

Background: Lipocalin-2 is induced in obesity/type 2 diabetes.
Results: TNFα and IFNγ induce LCN2 in vivo. STAT1 and NF-κB are required for LCN2 induction and bind the human LCN2 promoter.
Conclusion: An interplay between ERKs, STAT1, and NF-κB signaling pathways mediates the IFNγ and TNFα induction of LCN2.
Significance: Cytokine modulation of LCN2 increases our understanding of gene regulation in obesity/type 2 diabetes.
Keywords: Adipocyte, Adipose Tissue, ERK, Interferon, Tumor Necrosis Factor (TNF), NF-κB, STAT
Abstract
Lipocalin-2 (LCN2) is secreted from adipocytes, and its expression is up-regulated in obese and diabetic mice and humans. LCN2 expression and secretion have been shown to be induced by two proinflammatory cytokines, IFNγ and TNFα, in cultured murine and human adipocytes. In these studies, we demonstrated that IFNγ and TNFα induced LCN2 expression and secretion in vivo. Although we observed a strong induction of LCN2 expression and secretion from white adipose tissue (WAT) depots, the induction of LCN2 varied among different insulin-sensitive tissues. Knockdown experiments also demonstrated that STAT1 is required for IFNγ-induced lipocalin-2 expression in murine adipocytes. Similarly, knockdown of p65 in adipocytes demonstrated the necessity of the NF-κB signaling pathway for TNFα-mediated effects on LCN2. Activation of ERKs by IFNγ and TNFα also affected STAT1 and NF-κB signaling through modulation of serine phosphorylation. ERK activation-induced serine phosphorylation of both STAT1 and p65 mediated the additive effects of IFNγ and TNFα on LCN2 expression. Our results suggest that these same mechanisms occur in humans as we observed STAT1 and NF-κB binding to the human LCN2 promoter in chromatin immunoprecipitation assays performed in human fat cells. These studies substantially increase our knowledge regarding the requirements and mechanisms used by proinflammatory cytokines to induce LCN2 expression.
Introduction
Adipose tissue produces cytokines and hormones that modulate energy metabolism and systematic insulin sensitivity (1). Alterations in expression or secretion of these secreted proteins from adipocytes can contribute to insulin resistance. Chronic inflammation accompanied by immune cell infiltration into adipose tissue has been shown to play an important role in the pathogenesis of insulin resistance (2, 3). Proinflammatory cytokines secreted by adipose tissue immune cells can affect properties of adipocytes. Two major proinflammatory cytokines, IFNγ and TNFα, can induce insulin resistance in adipocytes (4–7) and modulate the expression and secretion of adipokines (8–10), including adiponectin (9, 11), which is exclusively expressed in mature adipocytes.
Lipocalin-2 (LCN2)2 belongs to the lipocalin family, a group of well known transporters of small hydrophobic molecules in circulation (12). LCN2 was initially characterized as a protein that interacts with and stabilizes matrix metalloproteinase 9 (13). LCN2 plays important roles in innate immune response by limiting bacterial growth (14–16). More recent studies have shown that LCN2 is a novel adipokine that is up-regulated in obese and type 2 diabetic subjects (17). However, the roles of LCN2 in modulating insulin sensitivity are still unclear. Studies of global LCN2 knock-out mice from separate groups have reported divergent phenotypes (18–20). Given the versatile functions of LCN2, it is not surprising that the phenotypes of global knock-out mice are inconsistent. Although the role of LCN2 in modulation of insulin action is not clear, it is clear that LCN2 is produced in adipocytes and regulated in murine and human obesity (17, 21, 22). In one study, administration of LCN2 induced peroxisome proliferator-activated receptor γ and adiponectin expression and ameliorated the negative effects of TNFα in 3T3-L1 adipocytes (23). Recently, LCN2 has been shown to be highly expressed in inguinal fat and to significantly affect high fat diet-induced adipose tissue expandability in a depot-specific manner (24). In this study, the absence of LCN2 expression significantly enhanced high fat diet-induced inguinal fat expansion. However, compared with wild type mice, high fat diet-induced epididymal fat expansion in LCN2-deficient mice was significantly decreased (24). Although some observations suggest a potentially metabolically protective role of LCN2, there are substantial inconsistent observations related to actions of LCN2 in relation to glucose tolerance and insulin sensitivity (18–20).
Despite the positive correlations of expression and circulating levels of LCN2 with obesity and insulin resistance (21, 22), the underlying mechanism mediating the induction of LCN2 is still unknown. Here, we report that IFNγ and TNFα induce LCN2 expression and secretion in vivo. Using knockdown methodology, we showed that STAT1 and NF-κB are required for the induction of LCN2 by proinflammatory cytokines. Our previous observations demonstrated that ERK signaling also contributes to the IFNγ- and TNFα-induced expression of LCN2 (25). Our new results reveal that ERK 1 and 2 activation affects the serine phosphorylation of STAT1 and NF-κB at specific sites. These studies suggest that STAT1Ser727 phosphorylation plays a role in the additive effects of IFNγ and TNFα on LCN2 expression in adipocytes. Moreover, we identified several STAT1 and NF-κB binding sites in the human LCN2 promoter, suggesting some translational relevance to our mechanistic studies. Collectively, our data demonstrate that IFNγ and TNFα are capable of inducing LCN2 from several insulin-sensitive tissues in vivo and provide insight into the interplay between ERKs and STAT1 or NF-κB signaling pathways in adipocytes.
EXPERIMENTAL PROCEDURES
Animals
Procedures for animal studies were approved by the Pennington Biomedical Research Center Institutional Animal Care and Use Committee. Adult male C57BL/6J mice were purchased from The Jackson Laboratory. All mice were housed in groups of three or four and had free access to water and standard chow (Purina LabDiet 5001). IFNγ and TNFα (R&D Systems) were dissolved in a sterile PBS with 0.1% BSA vehicle solution. 10-Week-old male mice (∼25 g) were injected intraperitoneally with 0.2 ml of vehicle, IFNγ (1.25 mg/kg of body weight), TNFα (0.05 mg/kg of body weight), or both cytokines. Mice were euthanized by CO2 gas inhalation 3 or 14 h postinjection. Tissues were removed, immediately frozen in liquid nitrogen, and stored at −80 °C. Blood was collected into serum separator tubes, allowed to clot at room temperature, and centrifuged at 1500 × g for 15 min at 4 °C. The supernatant (serum) was then transferred to a clean tube and stored at −80 °C for later analysis.
Cell Culture
Murine 3T3-L1 preadipocytes were grown in Dulbecco's modified Eagle's medium (DMEM) containing 10% bovine serum. Two days after confluence, a mixture of 0.5 mmol/liter 3-isobutyl-methylxanthine, 1 μmol/liter dexamethasone, and 1.7 μmol/liter insulin was added to induce preadipocyte differentiation in DMEM with 10% fetal bovine serum (FBS). Medium was changed by DMEM with 10% FBS 48 h later. DMEM was from Sigma. Bovine serum and FBS were from Hyclone. Human subcutaneous adipocytes in 6-well plates were from Zen-Bio. Recombinant mouse and human IFNγ was purchased from R&D Systems. Recombinant mouse and human TNFα was purchased from Invitrogen. UO126 was purchased from Promega.
Small Interfering RNA-mediated Knockdown
3T3-L1 adipocytes were trypsinized and replated in 24-well plates. Cells were transfected using DharmaFECT Duo (Thermo-Dharmacon) reagent and 100 nm siRNA (Thermo-Dharmacon; Non-targeting siRNA, catalog number D-001810-10-50; STAT1 siRNA, catalog number L-058881-00-0020; p65 siRNA, catalog number L-040776-00-0020). Non-targeting siRNA was used as a negative control. After 24 h, cells were harvested for RNA analysis and assessment of transfection efficiency.
RNA Analysis
Tissues were homogenized in TRIzol (Qiagen). Cell monolayers were harvested in RLT lysis buffer. Total RNA was isolated from homogenized tissues or harvested cells with an RNeasy mini kit (Qiagen). 10 μl of purified RNA was used for reverse transcription-PCR to generate cDNA. ΔΔCt real time PCR with SYBR Green Supermix reagent (Takara) and an Applied Biosystems 7900HT system were used to analyze cDNA. Cyclophilin A was used as an endogenous control. The following primers were used for real time PCR: mCyclophilin A: forward, CCACTGTCGCTTTTCGCCGC; reverse, TGCAAACAGCTCGAAGGAGACGC; mLipocalin-2: forward, TGCAAGTGGCCACCACGGAG; reverse, GCATTGGTCGGTGGGGACAGAGA.
Preparation of Whole Cell Extracts
3T3-L1 adipocytes were harvested in non-denaturing IP buffer containing 10 mmol/liter Tris (pH 7.4), 150 mmol/liter NaCl, 1 mmol/liter EGTA, 1 mmol/liter EDTA, 1% Triton X-100, and 0.5% Nonidet P-40 with protease inhibitors 1 μmol/liter phenylmethylsulfonyl fluoride, 1 μmol/liter pepstatin, 50 milliunits of trypsin inhibitory aprotinin, and 10 μmol/liter leupeptin and phosphatase inhibitor 2 mmol/liter sodium vanadate. Cell monolayers were scraped off the plates. Each extract was needled three times and centrifuged at 9500 × g for 10 min at 4 °C. Supernatants were then transferred to new tubes and analyzed with BCA (Pierce) to measure protein concentration.
Gel Electrophoresis and Immunoblotting
Proteins were separated on 7.5 or 10% gels by SDS-PAGE and transferred to nitrocellulose membranes. Membranes were blocked in 4% milk for 1 h and then incubated with primary antibody overnight at 4 °C. After incubation with HRP-conjugated secondary antibodies (Jackson ImmunoResearch Laboratories), enhanced chemiluminescence reagents (Pierce) were used to visualize results. The lipocalin-2 antibody was purchased from R&D Systems. ERK1/2, STAT5A, and p65Ser276 antibodies were from Santa Cruz Bioechnology. The adiponectin antibody was from Thermo-Pierce. STAT1, STAT1Tyr701, STAT1Ser727, STAT5, p65, p65Ser536, and MCP-1 antibodies were from Cell Signaling Technology. The active ERK1/2 antibody was from Promega.
Chromatin Immunoprecipitation (ChIP)
A SimpleChIP® enzymatic chromatin IP kit (Cell Signaling Technology) was used for the ChIP assay. Human subcutaneous adipocytes were serum-deprived overnight. Cells were treated with recombinant human cytokines for 30 min and then cross-linked with formaldehyde and glycine. Chromatin extracts were prepared according to the manufacturer's protocol. STAT1 and p65 antibodies for immunoprecipitation were purchased from Cell Signaling Technology. Purified DNA was quantified by real time PCR with SYBR Green Supermix and ROX buffer (Takara). Primer sequences for real time PCR were as follows: −176: forward, GAAACAGCACAAGGAAGGCACAGA; reverse, CCTGCGGAAACACTTGGCAAGATT; −856: forward, ATAACTGCTTCCCTGCTGGACAAG; reverse, GGCCTTATCCTTGAGGTCACTGAG; −1405: forward, CGGCCTGGCAGAGGATACTTTTTA; reverse, GTTGCACCAAAGCCTTCCCTTTCT; −1934: forward, GCCTCCCAGGTTCAAGCAATTCTT; reverse, CAAAGATTAGTTGGGCATGGTGGC; −3610: forward, CCACGCTGAACACTACTCACAAGA; reverse, TGCATGAAGGCAGCAAGATGCTTC. The formula used to calculate the percentage of input was as follows: Percentage of input = 2% × 2(C[T] 2% input sample − C[T] IP sample).
Adipose Tissue Explant Culture
Epididymal WAT and inguinal WAT (100 mg/well) were dissected, weighed, minced, and cultured in 6-well plates with low glucose DMEM containing 1% FBS. Minced tissues were treated or untreated with 0.5 nm TNFα. Total RNA was isolated after 3 h and analyzed by real time PCR.
Statistical Analysis
All data were analyzed by two-tailed unpaired Student's t test. Data from the animal experiments are presented as mean ± S.E. Results from studies of cultured adipocytes and ex vivo adipose tissues are shown as mean ± S.D. Results were considered statistically significant when p < 0.05 (#, p < 0.1; *, p < 0.05; **, p < 0.01; ***, p < 0.001 versus control).
RESULTS
Lipocalin-2 Expression and Secretion Are Induced by Proinflammatory Cytokines in Vivo
Several studies have shown that LCN2 is induced in obesity/type 2 diabetes mellitus (17, 21, 22), and we have shown that both IFNγ and TNFα induce LCN2 in cultured murine and human adipocytes (25). However, the ability of these cytokines to induce LCN2 in the whole animal has not been investigated. To determine the ability of these cytokines to modulate LCN2 in vivo, 10-week-old male C57BL/6J mice were injected intraperitoneally with IFNγ or TNFα. After 14 h, the mice were sacrificed, and epididymal fat pads were collected. As shown in Fig. 1A, both IFNγ and TNFα increased LCN2 mRNA levels in epididymal WAT. To determine whether these cytokines could modulate circulating levels of LCN2, we also collected serum from these mice. As shown in Fig. 1B, IFNγ and TNFα increased circulating levels of LCN2.
FIGURE 1.

IFNγ and TNFα induce lipocalin-2 expression and secretion in vivo. 10-Week-old male C57BL/6J mice were injected intraperitoneally with IFNγ (1.25 mg/kg of body weight) or TNFα (0.05 mg/kg of body weight) and euthanized 14 h later. A, total RNA in epididymal WAT was purified and analyzed by real time PCR. Cyclophilin A (PPIA) was used as an endogenous control. B, 250 μg of serum protein was subjected to Western blot analysis. Western blot results were quantified with ImageJ. The quantification is in the bottom panel. Data are shown as mean ± S.E., n = 4 mice/condition. *, p < 0.05; **, p < 0.01; ***, p < 0.001. Error bars represent S.E. AU, arbitrary units.
LCN2 is an adipokine but is also known to be expressed in other tissues. Therefore, we examined the ability of IFNγ and TNFα to induce LCN2 expression in other insulin-sensitive tissues. In this experiment, C57BL/6J mice were injected intraperitoneally with IFNγ, TNFα, or both of these cytokines. Our previous studies demonstrated that IFNγ and TNFα had an additive effect on the induction of LCN2 in cultured adipocytes (25). As shown in Fig. 2, treatments with IFNγ, TNFα, and the combination of both cytokines significantly induced LCN2 expression in epididymal WAT (Fig. 2A), liver (Fig. 2C), and heart (Fig. 2D) 3 h after cytokine injection. The combination of both cytokines caused a more robust elevation of LCN2 mRNA and provides novel data demonstrating that IFNγ and TNFα have additive effects on LCN2 expression in vivo. Treatment with either IFNγ or TNFα did not significantly induce LCN2 expression in inguinal WAT (Fig. 2B) or medial gastrocnemius (Fig. 2E). However, the combination treatment did significantly increase LCN2 in both of these tissues. As shown in Table 1, epididymal WAT is the tissue that primarily responds to IFNγ treatment as indicated by an increase in the LCN2 mRNA level of ∼15-fold. However, liver was the most sensitive to TNFα treatment, which resulted in an elevation of LCN2 mRNA levels by more than 2000-fold. Collectively, these experiments indicate that IFNγ and TNFα differentially modulate LCN2 expression in a tissue-specific manner and have additive effects in vivo.
FIGURE 2.

IFNγ and TNFα differentially induce lipocalin-2 expression in different tissues. 10-Week-old male C57BL/6J mice were injected intraperitoneally with IFNγ (1.25 mg/kg of body weight), TNFα (0.05 mg/kg of body weight), or both (Combo) and euthanized 3 h later. Total RNA in epididymal WAT (A), inguinal WAT (B), liver (C), heart (D), and gastrocnemius (E) was isolated and analyzed by real time PCR. Cyclophilin A (PPIA) was used as an endogenous control. Data are presented as mean ± S.E., n = 3 mice/condition. F, 100 mg of epididymal WAT and inguinal WAT from 9-week-old male C57BL/6J mice was treated with 0.5 nm TNFα for 3 h. Total RNA was isolated and analyzed by real time PCR. Cyclophilin A was used as an endogenous control. For each tissue, the results were normalized to the control condition. The figure is representative of an experiment performed three times on at least two mice. Error bars represent S.D. #, p < 0.1; *, p < 0.05; **, p < 0.01; ***, p < 0.001. AU, arbitrary units.
TABLE 1.
IFNγ and TNFα induced lipocalin-2 expression in different tissues
| -Fold |
|||
|---|---|---|---|
| IFNγ | TNFα | IFNγ + TNFα | |
| Epididymal WAT | 17a | 26a | 86b |
| Inguinal WAT | n.s.c | n.s.d (p = 0.0875) | 44a |
| Liver | 7e | 2175e | 4527a |
| Heart | 3e | 14b | 67b |
| Gastrocnemius | n.s. | n.s. | 9a |
a p < 0.01.
b p < 0.001.
c n.s., not significant.
d p < 0.1.
e p < 0.05.
To determine whether the site of injection contributed to the different responses of epididymal WAT and inguinal WAT, we dissected both adipose tissues depots from the same mouse and treated them with TNFα or vehicle in ex vivo experiments. As shown in Fig. 2F, expression of LCN2 increased by 8-fold in epididymal WAT but only by 3-fold in inguinal WAT following TNFα treatment. This observation suggests that proinflammatory cytokines regulate LCN2 expression in a tissue-dependent manner. However, the results also demonstrate that TNFα can significantly induce LCN2 expression in inguinal WAT. Considering our observation that intraperitoneal injection of TNFα did not significantly induce LCN2 expression in inguinal WAT (Fig. 2B), these ex vivo studies indicate that the site of injection can contribute to the response of proinflammatory cytokines in inguinal WAT.
NF-κB Is Required for TNFα-induced Lipocalin-2 Expression
Recent studies by our laboratory have shown that TNFα-induced NF-κB activation results in binding of p65 to one site in the murine LCN2 promoter and correlates with the induction of LCN2 expression (25). To determine the importance of NF-κB signaling in the ability of TNFα to induce LCN2 expression in adipocytes, we used a knockdown approach. Small interfering RNA targeting the p65 subunit of NF-κB was transfected into mature 3T3-L1 adipocytes. As shown in Fig. 3A, p65 mRNA levels were reduced by 60–70% with p65 siRNA transfection. Non-targeting siRNA was used as a negative control and had no effect on p65 mRNA expression. Two days after transfection, adipocytes were serum-deprived overnight and then treated with TNFα for 16 (for RNA analysis) or 24 h (for protein analysis). As expected, TNFα induced LCN2 mRNA and protein in control adipocytes and in adipocytes transfected with non-targeting siRNA. However, the ability of TNFα to induce LCN2 mRNA (Fig. 3B) and protein (Fig. 3C) was substantially reduced in cells transfected with p65 siRNA. STAT5A was used as a protein loading control. We also examined the expression level of MCP-1, an inflammatory marker that can be induced by TNFα and secreted by adipocytes. The TNFα-mediated induction of MCP-1 was also substantially decreased by p65 knockdown.
FIGURE 3.

NF-κB is required for TNFα-induced lipocalin-2 expression in adipocytes. 3T3-L1 adipocytes were transfected with 100 nm non-targeting siRNA or p65 siRNA. A, 24 h later, total RNA was purified and analyzed by real time PCR. B, after 40 h, cells were serum-deprived for 24 h and treated with 0.5 nm TNFα. Total RNA was isolated 16 h later and subjected to quantitative real time PCR. Cyclophilin A (PPIA) was used as an endogenous control. C, 40 h after transfection, adipocytes were serum-deprived for 24 h and treated with 0.5 nm TNFα. Whole cell extract was harvested after 24 h, and 150 μg of protein was analyzed by Western blot. Western blots were quantified with ImageJ. Data are from representative experiments that were independently performed three times. ***, p < 0.001; n.s., not significant. Error bars represent S.D. AU, arbitrary units; CTL, control; NT, non-targeting; KD, knockdown.
STAT1 Is Required for IFNγ-induced Lipocalin-2 Expression
Our recent studies have also shown that IFNγ-induced LCN2 expression correlates with the binding of STAT1 to five sites in the murine LCN2 promoter (25). To determine whether IFNγ-activated STAT1 was required for the induction of LCN2 in adipocytes, we used an siRNA knockdown approach. STAT1 mRNA was decreased by ∼60% with STAT1 knockdown (Fig. 4A). Transfection of non-targeting siRNA had no effect on the STAT1 mRNA level. Two days after transfection, mature adipocytes were serum-deprived and treated with IFNγ for 16 (for RNA analysis) or 24 h (for protein analysis). As reported previously, IFNγ treatment induced LCN2 mRNA (Fig. 4B) and protein levels (Fig. 4C) in 3T3-L1 adipocytes. Moreover, the induction of LCN2 was significantly reduced in adipocytes that had less STAT1. STAT1 knockdown had no effect on STAT5 proteins in adipocytes (Fig. 4C). It is known that IFNγ can induce MCP-1 in human mesangial cells (26); we observed a similar induction of MCP-1 in 3T3-L1 adipocytes following IFNγ treatment. However, STAT1 knockdown did not affect IFNγ-induced MCP-1 expression.
FIGURE 4.

STAT1 is necessary for IFNγ-induced lipocalin-2 expression in adipocytes. 3T3-L1 adipocytes were transfected with 100 nm STAT1 siRNA or non-targeting siRNA. A, 24 h later, total RNA was purified and analyzed by real time PCR. B, after 40 h, cells were serum-deprived for 24 h and treated with 100 ng/ml IFNγ. Total RNA was isolated 16 h later and subjected to quantitative real time PCR. Cyclophilin A (PPIA) was used as an endogenous control. C, 40 h after transfection, adipocytes were serum-deprived for 24 h and treated with 100 ng/ml IFNγ. Whole cell extracts were obtained after 24 h, and 150 μg of protein was analyzed by Western blot. Western blots were quantified with ImageJ. Each panel is representative of an experiment that was independently performed three times. ***, p < 0.001. Error bars represent S.D. AU, arbitrary units; CTL, control; NT, non-targeting; KD, knockdown.
TNFα- and IFNγ-induced ERK Signaling Modulates the Serine Phosphorylation of STAT1 and p65
Our recent studies on LCN2 have shown that the induction of this adipokine by both IFNγ and TNFα is dependent on ERK activation (25). Hence, we wanted to investigate how ERK signaling contributes to the effects of these cytokines. There is evidence to suggest that serine phosphorylation of NF-κB (27–29) and STAT 1 (30, 31) plays a role in the transcriptional activity of these signaling proteins. Therefore, we examined the phosphorylation of STAT1 at Ser727 and p65 at Ser536 to determine the effects of IFNγ or TNFα treatment on phosphorylation. Fully differentiated 3T3-L1 adipocytes were acutely treated (15 min) with IFNγ, TNFα, or both cytokines. As shown in Fig. 5A, IFNγ and TNFα activated the ERK 1 and 2 signaling pathways in adipocytes. Of note, TNFα treatment in the presence or absence of IFNγ induced phosphorylation of Ser536 on p65. However, IFNγ did not modulate p65 serine phosphorylation at this site. IFNγ did induce STAT1 tyrosine phosphorylation. All treatments induced STAT1Ser727 phosphorylation. The data clearly indicate an additive effect of IFNγ and TNFα on STAT1Ser727 phosphorylation.
FIGURE 5.
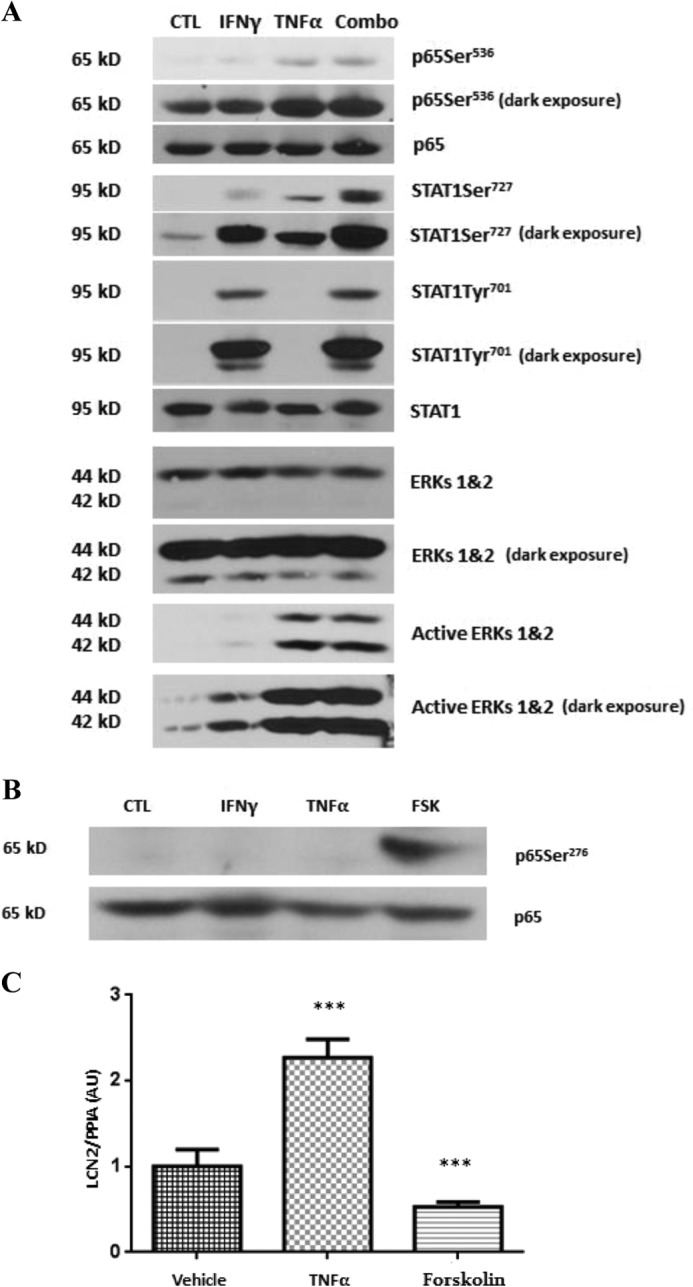
Regulation of the ERK signaling pathway and STAT1 and NF-κB serine phosphorylation by IFNγ and TNFα. A, mature adipocytes were treated with IFNγ (100 ng/ml), TNFα (0.5 nm), or the combination of both (Combo) for 15 min. Whole cell extracts were prepared, and 150 μg of protein was subjected to Western blot analysis. B, cells were treated with IFNγ (100 ng/ml), TNFα (0.5 nm), or forskolin (FSK) (20 μm) for 15 min. Whole cell extracts were harvested, and 150 μg of protein was analyzed by Western blot. C, 3T3-L1 adipocytes were treated with TNFα (0.5 nm) or forskolin (20 μm) for 16 h. Total RNA was isolated and subjected to real time PCR analysis. Cyclophilin A (PPIA) was used as an endogenous control. These data are from representative experiments performed with three different batches of cells. ***, p < 0.001. Error bars represent S.D. AU, arbitrary units; CTL, control.
NF-κB signaling studies have also revealed that p65 can be phosphorylated on Ser276 by activation of PKA (29). Therefore, we also examined p65Ser276 phosphorylation. Forskolin, a PKA activator, was used as a positive control in these experiments. As shown in Fig. 5B, neither IFNγ nor TNFα had an effect on p65Ser276. Nonetheless, we examined the ability of p65Ser276 phosphorylation to influence LCN2 expression. Mature adipocytes were treated with TNFα or forskolin for 16 h. TNFα induced LCN2 expression, whereas forskolin decreased LCN2 expression (Fig. 5C). These results suggest that phosphorylation of p65Ser276 does not participate in the induction of LCN2 expression.
Because our results suggested that serine phosphorylation may play a role in the additive effects of TNFα or IFNγ on LCN2 expression, we assessed whether the activation of ERKs was required for serine phosphorylation of p65 and STAT1. Mature 3T3-L1 adipocytes were pretreated with the highly specific ERK inhibitor UO126 for 30 min and then exposed to TNFα or IFNγ for an additional 15 min. As expected, ERK activation was inhibited by UO126 pretreatment. We observed that TNFα-induced p65Ser536 phosphorylation was eliminated by inhibition of ERKs (Fig. 6A). These data suggest that ERKs directly mediate Ser536 phosphorylation of p65. Although UO126 pretreatment had no effect on IFNγ-induced STAT1 tyrosine phosphorylation, both IFNγ- and TNFα-induced serine phosphorylation of STAT1 was abolished by ERK inhibition (Fig. 6B).
FIGURE 6.
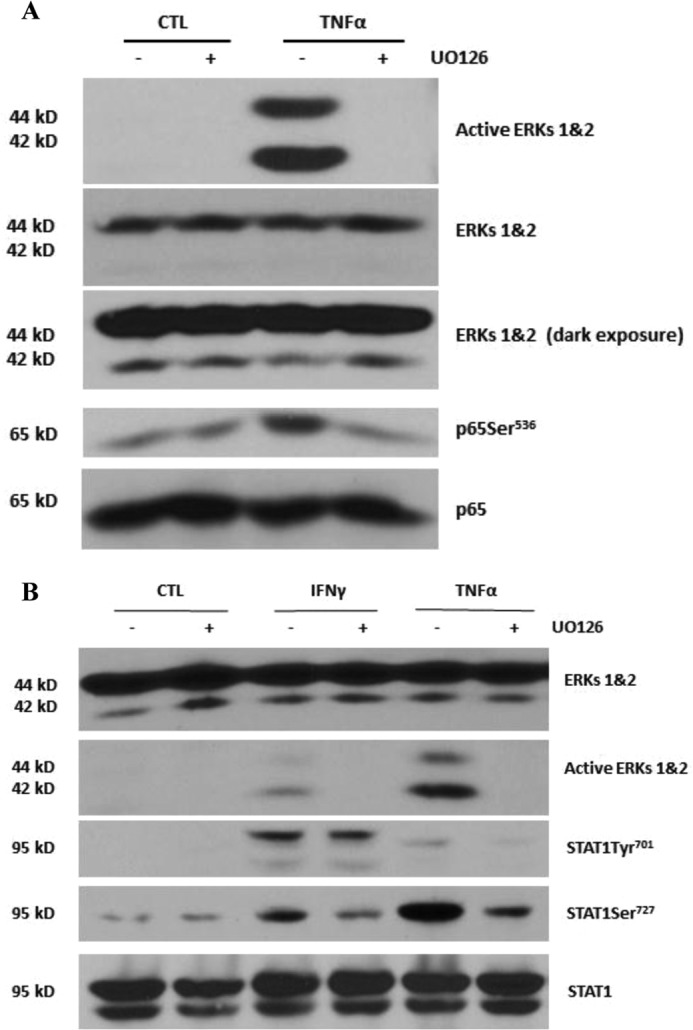
Activation of ERKs is required for serine phosphorylation of NF-κB p65Ser536 and STAT1Ser727. A, 3T3-L1 adipocytes were pretreated with 50 μm ERK inhibitor UO126 for 30 min and then treated with 0.5 nm TNFα for 15 min. Whole cell extracts were isolated, and 150 μg of protein was analyzed by Western blot. B, adipocytes were treated with 100 ng/ml IFNγ or 0.5 nm TNFα in the presence or absence of 50 μm UO126. Whole cell extracts were harvested, and 150 μg of protein was subjected to Western blot analysis. Data are from representative experiments that were independently performed three times. CTL, control.
Identification of STAT1 and NF-κB Binding Sites in Human LCN2 Promoter
As indicated above, our recent efforts have identified one NF-κB and several STAT1 binding sites in the murine LCN2 promoter (25). Hence, we performed an in silico analysis to identify potential STAT1 and NF-κB binding sites in human LCN2 promoter (Table 2). The four potential STAT1 binding sites and one potential NF-κB binding site were examined by ChIP assays. Human subcutaneous adipocytes were treated with recombinant human IFNγ or TNFα for 30 min. ChIP assays were performed with specific STAT1 and p65 antibodies. As shown in Fig. 7A, we observed that all four potential STAT binding sites were capable of interacting with STAT1 in an IFNγ-induced manner in human fat cells in vivo. In addition, we observed TNFα-induced p65 binding to an NF-κB binding site in the human LCN2 promoter in human adipocytes (Fig. 7B). These results are consistent with our previous observations that demonstrate that IFNγ or TNFα can induce LCN2 expression in human subcutaneous adipocytes (25).
TABLE 2.
STAT1 and NF-κB binding sites in human lipocalin-2 promoter
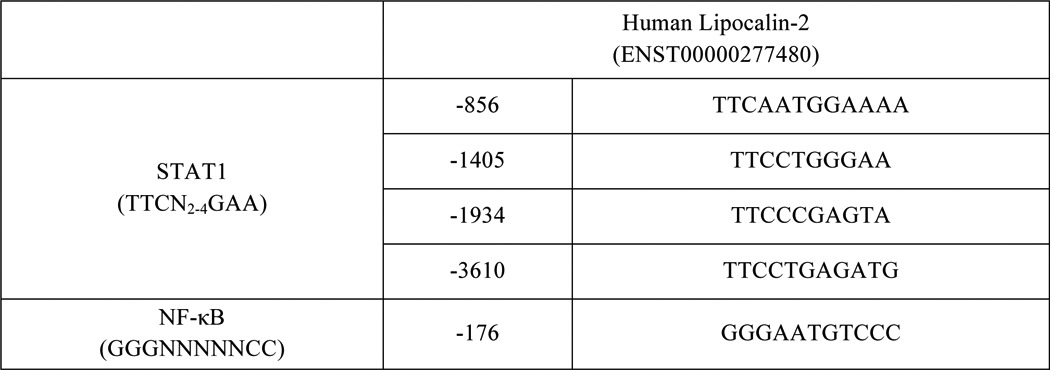
FIGURE 7.
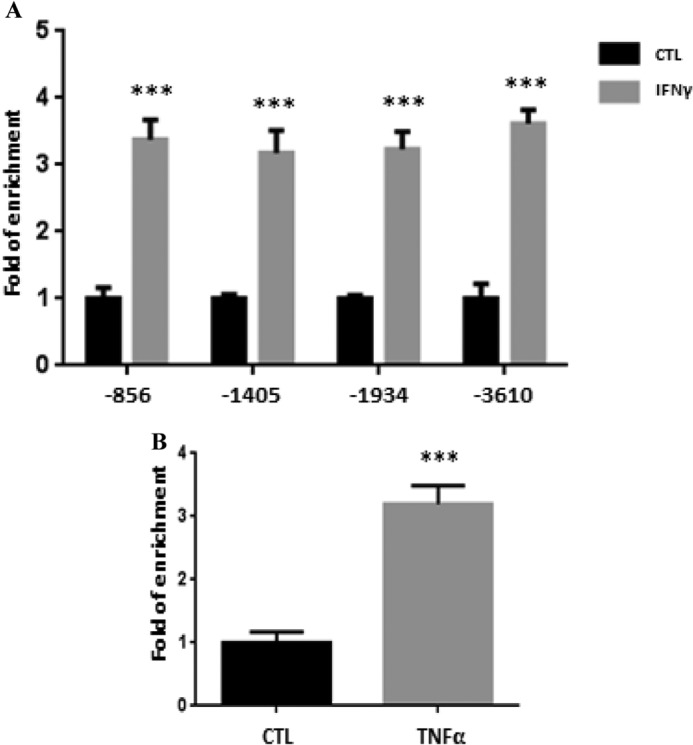
Identification of STAT1 and NF-κB binding sites in human lipocalin-2 promoter. A, human subcutaneous adipocytes were untreated or treated with 100 ng/ml recombinant human IFNγ for 30 min and then cross-linked with formaldehyde. Chromatin extracts were prepared and immunoprecipitated with STAT1 antibody. IP products were subjected to quantitative real time PCR analysis. The percentage of input was calculated and normalized to the control group. B, human subcutaneous adipocytes were untreated or treated with 0.5 nm recombinant human TNFα for 30 min and cross-linked with formaldehyde. Chromatin extracts were isolated and subjected to immunoprecipitation with NF-κB p65 antibody. IP products were analyzed by real time PCR. The percentage of input was calculated and normalized to the control. ***, p < 0.001. Error bars represent S.D. CTL, control.
DISCUSSION
Chronic inflammation in adipose tissue is associated with insulin resistance (2, 3). Proinflammatory cytokines secreted by immune cells present in adipose tissue can affect the function of adipocytes, including insulin sensitivity and endocrine functions. The adipokine LCN2 is significantly elevated in obese and diabetic subjects (21, 22), and studies in animal models demonstrate increased LCN2 in the adipose tissue of ob/ob and db/db mice (17). Despite the profound induction of LCN2 in obesity in mice and man, the underlying mechanisms mediating these effects are largely unknown. Our studies show that IFNγ and TNFα are able to induce LCN2 expression in vitro (25), and we have now demonstrated profound modulation of LCN2 in vivo. We observed that IFNγ and TNFα induced LCN2 in the primary insulin-sensitive tissues: adipose tissue, skeletal muscle, and liver. These studies also provide the first in vivo evidence to support the hypothesis that immune cell-derived inflammatory cytokines may be responsible for the increase of LCN2 expression in obesity and type 2 diabetes. Our studies also revealed tissue-specific responses. IFNγ treatment affected LCN2 expression primarily in epididymal WAT, whereas LCN2 modulation in liver was most sensitive to TNFα. We did not observe a significant increase of LCN2 in inguinal WAT or gastrocnemius in mice treated with IFNγ or TNFα. However, there was a significant cooperative effect of IFNγ and TNFα on LCN2 induction in most tissues examined. These are the first studies to demonstrate such an effect in vivo. In our studies, we delivered cytokines via intraperitoneal injection. Because inguinal WAT and gastrocnemius are not in the abdominal cavity, the lack of response in these tissues could be due to their physical distance from the injection site, which may lead to limited responsiveness to this specific method of cytokine treatment. We predicted that intravenous administration of the cytokines could produce different responses in these two tissues. The results from our ex vivo experiments clearly show that TNFα induced LCN2 expression in a fat depot-specific manner. Nonetheless, the significant up-regulation of LCN2 in TNFα-treated inguinal WAT that we observed ex vivo suggests that the injection site likely contributed to the lack of TNFα response in inguinal fat in our whole animal studies.
Our previous studies suggested that STAT1 mediates IFNγ-induced LCN2 expression and that NF-κB plays a role in TNFα-mediated induction of LCN2 in cultured adipocytes (25). However, both of these cytokines activate other signaling pathways, so we investigated the dependence on STAT1 or the p65 subunit of NF-κB by performing siRNA-mediated knockdown of these two transcription factors. Our results demonstrated that STAT1 knockdown significantly attenuated IFNγ-induced LCN2 expression, whereas p65 knockdown ameliorated the induction effects of TNFα. These observations demonstrate that IFNγ-induced LCN2 expression is largely mediated by STAT1 and that TNFα requires NF-κB to induce LCN2 expression in adipocytes. There is some additional specificity in our observations as the knockdown of p65 also inhibited TNFα-induced MCP-1 expression, but STAT1 knockdown did not affect the ability of IFNγ to induce MCP-1. It is not surprising that loss of STAT1 or p65 did not completely abolish the induction of LCN2 expression because siRNA-mediated knockdown of these transcription factors reduced, but did not abolish, their expression levels. Although LCN2 expression is likely affected by other transcription factors, our observations strongly suggest that STAT1 and p65 are critical for the induction of LCN2 by IFNγ and TNFα.
Our previous studies on LCN2 revealed that ERK 1 and 2 signaling was required for the induction of LCN2 expression by IFNγ and TNFα in adipocytes (25). To follow up on these studies, we examined serine phosphorylation of STAT1 and the p65 subunit of NF-κB. Some studies suggest that STAT1Ser727 phosphorylation contributes to the maximal transactivation activity of STAT1 (30). The NF-κB p65 subunit can be phosphorylated at Ser276 (29, 32) and Ser536 (27, 28). However, the functions of these phosphorylated serines are controversial. ERKs 1 and 2 are serine/threonine kinases activated by both IFNγ (33) and TNFα (34). We hypothesized that IFNγ- and TNFα-induced activation of ERKs may contribute to the serine phosphorylation of both STAT1 and NF-κB in adipocytes. Our results clearly show that phosphorylation of STAT1Ser727 and p65Ser536 was mediated by activation of ERKs 1 and 2. However, neither IFNγ nor TNFα treatment affected phosphorylation of p65Ser276. As expected, the PKA activator forskolin induced p65Ser276 phosphorylation, but it did not increase LCN2 expression. Forskolin treatment decreased LCN2 expression in adipocytes. These results indicate that p65Ser276 phosphorylation does not participate in the profound stimulatory effects of IFNγ and TNFα on LCN2 expression in adipocytes. The phosphorylation of p65Ser536 was induced by TNFα but was not enhanced in the presence of IFNγ. However, STAT1Ser727 phosphorylation was much greater in the presence of IFNγ and TNFα when compared with individual cytokine treatments. Our study also supports data showing that IFNγ-induced STAT1 serine phosphorylation is not dependent on tyrosine phosphorylation (35). Although we observed TNFα-induced STAT1Ser727 phosphorylation, we did not observe any effect on STAT1 tyrosine phosphorylation under these conditions. There is no doubt that the combination of IFNγ and TNFα had a cooperative effect on STAT1Ser727 phosphorylation and LCN2 expression. Although TNFα did not result in the tyrosine phosphorylation of STAT1, our results strongly suggest that TNFα induces ERK 1 and 2 phosphorylation of STAT1Ser727 that mediates a cooperative effect between IFNγ and TNFα on LCN2 gene expression. It should be noted that these cytokines can modulate other signaling proteins, and the cooperative effects on LCN2 expression may be influenced by ERK-independent pathways or other phosphorylation events. However, based on our previous studies on LCN2 (25) and our current results, we propose that STAT1Ser727 mediates cytokine cooperativity and that there is minimal contribution from other signaling pathways in the regulation of LCN2 in fat cells.
In summary, our study showed that the proinflammatory cytokines IFNγ and TNFα induce LCN2 expression and secretion in vivo. The presence of both cytokines resulted in an additive effect on LCN2 expression. STAT1 was required for IFNγ-induced LCN2 expression, whereas NF-κB was necessary for the induction of both LCN2 and MCP-1 by TNFα. ERK signaling appeared to contribute to the stimulatory effects of IFNγ and TNFα via modulating serine phosphorylation of STAT1 and the p65 subunit of NF-κB. Four STAT1 binding sites and one NF-κB binding site were also identified in the human LCN2 promoter, which suggests that our observations have translational relevance. These studies provide insight into the ability of proinflammatory cytokines to regulate adipocyte function. Although the roles of LCN2 in insulin resistance are still unclear, our mechanistic studies on LCN2 expression do increase our understanding of the modulation of this adipokine in conditions of obesity and type 2 diabetes.
This work was supported, in whole or in part, by National Institutes of Health Grant R01DK052968-15 (to J. M. S.).
- LCN2
- lipocalin-2
- WAT
- white adipose tissue
- IP
- immunoprecipitation.
REFERENCES
- 1. Kershaw E. E., Flier J. S. (2004) Adipose tissue as an endocrine organ. J. Clin. Endocrinol. Metab. 89, 2548–2556 [DOI] [PubMed] [Google Scholar]
- 2. Weisberg S. P., McCann D., Desai M., Rosenbaum M., Leibel R. L., Ferrante A. W., Jr. (2003) Obesity is associated with macrophage accumulation in adipose tissue. J. Clin. Investig. 112, 1796–1808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Xu H., Barnes G. T., Yang Q., Tan G., Yang D., Chou C. J., Sole J., Nichols A., Ross J. S., Tartaglia L. A., Chen H. (2003) Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J. Clin. Investig. 112, 1821–1830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hotamisligil G. S., Shargill N. S., Spiegelman B. M. (1993) Adipose expression of tumor necrosis factor-α: direct role in obesity-linked insulin resistance. Science 259, 87–91 [DOI] [PubMed] [Google Scholar]
- 5. Hotamisligil G. S., Spiegelman B. M. (1994) Tumor necrosis factor α: a key component of the obesity-diabetes link. Diabetes 43, 1271–1278 [DOI] [PubMed] [Google Scholar]
- 6. Hotamisligil G. S., Murray D. L., Choy L. N., Spiegelman B. M. (1994) Tumor necrosis factor α inhibits signaling from the insulin receptor. Proc. Natl. Acad. Sci. U.S.A. 91, 4854–4858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Stephens J. M., Lee J., Pilch P. F. (1997) Tumor necrosis factor-α-induced insulin resistance in 3T3-L1 adipocytes is accompanied by a loss of insulin receptor substrate-1 and GLUT4 expression without a loss of insulin receptor-mediated signal transduction. J. Biol. Chem. 272, 971–976 [DOI] [PubMed] [Google Scholar]
- 8. Stephens J. M., Pekala P. H. (1991) Transcriptional repression of the GLUT4 and C/EBP genes in 3T3-L1 adipocytes by tumor necrosis factor-α. J. Biol. Chem. 266, 21839–21845 [PubMed] [Google Scholar]
- 9. Waite K. J., Floyd Z. E., Arbour-Reily P., Stephens J. M. (2001) Interferon-γ-induced regulation of peroxisome proliferator-activated receptor γ and STATs in adipocytes. J. Biol. Chem. 276, 7062–7068 [DOI] [PubMed] [Google Scholar]
- 10. Zhao P., Stephens J. M. (2013) Identification of STAT target genes in adipocytes. JAKSTAT 2, e23092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ruan H., Hacohen N., Golub T. R., Van Parijs L., Lodish H. F. (2002) Tumor necrosis factor-α suppresses adipocyte-specific genes and activates expression of preadipocyte genes in 3T3-L1 adipocytes: nuclear factor-κB activation by TNF-α is obligatory. Diabetes 51, 1319–1336 [DOI] [PubMed] [Google Scholar]
- 12. Cowan S. W., Newcomer M. E., Jones T. A. (1990) Crystallographic refinement of human serum retinol binding protein at 2Å resolution. Proteins 8, 44–61 [DOI] [PubMed] [Google Scholar]
- 13. Kjeldsen L., Johnsen A. H., Sengeløv H., Borregaard N. (1993) Isolation and primary structure of NGAL, a novel protein associated with human neutrophil gelatinase. J. Biol. Chem. 268, 10425–10432 [PubMed] [Google Scholar]
- 14. Flo T. H., Smith K. D., Sato S., Rodriguez D. J., Holmes M. A., Strong R. K., Akira S., Aderem A. (2004) Lipocalin 2 mediates an innate immune response to bacterial infection by sequestrating iron. Nature 432, 917–921 [DOI] [PubMed] [Google Scholar]
- 15. Goetz D. H., Holmes M. A., Borregaard N., Bluhm M. E., Raymond K. N., Strong R. K. (2002) The neutrophil lipocalin NGAL is a bacteriostatic agent that interferes with siderophore-mediated iron acquisition. Mol. Cell 10, 1033–1043 [DOI] [PubMed] [Google Scholar]
- 16. Nelson A. L., Barasch J. M., Bunte R. M., Weiser J. N. (2005) Bacterial colonization of nasal mucosa induces expression of siderocalin, an iron-sequestering component of innate immunity. Cell. Microbiol. 7, 1404–1417 [DOI] [PubMed] [Google Scholar]
- 17. Yan Q. W., Yang Q., Mody N., Graham T. E., Hsu C. H., Xu Z., Houstis N. E., Kahn B. B., Rosen E. D. (2007) The adipokine lipocalin 2 is regulated by obesity and promotes insulin resistance. Diabetes 56, 2533–2540 [DOI] [PubMed] [Google Scholar]
- 18. Guo H., Jin D., Zhang Y., Wright W., Bazuine M., Brockman D. A., Bernlohr D. A., Chen X. (2010) Lipocalin-2 deficiency impairs thermogenesis and potentiates diet-induced insulin resistance in mice. Diabetes 59, 1376–1385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jun L. S., Siddall C. P., Rosen E. D. (2011) A minor role for lipocalin 2 in high-fat diet-induced glucose intolerance. Am. J. Physiol. Endocrinol. Metab. 301, E825–E835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Law I. K., Xu A., Lam K. S., Berger T., Mak T. W., Vanhoutte P. M., Liu J. T., Sweeney G., Zhou M., Yang B., Wang Y. (2010) Lipocalin-2 deficiency attenuates insulin resistance associated with aging and obesity. Diabetes 59, 872–882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Catalán V., Gómez-Ambrosi J., Rodríguez A., Ramírez B., Silva C., Rotellar F., Gil M. J., Cienfuegos J. A., Salvador J., Frühbeck G. (2009) Increased adipose tissue expression of lipocalin-2 in obesity is related to inflammation and matrix metalloproteinase-2 and metalloproteinase-9 activities in humans. J. Mol. Med. 87, 803–813 [DOI] [PubMed] [Google Scholar]
- 22. Wang Y., Lam K. S., Kraegen E. W., Sweeney G., Zhang J., Tso A. W., Chow W. S., Wat N. M., Xu J. Y., Hoo R. L., Xu A. (2007) Lipocalin-2 is an inflammatory marker closely associated with obesity, insulin resistance, and hyperglycemia in humans. Clin. Chem. 53, 34–41 [DOI] [PubMed] [Google Scholar]
- 23. Zhang J., Wu Y., Zhang Y., Leroith D., Bernlohr D. A., Chen X. (2008) The role of lipocalin 2 in the regulation of inflammation in adipocytes and macrophages. Mol. Endocrinol. 22, 1416–1426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Guo H., Bazuine M., Jin D., Huang M. M., Cushman S. W., Chen X. (2013) Evidence for the regulatory role of lipocalin 2 in high-fat diet-induced adipose tissue remodeling in male mice. Endocrinology 154, 3525–3538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhao P., Stephens J. M. (2013) STAT1, NF-κB and ERKs play a role in the induction of lipocalin-2 expression in adipocytes. Mol. Metab. 2, 161–170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Grandaliano G., Valente A. J., Rozek M. M., Abboud H. E. (1994) γ interferon stimulates monocyte chemotactic protein (MCP-1) in human mesangial cells. J. Lab. Clin. Med. 123, 282–289 [PubMed] [Google Scholar]
- 27. Hu J., Nakano H., Sakurai H., Colburn N. H. (2004) Insufficient p65 phosphorylation at S536 specifically contributes to the lack of NF-κB activation and transformation in resistant JB6 cells. Carcinogenesis 25, 1991–2003 [DOI] [PubMed] [Google Scholar]
- 28. Mattioli I., Sebald A., Bucher C., Charles R. P., Nakano H., Doi T., Kracht M., Schmitz M. L. (2004) Transient and selective NF-κB p65 serine 536 phosphorylation induced by T cell costimulation is mediated by IκB kinase β and controls the kinetics of p65 nuclear import. J. Immunol. 172, 6336–6344 [DOI] [PubMed] [Google Scholar]
- 29. Zhong H., SuYang H., Erdjument-Bromage H., Tempst P., Ghosh S. (1997) The transcriptional activity of NF-κB is regulated by the IκB-associated PKAc subunit through a cyclic AMP-independent mechanism. Cell 89, 413–424 [DOI] [PubMed] [Google Scholar]
- 30. Wen Z., Zhong Z., Darnell J. E., Jr. (1995) Maximal activation of transcription by Stat1 and Stat3 requires both tyrosine and serine phosphorylation. Cell 82, 241–250 [DOI] [PubMed] [Google Scholar]
- 31. Varinou L., Ramsauer K., Karaghiosoff M., Kolbe T., Pfeffer K., Müller M., Decker T. (2003) Phosphorylation of the Stat1 transactivation domain is required for full-fledged IFN-γ-dependent innate immunity. Immunity. 19, 793–802 [DOI] [PubMed] [Google Scholar]
- 32. Vermeulen L., De Wilde G., Van Damme P., Vanden Berghe W., Haegeman G. (2003) Transcriptional activation of the NF-κB p65 subunit by mitogen- and stress-activated protein kinase-1 (MSK1). EMBO J. 22, 1313–1324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hu J., Roy S. K., Shapiro P. S., Rodig S. R., Reddy S. P., Platanias L. C., Schreiber R. D., Kalvakolanu D. V. (2001) ERK1 and ERK2 activate CCAAAT/enhancer-binding protein-β-dependent gene transcription in response to interferon-γ. J. Biol. Chem. 276, 287–297 [DOI] [PubMed] [Google Scholar]
- 34. Vietor I., Schwenger P., Li W., Schlessinger J., Vilcek J. (1993) Tumor necrosis factor-induced activation and increased tyrosine phosphorylation of mitogen-activated protein (MAP) kinase in human fibroblasts. J. Biol. Chem. 268, 18994–18999 [PubMed] [Google Scholar]
- 35. Zhu X., Wen Z., Xu L. Z., Darnell J. E., Jr. (1997) Stat1 serine phosphorylation occurs independently of tyrosine phosphorylation and requires an activated Jak2 kinase. Mol. Cell. Biol. 17, 6618–6623 [DOI] [PMC free article] [PubMed] [Google Scholar]