

Abstract
The open reading frame PA1242 in the genome of Pseudomonas aeruginosa PAO1 encodes a putative protease belonging to the peptidase S8 family of subtilases. The respective enzyme termed SprP consists of an N-terminal signal peptide and a so-called S8 domain linked by a domain of unknown function (DUF). Presumably, this DUF domain defines a discrete class of Pseudomonas proteins as homologous domains can be identified almost exclusively in proteins of the genus Pseudomonas. The sprP gene was expressed in Escherichia coli and proteolytic activity was demonstrated. A P. aeruginosa ΔsprP mutant was constructed and its gene expression pattern compared to the wild-type strain by genome microarray analysis revealing altered expression levels of 218 genes. Apparently, SprP is involved in regulation of a variety of different cellular processes in P. aeruginosa including pyoverdine synthesis, denitrification, the formation of cell aggregates, and of biofilms.
Keywords: Biofilm, microarray, motility, orf PA1242, protease, Pseudomonas aeruginosa, Pyoverdine
Introduction
Pseudomonas aeruginosa is an opportunistic aerobic Gram-negative bacterium regarded as the major cause of death in cystic fibrosis patients (Murray et al. 2007). It causes both community-and hospital-acquired infections including ulcerative keratitis, skin and soft tissue infections, pneumonia, and infection after burn injuries (Zegans et al. 2002; Fujitani et al. 2011; Rezaei et al. 2011; Wu et al. 2011) and is the leading cause of respiratory tract infections in patients intubated during surgery, with a high mortality rate (Chastre and Fagon 2002). Epidemic increases in bacterial multidrug resistance and the occurrence of truly pan-resistant Gram-negative pathogens (e.g., P. aeruginosa, Acinetobacter baumannii, Klebsiella pneumonia) are major concerns in clinical microbiology, and thus, novel antibiotics are urgently needed (Payne 2008; Rasko and Sperandio 2010). However, the identification of potential drug targets and the success of novel antimicrobial agents proves to be difficult (Maeda et al. 2012). Part of the P. aeruginosa pathogenic potential is an impressive arsenal of virulence factors, like flagella and type IV pili, exopolysaccharides, lipopolysaccharides, and several secreted factors (Ramphal and Pier 1985; Nicas and Iglewski 1986; Drake and Montie 1988; Sato et al. 1988; Gupta et al. 1994). All these virulence factors are regulated by several complex regulatory systems including the quorum sensing (QS) system (Pesci et al. 1999; Venturi 2006). The yellow–green siderophore pyoverdine is also required for pathogenicity of P. aeruginosa as it is part of the major iron uptake system which is controlled by the iron starvation (IS) sigma factor PvdS (Ochsner et al. 2002; Tiburzi et al. 2008). Several other virulence factors including the proteases PrpL and AprA, are also regulated by PvdS (Shigematsu et al. 2001; Wilderman et al. 2001; Lamont et al. 2002). Two different surface organelles, namely a single polar flagellum and polar type IV pili, are responsible for P. aeruginosa motility. In liquid environments, P. aeruginosa can swim using flagella, whereas the type IV pili enable twitching motility on solid surfaces (Henrichsen 1972; Mattick 2002). On semisolid surfaces, P. aeruginosa moves by swarming via flagella and type IV pili (Köhler et al. 2000). These cell surface structures are also involved in biofilm formation (O'Toole and Kolter 1998; Klausen et al. 2003). P. aeruginosa biofilms pose a significant problem in both industrial and medical settings. Biofilms can form on medical devices like intravenous catheters and contact lenses and they are causative for the high resistance of P. aeruginosa against many antibiotics as well as the host immune system (Zegans et al. 2002). Biofilms exist in a complex extracellular matrix consisting of DNA, polysaccharides, and proteins. Their formation is a highly regulated process depending, among other factors, on cell motility and complex QS systems stabilizing such biofilms (Harmsen et al. 2010; Ghafoor et al. 2011; Mikkelsen et al. 2011).
Proteases represent a very diverse group of hydrolases which are found in all kingdoms of life (Rawlings and Barrett 1994). In bacteria, they are often involved in essential regulatory processes, for example, via degradation of abnormally folded proteins or by controlling the intracellular amounts of sigma factors and chaperones (Gottesman 1996). They catalyze the cleavage of peptide bonds in proteins and either show high specificity for defined amino acid (aa) sequences, or unspecifically hydrolyze proteins to yield oligopeptides or aa. Serine proteases contain serine as the active site residue and are grouped into 13 clans in the MEROPS database (Rawlings et al. 2012). Among them, family S8 of peptidases, also known as “subtilisin-like” or “subtilase” family, harbors the serine endopeptidase subtilisin and related enzymes with a characteristic Asp/His/Ser catalytic triad and represents the second largest family of serine proteases (Siezen and Leunissen 1997; Siezen et al. 2007; Rawlings et al. 2012). Subtilases are found in archaea, bacteria, viruses, fungi, yeast, and higher eukaryotes (Siezen and Leunissen 1997; Bergeron et al. 2000; Antao and Malcata 2005; Poole et al. 2007). The majority of subtilases is secreted but several members are localized intracellularly (Siezen and Leunissen 1997). In general, subtilases play a key role in protein maturation processes and precursor processing (Siezen et al. 2007). Subtilases typically show a multi-domain structure containing a signal sequence for translocation, a pro-domain for maturation, and a protease domain (Siezen and Leunissen 1997). The N-terminal propeptide of subtilases usually acts as an intramolecular chaperone (Shinde and Inouye 1993) that ensures correct folding of the enzyme, inhibits premature proteolytic activity (Kojima et al. 1997), and is autocatalytically cleaved off during enzyme maturation (Gallagher et al. 1995; Yabuta et al. 2001).
We have identified in the genome sequence of P. aeruginosa PAO1 (Stover et al. 2000), the open reading frame sprP (PA1242) encoding a so far hypothetical protein with homology to enzymes of the subtilisin-like peptidase family S8. The predicted protein SprP contains a signal sequence and a peptidase S8 domain. We have demonstrated that SprP exhibits protease activity and further report on the role of SprP for various important cellular functions in P. aeruginosa including growth, cellular motility, biofilm formation, and pyoverdine production.
Experimental Procedures
Bacterial strains, plasmids, media, and culture conditions
The strains and plasmids used in this study are listed in Table 1. Escherichia coli DH5α was used as host for cloning and expression; E. coli S17-1 was used for conjugal transfer. All strains were grown in lysogeny broth (LB) medium (10 g/L tryptone, 5 g/L yeast extract, 10 g/L NaCl). For anaerobic growth conditions, 50 mmol/L KNO3 was added to the medium. Cultures grown for 16 h at 37°C in LB medium (volume: 10 mL) in Erlenmeyer flasks were used to inoculate main cultures (volume: 25 mL) to an initial optical density of OD580nm = 0.1. Cultures were grown at 37°C in a rotating shaker at 150 rpm until they reached an optical density of OD580nm = 2.5. Sodium nitroprusside (SNP) was added to LB-medium at a concentration of 2 mmol/L. For E. coli, chloramphenicol (50 μg/mL) was used and for P. aeruginosa, chloramphenicol (300 μg/mL), and gentamycin (30 μg/mL) was added.
Table 1.
Strains, plasmids, and primers used in this study.
| Strain or plasmid | Genotype/Phenotype | Reference or source |
|---|---|---|
| Strains | ||
| Pseudomonas aeruginosa | ||
| PAO1 | Wild type | Holloway et al. (1979) |
| ΔsprP | ΔsprP (sprP::ΩGmr) | This work |
| Escherichia coli | ||
| DH5α | supE44 Δ(lacZYA-argF)U196 (Δ80ΔlacZM15) hsdR17 recA1 endA1 gyrA96 thi-1 relA1 | Woodcock et al. (1989) |
| S17-1 | Ec294::[RP4-2 (Tc::Mu) (Km::Tn7)], pro, res, recA, tra+, Tpr, Smr | Simon et al. (1983) |
| Plasmids | ||
| pBBR1MCS | Cmr mob lacZα Plac PT7 | Kovach et al. (1994) |
| pSUP202 | pBR325, Apr Cmr Tcr mob | Simon et al. (1983) |
| pBSL142 | ColE1 Apr Gmr | Alexeyev et al. (1995) |
| pBCSK | P37 PT3 Plac lacZ' Cmr ColE1 | Stratagene |
| pTZ110 | Cbr; promoterless lacZ | Schweizer and Chuanchuen (2001) |
| Primer | Cloning site | |
| sprP_up | CCGAAGCTTCCGGAAGCCGAGTCCATGCCG | HindIII |
| sprP_dw | AAGTGCGGATCCTCAGCGCACGCGCTC | BamHI |
| sprP_lacZup | GGGAATTCGCGCCTGCGGCTTGA | EcoRI |
| sprP_lacZdown | AAGGATCCGGACTCGGCTTCCGGAAC | BamHI |
| USTUP | AAATCTAGATGGAGCGGAAACTTCTAGTTA | XbaI |
| USTDW | GGGAATTCACGCGTGGACTCGGCTTCC | EcoRI |
| DSTUP | GGGAATTCACGCGTGGCGTTCCGCCGGCC | EcoRI |
| DSTDW | GGAAGCTTGGTGGTGGATCCGCACCAGTTCGA | HindIII |
Recombinant DNA techniques, gene cloning, and mutant construction
Recombinant DNA techniques were performed essentially as described by Sambrook et al. (1989). DNA fragments were amplified by polymerase chain reaction (PCR) standard methods. DNA modifying enzymes (Fermentas, St-Leon-Roth, Germany) were used according to manufacturer's instructions. Plasmid DNA was prepared as described by Birnboim and Doly (1979) and by using the innuPREP Plasmid Mini Kit (Analytik Jena, Jena, Germany) or, for genomic DNA from P. aeruginosa, the DNeasy Blood & Tissue Kit (Qiagen, Hilden, Germany).
The gene sprP was amplified by PCR using primers sprP_up and sprP_dw (Table 1) and chromosomal DNA of P. aeruginosa PAO1 as the template. The PCR products were digested with HindIII and BamHI, ligated into plasmid pBBR1MCS resulting in plasmid pBBRSP.
Construction of sprP deletion mutant was performed as described by Tielker et al. (2005). The plasmids for genomic deletion of sprP were constructed by PCR amplification of the regions located upstream and downstream of the sprP gene by use of the primers USTUP + USTDW and DSTUP + DSTDW. The resulting fragments were cloned into the XbaI/EcoRI and EcoRI/HindIII sites of plasmid pBCSK giving pUST and pDST, respectively. An Ω-gentamicin cassette was isolated as a 1.6 kb MluI fragment from plasmid pBSL142 and was subsequently cloned into the MluI site of pUST resulting in pUSTGm. The downstream fragment was then cloned into pUSTGm via EcoRI/HindIII to give pSPGm. pSPGm was digested with XbaI/HindIII and blunted with T4-polymerase. The fragment harboring the Ω-gentamicin cassette was ligated into ScaI-digested pSUP202 resulting in the suicide vector pSUSPGm. For the generation of the transcriptional fusion plasmid harboring the promoter region of sprP fused to a promoterless lacZ gene (encoding β-galactosidase), the upstream region of the sprP gene was amplified by use of the primers sprP_lacZup and sprP_lacZdw (Table 1). The fragment was digested with EcoRI/BamHI and cloned into plasmid pTZ110. The resulting plasmid pTZsprP contains the promoter region (556 bp) of sprP fused to the promoterless lacZ gene.
Construction of a P. aeruginosa ΔsprP
A P. aeruginosa sprP negative mutant was constructed by successively using the plasmid pSUSPGm. Replacement of the chromosomal copy of sprP was achieved after conjugation and homologous recombination with pSUSPGm. The sprP gene was deleted after homologous recombination to create an in-frame deletion and resulted in P. aeruginosa ΔsprP as confirmed by PCR. For complementation studies, plasmids pBBR1MCS and pBBRSP were introduced into P. aeruginosa ΔsprP by biparental mating.
Determination of protease activity
Agar plate assay
Bacteria grown as described above were plated on LB agar containing 3% (w/v) skim milk, plates were incubated for 16 h at 37°C and afterward overnight at 4°C to reduce bacterial growth. Clear zones (halos) around the colonies indicate protease activity.
Liquid assay
Whole cell extracts were prepared from cells grown to an OD580nm = 3. Cells were centrifuged at 21,500g for 20 min, resuspended in 0.2 mol/L Tris-HCl buffer, pH 8 (final OD580nm = 10) and sonicated for 2 × 4 min at 25% power. The substrate Suc-Ala-Ala-Pro-Phe-para-nitroanilide (Sigma-Aldrich, Seelze, Germany) was dissolved in 0.2 mol/L Tris-HCl buffer, pH 8, to a final concentration of 10 mmol/L (DelMar et al. 1979). A volume of 30 μL of whole cell extract was added to 35 μL of substrate solution, the reaction tubes were incubated for 16 h at 37°C, and the absorption was determined at 410 nm indicating the release of para-nitroanilide from the substrate.
Determination of β-galactosidase activity
Pseudomonas aeruginosa PAO1 was transformed with plasmid pTZsprP and promoter activity of sprP was monitored by determination of β-galactosidase activity according to the method of Miller (1972). The cultures were grown in 100 mL LB medium and cell growth (OD580nm) was determined. β-galactosidase activity was measured for each sample after cell lysis by sodium dodecyl sulfate (SDS)/chloroform permeabilization. After quenching of the reaction using 1 mol/L Na2CO3 and centrifugation to remove the cell debris, 1 mL of each sample was transferred to a cuvette and the absorption at 420 nm was determined.
Motility assays
The agar plate assays to analyze swarming, swimming, and twitching motilities were performed as described before (O'Toole and Kolter 1998; Rashid and Kornberg 2000; Tremblay and Deziel 2008). M9 minimal broth medium was used for swimming analysis. The swim plates contained, per liter, 4 g glucose, 0.25 g MgSO4, 0.02 g CaCl2, 7 g Na2HPO4, 3 g KH2PO4, 0.5 g NaCl, 1 g NH4Cl, and 0.3% w/v select agar (Invitrogen, Darmstadt, Germany). Swim plates were inoculated with 5 μL bacteria from overnight cultures (OD580nm = 3) grown in LB medium at 37°C. Plates were then incubated at 37°C for 16 h. The swarm plates contained, per liter, 1.07 g NH4Cl, 2.14 g Na2HPO4 × H2O, 2.99 g KH2PO4, 0.5 g NaCl, 0.25 g MgSO4, 0.15 g CaCl2 × 2 H2O, 1.98 g glucose, 5 g casamino acids, and 5 g select agar. Plates were dried for 30 min before use, inoculated with 5 μL bacteria from overnight cultures (OD580nm = 3) and incubated at 37°C for 16 h. The LB medium used to analyze the twitching motility contained, per liter, 10 g tryptone, 5 g yeast extract, 10 g NaCl, and 1.5% w/v agar. Plates were inoculated from overnight cultures of bacteria that grew on LB plates (1.5% w/v agar) using a sharp toothpick stabbed through the LB agar onto the bottom of the petri dish. The zone of motility, visible at the interface between agar and petri dish, was documented after incubation for 48 h at 37°C.
Aggregate formation assay
Different bacterial strains were transferred into 25 mL LB medium to an initial optical density of 0.1 (OD580nm) and the cultures were cultivated at 37°C until a cell density of OD580nm = 2.5 was reached. The formed aggregates were documented using photography.
Biofilm formation
The tests were adapted from the method originally described by O'Toole and Kolter (1998). Bacteria were grown for 16 h at 37°C in LB medium in 24-well microtiter-plates. Cells attached to the plates were subsequently stained by incubation with 200 μL 1% crystal violet (CV) for 20 min. Unattached cells were removed by rinsing the wells with water. The water remaining in the wells was evaporated at room temperature. The biofilm formation was photo documented. After addition of 1 mL ethanol the absorption at 590 nm was determined to quantify biofilm formation.
Quantification of pyoverdine production
Pseudomonas aeruginosa strains were grown in 10 mL LB medium to an OD580nm = 2.5 and pyoverdine concentrations in cell-free culture supernatants, filtered with 0.22 μm syringe filter (VWR International, Langenfeld, Germany), were determined as described (Meyer and Abdallah 1978).
Anaerobic growth
Pseudomonas aeruginosa strains were grown anaerobically in 20 mL LB medium containing 50 mmol/L KNO3 as electron acceptor. Prior to inoculation, the Erlenmeyer flasks were rubber plugged and flushed with N2 to create an oxygen free atmosphere; residual oxygen is rapidly consumed by growing bacteria. As a control, cells were grown under identical conditions, but aerobically, that is without rubber-plugged flasks and exposure to N2. Cell growth was determined as OD580nm during incubation at 37°C and rotary shaking after 6 h.
DNA microarray analysis
Array preparation
DNA microarrays were obtained from Agilent Technologies (Waldbronn, Germany). Agilent's eArray platform was used to design oligonucleotide probes and assemble the custom 4 × 44 K 60mer microarray (https://earray.chem.agilent.com/earray/). For genome-wide gene expression analysis of P. aeruginosa PAO1, the cDNA sequences of the genome annotation from National Center for Biotechnology Information (NCBI) (NC_002516) listing 5571 protein coding genes and 106 structural RNA coding genes were used as input in eArray to design one 60-mer probe for each gene with the best probe methodology. Furthermore, the cDNA sequences representing the 200 bp upstream region relative to the annotated start of each gene was used to design one 60-mer probe for each gene using the best probe methodology. For P. aeruginosa PAO1, 5636 specific probes for the annotated genes and 5649 specific probes for the 200 bp upstream region of the annotated genes were designed by eArray. The custom array design also included Agilent's control spots.
cDNA synthesis and hybridization
Pseudomonas aeruginosa PAO1 and ΔsprP were grown in LB medium until cell growth reached an OD580nm of 2.5. Total RNA was isolated by using the RNeasy kit (Qiagen) and treated with Ambion® DNase I (RNase-free) (Invitrogen, Darmstadt, Germany). cDNA synthesis for DNA microarray analysis was performed as described (Polen et al. 2007). A quantity of 25 μg of RNA was used for random hexamer-primed synthesis of fluorescently labeled cDNA with the fluorescent nucleotide analogues Cy3-dUTP and Cy5-dUTP (GE Healthcare, Freiburg, Germany). The mixture contained 3 μL of Cy3-dUTP or Cy5-dUTP (1 mmol/L), 3 μL of DTT (100 mmol/L), 6 μL of 5× first strand buffer (Invitrogen), 0.6 μL of dNTP-mix (25 mmol/L each of dATP, dCTP, and dGTP and 10 mmol/L dTTP), and 2 μL of Superscript II reverse transcriptase (Invitrogen, Darmstadt, Germany). The mixture was incubated for 10 min at room temperature and 110 min at 42°C, stopped by addition of 10 μL of 0.1 N NaOH, incubated for 10 min at 70°C, and then neutralized by addition of 10 μL of 0.1 N HCl. cDNA samples were purified by washing and centrifugation three times with water using Microcon columns (Merck, Darmstadt, Germany, YM-30). Purified cDNA samples to be compared were pooled and the prepared two-color samples (wild-type control vs. sprP-mutant strain) were hybridized at 65°C for 17 h using Agilent's Gene Expression Hybridization Kit and hybridization chamber. After hybridization the arrays were washed using Agilent's Wash Buffer Kit according to the manufacturer's instructions. Fluorescence of hybridized DNA microarrays was determined at 532 nm (Cy3-dUTP) and 635 nm (Cy5-dUTP) at 5 μm resolution with a GenePix 4000B laser scanner and GenePix Pro 6.0 software (Molecular Devices, Sunnyvale, CA). Fluorescence images were saved to raw data files in TIFF format (GenePix Pro 7.0).
Data analysis
Quantitative TIFF image analysis was carried out using GenePix image analysis software and results were saved as GPR-file (GenePix Pro 7.0). For background correction of spot intensities, ratio calculation and ratio normalization, GPR-files were processed using the BioConductor R-packages limma and marray (http://www.bioconductor.org). For further analysis, the processed and loess-normalized data as well as detailed experimental information according to MIAME (Brazma 2009) were stored in the DNA microarray database of the Center of Molecular Biotechnology in the Forschungszentrum Jülich GmbH (Polen and Wendisch 2004). The DNA microarray analysis was repeated independently five times by biological replicates. To search the data for differentially expressed genes by the processed Cy5/Cy3 ratio reflecting the relative RNA level, the criteria flags ≥0 (GenePix Pro 7.0) and signal/noise ≥3 for Cy5 (F635Median/B635Median) or Cy3 (F532Median/B532Median) were used. If the signal/noise of Cy5 and of Cy3 were <3 then signals were considered as to weak to analyze the Cy5/Cy3 ratio of a gene. Furthermore, P-values were calculated by a paired Student's t-test comparing the relative RNA levels of a gene in the replicates to the relative RNA levels of all other genes in the replicates. In addition, we also calculated adjusted P-values according to the method of Benjamini and Hochberg (1995), which is implemented in the R-package limma in the p.adjust function.
Computer analysis
The sprP gene sequence, the SprP protein sequence, and the downstream/upstream regions of sprP were retrieved from the pseudomonas.com database (Winsor et al. 2011). The prediction of the peptidase S8 domain and catalytic aa was performed with the conserved domain sequence (CDS) alignment search tool (Marchler-Bauer et al. 2011) offered by NCBI. The SprP protein sequence was used as a template and an E-value threshold of 0.01 was set. The signal peptide prediction was performed by using SignaP 4.0 (Petersen et al. 2011). General information about subtilisin-like serine proteases and protein classification was done with MEROPS (Rawlings et al. 2012) and the Prokaryote Subtilase Database (Siezen et al. 2007).
Results
SprP sequence analysis
A similarity search within the P. aeruginosa PAO1 genome using the aa sequence of Bacillus licheniformis subtilisin Carlsberg (Jacobs et al. 1985) revealed only two proteins with low but significant identities of 29% (PA1242) and 27% (EprS), respectively. These proteases were further analyzed for conserved domains (Marchler-Bauer et al. 2011) and orf PA1242 was identified to encode a putative protease previously annotated as a hypothetical protein in the Pseudomonas Genome Database (Winsor et al. 2011). It consists of 590 aa with a deduced molecular mass of ca. 64.9 kDa. The newly identified protein harbors an N-terminal type I export signal peptide of 21 aa followed by a 233 aa domain without any similarity to known proteins which was thus qualified as domain of unknown function (DUF). The C-terminal catalytic domain (aa 256–531) contains a putative protease motif of the MEROPS peptidase family S8 (Fig. 1). Given this similarity to the subtilisin-like family of proteases, we have named the protein encoded by PA1242 SprP (subtilisin-like protease P). A database search using the aa sequence of the DUF domain as template revealed 69 proteins with homology to hypothetical proteins and subtilases from various Pseudomonas species, namely P. aeruginosa, Pseudomonas alcaliphila, Pseudomonas mendocina, Pseudomonas brassicacearum, Pseudomonas fluorescens, Pseudomonas poae, Pseudomonas putida, Pseudomonas fulva, Pseudomonas viridiflava, and Pseudomonas stutzeri with identities varying from 100% to 53.4%. Interestingly, only three of the identified proteins belong to species other than Pseudomonas: a hypothetical protein from Halomonas boliviensis LC1 (58.7%), a predicted subtilase from Halomonas sp. HAL1 (58.6%) and a predicted subtilase from Halomonas zhanjiangensis (56.4%).
Figure 1.
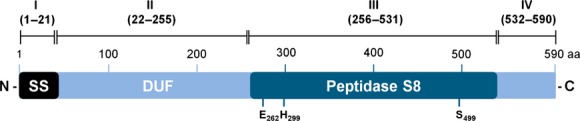
SprP domain composition. SprP consists of 590 amino acids (aa) with an N-terminal signal sequence (I, SS), a domain of unknown function (II, DUF), a peptidase S8 domain (III), and a C-terminal extension (IV). Numbers indicate the amino acids flanking the respective domains; putative catalytic triad residues E262, H299, and S499 are labeled.
SprP is a serine protease
Escherichia coli cells harboring the sprP gene on plasmid pBBRSP were grown on agar plates containing skim milk as the protease substrate. Protease activity indicated by the formation of clear halos around the colonies was exclusively detectable with E. coli harboring the sprP expression plasmid whereas the vector control did not show halo formation. SprP variant S499A which had the catalytic serine replaced by alanine also did not show protease activity (Fig. 2A). Protease activity was quantified using the synthetic substrate N-succinyl–L-Ala–L-Ala–L-Pro–L-Phe–p-nitroanilide which is specifically hydrolyzed by serine proteases (Brode et al. 1996) (Fig. 2B).
Figure 2.

SprP is a protease. (A) Escherichia coli DH5α was transformed with plasmid pBBRSP harboring sprP (right) or empty vector (left), plated on agar containing 3% skim milk and plates were incubated for 16 h at 37°C. (B) Cell extracts of strains shown in (A) were prepared after centrifugation and subsequent sonication and tested for proteolytic activity with Suc-Ala-Ala-Pro-Phe-p-nitroanilide as the substrate. Relative activity of 100% corresponds to 0.308; error bars indicate standard deviation.
The sprP gene is expressed during onset of the stationary phase
Plasmid pTZsprP was constructed harboring a transcriptional fusion of a 556 bp DNA fragment located upstream the sprP start codon with lacZ and transformed into P. aeruginosa PAO1. Bacteria were grown in LB medium and β-galactosidase activity was determined (Fig. 3). Activity of β-galactosidase steadily increased until it reached 1650 Miller units after 9 h to 12 h of growth at a cell density corresponding to an OD580nm of 2.5. During the period from 9 h to 14 h, β-galactosidase activity remained constant and then started to decrease after 14 h. Apparently, the highest expression level of sprP is reached at an optical density of OD580nm = 2.5; therefore, all further experiments were carried out with cells harvested at this optical density.
Figure 3.
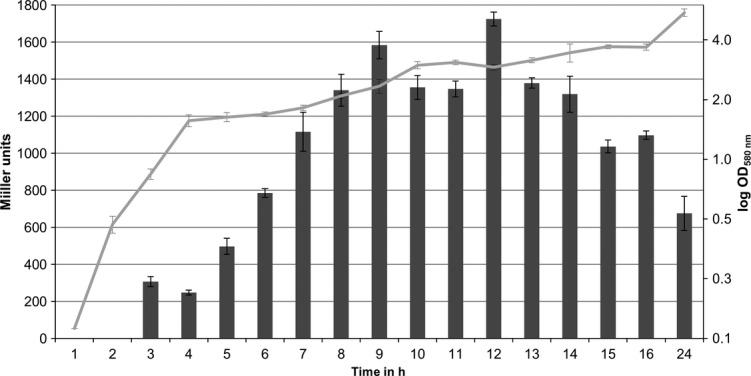
Promoter activity of sprP. Pseudomonas aeruginosa PAO1 containing plasmid pTZsprP, which harbors a 556 bp DNA fragment upstream of sprP fused to lacZ was inoculated to an initial cell density of OD580nm = 0.1 and incubated for 24 h at 37°C. Activity of β-galactosidase (bars) was determined as described by Miller (1972) and the corresponding growth of the culture (line) was determined as absorbance of the bacterial culture at 580 nm. Error bars indicate standard deviation.
Deletion of sprP promotes formation of cell aggregates and biofilms but abolishes cell motility
A sprP-negative P. aeruginosa mutant was constructed to elucidate the physiological role of SprP. Upon growth of P. aeruginosa ΔsprP, extensive cell aggregation was observed during the entire cultivation period (Fig. 4A). Apparently, cell aggregation reached a maximum at maximal sprP expression; furthermore, aggregation of P. aeruginosa ΔsprP could be prevented by providing sprP on plasmid pBBRSP. Cell aggregation was shown to be linked to biofilm formation (Schleheck et al. 2009), we therefore analyzed biofilm formation using polystyrene microtiter-plates. P. aeruginosa ΔsprP showed an almost sixfold increased biofilm formation as compared to the wild-type strain P. aeruginosa PAO1 (Fig. 4B). Biofilm formation could also be reduced to almost wild-type level by complementation in trans of P. aeruginosa ΔsprP with sprP.
Figure 4.
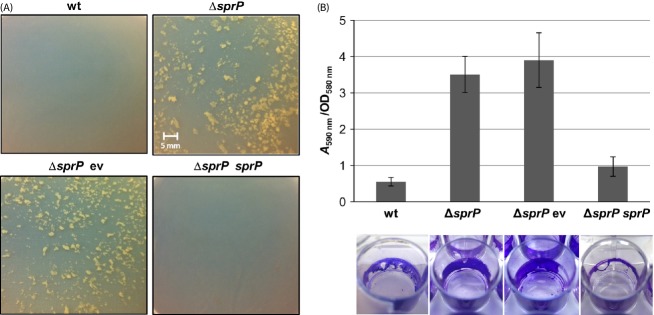
SprP affects cell aggregation and biofilm formation. Pseudomonas aeruginosa wild-type, ΔsprP mutant and ΔsprP mutant complemented with plasmid pBBRSP were inoculated to an initial cell density of OD580nm = 0.1 and incubated until the cultures reached an OD580nm of 2.5. (A) Cell aggregation was examined, cells transferred to petri dishes, and photographed. (B) Biofilm formation was analyzed in microtiter-plates inoculated with different strains and cultivated for 16 h at 37°C. Biofilms were stained with crystal violet, photographed, and dye was extracted with 100% ethanol and quantified photometrically at 590 nm. Biofilm formation is indicated as bound dye (A590nm) per cell density (OD580nm). Error bars indicate standard deviation. wt = P. aeruginosa PAO1, ΔsprP = P. aeruginosa ΔsprP harboring either ev = empty vector or sprP = plasmid pBBRSP.
Furthermore, biofilm formation by P. aeruginosa is linked to cell motility (Wilhelm et al. 2007; Rosenau et al. 2010; Tielen et al. 2010). Examination of P. aeruginosa ΔsprP for its swimming, swarming, and twitching motility revealed that all three types of motility were virtually abolished, but they could be reconstituted by complementation with plasmid-encoded sprP (Fig. 5).
Figure 5.

Deletion of sprP abolishes cell motility of Pseudomonas aeruginosa. Swarming and swimming agar plates contained M9 minimal medium with 0.5% agar for swarming and 0.3% agar for swimming. 5 μL of bacterial culture at OD580nm = 3 were added. Twitching motility was assessed after stabbing cells through 3 mm-thick LB agar plates onto the ground of the petri dishes. All plates were incubated for 16 h at 37°C. wt = P. aeruginosa PAO1, ΔsprP = P. aeruginosa ΔsprP harboring either ev = empty vector or sprP = plasmid pBBRSP.
Deletion of sprP results in pleiotropic effects at the level of transcription
The transcriptomes of P. aeruginosa wild-type and ΔsprP mutant were comparatively analyzed (Tables S1 and S2). The strains were grown in LB medium until cell growth reached an OD580nm of 2.5. A microarray analysis revealed the downregulation of 102 genes (Table S1) and upregulation of 116 genes (Table S2) in P. aeruginosa ΔsprP. Conditions were set such that regulation is ≥ twofold, P-value is ≤0.05, and the respective gene shows regulation in ≥3 independent experiments. Among the downregulated genes, 36 encode hypothetical proteins, five probable transcriptional regulators, and several genes are involved in the denitrification system. Upregulated genes include 46 encoding hypothetical proteins, three encoding probable transcriptional regulators, three sigma factors, and several genes belonging to the iron uptake system. A subset of the genes identified by transcriptome analysis was also tested for differential gene expression by quantitative PCR. The results confirmed the transcriptome data (Table S3).
Deletion of sprP increases pyoverdine production and reduces growth under anaerobic conditions
In P. aeruginosa ΔsprP several genes involved in pyoverdine production were strongly upregulated. These results are consistent with the observation that a P. aeruginosa ΔsprP culture showed an intense green color indicating strongly increased production of pyoverdines. The culture supernatant of P. aeruginosa ΔsprP contained almost fivefold the amount of pyoverdine as compared to the wild-type supernatant, complementation with the sprP gene reduced pyoverdine concentration to about wild-type level (Fig. 6).
Figure 6.
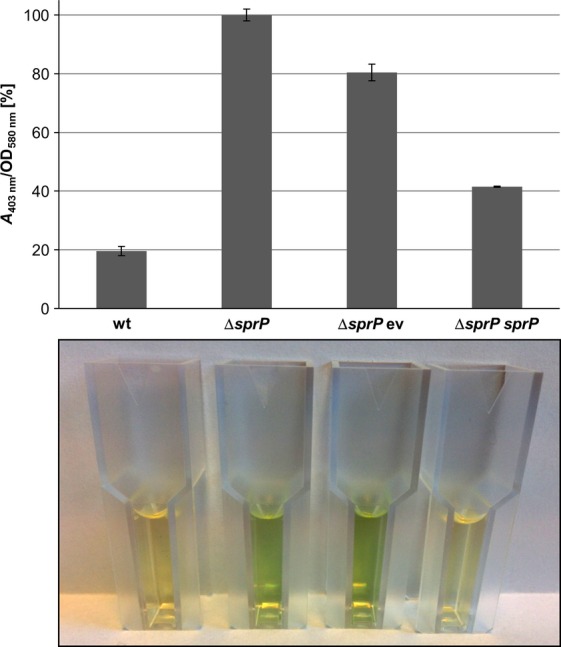
Deletion of sprP increases pyoverdine production by Pseudomonas aeruginosa. Strains were incubated at 37°C until cell growth reached an OD580nm of 2.5 and pyoverdine in cell-free culture supernatants was determined by absorbance at 403 nm (Meyer and Abdallah 1978). Pyoverdine production is indicated as pyoverdine absorbance (A403nm) per cell density (OD580 nm). Error bars indicate standard deviation calculated from biological triplicates. Cuvettes containing culture supernatants are shown underneath the diagram. wt = P. aeruginosa PAO1, ΔsprP = P. aeruginosa ΔsprP harboring either ev = empty vector or sprP = plasmid pBBRSP.
Additionally, we have analyzed growth of P. aeruginosa wild-type and ΔsprP under anaerobic conditions (Fig. 7A) by incubation at 37°C with rotary shaking after 3, 6, and 8 h. The most striking difference in cell growth was observed after 6 h when the cell density was 2.5 times lower under anaerobic than under aerobic conditions for the wild type and 3.8-fold lower for the ΔsprP mutant. The deficiency in anaerobic growth can largely be reverted by expression of the sprP gene in P. aeruginosa ΔsprP. Transcriptome analysis had shown that deletion of sprP lead to a downregulation of genes involved in the denitrification system. Hence, we assumed that the level of the denitrification product nitric oxide (NO) may be reduced. Thus, we added the synthetic NO donor SNP to the growth medium and cultivated the strains until a cell density of OD580nm = 2.5 was reached. Interestingly, addition of 2 mmol/L SNP to the growth medium largely abrogated the cell aggregation phenotype of P. aeruginosa ΔsprP (Fig. 7B).
Figure 7.

SprP affects anaerobic growth of Pseudomonas aeruginosa. (A) Bacterial cultures were incubated at 37°C for 6 h and growth differences under anaerobic and aerobic conditions are illustrated. (B) The cell aggregation phenotype of P. aeruginosa ΔsprP after cultivation in LB-medium and LB-medium supplemented with the nitric oxide donor sodium nitroprusside (SNP). wt = P. aeruginosa PAO1, ΔsprP = P. aeruginosa ΔsprP harboring either ev = empty vector or sprP = plasmid pBBRSP.
Discussion
Proteases are important bacterial virulence factors and several of them are well known for their interaction with host cells during the infection process (Hoge et al. 2010; Pearson et al. 2011; Kida et al. 2013; Tang et al. 2013). The MEROPS peptidase database lists 284 known and putative proteases and 135 non-peptidase homologues for P. aeruginosa (Rawlings et al. 2012). We have newly identified the subtilase SprP of P. aeruginosa which consists of three distinct domains. The large N-terminal (DUF, 233 aa) shows high homology to several hypothetical proteins and putative subtilases. Remarkably, these homologous proteins are almost exclusively encoded in genomes of bacteria belonging to the genus Pseudomonas suggesting a Pseudomonas-specific function. Deletion of domains DUF and S8 both lead to the loss of protease activity of SprP. Also, both constructs were unable to complement the phenotypes observed for P. aeruginosa ΔsprP (data not shown) indicating that full-length SprP is required. Most bacterial subtilases possess aspartic acid, histidine, and serine as catalytic residues thus belonging to the large group of D-H-S family subtilases. For SprP, glutamic acid, histidine, and serine were predicted as catalytic residues thereby assigning SprP to the E-H-S family of subtilases based on the Prokaryotic Subtilase Database (Siezen et al. 2007). SprP variant S499A which had the catalytic serine replaced by alanine did not show protease activity thus corroborating this assumption.
The predicted protease function of PA1242 was demonstrated after expression in E. coli on skim milk agar plates and by using the subtilisin-specific substrate Suc-AAPF-pNA (Brode et al. 1996) (Fig. 2). Experiments using a sprP::lacZ fusion demonstrated maximal expression of sprP at the onset of the stationary growth phase (Fig. 3). However, the absolute promoter activity (indicated in Miller units) was comparatively low (Morabbi Heravi et al. 2011; Salto et al. 1998) suggesting a low expression level of sprP in P. aeruginosa.
Transcriptome analyses revealed the highest level of upregulation for the genes antABC (max. 44.8-fold) and catBCA (max 20-fold) (Table S2). Both operons are involved in the degradation of anthranilate into TCA cycle intermediates (Chang et al. 2003; Urata et al. 2004; Choi et al. 2011). antA is directly activated by the activator AntR (Oglesby et al. 2008; Kim et al. 2012), which is shown to be upregulated after sprP deletion as well (3.9-fold) (Table S2). We did not analyze these effects in more detail but this study indicates that SprP could be of interest for the identification of regulators for the partly unknown regulation of the anthranilate metabolism (Choi et al. 2011). A P. aeruginosa ΔsprP mutant showed strong cell aggregation throughout the entire growth period (Fig. 4A). We also observed that these cell aggregates attach to a polystyrene surface resulting in significantly increased biofilm formation (Fig. 4B). Previously, it was shown that P. aeruginosa PAO1 cells also form aggregates in batch cultures (Schleheck et al. 2009). These aggregates are comparatively small with up to 600 μm in relation to the aggregates formed by the ΔsprP mutant. Furthermore, it was demonstrated that the expression of cdrAB genes resulted in increased biofilm formation and auto-aggregation in liquid cultures (Borlee et al. 2010). Interestingly, both genes are also upregulated in the P. aeruginosa ΔsprP strain (cdrA 3.3-fold, cdrB 4.1-fold; Table S2). Proteins encoded by the cdrAB operon regulate the intracellular level of c-di-GMP in P. aeruginosa that is linked to the production of matrix components and biofilm formation (Tischler and Camilli 2004; Hickman et al. 2005; Borlee et al. 2010; Povolotsky and Hengge 2012). The loss of motility observed for P. aeruginosa ΔsprP may represent another reason for cellular aggregation and increased biofilm formation. Transcriptome analyses of P. aeruginosa ΔsprP revealed a downregulation of pilA gene (2.5-fold) expression (Table S1). Previous studies have demonstrated that a pilA mutant showed a non-twitching phenotype (Chiang and Burrows 2003; Shrout et al. 2006) and may also be defective for swarming (Köhler et al. 2000).
Another surprising observation resulting from transcriptome analysis of P. aeruginosa ΔsprP refers to downregulation of the genes narK1 (21.3-fold), narK2 (5.9-fold), narJ (5.8-fold), narH (3.5-fold), narG (7.2-fold), nirN (2.8-fold), nirJ (2.1-fold), nirL (2.5-fold), nirF (3.9-fold), nirS (fivefold), nosZ (14.5-fold), nosD (4.5-fold) (Table S1), which are organized in five operons (Schobert and Jahn 2010) and are involved in denitrification and anaerobic growth of P. aeruginosa. Indeed, we observed a significantly impaired growth of P. aeruginosa ΔsprP under anaerobic conditions as compared to the wild-type strain (Fig. 7A). NO, a side product of denitrification, can induce dispersal of P. aeruginosa biofilms (Barraud et al. 2006, 2009). We found that genes encoding nitrate reductase (NarGHI) and nitrite reductase (NirS), which are involved in NO production were downregulated in P. aeruginosa ΔsprP. Thus, we speculated that the level of NO might be reduced in the mutant. As expected, addition of nitroprusside as an alternative source of NO significantly reduced cell aggregation of P. aeruginosa ΔsprP (Fig. 7B). Another identified probable protease PA3913 was already linked to anaerobic growth of P. aeruginosa (Filiatrault et al. 2006). It was shown that transposon mutants of PA3913 are unable to grow anaerobically for unknown reasons. This study shows that the putative protease PA3913 is also downregulated 6.4-fold in P. aeruginosa ΔsprP (Table S1). Presumably, both SprP and PA3913 are involved in regulation of anaerobic growth in P. aeruginosa.
The pyoverdine system is well characterized in P. aeruginosa (Ochsner et al. 2002; Lamont and Martin 2003; Cornelis 2010; Funken et al. 2011). We found significant upregulation of expression levels of regulatory genes fpvA (2.4-fold), fpvR (twofold), and pvdS (2.1-fold) as well as genes pvdA (sevenfold), pvdN (fivefold), pvdO (4.3-fold), pvdL (2.8-fold), and pvdH (2.1-fold) (Table S2) resulting in significantly increased pyoverdine production by P. aeruginosa ΔsprP. Increased pyoverdine synthesis and formation of cell aggregates were also observed when P. aeruginosa was grown under iron-limitation in M9 minimal media with succinate as sole carbon source (data not shown).
The relevance of proteases as part of regulatory networks and for virulence of P. aeruginosa was recently demonstrated (Breidenstein et al. 2012; Fernandez et al. 2012). Apparently, several intracellular proteases are involved in the regulation of different phenotypes like biofilm formation, motility, and antibiotic resistance. The exact cellular location of SprP is presently unknown, but an N-terminal signal sequence was predicted and results of Blonder et al. 2004 indicated that SprP is located in the membrane fraction isolated from P. aeruginosa suggesting that SprP may be located in the periplasm associated with the cytoplasmic or the outer membrane. This study adds protease SprP to the collection of P. aeruginosa proteases with an important regulatory function. As a next step, intracellular substrates for SprP need to be identified, for example, by isotope labeling and subsequent mass spectroscopic analyses.
Acknowledgments
As a member of the CLIB2021-Graduate Cluster for Industrial Biotechnology Alexander Pelzer received a scholarship financed by the Ministry of Innovation, Science and Research (MIWF) of North Rhine-Westphalia and the Heinrich-Heine-University of Duesseldorf.
Conflict of Interest
None declared.
Funding Information
As a member of the CLIB2021-Graduate Cluster for Industrial Biotechnology Alexander Pelzer received a scholarship financed by the Ministry of Innovation, Science and Research (MIWF) of North Rhine-Westphalia and the Heinrich-Heine-University of Duesseldorf.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Table S1. Selected genes downregulated in Pseudomonas aeruginosa ΔsprP in comparison to the wild-type strain.
Table S2. Selected genes upregulated in Pseudomonas aeruginosa ΔsprP in comparison to the wild-type strain.
Table S3. Validation of selected genes from microarray analysis by qPCR.
Table S4. Primer used for qPCR in this study.
References
- Alexeyev MF, Shokolenko IN, Croughan TP. Improved antibiotic-resistance gene cassettes and omega elements for Escherichia coli vector construction and in vitro deletion/insertion mutagenesis. Gene. 1995;160:63–67. doi: 10.1016/0378-1119(95)00108-i. [DOI] [PubMed] [Google Scholar]
- Antao CM, Malcata FX. Plant serine proteases: biochemical, physiological and molecular features. Plant Physiol. Biochem. 2005;43:637–650. doi: 10.1016/j.plaphy.2005.05.001. [DOI] [PubMed] [Google Scholar]
- Barraud N, Hassett DJ, Hwang SH, Rice SA, Kjelleberg S, Webb JS. Involvement of nitric oxide in biofilm dispersal of Pseudomonas aeruginosa. J. Bacteriol. 2006;188:7344–7353. doi: 10.1128/JB.00779-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barraud N, Schleheck D, Klebensberger J, Webb JS, Hassett DJ, Rice SA, et al. Nitric oxide signaling in Pseudomonas aeruginosa biofilms mediates phosphodiesterase activity, decreased cyclic di-GMP levels, and enhanced dispersal. J. Bacteriol. 2009;191:7333–7342. doi: 10.1128/JB.00975-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. Controlling the fasle discovery rate – a practical and powerful approach to multiple testing. J. Roy. Stat. Soc. 1995;57:289–300. [Google Scholar]
- Bergeron F, Leduc R, Day R. Subtilase-like pro-protein convertases: from molecular specificity to therapeutic applications. J. Mol. Endocrinol. 2000;24:1–22. doi: 10.1677/jme.0.0240001. [DOI] [PubMed] [Google Scholar]
- Birnboim HC, Doly J. A rapid alkaline extraction procedure for screening recombinant plasmid DNA. Nucleic Acids Res. 1979;7:1513–1523. doi: 10.1093/nar/7.6.1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blonder J, Goshe MB, Xiao W, Camp DG, Wingerd M, Davis RW, et al. Global analysis of the membrane subproteome of Pseudomonas aeruginosa using liquid chromatography-tandem mass spectrometry. J. Proteome Res. 2004;3:434–444. doi: 10.1021/pr034074w. [DOI] [PubMed] [Google Scholar]
- Borlee BR, Goldman AD, Murakami K, Samudrala R, Wozniak DJ, Parsek MR. Pseudomonas aeruginosa uses a cyclic-di-GMP-regulated adhesin to reinforce the biofilm extracellular matrix. Mol. Microbiol. 2010;75:827–842. doi: 10.1111/j.1365-2958.2009.06991.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brazma A. Minimum Information About a Microarray Experiment (MIAME) – successes, failures, challenges. ScientificWorldJournal. 2009;9:420–423. doi: 10.1100/tsw.2009.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breidenstein EB, Janot L, Strehmel J, Fernandez L, Taylor PK, Kukavica-Ibrulj I, et al. The Lon protease is essential for full virulence in Pseudomonas aeruginosa. PLoS ONE. 2012;7:e49123. doi: 10.1371/journal.pone.0049123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brode PF, III, Erwin CR, Rauch DS, Barnett BL, Armpriester JM, Wang ES, et al. Subtilisin BPN' variants: increased hydrolytic activity on surface-bound substrates via decreased surface activity. Biochemistry. 1996;35:3162–3169. doi: 10.1021/bi951990h. [DOI] [PubMed] [Google Scholar]
- Chang HK, Mohseni P, Zylstra GJ. Characterization and regulation of the genes for a novel anthranilate 1,2-dioxygenase from Burkholderia cepacia DBO1. J. Bacteriol. 2003;185:5871–5881. doi: 10.1128/JB.185.19.5871-5881.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chastre J, Fagon JY. Ventilator-associated pneumonia. Am. J. Respir. Crit. Care Med. 2002;165:867–903. doi: 10.1164/ajrccm.165.7.2105078. [DOI] [PubMed] [Google Scholar]
- Chiang P, Burrows LL. Biofilm formation by hyperpiliated mutants of Pseudomonas aeruginosa. J. Bacteriol. 2003;185:2374–2378. doi: 10.1128/JB.185.7.2374-2378.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi Y, Park HY, Park SJ, Park SJ, Kim SK, Ha C, et al. Growth phase-differential quorum sensing regulation of anthranilate metabolism in Pseudomonas aeruginosa. Mol. Cells. 2011;32:57–65. doi: 10.1007/s10059-011-2322-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornelis P. Iron uptake and metabolism in pseudomonads. Appl. Microbiol. Biotechnol. 2010;86:1637–1645. doi: 10.1007/s00253-010-2550-2. [DOI] [PubMed] [Google Scholar]
- DelMar EG, Largman C, Brodrick JW, Geokas MC. A sensitive new substrate for chymotrypsin. Anal. Biochem. 1979;99:316–320. doi: 10.1016/s0003-2697(79)80013-5. [DOI] [PubMed] [Google Scholar]
- Drake D, Montie TC. Flagella, motility and invasive virulence of Pseudomonas aeruginosa. J. Gen. Microbiol. 1988;134:43–52. doi: 10.1099/00221287-134-1-43. [DOI] [PubMed] [Google Scholar]
- Fernandez L, Breidenstein EB, Song D, Hancock RE. Role of intracellular proteases in the antibiotic resistance, motility, and biofilm formation of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2012;56:1128–1132. doi: 10.1128/AAC.05336-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filiatrault MJ, Picardo KF, Ngai H, Passador L, Iglewski BH. Identification of Pseudomonas aeruginosa genes involved in virulence and anaerobic growth. Infect. Immun. 2006;74:4237–4245. doi: 10.1128/IAI.02014-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujitani S, Sun HY, Yu VL, Weingarten JA. Pneumonia due to Pseudomonas aeruginosa: part I: epidemiology, clinical diagnosis, and source. Chest. 2011;139:909–919. doi: 10.1378/chest.10-0166. [DOI] [PubMed] [Google Scholar]
- Funken H, Knapp A, Vasil ML, Wilhelm S, Jaeger KE, Rosenau F. The lipase LipA (PA2862) but not LipC (PA4813) from Pseudomonas aeruginosa influences regulation of pyoverdine production and expression of the sigma factor PvdS. J. Bacteriol. 2011;193:5858–5860. doi: 10.1128/JB.05765-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher T, Gilliland G, Wang L, Bryan P. The prosegment-subtilisin BPN' complex: crystal structure of a specific ‘foldase’. Structure. 1995;3:907–914. doi: 10.1016/S0969-2126(01)00225-8. [DOI] [PubMed] [Google Scholar]
- Ghafoor A, Hay ID, Rehm BH. Role of exopolysaccharides in Pseudomonas aeruginosa biofilm formation and architecture. Appl. Environ. Microbiol. 2011;77:5238–5246. doi: 10.1128/AEM.00637-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottesman S. Proteases and their targets in Escherichia coli. Annu. Rev. Genet. 1996;30:465–506. doi: 10.1146/annurev.genet.30.1.465. [DOI] [PubMed] [Google Scholar]
- Gupta SK, Berk RS, Masinick S, Hazlett LD. Pili and lipopolysaccharide of Pseudomonas aeruginosa bind to the glycolipid asialo GM1. Infect. Immun. 1994;62:4572–4579. doi: 10.1128/iai.62.10.4572-4579.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harmsen M, Yang L, Pamp SJ, Tolker-Nielsen T. An update on Pseudomonas aeruginosa biofilm formation, tolerance, and dispersal. FEMS Immunol. Med. Microbiol. 2010;59:253–268. doi: 10.1111/j.1574-695X.2010.00690.x. [DOI] [PubMed] [Google Scholar]
- Henrichsen J. Bacterial surface translocation: a survey and a classification. Bacteriol. Rev. 1972;36:478–503. doi: 10.1128/br.36.4.478-503.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heravi KM, Wenzel M, Altenbuchner J. Regulation of mtl operon promoter of Bacillus subtilis: requirements of its use in expression vectors. Microb. Cell Fact. 2011;10:83. doi: 10.1186/1475-2859-10-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickman JW, Tifrea DF, Harwood CS. A chemosensory system that regulates biofilm formation through modulation of cyclic diguanylate levels. Proc. Natl. Acad. Sci. USA. 2005;102:14422–14427. doi: 10.1073/pnas.0507170102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoge R, Pelzer A, Rosenau F, Wilhelm S, Méndez-Vilas A. Current research, technology and education topics in applied microbiology and microbial biotechnology. Badajoz, Spain: Formatex Research Center; 2010. Weapons of a pathogen: proteases and their role in virulence of Pseudomonas aeruginosa; pp. 383–395. [Google Scholar]
- Holloway BW, Krishnapillai V, Morgan AF. Chromosomal genetics of Pseudomonas. Microbiol. Rev. 1979;43:73–102. doi: 10.1128/mr.43.1.73-102.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs M, Eliasson M, Uhlen M, Flock JI. Cloning, sequencing and expression of subtilisin Carlsberg from Bacillus licheniformis. Nucleic Acids Res. 1985;13:8913–8926. doi: 10.1093/nar/13.24.8913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kida Y, Taira J, Yamamoto T, Higashimoto Y, Kuwano K. EprS, an autotransporter protein of Pseudomonas aeruginosa, possessing serine protease activity induces inflammatory responses through protease-activated receptors. Cell. Microbiol. 2013;15:1168–1181. doi: 10.1111/cmi.12106. [DOI] [PubMed] [Google Scholar]
- Kim SK, Im SJ, Yeom DH, Lee JH. AntR-mediated bidirectional activation of antA and antR, anthranilate degradative genes in Pseudomonas aeruginosa. Gene. 2012;505:146–152. doi: 10.1016/j.gene.2012.05.004. [DOI] [PubMed] [Google Scholar]
- Klausen M, Heydorn A, Ragas P, Lambertsen L, Aaes-Jorgensen A, Molin S, et al. Biofilm formation by Pseudomonas aeruginosa wild type, flagella and type IV pili mutants. Mol. Microbiol. 2003;48:1511–1524. doi: 10.1046/j.1365-2958.2003.03525.x. [DOI] [PubMed] [Google Scholar]
- Köhler T, Curty LK, Barja F, van Delden C, Pechere JC. Swarming of Pseudomonas aeruginosa is dependent on cell-to-cell signaling and requires flagella and pili. J. Bacteriol. 2000;182:5990–5996. doi: 10.1128/jb.182.21.5990-5996.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kojima S, Minagawa T, Miura K. The propeptide of subtilisin BPN' as a temporary inhibitor and effect of an amino acid replacement on its inhibitory activity. FEBS Lett. 1997;411:128–132. doi: 10.1016/s0014-5793(97)00678-9. [DOI] [PubMed] [Google Scholar]
- Kovach ME, Phillips RW, Elzer PH, Roop RM, II, Peterson KM. pBBR1MCS: a broad-host-range cloning vector. Biotechniques. 1994;16:800–802. [PubMed] [Google Scholar]
- Lamont IL, Beare PA, Ochsner U, Vasil AI, Vasil ML. Siderophore-mediated signaling regulates virulence factor production in Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. USA. 2002;99:7072–7077. doi: 10.1073/pnas.092016999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamont IL, Martin LW. Identification and characterization of novel pyoverdine synthesis genes in Pseudomonas aeruginosa. Microbiology. 2003;149:833–842. doi: 10.1099/mic.0.26085-0. [DOI] [PubMed] [Google Scholar]
- Maeda T, Garcia-Contreras R, Pu M, Sheng L, Garcia LR, Tomas M, et al. Quorum quenching quandary: resistance to antivirulence compounds. ISME J. 2012;6:493–501. doi: 10.1038/ismej.2011.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchler-Bauer A, Lu S, Anderson JB, Chitsaz F, Derbyshire MK, DeWeese-Scott C, et al. CDD: a Conserved Domain Database for the functional annotation of proteins. Nucleic Acids Res. 2011;39(Database issue):D225–D259. doi: 10.1093/nar/gkq1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattick JS. Type IV pili and twitching motility. Annu. Rev. Microbiol. 2002;56:289–314. doi: 10.1146/annurev.micro.56.012302.160938. [DOI] [PubMed] [Google Scholar]
- Meyer JM, Abdallah MA. The Fluorescent Pigment of Pseudomonas fluorescens: biosynthesis, Purification and Physicochemical Properties. J. Gen. Microbiol. 1978;107:319–328. [Google Scholar]
- Mikkelsen H, Sivaneson M, Filloux A. Key two-component regulatory systems that control biofilm formation in Pseudomonas aeruginosa. Environ. Microbiol. 2011;13:1666–1681. doi: 10.1111/j.1462-2920.2011.02495.x. [DOI] [PubMed] [Google Scholar]
- Miller JH. Experiments in molecular genetics. Cold Spring Harbour, New York, NY: Cold Spring Harbour Laboratory; 1972. [Google Scholar]
- Murray TS, Egan M, Kazmierczak BI. Pseudomonas aeruginosa chronic colonization in cystic fibrosis patients. Curr. Opin. Pediatr. 2007;19:83–88. doi: 10.1097/MOP.0b013e3280123a5d. [DOI] [PubMed] [Google Scholar]
- Nicas TI, Iglewski BH. Production of elastase and other exoproducts by environmental isolates of Pseudomonas aeruginosa. J. Clin. Microbiol. 1986;23:967–969. doi: 10.1128/jcm.23.5.967-969.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Toole GA, Kolter R. Flagellar and twitching motility are necessary for Pseudomonas aeruginosa biofilm development. Mol. Microbiol. 1998;30:295–304. doi: 10.1046/j.1365-2958.1998.01062.x. [DOI] [PubMed] [Google Scholar]
- Ochsner UA, Wilderman PJ, Vasil AI, Vasil ML. GeneChip expression analysis of the iron starvation response in Pseudomonas aeruginosa: identification of novel pyoverdine biosynthesis genes. Mol. Microbiol. 2002;45:1277–1287. doi: 10.1046/j.1365-2958.2002.03084.x. [DOI] [PubMed] [Google Scholar]
- Oglesby AG, Farrow JM, III, Lee JH, Tomaras AP, Greenberg EP, Pesci EC, et al. The influence of iron on Pseudomonas aeruginosa physiology: a regulatory link between iron and quorum sensing. J. Biol. Chem. 2008;283:15558–15567. doi: 10.1074/jbc.M707840200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payne DJ. Microbiology. Desperately seeking new antibiotics. Science. 2008;321:1644–1645. doi: 10.1126/science.1164586. [DOI] [PubMed] [Google Scholar]
- Pearson JS, Riedmaier P, Marches O, Frankel G, Hartland EL. A type III effector protease NleC from enteropathogenic Escherichia coli targets NF-kappaB for degradation. Mol. Microbiol. 2011;80:219–230. doi: 10.1111/j.1365-2958.2011.07568.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pesci EC, Milbank JB, Pearson JP, McKnight S, Kende AS, Greenberg EP, et al. Quinolone signaling in the cell-to-cell communication system of Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. USA. 1999;96:11229–11234. doi: 10.1073/pnas.96.20.11229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen TN, Brunak S, von Heijne G, Nielsen H. SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat. Methods. 2011;8:785–786. doi: 10.1038/nmeth.1701. [DOI] [PubMed] [Google Scholar]
- Polen T, Wendisch VF. Genomewide expression analysis in amino acid-producing bacteria using DNA microarrays. Appl. Biochem. Biotechnol. 2004;118:215–232. doi: 10.1385/abab:118:1-3:215. [DOI] [PubMed] [Google Scholar]
- Polen T, Schluesener D, Poetsch A, Bott M, Wendisch VF. Characterization of citrate utilization in Corynebacterium glutamicum by transcriptome and proteome analysis. FEMS Microbiol. Lett. 2007;273:109–119. doi: 10.1111/j.1574-6968.2007.00793.x. [DOI] [PubMed] [Google Scholar]
- Poole CB, Jin J, McReynolds LA. Subtilisin-like proteases in nematodes. Mol. Biochem. Parasitol. 2007;155:1–8. doi: 10.1016/j.molbiopara.2007.05.001. [DOI] [PubMed] [Google Scholar]
- Povolotsky TL, Hengge R. ‘Life-style’ control networks in Escherichia coli: signaling by the second messenger c-di-GMP. J. Biotechnol. 2012;160:10–16. doi: 10.1016/j.jbiotec.2011.12.024. [DOI] [PubMed] [Google Scholar]
- Ramphal R, Pier GB. Role of Pseudomonas aeruginosa mucoid exopolysaccharide in adherence to tracheal cells. Infect. Immun. 1985;47:1–4. doi: 10.1128/iai.47.1.1-4.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rashid MH, Kornberg A. Inorganic polyphosphate is needed for swimming, swarming, and twitching motilities of Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. USA. 2000;97:4885–4890. doi: 10.1073/pnas.060030097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasko DA, Sperandio V. Anti-virulence strategies to combat bacteria-mediated disease. Nat. Rev. Drug Discovery. 2010;9:117–128. doi: 10.1038/nrd3013. [DOI] [PubMed] [Google Scholar]
- Rawlings ND, Barrett AJ. Families of serine peptidases. Methods Enzymol. 1994;244:19–61. doi: 10.1016/0076-6879(94)44004-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rawlings ND, Barrett AJ, Bateman A. MEROPS: the database of proteolytic enzymes, their substrates and inhibitors. Nucleic Acids Res. 2012;40(Database issue):343–350. doi: 10.1093/nar/gkr987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rezaei E, Safari H, Naderinasab M, Aliakbarian H. Common pathogens in burn wound and changes in their drug sensitivity. Burns. 2011;37:805–807. doi: 10.1016/j.burns.2011.01.019. [DOI] [PubMed] [Google Scholar]
- Rosenau F, Isenhardt S, Gdynia A, Tielker D, Schmidt E, Tielen P, et al. Lipase LipC affects motility, biofilm formation and rhamnolipid production in Pseudomonas aeruginosa. FEMS Microbiol. Lett. 2010;309:25–34. doi: 10.1111/j.1574-6968.2010.02017.x. [DOI] [PubMed] [Google Scholar]
- Salto R, Delgado A, Michan C, Marques S, Ramos JL. Modulation of the function of the signal receptor domain of XylR, a member of a family of prokaryotic enhancer-like positive regulators. J. Bacteriol. 1998;180:600–604. doi: 10.1128/jb.180.3.600-604.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T. Molecular cloning: a laboratory manual. New York, NY: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- Sato H, Okinaga K, Saito H. Role of pili in the pathogenesis of Pseudomonas aeruginosa burn infection. Microbiol. Immunol. 1988;32:131–139. doi: 10.1111/j.1348-0421.1988.tb01372.x. [DOI] [PubMed] [Google Scholar]
- Schleheck D, Barraud N, Klebensberger J, Webb JS, McDougald D, Rice SA, et al. Pseudomonas aeruginosa PAO1 preferentially grows as aggregates in liquid batch cultures and disperses upon starvation. PLoS ONE. 2009;4:e5513. doi: 10.1371/journal.pone.0005513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schobert M, Jahn D. Anaerobic physiology of Pseudomonas aeruginosa in the cystic fibrosis lung. Int. J. Med. Microbiol. 2010;300:549–556. doi: 10.1016/j.ijmm.2010.08.007. [DOI] [PubMed] [Google Scholar]
- Schweizer HP, Chuanchuen R. Small broad-host-range lacZ operon fusion vector with low background activity. Biotechniques. 2001;31:1258. doi: 10.2144/01316bm06. 1260–1262. [DOI] [PubMed] [Google Scholar]
- Shigematsu T, Fukushima J, Oyama M, Tsuda M, Kawamoto S, Okuda K. Iron-Mediated regulation of alkaline proteinase production in Pseudomonas aeruginosa. Microbiol. Immunol. 2001;45:579–590. doi: 10.1111/j.1348-0421.2001.tb01289.x. [DOI] [PubMed] [Google Scholar]
- Shinde U, Inouye M. Intramolecular chaperones and protein folding. Trends Biochem. Sci. 1993;18:442–446. doi: 10.1016/0968-0004(93)90146-e. [DOI] [PubMed] [Google Scholar]
- Shrout JD, Chopp DL, Just CL, Hentzer M, Givskov M, Parsek MR. The impact of quorum sensing and swarming motility on Pseudomonas aeruginosa biofilm formation is nutritionally conditional. Mol. Microbiol. 2006;62:1264–1277. doi: 10.1111/j.1365-2958.2006.05421.x. [DOI] [PubMed] [Google Scholar]
- Siezen RJ, Leunissen JA. Subtilases: the superfamily of subtilisin-like serine proteases. Protein Sci. 1997;6:501–523. doi: 10.1002/pro.5560060301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siezen RJ, Renckens B, Boekhorst J. Evolution of prokaryotic subtilases: genome-wide analysis reveals novel subfamilies with different catalytic residues. Proteins. 2007;67:681–694. doi: 10.1002/prot.21290. [DOI] [PubMed] [Google Scholar]
- Simon R, Priefer U, Puhler A. A broad host range mobilization system for in vivo genetic engineering: transposon mutagenesis in gram negative bacteria. Nat. Biotechnol. 1983;1:784–791. [Google Scholar]
- Stover CK, Pham XQ, Erwin AL, Mizoguchi SD, Warrener P, Hickey MJ, et al. Complete genome sequence of Pseudomonas aeruginosa PAO1, an opportunistic pathogen. Nature. 2000;406:959–964. doi: 10.1038/35023079. [DOI] [PubMed] [Google Scholar]
- Tang A, Caballero AR, Marquart ME, O'Callaghan RJ. Pseudomonas aeruginosa small protease (PASP), a keratitis virulence factor. Invest. Ophthalmol. Vis. Sci. 2013;54:2821–2828. doi: 10.1167/iovs.13-11788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiburzi F, Imperi F, Visca P. Intracellular levels and activity of PvdS, the major iron starvation sigma factor of Pseudomonas aeruginosa. Mol. Microbiol. 2008;67:213–227. doi: 10.1111/j.1365-2958.2007.06051.x. [DOI] [PubMed] [Google Scholar]
- Tielen P, Rosenau F, Wilhelm S, Jaeger KE, Flemming HC, Wingender J. Extracellular enzymes affect biofilm formation of mucoid Pseudomonas aeruginosa. Microbiology. 2010;156(Pt. 7):2239–2252. doi: 10.1099/mic.0.037036-0. [DOI] [PubMed] [Google Scholar]
- Tielker D, Hacker S, Loris R, Strathmann M, Wingender J, Wilhelm S, et al. Pseudomonas aeruginosa lectin LecB is located in the outer membrane and is involved in biofilm formation. Microbiology. 2005;151(Pt. 5):1313–1323. doi: 10.1099/mic.0.27701-0. [DOI] [PubMed] [Google Scholar]
- Tischler AD, Camilli A. Cyclic diguanylate (c-di-GMP) regulates Vibrio cholerae biofilm formation. Mol. Microbiol. 2004;53:857–869. doi: 10.1111/j.1365-2958.2004.04155.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tremblay J, Deziel E. Improving the reproducibility of Pseudomonas aeruginosa swarming motility assays. J. Basic Microbiol. 2008;48:509–515. doi: 10.1002/jobm.200800030. [DOI] [PubMed] [Google Scholar]
- Urata M, Miyakoshi M, Kai S, Maeda K, Habe H, Omori T, et al. Transcriptional regulation of the ant operon, encoding two-component anthranilate 1,2-dioxygenase, on the carbazole-degradative plasmid pCAR1 of Pseudomonas resinovorans strain CA10. J. Bacteriol. 2004;186:6815–6823. doi: 10.1128/JB.186.20.6815-6823.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venturi V. Regulation of quorum sensing in Pseudomonas. FEMS Microbiol. Rev. 2006;30:274–291. doi: 10.1111/j.1574-6976.2005.00012.x. [DOI] [PubMed] [Google Scholar]
- Wilderman PJ, Vasil AI, Johnson Z, Wilson MJ, Cunliffe HE, Lamont IL, et al. Characterization of an endoprotease (PrpL) encoded by a PvdS-regulated gene in Pseudomonas aeruginosa. Infect. Immun. 2001;69:5385–5394. doi: 10.1128/IAI.69.9.5385-5394.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilhelm S, Gdynia A, Tielen P, Rosenau F, Jaeger KE. The autotransporter esterase EstA of Pseudomonas aeruginosa is required for rhamnolipid production, cell motility, and biofilm formation. J. Bacteriol. 2007;189:6695–6703. doi: 10.1128/JB.00023-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winsor GL, Lam DK, Fleming L, Lo R, Whiteside MD, Yu NY, et al. Pseudomonas Genome Database: improved comparative analysis and population genomics capability for Pseudomonas genomes. Nucleic Acids Res. 2011;39(Database issue):D596–D600. doi: 10.1093/nar/gkq869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodcock DM, Crowther PJ, Doherty J, Jefferson S, DeCruz E, Noyer-Weidner M, et al. Quantitative evaluation of Escherichia coli host strains for tolerance to cytosine methylation in plasmid and phage recombinants. Nucleic Acids Res. 1989;17:3469–3478. doi: 10.1093/nar/17.9.3469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu DC, Chan WW, Metelitsa AI, Fiorillo L, Lin AN. Pseudomonas skin infection: clinical features, epidemiology, and management. Am. J. Clin. Dermatol. 2011;12:157–169. doi: 10.2165/11539770-000000000-00000. [DOI] [PubMed] [Google Scholar]
- Yabuta Y, Takagi H, Inouye M, Shinde U. Folding pathway mediated by an intramolecular chaperone: propeptide release modulates activation precision of pro-subtilisin. J. Biol. Chem. 2001;276:44427–44434. doi: 10.1074/jbc.M107573200. [DOI] [PubMed] [Google Scholar]
- Zegans ME, Becker HI, Budzik J, O'Toole G. The role of bacterial biofilms in ocular infections. DNA Cell Biol. 2002;21:415–420. doi: 10.1089/10445490260099700. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Selected genes downregulated in Pseudomonas aeruginosa ΔsprP in comparison to the wild-type strain.
Table S2. Selected genes upregulated in Pseudomonas aeruginosa ΔsprP in comparison to the wild-type strain.
Table S3. Validation of selected genes from microarray analysis by qPCR.
Table S4. Primer used for qPCR in this study.