doi: 10.1073/pnas.0604681103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brouwer N., Zuurman M.W., Wei T., Ransohoff R.M., Boddeke H.W., Biber K. Induction of glial L-CCR mRNA expression in spinal cord and brain in experimental autoimmune encephalomyelitis. Glia. 2004;46:84–94. doi: 10.1002/glia.10352. [DOI] [PubMed] [Google Scholar]
- 32.Hanamsagar R., Hanke M.L., Kielian T. Toll-like receptor (TLR) and inflammasome actions in the central nervous system. Trends Immunol. 2012;33:333–342. doi: 10.1016/j.it.2012.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hanke M.L., Kielian T. Toll-like receptors in health and disease in the brain: mechanisms and therapeutic potential. Clin Sci (Lond) 2011;121:367–387. doi: 10.1042/CS20110164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu T., Gao Y.J., Ji R.R. Emerging role of Toll-like receptors in the control of pain and itch. Neurosci Bull. 2012;28:131–144. doi: 10.1007/s12264-012-1219-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Loser K., Vogl T., Voskort M., Lueken A., Kupas V., Nacken W., Klenner L., Kuhn A., Foell D., Sorokin L., Luger T.A., Roth J., Beissert S. The Toll-like receptor 4 ligands Mrp8 and Mrp14 are crucial in the development of autoreactive CD8+ T cells. Nat Med. 2010;16:713–717. doi: 10.1038/nm.2150. [DOI] [PubMed] [Google Scholar]
- 36.Ueno M., Fujita Y., Tanaka T., Nakamura Y., Kikuta J., Ishii M., Yamashita T. Layer V cortical neurons require microglial support for survival during postnatal development. Nat Neurosci. 2013;16:543–551. doi: 10.1038/nn.3358. [DOI] [PubMed] [Google Scholar]
- 37.Butovsky O., Ziv Y., Schwartz A., Landa G., Talpalar A.E., Pluchino S., Martino G., Schwartz M. Microglia activated by IL-4 or IFN-gamma differentially induce neurogenesis and oligodendrogenesis from adult stem/progenitor cells. Mol Cell Neurosci. 2006;31:149–160. doi: 10.1016/j.mcn.2005.10.006. [DOI] [PubMed] [Google Scholar]
- 38.Parachikova A., Agadjanyan M.G., Cribbs D.H., Blurton-Jones M., Perreau V., Rogers J., Beach T.G., Cotman C.W. Inflammatory changes parallel the early stages of Alzheimer disease. Neurobiol Aging. 2007;28:1821–1833. doi: 10.1016/j.neurobiolaging.2006.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Heneka M.T., Wiesinger H., Dumitrescu-Ozimek L., Riederer P., Feinstein D.L., Klockgether T. Neuronal and glial coexpression of argininosuccinate synthetase and inducible nitric oxide synthase in Alzheimer disease. J Neuropathol Exp Neurol. 2001;60:906–916. doi: 10.1093/jnen/60.9.906. [DOI] [PubMed] [Google Scholar]
- 40.Lüth H.J., Münch G., Arendt T. Aberrant expression of NOS isoforms in Alzheimer's disease is structurally related to nitrotyrosine formation. Brain Res. 2002;953:135–143. doi: 10.1016/s0006-8993(02)03280-8. [DOI] [PubMed] [Google Scholar]
- 41.Westin K., Buchhave P., Nielsen H., Minthon L., Janciauskiene S., Hansson O. CCL2 is associated with a faster rate of cognitive decline during early stages of Alzheimer's disease. PLoS One. 2012;7:e30525. doi: 10.1371/journal.pone.0030525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sokolova A., Hill M.D., Rahimi F., Warden L.A., Halliday G.M., Shepherd C.E. Monocyte chemoattractant protein-1 plays a dominant role in the chronic inflammation observed in Alzheimer's disease. Brain Pathol. 2009;19:392–398. doi: 10.1111/j.1750-3639.2008.00188.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Galimberti D., Schoonenboom N., Scarpini E., Scheltens P., Dutch-Italian Alzheimer Research Group Chemokines in serum and cerebrospinal fluid of Alzheimer's disease patients. Ann Neurol. 2003;53:547–548. doi: 10.1002/ana.10531. [DOI] [PubMed] [Google Scholar]
- 44.Jankowsky J.L., Slunt H.H., Gonzales V., Jenkins N.A., Copeland N.G., Borchelt D.R. APP processing and amyloid deposition in mice haplo-insufficient for presenilin 1. Neurobiol Aging. 2004;25:885–892. doi: 10.1016/j.neurobiolaging.2003.09.008. [DOI] [PubMed] [Google Scholar]
- 45.Jimenez S., Baglietto-Vargas D., Caballero C., Moreno-Gonzalez I., Torres M., Sanchez-Varo R., Ruano D., Vizuete M., Gutierrez A., Vitorica J. Inflammatory response in the hippocampus of PS1M146L/APP751SL mouse model of Alzheimer's disease: age-dependent switch in the microglial phenotype from alternative to classic. J Neurosci. 2008;28:11650–11661. doi: 10.1523/JNEUROSCI.3024-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhong Z., Deane R., Ali Z., Parisi M., Shapovalov Y., O'Banion M.K., Stojanovic K., Sagare A., Boillee S., Cleveland D.W., Zlokovic B.V. ALS-causing SOD1 mutants generate vascular changes prior to motor neuron degeneration. Nat Neurosci. 2008;11:420–422. doi: 10.1038/nn2073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Henkel J.S., Beers D.R., Wen S., Bowser R., Appel S.H. Decreased mRNA expression of tight junction proteins in lumbar spinal cords of patients with ALS. Neurology. 2009;72:1614–1616. doi: 10.1212/WNL.0b013e3181a41228. [DOI] [PubMed] [Google Scholar]
- 48.Kihira T., Yoshida S., Hironishi M., Miwa H., Okamoto K., Kondo T. Changes in the incidence of amyotrophic lateral sclerosis in Wakayama, Japan. Amyotroph Lateral Scler Other Motor Neuron Disord. 2005;6:155–163. doi: 10.1080/14660820510030031. [DOI] [PubMed] [Google Scholar]
- 49.Kuzuhara S., Kokubo Y. Atypical parkinsonism of Japan: amyotrophic lateral sclerosis-parkinsonism-dementia complex of the Kii peninsula of Japan (Muro disease): an update. Mov Disord. 2005;20:S108–S113. doi: 10.1002/mds.20548. [DOI] [PubMed] [Google Scholar]
- 50.Kihira T., Suzuki A., Kondo T., Wakayama I., Yoshida S., Hasegawa K., Garruto R.M. Immunohistochemical expression of IGF-I and GSK in the spinal cord of Kii and Guamanian ALS patients. Neuropathology. 2009;29:548–558. doi: 10.1111/j.1440-1789.2009.01010.x. [DOI] [PubMed] [Google Scholar]