

Abstract
Objective:
To clarify the genetic, clinicopathologic, and neuroimaging characteristics of patients with hereditary diffuse leukoencephalopathy with spheroids (HDLS) with the colony stimulating factor 1 receptor (CSF-1R) mutation.
Methods:
We performed molecular genetic analysis of CSF-1R in patients with HDLS. Detailed clinical and neuroimaging findings were retrospectively investigated. Five patients were examined neuropathologically.
Results:
We found 6 different CSF-1R mutations in 7 index patients from unrelated Japanese families. The CSF-1R mutations included 3 novel mutations and 1 known missense mutation at evolutionarily conserved amino acids, and 1 novel splice-site mutation. We identified a novel frameshift mutation. Reverse transcription PCR analysis revealed that the frameshift mutation causes nonsense-mediated mRNA decay by generating a premature stop codon, suggesting that haploinsufficiency of CSF-1R is sufficient to cause HDLS. Western blot analysis revealed that the expression level of CSF-1R in the brain from the patients was lower than from control subjects. The characteristic MRI findings were the involvement of the white matter and thinning of the corpus callosum with signal alteration, and sequential analysis revealed that the white matter lesions and cerebral atrophy relentlessly progressed with disease duration. Spotty calcifications in the white matter were frequently observed by CT. Neuropathologic analysis revealed that microglia in the brains of the patients demonstrated distinct morphology and distribution.
Conclusions:
These findings suggest that patients with HDLS, irrespective of mutation type in CSF-1R, show characteristic clinical and neuroimaging features, and that perturbation of CSF-1R signaling by haploinsufficiency may play a role in microglial dysfunction leading to the pathogenesis of HDLS.
Hereditary diffuse leukoencephalopathy with spheroids (HDLS) is a progressive dementing disorder that predominantly affects the cerebral white matter.1 Patients with HDLS are clinically characterized by a gradual onset of cognitive and behavioral dysfunction, followed by motor impairments such as gait disturbance and bradykinesia.1–3 Recently, a gene encoding the colony stimulating factor 1 receptor (CSF-1R) has been identified as a causative gene in patients with HDLS.4 Mutations previously described in patients with HDLS are located within the kinase domain of CSF-1R.4–7 A previous study has shown that cells expressing mutant CSF-1R are defective in autophosphorylation of CSF-1R owing to CSF-1 stimulation.4 This finding suggests that abnormality in CSF-1R signaling is relevant to the pathogenesis of HDLS; however, the precise pathologic mechanism by which perturbation of CSF-1R signaling leads to HDLS has remained elusive.
Gross neuropathologic features of HDLS are characterized by prominent degenerative changes of the cerebral white matter with frontal prominence and the corpus callosum.1 Microscopic examination revealed loss of myelin sheaths and axons and the presence of abundant neuroaxonal spheroids.1,8–11 Another distinctive neuropathologic feature is the presence of pigmented macrophages in a background of white matter destruction. These pathologic features have been commonly described in patients with pigmented orthochromatic leukodystrophy (POLD).12,13 Patients with POLD have recently been shown to carry CSF-1R mutations.14
In this study, we identified 7 index patients from unrelated pedigrees with or without a family history who were found to carry various types of CSF-1R mutation. We attempted to characterize the molecular genetic, clinical, neuroimaging, and neuropathologic findings of these patients.
METHODS
Standard protocol approvals, registrations, and patient consents.
We enrolled 7 probands from 7 unrelated Japanese families. Genomic DNA was isolated from peripheral leukocytes from the patients. This study was approved by the institutional review board of Niigata University, and written informed consent was obtained from all the patients or their caregivers. Patients clinically suspected of having HDLS were referred to our laboratory for genetic testing for CSF-1R. Clinical presentations and neuroimaging findings of the patients were retrospectively evaluated by board-certified neurologists.
Genetic analyses.
Mutational analysis of CSF-1R was performed using sequences of both strands of all PCR-amplified coding exons and flanking intronic sequences as previously described.4 When the mutations were identified, we confirmed that the mutations were not found in known single nucleotide polymorphisms (SNPs) based on dbSNPs, and determined the absence of the mutations in normal controls by custom TaqMan SNP genotyping assay (Applied Biosystems, Foster City, CA). To predict the pathogenicity of amino acid substitutions caused by missense mutations, we conducted in silico analysis using the PolyPhen-2 and SIFT algorithms.15,16
Total RNA was extracted from autopsied brain tissues from the 2 patients with CSF-1R mutations (c.2442+1G>C and p.S688EfsX13) and from peripheral leukocytes from the patient with p.I794T mutation. Complementary DNA was synthesized using a high-capacity cDNA reverse transcription kit (Applied Biosystems).
Western blot analysis.
Proteins from the frontal cortex of the autopsied cases (c.2442+1G>C and p.S688EfsX13) and control subjects without neurologic disorders were extracted and fractionated as previously described.17 Detergent-extracted lysates were subjected to sodium dodecyl sulfate polyacrylamide gel electrophoresis followed by immunoblotting. A polyclonal anti-CSF-1R antibody that recognized the C-terminus of CSF-1R (C-20, Santa Cruz Biotechnology, Dallas, TX) and a monoclonal antibody that recognized the N-terminus of CSF-1R (B8, Santa Cruz Biotechnology) were used for detection of total CSF-1R. CSF-1R phosphorylated at Tyr546, Tyr723, and Tyr809 was detected using specific antiphosphorylated CSF-1R antibodies (Cell Signaling Technology, Beverly, MA). Methods for cell culture experiments are described in the e-Methods on the Neurology® Web site at www.neurology.org.
Analysis of MRI and CT.
We examined a total of 23 MRIs of 7 patients with HDLS with a CSF-1R mutation. MRI was conducted for diagnostic purposes using a 1.5T MRI system. Axial T1- and T2-weighted and fluid-attenuated inversion recovery (FLAIR) images were obtained from all the patients. Longitudinal MRI studies of the 7 patients with HDLS were carried out using a previously reported semiquantitative rating scale.18 The semiquantitative rating was carried out by 2 expert examiners. The scores between the 2 examiners were highly in agreement with a significant correlation (intraclass correlation coefficient = 0.98, 95% confidence interval 97–99). Six out of the 7 patients were evaluated by brain CT.
Neuropathologic techniques and immunohistochemistry.
Neuropathologic examination was performed on a biopsied specimen taken from the frontal white matter of patient III, and 4 autopsied brains of patient I,19 patient VI, a grandfather of patient II (patient IHC1 in table e-1),20 and a patient who is not included in table 1 (patient IHC2 in table e-1). The clinicopathologic findings of patient IHC2 who was found to carry the CSF-1R mutation of p.I794T were reported in detail elsewhere.9 Formalin-fixed, paraffin-embedded sections were prepared, and stained with hematoxylin & eosin and by the Klüver-Barrera method. Serial sections of the biopsied specimen and the frontal and occipital lobes of the autopsied brains were also immunostained with polyclonal antibodies against Iba1 (Wako, Richmond, VA; 1:2,000), CSF-1R (C-20) (1:100), and glucose transporter-5 (GLUT-5) (IBL, Minneapolis, MN; 1:50), and monoclonal antibodies against CD68 (Dako, Carpinteria, CA; 1:200). As autopsy controls, sections from the frontal lobe of 3 patients without any neurologic diseases and 2 patients with either Alzheimer disease or adrenoleukodystrophy were immunostained. Detailed information on the control cases is available in table e-1.
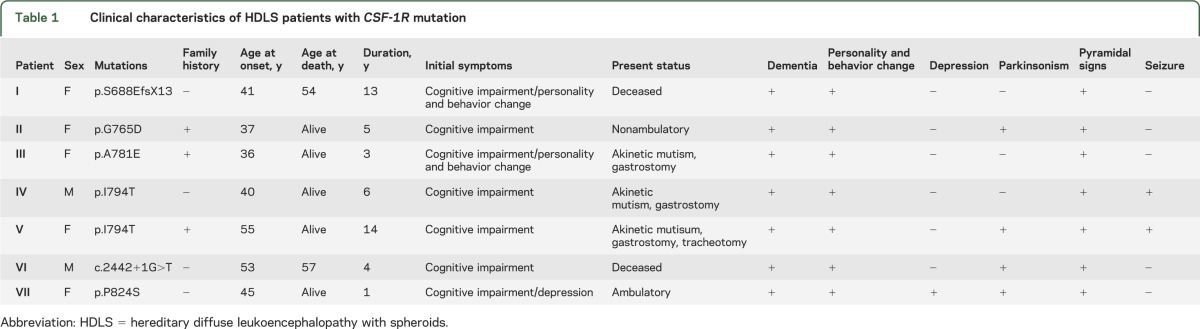
Table 1.
Clinical characteristics of HDLS patients with CSF-1R mutation
RESULTS
Identification of CSF-1R mutations.
We identified 6 different mutations of CSF-1R in 7 probands with HDLS of Japanese origin (figure 1A). These mutations included 3 novel missense mutations (c.2294G>A/p.G765D, c.2342C>A/p.A781E, and c.2470C>T/p.P824S), one novel splice-site mutation (c.2442+1G>T), and one novel single nucleotide insertion generating a premature stop codon (c.2060_2061insT/p.S688EfsX13). We detected one known missense mutation (c.2381T>C/p. I794T)4,6,7 in 2 patients from apparently unrelated pedigrees. All missense mutations were located in the tyrosine kinase domain of CSF-1R. These mutations were not described in the dbSNP database and not present in 124 healthy controls. The amino acids substituted by the missense mutations were highly conserved across species (figure e-1A). In silico analysis of the missense mutations using the PolyPhen-2 and SIFT programs revealed that these mutations are predicted to be pathogenic with high probability.
Figure 1. Identification of CSF-1R mutations, and mRNA and protein expressions of mutant CSF-1R.

(A) Schematic illustration of CSF-1R structure. Six different mutations identified in this study are shown below the diagram of electropherograms. The tyrosine kinase domain of CSF-1R is shown in gray. Numbers represent exons in which mutations were identified. Positions of previously reported mutations are shown as triangles above the diagram. IVS = intervening sequence. (B) Sequencing electropherogram of amplified genomic DNA and reverse transcription PCR amplicons of patient with frameshift mutation (p.S688EfsX13), which was predicted to undergo nonsense-mediated mRNA decay. The expression of the mutant allele was hardly detectable, suggesting that this frameshift mutation results in nonsense-mediated decay of mutant mRNA. The predicted amino acid sequences followed by a stop codon are shown in red. (C) Immunoblot analysis of CSF-1R protein using anti-CSF-1R antibody (C-20) in Triton X-100 soluble fraction from frontal cortex of autopsied cases (c.2442+1G>T and p.S688EfsX13) and 4 control subjects (1–4) without neurologic disorder. Note that the full-length CSF-1R (150 kDa and 130 kDa, representing mature protein and immature full-length protein, respectively) and proteolytically cleaved C-terminal fragment of CSF-1R (55 kDa) showed markedly decreased expression levels. The equivalency of protein loading is shown in the actin blot (bottom).
Clinical presentations.
Familial occurrence was observed in 3 probands, in whom autosomal dominant inheritance was suspected. Four probands had no family history of neurologic diseases; hence, these patients apparently represent sporadic cases, although the possibility remains that incomplete penetrance or other factors may account for the lack of familial history. Details of clinical presentations of the patients with CSF-1R mutations are summarized in table 1. The ages at onset of the patients ranged from 36 to 55 years with the mean age at onset of 44 years. Cognitive impairment is an initial symptom in all the patients. Cognitive impairment as well as behavioral and personality changes were the cardinal clinical features in all the patients. Parkinsonian symptoms such as bradykinesia and gait disturbance, pyramidal signs, and seizures were the frequent accompanying clinical manifestations. Six of the 7 patients progressed into being wheelchair-bound and having severe dementia with communication problems within 5 years after onset. One patient at 1 year after onset remains ambulatory and capable of verbal conversation.
mRNA expression of mutant CSF-1R.
We sought to ascertain whether the mutations found in this study result in alteration of mRNA expression or aberrant splicing. To clarify these issues, we performed reverse transcription (RT)-PCR analysis using mRNA specimens obtained from peripheral leukocytes or frozen brain tissues. Sequence analysis of the amplified CSF-1R mRNA from the patient with p.I794T mutation revealed that the mutant CSF-1R allele is expressed at a level comparable to the normal allele (figure e-1B). We next investigated mRNA expression in the cerebral cortex of the patient with the single nucleotide insertion that generates a premature stop codon (p.S688EfsX13). This premature stop codon is predicted to cause nonsense-mediated mRNA decay.21 To test this prediction, we amplified exon 15 of CSF-1R by RT-PCR. This analysis revealed that the expression level of the mutant allele is markedly decreased compared with that of the normal allele (figure 1B). We further attempted to determine the aberrant splice variants in the patient with splice-site mutation (c.2442+1G>T) involving exon 18. RT-PCR analysis using primers designed to amplify exons 17 to 20 revealed that 2 additional amplified fragments were detected in the patient’s mRNA in addition to the normal fragment (figure e-1C). We subcloned these aberrant splice transcripts and identified 3 aberrant splice variants with skipped exon 18 (figure e-1C). These splice transcripts are translated into in-frame truncated CSF-1R proteins.
Protein expression of CSF-1R in brain of patients with HDLS.
Having established that alteration of mRNA expression and aberrant splicing of CSF-1R occur in the brains of the patients, we next examined the protein expression of CSF-1R in the brain. We extracted the protein from the frontal cortex of 2 patients with the frameshift mutation (p.S688EfsX13) and the splice site mutation (c.2442+1G>T) as well as from the controls without neurologic disorder. Western blot analysis using the anti-CSF-1R antibody revealed the full-length CSF-1R migrating at ∼150 kDa as a mature form and ∼130 kDa as an immature form, and the cleaved C-terminal fragment (CTF) migrating at ∼55 kDa (figure 1C). The expression levels of full-length and cleaved CTF of CSF-1R in the patients were markedly lower than those in control subjects (figure 1C).
Defective autophosphorylation of CSF-1R in cells expressing mutant CSF-1R.
To address the functional properties of mutant CSF-1R identified in patients with HDLS, we transiently transfected wild-type or mutant CSF-1Rs cDNA into HEK293T cells. Western blot analysis using anti-CSF-1R antibodies revealed comparable expression levels between wild-type and mutant CSF-1Rs (figure e-2A). Under incessant stimulation with CSF-1 in a medium containing serum, wild-type CSF-1R showed constitutive autophosphorylation of CSF-1R at Tyr 546 and 723, whereas none of the 8 mutants found in our patients (S688EfsX13, G765D, A781E, I794T, aberrant splice variant [ASV] 1, ASV2, ASV3, and P824S) and 2 previously reported mutants (M766T and M875T)4 revealed detectable autophosphorylation of CSF-1R (figure e-2A). We next examined ligand-induced autophosphorylation of CSF-1R by CSF-1 stimulation (25 ng/mL) after removal of serum from the medium. Upon stimulation with CSF-1, we observed autophosphorylation of wild-type CSF-1R at Tyr 723 and 809 in a time-dependent manner, whereas neither ASV1 nor I794T CSF-1R showed autophosphorylation (figure e-2B). To extend this finding, we transiently transfected 10 mutant CSF-1Rs and examined ligand-induced autophosphorylation of CSF-1R by stimulation with CSF-1 or IL-34. None of the mutant CSF-1Rs underwent autophosphorylation of CSF-1R (figure e-2C).
We then determined whether the missense mutant affects autophosphorylation of wild-type CSF-1R in a dominant-negative manner. HEK293 cells inducibly expressing FLAG-tagged wild-type CSF-1R were further transfected with myc-His-tagged wild-type or mutant CSF-1R. Coexpression of wild-type CSF-1R increased the level of autophosphorylation of CSF-1R as compared with that observed in mock-transfected cells expressing FLAG-tagged wild-type CSF-1R (figure e-2D). Coexpression of mutant CSF-1Rs did not suppress the level of autophosphorylation occurring in FLAG-tagged wild-type CSF-1R (figure e-2D). These findings suggest that the missense mutants cause a loss of autophosphorylation of own CSF-1R, but do not suppress the autophosphorylation of wild-type CSF-1R by the dominant-negative mechanism.
Characteristic MRI findings.
MRIs of each patient are shown in figure 2A and figure e-3. All the patients showed bilateral hyperintensities in the white matter with frontal predominance visualized by FLAIR or T2-weighted imaging. Thinning of the corpus callosum with hyperintensity lesions were noted from the early phase of the disease (figure 2A). The changes of the white matter and the corpus callosum were already detectable 5 years prior to the onset in one patient, who underwent MRI for evaluation of his headache at age 48 (figure 2A). This finding suggests that MRI alterations precede the clinical symptoms of HDLS.
Figure 2. Longitudinal MRI changes of patients with hereditary diffuse leukoencephalopathy with spheroids.

(A) Sequential MRI studies of patient VI using fluid-attenuated inversion recovery (FLAIR) images. At the early stage of the disease, white matter hyperintensities were often found in the periventricular area, surrounding the anterior and posterior horns with a tendency to confluence, and in the fiber tract in the internal capsule. Enlargement of the lateral ventricles was also noticeable. Notably, the corpus callosum showed hyperintensities and thinning at the time of onset. The progression was relatively rapid and cortical atrophy became evident as the disease progressed. The MRI taken 5 years before the onset for the evaluation of headache showed subtle asymmetric white matter hyperintensities surrounding the anterior horns and faint signal changes and mild thinning of the corpus callosum. (B–E) Chronological changes in semiquantitative MRI scores in 7 patients with hereditary diffuse leukoencephalopathy with spheroids. MRI finding severity was evaluated from the total score (B, scores 0–57), which combines the white matter lesion (WML) score (C, 1–42) and the atrophy score (D, 0–13), and the presence of lesions in the thalamus and basal ganglia. (E) Correlation analysis between WML score and atrophy score.
Longitudinal MRI changes.
Longitudinal MRI changes were determined in 7 patients with HDLS using the MRI rating scale.18 The total MRI scores ranged from 12 to 44, the white matter lesion (WML) scores ranged from 11 to 32, and atrophy scores ranged from 1 to 12. These MRI scores increased with disease duration, and the average changes in scores per year were 3.7 ± 1.5 for total score, 2.5 ± 1.1 for WML score, and 1.3 ± 0.5 for atrophy score. There were statistically significant correlations of the disease duration with total (r = 0.94, p < 0.01), WML (r = 0.94, p < 0.01), and atrophy (r = 0.92, p < 0.01) scores (figure 2, B–D). We found a statistically significant correlation between WML score and atrophy score (r = 0.94, p < 0.01) (figure 2E).
Characteristic brain CT findings.
Five out of 6 patients who underwent brain CT demonstrated multiple spotty calcifications in the affected white matter (figure 3A and figure e-4A). The calcifications preferentially developed in the frontal white matter adjacent to the anterior horn of the lateral ventricles. Histopathologic analysis by von Kóssa staining of the lesions of calcifications in patient IV revealed that the lesions contained calcium deposits (figure 3, B and C, and figure e-4, B–D).
Figure 3. Spotty calcifications in white matter on CT images.

(A) Multiple lesions caused by calcifications in the brain as revealed by CT. The boxed area is enlarged at the right bottom of the panel. Small spotty calcifications were observed in the affected white matter. (B, C) Histopathologic findings of small lesions in frontal white matter close to the corpus callosum of patient VI carrying splice-site mutation. Calcium deposition and fibrillary gliosis were evident. (B) Hematoxylin & eosin, (C) von Kóssa reaction. Bar = 100 μm for B and C.
Neuropathologic findings of patients with HDLS.
Histopathologically, the brains of all patients with HDLS showed diffuse loss of myelin sheaths and axons in the white matter with severe gliosis prominent in the frontal lobe (figure 4A). Within the white matter lesions, there were scattered axonal spheroids (figure 4B) showing immunoreactivity for phosphorylated neurofilaments and numerous phagocytic cells with brownish granules (figure 4C). Interestingly, immunohistochemistry using microglia/macrophage markers, including Iba1, CD68, and GLUT-5, revealed microglia with characteristic features. For example, despite diffuse white matter degeneration and astrogliosis, activated microglia were spatially restricted rather than distributed diffusely (figure 4D). The activated microglia (Iba1-, CD68-, and GLUT-5-immunopositive cells) and phagocytic cells (Iba1- and CD68-immunopositive, but GLUT-5-negative cells) appeared to be segregated (data not shown). In human tissue, GLUT-5 immunopositivity is selectively observed in microglia rather than in other mononuclear phagocytic cells.22,23 Moreover, individual microglia demonstrated their characteristic morphology with thin processes and many knotlike structures (figure 4, E–H). Furthermore, in the white matter of patients with HDLS, the number of activated microglia immunopositive for CSF-1R (figure 4, I and J) was apparently smaller than those observed in brains affected by Alzheimer disease and adrenoleukodystrophy (figure 4, K and L).
Figure 4. Histopathologic features of patients with hereditary diffuse leukoencephalopathy with spheroids.

(A–C) White matter lesions of frontal lobe of patient VI. (A) Marked myelin loss of white matter with U-fibers spared. (B) Axonal spheroids (arrows) in white matter. (C) Abundant macrophages (arrowheads) in white matter. (D–L) Microglia in white matter of patients with hereditary diffuse leukoencephalopathy with spheroids and control brains. (D-H) Immunohistochemistry of Iba1 in degenerative white matter. (D, E) Patient VI. (F) Patient III. (G) Patient IHC1. (H) Patient IHC2. (D) The boxed area in (A) is enlarged. Spatially restricted appearance of Iba1-immunopositive activated microglia (upper right corner). (E–H) Characteristic features of microglia. (I–L) CSF-1R immunohistochemistry in degenerative white matter. (I) Patient VI. (J) Patient III. (K) Patient with Alzheimer disease. (L) Patient with adrenoleukodystrophy. Images in I and J were taken from serial sections of images in E and F, respectively. Note very faint or no CSF-1R immunopositivity in activated microglia in images I and J. (A) Klüver-Barrera staining, (B, C), hematoxylin & eosin staining, (D–L) immunohistochemistry of Iba1 (D–H) and CSF-1R (I–L). Bar = 7 mm for A, 33 μm for B and C, 306 μm for D, 50 μm for E, F, and I–L, and 25 μm for G and H.
DISCUSSION
The clinical presentations of our patients are essentially similar to those reported previously.2–4 Taken together with the previous reports,5,6 this study suggests that HDLS is relatively common in the Japanese population. We identified a novel mutation of single nucleotide insertion (c.2060_ 2061insT) that causes a frameshift generating a premature stop codon (p.S688EfsX13). This premature stop codon fulfills the criterion of nonsense-mediated mRNA decay.21 Supporting this notion, mRNA analysis revealed that the mutant allele is poorly expressed in the brain tissue of the patient. This finding indicates that haploinsufficiency of CSF-1R is sufficient for developing HDLS. It has not been determined whether missense mutations of CSF-1R cause haploinsufficiency or a dominant-negative effect of CSF-1R. Our cell culture experiments revealed that additional expression of mutant CSF-1R in cells inducibly expressing wild-type CSF-1R did not suppress the autophosphorylation of CSF-1R occurring in wild-type CSF-1R. These findings suggest that the dominant-negative effect of the missense mutant CSF-1R is not likely to be the mechanism underlying missense mutations in HDLS.
MRI of the patients in this study revealed relatively uniform findings characterized by frontoparietal white matter changes with corpus callosum involvement. Thinning of the corpus callosum often accompanied by signal intensity changes is exclusively observed from the very early stage. On the basis of our experience, the sagittal views of brain MRI with FLAIR or T2-weighted images are useful for detecting such early characteristic changes of the corpus callosum. Enlargement of lateral ventricles without cortical atrophy is also an early characteristic MRI feature, which may reflect a decrease in white matter volume. We quantified MRI abnormalities using a rating scale as previously reported.18 The MRI scores increased with increasing disease duration, and there was a significant correlation between MRI score and disease duration. This suggests that the MRI rating scale could be a useful tool for monitoring disease progression.
Calcifications in the frontal white matter were frequently observed by CT in our patients with CSF-1R mutations. These calcifications on CT images have not been paid much attention in patients with HDLS except for a single patient pathologically diagnosed with HDLS in whom similar calcification was observed.24 Conventional CT images often fail to detect small calcifications in the white matter; thus, we recommend the use of the thin-slice CT technique that reliably detects such small lesions. The calcifications in the white matter may be a disease-specific feature in patients with HDLS because they are not usually observed in patients with other neurologic diseases or healthy aged individuals. It is of note that CSF-1R signaling plays a role in the regulation of osteoclast cytoskeletal reorganization25,26; hence, the calcifications in patients might be directly related to the pathophysiology of CSF-1R mutations.
Histopathologic examinations of the brains of patients with HDLS showed the abnormal appearance of activated microglia in a spatially restricted manner and morphologic alteration of microglia, despite diffuse white matter degeneration and astrogliosis. Moreover, CSF-1R immunopositivity on remaining activated microglia was clearly weaker than that in brains with other diseases. In mice homozygous for a null mutation in Csf1r and in the CSF-1 ligand gene (Csf1op/op), a reduced number and morphologic alteration of microglia have been reported.27–31 The histopathologic features of microglia in the present patients appear to be similar to those of the mutant mice, even though the degree of abnormality is different between the 2 species. Moreover, macroscopic findings in brains of Csf1r-deficient mice such as reduced brain size, ventricular enlargement, and defects in the corpus callosum are also commonly observed in patients with HDLS.31 Impairment of the CSF-1R-mediated microglial repair of axonal degeneration may contribute to the development of HDLS, because mice showing defective microglia/macrophages in the brain appeared to show impaired remyelination.32 In contrast, a recent study has shown that CSF-1R is expressed in neurons and CSF-1 administration improves neuronal cell survival without significant microglial activation in a mouse model of neurodegeneration.33 Moreover, it was shown that CSF-1R is expressed in neuronal stem cells and the CSF-1R ligands directly stimulate neuronal differentiation and neuronal precursor cell survival.31 Further investigations are warranted to clarify whether microglia or neurons play a predominant role in the pathogenesis of HDLS.
Supplementary Material
ACKNOWLEDGMENT
The authors thank the patients and their relatives for participating in this study and Drs. Tokunaga, Kawachi, Sugai, and Toyoshima for assistance with data collection.
GLOSSARY
- ASV
aberrant splice variant
- CSF-1R
colony stimulating factor 1 receptor
- CTF
C-terminal fragment
- FLAIR
fluid-attenuated inversion recovery
- GLUT-5
glucose transporter-5
- HDLS
hereditary diffuse leukoencephalopathy with spheroids
- POLD
pigmented orthochromatic leukodystrophy
- RT
reverse transcription
- SNP
single nucleotide polymorphism
- WML
white matter lesion
Footnotes
Editorial, page 102
Supplemental data at www.neurology.org
AUTHOR CONTRIBUTIONS
Dr. Konno performed drafting the manuscript, study concept, acquisition of data, and analysis of data. Dr. Masayoshi Tada performed drafting the manuscript, study concept, acquisition of data, and analysis of data. Dr. Mari Tada performed study concept, acquisition of data, and analysis of data. Dr. Koyama performed acquisition of data and analysis of data. Dr. Nozaki performed acquisition of data and analysis of data. Dr. Harigaya performed acquisition of data. Dr. Nishimiya performed acquisition of data. Dr. Matsunaga performed acquisition of data. Dr. Yoshikura performed acquisition of data. Dr. Ishihara provided vital reagents and performed acquisition of data. Dr. Arakawa performed acquisition of data. A. Isami performed acquisition of data. Dr. Okazaki performed acquisition of data. Dr. Yokoo provided vital reagents and performed acquisition of data. Dr. Itoh provided vital reagents and performed acquisition of data. Dr. Yoneda performed acquisition of data. Dr. Kawamura performed acquisition of data. Dr. Inuzuka performed acquisition of data. Dr. Takahashi performed acquisition of data and study supervision. Dr. Nishizawa performed acquisition of data and study supervision. Dr. Onodera performed acquisition of data and study supervision. Dr. Kakita performed acquisition of data, interpretation of data, and study supervision. Dr. Ikeuchi performed drafting the manuscript, study concept, interpretation of data, and obtaining funding.
STUDY FUNDING
Supported in part by Grants-in-aid for Scientific Research (21200041 to T.I.) from the Japan Society of Promotion of Science, a Grant-in-aid from the Ministry of Health, Labour and Welfare of Japan, and a grant from the Tsubaki Memorial Foundation (to T.K.).
DISCLOSURE
T. Konno received research support from the Tsubaki Memorial Foundation. M. Tada is funded by Grants-in-aid for Scientific Research from the Japan Society of Promotion of Science. A. Koyama reports no disclosures. H. Nozaki is funded by a Grant-in-aid from the Ministry of Health, Labour and Welfare of Japan. Y. Harigaya, J. Nishimiya, A. Matsunaga, N. Yoshikura, K. Ishihara, A. Isami, K. Okazaki, H. Yokoo, K. Itoh, M. Yoneda, and M. Kawamura report no disclosures. T. Inuzuka is funded by Grants-in-aid from the Ministry of Health, Labour and Welfare of Japan. H. Takahashi is funded by Grants-in-aid for Scientific Research from the Japan Society of Promotion of Science and a Grant-in-aid from the Ministry of Health, Labour and Welfare of Japan. M. Nishizawa is funded by Grants-in-aid for Scientific Research from the Japan Society of Promotion of Science and a Grant-in-aid from the Ministry of Health, Labour and Welfare of Japan. O. Onodera is funded by Grants-in-aid for Scientific Research from the Japan Society of Promotion of Science and a Grant-in-aid from the Ministry of Health, Labour and Welfare of Japan. A. Kakita is funded by Grants-in-aid for Scientific Research from the Japan Society of Promotion of Science and a Grant-in-aid from the Ministry of Health, Labour and Welfare of Japan. T. Ikeuchi is funded by Grants-in-aid for Scientific Research from the Japan Society of Promotion of Science and a Grant-in-aid from the Ministry of Health, Labour and Welfare of Japan. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Axelsson R, Röyttä M, Sourander P, Akesson HO, Andersen O. Hereditary diffuse leukoencephalopathy with spheroids. Acta Psychiatr Scand Suppl 1984;314:1–65 [PubMed] [Google Scholar]
- 2.Wider C, Van Gerpen JA, DeArmond S, Shuster EA, Dickson DW, Wszolek ZK. Leukoencephalopathy with spheroids (HDLS) and pigmentary leukodystrophy (POLD): a single entity? Neurology 2009;72:1953–1959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wong JC, Chow TW, Hazrati LN. Adult-onset leukoencephalopathy with axonal spheroids and pigmented glia can present as frontotemporal dementia syndrome. Dement Geriatr Cogn Disord 2011;32:150–158 [DOI] [PubMed] [Google Scholar]
- 4.Rademakers R, Baker M, Nicholson AM, et al. Mutations in the colony stimulating factor 1 receptor (CSF1R) gene cause hereditary diffuse leukoencephalopathy with spheroids. Nat Genet 2012;44:200–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kinoshita M, Yoshida K, Oyanagi K, Hashimoto T, Ikeda S. Hereditary diffuse leukoencephalopathy with axonal spheroids caused by R782H mutation in CSF1R: case report. J Neurol Sci 2012;318:115–118 [DOI] [PubMed] [Google Scholar]
- 6.Mitsui J, Matsukawa T, Ishiura H, et al. CSF1R mutations identified in three families with autosomal dominantly inherited leukoencephalopathy. Am J Med Genet B Neuropsychiatr Genet 2012;159B:951–957 [DOI] [PubMed] [Google Scholar]
- 7.Kleinfeld K, Mobley B, Hedera P, Wegner A, Sriram S, Pawate S. Adult-onset leukoencephalopathy with neuroaxonal spheroids and pigmented glia: report of five cases and a new mutation. J Neurol 2013;260:558–571 [DOI] [PubMed] [Google Scholar]
- 8.Freeman SH, Hyman BT, Sims KB, et al. Adult onset leukodystrophy with neuroaxonal spheroids: clinical, neuroimaging and neuropathologic observations. Brain Pathol 2009;19:39–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Itoh K, Shiga K, Shimizu K, Muranishi M, Nakagawa M, Fushiki S. Autosomal dominant leukodystrophy with axonal spheroids and pigmented glia: clinical and neuropathological characteristics. Acta Neuropathol 2006;111:39–45 [DOI] [PubMed] [Google Scholar]
- 10.Van Gerpen JA, Wider C, Broderick DF, Dickson DW, Brown LA, Wszolek ZK. Insights into the dynamics of hereditary diffuse leukoencephalopathy with axonal spheroids. Neurology 2008;71:925–929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Baba Y, Ghetti B, Baker MC, et al. Hereditary diffuse leukoencephalopathy with spheroids: clinical, pathologic and genetic studies of a new kindred. Acta Neuropathol 2006;111:300–311 [DOI] [PubMed] [Google Scholar]
- 12.Gray F, Destee A, Bourre JM, et al. Pigmentary type of orthochromatic leukodystrophy (OLD): a new case with ultrastructural and biochemical study. J Neuropathol Exp Neurol 1987;46:585–596 [DOI] [PubMed] [Google Scholar]
- 13.Tuñón T, Ferrer I, Gállego J, Delgado G, Villanueva JA, Martinez-Peñuela JM. Leucodystrophy with pigmented glial and scavenger cells (pigmentary type of orthochromatic leucodystrophy). Neuropathol Appl Neurobiol 1988;14:337–344 [PubMed] [Google Scholar]
- 14.Nicholson AM, Baker MC, Finch NA, et al. CSF1R mutations link POLD and HDLS as a single disease entity. Neurology 2013;80:1033–1040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Adzhubei IA, Schmidt S, Peshkin L, et al. A method and server for predicting damaging missense mutations. Nat Methods 2010;7:248–249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc 2009;4:1073–1081 [DOI] [PubMed] [Google Scholar]
- 17.Kaneko H, Kakita A, Kasuga K, et al. Enhanced accumulation of phosphorylated α-synuclein and elevated β-amyloid 42/40 ratio caused by expression of the presenilin-1 ΔT440 mutant associated with familial Lewy body disease and variant Alzheimer's disease. J Neurosci 2007;27:13092–13097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sundal C, Van Gerpen JA, Nicholson AM, et al. MRI characteristics and scoring in HDLS due to CSF1R gene mutations. Neurology 2012;79:566–574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ishihara K, Horibe Y, Ohno H, et al. A clinicopathological study of young-onset dementia: report of 2 autopsied cases [in Japanese]. Brain Nerve 2011;63:1117–1123 [PubMed] [Google Scholar]
- 20.Ishida Y, Narita T, Kawarai M. An autopsy case of late-life nonmetachromatic leukodystrophy [In Japanese]. Brain Nerve 1967;19:835–842 [PubMed] [Google Scholar]
- 21.Kuzmiak HA, Maquat LE. Applying nonsense-mediated mRNA decay research to the clinic: progress and challenges. Trends Mol Med 2006;12:306–316 [DOI] [PubMed] [Google Scholar]
- 22.Sasaki A, Horikoshi Y, Yokoo H, Nakazato Y, Yamaguchi H. Antiserum against human glucose transporter 5 is highly specific for microglia among cells of the mononuclear phagocyte system. Neurosci Lett 2003;338:17–20 [DOI] [PubMed] [Google Scholar]
- 23.Horikoshi Y, Sasaki A, Taguchi N, et al. Human GLUT5 immunolabeling is useful for evaluating microglial status in neuropathological study using paraffin sections. Acta Neuropathol 2003;105:157–162 [DOI] [PubMed] [Google Scholar]
- 24.Martinez-Saez E, Shah S, Costa C, Fleminger S, Connor S, Bodi I. Adult onset leukodystrophy with neuroaxonal spheroids and demyelinating plaque-like lesions. Neuropathology 2012;32:285–292 [DOI] [PubMed] [Google Scholar]
- 25.Insogna KL, Sahni M, Grey AB, et al. Colony-stimulating factor-1 induces cytoskeletal reorganization and c-src-dependent tyrosine phosphorylation of selected cellular proteins in rodent osteoclasts. J Clin Invest 1997;100:2476–2485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dai XM, Ryan GR, Hapel AJ, et al. Targeted disruption of the mouse colony-stimulating factor 1 receptor gene results in osteopetrosis, mononuclear phagocyte deficiency, increased primitive progenitor cell frequencies, and reproductive defects. Blood 2002;99:111–120 [DOI] [PubMed] [Google Scholar]
- 27.Wegiel J, Wiśniewski HM, Dziewiatkowski J, et al. Reduced number and altered morphology of microglial cells in colony stimulating factor-1-deficient osteopetrotic op/op mice. Brain Res 1998;804:135–139 [DOI] [PubMed] [Google Scholar]
- 28.Sasaki A, Yokoo H, Naito M, Kaizu C, Shultz LD, Nakazato Y. Effects of macrophage-colony-stimulating factor deficiency on the maturation of microglia and brain macrophages and on their expression of scavenger receptor. Neuropathology 2000;20:134–142 [DOI] [PubMed] [Google Scholar]
- 29.Erblich B, Zhu L, Etgen AM, Dobrenis K, Pollard JW. Absence of colony stimulation factor-1 receptor results in loss of microglia, disrupted brain development and olfactory deficits. PLoS One 2011;6:e26317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ginhoux F, Greter M, Leboeuf M, et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 2010;330:841–845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nandi S, Gokhan S, Dai XM, et al. The CSF-1 receptor ligands IL-34 and CSF-1 exhibit distinct developmental brain expression patterns and regulate neural progenitor cell maintenance and maturation. Dev Biol 2012;367:100–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kondo Y, Adams JM, Vanier MT, Duncan ID. Macrophages counteract demyelination in a mouse model of globoid cell leukodystrophy. J Neurosci 2011;31:3610–3624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Luo J, Elwood F, Britschgi M, et al. Colony-stimulating factor 1 receptor (CSF1R) signaling in injured neurons facilitates protection and survival. J Exp Med 2013;210:157–172 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.