

Background: Flap endonucleases remove 5′-single-stranded DNA termini.
Results: T7 gene 6 exonuclease is a flap endonuclease that cleaves 5′-single-stranded termini one nucleotide into the duplex.
Conclusion: The flap endonuclease activity catalyzes the removal 5′-single-stranded tails arising from duplex DNA.
Significance: The specificity of gene 6 protein identifies it as a flap endonuclease that can remove unusual structures of recombination and replication.
Keywords: Bacteriophage, DNA Damage, DNA Repair, DNA Replication, DNA-Protein Interaction, RNA Processing, Bacteriophage T7, Gene 6 Exonuclease, Okazaki Fragment, Flap Endonuclease
Abstract
Flap endonucleases remove flap structures generated during DNA replication. Gene 6 protein of bacteriophage T7 is a 5′–3′-exonuclease specific for dsDNA. Here we show that gene 6 protein also possesses a structure-specific endonuclease activity similar to known flap endonucleases. The flap endonuclease activity is less active relative to its exonuclease activity. The major cleavage by the endonuclease activity occurs at a position one nucleotide into the duplex region adjacent to a dsDNA-ssDNA junction. The efficiency of cleavage of the flap decreases with increasing length of the 5′-overhang. A 3′-single-stranded tail arising from the same end of the duplex as the 5′-tail inhibits gene 6 protein flap endonuclease activity. The released flap is not degraded further, but the exonuclease activity then proceeds to hydrolyze the 5′-terminal strand of the duplex. T7 gene 2.5 single-stranded DNA-binding protein stimulates the exonuclease and also the endonuclease activity. This stimulation is attributed to a specific interaction between the two proteins because Escherichia coli single-stranded DNA binding protein does not produce this stimulatory effect. The ability of gene 6 protein to remove 5′-terminal overhangs as well as to remove nucleotides from the 5′-termini enables it to effectively process the 5′-termini of Okazaki fragments before they are ligated.
Introduction
Bacteriophage T7 gene 6 protein (gp6)2 is an exonuclease that hydrolyzes duplex DNA non-processively in a 5′ to 3′ direction and liberates nucleoside 5′-monophosphates (1). gp6 is involved in many aspects of DNA metabolism in phage-infected cells and is essential for T7 growth. In cells infected with mutant phage lacking a functional gp6, the host DNA is not fully degraded, and T7 DNA synthesis stops prematurely (1–3). The low processivity of gp6, its specificity for duplex DNA, and its ability to initiate hydrolysis at nicks in duplex DNA provide wide applicability for biochemical studies. These applications include the preparation of duplex DNA containing 3′-single-stranded DNA (ssDNA) overhangs and gaps as well as the preparation of ssDNA templates for DNA sequencing.
A major role, described early in studies on T7 DNA replication, is the degradation of the host DNA to generate nucleoside 5′-monophosphates, precursors for the synthesis of T7 DNA (4). Most of the nucleotides in the DNA of phage progeny produced per infection cycle are derived from the Escherichia coli chromosome. The T7-encoded gene 3 endonuclease participates in this degradation of host DNA by attacking the DNA endonucleolytically to generate 5′-monophosphate-terminated fragments (4, 5). gp6 then degrades these fragments to yield ssDNA and nucleoside 5′-monophosphates (1, 3, 6).
gp6 plays an important role in the processing of Okazaki fragments. Lagging strand DNA synthesis requires the synthesis of short oligoribonucleotides that serve as primers for the lagging strand DNA polymerase. In the T7 replication system, the T7 gene 4 helicase-primase catalyzes the template-directed synthesis of tetraribonucleotides and transfers them to T7 DNA polymerase for the initiation of synthesis of Okazaki fragments by T7 DNA polymerase (7). The RNA-terminated Okazaki fragments must eventually be processed to remove the RNA termini in preparation for joining to an adjacent Okazaki fragment by DNA ligase to create a continuous lagging strand. In T7 phage-infected cells, two enzymes can remove the RNA primers: the 5′–3′-exonucleolytic activity of E. coli DNA polymerase I and T7 gp6. When gp6 is defective, there is an accumulation of RNA-terminated Okazaki fragments, but maximum accumulation occurs in the absence of both gp6 and E. coli DNA polymerase I (8). The absence of DNA polymerase I does not result in detectable accumulation of RNA-primed DNA fragments. Thus, it is likely that gp6 plays the major role in primer removal during T7 replication (8). In fact, gp6 can hydrolyze a RNA strand in an RNA/DNA hybrid (9, 10), and purified gp6 catalyzes the removal of the tetraribonucleotide primers at the 5′-termini of fragments arising from the extension of RNA primers synthesized by gene 4 DNA primase (11). The products of this reaction are ATP and nucleoside 5′-monophosphates. gp6 also plays a role in the processing of T7 DNA concatemers prior to packaging. In phage-infected cells lacking gp6, concatemeric DNA is processed in an aberrant or uncontrolled manner, giving rise to short concatemers. Models have been proposed to explain the origin of the abnormal concatemers found in phage-infected cells deficient in gp6 (12, 13).
Recombination (2) and DNA repair (14, 15) are extremely efficient in T7-infected cells even in the absence of the host RecA pathway (16). gp6 plays a critical role in recombination as evidenced by the absence of DNA exchange when E. coli is infected with bacteriophage T7 deficient in gp6; using density labeling, no hybrid T7 molecules are observed (17). Likewise, no recombinant molecules are formed in vitro using extracts of E. coli infected with gene 6-deficient T7 (17). The role of gp6 in recombination may involve the hydrolysis of duplex DNA to generate a 3′-single-stranded tail for invading another duplex molecule (17–19).
Many 5′–3′-exonucleases catalyze the removal of a ssDNA 5′-tail on duplex DNA arising from a nick in the DNA (20, 21). These ssDNA tails are often referred to as “flaps” or “overhangs,” terms that we will use interchangeably. The initial cleavage of ssDNA by these enzymes often takes place within the duplex region of the DNA at the ssDNA-dsDNA junction with the release of the intact ssDNA fragment. The enzymes are particularly important in eukaryotic DNA replication when the synthesis of an Okazaki fragment encounters the 5′-terminus of another Okazaki fragment. Continued synthesis by the lagging strand DNA polymerase displaces the 5′-region of the downstream Okazaki fragments, resulting in a 5′-ssDNA segment or “flap” containing the initial RNA primer. Flap endonucleases (FENs) perform the removal of the flap so that ligation to the adjacent fragment will yield a continuous lagging strand (20). In addition to their role in Okazaki fragment processing, flap endonucleases have been shown to play critical roles in recombination, repair, and genome stability. They are found in bacteria as well in eukaryotes, E. coli DNA polymerase I being a good example of the former, where there is both a proofreading 3′ to 5′ activity and a 5′ to 3′ flap endonuclease activity (22). Although many of these enzymes are, in reality, exonucleases that simply make an initial cleavage distal to the 5′-terminus, the term “endonuclease” is now entrenched.
T7 DNA polymerase does not normally catalyze strand displacement synthesis (23). Upon encountering a downstream dsDNA fragment, T7 DNA polymerase halts synthesis and “idles” as its 3′ to 5′-proofreading exonuclease removes nucleotides, and the polymerase replaces them. The RNA primer is therefore not normally displaced by strand displacement synthesis catalyzed by the lagging strand DNA polymerase. Rather, the RNA primer is removed by the 5′–3′-exonuclease activity of T7 encoded gp6 or the 5′–3′-exonuclease activity of E. coli DNA polymerase I. Therefore, at first consideration, there does not appear to be a need for a flap endonuclease in the T7 replication system, although such an activity may be important in recombination. However, a number of circumstances can arise in T7-infected cells where flap endonuclease activity may play a critical role. We recently reported that the primase domain of the 56-kDa T7 gene 4 protein can use preformed oligonucleotides and even tRNA as functional primers for T7 DNA polymerase (24). In this reaction, the primase anneals the preformed primer (the 3′-end of tRNA) to the primase recognition site provided the four terminal 3′-nucleotides of the preformed RNA fragment are complementary to those of the recognition site. The 5′-terminal region of the fragment need not be complementary to the DNA, thus leading to a flap. Because oligonucleotides and tRNA appear to prime in vivo under certain conditions, there may be a need for a flap endonuclease. In addition, T7 DNA polymerase deficient in the 3′–5′-exonuclease activity does catalyze rather extensive strand displacement synthesis leading to 5′-single-stranded termini when synthesis encounters a duplex structure (23). The exonuclease activity of T7 DNA polymerase is selectively inactivated by an oxidation reaction that requires molecular oxygen, a reducing agent, and iron (25). Whether or not exonuclease-deficient DNA polymerase exists in T7 phage-infected cells is not known. E. coli ssDNA binding protein (SSB protein), which exists abundantly in phage-infected cells, enables gp5 to catalyze strand displacement synthesis (26, 27). Finally, T7 gene 2.5 ssDNA-binding protein enables T7 DNA polymerase to catalyze strand displacement synthesis at a nick in duplex DNA sufficient for the loading of T7 DNA helicase (27, 28).
The crystal structure of gp6 has not yet been solved, but a predicted structure of gp6 was generated with the I-TASSER server by exploiting multiple solved crystal structures that are selected based on a sequence profile and a predicted secondary structure of gp6 (Fig. 1) (29–31). The predicted model reveals characteristic structures observed in the FEN family: a helical arch structure forming a gateway to select ssDNA and a helix-three-turn-helix (H3TH) motif known to bind dsDNA in a nonspecific manner (32). Moreover, the carboxylate residues constituting the active site are highly conserved and are located in positions found in the FEN family (Fig. 1B). T5 exonuclease, based on its structure, is a close match to the predicted gp6 structure; the root mean square deviation is 1.84 Å over 272 Cαs. However, the predicted gp6 structure does not have a binding pocket for a single-nucleotide 3′-flap that is found in hFEN1 and is a part of the cleavage site (33–35).
FIGURE 1.
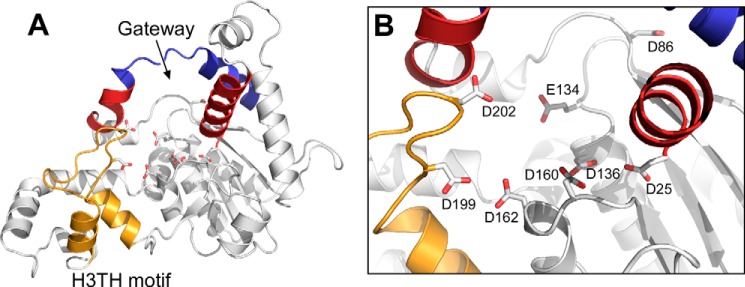
Predicted structure of gp6. A, the predicted structure of gp6 was generated by the I-TASSER server (29–31). A helical gateway and a helical cap in the helical arch are shown in red and blue, respectively. The H3TH motif involved in DNA binding is shown in yellow. Conserved carboxylates in the postulated active site are shown in stick representation. B, close-up of the postulated active site of gp6. The highly conserved carboxyl groups of the active site are labeled.
On the basis of the above observations we have examined the properties of gp6 to determine if it has flap endonuclease activity in addition to its 5′–3′-exonuclease activity. In the present study, we show that gp6 does indeed have a flap endonuclease activity that removes 5′-ssDNA termini on duplex DNA as well as ssDNA flaps of the type that arise from strand displacement synthesis. The finding that gp6 has flap endonuclease activity provides an explanation for the early observation that it can degrade phage λ DNA, although λ DNA has an ssDNA tail, and gp6 does not normally hydrolyze ssDNA (1).
EXPERIMENTAL PROCEDURES
Overproduction and Purification of gp6
T7 gene 6 was amplified by PCR from genomic DNA using primers that incorporated NdeI and HindIII for cloning. The PCR fragment was digested with NdeI and HindIII and inserted into the NdeI-HindIII sites of pET-28b (EMD Bioscience). The nucleotide sequence of the gene 6 insert was confirmed by DNA sequencing. The resulting plasmid was transformed into E. coli BL21 (DE3), and gp6 was overproduced as a recombinant protein containing a six-histidine tag at the N terminus. The bacterial culture was grown at 37 °C until it reached an A600 of around 1, at which time the temperature was decreased to 28 °C. Expression was induced with isopropyl β-d-thiogalactopyranoside at a final concentration of 0.5 mm at 28 °C for 4 h. The cells were harvested, resuspended in lysis buffer (50 mm sodium phosphate, pH 8.0, and 0.1 m NaCl), and ruptured by three cycles of freeze-thaw in the presence of lysozyme (0.5 mg/ml). Clear lysate was collected by centrifugation at 35,000 rpm for 1 h and loaded onto an Ni-NTA affinity column (Qiagen) in the presence of 50 mm imidazole. gp6 bound to the Ni-NTA column was eluted using 500 mm imidazole.
The Ni-NTA eluate was dialyzed against dialysis buffer (20 mm Tris-HCl, pH 7.5, 20 mm NaCl, and 0.1 mm DTT) to remove the imidazole. gp6 was further purified using an anion exchange column (HiTrapQ FF, GE Healthcare). Proteins were eluted with a 120-ml linear gradient from 50 mm to 1 m NaCl in buffer D (20 mm Tris-HCl, pH 7.5, 1 mm EDTA, 1 mm DTT, and 10% glycerol). Fractions containing gp6 were pooled, and ammonium sulfate was added with stirring until the mixture was saturated. The resulting precipitate was collected by centrifugation at 10,000 rpm and dissolved in 0.5 ml of 20 mm Tris-HCl, pH 7.5, 50 mm NaCl, 0.5 mm EDTA, and 0.5 mm DTT, followed by gel filtration chromatography (Sephacryl S-200 HR, GE Healthcare).
Fractions containing gp6 were combined and dialyzed against two changes of 1 liter of the dialysis buffer containing 5% glycerol. The dialyzed sample was applied to a column of Whatman P-11 phosphocellulose equilibrated with 20 mm potassium phosphate, pH 7.0, 20 mm KCl, 2 mm EDTA, 1 mm DTT, and 10% glycerol. The column was washed with the same buffer containing 20 mm KCl, followed by a 120-ml linear gradient of 20 mm to 2 m KCl in the same buffer. Fractions containing gp6 were pooled and concentrated by using Amicon 15 (Millipore). The purified protein was greater than 95% pure as determined by SDS-PAGE and Coomassie Blue staining.
Oligonucleotide Substrates
Oligonucleotides were purchased from Integrated DNA Technologies (sequences are listed in Table 1). Prior to annealing, oligonucleotides (100 pmol) were radiolabeled at either a 5′- or 3′-end using T4 polynucleotide kinase (New England Biolabs) and [γ-32P]ATP (PerkinElmer Life Sciences) or terminal transferase (New England Biolabs) and [α-32P]dCTP (PerkinElmer Life Sciences), respectively, in accordance with the manufacturer's instructions. Unincorporated radionucleotides were removed using the QIAquick nucleotide removal kit (Qiagen). The radiolabeled strand was annealed to a complementary strand at a molar ratio of 1:1.25, respectively, to ensure complete annealing of the radiolabeled strands. The two strands were placed in a buffer containing 20 mm Tris-HCl, pH 7.5, and 50 mm NaCl, heated at 95 °C for 5 min, and then slowly cooled to 25 °C. Minicircular DNA substrates were prepared as described previously (36). For an internally radiolabeled minicircular, 70-mer oligonucleotide (B3) was labeled with 32P at the 5′-end using polynucleotide kinase and then annealed with a splint DNA (5′-GGATA TGGTC GAGTG GTATA-3′). The ends of the 70-nt oligonucleotide were ligated by T4 DNA ligase (New England Biolabs), resulting in an internally labeled single-stranded circular DNA. The circular molecule was isolated from a denaturing gel and then annealed to a complementary strand (A10).
TABLE 1.
Oligonucleotides used in this study

a The nucleotides shown in lowercase letters indicate ribonucleotides.
For the substrate bearing a tRNA-like structure, the 3′-terminus of the oligonucleotide containing the RNA was labeled with 32P using terminal transferase. The radiolabeled oligonucleotide was heated at 95 °C and cooled slowly to form an RNA-hairpin structure and then annealed with a complementary DNA oligonucleotide at 37 °C.
gp6 Enzyme Assays
gp6 activity was measured in reaction mixtures (20 μl) containing 50 mm Tris-HCl, pH 8.0, 1 mm MgCl2, 20 mm KCl, 1 mm dithiothreitol (DTT), the indicated amount of DNA substrates, and the indicated amount of enzyme. Enzyme stocks were diluted in standard enzyme diluent containing 50 mm Tris-HCl, pH 7.5, 1 mm DTT, and 0.5 mg/ml bovine serum albumin. The reaction was initiated by the addition of gp6 diluted in standard enzyme diluent buffer and incubated at 37 °C for 10 min. Reactions were terminated by the addition of 5 μl of 95% formamide dye containing 100 mm EDTA. Samples were applied to a denaturing polyacrylamide gel containing 7 m urea, and electrophoresis was carried out in 1× TBE at 9 watts for 1.5–2 h. Subsequently, gels were dried and scanned by a Fuji BAS 1000 Bioimaging analyzer (Fujifilm Corp.). Data were analyzed using Image gauge (Fujifilm Corp.) and GraphPad Prism software (GraphPad Software, Inc.). The amounts of substrate and product were measured, and the percentage of product formed was determined by calculating the ratio of the product to substrate plus product. This method allows for the correction of loading variation among lanes. All assays were performed at least in triplicate, and representative data are shown.
Kinetic Analysis
gp6 cleavage kinetics were carried out in the same condition as the enzyme assay. Reaction mixtures were prepared with varying concentrations of 5′-overhang substrates (20, 100, 200, 400, and 800 nm). The substrates have a 48-bp duplex and 5′-ssDNA tails at both ends, one with a 22-nt ssDNA tail and the other with ssDNA of different lengths (0, 2, 6, 10, and 20 nt). The 5′-termini of the later ssDNA were labeled with 32P. Final concentrations of gp6 were 5, 10, 50, 50, and 100 nm for 0-, 2-, 6-, 10-, and 20-nucleotide 5′-overhang substrates, respectively. Assays were performed in triplicate. The general expression for the velocity of the reaction is v = d[P]/dt, where [P] = product concentration in nm. The product concentration was calculated using the equation, [P] = (Ip/(Is + Ip)) × [substrate], where Ip represents product intensity, Is is starting material intensity, and [substrate] is substrate concentration in nm. Velocity is expressed as converted substrate concentration (nm)/time (min). Km and Vmax values were calculated by directly fitting the data to the Michaelis-Menten equation.
RESULTS
gp6 Removes a 5′-ssDNA Tail on Duplex DNA
An old, puzzling observation was the ability of gp6 to degrade phage λ DNA, a duplex molecule that has 12-base single-stranded segments at both 5′-ends (6). gp6 does not hydrolyze ssDNA (6, 10), a finding that we have confirmed (data not shown). We determined whether gp6 would degrade ssDNA protruding from a DNA duplex using the two substrates depicted schematically in Fig. 2A. Substrate A consists of a duplex of 48 bp with one blunt end and a 22-nt single-stranded 5′-overhang at the other end. The 5′-terminus at the blunt end is labeled with 32P (Substrate A 5′-32P). This molecule is a suitable substrate for the well characterized 5′–3′-exonuclease activity of gp6 that sequentially releases nucleoside 5′-monophosphates. As will be shown below, the ssDNA tail on the unlabeled strand significantly slows the action of gp6 so that the kinetics of the exonuclease activity initiated at the blunt end can be accurately determined. Substrate B is similar to Substrate A except that both ends have 5′-ssDNA tails, one with the 22-nt ssDNA tail and the other with a 20-nt tail. The latter tail bears a 5′-32P (Substrate B 5′-32P). Substrate B 5′-32P addresses the ability of gp6 to initiate hydrolysis of a 5′-ssDNA tail on duplex DNA.
FIGURE 2.

gp6 removes a 5′-ssDNA tail on duplex DNA. A, Substrate A consists of a duplex of 48 bp with a 22-nt ssDNA tail attached to the 5′-terminus of one of the strands (A1:B1). The 48-nt strand was labeled with a 32P at either the 5′- or 3′-end. Substrate B is similar to Substrate A except that both ends have 5′-ssDNA tails, one with a 22-nt ssDNA tail and the other with a 20-nt tail. The strand bearing the 20-nt tail was labeled with a 32P at either the 5′- or 3′-terminus. The preparation of these substrates is described under “Experimental Procedures.” The 5′-overhang at the unlabeled end prevents rapid degradation of the duplex region from that terminus. B, Substrates A and B were labeled with 32P at their 5′-termini and incubated with gp6 as described under “Experimental Procedures.” Reactions (20 μl) were initiated by incubating increasing concentrations of gp6 (0.8, 4, 20, 100, and 500 nm) with a 50 nm concentration of the indicated DNA substrate. The radioactive oligoribonucleotide products were separated on an 18% polyacrylamide gel containing 7 m urea. The numbers on the left indicate the position and size of oligonucleotide standards. C, both Substrates A and B were labeled with 32P at their 3′- instead of their 5′-termini. Each substrate was incubated with gp6 and analyzed as described in B. The numbers on the left indicate the position and size of oligonucleotide standards.
As expected, gp6 hydrolyzes Substrate A 5′-32P effectively to release the 5′-32P-labeled mononucleotide (Fig. 2B). With Substrate B 5′-32P, the radioactive product of hydrolysis by gp6 migrates on the gel as a fragment of 21 nt. The length of the product indicates that gp6 cleaves the substrate at a position one nucleotide into the duplex on the labeled strand at the ssDNA-dsDNA junction. This result clearly demonstrates that gp6 has a structure-specific endonucleolytic activity similar to that observed with several other 5′-3′ nucleases, including T5 exonuclease, the 5′–3′-exonuclease domain of E. coli DNA polymerase I, and eukaryotic FEN proteins (20). The steady-state kinetic parameters for the two substrates were determined, measuring rates of initial cleavages over a range of substrate concentrations. The concentration of products, mononucleotides and 21-nt fragments, was determined for the exonuclease and endonuclease activities of gp6, respectively, by phosphorimaging analysis of the radioactive products after their separation by denaturing gel electrophoresis. The kcat of the exonuclease activity of gp6 with blunt-ended substrate is 2.8 min−1. On the other hand, the kcat of the endonuclease activity on Substrate B is 0.023 min−1 (Table 2). Thus, the exonuclease activity of gp6 is ∼120-fold greater than the activity (flap endonuclease) on DNA bearing a 20-nt 5′-ssDNA tail. However, once the 5′-flap is removed, the substrate is essentially identical to Substrate A, and the exonucleolytic cleavage should proceed at the more rapid rate. This observation distinguishes gp6 from other flap endonucleases in that the exonuclease activity of gp6 is much stronger than the endonuclease activity (37, 38). The absence of radioactive nucleotides with Substrate B attests to the insusceptibility of ssDNA to gp6.
TABLE 2.
Effect of the length of 5′-overhang on kcat and Km of the gp6 flap endonuclease
The kcat and Km for the flap endonuclease were determined using substrates having different lengths of the 5′-overhang. The substrates have a common structure shown in Fig. 2B. The kinetic parameters were determined as described under “Experimental Procedures.”
| Overhang length | kcat | Km |
|---|---|---|
| nt | min−1 | nm |
| 0 | 2.84 | 62.2 |
| 2 | 0.57 | 26.2 |
| 6 | 0.39 | 59.3 |
| 10 | 0.20 | 41.7 |
| 20 | 0.023 | 67.8 |
In order to determine if gp6 continues hydrolysis of the duplex DNA after removal of the ssDNA tail, the DNA strand bearing the 5′-ssDNA tail (Substrate B) was labeled with 32P at its 3′-terminus rather than at its 5′-terminus (Substrate B 3′-32P). As a control, Substrate A with the blunt end was also modified such that the 32P label was placed on the 3′-end of the strand (the 48-nt strand) rather than the 5′-end (Substrate A 3′-32P). With Substrate A 3′-32P, radioactive oligonucleotides ranging from 10 to 48 nt are observed by gel analysis indicative of the 5′–3′ hydrolysis by gp6 at a gp6 concentration of 4 nm (Fig. 2C). With Substrate B, 3′-32P hydrolysis of the labeled strand is not observed until concentrations of 20 nm or greater are present. gp6 continues hydrolysis of the duplex DNA after removal of the 5′-ssDNA tail but with a delay imposed by the rate-limiting step of first removing the 5′-ssDNA tail. The lack of a 48-bp intermediate is indicative of a far more rapid reaction on the substrate after removal of its 5′-ssDNA tail, due to the susceptibility of this intermediate to hydrolysis by the gp6 exonuclease. Furthermore, Substrate A was fully degraded by gp6 at the highest concentration (Fig. 2C), providing evidence that the method of preparation of substrates results in the complete annealing of the radiolabeled strands because gp6 cannot degrade unannealed ssDNA.
Effect of the 5′-Overhang Length on gp6 Flap Endonuclease Activity
In the above experiment, gp6 cleaves a 20-nt 5′-overhang at ∼0.8% of the rate of the exonuclease activity of gp6. We have examined the effect of the length of the 5′-overhang on the site of cleavage as well as the rate of cleavage by gp6. To examine this parameter, we have used DNA substrates having only a 2- or 6-nt 5′-overhang (Fig. 3A). As before, the 5′-overhangs have a 5′-terminal 32P label and the opposite end of the duplex bear a 5′-ssDNA tail to slow the exonuclease activity of gp6. gp6 also cleaves these shorter overhangs at the base of the flap, giving rise to fragments 3 and 7 nt, respectively, showing that gp6 cleaves one nucleotide into the duplex portion (Fig. 3A, closed arrowheads). The kinetic parameters for these substrates were determined by measuring the concentrations of the radioactive fragments produced by the endonuclease activity. The kcat values for gp6 endonuclease activity on 2- and 6-nt 5′-overhang substrates are 0.57 and 0.39 min−1, respectively (Table 2).
FIGURE 3.

Effect of the length of the 5′-overhang on flap endonuclease activity. A, the efficiency of flap endonuclease activity of gp6 on substrate having a 2-nt (A2:B1) or 6-nt (A3:B1) 5′-overhang was examined using the DNA substrates shown in the inset. The structures of the two substrates are identical to that of Substrate B shown in Fig. 2 except that the lengths of the 5′-overhang are either 2 or 6 nt. The 5′-terminus of the overhang was labeled with 32P. A 5′-ssDNA tail (22 nt) was also present at the other end of the duplex to prevent the degradation of the duplex portion of the substrate by the strong gp6 exonuclease activity. Reactions (20 μl) were carried out by incubating increasing concentrations of gp6 (0.8, 4, 20, 100, and 500 nm) with the indicated substrates (50 nm) at 37 °C for 10 min. The reaction products were separated on an 18% polyacrylamide gel containing 7 m urea and analyzed using a PhosphorImager. Closed arrowheads represent the products that are cleaved by the endonuclease activity of gp6 at one nucleotide into the duplex DNA. The numbers shown on the left of the autoradiograph indicate the size of the denatured intact labeled strands of the two substrates. B, the effect of different lengths of the 5′-overhang on gp6 endonuclease activity. The products generated by flap endonuclease activity of gp6 were measured on DNA substrates bearing different 5′-overhang length (0, 2, 6, 10, and 20 nt). The mononucleotide product was not included in the measurement in order to compare only the endonuclease activity. The 5′-termini of the overhangs were labeled with 32P, and a 22-nt 5′-overhang was present on the complementary strand to decrease the activity of the gp6 exonuclease activity on the 48-bp duplex region. Reactions were initiated by incubating gp6 (100 nm for the 20-nt overhang and 50 nm for the others) with substrates at 37 °C. The reaction products at the indicated time point were separated on a 20% polyacrylamide gel containing 7 m urea and analyzed using a PhosphorImager. In order to compare the rate of hydrolysis for each substrate, the product concentrations were normalized to the gp6 concentration needed to detect the product. The solid lines represent 5′-overhang substrates (A2–A5:B1), and the dashed line represents the blunt-ended substrate (A1:B1). Plots represent the mean, and error bars indicate S.D. from three independent experiments.
In Fig. 3B, the flap endonuclease activities on substrates having different lengths (0, 2, 6, 10, and 20 nt) of the 5′-overhang are compared. Substrates having shorter 5′-overhangs are cleaved more rapidly by the gp6 endonuclease activity. However, the 5′-terminus of a blunt-ended substrate is cleaved by gp6 10-fold more rapidly than the enzyme cleaves the 2-nt overhang.
Interestingly, with 2- and 6-nt overhangs, radioactive nucleoside monophosphates are observed in addition to the released fragment (Fig. 3 and Table 3). In the case of the 20-nt overhang substrate, essentially all of the radioactivity is released as a 21-nt fragment with no radioactive mononucleotides. The amount of nucleoside monophosphate is approximately equal to the amount of fragment released in the 2-nt overhang. With the 6-nt overhangs, ∼60% of the product is cleaved at the base and released as a 7-nt fragment, and 30% of the product radioactivity appears as nucleoside monophosphate. These results show that gp6 also hydrolyzes the ssDNA region prior to the endonucleolytic cleavage when the 5′-overhangs are short. It is unlikely that any of the radioactive nucleotides arose after the radioactive flap was released because gp6 does not hydrolyze ssDNA.
TABLE 3.
Effect of length of 5′-overhang on flap endonuclease activity and on the removal of a nucleotide from the 5′-overhang prior to endonucleolytic cleavage
Activities were measured with 100 nm gp6 and 50 nm DNA substrate as described under “Experimental Procedures.” Mononucleotides are products generated by the exonuclease activity of gp6, and oligonucleotides are products produced by the flap endonuclease activity of gp6. The exonuclease activity was determined by calculating the ratio of the radioactive mononucleotide products to the sum of the radioactive mononucleotide, oligonucleotide products, plus the nonhydrolyzed substrate based on an autoradiograph of a denaturing gel in each reaction. Likewise, the endonuclease activity was determined from the ratio of radioactive oligonucleotide products to radioactive mononucleotide and oligonucleotide products, plus the nonhydrolyzed substrate in each reaction. The ratio of the endonuclease activity to total nuclease activity was calculated by dividing the endonuclease activity by the total of both nuclease activities.
| Overhang length | Degradation |
Ratio of endonuclease to total nuclease activity | |
|---|---|---|---|
| Exonuclease | Endonuclease | ||
| nt | % | % | % |
| 0 | 96.0 | 0.0 | 0 |
| 2 | 51.0 | 47.7 | 48.3 |
| 6 | 28.8 | 60.3 | 67.7 |
| 10 | 3.8 | 78.8 | 95.4 |
| 20 | 0.5 | 25.8 | 98.0 |
The ability of gp6 to hydrolyze the 5′-terminal nucleotide on the flap in addition to releasing the flap is summarized in Table 3. Both of the shorter overhangs were cleaved at lower enzyme concentrations than that observed with the 20-nt overhang. The increase in the amount of mononucleotides released from the flap as its length decreases suggests that gp6 recognizes the very short overhang as a duplex structure. Unlike eukaryotic FEN proteins, the efficiency of the gp6 flap endonuclease activity is dependent on the length of the ssDNA overhang, with the activity decreasing with increasing length of the overhang (Fig. 3B and Table 3).
Effect of 3′-Extension near the Cleavage Site on Flap Endonuclease Activity
gp6 can cleave ssDNA endonucleolytically only when it extends from duplex DNA, resulting in cleavage at a position one nucleotide into the duplex at the ssDNA-dsDNA junction. This specificity implies that the junction is recognized by gp6. We examined the effect of an ssDNA extension (20 nt) of the 3′-end of the strand complementary to that bearing the 5′-flap on activity (see Fig. 4, inset). The 22-nt 5′-overhang is present on the complementary strand to decrease the activity of the gp6 exonuclease activity on the 48-bp duplex region. The presence of the ssDNA forming a Y-shaped structure depicted in the inset had an inhibitory effect on the flap endonuclease activity, decreasing activity ∼50% compared with a substrate lacking the 3′-extension (Fig. 4A). The 3′-extension also reduced the exonuclease activity by ∼20% as compared with a blunt-ended substrate (Fig. 4B). In control experiments, we confirmed that gp6 does not cleave the 3′-tail endonucleolytically using a substrate radiolabeled at the 3′-terminus on the tail (data not shown).
FIGURE 4.

Effect of 3′-extension near the cleavage site on flap endonuclease activity. A, a 3′-ssDNA extension of the complementary strand creates a Y-shaped structure (A4:B2). The efficiency of the flap endonuclease activity of gp6 was measured using a DNA substrate containing an extension (20 nt) of the 3′-end of the strand complementary to that bearing the 5′-flap. The 5′-terminus of the ssDNA extension of the 10-nt overhang was labeled with 32P, and a 22-nt 5′-overhang was present on the complementary strand to decrease the activity of the gp6 exonuclease activity on the 48-bp duplex region. A control DNA substrate lacks the 3′-ssDNA extension (A4:B1). Reactions were initiated by incubating gp6 (100 nm) with either substrate at 37 °C. The reaction products at the indicated time point were separated on a 20% polyacrylamide gel containing 7 m urea and analyzed using a PhosphorImager. The smearing observed above the uncut labeled strand (58 nt) is due to incomplete denaturation of DNA; the smear was considered as a non-cleaved substrate. The products of gp6 reactions containing the 10-nt 5′-overhang substrate with (closed circle) or without (open circle) the 3′-overhang were measured from three independent assays and are shown as plots of products versus time. Error bars, S.D. B, the same assay as in A was carried out except for using a substrate without a 5′-overhang (A1:B2) for measurement of the exonuclease activity. A blunt-ended substrate (A1:B1) served as a control. The products of gp6 reactions containing the 3′-extension substrate (closed circle) or the blunt-ended substrate (open circle) were measured from three independent assays and are shown as plots of products versus time. Error bars, S.D.
Flap Endonuclease Activity of gp6 at a Replication Fork
FEN proteins, the eukaryotic homologs of gp6, can remove the 5′-flap on a structure resembling a replication fork similar to the Y-shaped substrate used in Fig. 4A (38, 39). The presence of a duplex opposite to a 5′ flap strand enhances the efficiency of cleavage by FEN1 (21). In order to examine the effect of a duplex opposite to the flap on gp6 flap endonuclease activity, it is necessary to circumvent the potent 5′-exonuclease activity that would rapidly remove this annealed oligonucleotide used to construct this molecule. We have designed a DNA substrate using a minicircle DNA to eliminate this problem.
In the experiment shown in Fig. 5, we have used three different minicircle constructs. The chemically synthesized minicircle of 70 nt has no termini, thus eliminating any hydrolysis of this strand by the 5′–3′-exonuclease activity of gp6. The first minicircle DNA substrate consists of oligonucleotides fully complementary to the 70-nt minicircle and bears 10-nt 5′-single-stranded overhangs (Minicircle) (Fig. 5A). The second DNA consists of an oligonucleotide that forms 39 bp with the minicircle and a 10-nt 5′-overhang (minicircle with gap) (Fig. 5B). These molecules have a 31-nt gap, resulting in a Y-shaped structure similar to the substrate in Fig. 4A. gp6 releases the 5′-overhangs from the partially ssDNA minicircle substrate 1.3-fold more efficiently than from the fully duplex minicircle (Fig. 5, A and B); the flap endonuclease is less active when the flap arises from a nick. This conclusion is contrary to observations from homologs of FEN1 (38, 39). Equally important, the results obtained with the flap on minicircle DNA are identical to those obtained with linear DNA substrates, thus showing that the 5′-ssDNA tails on the non-radioactively labeled termini of the latter substrates are sufficient to eliminate most of the problems arising from the 5′–3′-exonuclease activity.
FIGURE 5.

Flap endonuclease activity of gp6 at a replication fork. Flap endonuclease assays were performed as described under “Experimental Procedures.” Reactions (10 μl) were carried out by incubating gp6 (100 nm) at 37 °C with the minicircle-based substrates (5 nm) depicted above (A). The minicircle substrate consists of an oligonucleotide having 70 nt 3′-complementary to the 70-nt minicircle and an additional portion of 10 nt 5′-non-complementary to the minicircle (A10:B5). The minicircle with gap substrate consists of an oligonucleotide having 39 nt 3′-complementary to the minicircle and 10 nt 5′-non-complementary to the minicircle (A7:B3). The minicircle with 3′-flap substrate has a 10-nt 5′-overhang and is identical to the one described in A except for an additional nucleotide at the 3′-end that results in a 1-nt 3′-flap (A12:B3). The reaction products were separated on an 18% denaturing polyacrylamide gel containing 7 m urea. Completely denatured strands are labeled on the right side of the autoradiograph. The smearing observed above those bands represents incomplete denaturation of substrates. B, the amount of oligoribonucleotide cleaved by gp6 in the assays in A was analyzed quantitatively using a Fuji BAS 1000 Bioimaging analyzer and presented in the graph. Plots represent the mean, and error bars indicate S.D. from three independent experiments.
The third DNA substrate has a 10-nt 5′-overhang and is identical to the first construct except for an additional nucleotide at the 3′-end of the annealed strand, resulting in a 1-nt 3′-overhang (minicircle with 3′-overhang) (Fig. 5, A and B). This substrate with a 1-nt 3′-overhang is an ideal substrate for eukaryotic homologs of gp6; a 3′ flap is known to stimulate the specificity for the cleavage site and efficiency of cleavage by FEN proteins (34, 40–43). Such a structure can occur, at least transiently, due to “breathing” of the DNA after strand displacement synthesis, where reannealing of the displaced strand results in displacement of the 3′-terminal nucleotide(s) (23, 44). The cleavage catalyzed by gp6 with this substrate occurs with similar efficiency compared with that observed with a substrate lacking the 3′-overhang.
Requirement for 5′-Terminus for gp6 Flap Endonuclease Activity
A junction between ssDNA and dsDNA is an absolute requirement for the endonucleolytic cleavage of a 5′-overhang by gp6; gp6 cannot degrade ssDNA alone. Can gp6 hydrolyze ssDNA at a gap in duplex DNA where a junction between ssDNA and dsDNA exists, but there is no free 5′-terminus? To address this point, we used a minicircle DNA of 70 nt annealed to a 49-nt oligonucleotide, resulting in a 39-bp duplex region (Fig. 6, inset). The 5′-terminus of the oligonucleotide is not complementary to the minicircle so that there is a 10-nt 5′-overhang to prevent hydrolysis of this strand by the 5′–3′-exonuclease activity of gp6. The resulting construct is a circular DNA molecule with a dsDNA (39-bp) and an ssDNA (31-nt) segment. The circular strand is internally labeled with 32P in order to detect cleavage of this strand. For comparison, we used the same construct bearing a label at the 5′-terminus of the overhang on the fragment annealed to the circular DNA (Fig. 6).
FIGURE 6.

Requirement of a 5′-terminus for gp6 flap endonuclease activity. The DNA substrate consists of a minicircle DNA of 70 nt annealed to a 49-nt oligonucleotide (see inset) (A7:B3). The 5′-terminus of the oligonucleotide is non-complementary to the minicircle so that there is a 10-nt 5′-overhang to prevent hydrolysis by the 5′–3′-exonuclease activity of gp6. The resulting construct is a circular DNA molecule with dsDNA (39-bp) and ssDNA (31-nt) segments. The potential site of cleavage by the gp6 flap endonuclease is shown by the dashed box and enlarged to show its identity to a flap cleavage site. The circular strand was labeled with 32P. The same molecule was used as a control, but the radioactive 32P was located at the 5′-terminus of the overhang and not on the circular strand (see inset with boxed cleavage site indicated). Reaction mixtures (10 μl) contained increasing concentrations of gp6 (0.8, 4, 20, 100, and 500 nm) and a 50 nm concentration of the DNA substrate with the radioactive circular strand or the substrate with the radioactively labeled 5′-overhang. The reaction was carried out at 37 °C for 10 min. gp3 (40 and 200 nm) was employed instead of gp6 in the same assay. For comparison, 100 nm gp6 was incubated with the 32P-labeled 5′-terminus of the 5′-ssDNA substrate at 37 °C for 10 min. The radioactive products formed in each reaction were identified by gel analysis on an 18% polyacrylamide gel containing 7 m urea and analyzed using a PhosphorImager. The radioactive mononucleotides arise from hydrolysis of non-ligated oligonucleotides used for construction of the circular strand that are annealed to the 39-nt complementary strand. The location of the marker, a 5′-radiolabeled 70-mer linear DNA consisting of the same sequence as the minicircle, is shown on the left. Closed arrowheads represent the products that are cleaved by the endonuclease activity of gp6 at one nucleotide into duplex DNA (11 nt).
The requirement for a 5′-terminus on the ssDNA for endonucleolytic cleavage by gp6 is apparent; no hydrolysis of the 32P-labeled circular strand is observed with the DNA substrate containing the 31-nt gap (Fig. 6, lanes 2–6). No decrease in signal intensity from the intact substrate was observed even at a concentration of 500 nm after electrophoresis of the products of the reaction on polyacrylamide gel containing 7 m urea. At the concentration of gp6 used in this assay, the 5′-32P labeled overhang in the internal control was efficiently removed, as evidenced by the release of a 32P-labeled 11-nt fragment (Fig. 6, lane 11). On the other hand, T7-encoded gene 3 endonuclease cleaves the circular strand and produces a wide range of fragments (Fig. 6, lanes 8 and 9). This result demonstrates that gp6 requires a 5′-end of the target strand for its endonucleolytic cleavage. Using the original definition of an exonuclease (45), the flap endonuclease activity of gp6 is actually an exonuclease that releases oligonucleotides by cleavage at sites distal from the 5′-terminus but requiring a 5′-terminus to initiate hydrolysis.
Stimulation of gp6 Activity by T7 Gene 2.5 ssDNA-binding Protein
The ssDNA-binding protein encoded by gene 2.5 (gp2.5) of bacteriophage T7 is essential for T7 growth (46). gp2.5 not only removes secondary structure in ssDNA; it also physically interacts with other T7 replication proteins to coordinate the multiple reactions occurring at the replication fork (7, 36). Previous studies found that gp2.5 stimulates the exonuclease activity of gp6 on double-stranded DNA, particularly in the presence of 100 mm NaCl, a salt concentration that inhibits gp6 activity (47). The finding that E. coli ssDNA protein did not stimulate gp6 implies that there is an interaction of gp2.5 with gp6. However, no physical interaction between the two proteins was detected (47).
We first measured the effect of gp2.5 on the 5′–3′-exonuclease activity on gp6 with double-stranded DNA (Fig. 7A). The DNA substrate, a 500-bp duplex DNA molecule with a 32P label at the 3′-terminus of one of the two strands, is hydrolyzed by gp6, as evidenced by the presence of labeled DNA fragments (lane 3). gp2.5 stimulates the hydrolysis of the DNA (lanes 4–6). With increasing amounts of gp2.5, there is a further increase in substrate hydrolysis. Densitometric analysis of the products indicates a stimulation of ∼3.5-fold. Interestingly, the presence of gp2.5 results in the accumulation of many products of 15 nt or less in length. Those 15-nt fragments might be the threshold of duplex stability under this condition. One possible explanation for the stimulation is the ability of gp2.5 to promote the formation of duplex regions by intra- or intermolecular base pairing, creating a preferable substrate for gp6. gp2.5 is known to facilitate greatly this type of annealing (48). NaCl (100 mm) drastically inhibits gp6 (Fig. 7A, compare lane 9 with lane 3). In confirmation of earlier studies (47), gp2.5 partially overcomes this inhibition (lanes 10–12). Also consistent with the previous study, this stimulatory effect is specific for gp2.5; E. coli ssDNA-binding protein does not stimulate gp6 (data not shown).
FIGURE 7.

Stimulation of gp6 activity by T7 gene 2.5 ssDNA-binding protein. Exonuclease and flap endonuclease assays were carried out as described under “Experimental Procedures.” A, 5′–3′-exonuclease assays were carried out using a 500-bp blunt-ended double-stranded DNA molecule with the 3′-terminus of one strand labeled with 32P. The asterisk shown in the inset represents the radiolabeled position. The reaction contained 100 nm gp6 and increasing concentrations of gp2.5 (0.2, 1, and 2 μm) with 5 nm DNA substrate. The reactions were performed at 37 °C for 10 min. The same assay was carried out in the presence of 100 mm NaCl. The reaction products were separated on an 18% polyacrylamide gel containing 7 m urea and analyzed using a PhosphorImager. B, the flap endonuclease activity was measured using the minicircle substrates having either a 5-, 10-, or 20-nt 5′-32P-ssDNA tail (A11:B5, A10:B5, and A9:B5). Reactions (20 μl) were performed by incubating 100 nm gp6 and increasing concentrations of gp2.5 (40, 200, and 1000 nm) with a 5 nm concentration of the indicated DNA substrate at 37 °C for 10 min. gp2.5 is represented by a black circle, and 32P is shown by an asterisk. The reaction products were separated on an 18% polyacrylamide gel containing 7 m urea and analyzed using a PhosphorImager. Closed arrowheads represent the products that are cleaved by the endonuclease activity of gp6 at one nucleotide into the duplex DNA. C, the same assay as in B was carried out except for using a minicircle substrate (see inset) that not only has a 5′-ssDNA overhang of 5, 10, or 20 nt but also a 31-nt ssDNA region adjacent to the overhang on the circular strand (A8:B5, A7:B5, and A6:B5). D, the activity of gp6 was measured in the presence or absence of NaCl (100 mm), shown as NaCl and Opt, respectively. In addition, the effect of gp2.5 on the reaction was examined in the presence of NaCl. The reaction was carried out by incubating 100 nm gp6 and 1000 nm gp2.5 with the 10-nt 5′-overhang substrates (5 nm) that are either fully or partially complementary to the 70-nt minicircle (A10:B3 or A7:B3) at 37 °C for 10 min. The reaction products were separated on an 18% polyacrylamide gel containing 7 m urea and analyzed using a PhosphorImager. Closed arrowheads represent the products that are cleaved by the endonuclease activity of gp6 at one nucleotide into the duplex DNA.
We have tested the effect of gp2.5 on the flap endonuclease of gp6 (Fig. 7, B–D). The minicircle substrates depicted in Fig. 7B have either a 5-, 10-, or 20-nt 5′-ssDNA tail that can be cleaved by the flap endonuclease. gp2.5 can stably bind to the 20-nt 5′-ssDNA tail, but the 5 nt ssDNA is not sufficient for binding. gp2.5 can bind to the 10-nt 5′-ssDNA, but the association is not stable (49). The flap endonuclease activity of gp6 on any of these substrates was not stimulated by gp2.5 over a range of concentrations (40, 200, and 1000 nm) (Fig. 7B). The minicircle substrates shown in Fig. 7C have not only a 5′-ssDNA overhang of 5, 10, or 20 nt but also a 31-nt ssDNA region adjacent to the overhang on the circular strand. gp2.5 can bind to this 31-nt ssDNA region. With the last two substrates, the amounts of cleavage of 5′-overhang are slightly increased at the highest concentration of gp2.5 (1000 nm). We conclude that the ssDNA region adjacent to the junction is important for the stimulatory effect of gp2.5. This result is still consistent with an earlier study (47) that showed that the first cleavage made by the exonuclease activity of gp6 at the 5′-terminus is not stimulated by gp2.5 because the substrate used in that study did not have an adjacent ssDNA region (47).
As shown in Fig. 7D (lanes 6 and 14), the flap endonuclease activity is, like the exonuclease activity, reduced significantly by the presence of NaCl. Whereas no stimulation of activity is observed with gp2.5 using the substrate with a 5′-overhang arising from nick (lanes 2, 3, 6, and 7), gp6 is stimulated slightly by gp2.5 using the substrate that has an ssDNA gap opposite to the 5′-flap (lanes 10, 11, 14, and 15). This stimulation by gp2.5 is observed regardless of the presence of salt. This result suggests that a single-strand region upstream of the strand to be hydrolyzed is required for gp2.5 stimulation.
Hydrolysis of RNA-terminated Flaps by gp6
We recently reported that under certain circumstances, preformed oligonucleotides and even tRNA could serve as primers for T7 DNA polymerase in conjunction with the 56-kDa T7 gene 4 primase/helicase (24). The functional oligoribonucleotides synthesized by the T7 DNA primase are pppACCC, pppACCA, and pppACAC. These oligoribonucleotides, synthesized in a template-directed reaction, serve as primers for T7 DNA polymerase. A large percentage of the tRNAs of E. coli have ACCA at their 3′-termini. A truncated form of the primase, 56-kDa gene 4 protein, found in vivo can stabilize this 3′-terminus at primase recognition sites on the template and transfer the tRNA to T7 DNA polymerase for extension into an Okazaki fragment. This tRNA region must then be removed to yield Okazaki fragments suitable for ligation into a continuous lagging strand.
We examined the ability of gp6 to remove the tRNA primer as it does with normal tetraribonucleotide RNA primers (8, 11). A tRNA-terminated Okazaki fragment was imitated by using an RNA hairpin loop whose duplex part has the same sequence as a stem region of tRNA and with ACCA at the 3′-terminus (see Fig. 8A, inset). This tRNA-like structure is located at the 5′-terminus of a DNA strand that is annealed to a complementary strand; the 3′-terminus of the strand bearing the RNA is labeled with 32P. As shown in Fig. 8A, gp6, even at high concentrations, does not hydrolyze this substrate bearing the tRNA-like structure. The mononucleotides observed at higher concentrations of gp6 are derived from degradation of the 3′-terminus of the labeled strand by the weak 3′–5′-exonuclease activity of gp6 (6). This result is consistent with those found for other FEN homologs that are not able to cleave a flap strand that has secondary structures or adducts on the flap region. On the other hand, gp3 endonuclease degrades the RNA hairpin strands, resulting in oligonucleotide fragments. This result indicates that gp3 and not gp6 could play a role in the removal of tRNA primers. gp3 endonuclease has been implicated in the use of tRNA as primer in T7 phage-infected cells (24).
FIGURE 8.

Flap endonuclease activity of gp6 on tRNA-terminated substrates. A, a tRNA-like primer attached to a DNA strand. The DNA substrate consists of a DNA strand (27 nt) with an RNA hairpin (7-bp duplex and 6-nt loop) annealed to a 70-nt oligonucleotide (see inset) (A13:B4). The duplex region is 31 bp and contains four ribonucleotides, ACCA, next to the RNA hairpin structure. 5′- and 3′-ssDNA are present at both ends of the strand complementary to that bearing the RNA hairpin. The 26-nt 5′-overhang prevents hydrolysis by the 5′–3′-exonuclease activity of gp6. The RNA hairpin strand is labeled with 32P at the 3′-end. Reaction mixtures (10 μl) contained increasing concentrations of gp6 (0.8, 4, 20, 100, and 500 nm) and a 25 nm concentration of the substrate. The reaction was carried out at 37 °C for 10 min. gp3 (40 and 200 nm) was employed instead of gp6 in the same assay. The radioactive products formed in each reaction were identified by gel analysis on a 20% polyacrylamide gel containing 7 m urea and analyzed using a PhosphorImager. The number shown on the left of the autoradiograph indicates the size of the oligonucleotide. B, RNA-terminated 5′-overhangs. The efficiency of the flap endonuclease activity of gp6 was measured using four kinds of RNA-terminated 5′-overhang substrates (see inset). The first substrate (A14:B5) is the RNA-terminated strand that is completely annealed to the complementary strand (0 nt). The second one (A14:B6) has a 2-nt 5′-overhang resulting in half of the RNA being single-stranded and the another half in the duplex (2 nt). The third substrate (A14:B7) has the whole RNA part single-stranded (4 nt). The last one (A14:B8) has a 6-nt 5′-overhang containing RNA and DNA segments (6 nt). The RNA segment of each substrate is shown with an open box. Almost the same molecules were used for comparison in which the RNA segment was replaced with DNA (A15:B5–8). The reaction (10 μl) was initiated by incubating 100 nm gp6 with the indicated substrate (10 nm) at 37 °C for 10 min. The radioactive products formed in each reaction were identified by gel analysis on a 22% polyacrylamide gel containing 7 m urea and analyzed using a PhosphorImager. The sequences of the major product are shown on the right of the autoradiograph, and the letters r and d represent ribo- and deoxyribonucleotide, respectively.
T7 DNA polymerase does not catalyze strand displacement synthesis when it encounters a duplex region (23). However, the polymerase has an intrinsic ability to displace strands under some conditions. T7 DNA polymerase deficient in the 3′–5′-exonuclease activity catalyzes strand displacement synthesis for several hundred nucleotides (23), and the presence of E. coli SSB proteins enables this reaction to occur (26, 27). “Breathing” of the 5′-terminus of the strand to be displaced most likely precedes the initiation of strand displacement synthesis. If this “breathing” occurs with an Okazaki fragment, then the tetraribonucleotide would be exposed as a flap. Inasmuch as strand displacement synthesis arising by such an event would be undesirable on the lagging strand, we examined the ability of gp6 to cleave an RNA flap of this size.
We prepared substrates in which one strand bears 5′-32P-labeled 5′-overhangs of varying length (0, 2, 4, and 6 nt) but with the ribonucleotide 5′-sequence, 5′-rACCA-3′. The complementary strand contains a 3′-overhang (see Fig. 8B, insets). Consequently, one substrate has the RNA sequence annealed to the complementary strand, one has a 5′-flap consisting of 5′-rAC-3′, one has a tetraribonucleotide flap (5′-rACCA-3′), and one has a 6-nt flap containing the RNA sequence. For comparison, similar substrates were prepared with the same structure and sequence except for replacing the four ribonucleotides with deoxyribonucleotides. With the 5′-recessed substrate lacking a flap, gp6 hydrolyzes the RNA-terminated strand with the same efficiency as it does the DNA substrate, resulting in nucleoside 5′-monophosphate (Fig. 8B, lanes 1 and 5). With a 2-nt flap, the products are trinucleotide and nucleoside monophosphate (Fig. 8B, lanes 2 and 6). The product, a trinucleotide, indicates that gp6 cleaves one nucleotide into the duplex region, although the region around the junction is RNA. The mononucleotides arise from hydrolysis of the ssDNA region by gp6 prior to the endonucleolytic cleavage, as observed with short 5′-flap DNA substrates. Intriguingly, with the 4-nt 5′-overhang, gp6 cleaves the phosphodiester linkage of the junction between ssRNA and dsDNA, releasing a 4-nt oligonucleotide product (Fig. 8B, lane 7). Recall that with a 4-nt 5′-DNA overhang, gp6 cleaved one nucleotide into the duplex, generating 5-nt oligonucleotide (Fig. 8B, lane 3). With the 6-nt overhang, gp6 mainly cleaved one nucleotide into the duplex region. In addition, gp6 hydrolyzed the phosphodiester linkage of the junction between RNA and DNA, resulting in more 4-nt fragments compared with that with the 6-nt 5′-DNA overhang. The results suggest that gp6 can recognize the junction between RNA and DNA when the RNA portion is unannealed. Clearly, however, gp6 can remove an RNA primer flap if strand displacement is initiated.
DISCUSSION
gp6 is an essential protein for T7 DNA replication in E. coli infected with bacteriophage T7. In vivo, gp6 is known to play a role in the degradation of host DNA, in the removal of the RNA primers from Okazaki fragments, and in recombination (4, 8, 17). Previous biochemical studies have shown that it is a 5′–3′-exonuclease specific for duplex DNA, and this activity is most likely important in all of the processes. Indeed, the 5′–3′-exonuclease activity can degrade E. coli DNA and also remove the 5′-terminal RNA primers from Okazaki fragments synthesized in vitro (6, 50). gp6 exhibits a moderate sequence homology to bacteriophage T5 gene D15 nuclease and the 5′–3′-exonuclease domain of E. coli DNA polymerase I. These two proteins have not only a 5′–3′-exonuclease activity on double-stranded DNA but also a structure-specific endonuclease activity similar to the eukaryotic FEN proteins, an activity designated flap endonuclease. This latter activity catalyzes the release 5′-ssDNA overhangs from duplex DNA (51, 52). In the present study, we have shown that gp6 is also a flap endonuclease with properties similar to these related enzymes.
Although gp6 does not degrade ssDNA, it does hydrolyze a 5′-ssDNA overhang on duplex DNA. However, in keeping with its specificity for double-stranded DNA, it does not introduce phosphodiester cleavages within the ssDNA itself. Rather, cleavage occurs one nucleotide into the duplex region at the ssDNA-dsDNA junction. The sequence of the duplex does not affect the activity, because gp6 exhibits the same behavior with the linear substrates and minicircle-based substrates as long as their 5′-flap lengths are the same. In contrast to its homologs, the flap endonuclease activity of gp6 is much weaker than the 5′–3′-exonuclease activity on overhangs; it hydrolyzes a blunt end DNA 10 times more efficiently than even a 2-nt 5′-overhang. The rate of hydrolysis of blunt-ended substrate ranges from 10- to 120-fold faster than that with 5′-overhang ones. This wide range of activity on flap substrates is due to the effect of the flap length and the surrounding structures on the flap endonuclease activity. Unlike the eukaryotic FEN proteins, the efficiency of gp6 flap endonuclease activity depends on the length of the ssDNA overhang (38, 42, 53).
The flap endonuclease activity of gp6, like the 5′–3′-exonuclease activity, requires a 5′-terminus. It does not release a 3′-overhang on DNA and does not hydrolyze an ssDNA flap if the 5′-terminus is sequestered within a duplex region. This requirement for a 5′-terminus is reflected in its inability to cleave at a single-stranded gap in duplex DNA. In this regard, the flap endonuclease activity is, in fact, an exonuclease activity, as defined by a nuclease that initiates a stepwise hydrolysis from the terminus of a DNA strand. For example, exonuclease VII of E. coli removes relatively large segments of DNA from 3′- or 5′-ssDNA overhangs on duplex DNA in a stepwise manner (54, 55). Similar to its exonuclease activity, the structure-specific endonuclease activity of gp6 is inhibited by NaCl at essentially identical salt concentrations. Importantly, both nuclease activities cleave one nucleotide into the duplex region. This similarity in location of the cleavage site, salt sensitivity, and the requirement for a 5′-terminus suggests that both activities occur on the same active site using similar catalytic mechanism. Only the sizes of products differ.
The structures of flap endonuclease proteins from multiple organisms from bacteriophage through archea to humans reveal a shared protein architecture that enables their endonuclease activity. A structural motif called a helical gateway or a helical arch is highly conserved in FEN proteins, forming an aperture on the active site through which the 5′-flap of a substrate is threaded for cleavage. A helix-two/three-turn-helix motif is also highly conserved, and this is involved in the non-sequence-specific binding of the duplex portion of the substrate containing the 3′-terminus of the 5′-flap (35). This motif also binds a potassium ion that plays a role in its interaction with the DNA. On the other hand, the other duplex region of the substrate opposite to the 5′-flap is bound to the N-terminal region of FEN proteins. This region has a helix called the wedging helix such that the DNA is bent at a 100° angle between the two duplex regions. In eukaryote and archeal FENs, this helix forms a 3′-flap binding pocket with the C-terminal region of the proteins, providing high selectivity for a substrate bearing a single-nucleotide 3′-flap (35, 56, 57). However, this binding pocket is not present in bacteriophage FEN homologs (32, 58, 59).
Because gp6 is homologous in sequence to T5 exonuclease (identity of 17.9% and similarity of 31.0%), a flap endonuclease whose structure is known, we hypothesize that their structures and mechanisms for flap cleavage are similar. A structural prediction for gp6 by the I-TASSER server (Fig. 1) shows high a similarity to the T5 exonuclease structure. The predicted model shows that the helical gateway and H3TH appear to be conserved in gp6 (33, 35, 59–62). The activity of gp6 is stimulated (∼2-fold) by the presence of potassium ions (data not shown). This stimulatory effect suggests that gp6 interacts with the duplex portion of DNA substrates via the H3TH motif in a manner analogous to that observed with hFEN1. In the predicted gp6 model, the wedging helix is seen, whereas like other bacteriophage homologs, the 3′-flap binding pocket is not present. This region of the protein interacts with the strands opposite to a 5′-flap. In contrast to eukaryotic FENs, gp6 hydrolyzes substrates containing a duplex opposite to a 5′-flap regardless of the presence of a 3′-flap with lower efficiency compared with a Y-shaped substrate. Moreover, the gp6-preferred substrate is a 5′-overhang but lacks a 3′-extension better than the Y-shaped substrates. Therefore, a lack of the 3′-flap binding pocket in the region explains these different substrate preferences between gp6 and eukaryotic FEN proteins.
Exonuclease I (EXO1), found in eukaryotes, is a close FEN1 paralogue and plays a critical role in DNA repair and recombination processes. EXO1 exhibits exonuclease activity at DNA nicks, gaps, and blunt ends. It can also endonucleolytically cleave 5′-flaps. Moreover, EXO1 activity is not stimulated by a 3′-flap of a substrate, unlike FEN1. These properties are similar to those of gp6, suggesting that gp6 could be involved in DNA repair in T7 bacteriophage-infected cells.
The predicted structure shows a gateway on the active site suggesting a mechanism to select and cleave 5′-flaps that is similar to other FEN proteins by which a ssDNA overhang is threaded through the helical gateway of FEN proteins to allow for phosphodiester bond cleavage at the proper position (32, 35, 53, 57, 63). Unlike eukaryotic FEN proteins, the flap endonuclease activity of gp6 is less efficient as the length of the 5′-overhang increases. This effect of the length of the 5′-flap is also observed with T5 exonuclease (53). The helical arch region of hFEN1 is disordered in the absence of a DNA substrate but becomes structured when DNA binds to the protein (35, 64). This disordered state of hFEN1 can thread a flap containing a short duplex and then cleave the flap, suggesting the ability to thread DNA containing 5′-flaps with adducts or DNA with long 5′-flaps (42, 43, 53). However, the structure of T5 exonuclease, whose structure is close to the gp6 model, shows that the helical arch is in the substrate-free state, suggesting that this conformational transition does not occur upon binding the DNA substrate. In this case, threading of longer DNA through the arch would be impeded because the size of the structured gateway can accommodate only ssDNA. Thus, the efficiency of flap endonucleases to cleave long DNA flaps may be dependent on the ability of the conformational transition of the helical gateway to accommodate them upon DNA binding.
gp2.5 stimulates the exonuclease and flap endonuclease activities of gp6 only when there is a 3′-ssDNA region opposite to a flap strand. gp2.5 is known to remove secondary structures in ssDNA. However, because E. coli ssDNA binding protein does not stimulate gp6, the stimulation is probably due to a specific interaction between gp2.5 and gp6. The presence of gp2.5 on the ssDNA region opposite to the flap strand could enhance the affinity of gp6 to its substrate. However, in the reaction with a 5′-32P-labeled duplex containing a 3′-overhang, stimulation by gp2.5 was not observed despite the presence of ssDNA (data not shown). It seems likely that gp6 performs the initial cleavage of this 5′-recessed substrate faster than that of a 5′-overhang because the 5′-tail need not be threaded through the gateway. In this situation, gp6 would be able to bind sufficiently long to the substrate for catalysis to occur. On the other hand, the binding period is not sufficiently long for cleavage of the 5′-overhangs. The presence of gp2.5 enables gp6 to bind more stably to a substrate, providing sufficient time to thread and cleave the 5′-overhang. The effect most likely involves a specific interaction between the two proteins. Moreover, the stimulatory effect of gp2.5 is detected in the presence of NaCl. The inhibition of gp6 by salt may arise from its interference in the binding of gp6 to a substrate. gp2.5 could diminish this inhibitory effect by retaining gp6 on the junction via a specific interaction with gp6. In addition, gp2.5 could increase the rate of hydrolysis by gp6 either by increasing the turnover rate or the processivity of the reaction, parameters that are difficult to measure with the relatively short DNA substrates used in these experiments. gp2.5 mediates the annealing of homologous DNA strands (65, 66). This property could provide a suitable DNA structure for gp6 by promoting duplex formation at termini where breathing of the DNA occurs.
One of the roles of gp6 in vivo is the removal of RNA primers from Okazaki fragments on the lagging strand (8). In this regard, it serves the same function as FEN proteins in eukaryotic DNA replication. However, a major difference between prokaryotic and eukaryotic systems is the mechanism by which the primers are recognized. In prokaryotic systems, the 5′–3′-exonuclease activity of proteins such as E. coli DNA polymerase I or T7 gp6 simply initiates hydrolysis of the hydrogen bonded primer from the 5′-terminus of the Okazaki fragment, releasing the 5′-nucleoside triphosphate as either pppApC or ATP and nucleoside 5′-monophosphate (11). In eukaryotic systems, the lagging strand DNA polymerase continues DNA synthesis when it encounters the 5′-terminus of a previously synthesized Okazaki fragment and displaces the 5′-terminus by strand displacement synthesis to produce a 5′-displaced strand (20, 67). The 5′-overhang is then removed by FEN1 (68, 69). Because T7 DNA polymerase does not normally catalyze strand displacement synthesis, it would appear that a flap endonuclease activity would not be required. However, it is important to note that the lack of strand displacement synthesis is due to the high 3′ to 5′ proofreading exonuclease activity of T7 DNA polymerase. Removal of the 3′–5′-exonuclease activity of T7 DNA polymerase enables it to efficiently mediate strand displacement synthesis (23). Strand displacement synthesis may actually occur quite frequently. For example, E. coli ssDNA-binding protein, an abundant protein in T7 phage-infected cells, enables T7 DNA polymerase to catalyze strand displacement synthesis (26, 27). As we show here, the RNA primers are removed efficiently by gp6. The efficiency of these reactions is almost identical to those of reactions with substrates not containing RNA. Interestingly, gp6 is capable of cleaving a junction between RNA and DNA especially when the junction exists in a displaced strand. There is no chemical difference in the backbone of RNA and DNA, but slight differences in the conformation of the backbone or the sugar pucker may be involved in substrate recognition of gp6. In T7 DNA replication, in contrast to eukaryotic DNA replication, the leading and lagging strand polymerases are identical. The eukaryotic polymerase (polymerase α) that extends RNA primers has low fidelity, a property that may lead to misincorporation of nucleotides in the lagging strand. Therefore, it is advantageous to remove the DNA region along with the RNA primers. On the other hand, in the T7 replication system, it is not necessary to remove the DNA portion on a lagging strand because it is synthesized by the same polymerase used for leading strand synthesis.
Even more intriguing is our recent finding that the T7 DNA primase, under circumstances where it cannot itself synthesize RNA primers, can use preformed oligonucleotides and even tRNA as primers for lagging strand DNA synthesis (24). In this reaction, 3′-sequences in the oligonucleotides complementary to those in the primase recognition sequence are annealed to the primase recognition sequence and are extended by T7 DNA polymerase. The resulting Okazaki fragments bear 5′-overhangs resulting from non-complementary sequences in the 5′-terminus of the oligonucleotide. Whereas these smaller oligonucleotides can be removed by gp6 flap endonuclease, the unusually large overhangs, such as tRNA, cannot but require gene 3 endonuclease for removal. gp6 may, however, be involved in the processing of the residual RNA remaining after gp3 cleavage. Finally, T7 DNA undergoes extensive recombination in phage-infected cells, and it is likely that many of the recombinant intermediates have 5′-overhangs.
Acknowledgments
We are grateful to Alfredo Hernandez for helpful discussion and constructive comments. We also thank all members of the Richardson laboratory for many discussions and Steven Moskowitz (Advanced Medical Graphics) for illustrations.
This work was supported, in whole or in part, by National Institutes of Health Grant GM54397 (to C. C. R.)
- gp6
- gp2.5, gp3, and gp5, gene 6, gene 2.5, gene 3, and gene 5 protein, respectively
- FEN
- flap endonuclease
- H3TH
- helix-three-turn-helix
- Ni-NTA
- nickel-nitrilotriacetic acid
- nt
- nucleotide(s).
REFERENCES
- 1. Kerr C., Sadowski P. D. (1972) Gene 6 exonuclease of bacteriophage T7. I. Purification and properties of the enzyme. J. Biol. Chem. 247, 305–310 [PubMed] [Google Scholar]
- 2. Studier F. W. (1969) The genetics and physiology of bacteriophage T7. Virology 39, 562–574 [DOI] [PubMed] [Google Scholar]
- 3. Center M. S., Studier F. W., Richardson C. C. (1970) The structural gene for a T7 endonuclease essential for phage DNA synthesis. Proc. Natl. Acad. Sci. U.S.A. 65, 242–248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sadowski P. D., Kerr C. (1970) Degradation of Escherichia coli B deoxyribonucleic acid after infection with deoxyribonucleic acid-defective amber mutants of bacteriophage T7. J. Virol. 6, 149–155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sadowski P. D. (1971) Bacteriophage T7 endonuclease. I. Properties of the enzyme purified from T7 phage-infected Escherichia coli B. J. Biol. Chem. 246, 209–216 [PubMed] [Google Scholar]
- 6. Kerr C., Sadowski P. D. (1972) Gene 6 exonuclease of bacteriophage T7. II. Mechanism of the reaction. J. Biol. Chem. 247, 311–318 [PubMed] [Google Scholar]
- 7. Hamdan S. M., Richardson C. C. (2009) Motors, switches, and contacts in the replisome. Annu. Rev. Biochem. 78, 205–243 [DOI] [PubMed] [Google Scholar]
- 8. Shinozaki K., Okazaki T. (1977) RNA-linked nascent DNA pieces in T7 phage-infected Escherichia coli cells. I. Role of gene 6 exonuclease in removal of the linked RNA. Mol. Gen. Genet. 154, 263–267 [DOI] [PubMed] [Google Scholar]
- 9. Serwer P., Watson R. H., Son M. (1990) Role of gene 6 exonuclease in the replication and packaging of bacteriophage T7 DNA. J. Mol. Biol. 215, 287–299 [DOI] [PubMed] [Google Scholar]
- 10. Shinozaki K., Okazaki T. (1978) T7 gene 6 exonuclease has an RNase H activity. Nucleic Acids Res. 5, 4245–4261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Engler M. J., Richardson C. C. (1983) Bacteriophage T7 DNA replication. Synthesis of lagging strands in a reconstituted system using purified proteins. J. Biol. Chem. 258, 11197–11205 [PubMed] [Google Scholar]
- 12. Miller R. C., Jr., Lee M., Scraba D. G., Paetkau V. (1976) The role of bacteriophage T7 exonuclease (gene 6) in genetic recombination and production of concatemers. J. Mol. Biol. 101, 223–234 [DOI] [PubMed] [Google Scholar]
- 13. White J. H., Richardson C. C. (1987) Processing of concatemers of bacteriophage T7 DNA in vitro. J. Biol. Chem. 262, 8851–8860 [PubMed] [Google Scholar]
- 14. Masker W. (1992) In vitro repair of double-strand breaks accompanied by recombination in bacteriophage T7 DNA. J. Bacteriol. 174, 155–160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kong D., Masker W. (1994) Deletion between direct repeats in T7 DNA stimulated by double-strand breaks. J. Bacteriol. 176, 5904–5911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Powling A., Knippers R. (1974) Some functions involved in bacteriophage T7 genetic recombination. Mol. Gen. Genet. 134, 173–180 [DOI] [PubMed] [Google Scholar]
- 17. Lee M., Miller R. C., Jr. (1974) T7 exonuclease (gene 6) is necessary for molecular recombination of bacteriophage T7. J. Virol. 14, 1040–1048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kerr C., Sadowski P. D. (1975) The involvement of genes 3, 4, 5 and 6 in genetic recombination in bacteriophage T7. Virology 65, 281–285 [DOI] [PubMed] [Google Scholar]
- 19. Alberts B., Stusser F. J. (1980) Mechanistic studies of DNA replication and genetic recombination. Academic Press, Inc., New York [Google Scholar]
- 20. Liu Y., Kao H. I., Bambara R. A. (2004) Flap endonuclease 1. A central component of DNA metabolism. Annu. Rev. Biochem. 73, 589–615 [DOI] [PubMed] [Google Scholar]
- 21. Finger L. D., Atack J. M., Tsutakawa S., Classen S., Tainer J., Grasby J., Shen B. (2012) The wonders of flap endonucleases. Structure, function, mechanism and regulation. Subcell. Biochem. 62, 301–326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Brutlag D., Kornberg A. (1972) Enzymatic synthesis of deoxyribonucleic acid. 36. A proofreading function for the 3′ leads to 5′ exonuclease activity in deoxyribonucleic acid polymerases. J. Biol. Chem. 247, 241–248 [PubMed] [Google Scholar]
- 23. Lechner R. L., Engler M. J., Richardson C. C. (1983) Characterization of strand displacement synthesis catalyzed by bacteriophage T7 DNA polymerase. J. Biol. Chem. 258, 11174–11184 [PubMed] [Google Scholar]
- 24. Zhu B., Lee S. J., Tan M., Wang E. D., Richardson C. C. (2012) Gene 5.5 protein of bacteriophage T7 in complex with Escherichia coli nucleoid protein H-NS and transfer RNA masks transfer RNA priming in T7 DNA replication. Proc. Natl. Acad. Sci. U.S.A. 109, 8050–8055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tabor S., Richardson C. C. (1987) Selective oxidation of the exonuclease domain of bacteriophage T7 DNA polymerase. J. Biol. Chem. 262, 15330–15333 [PubMed] [Google Scholar]
- 26. He Z. G., Rezende L. F., Willcox S., Griffith J. D., Richardson C. C. (2003) The carboxyl-terminal domain of bacteriophage T7 single-stranded DNA-binding protein modulates DNA binding and interaction with T7 DNA polymerase. J. Biol. Chem. 278, 29538–29545 [DOI] [PubMed] [Google Scholar]
- 27. Nakai H., Richardson C. C. (1988) The effect of the T7 and Escherichia coli DNA-binding proteins at the replication fork of bacteriophage T7. J. Biol. Chem. 263, 9831–9839 [PubMed] [Google Scholar]
- 28. Ghosh S., Marintcheva B., Takahashi M., Richardson C. C. (2009) C-terminal phenylalanine of bacteriophage T7 single-stranded DNA-binding protein is essential for strand displacement synthesis by T7 DNA polymerase at a nick in DNA. J. Biol. Chem. 284, 30339–30349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhang Y. (2008) I-TASSER server for protein 3D structure prediction. BMC Bioinformatics 9, 40–2105-9–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Roy A., Kucukural A., Zhang Y. (2010) I-TASSER. A unified platform for automated protein structure and function prediction. Nat. Protoc. 5, 725–738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Roy A., Yang J., Zhang Y. (2012) COFACTOR. An accurate comparative algorithm for structure-based protein function annotation. Nucleic Acids Res. 40, W471–W477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Grasby J. A., Finger L. D., Tsutakawa S. E., Atack J. M., Tainer J. A. (2012) Unpairing and gating. Sequence-independent substrate recognition by FEN superfamily nucleases. Trends Biochem. Sci. 37, 74–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chapados B. R., Hosfield D. J., Han S., Qiu J., Yelent B., Shen B., Tainer J. A. (2004) Structural basis for FEN-1 substrate specificity and PCNA-mediated activation in DNA replication and repair. Cell 116, 39–50 [DOI] [PubMed] [Google Scholar]
- 34. Friedrich-Heineken E., Hübscher U. (2004) The Fen1 extrahelical 3′-flap pocket is conserved from archaea to human and regulates DNA substrate specificity. Nucleic Acids Res. 32, 2520–2528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tsutakawa S. E., Classen S., Chapados B. R., Arvai A. S., Finger L. D., Guenther G., Tomlinson C. G., Thompson P., Sarker A. H., Shen B., Cooper P. K., Grasby J. A., Tainer J. A. (2011) Human flap endonuclease structures, DNA double-base flipping, and a unified understanding of the FEN1 superfamily. Cell 145, 198–211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lee J., Chastain P. D., 2nd, Kusakabe T., Griffith J. D., Richardson C. C. (1998) Coordinated leading and lagging strand DNA synthesis on a minicircular template. Mol. Cell 1, 1001–1010 [DOI] [PubMed] [Google Scholar]
- 37. Lee B. I., Wilson D. M., 3rd (1999) The RAD2 domain of human exonuclease 1 exhibits 5′ to 3′ exonuclease and flap structure-specific endonuclease activities. J. Biol. Chem. 274, 37763–37769 [DOI] [PubMed] [Google Scholar]
- 38. Harrington J. J., Lieber M. R. (1994) The characterization of a mammalian DNA structure-specific endonuclease. EMBO J. 13, 1235–1246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Murante R. S., Huang L., Turchi J. J., Bambara R. A. (1994) The calf 5′- to 3′-exonuclease is also an endonuclease with both activities dependent on primers annealed upstream of the point of cleavage. J. Biol. Chem. 269, 1191–1196 [PubMed] [Google Scholar]
- 40. Friedrich-Heineken E., Henneke G., Ferrari E., Hübscher U. (2003) The acetylatable lysines of human Fen1 are important for endo- and exonuclease activities. J. Mol. Biol. 328, 73–84 [DOI] [PubMed] [Google Scholar]
- 41. Kao H. I., Henricksen L. A., Liu Y., Bambara R. A. (2002) Cleavage specificity of Saccharomyces cerevisiae flap endonuclease 1 suggests a double-flap structure as the cellular substrate. J. Biol. Chem. 277, 14379–14389 [DOI] [PubMed] [Google Scholar]
- 42. Murante R. S., Rust L., Bambara R. A. (1995) Calf 5′ to 3′ exo/endonuclease must slide from a 5′ end of the substrate to perform structure-specific cleavage. J. Biol. Chem. 270, 30377–30383 [DOI] [PubMed] [Google Scholar]
- 43. Finger L. D., Blanchard M. S., Theimer C. A., Sengerová B., Singh P., Chavez V., Liu F., Grasby J. A., Shen B. (2009) The 3′-flap pocket of human flap endonuclease 1 is critical for substrate binding and catalysis. J. Biol. Chem. 284, 22184–22194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Masamune Y., Richardson C. C. (1971) Strand displacement during deoxyribonucleic acid synthesis at single strand breaks. J. Biol. Chem. 246, 2692–2701 [PubMed] [Google Scholar]
- 45. Kornberg A., Baker T. A. (1992) DNA Replication, 2nd Ed., pp. 403–437, W. H. Freeman, New York [Google Scholar]
- 46. Kim Y. T., Richardson C. C. (1993) Bacteriophage T7 gene 2.5 protein. An essential protein for DNA replication. Proc. Natl. Acad. Sci. U.S.A. 90, 10173–10177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Roberts L., Sadowski P., Wong J. T. (1982) Specific stimulation of the T7 gene 6 exonuclease by the phage T7 coded deoxyribonucleic acid binding protein. Biochemistry 21, 6000–6005 [DOI] [PubMed] [Google Scholar]
- 48. Rezende L. F., Willcox S., Griffith J. D., Richardson C. C. (2003) A single-stranded DNA-binding protein of bacteriophage T7 defective in DNA annealing. J. Biol. Chem. 278, 29098–29105 [DOI] [PubMed] [Google Scholar]
- 49. Marintcheva B., Hamdan S. M., Lee S. J., Richardson C. C. (2006) Essential residues in the C terminus of the bacteriophage T7 gene 2.5 single-stranded DNA-binding protein. J. Biol. Chem. 281, 25831–25840 [DOI] [PubMed] [Google Scholar]
- 50. Engler M. J., Lechner R. L., Richardson C. C. (1983) Two forms of the DNA polymerase of bacteriophage T7. J. Biol. Chem. 258, 11165–11173 [PubMed] [Google Scholar]
- 51. Lyamichev V., Brow M. A., Dahlberg J. E. (1993) Structure-specific endonucleolytic cleavage of nucleic acids by eubacterial DNA polymerases. Science 260, 778–783 [DOI] [PubMed] [Google Scholar]
- 52. Garforth S. J., Sayers J. R. (1997) Structure-specific DNA binding by bacteriophage T5 5′ → 3′ exonuclease. Nucleic Acids Res. 25, 3801–3807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Patel N., Atack J. M., Finger L. D., Exell J. C., Thompson P., Tsutakawa S., Tainer J. A., Williams D. M., Grasby J. A. (2012) Flap endonucleases pass 5′-flaps through a flexible arch using a disorder-thread-order mechanism to confer specificity for free 5′-ends. Nucleic Acids Res. 40, 4507–4519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Chase J. W., Richardson C. C. (1974) Exonuclease VII of Escherichia coli. Mechanism of action. J. Biol. Chem. 249, 4553–4561 [PubMed] [Google Scholar]
- 55. Chase J. W., Richardson C. C. (1974) Exonuclease VII of Escherichia coli. Purification and properties. J. Biol. Chem. 249, 4545–4552 [PubMed] [Google Scholar]
- 56. Williams R., Sengerová B., Osborne S., Syson K., Ault S., Kilgour A., Chapados B. R., Tainer J. A., Sayers J. R., Grasby J. A. (2007) Comparison of the catalytic parameters and reaction specificities of a phage and an archaeal flap endonuclease. J. Mol. Biol. 371, 34–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Sobhy M. A., Joudeh L. I., Huang X., Takahashi M., Hamdan S. M. (2013) Sequential and multistep substrate interrogation provides the scaffold for specificity in human flap endonuclease 1. Cell Rep. 3, 1785–1794 [DOI] [PubMed] [Google Scholar]
- 58. Ceska T. A., Sayers J. R., Stier G., Suck D. (1996) A helical arch allowing single-stranded DNA to thread through T5 5′-exonuclease. Nature 382, 90–93 [DOI] [PubMed] [Google Scholar]
- 59. Devos J. M., Tomanicek S. J., Jones C. E., Nossal N. G., Mueser T. C. (2007) Crystal structure of bacteriophage T4 5′ nuclease in complex with a branched DNA reveals how flap endonuclease-1 family nucleases bind their substrates. J. Biol. Chem. 282, 31713–31724 [DOI] [PubMed] [Google Scholar]
- 60. Dervan J. J., Feng M., Patel D., Grasby J. A., Artymiuk P. J., Ceska T. A., Sayers J. R. (2002) Interactions of mutant and wild-type flap endonucleases with oligonucleotide substrates suggest an alternative model of DNA binding. Proc. Natl. Acad. Sci. U.S.A. 99, 8542–8547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Allawi H. T., Kaiser M. W., Onufriev A. V., Ma W. P., Brogaard A. E., Case D. A., Neri B. P., Lyamichev V. I. (2003) Modeling of flap endonuclease interactions with DNA substrate. J. Mol. Biol. 328, 537–554 [DOI] [PubMed] [Google Scholar]
- 62. Doherty A. J., Serpell L. C., Ponting C. P. (1996) The helix-hairpin-helix DNA-binding motif. A structural basis for non-sequence-specific recognition of DNA. Nucleic Acids Res. 24, 2488–2497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Gloor J. W., Balakrishnan L., Bambara R. A. (2010) Flap endonuclease 1 mechanism analysis indicates flap base binding prior to threading. J. Biol. Chem. 285, 34922–34931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Sakurai S., Kitano K., Yamaguchi H., Hamada K., Okada K., Fukuda K., Uchida M., Ohtsuka E., Morioka H., Hakoshima T. (2005) Structural basis for recruitment of human flap endonuclease 1 to PCNA. EMBO J. 24, 683–693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Kong D., Nossal N. G., Richardson C. C. (1997) Role of the bacteriophage T7 and T4 single-stranded DNA-binding proteins in the formation of joint molecules and DNA helicase-catalyzed polar branch migration. J. Biol. Chem. 272, 8380–8387 [DOI] [PubMed] [Google Scholar]
- 66. Rezende L. F., Hollis T., Ellenberger T., Richardson C. C. (2002) Essential amino acid residues in the single-stranded DNA-binding protein of bacteriophage T7. Identification of the dimer interface. J. Biol. Chem. 277, 50643–50653 [DOI] [PubMed] [Google Scholar]
- 67. Burgers P. M. (2009) Polymerase dynamics at the eukaryotic DNA replication fork. J. Biol. Chem. 284, 4041–4045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Garg P., Stith C. M., Sabouri N., Johansson E., Burgers P. M. (2004) Idling by DNA polymerase δ maintains a ligatable nick during lagging-strand DNA replication. Genes Dev. 18, 2764–2773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Stith C. M., Sterling J., Resnick M. A., Gordenin D. A., Burgers P. M. (2008) Flexibility of eukaryotic Okazaki fragment maturation through regulated strand displacement synthesis. J. Biol. Chem. 283, 34129–34140 [DOI] [PMC free article] [PubMed] [Google Scholar]