

Background: PKR regulates many biological processes including stress response.
Results: An activation-loop-phosphorylation bypass mutant of PKR functions without a key dimer-interface residue. The PKR kinase domain together with a kinase-dead partner, but not alone, activates the catalytic function.
Conclusion: Activation loop phosphorylation occurs in cis following dimerization.
Significance: PKR autophosphorylates in the activation loop in cis to initiate the antiviral signaling cascade.
Keywords: Phosphorylation, Protein Kinase RNA (PKR), Protein Kinases, Protein Phosphorylation, Stress Response, Activation Loop, eIF2α
Abstract
Protein kinase R (PKR) functions in a plethora of cellular processes, including viral and cellular stress responses, by phosphorylating the translation initiation factor eIF2α. The minimum requirements for PKR function are homodimerization of its kinase and RNA-binding domains, and autophosphorylation at the residue Thr-446 in a flexible loop called the activation loop. We investigated the interdependence between dimerization and Thr-446 autophosphorylation using the yeast Saccharomyces cerevisiae model system. We showed that an engineered PKR that bypassed the need for Thr-446 autophosphorylation (PKRT446∼P-bypass mutant) could function without a key residue (Asp-266 or Tyr-323) that is essential for PKR dimerization, suggesting that dimerization precedes and stimulates activation loop autophosphorylation. We also showed that the PKRT446∼P-bypass mutant was able to phosphorylate eIF2α even without its RNA-binding domains. These two significant findings reveal that PKR dimerization and activation loop autophosphorylation are mutually exclusive yet interdependent processes. Also, we provide evidence that Thr-446 autophosphorylation during PKR activation occurs in a cis mechanism following dimerization.
Introduction
The kinase domain (KD)2 in most eukaryotic protein kinases remains in an inactive precursor state while associated with another domain in the same protein (intradomain) or with another partner protein (intersubunit)(1). Inactive-to-active transition of the KD requires binding of a specific modulator to the respective domain/or subunit or phosphorylation by an upstream kinase, or both (2–5). The KD then adopts a core catalytic structure composed of a smaller N-terminal lobe (N-lobe) and the larger C-terminal lobe (C-lobe), creating an active site at the interface of these lobes (1). While adopting the core structure, an internal loop called the activation loop is often phosphorylated. The activation loop contains one or two regulatory phospho-acceptor residues that are phosphorylated by another kinase (trans-phosphorylation) or by the kinase per se (autophosphorylation). For example, the protein kinase PDK1 (phosphoinositide-dependent protein kinase 1) trans-phosphorylates the activation loop of PKA (cyclic AMP-dependent protein kinase A) (6), whereas DYRK1 (dual specificity tyrosine-regulated kinase 1) autophosphorylates its own activation loop (7). However, the mechanism of activation loop phosphorylation is still not clear in many kinases.
Regardless of the mechanism of phosphorylation, the phosphorylated activation loop changes its conformation and bridges with both N- and C-lobes of the KD, thus adopting a catalytically competent conformation for substrate phosphorylation. Like a typical KD, the KD of PKR is activated by autophosphorylation in the activation loop (8–10). In PKR, two double-stranded RNA-binding domains (RBDs) at the N terminus serve as docking sites for virally produced dsRNA (11, 12) or PKR-activating protein PACT/RAX (13, 14). Binding of dsRNA or PACT/RAX to these RBDs releases the intramolecular auto-inhibition and brings two PKR molecules together (15–19). Such effector-induced PKR dimerization promotes autophosphorylation at several residues, especially the residue Thr-446 (8, 20, 21). The phosphorylated KD then phosphorylates the translation initiation factor eIF2α (9, 22–24) or nuclear factors NFAR1/2 (25) and elicits antiviral defense response, in part, by inhibiting protein synthesis (26), activating NFκB (27), and/or enhancing interferon production (28).
X-ray crystal structures have been resolved for the truncated PKR lacking the RBDs in apo form (i.e. K296R mutant) and in a complex with eIF2α (29). Several mechanistic details of PKR activation have been inferred from these structures although both are inactive under physiological conditions (20). In both structures, the KDs exist as dimers in which protomers associate with each other in a back-to-back orientation so that the catalytic sites face outward. We analyzed these structures and observed that the overall architecture of the apo-KD dimer was similar to the KD dimer complexed with eIF2α except at the region of the activation loop. This loop in the eIF2α-bound form adopts an extended conformation with a phosphorylated residue Thr-446 (Fig. 1A), whereas it remains largely unstructured in the apo form (Fig. 1A). This structural difference indicates that a Thr-446 phosphorylation-dependent conformational transition must occur within the activation loop during PKR activation. Previously, we have shown that Thr-446 autophosphorylation is directly related to PKR activation and coupled with KD dimerization (20, 30). Such coupled processes lead us to propose three different possible models for Thr-446 autophosphorylation (20). In the first model, the dimers phosphorylate activation loops on other dimers (trans-interdimer). Second, each protomer in a dimer phosphorylates its own activation loop (cis-intradimer). Third, one protomer in a dimer phosphorylates the activation loop of its partner (trans-intradimer). Several additional questions about the PKR activation mechanism remain unclear: what are the exact mechanisms and roles of Thr-446 autophosphorylation? How does dimerization promote Thr-446 autophosphorylation?
FIGURE 1.

Functional coupling between the phosphorylated activation loop and the helix-αC. A, analyses of PKR KD structures. The structural coordinates of PKR-KD-K296R (PDB ID: 3UIU) and PKR- KD bound to AMP-PNP and eIF2α (PDB ID: 2A19) were aligned in a single PyMol file by pairwise alignment. Proteins of the PKR-KD (gray) and the PKR-KD-K296R (green) are represented as ribbons. For clarity, the entire structure of eIF2α and several structural elements in PKR are omitted. The conserved residues at the helix-αC (E308, R307, and K304), β-strand 3 (β-3, K296), hinge (E367), activation loop (T446), and catalytic loop (R413 and D414) are shown. The activation loop (region between the DFG and SPE motifs) is colored in purple. The phosphate moiety (red) of the phosphorylated Thr-446 salt-bridges (thin dotted line in gray) with the residue Arg-413 from the catalytic loop and residues Lys-304 and Arg-307 from the helix-αC. The thick purple dotted line in the PKR-KD-K296R structure represents the unresolved portion of the activation loop. The K296R mutation is shaded yellow. B, in vivo analysis of PKR mutants by growth in yeast. The yeast strain H17 (eIF2B-desensitive) expressing WT PKR or indicated mutants were serially diluted and spotted on SD and SGal media. The Slg− phenotype on SGal medium indicates the functionality of PKR alleles. C, in vivo analysis of PKR and eIF2α phosphorylation. Whole cell extracts were prepared from yeast cells indicated in B and subjected to Western blot analysis using antibodies of phosphorylated eIF2α (eIF2α∼P), eIF2α, and phosphorylated PKR (PKR∼P) and PKR.
We showed that an engineered PKR that bypassed the need for Thr-446 autophosphorylation could function without a key residue (Asp-266 or Tyr-323) that is essentially required for the PKR dimerization; supporting the conclusion that dimerization precedes and stimulates activation loop autophosphorylation. Then, we showed that the Thr-446 phosphorylation occurs in a mechanism where each protomer within a dimer phosphorylates on its own (cis-intradimer).
EXPERIMENTAL PROCEDURES
Yeast Strains and Growth Condition
Four yeast Saccharomyces cerevisiae strains were used in this study: 1) H2557: MATα ura3-52 leu2-3 leu2-112 gcn2Δ, 2) H17 (eIF2B desensitive): Matα gcn3-102 leu2-3 leu2-112 ura3-52, 3) H1894: MATa ura3-52 leu2-3 leu2-112 trp1-63 gcn2Δ, and 4) MY71 in which chromosomal SUI2 gene in H2557 was replaced by a SUI2-S51A allele. These strains were obtained from Dr. Thomas E. Dever (NICHD, NIH). Synthetic dextrose (SD) minimal medium supplemented with appropriate nutrients was used to maintain the plasmids containing wild type PKR or its derivatives.
Plasmids
A wild-type (WT) Flag-(His)6-tagged PKR was used (9). Mutations were introduced in the WT PKR gene by fusion PCR. WT PKR and its site-directed mutants were expressed from a galactose-inducible promoter built into the vector plasmid p1079 (pEMLyex4) or p2444 (pEMLyex4-based vector with a TRP selectable marker). The plasmids used in this report are listed in the supplemental Table S1.
Western Blot Analysis
Yeast cells were grown in a synthetic dextrose (SD) medium supplemented with appropriate nutrients until the A600 reached 0.6 -0.7. Cells were collected and resuspended in 10% galactose (SGal) medium and allowed to grow for another 4 h at 30 °C. Whole cell extracts were prepared and subjected to Western blot analysis using an appropriate antibody. Four antibodies were used in these studies: 1) a phosphospecific antibody against eIF2α phosphorylated on Ser-51 (gift from Dr. Thomas E. Dever, NICHD, NIH), 2) a Thr-446 phosphospecific antibody of PKR (Cell Signaling), 3) polyclonal antibody of total eIF2α (gift from Dr. Thomas E. Dever, NICHD, NIH), and 4) polyclonal antibody of the C-terminal end of PKR (Cell Signaling).
Protein Expression, Purification, and Kinase Assay
Flag-tagged PKR and its derivatives were introduced into the S51A strain MY71. Yeast cells were then grown with appropriate nutrient in the presence of 10% galactose until the A600 reached 4.0. Cells were harvested, and PKR protein was purified by ANTI-FLAG M2-agarose (Sigma) as described previously (9). The GST-eIF2α (1–180) and GST-eIF2α-S51A (1–180) fusion constructs were introduced in Escherichia coli BL21 (DE3) plus cells (NOVAGEN). A culture of these cells (A600 = 0.5–0.8) was induced with 0.5 mm IPTG (isopropyl-β-d-thio-galactoside) for 6 h at 37 °C. Cells were collected, and the recombinant GST-eIF2α fusion protein was purified by glutathione-agarose resin (Roche), using the standard manufacturer's protocol. Phosphorylation of eIF2α by PKR was performed in a kinase buffer containing 20 mm Tris-HCl (pH 8.0), 50 mm KCl, 25 mm MgCl2, and 1 μm PMSF (phenylmethylsulfonyl fluoride) with 10 μCi of [γ-33P]ATP. Reactions were quenched after 20 min by addition of 2× SDS gel loading buffer, and the reaction products were separated by SDS-PAGE. The gel was stained with Coomassie blue, and the dried gel was subjected to autoradiography.
Structure Analysis
We analyzed protein structure using computer software PyMol (54).
RESULTS
Functional Coupling between the Phosphorylated Activation Loop and the Helix-αC of PKR
To gain an insight into how PKR transitions from an inactive-to-active state, we analyzed two x-ray crystal structures of the PKR-KD: 1) one is complexed with eIF2α (PDB ID: 2A1A) (29) and 2) the other is in the inactive apo form (K296R mutant, PDB ID: 3U1U). In both structures, each kinase domain is folded into a bi-lobal structure, and two kinase domains orient in parallel to form a back-to-back dimer. We observed that these two KD dimers superimpose well on each other (root mean square deviation = 1.54; supplemental Fig. S1), suggesting that the overall architecture of these proteins is similar. However, a prominent difference was observed in the region of activation loop (residues from Leu-435 to Lys-449) (Fig. 1A). In the apo-form, this loop remains largely unresolved and only three residues Val-Thr-Ser436–438 have been resolved as a β-strand. In the PKR-KD bound to eIF2α, the activation loop adopts an extended conformation with the residue Thr-446 being phosphorylated. These structural differences suggest that, even though the activation loop is unresolved in PDB 3U1U, the inactive-to-active transition likely requires a movement of the activation loop, as observed in numerous other protein kinases (2) so that the substrate, like eIF2α, can bind and be phosphorylated.
The structural analyses further revealed that two helix-αC residues Lys-304 and Arg-307 co-ordinate with the phosphate moiety of Thr-446 in the activation loop and the spatial positions of the helix-αC element remain essentially unchanged in both structures (Fig. 1A). This indicates that the inactive-to-active catalytic transition might not require re-positioning of the helix-αC, unlike what has been observed in most active protein kinase domains (3). However, a coupling between the helix-αC and the phosphorylated activation loop might be crucial. To determine whether these prominent structural interactions between the helix-αC and the phosphorylated activation loop are likely to be physiologically relevant, we replaced residues Lys-304 with alanine and Arg-307 with alanine or lysine. The alanine substitution was expected to reduce the PKR function, whereas the more conservative lysine substitution might be able to retain the needed hydrogen bond.
We expressed the wild-type (WT) human PKR and its mutants K304A, R307A, and R307K in yeast Saccharomyces cerevisiae from a galactose-inducible promoter. WT PKR in yeast cells expressed poorly from a galactose-inducible promoter when grown on a synthetic dextrose (SD) medium (Fig. 1B). The poor PKR expression did not affect regular cell function and physiological processes, and a normal yeast growth phenotype was observed (Fig. 1B, SD). On a synthetic galactose medium (SGal), PKR expression was induced. This induction interfered with normal cell functions, resulting in a slow-growth phenotype (Slg−) in an eIF2B-desensitive strain (Fig. 1B, SGal, column 1) because PKR excessively phosphorylated eIF2α, leading to partial inhibition of the eIF2B function (20). However, expression of a catalytically inactive PKR-K296R mutant showed no growth defect (column 2). Whole cell extracts from those cells were subjected to Western blot analyses, which showed that both WT and K296R mutant proteins were expressed under inducible conditions (Fig. 1C, PKR). Phosphorylations were observed on the residues PKR-T446 (Fig. 1C, PKR∼P lane) and eIF2α-Ser51 (eIF2α∼P, lane 1) in cells expressing WT PKR, but not in cells expressing K296R mutant (PKR∼P, lane 2 and eIF2α∼P, lane 2). These data confirmed that the Slg− phenotype was correlated with eIF2α phosphorylation and that the K296R substitution blocks both PKR and eIF2α phosphorylation, restoring rapid yeast growth (see Refs. 8–10 for other PKR assays in yeast where PKR induction causes a lethal phenotype).
An alanine substitution of the phosphate acceptor residue Thr-446 suppressed the Slg− phenotype (Fig. 1B, SGal, T446A, column 6) and reduced eIF2α phosphorylation (Fig. 1C, T446A, lane 6), supporting our previous observation that residue Thr-446 is a physiologically relevant phosphorylation site. Like the T446A substitution, the alanine substitution of residue Arg-307 (R307A), which was designed to destroy the observed salt-bridge interaction between the helix-αC and the activation loop (Fig. 1A), suppressed the Slg− phenotype (Fig. 1B, SGal, R307A, column 3) and decreased both Thr-446 and eIF2α phosphorylations (Fig. 1C, eIF2α∼P, lane 3). These data suggested that a productive interaction between the residue Arg-307 and the phosphorylated Thr-446 was required for PKR function. Unlike PKR-R307A, the PKR-R307K and PKR-K304A mutants retained the Slg− phenotype (Fig. 1B, SGal, columns 4 and 5), autophosphorylated on the residue Thr-446 (Fig. 1C, PKR∼P, lanes 4 and 5) and phosphorylated eIF2α (Fig. 1C, eIF2α∼P, lanes 4 and 5), indicating that the lysine substitution at position 307 enables formation of a salt-bridge with phosphorylated Thr-446, and that the interaction between residues Lys-304 and phosphorylated Thr-446 are not critical for PKR activity. Collectively, these structure-based mutational analyses reveal that a productive interaction between the Thr-446-phosphorylated activation loop and the helix-αC is important for the PKR catalytic function.
Thr-446 Phosphorylation Occurs in a cis Mechanism following Dimerization
To determine the mechanisms of Thr-446 autophosphorylation during the KD dimerization, we used a LIM-Ldb dimerization system (31). In this system, only the kinase domain of PKR was expressed in yeast cells as a monomer or forced to form a homodimer. The KD (residues 258 to 551) was fused to the Xenopus protein LIM domain (residues 1 to 58) and to LIM-domain binding protein Ldb (residues 290 to 350) creating LIM-PKRKD and Ldb-PKRKD fusion proteins, respectively. Yeast cells expressing either LIM-PKRKD (Fig. 2, yeast growth, row 2) or Ldb-PKRKD (row 3) alone, or neither one (row 1) showed no growth defect on SD and SGal media. However, expression of LIM- and Ldb-PKRKD fusion proteins together was lethal on SGal medium (Fig. 2, yeast growth, SGal, row 4). The lethality was suppressed in an isogenic eIF2α-S51A strain (Fig. 2A, right panel), supporting the conclusion that the lethality was due to activation of the PKR that phosphorylated eIF2α (31).
FIGURE 2.

Thr-446 phosphorylation occurs in a cis mechanism following dimerization. A, heterologous dimerization domains activate the PKR KD. The LIM-PKRKD fusion gene in the plasmid pC901 with a URA3 selectable marker and the Ldb-PKRKD fusion gene in the plasmid pC903 with a TRP selectable marker were introduced into WT yeast strain H2557 as well as S51A strain. The sign (+) means the presence, whereas the sign (−) means the absence of pC901/pC903 plasmids but contains the respective vector plasmid. The indicated D414A mutation was introduced in to the LIM-PKRKD fusion construct. Transformants were tested for growth on SD and SGal media. The PKR was inactive when the yeast grew on SGal medium, but active if the yeast did not grow. B, analysis of Thr-446 autophosphorylation and eIF2α phosphorylation. Yeast strains expressing indicated LIM- PKRKD (WT or D414A) and Ldb-PKRKD fusion protein were grown in the SGal medium and whole cell extracts were subjected to Western blot analysis using either a Thr-446-phosphospecific (T446∼P) or Ser-51 phosphospecific (eIF2α∼P) antibody followed by a polyclonal antibody of PKR or eIF2α. C, models represent mono- or dimeric form of LIM- and Ldb-PKRKD fusion proteins. The bi-lobal PKR-KD is shown in gray whereas the activation loops in purple solid lines with a phosphorylated residue (red circle). The inactive form of the kinase domain is colored orange.
The activation loop phosphorylation is a molecular marker of the active state of a protein kinase domain, including PKR (2, 20). Thus, we tested the phosphorylation status of the activation loops in LIM- and Ldb-PKRKD fusion proteins. Whole cell extracts from those yeast cells were subjected to Western blot analysis using a Thr-446-phosphospecific antibody and a polyclonal antibody to total PKR (Fig. 2B). The Ldb-PKRKD fusion protein expressed better than the LIM-PKRKD fusion protein (Fig. 2B, lower panel, compare lanes 2 and 3), as a monomer as depicted in Fig. 2C. The residue Thr-446 in either fusion protein was un-phosphorylated (Fig. 2B, lanes 2 and 3; compare with total PKR at the lower panel), suggesting that Thr-446 autophosphorylation does not occur spontaneously. In contrast, Thr-446 was phosphorylated in both LIM- and Ldb-PKRKD fusion proteins was phosphorylated when two proteins were expressed together in a same cell (Fig. 2B, lane 4 and in the right panel, lane 2), suggesting that dimerization resulting from LIM·Ldb interaction (Fig. 2C) resulted in Thr-446 phosphorylation.
We propose three models that might explain coupling of PKR dimerization and Thr-446 phosphorylation: (I) trans-intradimer, (II) cis-intradimer, or (III) trans-interdimer. To test the first two models, we introduced a kinase-inactivating mutation in the LIM-PKRKD (D414A mutation in the conserved HRD414 motif, data not shown) and expressed it alone or co-expressed with the Ldb-PKRKD. Expression of LIM-PKRKD-D414A alone did not affect yeast cell growth on SD and SGal media (Fig. 2A, yeast growth, SD, and SGal, row 5), whereas co-expression with the Ldb-PKRKD fusion protein exhibited an Slg− phenotype on SGal medium (Fig. 2A, rows 6), but not in the isogenic eIF2α-S51A strain (phosphorylation site Ser-51 mutated to an Ala, Fig. 2A, right panel), suggesting that the reduced growth was due to eIF2α phosphorylation. These data suggested that the LIM-PKRKD-D414A derivative retained the ability to activate the partner domain, even though it lacked the kinase activity. The residue Thr-446 in LIM-PKRKD-D414A fusion protein (Fig. 2B, Western blots, right panel, LIM-PKRKD∼T446P, lane 3) remained almost un-phosphorylated, ruling out the possibility of a trans-intradimer model of Thr-446 autophosphorylation. Phosphorylation of the residue Thr-446 in Ldb-PKRKD protein was observed when co-expressed with the LIM-PKRKD-D414A partner (Fig. 2B, Western blots, right panel, Ldb-PKRKD∼T446P, compare lanes 1 and 3). Consistent with the Thr-446 autophosphorylation, eIF2α was phosphorylated in yeast cells expressing LIM-PKRKD together with Ldb-PKRKD or Ldb-PKRKD-D414A (Fig. 2B, Western blots, right panel, eIF2α∼P, lanes 2 and 3). These results suggest that phosphorylation on residue Thr-446 in Ldb-PKRKD protein occurs in a cis mechanism following dimerization (see Fig. 2C).
Thr-446 Phosphorylation Does Not Occur in a trans Mechanism
To determine whether or not Thr-446 phosphorylation occurs in a trans-interdimer mechanism, a homodimeric GST-PKRKD fusion protein (a functional homodimer, see Ref. 31) was mixed with a kinase-dead PKR-K296R protein in a reaction buffer (Fig. 3A, Anti-PKR, 1×). Then, the phosphorylation status of the activation loops of both proteins was examined by Western blot analysis using a Thr-446 phosphospecific antibody of PKR (Fig. 3A, Anti-T446∼P). The same experiment was repeated with 5-fold higher protein concentration to examine any positive effect on Thr-446 phosphorylation (Fig. 3A, Anti-PKR, 5×). The residue Thr-446 was phosphorylated in the GST-PKRKD fusion protein in the presence or absence of PKR-K296R protein (Fig. 3A, GST-PKRKD∼P, lanes 1 and 3; and also in 5×, lanes 4 and 6); however, the residue Thr-446 was un-phosphorylated in the PKR-K296R protein (lanes 2, and also in 5× lane 5) even when it was mixed with GST-PKRKD fusion protein (lane 3 and in 5× lane 6); arguing against the trans mechanism of Thr-446 phosphorylation.
FIGURE 3.

The Thr-446 phosphorylation does not occur in trans mechanism. A, in vitro analysis of PKR autophosphorylation. Purified PKR-K296R and GST-PKRKD fusion proteins were incubated in a kinase reaction buffer. The reaction products were then separated by SDS-PAGE and subjected to Western blot analyses using phosphospecific antibody of Thr-446 followed by a polyclonal antibody of PKR. B, analysis of Thr-446 phosphorylation. GST-PKRKD co-expressed with WT PKR or PKR-K296R mutant in yeast. Whole cell extracts were prepared from these cells and subjected to Western blot analysis using a Thr-446 phosphospecific antibody of PKR (T446∼P). The membrane was stripped and re-probed with a polyclonal antibody of PKR.
To confirm these results under a more physiological cellular environment, we co-expressed the GST-PKRKD fusion protein and WT PKR or PKR-K296R mutant in yeast. Whole cell extracts were subjected to Western blot analysis using a Thr-446 phosphospecific antibody of PKR followed by an antibody against PKR. The residue Thr-446 in WT PKR (Fig. 3B, Anti-T446∼P, lane 1) or GST-PKRKD fusion protein (lanes 1 and 2) was phosphorylated. However, the residue Thr-446 in the PKR-K296R protein (Fig. 3B, lane 2) was not phosphorylated. Expressions of both of these proteins (Fig. 3B, Anti-PKR) were nearly equal, indicating that the trans phosphorylation of the Thr-446 residue is not a likely mechanism.
A PKRphk1 Chimera Bypasses the Requirement for Activation Loop Phosphorylation
PKR activation requires both dimerization and Thr-446 autophosphorylation. To further understand whether these processes are dependent, independent, and interdependent, we engineered the PKR gene so that it could bypass the requirement for activation loop phosphorylation. Initially, two PKR mutants (PKR-T446D and PKR-T446E) were created with the expectation that aspartate/glutamate could function as a phospho-mimetic substitution. As shown in Fig. 4A, yeast cells expressing the PKR-T446D mutant (column 4), like PKR-K296R (column 2), and PKR-T446A mutants (column 3), grew well on SGal medium. Western blot analysis showed that phosphorylation of eIF2α was substantially reduced in those cells (Fig. 4B, eIF2α∼P, lanes 2, 3, and 4), although mutant PKR proteins expressed comparatively better than WT (Fig. 4B, Western blots, compare lanes 1–4, PKR). These results indicated that the ability of PKR-T446D or PKR-T446E (data not shown) mutant to phosphorylate eIF2α is substantially lower than WT PKR. Collectively, we conclude that phospho-mimetic aspartate or glutamate does not bypass the requirement for activation loop phosphorylation.
FIGURE 4.

Functional substitution of activation loop residues of PKR by residues from the activation loop of phosphorylase kinase 1 (Phk1). A, in vivo analysis of PKR mutants by growth in yeast. The yeast strain H17 (eIF2B desensitive) or MY71 (eIF2α-S51A) harboring indicated WT PKR and its derivatives were serially diluted and spotted on SD and SGal media. B, in vivo analysis of eIF2α phosphorylation by PKR mutants. Whole cell extracts were prepared from yeast cells indicated in A and subjected to Western blot analysis using phosphospecific antibodies against Ser-51 (eIF2α∼P). The membrane was stripped and re-probed with a polyclonal antibody of eIF2α and PKR. C, in vitro analysis of eIF2α phosphorylation by PKR mutants. Purified PKR protein (WT, K296R, or PKRphk1) was mixed with the recombinant eIF2α and [γ-33P]ATP in a reaction buffer for 10 min. The reaction products were then separated using SDS-PAGE. The gel was stained, dried, and subjected to autoradiography to monitor the incorporation of 33P in eIF2α proteins. D, catalytic function of PKRphk1 chimera requires an active interaction between the activation loop and the helix-αC. Left panels, eIF2B desensitive strain H17 expressing indicated PKR mutants were tested for growth on SD and SGal media. Right panels, yeast cells were grown in the presence of galactose (10%) and harvested after 2 h. Whole cell extracts were then prepared and subjected to Western blot analyses using antibodies of phosphorylated eIF2α (eIF2α∼P), eIF2α, and PKR.
Then we attempted to replace the activation loop by a loop that would mimic the phosphorylation state. Thus, we performed a global search of kinase domain sequences and structures and identified kinases Phk1 (phosphorylase kinase 1) (32) and Chk1 (checkpoint kinase 1) (33) in which the activation loop elements are devoid of any regulatory phospho-acceptor residues (Fig. 5A). Moreover, we observed that the structure of PKR kinase domain (PDB ID: 2A1A) superimposed well onto the structure of Phk1 kinase domain (PDB ID: 2PHK, Fig. 5B) as well as onto the structure of Chk1 kinase domain (PDB ID: 1IA8), and that the phosphorylated residue Thr-446 in PKR aligned with the phospho-mimetic residue E182 in Phk1, whereas the helix-αC residue R307 in PKR aligned with K72 (Lys-72) in Phk1 (Fig. 5B) or K54 (Lys-54) in Chk1 (not shown). These structural analyses indicated that activation loop residues of Phk1 or Chk1 might functionally substitute the corresponding residues of PKR.
FIGURE 5.
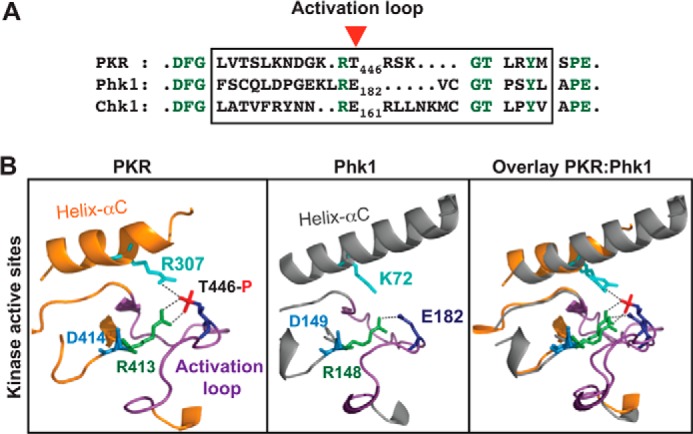
Kinase domains of PKR and Phk1 are structurally similar. A, residue E182 in Phk1 corresponds to the residue Thr-446 in PKR. The activation loop residues (boxed) of PKR, Phk1, and Chk1 were compared by pairwise alignment. The conserved residues are colored in green. A red arrowhead shows Thr-446 of PKR. B, kinase domains of PKR and Phk1 superimpose well on each other. Structural coordinates of the kinase domains of PKR (PDB ID: 2A1A) and Phk1 (PDB ID: 2PHK) were superimposed, using computer software PyMol. Proteins are shown as a ribbon presentation. For clarity, several structural elements are omitted. Only the helix-αC (colored orange in PKR whereas gray in Phk1) and the activation loop (colored purple both in PKR and Phk1) in the active site regions are shown. The conserved Arg-413 of the RD motif (R413 colored green) contacts with phosphorylated Thr-446 in PKR, whereas the corresponding Arg-148 in Phk1 (R148 colored green) contacts with phospho-mimetic E182. The catalytic base aspartate (D414 in PKR or D149 in Phk1) is colored in blue.
We replaced the activation loop residues of PKR by the respective residues from Phk1 or Chk1, generating PKRphk1 and PKRchk1chimera, and expressed these chimeras in yeast. Yeast cell expressing PKRphk1, but not PKRchk1, grew slowly like WT PKR on SGal medium (Fig. 4A, compare columns 1, 5, and 6). Western blot analysis showed that the slow-growth correlated with eIF2α phosphorylation (Fig. 4B, Western blots, eIF2α∼P, lanes 5 and 6). These results indicated that activation loop residues of Phk1, but not of Chk1, functionally substituted the activation loop residues of PKR. To confirm the in vivo results, we performed an in vitro kinase assay to test the ability of PKRphk1 to phosphorylate eIF2α. Purified PKRphk1 protein was mixed with the recombinant eIF2α and [γ-33P]ATP in a kinase reaction buffer. In parallel kinase assays that were performed under the same conditions, we used WT PKR and K296R protein (Fig. 4C, stain). As shown in Fig. 4C, WT PKR (lane 1) and the PKRphk1 chimera (lane 4) both autophosphorylated (PKR∼P) and phosphorylated the substrate eIF2α on the residue Ser-51 (eIF2α∼P), whereas the PKR-K296R protein neither autophosphorylated nor phosphorylated eIF2α (lane 3), confirming that the PKRPhk1 chimera is a functional eIF2α kinase.
To address the next obvious question of whether or not the PKRphk1 chimera required an active interaction between the activation loop and the helix-αC, in particular the interaction between the residues Glu-182 and Arg-307 (Fig. 5), we substituted each residue in PKRphk1 by alanine and expressed mutants PKRphk1E182A and PKRphk1R307A in yeast. Yeast cells expressing PKRphk1E182A (Fig. 4D, column 2) or PKRphk1R307A (column 4) grew rapidly like the PKR-R307A mutant (column 3) on SGal medium. Western blot analyses showed that rapid growth correlated with reduced eIF2α phosphorylation (Fig. 4D, Western blots, compare lanes 1–4) though expression of these PKR derivatives was better than WT (Fig. 4D, Western blot, PKR, lanes 1–4). These results suggest that, in the PKRPhk1 chimera, the positively charged residue Arg-307 in helix-αC and the phospho-mimetic glutamate in the activation loop (Glu-182 corresponding to Thr-446) play important roles in the PKR catalytic function. The residue Phk1-E182 aligns with the phosphorylated Thr-446 of PKR (Fig. 5); thus it appears that the residue Glu-182 in the PKRphk1 activation loop maintains the salt bridge interaction with the helix-αC.
Dimerization Precedes and Stimulates Activation Loop Phosphorylation
Though PKR has been reported to exist as a monomer (17, 34), numerous studies have challenged whether PKR functions as a monomeric protein. These studies include: 1) PKR forms a dimer at higher concentration (34) and 2) PKR exists as a monomer-dimer equilibrium that is enhanced by autophosphorylation (35). Furthermore, the crystal structure reveals that the PKR-KD forms a dimer by a back-to-back interaction between two N-lobes where several intra- and inter-molecular salt-bridges and hydrogen bonds stabilize the interaction. The prominent interactions include a salt-bridge between residues Arg-262 and Asp-266 and a hydrogen bond network between residues Asp-289, Tyr-293, and Tyr-323 (Fig. 6A). We have shown previously that mutations of residues Arg-262 and Asp-266 reduce the ability of PKR both to autophosphorylate Thr-446 and to phosphorylate eIF2α, whereas reciprocal exchange of these two residues restores both functions (30). We conclude that the KD dimerization is coupled with Thr-446 phosphorylation in the activation loop. But, it is still unclear how these processes are inter-related.
FIGURE 6.

Salt-bridge interactions at the dimer interface are partially important for PKRphk1 function. A, intra- and inter-molecular salt-bridge interactions at the PKR-KD dimer interface. A dimer of the PKR kinase domains (PDB ID: 2A19) is represented as a ribbon diagram. For clarity, only the interacting N-terminal lobes are shown. Several intra- and inter-molecular hydrophobic and salt-bridge interactions stabilize the dimer interface between protomer 1 (gray) and protomer 2 (green). Only two prominent salt-bridge interactions (R262·D266 and D289·Y293·Y323) are zoomed in a separate box. B, in vivo analysis of PKR mutants by growth in yeast and by eIF2α phosphorylation. The gcn2Δ yeast strains harboring indicated PKR mutants were tested for growth on SD and SGal media (upper panels). Whole cell extracts from these cells were subjected to Western blot analyses using antibodies of phosphorylated eIF2α (eIF2α∼P) and eIF2α.
We used the PKRphk1 chimera to understand the importance of dimerization on Thr-446 autophosphorylation. We hypothesized that PKRphk1 should be non-functional or partially functional if the RBD-mediated KD dimerization was required for Thr-446 autophosphorylation. To test this, residues Asp-266 and Tyr-323 in the PKRphk1 chimera were individually substituted to Arg and Ala, generating PKRphk1D266R and PKRphk1Y323A mutants, respectively. We then expressed these PKR derivatives in yeast, tested for growth on SGal medium, and examined eIF2α phosphorylation. The results were then compared with cells expressing WT PKR and PKR mutant K296R, D266R, or Y323A (Fig. 6B).
As expected, expression of WT PKR exhibited a lethal phenotype (Fig. 6B, SGal, column 1). The lethality was suppressed in cells expressing PKR mutants K296R, D266R or Y323A (Fig. 6B, SGal, columns 2, 3, and 4). Whole cell extracts from these cells were subjected to Western blot analyses using antibodies against phosphorylated eIF2α (eIF2α∼P) and eIF2α. Western blot showed that eIF2α was phosphorylated in cells expressing WT PKR (Fig. 7B, Western blots, eIF2α∼P, column 1), but not in cells expressing K296R mutant (Fig. 6B, Western blots, eIF2α∼P, column 2). Phosphorylation of eIF2α was substantially impaired in cells expressing PKR-D266R and PKR-Y323A mutants (Fig. 6B, Western blot, eIF2α∼P, columns 3 and 4). These results together with our previous observation that the reduced eIF2α phosphorylation was correlated with the reduced Thr-446 phosphorylation (20) suggest that KD dimerization is required for the catalytic activation of PKR.
FIGURE 7.
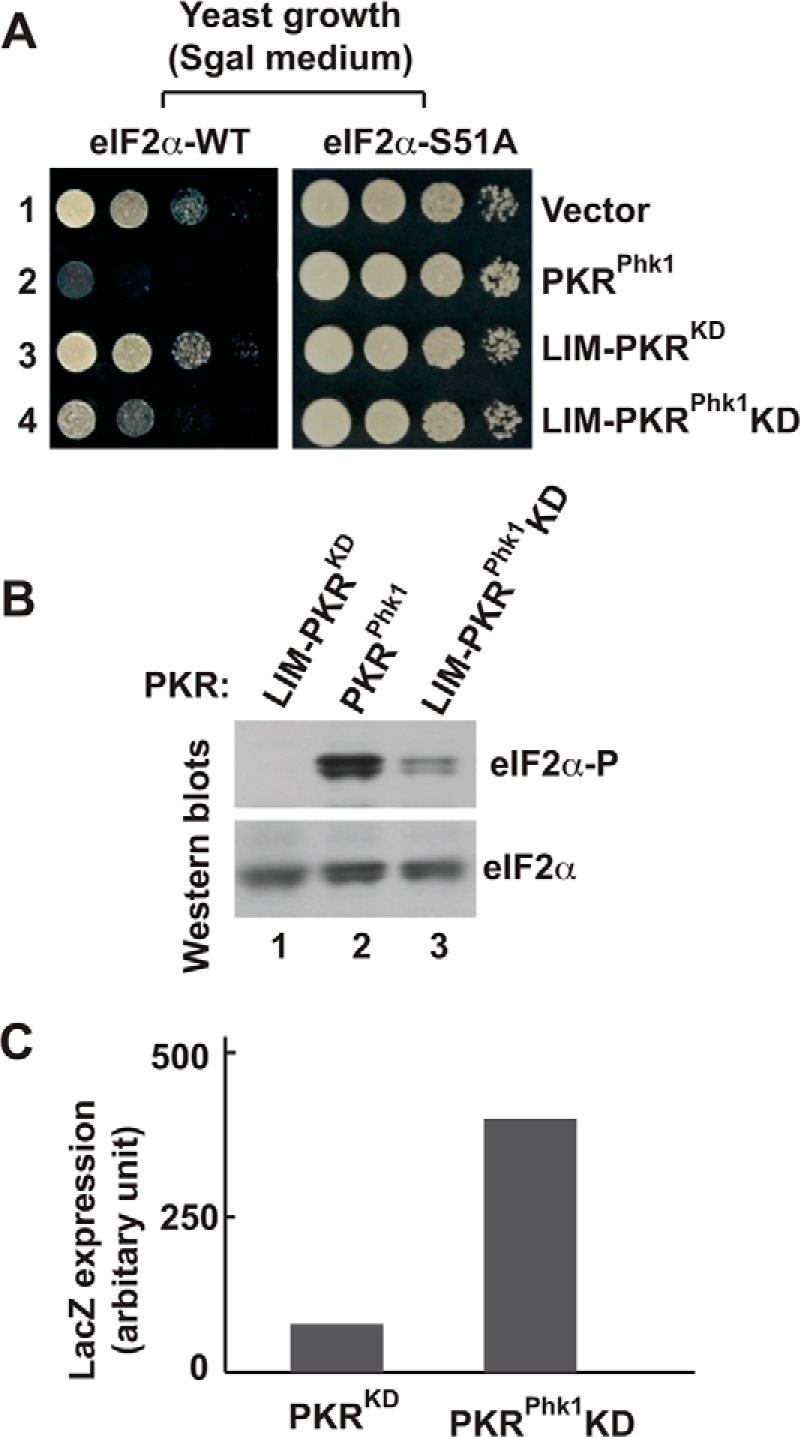
The kinase domain of PKRphk1 chimera is partially active. A, in vivo analysis of PKR mutants by growth in yeast. The WT (eIF2α-WT) and S51A (eIF2α-S51A) yeast strains harboring indicated PKR mutants were serially diluted and tested for growth on SGal medium. B, PKRKD-Phk1 phosphorylates eIF2α. Whole cell extracts from cells expressing indicated PKR derivatives were subjected to Western blot analyses using antibodies of phosphorylated eIF2α (eIF2α∼P) and eIF2α. C, PKRKD-Phk1 can activate the GCN4 translational control. Whole cell extracts of yeast cells carrying a GCN4 LacZ reporter plasmid p180 were prepared and β-gal activities were monitored as described previously (36).
Like WT PKR, expression of PKRphk1 exhibited a lethal phenotype on SGal and resulted in excessive phosphorylation of eIF2α (Fig. 6B, Western blot, eIF2α∼P, column 5). Expression of PKRphk1D266R or PKRphk1Y323A derivative increased the yeast growth (Fig. 6B, SGal, columns 6 and 7) when compared with PKRphk1 (column 5). The increased growth was correlated with a decreased eIF2α phosphorylation (Fig. 6B, Western blots, eIF2α∼P, compare columns 5, 6, and 7), but increased eIF2α phosphorylation when compared with PKR-D266R or PKR-Y323A derivative (compare columns 3, 4, 6, and 7). These increased eIF2α phosphorylation indicated the PKRphk1D266R or PKRphk1Y323A became activated without key dimer interface residues. Collectively, these data suggest that the RBD-mediated dimerization was partially important for PKR activation, even though activation loop autophosphorylation was bypassed. Furthermore, these data suggest that PKR dimerization precedes and stimulates the activation loop phosphorylation.
The Kinase Domain of PKRphk1 Functions without RNA Binding Domains
Next, to test the hypothesis that the kinase domain of PKRphk1 should be functional without its N-terminal RNA binding domains, we expressed only the kinase domain of the PKRphk1 in yeast, tested cell growth on SGal medium and examined eIF2α phosphorylation in those cells. Expression of LIM-PKRphk1-KD fusion (Fig. 7, eIF2α-WT, lane 4), like PKRphk1 (lane 2), substantially reduced yeast growth on SGal medium. However, no growth defect was observed in an isogenic S51A strain (Fig. 7, eIF2α-S51A), suggesting that growth reduction was due to eIF2α phosphorylation. As expected, expression of the PKR-KD only like a vector control (lane 1) showed no growth defect in both WT and S51A yeast strains (Fig. 7, lanes 1 and 4, see also Fig. 2A, column 2). As expected, Western blot analysis showed that PKRphk1 phosphorylated eIF2α (Fig. 7B, lane 2) and no phosphorylation of eIF2α was observed in yeast expressing PKR-KD (lane 1). Interestingly, we observed that the PKRphk1KD phosphorylated eIF2α (Fig. 7B, lane 3), suggesting that PKRphk1KD bypassed the requirement of absolute requirement for KD dimerization for its catalytic activity.
To test further that the PKRphk1KD actively phosphorylated eIF2α, yeast cell expressing PKR-KD or PKRphk1KD was transformed with a GCN4 LacZ reporter plasmid p180 (36). Transformants were grown till A600 reached ∼0.6. Then, cells were harvested, resuspended on a medium containing 10% galactose, and grown for another 6 h. Whole cell extracts were prepared, and β-gal activity was monitored (Fig. 7C). The LacZ expression was not induced in cells expressing PKR-KD, but was significantly induced in PKRphk1KD cell (5-fold), confirming that the PKRphk1KD is an active kinase and can activate the GCN4 translational control in yeast. Collectively, our data suggest that dimerization and KD activation are mutually exclusive yet interdependent events.
DISCUSSION
The mechanism of Thr-446 phosphorylation in the activation loop has been determined by expressing the PKRKD in yeast cells using a heterologous dimerization system. We showed that the PKRKD fused to LIM or Ldb is inactive when either one is present alone. When these fusion proteins are present together in the same cell, the PKRKD becomes functional because the LIM and Ldb domains form a heterodimer that holds two kinase domains together. Separation of LIM- and Ldb-PKRKD fusion proteins showed that each one was phosphorylated on the residue Thr-446, supporting that dimerization is required for Thr-446 autophosphorylation. Then, we showed that a kinase-dead isoform of LIM-PKRKD was able to activate the catalytic function of the partner Ldb-PKRKD fusion protein. Consistently, we showed that the residue Thr-446 in the Ldb-PKRKD, but not the kinase-dead isoform of LIM-PKRKD in the heterodimer, was phosphorylated. These results suggest that Thr-446 autophosphorylation does not occur via the trans-intradimer model, and must have occurred in mechanisms where 1) each protomer within a dimer phosphorylates on its own (cis-intradimer) and/or 2) an active partner in a dimer phosphorylates a monomeric protein or a protomer of the dimeric protein (trans-interdimer). To determine the trans-interdimer model of Thr-446 autophosphorylation, a homodimeric form of PKR (GST-PKRKD) was mixed with a kinase-dead PKR-K296R mutant. Absence of any detectable Thr-446 phosphorylation in the PKR-K296R protein (Fig. 3) suggests that Thr-446 phosphorylation in trans is not a likely mechanism.
In our earlier report (20), we ruled out that the cis-intradimer Thr-446 autophosphorylation is not a possible mechanism because the in vitro PKR autophosphorylation reaction shows second order kinetics with respect to PKR concentration (35, 37). The autophosphorylation reaction occurs at multiple residues other than Thr-446, including residues Ser-340, Ser-343, Ser-344, Ser-351, Ser-354, Ser-355, Ser-357, Thr-359, Ser-242, Thr-255, and Thr-258. Alanine mutation of these residues has no or minimum effect on the catalytic function of PKR (38), suggesting that those sites are not physiologically relevant phosphorylation sites. Therefore, we conclude that the in vitro PKR autophosphorylation reaction may not explain adequately the mechanism of Thr-446 phosphorylation.
Dar et al. reported that the PKR kinase domains form a back-to-back dimer (29). If this geometry needs to be maintained in an active PKR protein, the mechanism of trans-intradimer Thr-446 autophosphorylation is not a likely mechanism because a protomer in a dimer would not access the active site of the partner. Supporting this hypothesis, it was shown that the residue Thr-446 of LIM-PKRKD-D414A remained un-phosphorylated in an Ldb·LIM-PKRKD heterodimer (Fig. 2). However, the heterodimer was able to activate the catalytic function of its partner Ldb-PKRKD fusion protein while inducing Thr-446 phosphorylation (Fig. 2), fitting perfectly with the possibility that Thr-446 phosphorylation occurs in a cis mechanism following dimerization.
Although the structure of the PKR holoenzyme is still lacking, it has been shown that the isolated RBDs forms a dumbbell shaped monomer in solution (39). It has also been shown that the RDB2 interacts with the C-lobe of PKR-KD (40). Together with these results, we propose that RBD2 remains bound to the inactive kinase domain until an effector molecule (dsRNA, PACT, or RAX) binds and stimulates its release (Fig. 8). The allosteric interaction between the RBDs and the dsRNA, PACT, or RAX favors a conformation that triggers Thr-446 autophosphorylation in a cis mechanism. Then, PKR autophosphorylates in trans at multiple residues as reported earlier (38) and becomes fully active. The active PKR induces antiviral defense responses and functions in regulatory pathways of a plethora of biological processes, including myogenesis (41), cell cycle progression (42), and inflammation (43).
FIGURE 8.

Maturation and activation of PKR by autophosphorylation reactions. A conceptual model of the inactive PKR in which the RBD2 is bound to the kinase domain (40) and the activation loop (solid purple line) is un-phosphorylated. Binding of dsRNA or PACT (light blue star) to the RBD releases the autoinhibition (40). Consequently, the kinase domains of PKR adopt a favorable dimeric configuration (even with an inactive KD partner shown in gray) that facilitates autophosphorylation on the residue Thr-446 in cis. As shown by dotted lines, the phosphorylated Thr-446 couples with the helix-αC (orange cylinder) and an active closed conformation is achieved. Then PKR phosphorylates other residues in trans (shown in red circles) and becomes fully active.
PKR belongs to a family of eIF2α kinases that also contain GCN2, PERK, and HRI (44). Each of these kinases carries distinct regulatory domains that sense a specific cellular stress. HRI contains heme-binding domains that sense the intracellular heme deprivation (45), PERK contains a lumenal domain that senses the accumulation of unfolded protein in the endoplasmic reticulum (46), and GCN2 contains a HisRS-like (histidyl-tRNA synthetase) domain that senses the metabolite stress (47). Upon sensing stress through the sensor domain, each one activates its kinase domain and phosphorylates eIF2α to down-regulate protein synthesis. Each of these kinases functions as a dimer, and is regulated by autophosphorylation at a conserved threonine (corresponding to Thr-446), located in the activation loop. The kinase domains of PERK, similar to PKR, have been shown to form a dimer in a back-to-back orientation (48). Also, it has been shown that the phosphorylated activation loop in PERK kinase domain salt-bridges with the helix-αC. Thus, we propose that the activation of PERK-KD might resemble PKR-KD activation.
Approximately 10% of protein kinase domains encoded by the human genome lack either an N-lobe, a C-lobe, or one/two canonical kinase motifs (e.g. DFG and RD). They are unable to phosphorylate a substrate and are called pseudokinase domains (49). These pseudokinases positively or negatively regulate the functions of the partner domain (50). For example, a pseudokinase domain of human RNase L positively regulates its cognate RNase domain function (51), whereas a pseudokinase domain JH2 negatively regulates the tyrosine kinase activity of JAK2 (52). We showed that the kinase-dead PKR-D414A isoform (Fig. 2) activated the catalytic function of its partner, reminiscent of the functional interaction between the pseudokinase KSR1 (kinase suppressor of Ras) with Raf, MEK, or ERK (53). Thus, the PKR pseudokinase provides a molecular explanation of a pseudokinase domain function.
Supplementary Material
Acknowledgments
We thank Dr. Thomas E. Dever (NICHD, NIH) for reagents and helpful discussion throughout the project. We thank Profs. David Frick (UW-Milwaukee), Colin Scanes (UW-Milwaukee), and Adam Gaballe (Fred Hutchinson Cancer Research Center, Seattle, WA) for reading the manuscript and for useful suggestions.
This work was supported in part by National Institutes of Health Grant 1R15GM101575 (to M. D.).

This article contains supplemental Table S1 and Fig. S1.
- KD
- kinase domain
- PKR
- protein kinase R
- PDK
- phosphoinositide-dependent protein kinase
- DYRK
- dual specificity tyrosine-regulated kinase
- RBD
- RNA-binding domain
- AMP-PNP
- 5-adenylyl imidodiphosphonate.
REFERENCES
- 1. Hanks S. K., Hunter T. (1995) Protein kinases 6. The eukaryotic protein kinase superfamily: kinase (catalytic) domain structure and classification. FASEB J. 9, 576–596 [PubMed] [Google Scholar]
- 2. Huse M., Kuriyan J. (2002) The conformational plasticity of protein kinases. Cell 109, 275–282 [DOI] [PubMed] [Google Scholar]
- 3. Kornev A. P., Haste N. M., Taylor S. S., Eyck L. F. (2006) Surface comparison of active and inactive protein kinases identifies a conserved activation mechanism. Proc. Natl. Acad. Sci. U.S.A. 103, 17783–17788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Nolen B., Taylor S., Ghosh G. (2004) Regulation of protein kinases; controlling activity through activation segment conformation. Mol. Cell 15, 661–675 [DOI] [PubMed] [Google Scholar]
- 5. Endicott J. A., Noble M. E., Johnson L. N. (2012) The structural basis for control of eukaryotic protein kinases. Annu. Rev. Biochem. 81, 587–613 [DOI] [PubMed] [Google Scholar]
- 6. Biondi R. M., Cheung P. C., Casamayor A., Deak M., Currie R. A., Alessi D. R. (2000) Identification of a pocket in the PDK1 kinase domain that interacts with PIF and the C-terminal residues of PKA. EMBO J. 19, 979–988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lochhead P. A., Sibbet G., Morrice N., Cleghon V. (2005) Activation-loop autophosphorylation is mediated by a novel transitional intermediate form of DYRKs. Cell 121, 925–936 [DOI] [PubMed] [Google Scholar]
- 8. Romano P. R., Garcia-Barrio M. T., Zhang X., Wang Q., Taylor D. R., Zhang F., Herring C., Mathews M. B., Qin J., Hinnebusch A. G. (1998) Autophosphorylation in the activation loop is required for full kinase activity in vivo of human and yeast eukaryotic initiation factor 2α kinases PKR and GCN2. Mol. Cell Biol. 18, 2282–2297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Dey M., Trieselmann B., Locke E. G., Lu J., Cao C., Dar A. C., Krishnamoorthy T., Dong J., Sicheri F., Dever T. E. (2005) PKR and GCN2 kinases and guanine nucleotide exchange factor eukaryotic translation initiation factor 2B (eIF2B) recognize overlapping surfaces on eIF2α. Mol. Cell Biol. 25, 3063–3075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Feng G. S., Chong K., Kumar A., Williams B. R. (1992) Identification of double-stranded RNA-binding domains in the interferon-induced double-stranded RNA-activated p68 kinase. Proc. Natl. Acad. Sci. U.S.A. 89, 5447–5451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lemaire P. A., Anderson E., Lary J., Cole J. L. (2008) Mechanism of PKR Activation by dsRNA. J. Mol. Biol. 381, 351–360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Li S., Peters G. A., Ding K., Zhang X., Qin J., Sen G. C. (2006) Molecular basis for PKR activation by PACT or dsRNA. Proc. Natl. Acad. Sci. U.S.A. 103, 10005–10010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Patel R. C., Sen G. C. (1998) PACT, a protein activator of the interferon-induced protein kinase, PKR. EMBO J. 17, 4379–4390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ito T., Yang M., May W. S. (1999) RAX, a cellular activator for double-stranded RNA-dependent protein kinase during stress signaling. J. Biol. Chem. 274, 15427–15432 [DOI] [PubMed] [Google Scholar]
- 15. Wu S., Kaufman R. J. (1997) A model for the double-stranded RNA (dsRNA)-dependent dimerization and activation of the dsRNA-activated protein kinase PKR. J. Biol. Chem. 272, 1291–1296 [DOI] [PubMed] [Google Scholar]
- 16. Patel R. C., Stanton P., McMillan N. M., Williams B. R., Sen G. C. (1995) The interferon-inducible double-stranded RNA-activated protein kinase self-associates in vitro and in vivo. Proc. Natl. Acad. Sci. U.S.A. 92, 8283–8287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Langland J. O., Jacobs B. L. (1992) Cytosolic double-stranded RNA-dependent protein kinase is likely a dimer of partially phosphorylated Mr = 66,000 subunits. J. Biol. Chem. 267, 10729–10736 [PubMed] [Google Scholar]
- 18. Vattem K. M., Staschke K. A., Zhu S., Wek R. C. (2001) Inhibitory sequences in the N-terminus of the double-stranded-RNA-dependent protein kinase, PKR, are important for regulating phosphorylation of eukaryotic initiation factor 2α (eIF2α). Eur. J. Biochem. 268, 1143–1153 [DOI] [PubMed] [Google Scholar]
- 19. Vattem K. M., Staschke K. A., Wek R. C. (2001) Mechanism of activation of the double-stranded-RNA-dependent protein kinase, PKR: role of dimerization and cellular localization in the stimulation of PKR phosphorylation of eukaryotic initiation factor-2 (eIF2). Eur. J. Biochem. 268, 3674–3684 [DOI] [PubMed] [Google Scholar]
- 20. Dey M., Cao C., Dar A. C., Tamura T., Ozato K., Sicheri F., Dever T. E. (2005) Mechanistic link between PKR dimerization, autophosphorylation, and eIF2α substrate recognition. Cell 122, 901–913 [DOI] [PubMed] [Google Scholar]
- 21. Cole J. L. (2007) Activation of PKR: an open and shut case? Trends Biochem. Sci. 32, 57–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Dever T. E., Feng L., Wek R. C., Cigan A. M., Donahue T. F., Hinnebusch A. G. (1992) Phosphorylation of initiation factor 2α by protein kinase GCN2 mediates gene-specific translational control of GCN4 in yeast. Cell 68, 585–596 [DOI] [PubMed] [Google Scholar]
- 23. Lu J., O'Hara E. B., Trieselmann B. A., Romano P. R., Dever T. E. (1999) The interferon-induced double-stranded RNA-activated protein kinase PKR will phosphorylate serine, threonine, or tyrosine at residue 51 in eukaryotic initiation factor 2α. J. Biol. Chem. 274, 32198–32203 [DOI] [PubMed] [Google Scholar]
- 24. Dey M., Velyvis A., Li J. J., Chiu E., Chiovitti D., Kay L. E., Sicheri F., Dever T. E. (2011) Requirement for kinase-induced conformational change in eukaryotic initiation factor 2α (eIF2α) restricts phosphorylation of Ser51. Proc. Natl. Acad. Sci. U.S.A. 108, 4316–4321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Harashima A., Guettouche T., Barber G. N. (2010) Phosphorylation of the NFAR proteins by the dsRNA-dependent protein kinase PKR constitutes a novel mechanism of translational regulation and cellular defense. Genes Dev. 24, 2640–2653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hinnebusch A. G. (2011) Molecular mechanism of scanning and start codon selection in eukaryotes. Microbiol. Mol. Biol. Rev. 75, 434–467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kumar A., Haque J., Lacoste J., Hiscott J., Williams B. R. (1994) Double-stranded RNA-dependent protein kinase activates transcription factor NF-κB by phosphorylating I κB. Proc. Natl. Acad. Sci. U.S.A. 91, 6288–6292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sadler A. J., Williams B. R. (2008) Interferon-inducible antiviral effectors. Nat. Rev. Immunol. 8, 559–568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Dar A. C., Dever T. E., Sicheri F. (2005) Higher-order substrate recognition of eIF2α by the RNA-dependent protein kinase PKR. Cell 122, 887–900 [DOI] [PubMed] [Google Scholar]
- 30. Dey M., Cao C., Sicheri F., Dever T. E. (2007) Conserved intermolecular salt bridge required for activation of protein kinases PKR, GCN2, and PERK. J. Biol. Chem. 282, 6653–6660 [DOI] [PubMed] [Google Scholar]
- 31. Ung T. L., Cao C., Lu J., Ozato K., Dever T. E. (2001) Heterologous dimerization domains functionally substitute for the double-stranded RNA binding domains of the kinase PKR. EMBO J. 20, 3728–3737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Owen D. J., Noble M. E., Garman E. F., Papageorgiou A. C., Johnson L. N. (1995) Two structures of the catalytic domain of phosphorylase kinase: an active protein kinase complexed with substrate analogue and product. Structure 3, 467–482 [DOI] [PubMed] [Google Scholar]
- 33. Chen P., Luo C., Deng Y., Ryan K., Register J., Margosiak S., Tempczyk-Russell A., Nguyen B., Myers P., Lundgren K., Kan C. C., O'Connor P. M. (2000) The 1.7 A crystal structure of human cell cycle checkpoint kinase Chk1: implications for Chk1 regulation. Cell 100, 681–692 [DOI] [PubMed] [Google Scholar]
- 34. Carpick B. W., Graziano V., Schneider D., Maitra R. K., Lee X., Williams B. R. (1997) Characterization of the solution complex between the interferon-induced, double-stranded RNA-activated protein kinase and HIV-I trans-activating region RNA. J. Biol. Chem. 272, 9510–9516 [DOI] [PubMed] [Google Scholar]
- 35. Lemaire P. A., Lary J., Cole J. L. (2005) Mechanism of PKR activation: dimerization and kinase activation in the absence of double-stranded RNA. J. Mol. Biol. 345, 81–90 [DOI] [PubMed] [Google Scholar]
- 36. Mueller P. P., Hinnebusch A. G. (1986) Multiple upstream AUG codons mediate translational control of GCN4. Cell 45, 201–207 [DOI] [PubMed] [Google Scholar]
- 37. Kostura M., Mathews M. B. (1989) Purification and activation of the double-stranded RNA-dependent eIF-2 kinase DAI. Mol. Cell Biol. 9, 1576–1586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zhang F., Romano P. R., Nagamura-Inoue T., Tian B., Dever T. E., Mathews M. B., Ozato K., Hinnebusch A. G. (2001) Binding of double-stranded RNA to protein kinase PKR is required for dimerization and promotes critical autophosphorylation events in the activation loop. J. Biol. Chem. 276, 24946–24958 [DOI] [PubMed] [Google Scholar]
- 39. Nanduri S., Rahman F., Williams B. R., Qin J. (2000) A dynamically tuned double-stranded RNA binding mechanism for the activation of antiviral kinase PKR. EMBO J. 19, 5567–5574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Gelev V., Aktas H., Marintchev A., Ito T., Frueh D., Hemond M., Rovnyak D., Debus M., Hyberts S., Usheva A., Halperin J., Wagner G. (2006) Mapping of the auto-inhibitory interactions of protein kinase R by nuclear magnetic resonance. J. Mol. Biol. 364, 352–363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kronfeld-Kinar Y., Vilchik S., Hyman T., Leibkowicz F., Salzberg S. (1999) Involvement of PKR in the regulation of myogenesis. Cell Growth Differ. 10, 201–212 [PubMed] [Google Scholar]
- 42. Saelens X., Kalai M., Vandenabeele P. (2001) Translation inhibition in apoptosis: caspase-dependent PKR activation and eIF2-α phosphorylation. J. Biol. Chem. 276, 41620–41628 [DOI] [PubMed] [Google Scholar]
- 43. Lu B., Nakamura T., Inouye K., Li J., Tang Y., Lundbäck P., Valdes-Ferrer S. I., Olofsson P. S., Kalb T., Roth J., Zou Y., Erlandsson-Harris H., Yang H., Ting J. P., Wang H., Andersson U., Antoine D. J., Chavan S. S., Hotamisligil G. S., Tracey K. J. (2012) Novel role of PKR in inflammasome activation and HMGB1 release. Nature 488, 670–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Klann E., Dever T. E. (2004) Biochemical mechanisms for translational regulation in synaptic plasticity. Nat. Rev. Neurosci. 5, 931–942 [DOI] [PubMed] [Google Scholar]
- 45. Chen J. J., Throop M. S., Gehrke L., Kuo I., Pal J. K., Brodsky M., London I. M. (1991) Cloning of the cDNA of the heme-regulated eukaryotic initiation factor 2α (eIF-2α) kinase of rabbit reticulocytes: homology to yeast GCN2 protein kinase and human double-stranded-RNA-dependent eIF-2 alpha kinase. Proc. Natl. Acad. Sci. U.S.A. 88, 7729–7733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Harding H. P., Zhang Y., Ron D. (1999) Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 397, 271–274 [DOI] [PubMed] [Google Scholar]
- 47. Wek S. A., Zhu S., Wek R. C. (1995) The histidyl-tRNA synthetase-related sequence in the eIF-2α protein kinase GCN2 interacts with tRNA and is required for activation in response to starvation for different amino acids. Mol. Cell Biol. 15, 4497–4506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Cui W., Li J., Ron D., Sha B. (2011) The structure of the PERK kinase domain suggests the mechanism for its activation. Acta Crystallogr. D Biol. Crystallogr. 67, 423–428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kornev A. P., Taylor S. S. (2009) Pseudokinases: functional insights gleaned from structure. Structure 17, 5–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zeqiraj E., van Aalten D. M. (2010) Pseudokinases-remnants of evolution or key allosteric regulators? Curr. Opin. Struct. Biol. 20, 772–781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Dong B., Niwa M., Walter P., Silverman R. H. (2001) Basis for regulated RNA cleavage by functional analysis of RNase L and Ire1p. RNA 7, 361–373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ungureanu D., Wu J., Pekkala T., Niranjan Y., Young C., Jensen O. N., Xu C. F., Neubert T. A., Skoda R. C., Hubbard S. R., Silvennoinen O. (2011) The pseudokinase domain of JAK2 is a dual-specificity protein kinase that negatively regulates cytokine signaling. Nat. Struct. Mol. Biol. 18, 971–976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Hu J., Yu H., Kornev A. P., Zhao J., Filbert E. L., Taylor S. S., Shaw A. S. (2011) Mutation that blocks ATP binding creates a pseudokinase stabilizing the scaffolding function of kinase suppressor of Ras, CRAF and BRAF. Proc. Natl. Acad. Sci. U.S.A. 108, 6067–6072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. DeLano W. L. (2010) The PyMOL Molecular Graphics System, version 1.3r1, Schrödinger, LLC, New York [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.