

Abstract
Chemokines are small proteins best known for their role in controlling the migration of diverse cells, particularly leukocytes. Upon binding to their G-protein-coupled receptors on the leukocytes, chemokines stimulate the signaling events that cause cytoskeletal rearrangements involved in cell movement, and migration of the cells along chemokine gradients. Depending on the cell type, chemokines also induce many other types of cellular responses including those related to defense mechanisms, cell proliferation, survival, and development. Historically, most research efforts have focused on the interaction of chemokines with their receptors, where monomeric forms of the ligands are the functionally relevant state. More recently, however, the importance of chemokine interactions with cell surface glycosaminoglycans has come to light, and in most cases appears to involve oligomeric chemokine structures. This review summarizes existing knowledge relating to the structure and function of chemokine oligomers, and emerging methodology for determining structures of complex chemokine assemblies in the future.
1. INTRODUCTION
Oligomerization of proteins is widespread in biological systems, with estimates of homooligomerization occurring with up to 70% of all eukaryotic proteins.1,2 There are many benefits to this type of association; oligomerization can increase the efficiency of processes involving multiple proteins such as signaling and enzymatic reactions, it can help to stabilize and protect proteins against proteolytic processes, it can amplify affinities of proteins for their ligands by organizing them in polymeric constructs or surface arrays, and it can increase the complexity, diversity, and regulation of signaling events.1–3 In this review, we highlight the known roles that oligomerization plays in the chemokine-mediated regulation of cell migration and signaling in the immune system. We also highlight the structural features of chemokine oligomerization, and new methodology for the characterization of these oligomeric species.
1.1. The chemokine superfamily
Chemokines, or chemotactic cytokines, are small proteins (8–12 kDa) that provide directional signals for the migration of cells.4,5 Approximately 45 human chemokines have been identified and are classified into four families based on the sequence of conserved cysteines in their amino termini6: XC chemokines contain a single cysteine; CC chemokines have sequential cysteines; in CXC chemokines, the cysteines are separated by one amino acid; and CX3C chemokines have two cysteines separated by three amino acids. In early studies, the ligands were named with a nonsystematic nomenclature (e.g., RANTES, MIP-1α). However, a systematic nomenclature for chemokines was introduced in 2002 and uses the cysteine motif followed by L (for ligand) and a number, to enable assignment of chemokines to their subfamilies (e.g., CCL5, CXCL2, CX3CL1, XCL1).7 Twenty-two chemokine receptors have been identified and are similarly named by the cysteine motif, R (for receptor) and a number, for example, CCR5. The receptors belong to the G-protein-coupled receptor (GPCR) superfamily and interact predominantly with the Gαi subfamily of G proteins.
Many chemokine receptors bind to more than one ligand and similarly most chemokines bind to more than one receptor, leading to a significant level of potential cross-reactivity. The promiscuous pairing of chemokines and receptors was initially thought to provide redundancy in the system.8,9 However, the concept of functional selectivity or biased agonism has recently emerged, where different ligands bind the same receptor but activate different sets of signaling pathways, potentially providing for finely tuned signaling responses. It is also possible that the oligomeric properties of chemokines and their specificities for different glycosaminoglycans (GAGs; described later) may break the apparent redundancy by controlling chemokine localization and density, certain aspects of signaling, and ultimately the migration and functional responses of the chemokine receptor expressing cells.10
Chemokines are produced by, and act on, a wide variety of cells. In addition to their role in controlling leukocyte migration in the context of immune surveillance and inflammation, chemokines and their receptors are involved in the migration and signaling responses of cells during other processes such as development, angiogenesis, wound repair, and neural regeneration.11,12 Further, many diseases are associated with deregulated expression or utilization of chemokines and their receptors: For example, unresolved chemokine-mediated cell migration and activation contribute to many chronic inflammatory diseases such as multiple sclerosis and rheumatoid arthritis.13–15 Specific chemokines and receptors are also key mediators in the metastasis of cancer cells from primary tumors to secondary sites.16 And HIV utilizes primarily two chemokine receptors as portals for entry into cells.17,18 Accordingly, chemokines and their receptors have been high-priority therapeutic targets for many years.6,19
1.2. Chemokine-mediated cell migration
Despite the central role of chemokines and their receptors in cell migration, it is important to note that it is a complex multistep process, involving many proteins and interactions.20,21 For example, in the context of an inflammatory response, mucin-like selectin ligands on leukocytes (e.g., P-selectin glycoprotein ligand 1, PSGL-1, and L-selectin) interact with counter receptors (e.g., P- and E-selectins) on endothelial cells to capture the leukocytes from rapid diffusion through the vasculature and to promote slow rolling of the leukocytes along the blood vessel endothelium through labile interactions between the cell surfaces. The slow rolling of the leukocytes enables them to sample the endothelial surface where they encounter chemokines produced by or deposited on the endothelium. Interaction of receptors on the leukocytes with chemokines then triggers the activation of integrins into their high-affinity state, binding of the integrins to their ligands (e.g., ICAMs) on the endothelial cells, and rapid leukocyte arrest. The end result is the migration of the leukocytes through the vascular endothelial layer following a gradient of chemokines and adhesive molecules. While the selectins, integrins, their ligands, and the chemokine receptors are all membrane tethered, the immobilization of chemokines on the endothelial surface and the formation of chemokine gradients involve a different mechanism: interactions with GAGs. Thus, chemokines have two types of receptors: seven transmembrane GPCRs, and the long linear sulfated carbohydrate chains of proteoglycans. The ability of chemokines to interact with both of these macromolecules relies on the ability of chemokines to reversibly oligomerize as described in the next sections.
2. FUNCTIONAL EFFECTS OF CHEMOKINE OLIGOMERIZATION
As implied above and detailed in Section 3, most chemokines have a propensity to oligomerize; however, it is widely accepted that monomeric chemokines are the forms with the highest affinity for their receptors and the capacity to promote cell migration. This finding was demonstrated with mutant chemokines that were impaired in their ability to oligomerize, but were still capable of binding and activating their receptors with equivalent potencies as the WT proteins.22–24 Further, in some CC chemokines, the residues that are critical for receptor activation are buried in the dimers, and therefore it is predictable that CC dimers would not be capable of inducing cell migration.24 Beyond the knowledge that it is the monomeric form that activates the receptor, and in the absence of a crystal structure, the prevailing view of chemokine:receptor complexes is that of the so-called two-site model of receptor interaction in which the N-terminus of a chemokine monomer interacts with the helical bundle of the receptor (site 2) and the rest of the chemokine (the core domain), binds to the N-terminus of the receptor (site 1) (see figures in Refs. 25,26). The two-site model is supported by mutagenesis studies which indicate that the chemokine N-terminus is the key signaling domain, as modifications to the N-terminus can dramatically change the signaling response of the receptor to which it binds, often with only a small impact on binding affinity (see Section 3). Secondly, structural studies by solution NMR have demonstrated that peptides from the N-termini of chemokine receptors bind to the chemokine core domain.27,28
So the question is, if receptors are activated with chemokine monomers then what, if any, is the purpose of chemokine oligomerization? To address the relevance of oligomerization, monomeric variants of several chemokines (RANTES (CCL5), MCP-1 (CCL2), and MIP-1β (CCL4)) were used in in vivo models of cell migration where they were shown to be defective in promoting migration, in contrast to their WT counterparts and in contrast to their ability to induce migration in vitro.22 These data clearly demonstrated that oligomerization was required for some aspects of chemokine function. Similarly, it was shown that binding to GAGs was required in vivo, using GAG-binding deficient mutants.22 These two observations were then linked by in vitro biochemical studies, which demonstrated that chemokines oligomerize on GAG chains.29 In the context of cell migration, oligomerization of chemokines on GAGs is thought to concentrate them in localized areas, thereby forming a gradient, providing the affinity needed for retaining chemokines on cell surfaces particularly in the presence of shear forces from blood flow.30 Oligomerization and interactions with GAGs also protect chemokines from proteolysis and thus contribute to the duration of the “chemokine signal.”31
However, chemokine oligomerization has also been associated with other aspects of cell migration and chemokine biology. Oligomerization of RANTES (CCL5) has been shown to be necessary for monocyte arrest through CCR1-mediated activation of integrins; furthermore, the oligomers were proposed to bridge surface bound RANTES through GAGs on the endothelium with CCR1 on the monocytes.32 Oligomerization of IP-10 (CXCL10) was also shown to be required for transendothelial migration of T-cells and attributed to the fact that only the WT form, but not a monomeric form, could be retained on endothelial cell surfaces.30 GAG interactions, and in fact specific sulfation patterns of GAGs, were shown to be important for the pericellular transport and transcytosis of IL-8 (CXCL8) across endothelial cells, processes needed for the presentation of chemokines on the requisite cell surface.33 Similarly, heparin sulfate was shown to be required for binding of CCL21 to its receptor CCR7 and the induction of migration.34 While both of these examples were not specifically attributed to oligomerization, there is overwhelming evidence that chemokines oligomerize on GAGs and thus are likely required for these processes. Finally, an artificially stabilized “locked dimer” of SDF-1 (CXCL12) has been shown to bind CXCR4, but does not cause cell migration, perhaps suggesting that it could act as a “stop signal” for migrating cells.27 Indeed, it inhibited the metastasis of melanoma cells to the lung, as well as the growth of established pulmonary tumors.35 In addition to its antagonism of cell migration, part of the mechanism was shown to be the superior serum stability of the locked dimer compared to the WT protein, due to protection from proteolysis. These data also suggest the possibility that, at least in the case of CXC chemokines, different oligomeric forms of the same chemokine can show biased signaling.
Oligomeric chemokines have also been shown to be involved directly in alternative signaling processes. WT RANTES (CCL5) but not oligomerization-deficient variants induce apoptosis and T-cell activation through G-protein-independent protein tyrosine kinase activation and interactions with CD44.36,37 Additionally, early studies showed that MIP-1α (CCL3), MIP-1β (CCL4), and RANTES (CCL5) are secreted from the activated cytotoxic T cells as large 400–600 K complexes with proteoglycans, and importantly, that these macromolecular proteoglycan-associated complexes blocked HIV entry into monocytes and lysed virus producing cells.38 Many chemokine receptors have been demonstrated to form homodimers and possibly higher-order oligomers. However, whether oligomerized forms of chemokines bind receptor oligomers is currently unknown; it is not even clear whether chemokine monomers bind receptors in a 1:1 or 1:2 stoichiometry.39 Nevertheless, one could envision a great deal of regulated signaling that could arise from reversible oligomerization of both the chemokine and chemokine receptor as suggested above with the apparent-biased signaling of SDF-1 (CXCL12).27
Much less is known about chemokine hetero-oligomers in terms of their functional affects. However, many chemokines have been shown to hetero-oligomerize not only with other subfamily members but between subfamilies (e.g., CC and CXC).40 One of the best-studied examples is PF-4 (CXCL4), which associates with RANTES (CCL5).41 These heteromers are produced by activated platelets and deposited on endothelial cells, where the presence of PF-4 (CXCL4) has been shown to significantly enhance monocyte arrest induced by RANTES (CCL5). Further, peptides that block formation of the heteromers proved effective in inhibiting atherosclerosis in hyperlipidemic mice, underscoring the importance of these interactions.42
Despite the above examples, there is still limited information on the functional significance of chemokine oligomerization as well as their interactions with GAGs. Nevertheless, many studies have demonstrated that interfering with either oligomerization or GAG binding of chemokines can provide therapeutic benefit in a number of disease models.43–45 To pursue such strategies, a detailed understanding of the structures in question is required, as described in the next sections.
3. CHEMOKINE STRUCTURE
The structures of chemokine oligomers are key to the mechanistic understanding of their functions. There are now numerous structures of chemokines in monomeric and dimeric forms. However, structure elucidation of oligomeric forms, and particularly forms in complex with GAG chains, has been much more challenging. In fact, a number of the early structure determinations, as well as functional studies, were conducted on mutants that did not form structures of higher complexity than dimers (the E66S mutant in the case of RANTES (CCL5)) or were based on data collected at pH conditions that inhibited oligomer formation (pH ~3 in the case of RANTES (CCL5)). As the importance of chemokine oligomerization to function has become more clear, a few structures of higher oligomers have emerged. We review these structures here, and subsequently highlight the evolving techniques that promise to provide more structural information on these highly relevant macromolecular complexes in the future.
3.1. Tertiary structure
Despite high-sequence variability among chemokines, their tertiary structures are remarkably similar.46,47 More than 30 chemokine structures have been solved by high-resolution techniques so far (Table 20.1), and all adopt the “chemokine” fold (Fig. 20.1). The fold consists of an extended N-terminus followed by a core structure made up of a three-stranded beta sheet and a C-terminal helix arranged in a “Greek key” motif. The structure is usually stabilized by the presence of two disulfide bonds, connecting the N-terminus to the core structure. However, chemokines containing only one (lymphotactin/XCL1) as well as three (I-309/CCL1, HCC-2/CCL15, and Ckβ8/CCL23) disulfide bonds also exist. Besides these common features, chemokines also possess an irregular loop referred to as the “N-loop” which connects the N-terminus to the first beta strand, and a long linker connecting the first and second beta strands in the sheet, often referred to as the 30s loop. The 30s loop contains one of the cysteines involved in forming a disulfide bond and also plays a role in the formation of higher-order oligomers.
Table 20.1.
Chemokines with known structures and oligomerization states
| Subfamily | Name | Oligomeric state |
|---|---|---|
| CXC | MGSA (CXCL1) | Dimer (pH 5.5)48 |
| Gro-β (CXCL2) | Dimer (pH 5.5)49 | |
| PF4 (CXCL4) | Tetramer (pH 5.9)50,51 | |
| NAP-2 (CXCL7) | Tetramer (X-ray).52 Monomer(NMR, pH 3.6).53 | |
| IL-8 (CXCL8) | Dimer (pH 5.2)54 | |
| IP-10 (CXCL10) | Tetramer (pH 4.4)55 | |
| I-TAC (CXCL11) | Monomer (pH 5.0)56 | |
| SDF-1α (CXCL12) | Dimer (X-ray).57 Monomer (NMR, pH 4.9).58 | |
| CC | I-309 (CCL1) | Monomer (pH 5.0)59 |
| MCP-1 (CCL2) | Dimer (pH 5.4)60,61 | |
| MIP-1α (CCL3) | Dimer (pH 4.5)62 | |
| MIP-1β (CCL4) | Dimer (pH 2.5)63 | |
| RANTES (CCL5) | Dimer (pH 3.2)64 | |
| MCP-3 (CCL7) | Monomer (pH 5.1)65 | |
| MCP-2 (CCL8) | Monomer (pH 7.5)66 | |
| Eotaxin-1 (CCL11) | Monomer (pH 5.0)67 | |
| MCP-4 (CCL13) | Dimer (pH 6.5)68 | |
| HCC-1 (CCL14) | Dimer (pH 6.2)69 | |
| HCC-2 (CCL15) | Monomer (pH 3.0)70 | |
| TARC (CCL17) | Dimer(pH 5.6)71 | |
| Ckβ9 (CCL21) | Monomer (pH 5.9)72 | |
| MPIF-1 (CCL23) | Monomer (pH 5.2)73 | |
| Eotaxin-4 (CCL24) | Monomer (pH 4.0)74 | |
| Eotaxin-3 (CCL26) | Monomer (pH 5.0)75 | |
| CTACK (CCL27) | Monomer (pH 5.6)76 | |
| C | Lymphotactin (XCL1) | Monomer–dimer equilibrium (pH 6.0)77 |
| CX3C | Fractalkine (CX3CL1) | Monomer (NMR, pH 5.0)78/dimer (X-ray, pH 3.0)79 |
Figure 20.1.

Monomeric structure of the chemokine RANTES (CCL5) (PDB ID 1U4L).
The affinity of most chemokines for their receptors is quite high, with dissociation constants (Kds) in the low nanomolar range. Numerous mutagenesis studies have shown that the N-terminus (preceding the first cysteine) is the key segment for causing receptor activation. For example, many chemokines lacking all or a few amino acids from the N-terminus can still bind to the receptor, often with high affinity, but they no longer induce the conformational changes required for activation, thereby functioning as antagonists.80 Indeed, proteolytic processing of the N-terminus is a natural regulatory mechanism for turning chemokine signaling off and in some cases has been shown to alter receptor specificity.81 Other minor modifications such as extensions of the N-terminus with a single amino acid or single point mutations can convert chemokines from agonists into antagonists, or change the trafficking behavior of the receptors. For example, addition of a single methionine to the N-terminus of RANTES (CCL5) confers antagonist activity.82 Several groups have taken advantage of this general feature to produce high-affinity chemokine antagonists for proof of concept studies of the role of chemokine:receptor pairs in specific diseases.83 Further, variants of RANTES (CCL5) have been discovered that cause its receptor, CCR5, to internalize for prolonged periods of time, making for powerful agents that block HIV infection.84
While the chemokine N-terminus is important for receptor activation, several studies have confirmed that other segments are involved in controlling binding to receptors and likely the orientation of the N-terminal domain with respect to the receptor-binding pocket. The presence of the chemokine core domain (everything but the N-terminus) seems to facilitate a tighter interaction as peptides derived from only the N-termini of chemokines bind to receptors with affinities that can be many orders of magnitude weaker than wild-type proteins.85,86 Specific regions of the core domain involved in receptor interactions have been identified: The N-loop (residues 10–20) is a region that has been shown to be critical for receptor binding.87 In some cases, mutation of one residue in the N-loop can result in a three orders of magnitude decrease in the affinity of a chemokine for its receptor. Several studies involving engineered chimeric chemokines with core residues (beta strands and alpha helix) from one chemokine and N-terminal/N-loop residues from another chemokine also implicated the first 20 residues as the primary determinants of receptor binding and activation.88,89 The 30s and 40s loops, often rich in basic residues, have also been shown to be crucial to receptor interactions.90–93 One of the few structural studies done with whole receptor, as opposed to peptides, has produced evidence for a remote interaction with residues 59 and 63 of MIP1-α (CCL3),94 but generally the C-termini of chemokines are not thought to directly interact with the receptors. Despite the rich mutational data, theoretical prediction of chemokine-receptor specificity remains difficult. The only exception is the ELR sequence motif in the N-termini of CXC chemokines, which usually predestines the chemokines as ligands for CXCR1 and CXCR2, and confers them with the proangiogenic properties of stimulating endothelial cell proliferation and migration.95
3.2. Dimer structures
Many if not most chemokines exhibit tendencies to form dimers and higher-order oligomers. Although monomeric chemokines are believed to be the dominant high-affinity form capable of interacting and activating receptors, oligomeric forms are increasingly recognized as being important to function. Some chemokines such as RANTES (CCL5) readily oligomerize, forming high-order polymers under physiological conditions. Other chemokines, such as MCP-3 (CCL7) and fractalkine (CX3CL1), do not oligomerize, or oligomerize only at very high concentrations, respectively. For most dimer-forming chemokines, the Kd for self-association is in the low micromolar range; however, environmental conditions such as pH and ionic strength can strongly influence dimer stability. Although the physiological concentration of chemokine is believed to be in the nanomolar range, such that soluble oligomers of chemokines should be a small fraction of the total population in vivo, local concentrations of chemokines on the endothelium are likely high enough to induce formation of dimers and other higher-order oligomers. Moreover, the interactions of chemokines with GAGs, which cover the endothelial surface, are known to facilitate oligomer formation, even for chemokines that form weak dimers alone in solution. That these interactions are important has been convincingly demonstrated by several studies which showed that chemokine mutants devoid of their ability to oligomerize or interact with GAGS are functionally deficient in vivo.22,45,96
There is slightly more diversity in dimer structures compared to tertiary structures, and the mode of dimerization seems to be largely dictated by the chemokine subfamily. CC-type chemokines dimerize through residues near the N-terminus, encompassing the first cysteine. These residues in one monomer form an intermolecular antiparallel two-stranded beta-sheet with the same residues in another monomer, resulting in an elongated dumbbell-shaped structure (Fig. 20.2). The subunits are stabilized by a combination of hydrogen-bonding, hydrophobic, and electrostatic interactions. CXC-type chemokines form a distinctly different dimer structure. The dimerization of CXC-type chemokines relies on both the pairing of residues in the first strand of the core beta-sheet in each monomer, and the positioning of the C-terminal helices across beta-sheets from both monomers (Fig. 20.2). The net result is the formation of a large intermolecular beta-sheet of six strands with more contacts between the monomers than in CC-type dimers. The overall complex is also more globular and resembles major histocompatibility proteins.
Figure 20.2.

Dimer structures of chemokines. Monomeric units in each dimer are colored light or dark gray. The PDB ID of each structure is shown in parentheses.
CX3C and XC chemokines form unique dimer structures. The XC lymphotactins, which have only a single cystine at their N-terminus, adopt two conformations that are in slow exchange with one another. One conformation is monomeric and has the conventional chemokine fold. The other conformation contains a four-stranded beta-sheet, formed when the usually extended N-terminus pairs with the third strand in the sheet (Fig. 20.2). The expanded beta-sheet is also the dimerization domain for the dimer. The sheets are packed against one another in a beta-sandwich configuration and are held together by hydrophobic interactions. Interestingly, only the canonical chemokine conformation of lymphotactin is capable of binding the receptor and only the alternative oligomer is capable of binding heparin, suggesting that both conformations maybe functional.77 However, the conformational flexibility of the lymphotactins is likely the exception rather than the rule, since the lack of the second disulfide bond connecting the N-terminus to the 30s loop probably allows the alternative dimer to form.
The second unusual chemokine is fractalkine. In its intact form, fractalkine is a mucin-like membrane protein with an extracellular chemokine domain that overall can function as a tight adhesion molecule, independent of G-protein coupling. However, the chemokine domain can be liberated from the membrane anchor by proteases and then behaves like a traditional chemokine. Although it only dimerizes at high concentrations, the existence of dimers similar to CC-type dimers was observed in a crystal structure. One notable difference does exist between fractalkine and regular CC-type dimers: the unique positions of the disulfide bonds in fractalkine force the core of each monomer to be much closer to one another, thus adopting a much more compact shape.
3.3. Higher-order quaternary structures
For many chemokines, higher-order oligomer formation is common, and the variety of oligomer configurations appears to be extensive and unpredictable. Many of the oligomeric forms have been demonstrated to exist in solution by techniques such as size-exclusion chromatography, analytical ultracentrifugation, and small angle X-ray scattering (SAXS), either alone or induced by binding to GAGs. Others have been captured in crystal structures, suggesting a propensity to form under certain conditions such as bound to GAGs (described below). Among the best-characterized structures are those for MCP-1 (CCL2) and PF4 (CXCL4). MCP-1 (CCL2) is a dimer in solution but forms a tetramer in the presence of GAGs as small as octasaccharides. In the MCP1 (CCL2) tetramers, each monomer contacts one neighbor with a CC-type interface and the other neighbor with a CXC-type interface, giving the tetramer two CC-type dimers (the primary, more stable dimers), two CXC-type dimers, and an overall globular shape97 (Fig. 20.3). PF4 (CXCL4) also forms similar tetramers; however, it is a stable tetramer, even in the absence of GAGs.
Figure 20.3.

Known quaternary structures of chemokines. Each dimeric unit is colored in light and dark gray alternately. The PDB ID of each structure is shown in parentheses.
IP-10 (CXCL10) also forms tetramers. However, three types of tetramers were observed in crystals from different space groups. While one of the tetramers resembles the MCP-1 (CCL2) tetramer (M form), the other two (T and H forms) are more extended and involve a network of intermolecular contacts among beta-sheet residues and the formation of a large curved beta-sheet.55 The structure of murine IP-10 (CXCL10), determined by the same authors, adopts an even more extended form with a 12-stranded beta-sheet.98 Crystal structures of SDF-1α (CXCL12) also revealed an extended oligomer that contains CC-type dimer contacts as well as unique intermolecular contacts involving both the C-terminal helix and the first beta strand99 (Fig. 20.3). Of the chemokines that have been characterized, RANTES (CCL5) and MIP-1α/β (CCL3/4) are unique in their ability to form very large oligomers in solution at physiological pH. The oligomer distribution of RANTES (CCL5) has recently been characterized by native MS; even at the deaggregating pH of 4.5, octamers and hexamers have been observed (see Section 5.4).100 Oligomers of MIP-1 α/β (CCL3/4) have been crystallized and show that chemokines are oligomerized by stacking CC-type dimeric subunits in a helical fashion101 (Fig. 20.3). The oligomers are held together by several key interdimer electrostatic contacts. The tetramer structure of RANTES (CCL5) in solution has also been characterized using an integrated multitechnique approach combining solution NMR, SAXS, and mass spectrometry (MS). The oligomerization interface of the extensible model involves the same residues as the MIP-1α/β (CCL3/4) oligomerization interface, but the oligomer is much more extended because the interdimer interactions take place between only a single monomer in each neighboring dimeric unit, whereas the MIP-1 α/β (CCL3/4) oligomers utilize both monomers of each dimeric unit to contact adjacent dimers100 (Fig. 20.3).
Oligomers with exposed oligomerization surfaces at the ends of extended structures, as in the case of MIP-1α/β (CCL3/4), naturally lend themselves to formation of oligomers of ever increasing sizes. In fact, a simple model in which the free energy of association would be the same at each successive addition of a dimer would lead to an extremely broad distribution of oligomer sizes. For an association constant of 104 and monomer concentration of 10−4 M, concentrations of tetramer, hexamer, octamer, etc., would be 10−4, 10−4, 10−4, etc. That unrestricted oligomerization does not happen in nature very likely is the result of the high-charge density these molecules normally carry. RANTES (CCL5) at neutral pH would have a net charge of +12 for the dimer. Even if counter ion association reduced the effective charge to +3, the attenuation of high oligomer forms by charge repulsion would be substantial. With a simple charge repulsion model that assumes (i) the charge is effectively concentrated at the dimer center, (ii) that the oligomers are linear, and (iii) that addition of each subunit is characterized by a noncharge compensated association constant of 105, one can calculate that the teramer, hexamer, and octamer formation constants would be 1.67×104, 1.14×104, and 0.42×104. Studies characterizing the oligomerization interface of RANTES (CCL5) were done at pH of 4.5 where tetramers dominate. Partial protonation of acidic amino acids would artificially increase the effective positive charge. An effective charge of +4 on each dimer and an uncompensated association constant of 106 predicts that more than 90% of the species would be tetramers using this simple model. The actual in vivo situation is likely to be more complex for many reasons, including the presence of GAG interactions as discussed below. The highly negatively charged GAGs would further reduce the effective charge of a positively charged chemokine and shift the equilibrium to higher-order oligomers—as is observed.
4. CHEMOKINE OLIGOMERIZATION AND GAG BINDING
GAGs are linear polysaccharides that are found on cell surfaces or in the extracellular matrix. They consist of repeating disaccharide units with one saccharide being either galactosamine (chondroitin sulfate or dermatan sulfate) or glucosamine (hyaluronan, heparin, heparan sulfate, or keratin sulfate) and the other being either a uronic acid (all types except keratan sulfate) or galactose (keratan sulfate). GAGs are extremely anionic due to a high content of uronic acids and sulfation. The presence of the high charge density allows GAGs to interact strongly with many signaling molecules. The existence of chemokine: GAG interactions is not surprising given the highly basic nature of most chemokines and the need for some mechanism of cell surface immobilization; thus the importance of these interactions was recognized early on.102 Further, the interactions are by no means nonspecific. Studies comparing salt elution concentrations of different chemokines on Mono-S and heparin columns showed affinities for Mono-S resin and heparin resin do not correlate strictly with charge.103 Competitive adhesion assays with immobilized heparin or cells not expressing chemokine receptors also demonstrated the ability of chemokines to discriminate between different types of GAGs.103,104 Functionally, GAG–chemokine interactions are believed to be important in several aspects of chemotaxis, including prevention of chemokine proteolysis, and stabilization of chemokine gradients on the endothelial surfaces under blood flow, so that they can provide directional cues for migrating cells. The fact that models of leukocyte activation stipulate that leukocytes should only be activated by chemokines close to the endothelium surface also implies that chemokines bound to GAGs may in fact be active in vivo, making understanding the structures of GAG-bound chemokines a high priority.
GAG-binding proteins often oligomerize upon binding GAGs. This phenomenon has been shown to be crucial in the role of GAGs as coreceptors for a number of growth factors and as facilitators of amyloid fibril formation in vivo. The role of GAG-induced chemokine oligomerization in promoting localization of chemokines was revealed through in vitro studies conducted by Hoogewerf et al. They showed that binding of GAGs to chemokines induced oligomerization of a number of chemokines and that removal of GAGs from cell surfaces reduced the binding of chemokines to the cell surface significantly. Whether chemokines can bind simultaneously to GAGs and receptors is currently unknown. However, as described above, the functional requirement for the oligomerization of RANTES in inducing monocyte arrest was proposed to provide a mechanism for bridging surface bound RANTES through GAGs on the endothelium with CCR1 on the monocytes.32
Mutagenesis studies on a handful of chemokines have identified some motifs important for binding GAGs. In vivo assays designed by Proudfoot and coworkers were especially instrumental in establishing the fact that although GAG-binding deficient mutants were able to induce cell migration in vitro, they failed to do so in vivo.22 In keeping with early studies on the amino preferences for heparin, these studies identified arginine and lysines in chemokines as the residues most responsible for GAG binding, but histidines were also found in GAG-binding motifs of some chemokines. The BBXB motif known to bind GAGs has been found in MIP-1α/β (CCL3/4) (40s loop), RANTES (CCL5) (40s loop), and SDF1α (CXCL12) (20s loop). All are confirmed to be crucial to GAG binding.93,105–107 MCP-1(CCL2) and IL-8 (CXCL8) also contain clusters of basic amino acid that have been shown to be important for GAG interactions.91,104,108
Although no experimental structures of chemokines bound to anything longer than a disaccharide GAG exist, chemokine oligomer structures without GAGs do offer us some clues as to how GAGs may stabilize oligomerization. The MCP-1 (CCL2), PF-4 (CCL4), and IP-10 (CXCL10) M tetramer structures show basic residues in each monomer to be arranged on the surface to form a binding strip across the entire tetramer structure, prompting the proposal of a “beads-on-a-string” model for these GAG–chemokine interactions (Fig. 20.4). The overall acidic nature of GAGs should also neutralize charge repulsion among the basic chemokine monomers, thus stabilizing the oligomer structure. However, not all chemokines oligomerize in a globular fashion. MIP-1α/β (CCL3/4) oligomerizes in a linear fashion, forming a basic channel on the oligomer surface in which GAGs can bind. Surprisingly, SAX analysis of MIP-1α/β (CCL3/4) showed oligomers formed in the presence of heparin are smaller than without heparin,101 contrary to a study of the chemokine carried out with other techniques.29 This complex response may be the result of the rather unique charge distribution in MIP-1α/β (CCL3/4). In contrast to the high-positive charge of RANTES (CCL5), MIP-1α/β (CCL3/4) actually have a net negative charge (−1).
Figure 20.4.

GAG-binding motifs of chemokines. Amino acids known to bind GAGs are shown as black sticks. The same residues are also colored black on the oligomer surface depiction. For IL-8 (CXCL8), K20, R60, K64, K67, and R68 are shown. For MCP-1 (CCL2), R18, K19, R24, K49, K58, and H66 are shown. For MIP1α (CCL3), R18, K45, R46, and K48 are shown. For RANTES (CCL5), R17, K44, R45, and K47 are shown.
In the oligomer structure of RANTES (CCL5), the BBXB motifs form two regular arrays along the length of the oligomer, providing a possible GAG-binding site on the oligomer. However, RANTES (CCL5) is sufficiently basic that the formation of oligomers also creates large basic patches elsewhere in the structure due to close packing of other basic amino acids on the molecule. The potential of these basic patches as GAG-binding sites means RANTES (CCL5) may possess multiple GAG-binding sites as an oligomer.
The discovery of these linear chemokine oligomers prompts the question of their in vivo relevance. Although no chemokine oligomer structure has been solved in the presence of GAGs, the presentations of basic amino acid clusters on these linear oligomers are usually compatible with simultaneous interactions with both GAGs and receptors. Their existence produces interesting hypotheses for the chemotaxis process. The primary role of a chemokine concentration gradient is to provide directional cues for the migration of leukocytes and their activation. The random distribution of chemokines around the inflammatory site with concentration gradients controlled by surface diffusion still presents leukocytes with substantial translational migration freedom, which may translate to slower targeting efficiency. The formation of large linear oligomers could favor a more efficient leukocyte targeting process by limiting the movement of leukocytes to a one-dimensional process.
It is worth noting that residues identified as GAG-binding epitopes in several chemokines are also involved in receptor binding. R17 of RANTES (CCL5) has been identified as crucial to activation of CCR1, but strong evidence implicates the same residue in GAG-binding. Similarly, mutations in the BBXB motif of RANTES (CCL5) and MIP-1β (CCL4) reduced its affinity for CCR1 by 200-fold and identical changes in MIP-1β (CCL4) reduced its affinity for CCR5 by 77-fold.22 The recent crystal structure of CXCR4 showed a large acidic binding site for its ligand SDF-1 (CXCL12), providing a rationale for overlap in GAG and receptor-binding sites in chemokines. The functional consequence of competition between the two interactions for the shared binding motif has yet to be investigated, but it is reasonable to hypothesize that higher-order oligomerization may have evolved to allow simultaneous GAG- and receptor-interaction by displaying multiple binding sites in the same entity.109 The linear oligomerization states of chemokines are especially suited for this purpose.
Given the known tissue-specific variations in GAG species and sulfation patterns, it is tempting to speculate as to whether chemokines are capable of recognizing specific GAGs. Because of the difficulties in purifying structurally homogeneous GAGs of sufficient length, characterizing specific GAG–chemokine interactions remains difficult. However, current knowledge of chemokine oligomer structures already reveals much complexity in GAG–chemokine interactions: lymphotactin only interacts strongly with heparin as a noncanonical dimer while the monomer is potent in binding its receptor, thus excluding the possibility of simultaneous interactions with both GAGs and receptors. IP-10 (CXCL10) can adopt different tetramer forms with different displays of basic amino acid patterns on their surface and consequently different GAG specificities. SDF-1γ (CXCL12) possesses an extended basic C-terminal tail, which may endow it with considerable promiscuity, allowing it to access GAGs of different species without oligomerizing. These observations suggest flexibility in GAG–chemokine interactions, which may influence their specificity for GAGs under different conditions.93,105–107 Some attempts have been made to characterize the GAG length dependence of chemokine:GAG interactions, and a wide variety of affinities have been demonstrated. Spillmann et al. showed that IL-8 (CXCL8) dimers only bind to HS weakly and that a HS fragment 18 saccharides long was required to achieve measureable affinity.110 On the other end of the spectrum, RANTES (CCL5) binds to HS hexasaccharides with a Kd of less than 100 nM.111 In fact, it has been observed that GAG fragments as short as hexasaccharides are capable of inducing formation of higher-order oligomers in RANTES (CCL5), even though the size of the dimer is capable of accommodating a much longer fragment. While no Kd has been reported for MIP1α (CCL3) binding to GAGs, a heparinase protection study showed that a HS fragment with 14 saccharides seems to be the optimum size.112 Although the original study speculated that dimers were the dominant oligomeric forms in solution, in view of the work by Ren et al.,101 it is highly likely that HS fragments were binding a much larger oligomer.
5. STRUCTURAL METHODS FOR OLIGOMERS AND GAG INTERACTIONS
Given the importance of oligomerization and GAG-binding interactions, it is worth considering how data on these interactions will be acquired in the future. Much of the data presented above was obtained by X-ray crystallography. The resolution in most cases is at a level seldom obtained by other methodology. However, there are limitations. The oligomerization interactions, at least in the case of linear oligomers, have no definitive limits on oligomer size, increasing the likelihood that crystal packing will impose restrictions that do not exist in solution or in vivo. Also, the GAGs to which chemokines bind are extraordinarily heterogeneous. This makes homogeneous samples of any significant length difficult to obtain, and the use of heterogeneous samples inhibits crystallization of complexes. Furthermore, it is very likely that hetero-oligomers may be functionally relevant, as are complexes involving multiple receptor molecules, and receptors anchored in their native membrane environment. All of this demands a fresh look at options for structural characterization.
5.1. NMR of protein–protein complexes
NMR offers some advantages when it comes to examining interactions between multiple proteins (chemokine oligomers or chemokine–receptor interactions) or interactions between proteins and ligands (GAGs). It does not require crystallization and is reasonably tolerant of heterogeneity, at least in the case of rapidly exchanging heterogeneous ligands. It is also a solution-based technique applicable in aqueous buffers that approach physiological conditions or media that mimic membrane environments. However, it does have limitations in that the sensitivity of the technique requires solutions that are several hundred micromolar in the protein/complex of interest, enrichment with NMR observable isotopes (13C, 15N) is required, and investigations of large assemblies are limited by resonance broadening (this increases linearly with the molecular weight of the system and causes loss of both resolution and sensitivity). Nevertheless, NMR has made a substantial contribution to investigation of chemokines and their oligomerization properties.
NMR structures of monomeric and dimeric chemokines are abundant.35,60,64,67,113–117 The small size of monomeric chemokines, less than 10 kDa in most cases, has for years put chemokines well within resolution and sensitivity limits of NMR. Early structures were produced without isotopic labeling or labeling with 15N only. All relied on distance constraints from short-range 1H–1H nuclear Overhauser effects (NOEs). Later, structures, and particularly larger dimeric structures, relied on triple resonance methods for resonance assignment and resolution of larger numbers of NOE cross-peaks (13C and 15N labeling). All have produced useful atomic resolution structures.
The utility of NMR in detecting homotypic chemokine–chemokine interactions, as well as heterotypic interactions with other chemokines or receptors exists at many levels. The most widely practiced approach utilizes chemical shift perturbation. Chemical shifts of resonances, or more commonly the positions of cross-peaks in 15N–1H heteronuclear single quantum coherence (HSQC) spectra, change on contact with ligands or other proteins due to the magnetic susceptibility anisotropy of nearby groups such as phenyl rings and electric field effects of nearby charged groups. They also change (especially for heteronuclei such as 15N or 13C) because of induced distortions of bond angles in the peptide backbone. These multiple contributions make it difficult to quantitatively convert perturbations to distances of approach. Even occasional remote changes can occur due to allosteric effects on protein geometry, but when perturbations to resonances from several clustered residues are observed, identification of regions of contact can be done quite reliably. There are examples where likely dimer interfaces have been identified in this way. A good example is in the characterization of a dimeric variant of the viral analog of human MIP1-β (CCL4), vMIP-II,118 which is a broad-spectrum antagonist, produced to subvert the human immune system. Shifting the dominant monomer form toward a dimer through a single mutation alters both GAG affinity and binding geometry making identification of the dimer interaction surface particularly relevant. Chemical shift perturbations have also been used to identify chemokine sites that interact with receptors. Most notable is the early work on identification of the importance of N-terminal residues of RANTES (CCL5) on interaction of peptides from the CCR5 receptor.28 More recently perturbations of 15N–1H cross-peaks from the N-terminal portion of the CXCR4 receptor (1–38) have been used to infer different modes of interaction for monomeric and dimeric forms of the CXCL12 chemokine.35
There are also more quantitative methods for identifying interface residues that involve diffusion of perturbed magnetization across an interface (an NMR cross-saturation experiment).119 Here, perdeuterated proteins with amide protons at 15N-labeled sites (reintroduced by back-exchange) or having only 13C methyl groups protonated are mixed with fully protonated and nonisotope-labeled protein. Proton resonance characteristics of the non-labeled protonated species are saturated, saturation spreads across the protein–protein interface in a distance-dependent fashion and saturation is detected through the isotopically labeled sites at the interface. The very steep distance dependence (1/r6) ensures that sites at the interface are selectively detected. An example of this type of experiment can be found in the determination of a tetramer structure for CCL5 where both monomer–monomer and dimer–dimer interfaces were identified.100 There is also a very interesting application to the interaction of the CCR5 receptor with MIP1-α (CCL3).94 Chemokine receptors are integral membrane proteins and CCR5, in particular, seems to require more bilayer-like membrane mimetics to maintain a functional configuration. Nanodiscs are assemblies that fulfill this purpose, but they are very large.120 The Shimada group has used a modified version of the cross-saturation experiment (transferred cross-saturation), and the higher sensitivity protonated methyl labeling of the chemokine CCL3, to facilitate investigations of this very large complex. Unexpectedly, valines 59 and 63 were identified as components of the interaction site and add to the chemokine N-terminal regions normally implicated in interaction with receptors. The method does require rapid exchange on and off the receptor, so that detection of an excess of free chemokine population can be used to report on the complex; therefore, it is possible that the interactions arise from a secondary (weaker) binding event. Nevertheless, the application does illustrate the potential for applying NMR to some very large systems.
Actual structural characterization of oligomers and complexes is more of a challenge. Traditionally, this would be approached by seeking short-range NOEs connecting residues on different proteins of a complex. Acquiring these data is particularly challenging for symmetric homo-oligomers because resonances from partners in the complex are degenerate. However, there are strategies referred to as X-filtered experiments in which partners are labeled with 13C and 12C, respectively, and signals filtered for ones arising from 13C-labeled sites and detected on 12C sites or vice-versa.121 These are effective, but labor intensive in terms of the need for preparation of multiple samples and also sensitivity limited in the case of homo-oligomers due to detection of only half the complexes formed. As most interface NOEs involve interaction of protons on side chains, they also require complete side chain assignments, something that becomes more difficult as the size of complexes increases.
There are additional long range distance-dependent interactions that can be useful in the structural characterization of chemokine oligomers. In particular, paramagnetic relaxation enhancement (PRE) experiments, in which line broadening or spin relaxation times show a 1/r6 dependence, can be used. They do require the introduction of a paramagnetic site in a protein or in a GAG ligand. Most commonly a nitroxide-carrying TEMPO adduct is used. Because of the high-magnetic moment of the unpaired electron on the nitroxide (650 times larger than that of a proton) and the square dependence of relaxation enhancement on this moment, these effects extend over many tens of angstroms and are potentially effective in positioning one protein relative to another or positioning a ligand in its binding site. While there are few applications to positioning of chemokines in protein complexes at this point, there are applications to positioning GAGs in chemokine binding sites. These will be discussed in the following section.
Long-range NMR constraints can also come from orientation-dependent NMR measurements, the most common of which are residual dipolar coupling (RDC) measurements.84,122,123 RDCs are measured in orientable media (liquid crystals or stretched polyacrylamide gels) that can induce partial alignment of molecules of interest. They are usually measured as a change in couplings between directly bonded pairs of nuclei (e.g., 15N–1H amide bonds) when through space dipole–dipole contributions fail to average to zero. Their dependence on the average of the function, (3cos2θ−1), where θ is the angle between the spectrometer’s magnetic field and the internuclear (bond) vector, provides a powerful determinant of molecular geometry. When sufficient numbers of RDCs are measured for well-defined structures, alignment axes can be determined, and when two different proteins are involved superposition of alignment axes greatly restricts allowed structures.124,125 Typically, these RDC-based restrictions are combined with data on interfacial contacts and translational restraints to produce unique structures. For symmetric homo-oligomers, there are symmetry-based rules that allow identification of the symmetry axis in the coordinate frame of the monomer and generation of symmetry-related elements by rotation about this axis. Applications involving the determination of structures for symmetric dimers have appeared.124,126 RDCs are particularly advantageous when working with higher-order oligomers as only backbone resonance assignments are needed and RDCs can be measured with high precision in relatively large systems. This gives them an advantage over more traditional X-filtered experiments for determination of structures, particularly when structures of constituent monomers are known from other sources. A good example of an RDC application to a higher-order chemokine oligomer is the structure determination of a CCL5 tetramer.100 Previously available structures for CCL5 existed from both crystallography127 and solution NMR studies.64,115 These were produced at low pH (~3) where the structure is predominantly a dimer of the canonical CC form. At higher pH (4.5), the system is predominantly a tetramer as confirmed by light scattering and NMR diffusion studies. Comparing experimental RDCs to calculated RDCs based on a monomer from the crystal structure confirmed that the monomer structure was preserved in the tetramer. The appearance of just a single set of 15N–1H HSQC cross-peaks suggested a symmetric structure (at least as averaged over the timescale, ~1 ms, needed to resolve NMR resonances). More surprising was that the symmetry axis of the tetramer, as identified from RDCs, coincided with the symmetry axis of the dimer in the crystal structure. There are only a few possible ways to construct a tetramer while preserving this symmetry axis. Adding interface residue data and SAXS data as discussed below, an elongated structure was deduced. This is one in which higher concentrations and GAG interactions can promote the addition of other dimer units to make progressively longer hexamers, octamers, etc. These structures are in fact observed in subsequent mass spectrometric analysis of low-concentration samples prepared at higher pH.
In the future, residual chemical shift anisotropies (RCSAs) are likely to be added to RDCs for the determination of monomer orientations in oligomeric structures.128 These display identical angle dependencies to RDCs, but the vectors are principal axes of shift tensors and an average over the three axes are observed. Their advantage is that they depend only on measuring chemical shift changes for highly anisotropic sites such as a 13C carbonyl in a peptide or acetyl group, and can likely be measured in even larger systems. Again applications to chemokines are few at present, but there are applications to the determination of preferred conformations for GAG oligomers.129
5.2. NMR of GAG–protein complexes
Interest in structural aspects of GAG–protein complexes remains high both because of scientific curiosity and the potential application of GAGs in treatment of a range of diseases from cancer to atherosclerosis. However, structural characterization of GAG–protein complexes presents significant challenges. Among them is the tendency of GAGs to induce oligomerization of chemokines. Many of the NMR methods discussed above can be effective in the investigation of such complexes. However, significant technical challenges also arise from the lack of a library of structurally homogeneous GAG fragments large enough to reproduce conditions experienced in vivo, and the relatively small number of routes to the production of isotopically labeled forms of these fragments.
NMR methods
NMR has always been instrumental in the unambiguous determination of GAG structures, but its ability to solve high-resolution protein structures combined with its sensitivity to protein–ligand interactions and tolerances of heterogeneous GAG fragment mixtures has made it an excellent tool for characterizing GAG–chemokine interactions. The most popular use of NMR involves identifying the GAG-binding site of the protein using ligand-induced chemical shift changes for isotopically labeled sites in chemokine, much like the identification of interfaces in chemokine oligomers discussed above. Binding of GAGs to proteins often changes the magnetic environment in nearby atoms by electrostatic or structure perturbation mechanisms, thus generating a change in a labeled atom’s chemical shift. If the assignments of the resonances are known, residues most perturbed by ligand binding can be determined easily. Since both mechanisms tend to be short range, those perturbed most by the ligand are often deemed to be residues close to the GAG-binding site. Interactions between GAG fragments and proteins often fall in the fast exchange regime on the NMR time scale, resulting in a gradual shift of resonances with increasing ligand concentration (Fig. 20.5). This also allows the Kd of binding to be extracted easily and precisely. The most popular atoms monitored are the backbone amide nitrogen and protons, whose chemical shifts are sensitive even to minute perturbations in the magnetic environment. The two-dimensional NMR experiment, 1H–15N HSQC spectroscopy, allows simultaneous measurement of both chemical shifts easily.
Figure 20.5.

Chemical shift perturbation on addition of a four-sufated chondroitin sulfate hexamer to 15N-labeled RANTES (CCL5). A series of 15N–1H HSQC spectra are superimposed to show chemical shift changes as increasing amounts of the hexamer are added. Among cross-peaks shifting are those assigned to residues from the BBXB motif, R44, K45, N46, R47. Spectra are taken at 600 MHz for 1H on a ~400 μM, pH 3.5, sample of wild-type RANTES (CCL5).
If the changes in chemical shifts of the protein atoms are too small, or if the protein undergoes significant conformational changes upon binding, GAGs functionalized with paramagnetic ligands can be used to probe the protein–GAG interactions. Gemma et al. coupled amino TEMPO, a stable nitroxide radical, to the carboxyl group of uronic acid in a heparin disaccharide and successfully probed the GAG-binding sites on CCP module 7 of complement factor H using the adduct.130 Although this is the only reported case of a protein–GAG interaction study utilizing paramagnetic GAG ligands to date, the ease by which GAG fragments can be functionalized on the reducing end should encourage more examples in the future.
Despite the popularity of chemical shift mapping in GAG–protein studies and emerging paramagnetic methods, solving high-resolution structures of GAG–protein complexes remains a challenge for NMR. The dominant contributions to protein–GAG interactions are electrostatic interactions between sulfates on the GAG fragment and side chains of basic amino acids such as lysine, arginine, and histidine. The lack of intermolecular proton contacts in these interactions makes their detection through traditional homonuclear NOESY, which is sensitive to only proton–proton contacts, difficult. So far, the traditional approach of solving protein–ligand interactions has not produced a single structure of GAG–protein complexes. However, some innovative approaches are being developed. One method identifies the basic amino acid involved by combining cross-linking with solution NMR. By slowing down the exchange of protons between lysine side chains and solvent using low pH (3.0) and low temperature (10 °C), the proton signal from side chain amino groups of lysines can be observed. Cross-linking of the activated carboxyls in the GAG fragments to the lysines during the binding event produced significant changes to the spectrum, therefore allowing unambiguous identification of lysine residues involved in GAG binding.131 This covalent modification technique forms an excellent complement to the chemical shift mapping approach, which is usually carried out using only backbone amide protons that may or may not respond to GAG-binding.
Besides observing perturbation of protein by GAG ligands, NMR is also well suited for studying GAG conformation in the presence of protein. Interactions of chemokines and small GAG fragments usually occur on the fast exchange regime on the NMR time scale. This allows well-established ligand techniques such as saturation transfer difference (STD) and transferred NOE (trNOE) to be applied to the study of GAG conformations in the presence of proteins.103,132,133 The only limitation on these techniques is the insolubility of GAG–chemokine complexes at high chemokine concentrations (>100 μM). However, STD only requires low concentrations of protein (~10 μM), which can be a great advantage. The only published example where STD is utilized to investigate GAG–chemokine interactions is a study of CXCL12α with a 13C-labeled HS-like octasaccharide derived from E. coli K5 capsular polysaccharide.134 The labeling allowed the use of 2D methods to improve resolution, and the research identified many protons on both glucosamine and glucuronic acid as being in the interface. The information allowed the construction of a model of GAGs bound to the CXCL12α dimer. Another potential source of structural information obtainable from 13C-enriched GAG fragments is RCSA from carbonyl carbons. The amide bond in most N-acetyl hexosamines is quite rigid,135 and therefore orientation of the acetate group can serve as a good indicator of hexosamine ring orientation. In a study of chondroitin sulfate conformation, Yu et al. measured RCSA from 13C acetate-labeled CS hexasaccharides and used the information to supplement existing RDC data for polysaccharide conformation determination.129 Although not yet applied to GAG–chemokine interactions, the technique will be valuable in defining bound GAG conformation and orientation in GAG–chemokine complexes. So far, there is no reported example of trNOE applied to the study of GAG–chemokine interactions. But, the technique has been utilized to determine the bound conformations of GAG fragments in other systems, including the well-characterized GAG interactions of antithrombin136 and cobra cardiotoxin.137
Other than obtaining high-resolution structures for both chemokine and GAGs, NMR is also capable of studying conformational dynamics in these molecules, especially if appropriate isotopic labels are present. NMR signals are sensitive to motions of atoms at a wide range of time scales, allowing frequencies of rotational motions to be characterized in terms of the rotational correlation times. Protein dynamics can also be measured on a residue-specific basis if assignments of resonances from backbone amide nitrogens and protons are available. Such analysis has been applied to both FGF-2 and hepatocyte growth factor (HGF). In the cases of HGF, residues in the GAG-binding regions of the protein experienced significant increases in motions on micro-to-millisecond time scale.138 FGF-2 was shown to have an overall increase in rotational correlation time, indicative of GAG-induced oligomerization.139 However, no dynamic studies have been conducted on protein-induced changes in GAG dynamics. The ligand dynamics information is potentially valuable in understanding the kinetics and thermodynamics of protein–GAG interactions possessing multiple binding modes. The main technical barrier preventing this type of study lies in difficulties in obtaining isotopically labeled GAG fragments.
Sources of isotopically labeled GAGs
Traditionally, all homogeneous GAG fragments have been derived from natural GAG polysaccharides. Because of the complexity in GAG structures and challenges of the underlying carbohydrate chemistry, organic syntheses of GAG fragments have remained the privilege of a few synthetic chemists.140 The isolation of GAG fragments from natural sources usually follows a three step procedure141: Natural GAG polymers are partially fragmented using appropriate endohydrolases or endolyases. For instance, heparin is most efficiently depolymerized with heparinase I from Flavobacterium heparinum and chondroitin sulfate can be depolymerized with either chondroitinases ABC, or AC, or hyaluronidase. The digested fragments are then separated based on size using size-exclusion chromatography. Finally, fragments of similar sizes are further separated using strong-anion exchange (SAX) HPLC. The success of the last step relies on the fact that SAX HPLC columns are capable of distinguishing fragments possessing different sulfation patterns despite identical total charge on each fragment. Because of sulfation pattern uniformity in some GAGs, SAX HPLC is sufficient to purify short chondroitin sulfate A or DS fragments to homogeneity.142 However, rigorous separation of heparin beyond the size of octasaccharides continues to be challenging. Although many alternative analytical techniques for GAGs have been developed for GAG analysis, the above procedure has remained the only viable protocol for obtaining GAG fragments in preparative quantities.
Besides vertebrate sources of GAGs, GAG mimics produced using bacterial capusular polysaccharide (CPS), either as the final product or as the precursor for further chemical sulfation, are also becoming more prevalent. Many bacteria generate CPS that are similar to GAGs in carbohydrate composition. Streptococcus group C produces CPS that are identical to hyaluronan while CPS from E. coli strain K5 have the same carbohydrate structure as heparin and heparan sulfate.143 They can be used as GAG mimics either without modification or after N-deacetylation followed by N- and O-sulfation.144 So far, the sulfation methods used are limited to sulfur trioxide complexed with either trimethyl amine or pyridine, both of which have limited specificity.144,145 Although N-sulfation and 6-O-sulfation have proven to be quite easy, specific 2-O-sulfation on uronic acids or 4-O-sulfation on GalNAc has proven to be challenging145; furthermore, no chemical method to convert glucuronic acid to iduronic acid exists. Difficulties in specifically modifying GAGs can be partially lessened through chemoenzymatic approaches to GAG synthesis. In vitro production of GAGs using isolated bacterial enzymes are already feasible,146 and the technology is quite mature for production of hyaluronan molecular weight standards.147,148 A recent study produced the potent antithrombin-binding heparin pentasaccharide using a chemoenzymatic approach in vitro starting from disaccharide units.149 Such methods have the promise of producing size- and structure-defined fragments, therefore greatly simplifying purification procedures.
Despite its disadvantages, production of GAG mimics from bacterial CPS can provide new opportunities for the study of GAG–protein interactions using NMR. Lack of GAG fragments enriched in NMR active nuclei such as 13C and 15N has prevented unambiguous identification of protein-contacting GAG protons in cases of long fragments and made the study of GAG dynamics limited to proton relaxation measurements, which can be difficult to interpret because of the numerous factors involved in proton relaxation. The main reason for the deficiency is because vertebrate cells, which are often the sole source of GAGs, cannot yet be grown in isotopically enriched media in an economically feasible fashion. To overcome this deficiency, bacterial CPS grown in isotope-enriched media can be used as a substitute. This was the strategy used by Laguri et al. to obtain 13C-labeled heparin octasaccharide for studying the interaction of CXCL12 with GAGs,134 the only study of chemokine–GAG interaction using 13C-labeled GAG mimics so far. However, the procedure remains a complicated one and limited to heparin/HS mimics only. 15N enrichment of the same polysaccharide has also been carried out150 and was demonstrated to be of value in the study of GAG dynamics as interpretation of amide nitrogen relaxation is well established.
An alternative to metabolic incorporation of isotopic labels is through the use of chemical modification.129 N-acetyl groups can be chemically deacetylated and then reacetylated using 13C acetic anhydride. In the cited work on chondroitin sulfate, the identity of each carbonyl signal in the 13C NMR spectrum was determined based on correspondence between 13C NMR signal intensity and percentage isotopic enrichment as indicated by MS/MS analysis. The labeling allowed additional structural information to be gained using RCSAs and is potentially applicable to any GAG fragment containing N-acetyl hexosamine.
Although the production of isotopically enriched GAGs using mammalian systems is still difficult, one feature of mammalian carbohydrate metabolism has been explored to create 15N-labeled GAGs using mammalian cell culture. In vertebrates, the nitrogen in N-acetyl-hexosamine is derived mainly from the side chain amide of glutamine. Supplementing the media with 15N glutamine therefore offers an elegant and economical way to produce 15N-labeled GAGs in vertebrate cell cultures. Pomin et al. carried out such a study and was able to produce 15N-labeled CS and HS using both mouse lung endothelial and Chinese hamster ovary cells.151 As the 15N chemical shift of GAGs is sensitive to GAG types, labeling allowed accurate quantification of ratios of different GAGs existing on the cell surface. The application of such techniques can make quantification of cell surface GAGs possible.
5.3. Global structure of oligomers from SAXS
SAXS is an important technique in elucidating the structural characteristics of macromolecular complexes in solution.152,153 It utilizes interferences between X-ray photons coherently scattered by atoms to measure geometrical arrangements of atoms in a molecule. Unlike X-ray diffraction using crystals, SAXS samples are solutions of macromolecules with no orientational homogeneity, and thus scattering intensity is dependent on the scattering angle alone. The plot of the scattering intensity versus diffraction angle offers the main experimental data in SAX experiments, and intensities at smaller scattering angles are dependent on the global shape and size of the molecular complex in solution. Although the resolution of the structural information obtained is quite low, it is often valuable in determining the solution states of biomolecule complexes. The ease with which SAXS samples can be prepared and the advent of commercially available benchtop SAXS instruments also made the technique more accessible to researchers eager to combine SAXS data with structural information obtained from other sources, especially NMR spectroscopy. The theory of molecular scattering is well understood, and accurate prediction of the scattering curve of known molecular structures offers a powerful way to determine the solution state of many macromolecules. For samples containing a single species, extraction of the radius of gyration as well as inter-atomic distance distribution is routine. If the description of the geometrical shape of the molecule is simple, existing models can be also used to estimate a number of other important parameters, such as the lengths of rods or radii of spheres. Even a medium resolution shape envelope of the molecule can be obtained through sophisticated combinatorial search algorithms that generate the shape envelope best reproducing experimental results. Since intensity at zero angle is directly proportional to the molecular mass of the complex in solution, the total mass of a complex can be estimated using SAXS data, provided data acquired on samples of known molecular composition are also available. For samples containing a heterogeneous mixture of oligomerization states, which is often the case for many chemokine oligomers, the data interpretation is more challenging. However, if shapes of species in existence are available, scattering contribution from each species can be calculated and summed to produce the best fit to experimental data.
At least two studies on chemokine oligomerization have incorporated SAXS data. In the study by Ren et al., SAXS was used to estimate the size distributions of MIP1α and MIP1β (CCL3/4) in the presence and absence of heparin.101 The scattering curve of the two chemokines showed characteristics of rod-shaped molecules, which agreed with the crystal structures of the oligomeric chemokines. Fitting of the scattering curve to theoretical prediction of polymer size distributions using a reversible polymerization model allowed the distribution of polymers to be extracted. Interestingly, it was discovered that heparin reduced the size of wild-type MIP1α (CCL3) polymers, but promoted the oligomerization of a nonaggregating mutant of MIP1α (CCL3). The results were also confirmed using size-exclusion chromatography. In the study of the RANTES (CCL5) polymer, SAXS data also played a crucial role.100 In this study, the major oligomerization state of the chemokine was characterized using data from NMR relaxation and pulse field gradient diffusion experiments, and RDCs were used to determine the orientation of the dimeric units relative to one another. This information greatly reduced the possible structural configurations of a RANTES (CCL5) oligomer. Further analysis by SAXS showed the oligomer to adopt an irregular and extended shape. Using a simple two-dimensional search algorithm, a set of models satisfying the NMR data was generated. The final structural model was then selected based on the agreement between the theoretically predicted scattering curves of each model and the experimental scattering curve. A configuration with each monomeric unit of the dimer making contacts with adjacent dimers was found to fit both NMR and SAXS data well.
5.4. MS analysis of chemokine oligomerization
MS is a powerful and versatile tool for the analysis of protein oligomerization. Depending on the methods used, MS is capable of determining the stoichiometry of oligomerization (even for polydisperse oligomer mixtures) and analysis of the structure of the oligomer. MS is only capable of analyzing ions in the gas phase. Therefore, in order to use MS to study solution-phase oligomerization, it is either necessary to transfer the oligomer intact into the gas phase (referred to as “native spray”) or else to modify the protein in solution in a manner that transfers information regarding oligomerization into the gas phase. Both strategies can give valuable information on chemokine oligomerization, but each requires some thought regarding their strengths and limitations.
5.4.1 “Native spray” analysis
MS is capable of measuring the mass-to-charge ratio (m/z) of chemokine oligomers in the gas phase. Based on either high-resolution analysis of 13C isotopomers or analysis of the spacing between neighboring charge states of the same oligomeric state, the actual charge state (and therefore the actual mass) of each oligomer can be determined, allowing for the direct measurement of the size and dispersion of oligomers. However, in order for the information generated to be biologically relevant, the oligomer(s) that exist in solution must be transferred intact into the gas phase.
Fortunately, the development of electrospray allows for the intact transfer of noncovalent complexes from solution to the gas phase in many situations.154 In order to achieve these “native spray” conditions, several changes to the standard electrospray conditions most widely used for protein and peptide analysis must be allowed. The samples must be sprayed in solutions amenable to complex formation, which often eliminates the use of standard modifiers such as organic solvents and mild organic acids. These conditions often lead to decreased sensitivity compared to standard electrospray conditions. Additionally, the parameters of the ionization source must be tuned to make the transfer from solution to gas phase as gentle as possible. Typically, ions are allowed to undergo energetic collisions with neutral gas molecules to aid in the desolvation of the ion; however, such collisions will also disassemble noncovalent complexes in many cases. Lowering the energy of these collisions helps to maintain the native complex during the transfer to the gas phase, but decreases the efficiency of desolvation and generally reduces sensitivity. Lowering these desolvation potentials also can result in the gas phase transfer of partially desolvated protein-solvent “clusters” that retain various numbers of solvent molecules and/or buffer ions noncovalently bound to the protein. This added dispersity causes one individual protein complex to have multiple, closely-related m/z values consisting of the complex and different numbers and compositions of solvent clusters, leading to broad and poorly resolved peaks in the mass spectrum. Additionally, properly folded proteins and protein complexes usually retain less charge than unfolded proteins, resulting in signals at much higher m/z values than are typically seen in denatured protein analysis. These m/z values are above the scan range of many common commercially available mass spectrometers, and often require specialized or customized hardware to detect.155 In order to minimize the difficulties of desolvation and declustering, complexes are often ionized in very low-flow nanospray experiments. Nanospray tends to form smaller solvent droplets than pneumatically assisted electrospray processes, requiring less energy for desolvation even under aqueous conditions.156
Even with these added difficulties, MS can still be an invaluable tool for the analysis of chemokine oligomerization. In 2005, Yu et al. performed native spray analysis of various chemokine ligands of CCR2 in the presence and absence of heparin sulfate octasaccharides. In native spray analysis, MCP-1 (CCL2) and MCP-2 (CCL8) were found to exist in solution in an equilibrium between homodimer and monomer. However, using native spray MS, it was shown that only the dimer bound to the heparan sulfate octasaccharide. Similar studies of other CCR2 chemokines did not reveal any oligomers by native spray, and additionally showed heparan sulfate octasaccharide binding to the chemokine monomer. In all cases, the native spray MS results confirmed previous solution-phase analysis results.157 Further work by Crown et al. examined heterodimerization of these CCR2 chemokine ligands. Using native spray MS, the authors were able to measure apparent equilibriums between homo-dimer and heterodimer complexes of these CCR2 ligand chemokines. Heterodimerization dominated in mixtures of CCL2 and CCL8 in the absence of Arixtra, a heparan sulfate pentasaccharide derivative. All other combinations of CCL2, CCL8, and other CCR2 chemokine ligands examined revealed only homodimerization of CCL2 or CCL8, with the other paired chemokines existing only as monomers. However, upon addition of Arixtra, all pairs tested were observed to generate significant amounts of heterodimer, with the exception of CCL2–CCL13.158 More recently, Carlson et al. have examined the heterodimerization of CXCL4 and CXCL7 with other platelet-derived chemokines. Using native spray MS, the authors were able to screen 24 different combinations of CC and CXC chemokines and their ability to form heterodimers with CXCL4 and CXCL7.159 This screening process is made possible by the relative speed and sensitivity of native spray MS when compared to other analytical techniques used for oligomerization analysis, such as NMR and analytical ultracentrifugation.
The previous examples reveal how native spray MS can be used to study hetero- and homodimerization of chemokines; however, the analysis of polydisperse mixtures of oligomers can also be successfully analyzed by native spray MS. An example is shown in Fig. 20.6, where native spray MS was performed on CCL5, examining the oligomerization that occurred spontaneously in a 10 μM buffered aqueous solution of RANTES (CCL5) at pH 4.5. The peaks in the spectrum are broad, representing poor desolvation and declustering. The poor resolution makes it impossible to assign the charge state of any individual peak in isolation. However, as the homo-oligomer is made up of some unknown number of subunits of known size, and as incomplete desolvation and declustering will only add mass to the protein complex, we can algebraically determine which combinations of oligomer number and charge state would be consistent with each peak, by considering the low-mass edge of the peak as being closest to the fully desolvated mass of the complex. Some peaks, such as the peak at m/z 2019, can be fit to multiple combinations of oligomeric state and charge state; a monomer with four charges yields the same m/z as a homodimer with 8 charges, a homotetramer with 16 charges, etc. However, proteins and protein complexes in electrospray do not exist as a single charge state; they exist as a distribution of multiple charge states. Therefore, if we are detecting a homodimer with eight charges, we should also detect a homo-dimer with seven charges and/or a homodimer with nine charges, neither of which share an m/z ratio with any charge state of a monomer. The peaks at m/z 1795 and m/z 2334 clearly reveal that the dimer exists in the gas phase. Similar characteristic charge states can be identified in the mass spectrum in Fig. 20.6 for tetramer, hexamer, and octamer, but not for any odd-numbered oligomer. These data from this polydisperse collection of oligomerization states clearly demonstrate that RANTES (CCL5) oligomerizes by the sequential addition of dimers.100
Figure 20.6.
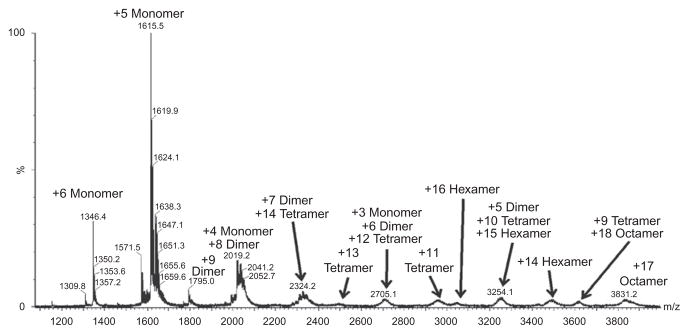
Native-spray analysis of 10 μM wild-type RANTES (CCL5) at pH 4.5. Only even numbered oligomers are observed from dimer to octamer, indicating that the RANTES (CCL5) oligomer is assembled via concatenation of the dimer subunit.
5.4.2 Hydroxyl radical protein footprinting analysis
In addition to transferring intact chemokine oligomers to the gas phase for analysis, it is also possible to use solution-phase protein chemistry to modify proteins in a structurally sensitive manner, and view how the chemistry changes upon oligomerization of the chemokine. A technique known as hydroxyl radical protein footprinting (HRPF) has previously been used by Wang et al. to study the structure of the RANTES (CCL5) tetramer. In HRPF, proteins in dilute aqueous solution are exposed to hydroxyl radicals formed in solution. These hydroxyl radicals can be formed by a wide variety of methods: chemical catalysis,160 photochemistry,161,162 electro-chemistry,163,164 and radiolysis.165–167 The hydroxyl radicals diffuse to the surface of the protein, where they react with the amino acid side chains present on the surface of the folded protein or protein complex. The modifications are stable, so after oxidation the protein is denatured, proteolytically digested, and the sites of oxidation are identified and quantified by LC-MS/MS. The LC-MS/MS is done either using an isotopically labeled internal standard to measure the loss of unoxidized peptide, or without the use of a standard by comparison of ion intensity of the unoxidized peptide to the intensity of all of the oxidized versions of that peptide.
The apparent rate of reaction has been shown to be a function of primarily two factors: the inherent chemical reactivity of the amino acid side chain168,169 and the solvent accessibility of that side chain.170,171 By comparing proteins of identical or very similar sequences but different conformations, changes in the apparent rate of oxidation of a side chain can be directly correlated with changes in the average solvent accessibility of that side chain. One confounding factor in HRPF is the fact that protein oxidation can trigger conformational changes, causing further oxidation to probe nonnative states. Therefore, in order to ensure that HRPF is probing native states, either the protein oxidation must be followed kinetically to ensure that oxidation follows a single rate constant,172 oxidation must be limited to a single oxidation event per protein,160 or oxidation must be completed on a timescale shorter than the protein can undergo large-scale oxidation-induced conformational changes.173 If these procedures are followed, along with proper quenching of secondary oxidants, HRPF data can accurately and reliably detect and describe changes in protein conformation.174
Wang et al. used HRPF, combined with NMR and SAXS, to compare the structure of the RANTES (CCL5) tetramer with that of the dimer in an attempt to determine the dimer–dimer interface by the protection upon oligomerization. At MS compatible concentrations (~20 μM), wild-type RANTES (CCL5) at pH 4.5 exists primarily as a tetramer, with a small amount of dimer and a small amount of higher-order oligomer also present. The E66S mutant of RANTES (CCL5) at pH 4.5 exists primarily as dimer, with some monomer present. Wang et al. used HRPF to compare the structures of wild-type RANTES (CCL5) to the E66S mutant at pH 4.5. The results of this analysis are shown in Fig. 20.7, mapped upon the final structure proposed for the RANTES (CCL5) tetramer. Protected sites can be divided into two broad quantitative categories: amino acids that exhibited strong protection (>2× decrease in oxidation) and sites that exhibited weak protection (<1.5× decrease in oxidation). Regions that exhibited weak protection were found to be present in the monomer–monomer interface, and could be attributed to the decrease in the concentration of monomer in the WT RANTES (CCL5) compared to the E66S mutant. The region of strong protection was determined to be the core of the tetramer interface, which was verified by NMR cross-saturation data.100
Figure 20.7.

Hydroxyl radical protein footprinting of wild-type RANTES (CCL5) at pH 4.5 (tetramer) compared to the E66S mutant at pH 4.5 (dimer) mapped on a model of the RANTES (CCL5) tetramer. Regions of weak protection (<1.5× reduction in oxidation) from oxidation in the wild-type compared to the E66S mutant (gray) are found in the monomer–monomer interface and can be explained by decrease in free monomer in the wild-type sample. Regions of strong protection (>2× reduction in oxidation) from oxidation in the wild-type compared to the E66S mutant (black) are not in the monomer–monomer interface, and are therefore protected by the dimer–dimer interaction.
Wild-type RANTES (CCL5) at pH 7 spontaneously forms a polydisperse mixture of very large oligomers. These oligomers are far too large to probe by NMR and exist as a complex mixture of different sizes of oligomer. However, they can be probed by HRPF by comparing wild-type RANTES (CCL5) at pH 4.5 (mostly tetramer) to wild-type RANTES (CCL5) at pH 7 (large oligomers). The inherent rate of reaction of hydroxyl radicals is very pH-insensitive, allowing for the ready comparison of pH-induced changes.168,175 The results of this analysis are shown in Fig. 20.8, mapped upon the structure of an octomer. The protection resulting from the transition from wild-type at pH 4.5–7 mirror the protection resulting from the transition from dimer to tetramer, only greater in scale. The region of “strong protection” in the dimer–dimer interface is actually completely protected from oxidation in the pH 7 sample, while the regions of “weak protection” at the monomer–monomer interface are still oxidized in the pH 7 sample, albeit at lower amounts than in the pH 4.5 sample. These results are consistent with the larger oligomers existing as linear expansions of the tetramer, as shown in Fig. 20.8. HRPF is one of the few techniques that can actually generate structural data on these polydisperse mixtures of chemokine oligomers, and could be quite valuable in the future for the analysis of these biologically critical structures.
Figure 20.8.

Hydroxyl radical protein footprinting of wild-type RANTES (CCL5) at pH 4.5 (tetramer) compared to pH 7 (large oligomers) mapped on a model of the RANTES (CCL5) octamer. Regions of complete protection from oxidation at pH 7 but found heavily oxidized at pH 4.5 (black) are found at the dimer–dimer interface, while regions of partial protection at pH 7 compared to pH 4.5 (gray) are found at the monomer–monomer interface. These pattern of protection mirrors the results found for the comparison of wild-type RANTES (CCL5) at pH 4.5 (tetramer) compared to the E66S mutant at pH 4.5 (dimer), with the extent of protection increasing dramatically.
6. CONCLUSIONS
It is clear that the complexities of chemokine oligomerization, interaction with GAGs, and interaction with receptors are just beginning to be appreciated. Nevertheless, there is substantial evidence of the importance of these interactions. For example, some chemokine–receptor pairs are believed involved in metastasis of malignant cells,16 and in the case of the CXCL12–CXCR4 pair, a constitutive dimeric form of the chemokine, as opposed to the monomeric form inhibited metastasis.35 Also, in the case of inhibition of HIV entry, some of the most effective designed RANTES (CCL5) mutants are know to have reduced GAG binding and oligomerization tendencies.176 We expect our understanding of oligomerization, GAG interaction, and receptor interaction, to have further impact on strategies to intervene in disease processes in the future.
Developing this understanding will be challenging. The systems of interest will be of increasing size and complexity. In the case of chemokine oligomerization, we have just begun to examine heteromeric complexes, but there are cases such as PF4 (CCL4)/RANTES (CCL5) complexes that are clearly biologically significant. In the case of GAG interactions, there are major issues with the availability of homogeneous GAG oligomers with which to carry out biophysical characterization. In the case of receptor interactions, there are additional challenges associated with the need to examine complexes in their native membrane associated environment.
There are, however, reasons to be optimistic about progress in this area. Technology suitable for the characterization of chemokine oligomers and chemokine complexes is rapidly evolving. NMR methods based on orientational and long-range paramagnetic constraints are well suited to the study of larger complexes assembled from monomers of known structure. MS methods for interface identification are now validated and applicable to complexes inaccessible to NMR because of size or concentration. Synthetic and enzymatic methods for the preparation of pure GAG oligomers are quickly advancing. We have not reviewed methods for macroscale structural characterization such as cryo-electron microscopy and atomic force microscopy, but these will clearly play a role in the future. The resulting improved view of the structure of complex assemblies involving chemokines, and the interactions that control their formation, will clearly have an impact on our ability to combat disease.
Acknowledgments
Financial support for this work was provided by NIH grants from the National Center for Research Resources (5 P41 RR005351-23) and the National Institute of General Medical Sciences (8 P41 GM103390-23), in support of the Resource for Integrated Glycotechnology at the University of Georgia and from the National Institute of General Medical Sciences (U01 GM094612) and the National Institute of Allergy and Infectious Diseases (R01 AI37113-14) in support of research at the University of California San Diego.
References
- 1.Goodsell DS, Olson AJ. Structural symmetry and protein function. Annu Rev Biophys Biomol Struct. 2000;29:105–53. doi: 10.1146/annurev.biophys.29.1.105. [DOI] [PubMed] [Google Scholar]
- 2.Levy ED, Pereira-Leal JB, Chothia C, Teichmann SA. 3D complex: a structural classification of protein complexes. PLoS Comput Biol. 2006;2(11):1395–406. doi: 10.1371/journal.pcbi.0020155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ali MH, Imperiali B. Protein oligomerization: how and why. Bioorg Med Chem. 2005;13 (17):5013–20. doi: 10.1016/j.bmc.2005.05.037. [DOI] [PubMed] [Google Scholar]
- 4.Sallusto F, Baggiolini M. Chemokines and leukocyte traffic. Nat Immunol. 2008;9 (9):949–52. doi: 10.1038/ni.f.214. [DOI] [PubMed] [Google Scholar]
- 5.Baggiolini M. Chemokines and leukocyte traffic. Nature. 1998;392(6676):565–8. doi: 10.1038/33340. [DOI] [PubMed] [Google Scholar]
- 6.Scholten DJ, Canals M, Maussang D, et al. Pharmacological modulation of chemokine receptor function. Br J Pharmacol. 2012;165(6):1617–43. doi: 10.1111/j.1476-5381.2011.01551.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bacon K, Baggiolini M, Broxmeyer H, et al. Chemokine/chemokine receptor nomenclature. J Interferon Cytokine Res. 2002;22(10):1067–8. doi: 10.1089/107999002760624305. [DOI] [PubMed] [Google Scholar]
- 8.Mantovani A. The chemokine system: redundancy for robust outputs. Immunol Today. 1999;20(6):254–7. doi: 10.1016/s0167-5699(99)01469-3. [DOI] [PubMed] [Google Scholar]
- 9.Devalaraja MN, Richmond A. Multiple chemotactic factors: fine control or redundancy? Trends Pharmacol Sci. 1999;20(4):151–6. doi: 10.1016/s0165-6147(99)01342-5. [DOI] [PubMed] [Google Scholar]
- 10.Johnson Z, Proudfoot AE, Handel TM. Interaction of chemokines and glycosaminoglycans: a new twist in the regulation of chemokine function with opportunities for therapeutic intervention. Cytokine Growth Factor Rev. 2005;16(6):625–36. doi: 10.1016/j.cytogfr.2005.04.006. [DOI] [PubMed] [Google Scholar]
- 11.Raman D, Sobolik-Delmaire T, Richmond A. Chemokines in health and disease. Exp Cell Res. 2011;317(5):575–89. doi: 10.1016/j.yexcr.2011.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li MZ, Hale JS, Rich JN, Ransohoff RM, Lathia JD. Chemokine CXCL12 in neurodegenerative diseases: an SOS signal for stem cell-based repair. Trends Neurosci. 2012;35(10):619–28. doi: 10.1016/j.tins.2012.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tuttolomondo A, Di Raimondo D, Pecoraro R, Arnao V, Pinto A, Licata G. Atherosclerosis as an inflammatory disease. Curr Pharm Des. 2012;18(28):4266–88. doi: 10.2174/138161212802481237. [DOI] [PubMed] [Google Scholar]
- 14.Gerard C, Rollins BJ. Chemokines and disease. Nat Immunol. 2001;2(2):108–15. doi: 10.1038/84209. [DOI] [PubMed] [Google Scholar]
- 15.Christopherson KW, 2nd, Hromas RA. Endothelial chemokines in autoimmune disease. Curr Pharm Des. 2004;10(2):145–54. doi: 10.2174/1381612043453405. [DOI] [PubMed] [Google Scholar]
- 16.O’Hayre M, Salanga CL, Handel TM, Allen SJ. Chemokines and cancer: migration, intracellular signalling and intercellular communication in the microenvironment. Biochem J. 2008;409:635–49. doi: 10.1042/BJ20071493. [DOI] [PubMed] [Google Scholar]
- 17.Wilen CB, Tilton JC, Doms RW. HIV: cell binding and entry. Cold Spring Harb Perspect Med. 2012;2(8):1–13. doi: 10.1101/cshperspect.a006866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wilen CB, Tilton JC, Doms RW. Molecular mechanisms of HIV entry. Adv Exp Med Biol. 2012;726:223–42. doi: 10.1007/978-1-4614-0980-9_10. [DOI] [PubMed] [Google Scholar]
- 19.Schall TJ, Proudfoot AE. Overcoming hurdles in developing successful drugs targeting chemokine receptors. Nat Rev Immunol. 2011;11(5):355–63. doi: 10.1038/nri2972. [DOI] [PubMed] [Google Scholar]
- 20.Springer TA. Traffic signals for lymphocyte recirculation and leukocyte emigration: the multistep paradigm. Cell. 1994;76(2):301–14. doi: 10.1016/0092-8674(94)90337-9. [DOI] [PubMed] [Google Scholar]
- 21.Springer TA. Traffic signals on endothelium for lymphocyte recirculation and leukocyte emigration. Annu Rev Physiol. 1995;57:827–72. doi: 10.1146/annurev.ph.57.030195.004143. [DOI] [PubMed] [Google Scholar]
- 22.Proudfoot AEI, Handel TM, Johnson Z, et al. Glycosaminoglycan binding and oligomerization are essential for the in vivo activity of certain chemokines. Proc Natl Acad Sci USA. 2003;100(4):1885–90. doi: 10.1073/pnas.0334864100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rajarathnam K, Sykes BD, Kay CM, et al. Neutrophil activation by monomeric interleukin-8. Science. 1994;264(5155):90–2. doi: 10.1126/science.8140420. [DOI] [PubMed] [Google Scholar]
- 24.Paavola CD, Hemmerich S, Grunberger D, et al. Monomeric monocyte chemo-attractant protein-1 (MCP-1) binds and activates the MCP-1 receptor CCR2B. J Biol Chem. 1998;273(50):33157–65. doi: 10.1074/jbc.273.50.33157. [DOI] [PubMed] [Google Scholar]
- 25.Blanpain C, Doranz BJ, Bondue A, et al. The core domain of chemokines binds CCR5 extracellular domains while their amino terminus interacts with the transmembrane helix bundle. J Biol Chem. 2003;278(7):5179–87. doi: 10.1074/jbc.M205684200. [DOI] [PubMed] [Google Scholar]
- 26.Stephens B, Handel TM. Chemokine receptor oligomerization and allostery. Prog in Mol Biol Transl Sci. 2013;115:375–420. doi: 10.1016/B978-0-12-394587-7.00009-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Veldkamp CT, Seibert C, Peterson FC, et al. Structural basis of CXCR4 sulfotyrosine recognition by the chemokine SDF-1/CXCL12. Sci Signal. 2008;1(37):ra4. doi: 10.1126/scisignal.1160755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Duma L, Haussinger D, Rogowski M, Lusso P, Grzesiek S. Recognition of RANTES by extracellular parts of the CCR5 receptor. J Mol Biol. 2007;365(4):1063–75. doi: 10.1016/j.jmb.2006.10.040. [DOI] [PubMed] [Google Scholar]
- 29.Hoogewerf AJ, Kuschert GS, Proudfoot AE, et al. Glycosaminoglycans mediate cell surface oligomerization of chemokines. Biochemistry. 1997;36(44):13570–8. doi: 10.1021/bi971125s. [DOI] [PubMed] [Google Scholar]
- 30.Campanella GS, Grimm J, Manice LA, et al. Oligomerization of CXCL10 is necessary for endothelial cell presentation and in vivo activity. J Immunol. 2006;177 (10):6991–8. doi: 10.4049/jimmunol.177.10.6991. [DOI] [PubMed] [Google Scholar]
- 31.Ellyard JI, Simson L, Bezos A, Johnston K, Freeman C, Parish CR. Eotaxin selectively binds heparin. An interaction that protects eotaxin from proteolysis and potentiates chemotactic activity in vivo. J Biol Chem. 2007;282(20):15238–47. doi: 10.1074/jbc.M608046200. [DOI] [PubMed] [Google Scholar]
- 32.Baltus T, Weber KS, Johnson Z, Proudfoot AE, Weber C. Oligomerization of RANTES is required for CCR1-mediated arrest but not CCR5-mediated transmigration of leukocytes on inflamed endothelium. Blood. 2003;102(6):1985–8. doi: 10.1182/blood-2003-04-1175. [DOI] [PubMed] [Google Scholar]
- 33.Wang L, Fuster M, Sriramarao P, Esko JD. Endothelial heparan sulfate deficiency impairs L-selectin- and chemokine-mediated neutrophil trafficking during inflammatory responses. Nat Immunol. 2005;6(9):902–10. doi: 10.1038/ni1233. [DOI] [PubMed] [Google Scholar]
- 34.Yin X, Truty J, Lawrence R, et al. A critical role for lymphatic endothelial heparan sulfate in lymph node metastasis. Mol Cancer. 2010;9:316. doi: 10.1186/1476-4598-9-316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Drury LJ, Ziarek JJ, Gravel S, et al. Monomeric and dimeric CXCL12 inhibit metastasis through distinct CXCR4 interactions and signaling pathways. Proc Natl Acad Sci USA. 2011;108(43):17655–60. doi: 10.1073/pnas.1101133108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Appay V, Dunbar PR, Cerundolo V, McMichael A, Czaplewski L, Rowland-Jones S. RANTES activates antigen-specific cytotoxic T lymphocytes in a mitogen-like manner through cell surface aggregation. Int Immunol. 2000;12(8):1173–82. doi: 10.1093/intimm/12.8.1173. [DOI] [PubMed] [Google Scholar]
- 37.Roscic-Mrkic B, Fischer M, Leemann C, et al. RANTES (CCL5) uses the proteoglycan CD44 as an auxiliary receptor to mediate cellular activation signals and HIV-1 enhancement. Blood. 2003;102(4):1169–77. doi: 10.1182/blood-2003-02-0488. [DOI] [PubMed] [Google Scholar]
- 38.Wagner L, Yang OO, Garcia-Zepeda EA, et al. Beta-chemokines are released from HIV-1-specific cytolytic T-cell granules complexed to proteoglycans. Nature. 1998;391(6670):908–11. doi: 10.1038/36129. [DOI] [PubMed] [Google Scholar]
- 39.Wu BL, Chien EYT, Mol CD, et al. Structures of the CXCR4 chemokine GPCR with small-molecule and cyclic peptide antagonists. Science. 2010;330(6007):1066–71. doi: 10.1126/science.1194396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kramp BK, Sarabi A, Koenen RR, Weber C. Heterophilic chemokine receptor interactions in chemokine signaling and biology. Exp Cell Res. 2011;317(5):655–63. doi: 10.1016/j.yexcr.2010.11.014. [DOI] [PubMed] [Google Scholar]
- 41.von Hundelshausen P, Koenen RR, Sack M, et al. Heterophilic interactions of platelet factor 4 and RANTES promote monocyte arrest on endothelium. Blood. 2005;105 (3):924–30. doi: 10.1182/blood-2004-06-2475. [DOI] [PubMed] [Google Scholar]
- 42.Koenen RR, von Hundelshausen P, Nesmelova IV, et al. Disrupting functional interactions between platelet chemokines inhibits atherosclerosis in hyperlipidemic mice. Nat Med. 2009;15(1):97–103. doi: 10.1038/nm.1898. [DOI] [PubMed] [Google Scholar]
- 43.Shahrara S, Proudfoot AE, Park CC, et al. Inhibition of monocyte chemoattractant protein-1 ameliorates rat adjuvant-induced arthritis. J Immunol. 2008;180(5):3447–56. doi: 10.4049/jimmunol.180.5.3447. [DOI] [PubMed] [Google Scholar]
- 44.Proudfoot AE, Power CA, Rommel C, Wells TN. Strategies for chemokine antagonists as therapeutics. Semin Immunol. 2003;15(1):57–65. doi: 10.1016/s1044-5323(02)00128-8. [DOI] [PubMed] [Google Scholar]
- 45.Johnson Z, Kosco-Vilbois MH, Herren S, et al. Interference with heparin binding and oligomerization creates a novel anti-inflammatory strategy targeting the chemokine system. J Immunol. 2004;173(9):5776–85. doi: 10.4049/jimmunol.173.9.5776. [DOI] [PubMed] [Google Scholar]
- 46.Allen SJ, Crown SE, Handel TM. Chemokine: receptor structure, interactions, and antagonism. Annu Rev Immunol. 2007;25:787–820. doi: 10.1146/annurev.immunol.24.021605.090529. [DOI] [PubMed] [Google Scholar]
- 47.Fernandez EJ, Lolis E. Structure junction, and inhibition of chemokines. Annu Rev Pharmacol. 2002;42:469–99. doi: 10.1146/annurev.pharmtox.42.091901.115838. [DOI] [PubMed] [Google Scholar]
- 48.Fairbrother WJ, Reilly D, Colby TJ, Hesselgesser J, Horuk R. The solution structure of melanoma growth-stimulating activity. J Mol Biol. 1994;242(3):252–70. doi: 10.1006/jmbi.1994.1577. [DOI] [PubMed] [Google Scholar]
- 49.Qian YQ, Johanson KO, McDevitt P. Nuclear magnetic resonance solution structure of truncated human GRO beta [5–73] and its structural comparison with CXC chemokine family members GRO alpha and IL-8. J Mol Biol. 1999;294(5):1065–72. doi: 10.1006/jmbi.1999.3333. [DOI] [PubMed] [Google Scholar]
- 50.Mayo KH, Roongta V, Ilyina E, et al. NMR solution structure of the 32-Kda platelet factor-4 Elr-motif N-terminal chimera—a symmetrical tetramer. Biochemistry. 1995;34 (36):11399–409. doi: 10.1021/bi00036a012. [DOI] [PubMed] [Google Scholar]
- 51.Zhang XH, Chen LQ, Bancroft DP, Lai CK, Maione TE. Crystal-structure of recombinant human platelet factor-4. Biochemistry. 1994;33(27):8361–6. doi: 10.1021/bi00193a025. [DOI] [PubMed] [Google Scholar]
- 52.Malkowski MG, Wu JY, Lazar JB, Johnson PH, Edwards BFP. The crystal-structure of recombinant human neutrophil-activating peptide-2 (M6l) at 1. 9-Angstrom resolution. J Biol Chem. 1995;270(13):7077–87. doi: 10.1074/jbc.270.13.7077. [DOI] [PubMed] [Google Scholar]
- 53.Young H, Roongta V, Daly TJ, Mayo KH. NMR structure and dynamics of monomeric neutrophil-activating peptide 2. Biochem J. 1999;338:591–8. [PMC free article] [PubMed] [Google Scholar]
- 54.Clore GM, Appella E, Yamada M, Matsushima K, Gronenborn AM. 3-Dimensional structure of Interleukin-8 in solution. Biochemistry. 1990;29(7):1689–96. doi: 10.1021/bi00459a004. [DOI] [PubMed] [Google Scholar]
- 55.Swaminathan GJ, Holloway DE, Colvin RA, et al. Crystal structures of oligomeric forms of the IP-10/CXCL10 chemokine. Structure. 2003;11(5):521–32. doi: 10.1016/s0969-2126(03)00070-4. [DOI] [PubMed] [Google Scholar]
- 56.Booth V, Clark-Lewis I, Sykes BD. NMR structure of CXCR3 binding chemokine CXCL11 (ITAC) Protein Sci. 2004;13(8):2022–8. doi: 10.1110/ps.04791404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dealwis C, Fernandez EJ, Thompson DA, Simon RJ, Siani MA, Lolis E. Crystal structure of chemically synthesized [N33A] stromal cell-derived factor 1 alpha, a potent ligand for the HIV-1 “fusin” coreceptor. Proc Natl Acad Sci USA. 1998;95 (12):6941–6. doi: 10.1073/pnas.95.12.6941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Crump MP, Gong JH, Loetscher P, et al. Solution structure and basis for functional activity of stromal cell-derived factor-1; dissociation of CXCR4 activation from binding and inhibition of HIV-1. EMBO J. 1997;16(23):6996–7007. doi: 10.1093/emboj/16.23.6996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Keizer DW, Crump MP, Lee TW, Slupsky CM, Clark-Lewis I, Sykes BD. Human CC chemokine I-309, structural consequences of the additional disulfide bond. Biochemistry. 2000;39(20):6053–9. doi: 10.1021/bi000089l. [DOI] [PubMed] [Google Scholar]
- 60.Handel TM, Domaille PJ. Heteronuclear (H-1, C-13, N-15) NMR assignments and solution structure of the monocyte chemoattractant protein-1 (MCP-1) dimer. Biochemistry. 1996;35(21):6569–84. doi: 10.1021/bi9602270. [DOI] [PubMed] [Google Scholar]
- 61.Lubkowski J, Bujacz G, Boque L, Domaille PJ, Handel TM, Wlodawer A. The structure of MCP-1 in two crystal forms provides a rare example of variable quaternary interactions. Nat Struct Biol. 1997;4(1):64–9. doi: 10.1038/nsb0197-64. [DOI] [PubMed] [Google Scholar]
- 62.Czaplewski LG, McKeating J, Craven CJ, et al. Identification of amino acid residues critical for aggregation of human CC chemokines macrophage inflammatory protein (MIP)-1 alpha, MIP-1 beta, and RANTES - Characterization of active disaggregated chemokine variants. J Biol Chem. 1999;274(23):16077–84. doi: 10.1074/jbc.274.23.16077. [DOI] [PubMed] [Google Scholar]
- 63.Lodi PJ, Garrett DS, Kuszewski J, et al. High-resolution solution structure of the beta-chemokine hMIP-1-beta by multidimensional NMR. Science. 1994;263(5154):1762–7. doi: 10.1126/science.8134838. [DOI] [PubMed] [Google Scholar]
- 64.Chung CW, Cooke RM, Proudfoot AEI, Wells TNC. The 3-dimensional solution structure of RANTES. Biochemistry. 1995;34(29):9307–14. doi: 10.1021/bi00029a005. [DOI] [PubMed] [Google Scholar]
- 65.Kim KS, Rajarathnam K, ClarkLewis I, Sykes BD. Structural characterization of a monomeric chemokine: monocyte chemoattractant protein-3. FEBS Lett. 1996;395 (2–3):277–82. doi: 10.1016/0014-5793(96)01024-1. [DOI] [PubMed] [Google Scholar]
- 66.Blaszczyk J, Van Coillie E, Proost P, et al. Complete crystal structure of monocyte chemotactic protein-2, a CC chemokine that interacts with multiple receptors. Biochemistry. 2000;39(46):14075–81. doi: 10.1021/bi0009340. [DOI] [PubMed] [Google Scholar]
- 67.Crump MP, Rajarathnam K, Kim KS, Clark-Lewis I, Sykes BD. Solution structure of eotaxin, a chemokine that selectively recruits eosinophils in allergic inflammation. J Biol Chem. 1998;273(35):22471–9. doi: 10.1074/jbc.273.35.22471. [DOI] [PubMed] [Google Scholar]
- 68.Barinka C, Prahl A, Lubkowski J. Structure of human monocyte chemoattractant protein 4 (MCP-4/CCL13) Acta Crystallogr D Biol Crystallogr. 2008;64:273–8. doi: 10.1107/S0907444907066164. [DOI] [PubMed] [Google Scholar]
- 69.Blain KY, Kwiatkowski W, Zhao Q, et al. Structural and functional characterization of CC chemokine CCL14. Biochemistry. 2007;46(35):10008–15. doi: 10.1021/bi700936w. [DOI] [PubMed] [Google Scholar]
- 70.Sticht H, Escher SE, Schweimer K, Forssmann WG, Rosch P, Adermann K. Solution structure of the human CC chemokine 2: a monomeric representative of the CC chemokine subtype. Biochemistry. 1999;38(19):5995–6002. doi: 10.1021/bi990065i. [DOI] [PubMed] [Google Scholar]
- 71.Asojo OA, Boulegue C, Hoover DM, Lu WY, Lubkowski J. Structures of thymus and activation-regulated chemokine (TARC) Acta Crystallogr D Biol Crystallogr. 2003;59:1165–73. doi: 10.1107/s0907444903009454. [DOI] [PubMed] [Google Scholar]
- 72.Love M, Sandberg JL, Ziarek JJ, et al. Solution Structure of CCL21 and Identification of a Putative CCR7 Binding Site. Biochemistry. 2012;51(3):733–5. doi: 10.1021/bi201601k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Rajarathnam K, Li YL, Rohrer T, Gentz R. Solution structure and dynamics of myeloid progenitor inhibitory factor-1 (MPIF-1), a novel monomeric CC chemokine. J Biol Chem. 2001;276(7):4909–16. doi: 10.1074/jbc.M005085200. [DOI] [PubMed] [Google Scholar]
- 74.Mayer KL, Stone MJ. NMR solution structure and receptor peptide binding of the CC chemokine eotaxin-2. Biochemistry. 2000;39(29):8382–95. doi: 10.1021/bi000523j. [DOI] [PubMed] [Google Scholar]
- 75.Ye J, Mayer KL, Mayer MR, Stone MJ. NMR solution structure and backbone dynamics of the CC chemokine eotaxin-3. Biochemistry. 2001;40(26):7820–31. doi: 10.1021/bi010252s. [DOI] [PubMed] [Google Scholar]
- 76.Jansma AL, Kirkpatrick JP, Hsu AR, Handel TM, Nietlispach D. NMR analysis of the structure, dynamics, and unique oligomerization properties of the chemokine CCL27. J Biol Chem. 2010;285(19):14424–37. doi: 10.1074/jbc.M109.091108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Tuinstra RL, Peterson FC, Kutlesa S, Elgin ES, Kron MA, Volkman BF. Interconversion between two unrelated protein folds in the lymphotactin native state. Proc Natl Acad Sci USA. 2008;105(13):5057–62. doi: 10.1073/pnas.0709518105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mizoue LS, Bazan JF, Johnson EC, Handel TM. Solution structure and dynamics of the CX3C chemokine domain of fractalkine and its interaction with an N-terminal fragment of CX(3)CR1. Biochemistry. 1999;38(5):1402–14. doi: 10.1021/bi9820614. [DOI] [PubMed] [Google Scholar]
- 79.Hoover DM, Mizoue LS, Handel TM, Lubkowski J. The crystal structure of the chemokine domain of fractalkine shows a novel quaternary arrangement. J Biol Chem. 2000;275(30):23187–93. doi: 10.1074/jbc.M002584200. [DOI] [PubMed] [Google Scholar]
- 80.Gong JH, Clarklewis I. Antagonists of monocyte chemoattractant protein-1 identified by modification of functionally critical NH2-terminal residues. J Exp Med. 1995;181 (2):631–40. doi: 10.1084/jem.181.2.631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Guan E, Wang JH, Roderiquez G, Norcross MA. Natural truncation of the chemokine MIP-1 beta/CCL4 affects receptor specificity but not Anti-HIV-1 activity. J Biol Chem. 2002;277(35):32348–52. doi: 10.1074/jbc.M203077200. [DOI] [PubMed] [Google Scholar]
- 82.Proudfoot AEI, Power CA, Hoogewerf AJ, et al. Extension of recombinant human RANTES by the retention of the initiating methionine produces a potent antagonist. J Biol Chem. 1996;271(5):2599–603. doi: 10.1074/jbc.271.5.2599. [DOI] [PubMed] [Google Scholar]
- 83.Williams SA, Harata-Lee Y, Comerford I, Anderson RL, Smyth MJ, McColl SR. Multiple functions of CXCL12 in a syngeneic model of breast cancer. Mol Cancer. 2010;9(250):1–10. doi: 10.1186/1476-4598-9-250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Gaertner H, Cerini F, Escola JM, et al. Highly potent, fully recombinant anti-HIV chemokines: reengineering a low-cost microbicide. Proc Natl Acad Sci USA. 2008;105(46):17706–11. doi: 10.1073/pnas.0805098105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Loetscher P, Gong JH, Dewald B, Baggiolini M, Clark-Lewis I. N-terminal peptides of stromal cell-derived factor-1 with CXC chemokine receptor 4 agonist and antagonist activities. J Biol Chem. 1998;273(35):22279–83. doi: 10.1074/jbc.273.35.22279. [DOI] [PubMed] [Google Scholar]
- 86.Luo JS, Luo ZW, Zhou NM, Hall JW, Huang ZW. Attachment of C-terminus of SDF-1 enhances the biological activity of its N-terminal peptide. Biochem Biophys Res Commun. 1999;264(1):42–7. doi: 10.1006/bbrc.1999.1476. [DOI] [PubMed] [Google Scholar]
- 87.Pakianathan DR, Kuta EG, Artis DR, Skelton NJ, Hebert CA. Distinct but overlapping epitopes for the interaction of a CC-chemokine with CCR1, CCR3, and CCR5. Biochemistry. 1997;36(32):9642–8. doi: 10.1021/bi970593z. [DOI] [PubMed] [Google Scholar]
- 88.Clarklewis I, Dewald B, Loetscher M, Moser B, Baggiolini M. Structural requirements for interleukin-8 function identified by design of analogs and CXC chemokine hybrids. J Biol Chem. 1994;269(23):16075–81. [PubMed] [Google Scholar]
- 89.Lowman HB, Slagle PH, DeForge LE, et al. Exchanging interleukin-8 and melanoma growth-stimulating activity receptor binding specificities. J Biol Chem. 1996;271 (24):14344–52. doi: 10.1074/jbc.271.24.14344. [DOI] [PubMed] [Google Scholar]
- 90.Beall CJ, Mahajan S, Kuhn DE, Kolattukudy PE. Site-directed mutagenesis of monocyte chemoattractant protein-1 identifies two regions of the polypeptide essential for biological activity. Biochem J. 1996;313:633–40. doi: 10.1042/bj3130633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hemmerich S, Paavola C, Bloom A, et al. Identification of residues in the monocyte chemotactic protein-1 that contact the MCP-1 receptor, CCR2. Biochemistry. 1999;38 (40):13013–25. doi: 10.1021/bi991029m. [DOI] [PubMed] [Google Scholar]
- 92.Martin L, Blanpain C, Garnier P, Wittamer V, Parmentier M, Vita C. Structural and functional analysis of the RANTES-glycosaminoglycans interactions. Biochemistry. 2001;40(21):6303–18. doi: 10.1021/bi002670n. [DOI] [PubMed] [Google Scholar]
- 93.Proudfoot AEI, Fritchley S, Borlat F, et al. The BBXB motif of RANTES is the principal site for heparin binding and controls receptor selectivity. J Biol Chem. 2001;276 (14):10620–6. doi: 10.1074/jbc.M010867200. [DOI] [PubMed] [Google Scholar]
- 94.Yoshiura C, Kofuku Y, Ueda T, et al. NMR Analyses of the Interaction between CCR5 and Its Ligand Using Functional Reconstitution of CCR5 in Lipid Bilayers. J Am Chem Soc. 2010;132(19):6768–77. doi: 10.1021/ja100830f. [DOI] [PubMed] [Google Scholar]
- 95.Strieter RM, Burdick MD, Gomperts BN, Belperio JA, Keane MP. CXC chemokines in angiogenesis. Cytokine Growth Factor Rev. 2005;16(6):593–609. doi: 10.1016/j.cytogfr.2005.04.007. [DOI] [PubMed] [Google Scholar]
- 96.Ali S, O’Boyle G, Hepplewhite P, Tyler JR, Robertson H, Kirby JA. Therapy with nonglycosaminoglycan-binding mutant CCL7: a novel strategy to limit allograft inflammation. Am J Transplant. 2010;10(1):47–58. doi: 10.1111/j.1600-6143.2009.02868.x. [DOI] [PubMed] [Google Scholar]
- 97.Lau EK, Paavola CD, Johnson Z, et al. Identification of the glycosaminoglycan binding site of the CC chemokine, MCP-1 - Implications for structure and function in vivo. J Biol Chem. 2004;279(21):22294–305. doi: 10.1074/jbc.M311224200. [DOI] [PubMed] [Google Scholar]
- 98.Jabeen T, Leonard P, Jamaluddin H, Acharya KR. Structure of mouse IP-10, a chemokine. Acta Crystallogr D Biol Crystallogr. 2008;64:611–9. doi: 10.1107/S0907444908007026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Murphy JW, Yuan H, Kong Y, Xiong Y, Lolis EJ. Heterologous quaternary structure of CXCL12 and its relationship to the CC chemokine family. Proteins. 2010;78 (5):1331–7. doi: 10.1002/prot.22666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Wang X, Watson C, Sharp JS, Handel TM, Prestegard JH. Oligomeric structure of the chemokine CCL5/RANTES from NMR, MS, and SAXS data. Structure. 2011;19 (8):1138–48. doi: 10.1016/j.str.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Ren M, Guo Q, Guo LA, et al. Polymerization of MIP-1 chemokine (CCL3 and CCL4) and clearance of MIP-1 by insulin-degrading enzyme. EMBO J. 2010;29 (23):3952–66. doi: 10.1038/emboj.2010.256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Handin RI, Cohen HJ. Purification and binding properties of human platelet factor 4. J Biol Chem. 1976;251(14):4273–82. [PubMed] [Google Scholar]
- 103.Handel TM, Johnson Z, Crown SE, Lau EK, Sweeney M, Proudfoot AE. Regulation of protein function by glycosaminoglycans—as exemplified by chemokines. Annu Rev Biochem. 2005;74:385–410. doi: 10.1146/annurev.biochem.72.121801.161747. [DOI] [PubMed] [Google Scholar]
- 104.Kuschert GSV, Coulin F, Power CA, et al. Glycosaminoglycans interact selectively with chemokines and modulate receptor binding and cellular responses. Biochemistry. 1999;38(39):12959–68. doi: 10.1021/bi990711d. [DOI] [PubMed] [Google Scholar]
- 105.Amara A, Lorthioir O, Valenzuela A, et al. Stromal cell-derived factor-1 alpha associates with heparan sulfates through the first beta-strand of the chemokine. J Biol Chem. 1999;274(34):23916–25. doi: 10.1074/jbc.274.34.23916. [DOI] [PubMed] [Google Scholar]
- 106.Koopmann W, Krangel MS. Identification of a glycosaminoglycan-binding site in chemokine macrophage inflammatory protein-1 alpha. J Biol Chem. 1997;272(15):10103–9. doi: 10.1074/jbc.272.15.10103. [DOI] [PubMed] [Google Scholar]
- 107.Sadir R, Baleux F, Grosdidier A, Imberty A, Lortat-Jacob H. Characterization of the stromal cell-derived factor-l alpha-heparin complex. J Biol Chem. 2001;276(11):8288–96. doi: 10.1074/jbc.M008110200. [DOI] [PubMed] [Google Scholar]
- 108.Jarnagin K, Grunberger D, Mulkins M, et al. Identification of surface residues of the monocyte chemotactic protein 1 that affect signaling through the receptor CCR2. Biochemistry. 1999;38(49):16167–77. doi: 10.1021/bi9912239. [DOI] [PubMed] [Google Scholar]
- 109.Salanga CL, Handel TM. Chemokine oligomerization and interactions with receptors and glycosaminoglycans: the role of structural dynamics in function. Exp Cell Res. 2011;317(5):590–601. doi: 10.1016/j.yexcr.2011.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Spillmann D, Witt D, Lindahl U. Defining the interleukin-8-binding domain of heparan sulfate. J Biol Chem. 1998;273(25):15487–93. doi: 10.1074/jbc.273.25.15487. [DOI] [PubMed] [Google Scholar]
- 111.Rek A, Brandner B, Geretti E, Kungl AJ. A biophysical insight into the RANTES-glycosaminoglycan interaction. Biochim Biophys Acta. 2009;1794(4):577–82. doi: 10.1016/j.bbapap.2009.01.001. [DOI] [PubMed] [Google Scholar]
- 112.Stringer SE, Forster MJ, Mulloy B, Bishop CR, Graham GJ, Gallagher JT. Characterization of the binding site on heparan sulfate for macrophage inflammatory protein 1 alpha. Blood. 2002;100(5):1543–50. [PubMed] [Google Scholar]
- 113.Kim S, Jao S, Laurence JS, LiWang PJ. Structural comparison of monomeric variants of the chemokine MIP-1 beta having differing ability to bind the receptor CCR5. Biochemistry. 2001;40(36):10782–91. doi: 10.1021/bi011065x. [DOI] [PubMed] [Google Scholar]
- 114.Meunier S, Bernassau JM, Guillemot JC, Ferrara P, Darbon H. Determination of the three-dimensional structure of CC chemokine monocyte chemoattractant protein 3 by H-1 two-dimensional NMR spectroscopy. Biochemistry. 1997;36(15):4412–22. doi: 10.1021/bi9627929. [DOI] [PubMed] [Google Scholar]
- 115.Skelton NJ, Aspiras F, Ogez J, Schall TJ. Proton NMR assignments and solution conformation of RANTES, a chemokine of the C-C type. Biochemistry. 1995;34(16):5329–42. doi: 10.1021/bi00016a004. [DOI] [PubMed] [Google Scholar]
- 116.Veldkamp CT, Ziarek JJ, Su JD, et al. Monomeric structure of the cardioprotective chemokine SDF-1/CXCL12. Protein Sci. 2009;18(7):1359–69. doi: 10.1002/pro.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Volkman BF, Liu TY, Peterson FC. Lymphotactin structural dynamics. In: Handel TM, Hamel DJ, editors. Chemokines, Part B. Methods in enzymology. Vol. 461. Elsevier; 2009. pp. 51–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Zhao B, LiWang PJ. Characterization of the Interactions of vMIP-II, and a Dimeric Variant of vMIP-II, with Glycosaminoglycans. Biochemistry. 2010;49(33):7012–22. doi: 10.1021/bi100549y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Shimada I, Ueda T, Matsumoto M, et al. Cross-saturation and transferred cross-saturation experiments. Prog Nucl Magn Reson Spectrosc. 2009;54(2):123–40. [Google Scholar]
- 120.Bayburt TH, Sligar SG. Membrane protein assembly into Nanodiscs. FEBS Lett. 2010;584(9):1721–7. doi: 10.1016/j.febslet.2009.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Greenfield NJ, Huang YJ, Palm T, et al. Solution NMR structure and folding dynamics of the N terminus of a rat non-muscle alpha-tropomyosin in an engineered chimeric protein. J Mol Biol. 2001;312(4):833–47. doi: 10.1006/jmbi.2001.4982. [DOI] [PubMed] [Google Scholar]
- 122.Bax A, Kontaxis G, Tjandra N. Dipolar couplings in macromolecular structure determination. Nuclear Magnetic Resonance of Biological Macromolecules, Part B. Meth Enzymol. 2001;339:127–74. doi: 10.1016/s0076-6879(01)39313-8. [DOI] [PubMed] [Google Scholar]
- 123.Prestegard JH, Bougault CM, Kishore AI. Residual dipolar couplings in structure determination of biomolecules. Chem Rev. 2004;104(8):3519–40. doi: 10.1021/cr030419i. [DOI] [PubMed] [Google Scholar]
- 124.Lee HW, Wylie G, Bansal S, et al. Three-dimensional structure of the weakly associated protein homodimer SeR13 using RDCs and paramagnetic surface mapping. Protein Sci. 2010;19(9):1673–85. doi: 10.1002/pro.447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Wang X, Lee HW, Liu YZ, Prestegard JH. Structural NMR of protein oligomers using hybrid methods. J Struct Biol. 2011;173(3):515–29. doi: 10.1016/j.jsb.2010.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Wang X, Bansal S, Jiang M, Prestegard JH. RDC-assisted modeling of symmetric protein homo-oligomers. Protein Sci. 2008;17(5):899–907. doi: 10.1110/ps.073395108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Shaw JP, Kryger G, Cleasby A, et al. Crystallization and preliminary-X-ray diffraction studies of human RANTES. J Mol Biol. 1994;242(4):589–90. doi: 10.1006/jmbi.1994.1605. [DOI] [PubMed] [Google Scholar]
- 128.Liu YZ, Prestegard JH. A device for the measurement of residual chemical shift anisotropy and residual dipolar coupling in soluble and membrane-associated proteins. J Biomol NMR. 2010;47(4):249–58. doi: 10.1007/s10858-010-9427-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Yu F, Wolff JJ, Amster IJ, Prestegard JH. Conformational preferences of chondroitin sulfate oligomers using partially oriented NMR spectroscopy of C-13-labeled acetyl groups. J Am Chem Soc. 2007;129(43):13288–97. doi: 10.1021/ja075272h. [DOI] [PubMed] [Google Scholar]
- 130.Gemma E, Hulme AN, Jahnke A, et al. DMT-MM mediated functionalisation of the non-reducing end of glycosaminoglycans. Chem Commun. 2007;26:2686–8. doi: 10.1039/b617038b. [DOI] [PubMed] [Google Scholar]
- 131.Blaum BS, Deakin JA, Johansson CM, et al. Lysine and arginine side chains in glycosaminoglycan-protein complexes Investigated by NMR, cross-linking, and mass spectrometry: a case study of the factor H-heparin Interaction. J Am Chem Soc. 2010;132 (18):6374–81. doi: 10.1021/ja1000517. [DOI] [PubMed] [Google Scholar]
- 132.Roldos V, Canada FJ, Jimenez-Barbero J. Carbohydrate-protein interactions: a 3D View by NMR. Chembiochem. 2011;12:990–1005. doi: 10.1002/cbic.201000705. [DOI] [PubMed] [Google Scholar]; 12(9):CP27–CP27. [Google Scholar]
- 133.Meyer B, Peters T. NMR Spectroscopy techniques for screening and identifying ligand binding to protein receptors. Angew Chem Int Ed. 2003;42(8):864–90. doi: 10.1002/anie.200390233. [DOI] [PubMed] [Google Scholar]
- 134.Laguri C, Sapay N, Simorre JP, et al. C-13-labeled heparan sulfate analogue as a tool to study protein/heparan sulfate interactions by NMR spectroscopy: application to the CXCL12 alpha chemokine. J Am Chem Soc. 2011;133(25):9642–5. doi: 10.1021/ja201753e. [DOI] [PubMed] [Google Scholar]
- 135.Davis JT, Hirani S, Bartlett C, Reid BR. H-1-NMR studies on an Asn-linked glycopeptide—Glcnac-1 C2-N2 bond is rigid in H2O. J Biol Chem. 1994;269 (5):3331–8. [PubMed] [Google Scholar]
- 136.Hricovini M, Guerrini M, Bisio A, Torri G, Petitou M, Casu B. Conformation of heparin pentasaccharide bound to antithrombin III. Biochem J. 2001;359:265–72. doi: 10.1042/0264-6021:3590265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Tjong SC, Chen TS, Huang WN, Wu WG. Structures of heparin-derived tetrasaccharide bound to cobra cardiotoxins: heparin binding at a single protein site with diverse side chain interactions. Biochemistry. 2007;46(35):9941–52. doi: 10.1021/bi700995v. [DOI] [PubMed] [Google Scholar]
- 138.Zhou HJ, Casas-Finet JR, Coats RH, et al. Identification and dynamics of a heparin-binding site in hepatocyte growth factor. Biochemistry. 1999;38(45):14793–802. doi: 10.1021/bi9908641. [DOI] [PubMed] [Google Scholar]
- 139.Moy FJ, Safran M, Seddon AP, et al. Properly oriented heparin-decasaccharide-induced dimers are the biologically active form of basic fibroblast growth factor. Biochemistry. 1997;36(16):4782–91. doi: 10.1021/bi9625455. [DOI] [PubMed] [Google Scholar]
- 140.Boons GJ. Abstracts of Papers of the American Chemical Society. Aug, 2010. Modular synthesis of heparan sulfate oligosaccharides for array development; p. 240. [Google Scholar]
- 141.Deepa SS, Yamada S, Fukui S, Sugahara K. Structural determination of novel sulfated octasaccharides isolated from chondroitin sulfate of shark cartilage and their application for characterizing monoclonal antibody epitopes. Glycobiology. 2007;17 (6):631–45. doi: 10.1093/glycob/cwm021. [DOI] [PubMed] [Google Scholar]
- 142.Pomin VH, Park Y, Huang RR, et al. Exploiting enzyme specificities in digestions of chondroitin sulfates A and C: production of well-defined hexasaccharides. Glycobiology. 2012;22(6):826–38. doi: 10.1093/glycob/cws055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.DeAngelis PL. Glycosaminoglycan polysaccharide biosynthesis and production: today and tomorrow. Appl Microbiol Biotechnol. 2012;94(2):295–305. doi: 10.1007/s00253-011-3801-6. [DOI] [PubMed] [Google Scholar]
- 144.Casu B, Grazioli G, Razi N, et al. Heparin-like compounds prepared by chemical modification of capsular polysaccharide from Escherichiacoli K5. Carbohydr Res. 1994;263 (2):271–84. doi: 10.1016/0008-6215(94)00172-3. [DOI] [PubMed] [Google Scholar]
- 145.Nagasawa K, Uchiyama H, Wajima N. Chemical sulfation of preparations of chondroitin 4-sulfate and 6-sulfate, and dermatan sulfate—preparation of chondroitin sulfate E-Like materials from chondroitin 4-sulfate. Carbohydr Res. 1986;158:183–90. [Google Scholar]
- 146.DeAngelis PL, Jing W, Tracy BS, Oatman LC, Gay DF. Chemoenzymatic synthesis of glycosaminoglycans with pasteurella synthases. Glycobiology. 2003;13(11):838–9. [Google Scholar]
- 147.DeAngelis PL, Oatman LC, Gay DF. Rapid chemoenzymatic synthesis of monodisperse hyaluronan oligosaccharides with immobilized enzyme reactors. J Biol Chem. 2003;278(37):35199–203. doi: 10.1074/jbc.M306431200. [DOI] [PubMed] [Google Scholar]
- 148.Jing W, Haller FM, Almond A, DeAngelis PL. Defined megadalton hyaluronan polymer standards. Anal Biochem. 2006;355(2):183–8. doi: 10.1016/j.ab.2006.06.009. [DOI] [PubMed] [Google Scholar]
- 149.Xu YM, Masuko S, Takieddin M, et al. Chemoenzymatic synthesis of homogeneous ultralow molecular weight heparins. Science. 2011;334(6055):498–501. doi: 10.1126/science.1207478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Mobli M, Nilsson M, Almond A. The structural plasticity of heparan sulfate NA-domains and hence their role in mediating multivalent interactions is confirmed by high-accuracy (15)N-NMR relaxation studies. Glycoconj J. 2008;25(5):401–14. doi: 10.1007/s10719-007-9081-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Pomin VH, Sharp JS, Li XY, Wang LC, Prestegard JH. Characterization of glycosaminoglycans by N-15 NMR spectroscopy and in vivo isotopic labeling. Anal Chem. 2010;82(10):4078–88. doi: 10.1021/ac1001383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Putnam CD, Hammel M, Hura GL, Tainer JA. X-ray solution scattering (SAXS) combined with crystallography and computation: defining accurate macromolecular structures, conformations and assemblies in solution. Q Rev Biophys. 2007;40(3):191–285. doi: 10.1017/S0033583507004635. [DOI] [PubMed] [Google Scholar]
- 153.Svergun DI. Small-angle X-ray and neutron scattering as a tool for structural systems biology. Biol Chem. 2010;391(7):737–43. doi: 10.1515/BC.2010.093. [DOI] [PubMed] [Google Scholar]
- 154.Rostom AA, Robinson CV. Disassembly of intact multiprotein complexes in the gas phase. Curr Opin Struct Biol. 1999;9(1):135–41. doi: 10.1016/s0959-440x(99)80018-9. [DOI] [PubMed] [Google Scholar]
- 155.Sobott F, Hernandez H, McCammon MG, Tito MA, Robinson CV. A tandem mass spectrometer for improved transmission and analysis of large macromolecular assemblies. Anal Chem. 2002;74(6):1402–7. doi: 10.1021/ac0110552. [DOI] [PubMed] [Google Scholar]
- 156.Chung EW, Henriques DA, Renzoni D, et al. Probing the nature of interactions in SH2 binding interfaces—evidence from electrospray ionization mass spectrometry. Protein Sci. 1999;8(10):1962–70. doi: 10.1110/ps.8.10.1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Yu Y, Sweeney MD, Saad OM, et al. Chemokine-glycosaminoglycan binding: specificity for CCR2 ligand binding to highly sulfated oligosaccharides using FTICR mass spectrometry. J Biol Chem. 2005;280(37):32200–8. doi: 10.1074/jbc.M505738200. [DOI] [PubMed] [Google Scholar]
- 158.Crown SE, Yu Y, Sweeney MD, Leary JA, Handel TM. Heterodimerization of CCR2 chemokines and regulation by glycosaminoglycan binding. J Biol Chem. 2006;281 (35):25438–46. doi: 10.1074/jbc.M601518200. [DOI] [PubMed] [Google Scholar]
- 159.Carlson J, Baxter SA, Dreau D, Nesmelova IV. The heterodimerization of platelet-derived chemokines. Biochim Biophys Acta. 2012;1834(1):158–68. doi: 10.1016/j.bbapap.2012.09.010. [DOI] [PubMed] [Google Scholar]
- 160.Sharp JS, Becker JM, Hettich RL. Protein surface mapping by chemical oxidation: structural analysis by mass spectrometry. Anal Biochem. 2003;313(2):216–25. doi: 10.1016/s0003-2697(02)00612-7. [DOI] [PubMed] [Google Scholar]
- 161.Sharp JS, Becker JM, Hettich RL. Topology characterization of model proteins by photochemical side chain oxidation and mass spectrometry. FASEB J. 2003;17(5):A988. [Google Scholar]
- 162.Hambly DM, Gross ML. Laser flash photolysis of hydrogen peroxide to oxidize protein solvent-accessible residues on the microsecond timescale. J Am Soc Mass Spectrom. 2005;16(12):2057–63. doi: 10.1016/j.jasms.2005.09.008. [DOI] [PubMed] [Google Scholar]
- 163.McClintock C, Kertesz V, Hettich RL. Development of an electrochemical oxidation method for probing higher order protein structure with mass spectrometry. Anal Chem. 2008;80(9):3304–17. doi: 10.1021/ac702493a. [DOI] [PubMed] [Google Scholar]
- 164.Maleknia SD, Chance MR, Downard KM. Electrospray-assisted modification of proteins: a radical probe of protein structure. Rapid Commun Mass Spectrom. 1999;13 (23):2352–8. doi: 10.1002/(SICI)1097-0231(19991215)13:23<2352::AID-RCM798>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 165.Maleknia SD, Brenowitz M, Chance MR. Millisecond radiolytic modification of peptides by synchrotron X-rays identified by mass spectrometry. Anal Chem. 1999;71 (18):3965–73. doi: 10.1021/ac990500e. [DOI] [PubMed] [Google Scholar]
- 166.Sharp JS, Sullivan DM, Cavanagh J, Tomer KB. Measurement of multisite oxidation kinetics reveals an active site conformational change in Spo0F as a result of protein oxidation. Biochemistry. 2006;45(20):6260–6. doi: 10.1021/bi060470r. [DOI] [PubMed] [Google Scholar]
- 167.Watson C, Janik I, Zhuang T, Charvatova O, Woods RJ, Sharp JS. Pulsed electron beam water radiolysis for submicrosecond hydroxyl radical protein footprinting. Anal Chem. 2009;81(7):2496–505. doi: 10.1021/ac802252y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Buxton GV, Greenstock CL, Helman WP, Ross AB. Critical-review of rate constants for reactions of hydrated electrons, hydrogen-atoms and hydroxyl radicals (.Oh/O-) in aqueous-solution. J Phys Chem Ref Data. 1988;17(2):513–886. [Google Scholar]
- 169.Xu G, Chance MR. Radiolytic modification and reactivity of amino acid residues serving as structural probes for protein footprinting. Anal Chem. 2005;77(14):4549–55. doi: 10.1021/ac050299+. [DOI] [PubMed] [Google Scholar]
- 170.Chance MR. Unfolding of apomyoglobin examined by synchrotron footprinting. Biochem Biophys Res Commun. 2001;287(3):614–21. doi: 10.1006/bbrc.2001.5628. [DOI] [PubMed] [Google Scholar]
- 171.Charvatova O, Foley BL, Bern MW, Sharp JS, Orlando R, Woods RJ. Quantifying protein interface footprinting by hydroxyl radical oxidation and molecular dynamics simulation: application to galectin-1. J Am Soc Mass Spectrom. 2008;19(11):1692–705. doi: 10.1016/j.jasms.2008.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Guan JQ, Vorobiev S, Almo SC, Chance MR. Mapping the G-actin binding surface of cofilin using synchrotron protein footprinting. Biochemistry. 2002;41(18):5765–75. doi: 10.1021/bi0121104. [DOI] [PubMed] [Google Scholar]
- 173.Gau BC, Sharp JS, Rempel DL, Gross ML. Fast photochemical oxidation of protein footprints faster than protein unfolding. Anal Chem. 2009;81(16):6563–71. doi: 10.1021/ac901054w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Watson C, Sharp JS. Conformational analysis of therapeutic proteins by hydroxyl radical protein footprinting. AAPS J. 2012;14(2):206–17. doi: 10.1208/s12248-012-9336-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Vahidi S, Stocks BB, Liaghati-Mobarhan Y, Konermann L. Mapping pH-induced protein structural changes under equilibrium conditions by pulsed oxidative labeling and mass spectrometry. Anal Chem. 2012;84(21):9124–30. doi: 10.1021/ac302393g. [DOI] [PubMed] [Google Scholar]
- 176.Jin HJ, Kagiampakis I, Li PW, LiWang PJ. Structural and functional studies of the potent anti-HIV chemokine variant P2-RANTES. Proteins. 2010;78(2):295–308. doi: 10.1002/prot.22542. [DOI] [PMC free article] [PubMed] [Google Scholar]