

Abstract
Three structurally defined QS-21-based immune adjuvant candidates (2a-2c) have been synthesized. Application of the two-stage activation glycosylation approach utilizing allyl glycoside building blocks improved the synthetic accessibility of the new adjuvants. The efficient synthesis and establishment of the stand-alone adjuvanticity of the examined synthetic adjuvant (2b) open the door to the pursuit of a new series of structurally defined QS-saponin based synthetic adjuvants.
Introduction
Naturally occurring saponin QS-21 (1, Figure 1) is one of the most potent vaccine adjuvants tested in both infectious disease and cancer settings.1-4 It stimulates both Th1 and Th2 immunity, which is important to developing subunit vaccines against cancers and intracellular pathogens (e.g., HIV, TB and malaria) when a sufficiently potent cytotoxic T lymphocyte (CTL) response is desired.3,5 QS-21 is a mixture of two saponin natural products (i.e., 1a and 1b in a ratio of 2:1) obtained from the tree bark of Quillaja saponaria (QS) Molina.6-8 These two isomers have the same adjuvanticity and toxicity.9,10 However, the natural saponin QS-21 has inherent drawbacks (such as chemical instability, limited supply, difficulty and low-yielding purification, and dose-limiting toxicity), which prevent it from wider use. The chemical instability of QS-21 originates from the hydrolytically labile ester linkage connecting the acyl side chain and the fucose moiety of the linear tetrasaccharide and the one embedded in the side chain. Structure-activity studies showed that loss of the lipophilic acyl chain results in loss of adjuvant activity of QS-21 in stimulating a lymphoproliferative response and CTL production.11-14 Gin and coworkers recently circumvented this chemical instability problem by replacing the ester linkage(s) with hydrolytically more stable amide bond(s).15,16 The analogs showed adjuvant activity similar to the natural products; however, the dose-limiting toxicity issue was not completely resolved and the synthesis is still not a trivial task. There remains an imperative need for new QS saponin-based structurally defined adjuvants with enhanced adjuvant activity, attenuated toxicity, and good synthetic accessibility.
Figure 1. Natural saponin immunoadjuvant QS-21 (1).

To pursue such an adjuvant, we need different QS-21 derivatizing strategies. An important clue emerged from the early work of Marciani and coworkers in developing the semi-synthetic saponin analog GPI-0100.11-14 GPI-0100 was prepared from the QS-21-containing QS tree bark extracts by complete removal of the acyl chain and subsequent incorporation of a dodecylamine chain via amide formation. The resulting complex mixture retained the capacity of stimulating humoral as well as T-cell immunity with the production of antigen-specific CTL. More importantly, the toxicity was reduced dramatically. However, GPI-0100's heterogeneity and variability in content and composition affect its efficacy, formulation, and clinical use in humans. Nevertheless, these results along with the early structure-function studies of Soltysik and coworkers 17 suggested that modifying the glucuronic acid moiety of the branched trisaccharide unit of QS saponins might result in chemically stable and structurally defined saponin analogs with maintained adjuvant activity and lowered toxicity.
To verify the feasibility of the new QS derivatizing strategy, we decided to synthesize the QS-21 analogs 2a and 2b (Scheme 1), derived from QS-21api (1a) and QS-21xyl (1b), respectively. Thus, instead of having a side chain connected to the linear tetrasaccharide, the new adjuvants have an aliphatic chain connected to the glucuronic acid unit of the branched trisaccharide domain through an amide bond. These two compounds were considered among the representative and immune active components of GPI-0100.11 Liu and coworkers first attempted synthesis of these compounds by derivatizing purified QS-21 in a way similar to making GPI-0100;18 however, the controversial immunological results raised questions about the nature of their products.19 Therefore, it is important to establish the identity of 2a and 2b unambiguously through organic synthesis and to re-evaluate the adjuvanticity of the pure compounds. Moreover, recent structure-activity-relationship studies by Gin and coworkers demonstrated that the synthetic QS-21-based adjuvant with a truncated linear trisaccharide segment is as active as the one with the full length linear tetrasaccharide.16 This discovery can be valuable to further simplifying the synthesis for better accessibility. To evaluate the immunological impact of a truncated linear saccharide segment in the new series of synthetic adjuvants, our synthetic targets also include 2c.
Scheme 1. Design and Retro-Synthesis of QS-21 Analogs.

Results and Discussion
In the retro-synthesis (Scheme 1), the saponin analogs 2a-2c can be prepared from the known quillaic acid-trisaccharide conjugate 3, the linear saccharide donor 4, and dodecylamine. The conjugate 3 was prepared in three steps from commercially available saponins.7,20 Synthesis of the fully protected tetra-and trisaccharide skeletons of the donor 4 can adopt the efficient two-stage activation approach using allyl glycosyl building blocks.21-24
For the linear tetrasaccharide synthesis, we adopted a 2+2 strategy. The synthesis began with the preparation of allyl rhamnoside donor 521,25 and the allyl fucoside acceptor 626 (Scheme 2). For synthesis of the allyl rhamnosyl-fucoside disaccharide 7, the anomeric allyl group of 5 was first isomerized to a prop-1-enyl group by using the hydrogen-activated catalyst [Ir(COD)(PMePh2)2]PF6.27,28 Subsequent treatment of the obtained prop-1-enyl donor with NIS/TfOH in the presence of the acceptor 6, followed by removal of the acetyl group at 4-O of the rhamnosyl unit provided the disaccharide 7 in 78% yield.21-24 For making the apiose-terminated tetrasaccharide as in 2a, the 2+2 strategy requires an allyl apiosyl xyloside disaccharide as the donor. The reaction of the peracetyl apioside donor 8 and the allyl xyloside acceptor 9 promoted by TESOTf led to the allyl disaccharide 10 in 88% yield.8 For making the xylose-terminated tetrasaccharide as in 2b, the two-stage activation approach using the allyl xyloside donor 1123 and the accepter 9 provided the disaccharide 12 in 71% yield. The 2-O pivaloyl group in 10-12 was used for directing the desired 1,2-trans selective glycosylation.29,30 The same two-stage activation procedure was then applied to make the linear saccharides 13a-13c through the reactions of the acceptor 7 with the donors 10, 12, and 11, respectively.
Scheme 2. Synthesis of Oligosaccharide Donorsa.

aReagents and conditions: (a) H2-activated [Ir(COD)(PMePh2)2]PF6, THF, 23°C; 6, NIS/TfOH, MeCN, 23 °C; NaOMe, 78%; (b) TESOTf, CH2Cl2, 0->23 °C, 88%; (c) H2-activated [Ir(COD)(PMePh2)2]PF6, THF, 23 °C; 9, NIS/TfOH, MeCN, 23 °C, 71%; (d) H2-activated [Ir(COD)(PMePh2)2]PF6, THF, 23 °C; 7, NIS/TfOH, MeCN, 23 °C, 76% for 13a, 69% for 13b, 83% for 13c; (e) NaOMe, MeOH, 23 °C; TFA/H2O (4:1); Ac2O, TEA, DMAP, CH2Cl2, 23 °C; (f) H2-activated [Ir(COD)(PMePh2)2]PF6, THF, 23 °C; NIS/H2O, 23 °C; (g) Cs2CO3, Cl3CCN, CH2Cl2, 23 °C, 78% for 4a, 85% for 4b, 73% for 4c.
The two-stage glycosylation conditions do not work well with carboxylic acid acceptors, thus we planned to convert the linear saccharide donors 13a-c to their corresponding trichloroacetimidate donors. However, we observed that for 13b, without any electron-withdrawing groups on the fucose unit, the corresponding imidate was unstable; the glycosylation reaction with the quillaic acid acceptor was not clean, leading to the undesired α glycosidic bond formation. This was not observed in the original QS-21 synthesis in which the fucosyl trichloroacetimidate donor has an ester moiety at its 4-O group.8 The protecting groups in the tetrasaccharides 13a-c were then replaced with acetyl groups and subsequently converted to the imidates 4a-c. The yields of 4a-c from the corresponding 13a-c were 78%, 85%, and 73%, respectively.
Glycosylation of the obtained imidates with the quillaic acid-trisaccharide conjugate (3) afforded the compounds 14a-c with the desired anomeric stereochemistry in 57%, 72%, and 60% yield, respectively (Scheme 3). Debenzylation under hydrogenolysis conditions followed by coupling of the released carboxyl group with dodecylamine using the coupling reagent HATU31 led to the fully protected product 15a-c in 85%, 97%, and 78% yield, respectively. The final global deprotection procedure consisted of two steps, i.e., removal of all the triethylsilyl groups with TFA/H2O (4:1 v/v) at 0 °C and removal of the acetyl groups with K2CO3 in methanol. The ester linkage between the linear oligosaccharide and the quillaic acid is known to be ruptured under basic conditions only at elevated temperature.8,32 Indeed, under the mild saponification conditions, there was no detectable cleavage of this ester linkage. The final products 2a-2c were obtained in 95%, 85%, and 92% yield, respectively. Interestingly, switching the sequence of these last two steps resulted in cleavage of the quillaic acid-fucoside ester linkage at the stage of TFA/water treatment.
Scheme 3. Synthesis of Adjuvantsa.

aReagents and conditions: (a) BF3.OEt2, CH2Cl2, -78 °C, 57% for 14a, 72% for 14b, 60% for 14c; (b) Pd/C, THF, H2, 55 psi; (c) dodecyl amine, HATU, DIPEA, CHCl3, 23 °C, 85% for 15a, 97% for 15b, 78% for 15c; (d) TFA/H2O (4:1), 0 °C; (e) K2CO3, MeOH, 23 °C, 95% for 2a, 85% for 2b, 92% for 2c.
With the adjuvants in hand, we first evaluated the adjuvant activity of 2b in order to determine the effect of derivatizing at the glucuronic acid unit of the branched trisaccharide domain on adjuvant activity and toxicity. We determined the effectiveness of 2b to potentiate immune responses to rHagB, a recombinant, non-fimbrial adhesion hemagglutinin B antigen from the periodontal pathogen Porphyromonas gingivalis.33-39 We also compared the immunopotentiating activity of 2b with that of GPI-0100. Groups of female BALB/c mice (8-10 weeks of age, six per group) were immunized by the subcutaneous route (s.c.) with rHagB (20 μg) alone or with GPI-0100 (100 μg) or 2b (100 μg) on days 0, 14 and 28.39 Mice were weighed and serum samples were collected prior to each immunization and at 6 and 12 weeks following the initial immunization. The levels of serum IgG antibody activity to rHagB were determined by enzyme-linked immunosorbent assays (ELISA). A serum IgG response was detected in all three groups by week 2 after the initial immunization. A significant increase was seen in the level of IgG anti-rHagB antibody activity in mice receiving rHagB+GPI-0100 (P < 0.001) or rHagB+2b (P < 0.05) following the second immunization. The magnitude of the response continued to increase following the third immunization in mice receiving rHagB alone or rHagB+2b and persisted through week 12. The responses induced following immunization with rHagB+2b were significantly higher than those induced with rHagB alone at weeks 6 (P < 0.01) and 12 (P < 0.05). GPI-0100 potentiated significantly higher (P < 0.001) responses to rHagB than seen with antigen alone at weeks 2, 4, 6 and 12. In addition, mice immunized with rHagB+GPI-0100 had significantly higher levels of IgG activity than mice receiving rHagB+2b at weeks 4 (P < 0.001) and 6 (P < 0.01). However, no further increase in antibody activity was seen following the third immunization in mice immunized with rHagB+GPI-0100, and the level of activity dropped significantly (P < 0.001) at week 12. No sign of significant toxicity was observed in all the mice based on weight monitoring (see details in Supporting Information). These results indicate that 2b is effective in potentiating and maintaining a serum IgG response to rHagB following systemic immunization.
The implication of these immunological studies is twofold. First, the establishment of the preclinical adjuvant activity of 2b proved that derivatizing at the glucuronic acid unit of the branched trisaccharide domain is a viable way to access new adjuvants with adjuvanticity and toxicity profiles different from those currently known, structurally defined, natural and un-natural QS products. The lower adjuvant activity of 2b than GPI-0100 in potentiating the IgG response to rHagB might be related to its low water-solubility compared to that of GPI-0100. The water-solubility of the other two synthetic adjuvants 2a and 2c is also low, and therefore, we did not proceed to do immunological evaluations on them. A rational step to improve adjuvanticity by increasing water-solubility of new synthetic adjuvants is to incorporate more hydrophilic side chains to the glucuronic acid unit. Second, the preferable adjuvanticity and toxicity profile of GPI-0100 can perhaps be attributed to components other than the QS-21 derivative. An identified major component of GPI-0100 originated from QS-17/18 and expected to be more water soluble than the QS-21 derivative due to an extra glucose attached to the O-3 of the rhamnose unit in the linear tetrasaccharide.11,14 Efforts to improve adjuvant activity along these two directions are current underway in our laboratory.
In summary, three structurally defined QS-21-based immune adjuvants were synthesized by using the efficient modular semi-synthetic strategy. Application of the two-stage activation approach of glycosylation simplified the linear saccharide synthesis and improved the overall synthetic efficiency. The verified synthetic strategy paves the way to accessing a variety of un-natural QS saponin derivatives for a thorough structure-activity exploration and for systematic and rational optimization of the QS family of immune adjuvants. The immunological results guide us to design new QS derivatives that can potentially have high adjuvanticity and low toxicity.
Experimental Section
General
Organic solutions were concentrated by rotary evaporation at ca. 12 Torr. Flash column chromatography was performed employing 230-400 mesh silica gel. Thin-layer chromatography was performed using glass plates pre-coated to a depth of 0.25 mm with 230-400 mesh silica gel impregnated with a fluorescent indicator (254 nm). Infrared (IR) data are presented as frequency of absorption (cm -1). Proton and carbon-13 nuclear magnetic resonance (1H NMR or 13C NMR) spectra were recorded on 300, 400 and 700 MHz NMR spectrometers; chemical shifts are expressed in parts per million (δ scale) downfield from tetramethylsilane and are referenced to residual protium in the NMR solvent (CHCl3: δ 7.26). Data are presented as follows: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet and/or multiple resonances), coupling constant in Hertz (Hz), integration. HRMS was conducted with either ESI or MALDI ionization method and with TOF mass analyser.
Materials
Tetrahydrofuran, toluene, dichloromethane, and acetonitrile were distilled from appropriate drying reagents under a nitrogen atmosphere at 760 torr. Other chemicals were obtained from commercial vendors and used without further purification.
Allyl 4-O-acetyl-2,3-O-isopropylidene-α-l-rhamnopyranoside (5)
l-(+)-rhamose (5 g, 27.5 mmol) was stirred in allyl alchol (30 mL, 0.55mol) in the presence of acetyl chloride (5 mL, 60 mmol) at room temperature. Upon completion of the reaction, the reaction mixture was concentrated to dryness under reduced pressure. The crude material was treated with 2,2-dimethoxypropane (40 mL) in the presence of TsOH (300 mg, 1.6 mmol) in 10 mL of dichloromethane and 5 mL of acetone. The reaction mixture was concentrated, and extracted with ethyl acetate. The combined organic layers were concentrated and the crude material was treated with triethylamine (5 mL), acetic anhydride (7.6 mL, 69 mmol), and dimethylaminopyridine (101 mg, 0.83 mmol) in 10 mL of dichloromethane. Upon completion of the reaction, the the reaction mixture was concentrated, and purified with flash column chromatography (petroleum ether/ethyl acetate 10:1) to afford the desired fucoside 5 (5.2 g, 67% over three steps) as a colorless oil.21,25
Allyl 3,4-O-isopropylidene-α-d-fucopyranoside (6)
d-(+)-fucose (0.99g, 5.9 mmol) was stirred in allyl alcohol (4mL, 59 mmol) in the presence of acetyl chloride (0.85mL, 11.8 mmol) at room temperature. Upon completion of the reaction after 1.5h, the reaction mixture was concentrated to dryness under reduced pressure. The crude material was treated with 2,2-dimethoxypropane (20mL, 161 mmol) in the presence of TsOH (100 mg, 0.52 mmol) in 10 mL of dichloromethane and 5 mL of acetone. After 3 h, the reaction was quenched with saturated K2CO3 solution. The reaction mixture was concentrated, partitioned in ethyl acetate and water, and extracted with etheyl acetate. The combined organic layers were concentrated and purified with flash column chromatography (petroleum ether/ethyl acetate 2:1) to afford the desired fucoside 6 (1.2 g, 85% over two steps) as a colorless oil.26
Allyl 2,3-O-isopropylidene-α-l-rhamnopyranosyl-(1→2)-3,4-O-isopropylidene-α-d-fucopyrano-side (7)
A solution of [Ir(COD)(PMePh2)2]PF6 (45 mg, 0.053 mmol) in THF was degassed and stirred under hydrogen atmosphere for 15 min at room temperature. The obtained clear solution of the activated catalyst was injected to 5 (600 mg, 2.11 mmol) in THF. The reaction mixture was stirred for 60 min, and THF was removed under vacu. The residue and the acceptor 6 (340 mg, 1.39 mmol) were dried together by azeotropic removal of water with toluene. The glycosylation partners were then dissolved in 80 mL of acetonitrile. To the reaction solution, NIS (475 mg, 2.11 mmol) was added, followed immediately by TfOH (1.8 μL, 0.042 mmol) at room temperature. The reaction was stirred for 5 min and quenched with 0.3 mL of triethyl amine. The solution was then concentrated and the residue was treated with 2 mL of methanol solution of sodium methoxide (25%). After 5 h, the reaction mixture was concentrated and purified with flash column chromatography on silica gel (eluted with petroleum ether/ethyl acetate 2:1) to afford 7 (471 mg, 78% yield) as a colorless oil. Rf 0.3 (petroleum ether/ethyl acetate 2:1); [α]D23 = +64.7 (c = 4.42, CHCl3); 1H NMR (400 MHz, CDCl3) δ 5.91 (dddd, J =17.2, 10.4, 6.2, 5.3 Hz, 1H), 5.34 (ddd, J =17.2, 1.6, 1.5 Hz, 1H), 5.27 (s, 1 H), 5.23 (dd, J =10.4, 1.5 Hz, 1 H), 4.8 (d, J =3.5 Hz, 1 H), 4.35-4.25 (m, 2 H), 4.17 (ABMX2, J =13.0, 5.2, 1.3 Hz, 1 H), 4.13-4.07 (m, 2 H); 4.06 (dd, J =5.3, 2.5 Hz, 1 H), 4.0 (ABMX2, J =13.0, 5.3, 1.2 Hz, 1 H), 3.81 (dd, J =8.3, 3.6 Hz, 1 H), 3.67 (dq, J =9.1, 6.1 Hz, 1 H), 3.35 (t, J =7.6 Hz, 1 H), 1.54 (s, 3 H), 1.52 (s, 3 H), 1.36-1.35 (m, 9 H), 1.25 (d, J =6.4 Hz, 3 H); 13C NMR (100 MHz, CDCl3) δ 133.3, 117.7, 109.0, 108.7, 98.2, 96.9, 78.0, 76.0, 75.9, 75.5, 75.3, 74.0, 68.2, 65.8, 62.9, 28.2, 27.8, 26.3, 25.9, 17.3, 16.0; IR (neat) 3482, 2987, 2937, 1559, 1382, 1078; HRMS (ESI-TOF) m/e: [M+Na]+ calcd. for C21H34NaO9 453.2101; found 453.2101.
Allyl 2,3,4-tri-O-pivaloyl-α-d-xylopyranoside (11) and allyl 2,4-di-O-pivaloyl-α-d-xylopyranoside (9)
d-(+)-xylose (1.5g, 10 mmol) was stirred in allyl alchol (11.6 g, 0.2 mol) in the presence of acetyl chloride (3.1 g, 40 mmol) at room temperature. Upon completion of the reaction, the reaction mixture was concentrated to dryness under reduced pressure. To a portion of the obtained allyl xyloside (1.2 g, 6.3 mmol) was added pivaloyl chloride (1.74 mL, 13.8 mmol) and 7 mL of pyridine. The reaction mixture was stirred for 2.5 h and then filtered through a silica gel plug. The obtained solution was partitioned in ethyl acetate and water. The organic layer was concentrated and subjected to column chromatography (petroleum ether/ethyl acetate 9:1 to 5:1) to afford the allyl 2,3,4-tri-O-pivaloyl-α-d- xyloside (11) (240 mg, 8%) and the allyl 2,4-di-O-pivaloyl-α-d-xyloside (9) (942 mg, 41%) as a colorless oil. For 9, Rf 0.4 (benzene/ethyl acetate 9:1); [α]D23 = +101 (c = 1.1, CHCl3); 1H NMR (400 MHz, CDCl3) δ 5.85 (dddd, J =17.2, 10.4, 6.0, 5.2 Hz, 1H), 5.32 (dq, J =17.2, 1.6 Hz, 1H), 5.21 (ddt, J =10.4, 1.6, 1.4 Hz, 1 H), 5.02 (d, J =3.7 Hz, 1 H), 4.84 (ddd, J =10.8, 10.0, 5.9 Hz, 1 H), 4.7 (dd, J =10.0, 3.7 Hz, 1 H), 4.2 (ABMX2, J =13.1, 5.2, 1.6 Hz, 1 H), 4.14 (t, J =10.0 Hz, 1 H), 3.95 (ABMX2, J =13.1, 6.0, 1.4 Hz, 1 H), 3.75 (dd, J =10.7, 5.9 Hz, 1 H), 3.58 (t, J =10.7 Hz, 1 H), 1.23 (s, 9 H), 1.21 (s, 9 H); 13C NMR (100 MHz, CDCl3) δ 178.27, 178.23, 133.4, 117,6, 95.2, 73.3, 71.5, 69.8, 68.6, 58.5, 38.87, 38.83, 27.05, 27.02; IR (neat) 2965, 1740, 1487, 1267, 1162, 1053; HRMS (ESI-TOF) m/e: [M+H]+ calcd. for C18H31O7 359.2070; found 359.2061.
Allyl 2,3,5-tri-O-acetyl-β-d-apiofuranosyl-(1→3)-2,4-di-O-pivaloyl-β-d-xylopyranoside (10)
The glycosyl donor 8 (500 mg, 1.57 mmol) and the acceptor 9 (731 mg, 2.0 mmol) were dried together by azeotropic removal of water with toluene. The glycosylation partners were then dissolved in 30 mL of dichloromethane and treated with TESOTf (41.5 mg, 0.157 mmol) at 0 °C. The reaction was then allowed to warm to room temperature. After 15 min, the reaction was quenched with triethylamine and concentrated for column purification on silica gel (eluted with petroleum ether/ethyl acetate 3:1) to afford 10 (857 mg, 88%) as a white amorphous solid. Rf 0.3 (petroleum ether/ethyl acetate 2:1); [α]D23 = +7.4 (c = 1.02, CHCl3); 1H NMR (400 MHz, CDCl3) δ 5.85 (dddd, J =16.6, 10.4, 6.0, 5.4 Hz, 1H), 5.33 (s, 1 H), 5.3 (ddt, J =17.2, 1.6, 1.5 Hz, 1H), 5.23-5.20 (m, 2 H), 5.05 (d, J =3.7 Hz, 1 H), 4.96 (ddd, J =10.6, 9.1, 6.1 Hz, 1 H), 4.83 (d, J =12.4 Hz, 1 H), 4.73 (dd, J =9.8, 3.7 Hz, 1 H), 4.34 (d, J = 12.4 Hz, 1 H), 4.22 (AB, J =10.6 Hz, 1 H), 4.20 (t, J =9.4 Hz, 1 H), 4.17 (ABMX2, J =12.9, 5.3, 1.4 Hz, 1 H), 4.08 (AB, J= 10.6 Hz, 1 H), 3.92 (ABMX2, J =13.0, 6.1, 1.3 Hz, 1 H), 3.70 (dd, J =10.9, 6.1 Hz, 1 H), 3.50 (t, J =10.8 Hz, 1 H), 2.10 (s, 3 H), 2.08 (s, 3 H), 1.98 (s, 3 H), 1.22 (s, 18 H); 13C NMR (75 MHz, CDCl3) δ 177.1, 177.0, 170.0, 169.3, 168.5, 133.1, 117.6, 106.0, 94.4, 83.4, 76.2, 75.8, 72.8, 71.8, 69.1, 68.4, 62.1, 58.0, 38.5, 38.4, 26.8, 26.7, 20.7, 20.3, 20.1; IR (neat) 2974, 1751,1146, 1050; HRMS (ESI-TOF) m/e: [M+Na]+ calcd. for C29H44NaO14 639.2629; found 639.2628.
Allyl 2,3,4-tri-O-pivaloyl-β-d-xylopyranosyl-(1→3)-2,4-di-O-pivaloyl-α-d-xylopyranoside (12)
A solution of [Ir(COD)(PMePh2)2]PF6 (8.5 mg, 10 μmol) in 1 mL of THF was degassed and stirred under hydrogen atmosphere for 15 min at room temperature. The obtained clear solution of the activated catalyst was injected to allyl 2,3,4-tri-O-pivaloyl-α-d-xyloside 11 (250 mg, 0.56 mmol) in 1 mL of THF. The reaction mixture was stirred for 60 min, and THF was removed under vacu. The residue and allyl 2,4-di-O-pivaloyl-α-d-xyloside 9 (281mg, 0.78 mmol) were dried together by azeotropic removal of water with toluene. The glycosylation partners were then dissolved in 30 mL of acetonitrile. To the reaction solution, NIS (126 mg, 0.56 mmol) was added, followed immediately by TfOH (25 μL, 0.28 mmol) at room temperature. The reaction was stirred for 10 min and quenched with triethyl amine. The solution was then concentrated and purified with flash column chromatography on silica gel (eluted with benzene/ethyl acetate 20:1) to afford 12 (298 mg, 71% yield) as a white amorphous solid. Rf 0.5 (benzene/ethyl acetate 20:1); [α]D22 = +27.5 (c = 1.3, CHCl3); 1H NMR (400 MHz, CDCl3) δ 5.85 (dddd, J =16.7, 11.4, 6.4, 5.6 Hz, 1H), 5.30 (dq, J =16.7, 1.5 Hz, 1H), 5.2 (ddt, J =11.4, 1.6, 1.2 Hz, 1 H), 5.15 (t, J =9.2 Hz, 1 H), 5.08 (d, J =3.7 Hz, 1 H), 5.0-4.7 (m, 4 H), 4.6 (dd, J =9.7, 3.7 Hz, 1 H), 4.33 (t, J =9.5 Hz, 1 H), 4.15 (ABMX2, J =12.7, 5.6, 1.4, 1.2 Hz, 1 H), 4.09 (dd, J =12.5, 5.6 Hz, 1 H), 3.90 (ABMX2, J =12.7, 6.4, 1.3, 1.2 Hz, 1 H); 3.76 (dd, J =10.8, 6.0 Hz, 1 H), 3.49 (t, J =10.8 Hz, 1 H), 3.24 (dd, J =11.5, 10.1 Hz, 1 H), 1.29 (s, 9 H), 1.21 (s, 9 H), 1.16 (s, 9 H), 1.114 (s, 9 H), 1.107 (s, 9 H); 13C NMR (100 MHz, CDCl3) δ 177.5, 177.3, 177.2, 177.1, 176.2, 133.24, 133.23, 118.2, 100.3, 94.3, 74.3, 72.8, 71.5, 70.9, 69.1, 68.9, 68.8, 62.5, 58.3, 38.74, 38.69, 38.68, 38.61, 38.60, 38.55, 38.54, 27.14, 27.11, 27.07, 27.03, 26.99, 26.97, 26.95, IR (neat) 2974, 1740, 1481, 1280, 1145, 912, 734; HRMS (ESI-TOF) m/e: [M+Na]+ calcd. for C38H62NaO14 765.4037; found 765.4043.
Allyl 2,3,5-tri-O-acetyl-β-d-apiofuranosyl-(1→3)-2,4-di-O-pivaloyl-β-d-xylopyranosyl-(1→4)-2,3-O-isopropylidene-α-l-rhamnopyranosyl-(1→2)-3,4-O-isopropylidene-α-d-fucopyranoside (13a)
A solution of [Ir(COD)(PMePh2)2]PF6 (2 mg, 2.4 μmol) in 1 mL of THF was degassed and stirred under hydrogen atmosphere for 15 min at room temperature. The obtained clear solution of the activated catalyst was injected to 10 (110 mg, 0.178 mmol) in THF. The reaction mixture was stirred overnight and then THF was removed under vacu. The residue and the acceptor 7 (43 mg, 0.1 mmol) were dried together by azeotropic removal of water with toluene. The glycosylation partners were then dissolved in 5 mL of acetonitrile. To the reaction solution, NIS (43 mg, 0.19 mmol) was added, followed immediately by TfOH (1.7 μL, 0.019 mmol) at room temperature. The reaction was stirred for 5 min and quenched with triethyl amine. The solution was then concentrated and purified with flash column chromatography on silica gel (eluted with petroleum ether/ethyl acetate 2:1) to afford the tetrasaccharide product 13a (75 mg, 76%) as a white amorphous solid. Rf 0.4 (petroleum ether/ethyl acetate 2:1); [α]D23 = -23.8 (c = 0.26, CHCl3); 1H NMR (400 MHz, CDCl3) δ 5.89 (dddd, J =17.2, 10.4, 6.3, 5.3 Hz, 1H), 5.35 (dq, J =17.2, 1.6 Hz, 1H), 5.274 (s, 1 H), 5.266 (s, 1 H), 5.25 (ddt, J =11.9, 1.5, 1.2 Hz, 1 H), 5.09 (s, 1 H), 5.0-4.9 (m, 3 H), 4.82 (d, J =3.6 Hz, 1 H), 4.77 (d, J =12.4 Hz), 4.45 (d, J = 12.4 Hz), 4.29 (dd, J =8.3, 5.3 Hz, 1 H), 4.23 (d, J =5.5 Hz, 1 H), 4.21 (d, J =12.6 Hz, 1 H), 4.18 (ABMX2, J =13.0, 5.3, 1.3 Hz, 1 H), 4.13 (dd, J =6.4, 2.5 Hz, 1 H), 4.11-4.03 (m, 4 H), 4.02 (ABMX2, J =13.0, 6.4, 1.2 Hz, 1 H), 3.92 (m, 1 H), 3.8 (dd, J =8.3, 3.6 Hz, 1 H), 3.63 (m, 1 H), 3.53 (dd, J =9.9, 7.6 Hz, 1 H), 3.24 (dd, J =12.1, 8.1 Hz, 1 H), 2.1 (s, 3 H), 2.09 (s, 3 H), 2.00 (s, 3 H), 1.55 (s, 3 H), 1.51 (s, 3 H), 1.368 (d, J =5.3 Hz, 3 H), 1.363 (s, 3 H), 1.33 (s, 3 H), 1.25 (s, 9 H), 1.23 (s, 9 H), 1.196 (d, J =6.2 Hz, 3 H); 13C NMR (176 MHz, CDCl3) δ 177.3, 176.5, 170.5, 169.7, 168.9, 133.4, 118.2, 109.1, 109.0, 106.2, 99.6, 98.3, 97.0, 83.8, 79.1, 78.0, 77.9, 76.6, 76.3, 76.2, 76.0, 75.5, 72.2, 70.7, 68.4, 64.3, 63.0, 62.6, 38.8, 28.4, 27.9, 27.17, 27.13, 27.10, 26.5, 26.4, 21.1, 20.7, 20.5, 17.4, 16.2; IR (neat) 2983, 2937, 1751, 1370, 1221; HRMS (ESI-TOF) m/e: [M+Na]+ calcd. for C47H72NaO22 1011.4413; found 1011.4414.
Allyl 2,3,4-tri-O-pivaloyl-β-d-xylopyranosyl-(1→3)-2,4-di-O-pivaloyl-β-d-xylopyranosyl-(1→4)-2,3-O-isopropylidene-α-l-rhamnopyranosyl-(1→2)-3,4-O-isopropylidene-α-d-fucopyranoside (13b)
A solution of [Ir(COD)-(PMePh2)2]PF6 (40 mg, 0,04 mmol) in THF was degassed and stirred under hydrogen atmosphere for 15 min at room temperature. The obtained clear solution of the activated catalyst was injected to 12 (380 mg, 0.5 mmol) in THF. The reaction mixture was stirred overnight, and THF was removed under vacu. The residue and the acceptor 7 (146 mg, 0.34 mmol) were dried together by azeotropic removal of water with toluene. The glycosylation partners were then dissolved in 30 mL of acetonitrile. To the reaction solution, NIS (115 mg, 0.51 mmol) was added, followed immediately by TfOH (4.4 μL, 0.05 mmol) at room temperature. The reaction was stirred for 1 min and quenched with triethyl amine. The solution was then concentrated and purified with flash column chromatography on silica gel (eluted with petroleum ether/ethyl acetate 5:1) to afford the tetrasaccharide product 13b (260 mg, 69% yield) as a white amorphous solid. Rf 0.34 (petroleum ether/ethyl acetate 5:1); [α]D23 = -17.2 (c = 0.76, CHCl3); 1H NMR (300 MHz, CDCl3) δ 5.86 (m, 1H), 5.3 (d, J =17.2 Hz, 1H), 5.23 (s, 1 H), 5.2 (d, J =10.9 Hz, 1 H), 5.15 (t, J =9.0 Hz, 1 H), 4.9-4.7 (m, 7 H), 4.24 (dd, J =8.3, 5.3 Hz, 1 H), 4.21 (d, J =5.9 Hz, 1 H), 4.17-3.94 (m, 8 H), 3.77 (dd, J =8.3, 3.5 Hz, 1 H), 3.63-3.48 (m, 2 H), 3.24 (d, J = 10.0 Hz, 1 H), 3.2 (dd, J =11.7, 2.3 Hz, 1 H), 1.5 (s, 3 H), 1.47 (s, 3 H), 1.33-1.32 (m, 6 H), 1.29 (s, 3 H), 1.25 (s, 9 H), 1.191 (s, 9 H), 1.16 (d, J =6.0 Hz, 3 H), 1.13 (s, 9 H), 1.12 (s, 9 H), 1.1 (s, 9 H); 13C NMR (75 MHz, CDCl3) δ 177.5, 177.3, 177.1, 176.5, 176.1, 133.5, 117.9, 109.0, 108.9, 99.9, 99.2, 98.1, 97.1, 77.8, 77.6, 77.2, 76.3, 76.03, 75.95, 75.6, 75.0, 73.2, 71.4, 70.9, 69.5, 69.0, 68.4, 64.1, 63.0, 62.7, 62.4, 38.74, 38.68, 38.63, 29.6, 28.4, 27.8, 27.2, 27.06, 27.03, 27.00, 26.44, 26.39, 17.4, 16.2; IR (neat) 2978, 2937, 1743, 1141; HRMS (ESI-TOF) m/e: [M+Na]+ calcd. for C56H90NaO22 1137.5821; found 1137.5823.
Allyl 2,3,4-tri-O-pivaloyl-β-d-xylopyranosyl-(1→4)-2,3-O-isopropylidene-α-l-rhamnopyranosyl-(1→2)-3,4-O-isopropylidene-α-d-fucopyranoside (13c)
A solution of [Ir(COD)(PMePh2)2]PF6 (2 mg, 0.0022 mmol) in THF (0.5 mL) was degassed and stirred under hydrogen atmosphere for 10 min at room temperature. The obtained clear solution of the activated catalyst was injected to 11 (99 mg, 0.22 mmol) in THF (1.5 mL). The reaction mixture was stirred for 2 hrs, and THF was removed in vacu. The residue and the acceptor 7 (64 mg, 0.149 mmol) were dried together by azeotropic removal of water with toluene. The glycosylation partners were then dissolved in 20 mL of acetonitrile. To the reaction solution, NIS (50.0 mg, 0.22 mmol) was added, followed immediately by TfOH (0.94 μL, 0.022 mmol) in acetonitrile (0.2 mL) at room temperature. The reaction was stirred for 5 min and quenched with 0.2 mL of triethyl amine. The solution was then concentrated and purified with flash column chromatography on silica gel (eluted with petroleum ether/ethyl acetate 8:1) to afford 13c (100.0 mg, 83% yield) as a white amorphous solid. Rf 0.4 (petroleum ether/ethyl acetate 6:1); [α]D23 = +9.8 (c = 0.95, CHCl3); 1H NMR (300 MHz, CDCl3) δ 5.83 (dddd, J =16.8, 10.4, 6.3, 5.2 Hz, 1H), 5.35-5.16 (m, 4 H), 5.0-4.89 (m, 3 H), 4.78 (d, J =3.5 Hz, 1H), 4.24 (dd, J =8.3, 5.3 Hz, 1 H), 4.20 (d, J =5.6 Hz, 1 H), 4.13 (ABMX2, J =13.2, 5.2, 1.4 Hz, 1 H), 4.09-4.00 (m, 4 H), 3.97 (ABMX2, J =13.0, 7.4, 1.3 Hz, 1 H), 3.77 (dd, J =8.3, 3.6 Hz, 1 H), 3.64-3.48 (m, 2 H), 3.23 (dd, J =11.4, 10.0 Hz, 1 H), 1.5 (s, 3 H), 1.49 (s, 3 H), 1.32 (d, J =6.6 Hz, 3 H), 1.322 (s, 3 H), 1.30 (s, 3H), 1.15 (s, 9 H), 1.12 (s, 9 H), 1.10 (s, 9 H); 13C NMR (75 MHz, CDCl3) δ 177.2, 177.0, 176.5, 133.4, 118.0, 99.8, 98.3, 97.0, 78.2, 77.8, 76.3, 76.2, 75.9, 75.5, 71.8, 71.2, 69.4, 68.3, 64.2, 63.0, 62.6, 38.7, 38.6, 28.4, 27.8, 27.1, 27.0, 26.43, 26.36, 17.3, 16.2; IR (neat) 2982, 2937, 1743, 1144; HRMS (ESI-TOF) m/e: [M+Na]+ calcd. for C41H66NaO16 837.4249; found 837.4239.
2,3,5-tri-O-acetyl-β-d-apiofuranosyl-(1→3)-2,4-di-O-acetyl-β-d-xylopyran-osyl-(1→4)-2,3-O-di-acetyl-α-L-rhamnopyranosyl-(1→2)-3,4-di-O-acetyl-α-d-fucopyranosyl trichloroacetimidate (4a)
The tetrasaccharide product 13a (200 mg, 0.2 mmol) in 3 mL of methanol was treated with 0.2 mL of sodium methoxide in methanol (25% w/w) for 24 hour at room temperature. Upon completion of the reaction, methanol was removed and in an ice bath, the residue was treated with 1 mL of pre-cooled TFA/H2O (4:1). The reaction was stirred at 0 °C for 45 min, and then the reaction solution was concentrated in vacu at 0 °C. The residue was dissolved in 3 mL of DCM and treated with triethyl amine (2 mL, 14 mmol), dimethylamino pyridine (5 mg, 0.041 mmol), and acetic anhydride (0.5 mL, 5.3 mmol). The reaction was stirred overnight and then concentrated. The residue was purified with column chromatography on silica gel (eluted with petroleum ether/ethyl acetate gradients) to afford the desired tetrasaccharide intermediate S1 (168 mg, 84%). Rf 0.33 (petroleum ether/ethyl acetate 1:2). A solution of [Ir(COD)(PMePh2)2]PF6 (6 mg, 7 μmol) in THF was degassed and stirred under hydrogen atmosphere for 15 min at room temperature. The obtained clear solution of the activated catalyst was added to the tetrasaccharide S1 (117 mg, 0.118 mmol) in 5 mL of THF. After stir overnight, the isomerization reaction was complete, and drops of water and NIS (27 mg, 0.12 mmol) were added. The deallylation reaction completed instantly. The reaction mixture was concentrated and purified with flash column chromatography (eluted with petroleum ether/ethyl acetate gradients). Rf 0.1 (petroleum ether/ethyl acetate 1:2). The obtained hemiacetal was treated with trichloroacetonitrile (1.0 mL, 10 mmol) and cesium carbonate (75 mg, 0.23 mmol) in dichloromethane (5 mL) at room temperature overnight. The reaction mixture was then concentrated and subjected to flash column chromatography (eluted with eluted with petroleum ether/ethyl acetate gradients) to afford the imidate 4a (120 mg, 93%) as a white amorphous solid. Rf = 0.3 (petroleum ether/ethyl acetate 1:2); [α]D22 = -13 (c = 2.0, CHCl3); 1H NMR (400 MHz, CDCl3) δ 8.75 (s, 1 H), 6.48 (d, J =3.75 Hz, 1 H), 5.39 (dd, J =3.2, 0.9 Hz, 1 H), 5.31 (dd, J =10.5, 3.2 Hz, 1 H), 5.13 (s, 1 H), 5.11 (dd, J =9.8, 3.4 Hz, 1 H), 4.98-4.97 (m, 2 H), 4.95-4.86 (m, 3 H), 4.61 (AB1, J =12.5 Hz, 1 H), 4.52 (AB1, J =12.5 Hz, 1 H), 4.48 (d, J =7.9 Hz, 1 H), 4.34 (q, J =7.2 Hz, 1 H), 4.21 (t, J =5.5 Hz, 1 H), 4.20 (AB2, J =10.4 Hz, 1 H), 4.07 (AB2, J =10.4 Hz, 1 H), 4.04 (m, 1 H), 3.86 (m, 1 H), 3.66 (t, J =9.4 Hz, 1 H), 3.62 (t, J =9.6 Hz, 1 H), 3.22 (dd, J =11.7, 9.8 Hz, 1 H), 2.2 (s, 3 H), 2.1 (s, 3 H), 2.086 (s, 3 H), 2.083 (s, 3 H), 2.080 (s, 3 H), 2.06 (s, 3 H), 2.05 (s, 3 H), 2.04 (s, 3 H), 2.01 (s, 3 H), 1.28 (d, J =6.2 Hz, 3 H), 1.16 (d, J =6.5 Hz, 3 H); 13C NMR (100 MHz, CDCl3) δ 170.32, 170.27, 170.05, 169.9, 169.7, 169.6, 169.1, 169.0, 168.97, 161.0, 107.0, 101.0, 98.3, 94.9, 90.7, 83.4, 79.7, 76.4, 75.4, 72.6, 71.8, 71.5, 70.8, 70.6, 70.2, 70.1, 69.8, 67.5, 67.4, 62.7, 62.6, 21.0, 20.858, 20.850, 20.84, 20.6, 20.56, 20.52, 20.50, 20.4, 17.5, 15.9; IR (neat) 3337, 2937, 2361, 2345, 1750, 1675, 1437, 1373, 1223, 1052, 932; HRMS (ESI-TOF) m/e: [M-C(NH)OCCl3+OMe+Na]+ calcd. for C41H58NaO26 989.3114; found 989.3117.
2,3,4-tri-O-acetyl-β-d-xylopyranosyl-(1→3)-2,4-di-O-acetyl-β-d-xylopyran-osyl-(1→4)-2,3-di-O-acetyl-α-l-rhamnopyranosyl-(1→2)-3,4-di-O-acetyl-a-d-fucopyranosyl trichloroacetimidate (4b)
The tetrasaccharide product 13b (397 mg, 0.356 mmol) in 15 mL of methanol was treated with 1 mL of methanol solution of sodium methoxide (25% w/w). The reaction was stirred at room temperature overnight. Upon completion of the reaction, methanol was removed and the residue was treated with TFA/H2O (4:1) at room temperature until completion. The reaction mixture was concentrated and pump-dried overnight. The residue was treated with acetic anhydride (1 mL, 10 mmol), triethyl amine (3.5 mL, 26 mmol) and dimethylamino pyridine (8 mg, 6.5 μmol). The reaction completed in 3 h, and then the reaction mixture was concentrated and subjected to flash column chromatography (eluted with petroleum ether/ethyl acetate 1:1) to afford the desired tetrasaccharide intermediate S2 (320 mg, 90%). Rf 0.1 (petroleum ether/ethyl acetate 1:1); [α]D23 = -13 (c = 1.61, CH2Cl2); 1H NMR (400 MHz, CDCl3) δ 5.9 (dddd, J =17.0, 10.4, 6.4, 5.1 Hz, 1H), 5.37-5.25 (m, 4 H), 5.19 (dd, J =9.6, 3.4 Hz, 1 H), 5.1-5.02 (m, 2 H), 4.95-4.82 (m, 5 H), 4.6 (d, J =6 Hz, 1 H), 4.54 (d, J =7.5 Hz, 1 H), 4.25-3.95 (m, 6 H), 3.81 (m, 1 H), 3.76 (t, J =8.9 Hz, 1 H), 3.65 (t, J =9.6 Hz, 1 H), 3.4 (dd, J =12.1, 7.4 Hz, 1 H), 3.31 (dd, J = 12.0, 9.2 Hz, 1 H), 2.14 (s, 3 H), 2.12 (s, 3 H), 2.09 (s, 3 H), 2.08 (s, 3 H), 2.07 (s, 3 H), 2.064 (s, 3 H), 2.060 (s, 3 H), 2.05 (s, 3 H), 2.03 (s, 3 H), 2.0 (s, 3 H), 1.26 (d, J =6.2 Hz, 3 H), 1.12 (d, J =6.6 Hz, 3 H); 13C NMR (100 MHz, CDCl3) δ 170.3, 169.9, 169.8, 169.78, 169.72, 169.6, 169.2, 169.1, 168.8, 133.2, 118.2, 100.7, 100.5, 98.5, 96.9, 78.7, 75.5, 73.3, 71.9, 71.4, 70.9, 70.3, 70.2, 69.8, 69.5, 69.4, 68.6, 68.5,67.2, 64.3, 62.4, 61.4, 20.79, 20.77, 20.72, 20.6, 20.53, 20.50, 20.46, 20.43, 20.40, 17.5, 15.7; IR (neat) 2983, 2939, 2876, 1750, 1634, 1431, 1372, 1223; HRMS (ESI-TOF) m/e: [M+H]+ calcd. for C43H61O26 993.3451; found 993.3448. A solution of [Ir(COD)(PMePh2)2]PF6 (4.6 mg, 5.4 μmol) in THF was degassed and stirred under hydrogen atmosphere for 15 min at room temperature. The obtained clear solution of the activated catalyst was added to the tetrasaccharide S2 (135 mg, 0.136 mmol) in 5 mL of THF. After 2 h, the isomerization reaction was complete, and drops of water and NIS (59 mg, 0.2 mmol) were added. The deallylation reaction completed instantly. The reaction mixture was concentrated and purified with flash column chromatography (eluted with dichloromethane/methanol 20:1, Rf 0.4). The obtained hemiacetal was treated with trichloroacetonitrile (1.2 mL, 12 mmol) and cesium carbonate (90 mg, 0.28 mmol) in dichloromethane (2 mL) at room temperature overnight. The reaction mixture was then concentrated and subjected to flash column chromatography (eluted with methanol/dichloromethane) to afford the imidate 4b (141 mg, 94%) as a white amorphous solid. Rf = 0.7 (dichloromethane/methanol 20:1); [α]D24 = -20.4 (c = 0.7, CHCl3); 1H NMR (400 MHz, CDCl3) δ 8.76 (s, 1 H), 6.48 (d, J =3.7 Hz, 1 H), 5.3 (dd, J =3.2, 0.9 Hz, 1 H), 5.32 (ABM, J =11.0, 3.2 Hz, 1 H), 5.31 (s, 1 H), 5.1 (dd, J =9.7, 3.4 Hz, 1 H), 5.05 (t, J =7.6 Hz, 1 H), 4.96 (dd, J =3.4, 1.8 Hz, 1 H), 4.95-4.85 (m, 4 H), 4.78 (dd, J =7.8, 6.0 Hz, 1 H), 4.6 (d, J =5.9 Hz, 1 H), 4.46 (d, J =7.8 Hz, 1 H), 4.35 (q, J =7.2 Hz, 1 H), 4.21 (dd, J =10.4, 3.7 Hz, 1 H), 4.11 (dd, J =11.9, 4.3 Hz, 1 H), 4.05 (dd, J = 11.8, 5.6 Hz, 1 H), 3.9 (dq, J =9.4, 6.1 Hz, 1 H), 3.73 (t, J =9.2 Hz, 1 H), 3.62 (t, J =9.6 Hz, 1 H), 3.4 (dd, J =12.2, 7.2 Hz, 1 H), 3.26 (dd, J =11.8, 9.7 Hz, 1 H), 2.2 (s, 3 H), 2.1 (s, 3 H), 2.06-2.03 (m, 21 H), 1.28 (d, J =6.3 Hz, 3 H), 1.16 (d, J =6.5 Hz, 3 H); 13C NMR (100 MHz, CDCl3) δ 170.3, 170.1, 170.0, 169.86, 169.82, 169.6, 169.3, 169.1, 168.8, 160.9, 101.0, 100.5, 98.2, 94.9, 90.8, 79.0, 75.4, 72.3, 71.3, 70.8, 70.6, 70.3, 70.22, 70.16, 69.8, 69.5, 68.6, 67.5, 67.4, 62.6, 61.4, 60.3, 53.4, 20.84, 20.80, 20.7, 20.63, 20.61, 20.56, 20.5, 20.4, 17.5, 15.9; IR (neat) 3156, 2977, 2931, 2867, 1755; HRMS (ESI-TOF) m/e: [M-C(NH)OCCl3+OMe+Na]+ calcd. for C41H58NaO26 989.3114; found 989.3121.
2,3,4-tri-O-acetyl-β-d-xylopyranosyl-(1→4)-2,3-di-O-acetyl-α-l-rhamno-pyranosyl-(1→2)-3,4-di-O-acetyl-α-d-fucopyranosyl trichloroacetimidate (4c)
The tetrasaccharide product 13c (90 mg, 0 11 mmol) in 2 mL of methanol was treated with 0.2 mL of methanol solution of sodium methoxide (25% w/w). The reaction was stirred at room temperature overnight. Upon completion of the reaction, methanol was removed and the residue was treated with TFA/H2O (4:1) at 0 °C until completion. The reaction mixture was concentrated and pump-dried. The residue was treated with acetic anhydride (0.4 mL, 4 mmol), triethyl amine (1.2 mL, 9 mmol) and dimethylamino pyridine (3 mg, 2.4 μmol). The reaction completed in 3 h, and then the reaction mixture was concentrated and subjected to flash column chromatography (eluted with petroleum ether/ethyl acetate 1:1) to afford the desired tetrasaccharide intermediate S3 (79 mg, 92%) as colorless oil. Rf 0.4 (petroleum ether/ethyl acetate 1:1). A solution of [Ir(COD)(PMePh2)2]PF6 (4 mg, 4.7 μmol) in 3.0 mL of THF was degassed and stirred under hydrogen atmosphere for 15 min at room temperature. The obtained clear solution of the activated catalyst was injected to S3 (72 mg, 0.0927 mmol). The reaction mixture was stirred for 80 min, and drops of water, NIS (23 mg, 0.1 mmol) was added at room temperature. The reaction was stirred until there was no starting material left. The solution was then concentrated and purified with flash column chromatography on silica gel (eluted with petroleum ether/ethyl acetate gradients 50%-85% EtOAc) to afford the intermediate hemiacetal. Rf 0.3 (petroleum ether/ethyl acetate gradients 1:2). The obtained hemiacetal was treated with trichloroacetonitrile (0.5 mL, 5 mmol) and cesium carbonate (37 mg, 0.113 mmol) in 2.5 mL of dichloromethane at room temperature overnight. The reaction mixture was then concentrated and purified with flash column chromatography (eluted with petroleum ether/ethyl acetate gradients 50%-85% EtOAc) to afford the imidate 4c (65 mg, 80%) as a white amorphous solid. Rf 0.5 (petroleum ether/ethyl acetate 1:2); [α]D23 = +5.2 (c = 1.95, CHCl3); 1H NMR (300 MHz, CDCl3) δ 8.71 (s, 1H), 6.47 (d, J =3.7 Hz, 1H), 5.39 (d, J =2.4 Hz, 1H), 5.32 (dd, J =10.5, 3.2 Hz, 1H), 5.18 – 5.05 (m, 2H), 5.00 – 4.83 (m, 4H), 4.57 (d, J =7.7 Hz, 1H), 4.34 (q, J =6.9 Hz, 1H), 4.20 (dd, J =10.4, 3.8 Hz, 1H), 4.11 (dd, J =11.7, 5.4 Hz, 1H), 3.84 (m, 1H), 3.64 (t, J =9.6 Hz, 1H), 3.31 (dd, J =11.7, 10.0 Hz, 1H), 2.16 (s, 3H), 2.12 (s, 3H), 2.06 (s, 3H), 2.04 (d, J =2.2 Hz, 6H), 2.01 (s, 3H), 1.98 (s, 3H), 1.30 (d, J =6.2 Hz, 3H), 1.16 (d, J =6.5 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 170.3, 170.2, 170.1, 169.9, 169.8, 169.3, 161.2, 101.0, 98.5, 95.1, 90.8, 77.2, 75.9, 72.1, 71.8, 71.0, 70.8, 70.6, 70.3, 70.1, 69.3, 67.6, 67.5, 62.5, 29.7, 20.9, 20.66, 20.63, 20.60, 20.54, 20.4, 17.5, 16.0; IR (neat): 2927, 1751, 1675, 1370, 1242, 1221. HRMS (ESI-TOF) m/e: [M+Na]+ calcd. for C33H44 Cl3NNaO20 902.1420, found 902.1423.
14a
The α-imidate 4a (43 mg, 0.04 mmol) was dried by azeotropic removal of water with toluene. The quillaic acid acceptor 3 (68 mg, 0.033 mmol) was then added, followed by 2.0 mL of DCM. The solution was then cooled in a dry ice-acetone bath. To the cooled solution was added a solution of boron trifluoride diethyl etherate (5 μL, 0.04 mmol) in 50 μL of DCM. The reaction solution was stirred at -78 °C for 5 h before quenched with triethyl amine. The reaction mixture was concentrated and purified with flash column chromatography on silica gel (eluted with petroleum ether/ethyl acetate gradients: 0% EtOAc to 50% EtOAc) to afford the desired conjugate 14a (56 mg, 57%) as a white amorphous solid. Rf = 0.4 (petroleum ether/ethyl acetate 1:1); [α]D23 = -21.0 (c = 1.25, CHCl3); Characteristic protons: 1H NMR (400 MHz, CDCl3) δ 9.7 (s, 1 H), 7.35-7.31 (m, 5 H), 5.4 (d, J =7.8 Hz, 1 H), 5.3-5.25 (m, 2 H), 5.21 (d, J =3.0 Hz, 1 H), 5.15 (s, 1 H), 5.13-4.8 (m, 8 H), 4.61 (AB1, J =12.5 Hz, 1 H), 4.57 (AB2, J =13.4 Hz, 1 H), 4.56 (s, 1 H), 4.52 (AB1, J =12.4 Hz, 1 H), 4.47 (s, 1 H), 4.42 (d, J =7.1 Hz, 1 H), 4.24-4.16 (m, 2 H), 4.1-4.0 (m, 2 H), 3.96-3.71 (m, 10 H), 3.68 (t, J =8.8 Hz, 1 H), 3.65-3.54 (m, 4 H), 3.46 (m, 1 H), 3.42-3.22 (m, 5 H), 3.12 (t, J =11.0 Hz, 1 H), 2.83 (dd, J =11.3, 2.4 Hz, 1 H), 2.25 (t, J =14.0 Hz, 1 H), 2.2 (s, 3 H), 2.1 (s, 3 H); 13C NMR (100 MHz, CDCl3) δ 212.6, 175.8, 170.52, 170.48, 170.3, 169.9, 169.7, 169.33, 169.31, 169.2, 168.5, 143.6, 135.4, 128.6, 128.4, 128.25, 128.2, 121.7, 107.0, 103.6, 101.5, 100.95, 100.89, 98.5, 94.0, 86.2, 83.57, 83.56, 79.3, 78.9, 78.8, 77.3, 76.6, 76.5, 76.3, 76.1, 75.9, 75.7, 75.2, 72.8, 72.7, 72.6, 71.8, 71.6, 71.5, 71.4, 71.2, 70.4, 70.2, 69.9, 69.3, 68.3, 66.9, 65.4, 64.5, 62.9, 62.6, 60.4, 54.0, 49.6, 49.5, 46.9, 46.3, 41.8, 41.1, 40.0, 38.2, 36.2, 35.2, 34.6, 32.9, 32.6, 30.7, 30.6, 29.8, 26.5, 25.5, 24.5, 23.6, 21.16, 21.11, 21.07, 21.02, 21.01, 20.82, 20.79, 20.70, 20.62, 20.60, 20.4, 19.23, 19.17, 17.73, 17.66, 16.03, 16.02, 13.8, 12.3, 7.65, 7.55, 7.3, 7.253, 7.246, 7.2, 7.1, 6.994, 6.88, 6.0, 5.7, 5.55, 5.47, 5.44, 5.36, 5.33, 5.07, 4.5; IR (neat) 2954, 2877, 1751, 1457, 1374, 1225; MS (MALDI) m/e: [M+Na]+ calcd. for C148H258NaO45Si9 3030.5721 (62.5%), 3031.5717 (28.5%), 3031.5755 (100.0%), 3032.5690 (18.8%), 3032.5750 (45.7%), 3032.5788 (79.5%), 3033.5685 (7.6%), 3033.5723 (30.1%), 3033.5746 (9.3%), 3033.5784 (36.3%), 3033.5797 (9.2%), 3033.5822 (41.9%), 3034.5719 (12.2%), 3034.5757 (23.9%), 3034.5780 (7.4%), 3034.5818 (19.1%), 3034.5831 (7.4%), 3034.5855 (16.4%), 3035.5752 (9.7%), 3035.5790 (12.6%), 3035.5851 (7.5%), found 3030.611, 3031.628, 3032.628, 3033.629, 3034.616, 3035.645.
14b
The α-imidate 4b (135 mg, 0.123 mmol) was dried by azeotropic removal of water with toluene. The quillaic acid acceptor 3 (222 mg, 0.106 mmol) was then added, followed by 3.0 mL of DCM. The solution was then cooled in a dry ice-acetone bath. To the cooled solution was added boron trifluoride diethyl etherate (16 μL, 0.123 mmol). The reaction solution was stirred at -78 °C for 7 h before quenched with triethyl amine. The reaction mixture was concentrated and taken into petroleum ether and water. The aqueous layer was concentrated and purified with flash column chromatography on silica gel (eluted with petroleum ether/ethyl acetate 1:1) to afford the desired conjugate 14b (232 mg, 72%) as a white amorphous solid. Rf = 0.4 (petroleum ether/ethyl acetate 1:1); [α]D23 = -22.3 (c = 0.79, CHCl3); Characteristic protons: 1H NMR (400 MHz, CDCl3) δ 9.7 (s, 1 H), 7.35-7.31 (m, 5 H), 5.4 (d, J =7.8 Hz, 1 H), 5.32-5.26 (m, 2 H), 5.2 (d, J =3.4 Hz, 1 H), 5.13-4.98 (m, 4 H), 4.94-4.84 (m, 3 H), 4.80 (dd, J =7.7, 5.9 Hz, 1 H), 4.58 (d, J =5.8 Hz, 1 H), 4.56 (d, J =7.4 Hz, 1 H), 4.48 (s, 1 H), 4.42 (d, J =7.2 Hz, 1 H), 4.18 (d, J =7.2 Hz, 1 H), 4.12 (dd, J =12.1, 4.4 Hz, 1 H), 4.04 (dd, J =11.5, 5.5 Hz, 1 H), 3.97-3.71 (m, 9 H), 3.63-3.52 (m, 3 H), 3.48 (m, 1 H), 3.42-3.22 (m, 5 H), 3.12 (t, J =11.0 Hz, 1 H), 2.83 (dd, J =11.2, 3.2 Hz, 1 H); 13C NMR (100 MHz, CDCl3) δ 212.9, 176.0, 170.8, 170.62, 170.60, 170.5, 170.3, 170.1, 169.8, 169.6, 169.4, 168.8, 143.9, 135.7, 128.87, 128.86, 128.85, 128.6, 128.52, 128.51, 122.0, 103.9, 101.8, 101.2, 101.1, 101.0, 98.7, 94.3, 86.6, 79.2, 79.1, 79.08, 77.64, 77.59, 77.4, 76.96, 76.8, 76.5, 76.4, 76.2, 75.9, 75.5, 73.0, 72.9, 72.4, 72.0, 71.82, 71.79, 71.5, 70.72, 70.68, 70.4, 70.2, 70.0, 69.7, 69.1, 68.6, 67.2, 65.7, 62.8, 61.9, 60.7, 54.3, 49.9, 49.8, 47.2, 46.6, 42.1, 41.4, 40.3, 38.5, 36.5, 35.5, 34.9, 33.2, 32.9, 31.0, 30.9, 26.7, 25.8, 24.8, 23.8, 21.4, 21.3, 21.2, 21.12, 21.10, 21.0, 20.96, 20.9, 20.7, 18.1, 17.9, 16.33, 16.31, 12.6, 7.95, 7.85, 7.6, 7.55, 7.54, 7.53, 7.52, 7.4, 7.24, 7.18, 6.3, 6.0, 5.84, 5.76, 5.73, 5.66, 5.63, 5.4, 4.8; IR (neat) 2950, 2886, 1755, 1460, 1375, 1247, 1226; MS (MALDI) m/e: [M+Na]+ calcd. for C148H258NaO45Si9 3030.5721 (62.5%), 3031.5717 (28.5%), 3031.5755 (100.0%), 3032.5690 (18.8%), 3032.5750 (45.7%), 3032.5788 (79.5%), 3033.5685 (7.6%), 3033.5723 (30.1%), 3033.5746 (9.3%), 3033.5784 (36.3%), 3033.5797 (9.2%), 3033.5822 (41.9%), 3034.5719 (12.2%), 3034.5757 (23.9%), 3034.5780 (7.4%), 3034.5818 (19.1%), 3034.5831 (7.4%), 3034.5855 (16.4%), 3035.5752 (9.7%), 3035.5790 (12.6%), 3035.5851 (7.5%); found 3030.602, 3031.611, 3032.605, 3033.613, 3034.619, 3035.619.
14c
The α-imidate 4c (53 mg, 0.06 mmol) and the quillaic acid acceptor 3 (104 mg, 0.05 mmol) were dried by azeotropic removal of water with toluene. DCM (2 mL) was added and the solution was then cooled in a dry ice-acetone bath. To the cooled solution was added boron trifluoride diethyl etherate (7.5 μL, 0.06 mmol). The reaction solution was stirred at -78 °C for 5 h before quenched with triethyl amine. The reaction mixture was concentrated and purified with flash column chromatography on silica gel (eluted with petroleum ether/ethyl acetate gradients: 25%-50% EtOAc) to afford the desired conjugate 14c (84 mg, 60%) as a white amorphous solid. Rf = 0.7 (petroleum ether/ethyl acetate 1:1); [α]D23 = -16.9 (c = 1.17, CHCl3); Characteristic protons: 1H NMR (400 MHz, CDCl3) δ 9.7 (s, 1 H), 7.36-7.34 (m, 5 H), 5.46 (d, J =7.9 Hz, 1 H), 5.32-5.28 (m, 2 H), 5.2 (d, J =3.7 Hz, 1 H), 5.15-5.06 (m, 4 H), 5.03 (dd, J =10.1, 3.4 Hz, 1 H), 4.98 (td, J =10.1, 5.4 Hz, 1 H), 4.90-4.85 (m, 2 H), 4.66 (d, J =7.5 Hz, 1 H), 4.58 (d, J =7.5 Hz, 1 H), 4.49 (s, broad, 1 H), 4.44 (d, J =7.3 Hz, 1 H), 4.2 (d, J =7.2 Hz, 1 H), 4.13 (dd, J =11.6, 5.2 Hz, 1 H), 3.97-3.74 (m, 11 H), 3.64-3.54 (m, 4 H), 3.49 (m, 1 H), 3.42-3.32 (m, 4 H), 3.26 (t, J =8.0 Hz, 1 H), 3.15 (t, J =11.0 Hz, 1 H), 2.85 (dd, J =13.6, 3.3 Hz, 1 H); 2.27 (t, J =13.1 Hz, 1 H), 2.19 (s, 3 H), 2.13 (s, 3 H), 2.09 (s, 3 H), 2.07 (s, 3 H), 2.05 (s, 3 H), 2.03 (s, 3 H), 1.99 (s, 3 H); 13C NMR (100 MHz, CDCl3) δ 212.5, 175.7, 170.35, 170.28, 170.23, 170.18; 169.7, 169.4, 169.3, 168.4, 143.4, 135.3, 128.4, 128.2, 128.1, 121.6, 103.5, 101.4, 100.8, 98.3, 93.9, 86.1, 78.8, 78.7, 76.8, 76.5, 76.4, 76.05, 75.98, 75.79, 75.1, 72.6, 72.5, 72.0, 71.6, 71.4, 71.1, 71.0, 70.9, 70.2, 70.0, 69.3, 69.1, 68.1, 66.8, 65.3, 62.4, 60.2, 58.5, 53.8, 49.4, 49.3, 46.8, 46.1, 41.7, 40.9, 39.9, 38.1, 36.0, 35.1, 34.5, 32.7, 32.5, 30.6, 30.4, 29.7, 27.5, 26.3, 25.3, 24.4, 23.4, 20.94,20.88, 20.68, 20.66, 20.5, 20.4, 20.2, 19.4, 17.6, 17.5, 15.89, 15.87, 12.2, 8.2, 7.5, 7.4, 7.2, 7.11, 7.09, 6.9, 6.8, 6.7, 5.9, 5.6, 5.41, 5.33, 5.30, 5.23, 5.19, 5.0, 4.9, 4.4; IR (neat) 2954, 2877, 1751, 1457, 1374, 1224, 1090; MS (MALDI) m/e: [M+Na]+ calcd. for C139H246NaO39Si9 2814.5087 (66.5%), 2815.5083 (30.4%), 2815.5121 (100.0%), 2816.5056 (20.0%), 2816.5117 (45.7%), 2816.5154 (74.6%), 2817.5089 (30.1%), 2817.5150 (34.1%), 2817.5188 (36.9%), 2818.5085 (12.2%), 2818.5123 (22.5%), 2818.5184 (16.8%), 2818.5222 (13.6%), 2819.5156 (11.1%), found 2814.361, 2815.399, 2816.401, 2817.400, 2818.419, 2819.421.
15a
The compound 14a (84 mg, 0.028 mmol) with Pd/C (24 mg, 10% w/w, 0.02 mmol) in 3.0 mL of THF were under hydrogen at 55 psi for 21 hours. The reaction mixture was filtered through a celite plug and concentrated. The residue was treated with dodecylamine (15 mg, 0.084 mmol), HATU (32 mg, 0.084 mmol) and N,N-diisopropylethylamine (12 μL, 0.07 mmol) in 3.0 mL of chloroform at room temperature for 2 h. The reaction mixture was then concentrated and purified directly with column chromatography on silica gel (eluted with petroleum ether/ethyl acetate gradients: 25%-40% EtOAc) to afford the amide 15a (74 mg, 86%) as a white amorphous solid. Rf = 0.53 (petroleum ether/ethyl acetate 1:1); [α]D23 = -27.7 (c = 0.94, CHCl3); Characteristic protons: 1H NMR (400 MHz, CDCl3) δ 9.7 (s, 1 H), 6.14 (t, J =5.7 Hz, 1 H), 5.46 (d, J =7.8 Hz, 1 H), 5.28 (s, broad, 1 H), 5.21 (d, J =3.5 Hz, 1 H), 5.15 (s, 1 H), 5.11 (m, 1 H), 5.06 (dd, J =9.8, 3.3 Hz, 1 H), 5.01 (dd, J =10.0, 3.3 Hz, 1 H), 4.98 (s, 1 H), 4.96-4.85 (m, 3 H), 4.62 (d, J =12.4 Hz, 1 H), 4.57 (d, J =7.4 Hz, 1 H), 4.54 (s, 1 H), 4.51 (d, J =3.6 Hz, 1 H), 4.49 (s, 1 H), 4.43 (d, J =7.2 Hz, 1 H), 4.26 (d, J =7.5 Hz, 1 H), 4.21 (AB, J =10.4 Hz, 1 H), 4.08 (AB, J =10.4 Hz, 1 H), 4.04 (m, 1 H), 3.93 (s, 1 H), 3.91 (t, J =8.1 Hz, 1 H), 2.16 (s, 3 H), 2.11 (s, 3 H), 2.09 (s, 3 H), 2.05 (s, 3 H), 2.04 (s, 3 H), 2.01 (s, 3 H); 13C NMR (100 MHz, CDCl3) δ 212.1, 175.7, 170.53, 170.47, 170.34, 170.32, 169.9, 169.8, 169.34, 169.32, 169.2, 168.3, 143.5, 121.7, 102.9, 101.53, 101.46, 100.9, 98.6, 94.0, 86.0, 83.6, 79.7, 79.4, 78.9, 77.6, 77.4, 77.3, 76.6, 76.5, 76.4, 76.3, 75.91, 75.87, 75.7, 75.2, 72.8, 72.7, 72.6, 71.8, 71.6, 71.51, 71.45, 70.4, 70.1, 70.0, 69.4, 68.3, 65.5, 62.9, 62.6, 60.6, 54.1, 49.6, 49.4, 46.9, 46.3, 41.8, 41.0, 40.0, 39.3, 38.2, 36.2, 35.3, 34.6, 32.9, 32.6, 32.0, 30.8, 30.6, 29.81, 29.76, 29.73, 29.72, 29.7, 29.5, 29.4, 27.1, 26.5, 25.5, 24.5, 23.5, 22.8, 21.17, 21.06, 21.03, 21.02, 20.8, 20.7, 20.63, 20.60, 20.4, 17.7, 17.6, 16.0, 14.2, 12.2, 7.7, 7.6, 7.35, 7.30, 7.26, 7.23, 7.14, 7.05, 6.95, 6.9, 6.0, 5.77, 5.68, 5.64, 5.55, 5.46, 5.39, 5.35, 5.1, 4.6; IR (neat) 2954, 2877, 1752, 1459, 1374, 1225, 1077, 1009; MS (MALDI) m/e: [M+K]+ calcd. for C153H277KNO44Si9 3123.7029 (60.4%), 3124.7025 (27.6%), 3124.7063 (100.0%), 3125.6997 (18.2%), 3125.7058 (45.7%), 3125.7096 (82.2%), 3126.7031 (30.1%), 3126.7092 (37.6%), 3126.7130 (44.8%), 3127.7027 (12.2%), 3127.7065 (24.8%), 3127.7125 (20.5%), 3127.7163 (18.2%), 3128.7098 (13.5%), 3128.7060 (10.1%), found 3123.608, 3124.604, 3125.604, 3126.603, 3127.607, 3128.605.
15b
The compound 14b (40 mg, 0.013 mmol) with Pd/C (16 mg, 10% w/w, 0.015 mmol) in 3.0 mL of THF were under hydrogen at 55 psi for 23 hours. The reaction mixture was filtered through a celite plug and concentrated. The residue was treated with dodecylamine (7.5 mg, 0.04 mmol), HATU (7 mg, 0.018 mmol) and N,N-diisopropylethylamine (6 μL, 0.03 mmol) in 1.0 mL of chloroform at room temperature overnight. The reaction mixture was then concentrated and purified directly with column chromatography on silica gel (eluted with petroleum ether/ethyl acetate gradients: 40%-50% EtOAc) to afford the amide 15b (39 mg, 95%) as a white amorphous solid. Rf = 0.6 (petroleum ether/ethyl acetate 1:1); [α]D22 = -27.4 (c = 5.0, CHCl3); Characteristic protons: 1H NMR (400 MHz, CDCl3) δ 9.69 (s, 1 H), 6.17 (t, J =5.4 Hz, 1 H), 5.4 (d, J =7.8 Hz, 1 H), 5.23 (s, broad, 1 H), 5.20 (d, J =3.3 Hz, 1 H), 5.1 (m, 1 H), 5.07-4.98 (m, 3 H), 4.92-4.85 (m, 4 H), 4.78 (dd, J =7.8, 5.9 Hz, 1 H), 4.59 (d, J =5.6 Hz, 1 H), 4.55 (d, J =7.3 Hz, 1 H), 4.52 (d, J =7.2 Hz, 1 H), 4.48 (s, 1 H), 4.42 (d, J =7.0 Hz, 1 H), 4.25 (d, J =7.6 Hz, 1 H), 4.11 (dd, J =12.1, 4.4 Hz, 1 H), 4.04 (dd, J =11.8, 5.1 Hz, 1 H), 3.93 (s, 1 H), 3.90 (t, J =8.0 Hz, 1 H), 3.83-3.70 (m, 6 H), 3.70-3.55 (m, 5 H), 3.49 (m, 1 H), 3.41-3.15 (m, 7 H), 3.12 (t, J =11.3 Hz, 1 H), 2.83 (dd, J =11.2, 3.2 Hz, 1 H), 2.16 (s, 3 H), 2.10 (s, 3 H), 2.07 (s, 3 H); 13C NMR (100 MHz, CDCl3) δ 212.0, 175.5, 170.3, 170.2, 170.0, 169.8, 169.6, 169.3, 169.2, 169.0, 168.1, 143.4, 121.5, 102.8, 101.4, 101.3, 100.7, 100.5, 98.3, 93.9, 85.9, 79.58, 79.56, 78.7, 78.6, 77.5, 77.2, 76.4, 76.2, 76.1, 75.74, 75.68, 75.4, 75.0, 72.6, 72.4, 71.9, 71.6, 71.4, 70.3, 70.2, 69.9, 69.8, 69.5, 69.2, 68.6, 68.1, 65.3, 62.4, 61.7, 61.5, 60.4, 53.9, 49.4, 49.2, 46.8, 46.1, 41.6, 40.9, 39.8, 39.2, 38.0, 36.0, 35.1, 34.4, 32.7, 32.4, 31.9, 30.6, 30.4, 29.65, 29.60, 29.57, 29.55, 29.54, 29.5, 29.31, 29.30, 29.27, 27.9, 27.3, 26.9, 26.3, 25.4, 24.3, 23.3, 22.6, 20.9, 20.8, 20.7, 20.6, 20.55, 20.51, 20.46, 20.3, 18.4, 17.6, 17.5, 15.86, 15.84, 14.1, 12.0, 8.1, 7.5, 7.4, 7.19, 7.163, 7.156, 7.148, 7.13, 7.09, 7.06, 7.03, 6.98, 6.8, 6.7, 5.8, 5.6, 5.51, 5.48, 5.38, 5.29, 5.22, 5.18, 4.9, 4.7, 4.4, 4.1; IR (neat) 2947, 1755, 1463, 1375, 1247, 1229; MS (MALDI) m/e: [M+Na]+ calcd. for C153H277 NNaO44Si9 3107.7290 (60.4%), 3108.7285 (27.6%), 3108.7323 (100.0%), 3109.7258 (18.2%), 3109.7319 (45.7%), 3109.7357 (82.2%), 3110.7292 (30.1%), 3110.7352 (37.6%), 3110.7390 (44.8%), 3111.7287 (12.2%), 3111.7325 (24.8%), 3111.7386 (20.5%), 3111.7424 (18.2%), 3112.7321 (10.1%), 3112.7359 (13.5%), found 3107.678, 3108.727, 3109.704, 3110.710, 3111.711, 3112.732.
15c
The compound 14c (55 mg, 0.0197 mmol) with Pd/C (12 mg, 10% w/w, 0.011 mmol) in 2.5 mL of THF under hydrogen at 55 psi for 24 hours. The reaction mixture was filtered through a celite plug and concentrated. The residue was treated with dodecylamine (11 mg, 0.06 mmol), HATU (23 mg, 0.06 mmol) and N,N-diisopropylethylamine (8.5 μL, 0.05 mmol) in 3.0 mL of chloroform at room temperature overnight. The reaction mixture was then concentrated and purified directly with column chromatography on silica gel (eluted with petroleum ether/ethyl acetate gradients: 25%-50% EtOAc) to afford the amide 15c (58 mg, 78%) as a white amorphous solid. Rf = 0.3 (petroleum ether/ethyl acetate 2:1); [α]D22 = -22.8 (c = 1.47, CHCl3); Characteristic protons: 1H NMR (400 MHz, CDCl3) δ 9.69 (s, 1 H), 6.11 (t, J =5.5 Hz, 1 H), 5.44 (d, J =7.9 Hz, 1 H), 5.28 (s, broad, 1 H), 5.20 (d, J =3.3 Hz, 1 H), 5.11- 5.09 (m, 2 H), 5.07 (dd, J =9.6, 3.3 Hz, 1 H), 5.0 (dd, J =10.1, 3.3 Hz, 1 H), 4.94 (td, J =9.2, 5.3 Hz, 1 H), 4.88-4.83 (m, 2 H), 4.64 (d, J =7.5 Hz, 1 H), 4.52 (d, J =7.3 Hz, 1 H), 4.48 (s, 1 H), 4.42 (d, J =7.2 Hz, 1 H), 4.25 (d, J =7.6 Hz, 1 H), 4.11 (dd, J =11.7, 5.5 Hz, 1 H), 3.93 (d, J =1.7 Hz, 1 H), 3.90 (t, J =7.4 Hz, 1 H), 3.83-3.78 (m, 5 H), 3.7 (t, J =7.4 Hz, 1 H), 3.64-3.57 (m, 5 H), 3.5 (m, 1 H), 3.4-3.2 (m, 7 H), 2.83 (dd, J =13.9, 3.2 Hz, 1 H), 2.25 (t, J =13.5 Hz, 1 H), 2.16 (s, 3 H), 2.11 (s, 3 H), 2.07 (s, 3 H), 2.05 (s, 3 H), 2.03 (s, 3 H), 2.01 (s, 3 H), 1.97 (s, 3 H); 13C NMR (176 MHz, CDCl3) δ 212.1, 175.6, 170.38, 170.32, 170.28, 170.21, 169.9, 169.5, 169.3, 168.2, 143.4, 121.6, 102.9, 101.4, 101.3, 100.9, 98.4, 93.9, 86.0, 79.6, 78.8, 77.5, 76.4, 76.2, 76.1, 76.0, 75.8, 75.7, 75.0, 72.6, 72.48, 72.46, 72.0, 71.7, 71.4, 71.2, 71.0, 70.3, 70.0; 69.3, 69.1, 68.2, 65.4, 62.4, 60.4, 58.6, 53.9, 49.5, 49.3, 46.8, 46.2, 41.7, 40.9, 39.9, 39.2, 38.1, 36.1, 35.1, 34.5, 32.8, 32.5, 31.9, 30.7, 30.5, 29.61, 29.60, 29.4, 29.3, 27.0, 26.4, 25.4, 24.4, 23.4, 22.7, 20.97, 20.92, 20.72, 20.70, 20.5, 20.4, 20.3, 17.6, 17.5, 15.92, 15.90, 14.1, 12.1, 8.3, 7.6, 7.5, 7.25, 7.16, 7.13, 7.0, 6.85, 6.80, 5.9, 5.8, 5.7, 5.4, 5.34, 5.27, 5.24, 5.0, 4.4; IR (neat) 2954, 2877, 1755, 1374, 1223, 1089; MS (MALDI) m/e: [M+Na]+ calcd. for C144H265NNaO38Si9 2891.6656 (64.2%), 2892.6651 (29.3%), 2892.6689 (100.0%), 2893.6624 (19.3%), 2893.6685 (45.7%), 2893.6723 (77.3%), 2894.6658 (30.1%), 2894.6718 (35.3%), 2894.6756 (39.6%), 2895.6653 (12.2%), 2895.6691 (23.3%), 2895.6752 (18.1%), 2895.6790 (15.1%), 2896.6725 (11.9%), found 2891.704, 2892.713, 2893.738, 2894.731, 2895.728, 2896.723.
2a
The fully protected compound 15a (58 mg, 0.0188 mmol) in 0.2 mL of dichloromethane was cooled in an ice bath. It was treated with 1.0 mL of trifluoroacetic acid in water (4:1, v/v) precooled in an ice bath. The reaction was stirred at 0 °C for 40 min and then the solution was concentrated to dryness at 0 °C. To the residue was added 5 drops of chloroform, 1.0 mL of MeOH, and K2CO3 (20 mg, 0.14 mmol). The reaction mixture was stirred at room temperature overnight. The white suspension was concentrated, and the residue was washed with water until pH ∼7. Lyophilization afforded the product 2a as a white powder (30 mg, 95%). Characteristic protons: 1H NMR (700 MHz, CDCl3) δ 9.49 (s, 1 H), 7.91(s, broad, 1 H), 5.25 (d, J =7.0 Hz, 1 H), 4.62 (d, J =6.3 Hz, 1 H), 4.48 (d, J =7.3 Hz, 1 H), 4.46 (d, J =2.1 Hz, 1 H), 4.39 (d, J =7.7 Hz, 1 H), 4.3 (s, broad, 1 H), 4.25 (d, J =7.2 Hz, 1 H), 2.85 (m, 1 H), 2.2 (m, 1 H); 13C NMR (176 MHz, DMSO) δ 210.1, 175.1, 167.3, 143.0, 121.3, 108.8, 105.0, 103.1, 102.5, 102.4, 102.2, 99.7, 93.2, 84.2, 83.7, 81.8, 81.5, 79.0, 76.8, 76.5, 75.9, 75.5, 74.8, 74.3, 74.1, 73.5, 73.4, 73.0, 72.5, 71.5, 71.2, 70.5, 70.2, 69.8, 69.4, 69.1, 68.0, 67.9, 67.0, 65.8, 65.7, 63.7, 59.8, 53.8, 48.5, 47.7, 47.6, 46.3, 45.9, 40.9, 40.5, 39.9, 38.1, 37.5, 35.4, 34.8, 34.6, 33.2, 32.7, 31.7, 31.21, 31.18, 30.4, 30.1, 29.3, 29.12, 29.06, 29.04, 28.96, 28.89, 28.86, 28.77, 28.74, 28.66, 28.58, 28.54, 28.3, 26.3, 26.1, 24.4, 24.3, 24.1, 22.7, 22.02, 21.98, 19.7, 17.8, 17.7, 16.6, 16.1, 15.3, 13.9, 10.3; HRMS (ESI-TOF) m/e: [M+Na]+ calcd. for C81H133NNaO35 1702.8556; found 1702.8528.
2b
The fully protected compound 15b (80 mg, 0.026 mmol) in 0.2 mL of dichloromethane was cooled in an ice bath. It was treated with 1.0 mL of trifluoroacetic acid in water (4:1, v/v) precooled in an ice bath. The reaction was stirred at 0 °C for 40 min and then the solution was concentrated to dryness at 0 °C. To the residue was added 5 drops of chloroform, 1.0 mL of MeOH, and K2CO3 (15 mg, 0.11 mmol). The reaction mixture was stirred at room temperature overnight. The white suspension was concentrated, and the residue was washed with water until pH ∼7. Lyophilization afforded the product 2b as a white powder (37 mg, 85%). Characteristic protons: 1H NMR (400 MHz, DMSO) δ 9.54 (s, 1 H), 7.91(s, broad, 1 H), 5.30 (d, J =7.2 Hz, 1 H), 5.26 (s, broad, 1 H), 5.19 (s, 1 H), 4.65 (t, J =3.3 Hz, 1 H), 4.7 (d, J =7.3 Hz, 1 H), 4.51 (d, J =7.9 Hz, 1 H), 4.48 (d, J =7.4 Hz, 1 H), 4.38 (s, broad, 1 H), 4.37 (d, J =7.2 Hz, 1 H), 2.9 (m, 1 H), 2.3 (m, 1 H); 13C NMR (176 MHz, DMSO) δ 210.9, 175.6, 167.9, 143.6, 121.8, 105.2, 104.8, 103.7, 103.0, 102.8, 102.3, 100.3, 93.7, 85.9, 84.8, 84.4, 82.7, 79.7, 77.4, 77.1, 76.6, 76.1, 75.3, 74.6, 74.20, 74.13, 74.09, 73.9, 73.6, 73.0, 72.0, 71.7, 71.1, 70.8, 70.1, 69.9, 69.8, 69.6, 68.6, 68.2, 67.5, 66.3, 66.2, 66.0, 60.3, 54.4, 48.3, 48.2, 46.8, 46.4, 41.4, 41.0, 40.9, 38.6, 38.1, 36.0, 35.3, 35.1, 33.3, 32.3, 31.81, 31.77, 31.1, 30.6, 29.74, 29.67, 29.65, 29.57, 29.47, 29.35, 29.27, 29.18, 29.1, 28.9, 26.8, 26.7, 25.0, 24.7, 23.3, 22.62, 22.58, 20.3, 18.4, 17.1, 16.7, 15.9, 14.5, 10.9; HRMS (ESI-TOF) m/e: [M+Na]+ calcd. for C81H133KNO35 1718.8290; found 1718.8269.
2c
The fully protected compound 15c (44 mg, 0.015 mmol) in 0.2 mL of dichloromethane was cooled in an ice bath. It was treated with 1.0 mL of trifluoroacetic acid in water (4:1, v/v) precooled in an ice bath. The reaction was stirred at 0 °C for 40 min and then the solution was concentrated to dryness at 0 °C. To the residue was added 5 drops of chloroform, 1.0 mL of MeOH, and K2CO3 (15 mg, 0.11 mmol). The reaction mixture was stirred at room temperature overnight. The white suspension was concentrated, and the residue was washed with water until pH ∼7. Lyophilization afforded the product 2c as a white powder (22 mg, 92%). Characteristic protons: 1H NMR (400 MHz, DMSO) δ 9.50 (s, 1 H), 7.92(m, 1 H), 5.22 (d, J =7.3 Hz, 1 H), 5.20 (s, 1 H), 4.48 (d, J =7.5 Hz, 1 H), 4.46 (s, 1 H), 4.35 (d, J =7.6 Hz, 1 H), 4.32 (s, 1 H), 4.25 (d, J =7.1 Hz, 1 H), 2.85 (m, 1 H), 2.24 (m, 1 H); 13C NMR (176 MHz, DMSO) δ 210.8, 175.5, 167.9, 143.7, 121.7, 105.66, 103.68, 103.0, 102.8, 100.2, 93.7, 84.8, 84.4, 82.5, 77.4, 77.0, 76.1, 75.3, 74.9, 74.8, 74.1, 73.9, 73.3, 73.0, 72.0, 71.8, 71.1, 70.9, 70.3, 69.9, 69.6, 69.5, 68.5, 67.5, 66.4, 66.3, 60.3, 60.2, 54.4, 48.4, 48.3, 46.9, 46.4, 41.5, 41.0, 40.5, 38.6, 38.5, 38.1, 36.0, 35.4, 35.1, 33.3, 32.3, 31.8, 31.1, 30.6, 29.73, 29.66, 29.64, 29.56, 29.45, 29.35, 29.26, 26.8, 26.7, 25.1, 24.6, 23.3, 22.6, 20.4, 18.4, 17.1, 16.7, 15.9, 14.5, 11.0; HRMS (ESI-TOF) m/e: [M+Na]+ calcd. for C76H125NNaO31 1570.8128; found 1570.8122.
Immunological evaluation of QS-21-based immune adjuvant 2b
Mice and immunization
BALB/c mice used in this study were purchased from Frederick Cancer Research (Fredrick, MD) and maintained within an environmentally controlled, pathogen-free animal facility at the University of Alabama at Birmingham (UAB). To assess the adjuvant activity of QS-21-based immune adjuvant 2b, groups of female mice (8-10 weeks of age; 6 mice per group) were immunized by the subcutaneous (s.c.) route with rHagB (20 μg) along or with GPI-0100 (100μg) or 2b (100 μg) on days 0, 14 and 28. Mice were weighed and blood samples were collected prior to and at various time points following the initial immunization. Blood samples were collected from the retro-orbital plexus by using heparinized capillary pipettes. The serum was obtained after centrifugation and stored at −20°C until assayed. All experiments were performed according to the National Institute of Health guidelines, and protocols were approved by the UAB Institutional Animal Care and Use Committee.
Elisa
The levels of specific serum IgG against rHagB in each group were determined by an enzyme-linked immunosorbent assay (ELISA). Maxisorp microtiter plates (NUNC International, Roskilde, Denmark) were coated with rHagB (1 μg/ml) or with goat anti-mouse IgG (Southern Biotechnology Associates, Inc., Birmingham, AL) in borate buffer saline (BBS; 100 mM NaCl, 50 mM boric acid, 1.2 mM Na2B4O7, pH 8.2) at 4°C overnight. Plates were blocked with 1% bovine serum albumin (BSA) and 0.02% sodium azide in BBS for 2 h at room temperature. Serial twofold dilutions of serum samples and a mouse immunoglobulin reference serum (MP Biomedicals, Solon, OH) were added in duplicate to the plates. After incubation (overnight at 4°C) and washing of the plates, horseradish peroxidaseconjugated goat anti-mouse IgG (Southern Biotechnology Associates, Inc.) was added to appropriate wells. After 4 h of incubation at room temperature, plates were washed and developed by o-phenylenediamine substrate with hydrogen peroxide. Color development was recorded at 490 nm. The concentrations of antibodies were determined by interpolation on standard curves generated by using the mouse immunoglobulin reference serum and constructed by a computer program based on four-parameter logistic algorithms (Softmax/Molecular Devices Corp., Menlo Park, CA).
Statistical analysis
Statistical significance in antibody responses and body weights between groups was evaluated by ANOVA and the Tukey multiple-comparisons test using the InStat program (Graph Pad Software, San Diego, CA). Differences were considered significant at a P value < 0.05.
Supplementary Material
Figure 2.
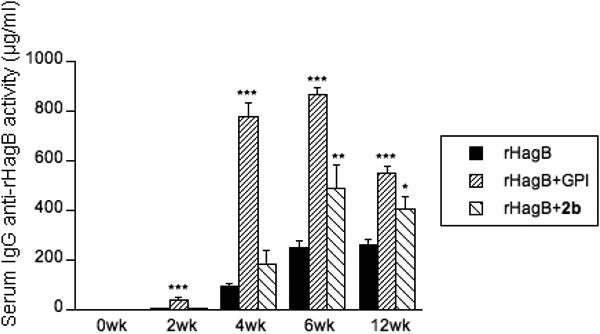
Serum IgG anti-rHagB response in mice immunized by the s.c. route with rHagB alone or with GPI-0100 or 2b. Mice were immunized on days 0, 14 and 28. Serum samples were collected prior to each immunization and at 6 and 12 weeks following the initial immunization. Values are expressed as mean ± SEM. * P < 0.05, ** P < 0.01 and *** P < 0.001 compared with mice immunized with rHagB alone.
Acknowledgments
This research was supported by the NIH (AI099407) and the UAB CAS Interdisciplinary Innovation Team Award. We also thank Dr. Michael J. Jablonsky for assistance with NMR spectroscopy, Dr. Jim Riordan for assistance with mass spectrometry, and F. Dong and X. Liang for their early contribution to the synthesis.
Footnotes
DEDICATION. Dedicated to the memory of Professor David Y. Gin.
Supporting Information Available: 1H and 13C NMR spectra of new compounds and 1H spectra of intermediates and known compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Ragupathi G, Gardner JR, Livingston PO, Gin DY. Expert Rev Vaccines. 2011;10:463. doi: 10.1586/erv.11.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brunner R, Jensen-Jarolim E, Pali-Scholl I. Immunol Lett. 2010;128:29. doi: 10.1016/j.imlet.2009.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kensil CR, Mo AX, Truneh A. Frontiers in Bioscience. 2004;9:2972. doi: 10.2741/1452. [DOI] [PubMed] [Google Scholar]
- 4.Leroux-Roels G. Vaccine. 2010;28(Suppl 3):C25. doi: 10.1016/j.vaccine.2010.07.021. [DOI] [PubMed] [Google Scholar]
- 5.Kensil CR, Liu G, Anderson C, Storey J. In: Vaccine Adjuvants: Immunological and Clinical Principles. Hackett CJ, Harn DAJ, editors. Humana Press Inc; Totowa, N. J.: 2005. p. 221. [Google Scholar]
- 6.Kensil CR, Patel U, Lennick M, Marciani D. J Immun. 1991;146:431. [PubMed] [Google Scholar]
- 7.Deng K, Adams MM, Damani P, Livingston PO, Ragupathi G, Gin DY. Angew Chem Int Ed. 2008;47:6395. doi: 10.1002/anie.200801885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang P, Kim YJ, Navarro-Villalobos M, Rohde BD, Gin DY. J Am Chem Soc. 2005;127:3256. doi: 10.1021/ja0422007. [DOI] [PubMed] [Google Scholar]
- 9.Ragupathi G, Damani P, Deng K, Adams MM, Hang J, George C, Livingston PO, Gin DY. Vaccine. 2010;28:4260. doi: 10.1016/j.vaccine.2010.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kensil CR. Critical Reviews in Therapeutic Drug Carrier Systems. 1996;13:1. [PubMed] [Google Scholar]
- 11.Marciani D, Press JB, Reynolds RC, Pathak AK, Pathak V, Gundy LE, Farmer JT, Koratich MS, May RD. Vaccine. 2000;18:3141. doi: 10.1016/s0264-410x(00)00118-3. [DOI] [PubMed] [Google Scholar]
- 12.Marciani DJ, Pathak AK, Reynolds RC, Seitz L, May RD. Inter Immunopharmacology. 2001;1:813. doi: 10.1016/s1567-5769(01)00016-9. [DOI] [PubMed] [Google Scholar]
- 13.Marciani DJ, Ptak RG, Voss TG, Reynolds RC, Pathak AK, Chamblin TL, Scholl DR, May RD. Inter Immunopharmacology. 2002;2:1703. doi: 10.1016/s1567-5769(02)00192-3. [DOI] [PubMed] [Google Scholar]
- 14.Marciani DJ, Reynolds RC, Pathak AK, Finley-Woodman K, May RD. Vaccine. 2003;21:3961. doi: 10.1016/s0264-410x(03)00298-6. [DOI] [PubMed] [Google Scholar]
- 15.Adams MM, Damani P, Perl NR, Won A, Hong F, Livingston PO, Ragupathi G, Gin DY. J Am Chem Soc. 2010;132:1939. doi: 10.1021/ja9082842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chea EK, Fernandez-Tejada A, Damani P, Adams MM, Gardner JR, Livingston PO, Ragupathi G, Gin DY. J Am Chem Soc. 2012;134:13448. doi: 10.1021/ja305121q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Soltysik S, Wu JY, Recchia J, Wheeler DA, Newman MJ, Coughlin RT, Kensil CR. Vaccine. 1995;13:1403. doi: 10.1016/0264-410x(95)00077-e. [DOI] [PubMed] [Google Scholar]
- 18.Liu G, Anderson C, Scaltreto H, Barbon J, Kensil CR. Vaccine. 2002;20:2808. doi: 10.1016/s0264-410x(02)00209-8. [DOI] [PubMed] [Google Scholar]
- 19.Marciani DJ, Pathak AK, Reynolds RC. Vaccine. 2002;20:3237. doi: 10.1016/s0264-410x(02)00298-0. [DOI] [PubMed] [Google Scholar]
- 20.Higuchi R, Tokimitsu Y, Fujioka T, Komori T, Kawasaki T, Oakenful DG. Phytochemistry. 1987;26:229. [Google Scholar]
- 21.Wang P, Haldar P, Wang Y, Hu H. J Org Chem. 2007;72:5870. doi: 10.1021/jo070512x. [DOI] [PubMed] [Google Scholar]
- 22.Wang Y, Liang X, Wang P. Tetrahedron Lett. 2011;52:3912. [Google Scholar]
- 23.Wang Y, Zhang X, Wang P. Org Biomol Chem. 2010;8:4322. doi: 10.1039/c002865g. [DOI] [PubMed] [Google Scholar]
- 24.Yang H, Wang P. J Org Chem. 2013;78:1858. doi: 10.1021/jo301664c. [DOI] [PubMed] [Google Scholar]
- 25.Mahajan R, Dixit S, Khare NK, Khare A. J Carb Chem. 1994;13:63. [Google Scholar]
- 26.Liu M, Yu B, Wu X, Hui Y, Fung KP. Carbohydr Res. 2000;329:745. doi: 10.1016/s0008-6215(00)00244-5. [DOI] [PubMed] [Google Scholar]
- 27.Baudry D, Ephritikhine M, Felkin HJ. Chem Commun. 1978:694. [Google Scholar]
- 28.Oltyoort JJ, Boeckel V, A CA, De Koning JH, Van Boom JH. Synthesis. 1981:305. [Google Scholar]
- 29.Harreus A, Kunz H. Lieb Ann Chem. 1986:717. [Google Scholar]
- 30.Kunz H, Sager W. Helv Chim Acta. 1985;68:283. [Google Scholar]
- 31.Carpino L. J Am Chem Soc. 1993;115:4397. [Google Scholar]
- 32.Pillion DJ, Amsden JA, Kensil CR, Recchia J. J Pharm Sci. 1996;85:518. doi: 10.1021/js9504651. [DOI] [PubMed] [Google Scholar]
- 33.Gaddis DE, Maynard CL, Weaver CT, Michalek SM, Katz J. J Leukoc Biol. 2013;93:21. doi: 10.1189/jlb.0512220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gaddis DE, Michalek SM, Katz J. Mol Immunol. 2009;46:2493. doi: 10.1016/j.molimm.2009.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gaddis DE, Michalek SM, Katz J. J Immunol. 2011;186:5772. doi: 10.4049/jimmunol.1003192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yang QB, Martin M, Michalek SM, Katz J. Infect Immun. 2002;70:3557. doi: 10.1128/IAI.70.7.3557-3565.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang P, Martin M, Michalek SM, Katz J. Infect Immun. 2005;73:3990. doi: 10.1128/IAI.73.7.3990-3998.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang P, Martin M, Yang QB, Michalek SM, Katz J. Infect Immun. 2004;72:637. doi: 10.1128/IAI.72.2.637-644.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang P, Yang QB, Marciani DJ, Martin M, Clements JD, Michalek SM, Katz J. Vaccine. 2003;21:4459. doi: 10.1016/s0264-410x(03)00438-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.