

Abstract
Non-alcoholic fatty liver disease (NAFLD) is one of the major health concerns worldwide. Farnesoid X receptor (FXR) is considered a therapeutic target for treatment of NAFLD. However, the mechanism by which activation of FXR lowers hepatic triglyceride (TG) levels remains unknown. Here we investigated the role of hepatic carboxylesterase 1 (CES1) in regulating both normal and FXR-controlled lipid homeostasis. Over-expression of hepatic CES1 lowered hepatic TG and plasma glucose levels in both wild-type and diabetic mice. In contrast, knockdown of hepatic CES1 increased hepatic TG and plasma cholesterol levels. These effects likely resulted from the TG hydrolase activity of CES1, with subsequent changes in fatty acid oxidation and/or de novo lipogenesis. Activation of FXR induced hepatic CES1, and reduced the levels of hepatic and plasma TG as well as plasma cholesterol in a CES1-dependent manner. Therefore, hepatic CES1 plays a critical role in regulating both lipid and carbohydrate metabolism and FXR-controlled lipid homeostasis.
Keywords: Carboxylesterase 1, FXR, triglycerides, cholesterol, glucose, NAFLD
Introduction
Nonalcoholic fatty liver disease (NAFLD), one of the most common liver diseases worldwide, encompasses a spectrum of liver disorders ranging from hepatic steatosis to nonalcoholic steatohepatitis (NASH), fibrosis, and cirrhosis. NAFLD is often associated with obesity, dyslipidemia, insulin resistance, and type 2 diabetes, and is recognized as the hepatic manifestation of the metabolic syndrome (1). By far, the molecular mechanisms underlying the development of NAFLD remain to be fully understood.
Hepatic triglyceride (TG) accumulation (steatosis), the hallmark of NAFLD, may result from dysfunctional regulation of hepatic TG synthesis, hydrolysis, and secretion. While TG synthesis and secretion in the liver have been well understood, hepatic TG hydrolysis is largely unknown. Very recently, adipose triglyceride lipase (ATGL), an adipose-enriched TG lipase, has been shown to play an important role in regulating hepatic TG turnover (2, 3). However, the expression of ATGL in the liver is low (4), thus raising the possibility that additional TG hydrolase(s) (TGH) may exist in the liver.
Carboxylesterase 1 (CES1) is highly expressed in the liver (5). Human CES1 is also called cholesteryl ester hydrolase (CEH), as it displays CEH activity (6). Consistent with its CEH activity, over-expression of human CES1 in macrophages attenuates the development of atherosclerosis in Ldlr−/− mice (7). Despite high expression in the liver, the role of hepatic CES1 in metabolic control is largely unknown. In an in vitro study, murine Ces1 was shown to prevent TG accumulation in rat McArdle-RH7777 cells (8). However, the physiological relevance of this finding remains unclear.
Nuclear receptors are ligand-activated transcription factors with important regulatory roles in both development and adult physiology. The farnesoid X receptor (FXR; NR1H4), a member of the nuclear receptor superfamily, is important for maintaining bile acid, lipid, and glucose homeostasis. Activation of FXR displays beneficial effects on lipid and carbohydrate metabolism (9). First, it lowers plasma TG and cholesterol levels (10–12) and improves insulin sensitivity (12–15). Second, it increases reverse cholesterol transport (16) and protects against atherosclerosis (17, 18). Lastly, it lowers hepatic TG level and protects against NAFLD (12, 19–22). The beneficial effects of FXR activation on hepatic TG homeostasis indicate that FXR is a therapeutic target for treatment of NAFLD (23, 24).
Mechanistically, Watanabe et al. attributed the FXR-mediated reduction in hepatic TG level to an FXR-SHP-SREBP-1c pathway (19), in which FXR induces small heterodimer partner (SHP) and SHP in turn represses sterol regulatory element binding protein 1c (SREBP-1c), a master regulator of genes involved in fatty acid (FA) biosynthesis. SHP is an atypical nuclear receptor that functions as a repressor by interacting with other nuclear receptors or transcription factors, and is a known FXR target gene (25, 26). In contrast with the data published by Watanabe et al. (19), subsequent studies from other laboratories failed to support the role of the FXR-SHPSREBP-1c pathway in FXR-mediated reduction in hepatic TG levels. First, transgenic expression of human SHP in mouse livers increases both SREBP-1c expression and TG level in the liver (27). Second, Shp−/− mice have reduced hepatic TG level when fed a high fat diet (28). Third, knockout of Shp in ob/ob mice prevents hepatic TG accumulation (29). Lastly, although activation of FXR represses SREBP-1c expression, activation of FXR does not suppress SREBP-1c target genes, such as fatty acid synthase (30). Collectively, these data strongly suggest that the FXR-SHP-SREBP-1c pathway is unlikely to be the underlying mechanism by which activation of FXR reduces hepatic TG levels.
In this study, we investigated the role of hepatic CES1 in both normal and FXR-controlled lipid metabolism. Over-expression of CES1 in the liver lowers hepatic TG levels and improves glucose tolerance in diabetic mice. In contrast, knockdown of CES1 in the liver increases the levels of hepatic TG and plasma non-HDL-C (non-high-density lipoprotein cholesterol) levels. The effect of hepatic CES1 on lipid metabolism likely results from its TGH activity, and subsequent changes in fatty acid oxidation (FAO) and/or lipogenesis. Hepatic CES1 is inducible by FXR and is a direct FXR target gene. Activation of FXR lowers the levels of hepatic TG, plasma TG, and plasma cholesterol in a CES1-dependent manner. Thus, our study reveals a critical role for hepatic CES1 in regulating both lipid and carbohydrate metabolism and in FXR-controlled lipid homeostasis.
Materials and Methods
Mice, Diets and Ligands
C57BL/6 mice, ob/ob mice, db/db mice, and Fxr−/− mice were purchased from the Jackson Laboratories (Bar Harbor, Maine, USA). All mice were fed a standard chow diet. Specific FXR agonists GW4064 (31) (30 mg/kg, twice a day) and OCA (INT-747) (32) (30 mg/kg/d) have been described previously and were administered by gavage. Unless otherwise stated, male mice were used and all mice were fasted for 5–6 h prior to euthanization. All the animal studies have been approved by the Institutional Animal Care and Use Committee at Northeast Ohio Medical University.
Real-Time PCR
RNA was isolated using TRIzol Reagent (Invitrogen, CA). mRNA levels were determined by quantitative reverse-transcription polymerase chain reaction (qRT-PCR) on a 7500 real-time PCR machine from Applied Biosystems (Foster City, CA) by using SYBR Green Supermix (Roche, Indianapolis, IN). Results were calculated using Ct values and normalized to 36B4 mRNA level.
Lipid and Lipoprotein Analysis
Approximately 100 mg liver was homogenized in methanol and lipids were extracted in chloroform/methanol (2:1 v/v) as described (33). Hepatic triglyceride and cholesterol levels were then quantified using Infinity reagents from Thermo Scientific (Waltham, MA). Hepatic fatty acid profile was quantified using gas chromatography (GC)-mass spectrometry at the Mouse Metabolic Phenotyping Center (MMPC) of Case Western Reserve University (Cleveland, OH). Hepatic total free fatty acids and free cholesterol were quantified using kits from BioVision (Milpitas, CA). Plasma lipid and glucose levels were also determined using Infinity reagents. Briefly, after 100 μl plasma was injected, lipoproteins were run at 0.5 ml/min in a buffer containing 0.15 M NaCl, 0.01 M Na2HPO4, 0.1 mM EDTA, pH 7.5, and separated on a Superose 6 10/300 GL column (GE Healthcare) by using BioLogic DuoFlow QuadTec 10 System (Bio-Rad, CA). 500 μl of sample per fraction was collected.
Hepatic Lipogenesis
Mice were fasted for 4 h and then injected intraperitoneally with 2H2O (20–30 μl/g). After 4 h, liver and plasma were snap-frozen in liquid nitrogen. The newly synthesized palmitate, triglycerides, and cholesterol were measured by mass spectrometry at MMPC of Case Western Reserve University.
Statistical Analysis
Statistical significance was analyzed using unpaired Student t test or ANOVA (GraphPad Prisim, CA). All values are expressed as mean±SEM. Differences were considered statistically significant at P<0.05.
Other detailed Materials and Method are provided in the Supplementary Information.
Results
Over-expression of hepatic CES1 lowers hepatic triglyceride levels and improves glucose homeostasis
Our data have shown that hepatic CES1 is down-regulated by fasting (Figure S1). To investigate whether hepatic CES1 plays a role in lipid and/or carbohydrate metabolism, we generated adenovirus expressing Ces1 (Ad-Ces1-GFP), which was subsequently injected intravenously (i.v.) to C57BL/6 mice. Hepatic expression of CES1 had no effect on plasma cholesterol or TG levels (Figure S2A), but significantly lowered plasma glucose levels by ~ 30% (Figure 1A, left panel) and hepatic TG levels by 60% (Figure 1A, right panel). Hepatic cholesterol levels also tended to decrease (p=0.14) (data not shown).
Figure 1. Hepatic expression of CES1 lowers hepatic triglyceride levels and improves glucose homeostasis.
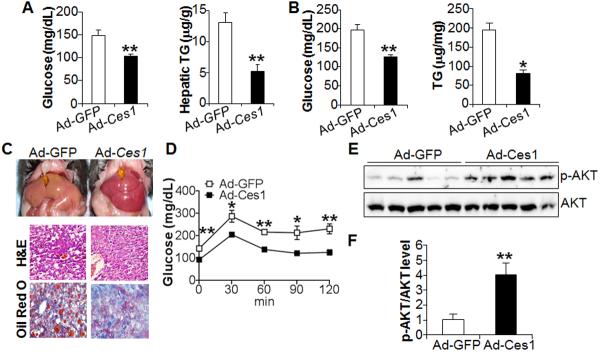
(A) C57BL/6 mice were i.v. injected with either Ad-GFP or Ad-Ces1 (n=7–8 mice per group). After 7 days, mice were fasted for 5 h. Plasma glucose (left panel) and hepatic TG (right panel) levels were determined. (B–F) ob/ob mice were i.v. injected with Ad-GFP or Ad-Ces1 (n=5 mice per group). After 7 days and a 5-h fast, mice were euthanized. Plasma glucose (B, left panel) and hepatic TG levels (B, right panel) were determined. Representative liver images are shown in (C, top panel) and representative H&E staining (C, middle panel) or oil red O staining (C, bottom panel) of the liver sections are shown in (C). Glucose tolerance test (GTT) was performed after a 16 h fast (D). Western blot assays were performed using liver lystates (E) and protein levels quantified using ImageJ software (F).
To determine whether hepatic CES1 has similar effects on lipid and glucose homeostasis in obese and diabetic mice, we over-expressed Ces1 in the liver of ob/ob mice. Hepatic expression of Ces1 in diabetic ob/ob mice lowered plasma glucose levels by 32% (Figure 1B, left panel) and hepatic TG levels by 60% (Figure 1B, right panel), which was also evidenced by the appearance of more reddish liver (Figure 1C, top panel) and smaller lipid droplets in hepatocytes (Figure 1C, lower panels). In addition, hepatic expression of Ces1 significantly improved glucose tolerance in a glucose tolerance test (Figure 1D). Consistent with the latter finding, hepatic expression of Ces1 increased hepatic p-AKT level by four fold (Figure 1E, 1F), indicating that hepatic CES1 expression enhances hepatic insulin signaling.
Over-expression of hepatic CES1 does not affect fatty acid lipogenesis or very low-density lipoprotein (VLDL) secretion
To determine the underlying mechanism by which over-expression of hepatic CES1 has beneficial effects on lipid and carbohydrate metabolism, we first determined hepatic gene expression. Hepatic expression of Ces1 in C57BL/6 mice selectively regulated the expression of only certain genes (Figure S2B and S2C). Consistent with hepatic cholesterol levels that tended to decrease (data not shown), genes involved in cholesterol biosynthetic pathways, including Srebp-2, HMG-CoA reductase (Hmgcr) and HMG-CoA synthase (Hmgcs), were reduced by 20–25% (Figure 2A). In contrast, hepatic genes involved in TG synthesis, including Srebp-1c, acetyl-coA carboxylesterase 1 (Acc1), Fas, acyl-CoA:diacylglycerol acyltransferase 1 (Dgat1) and Dgat2, were not significantly different between the two groups (Figure 2A). Interestingly, Acc2, which represses fatty acid oxidation (FAO), was inhibited, whereas a few peroxisome proliferator-activated receptor α (PPARα) target genes, including fatty acid translocase Cd36, pyruvate dehydrogenase kinase 4 (Pdk4), angiopoietin-like protein 4 (Angptl4) (34–37), were significantly induced (Figure 2A). Carnitine palmitoyltransferase 1b (Cpt1b), a rate-limiting enzyme in mitochondrial FAO, was also induced (Figure 2A). In addition, expression of hepatic Ces1 also induced glucokinase (Gck) expression and tended to reduce hepatic Pepck expression (p=0.06) (Figure 2A). Hepatic Gck, which facilitates glucose uptake by phosphorylating glucose to glucose-6-phosphate, is transcriptionally regulated by insulin (38). Finally, in ob/ob mice, hepatic expression of Ces1 reduced the expression of gluconeogenic genes Pepck and G6pase (Figure 2A, right panel), consistent with enhanced insulin signaling in these mice (Figure 1D–F). Thus, over-expression of hepatic Ces1 may result in increased PPARα activity and insulin sensitivity in the liver.
Figure 2. Hepatic expression of CES1 selectively regulates gene expression and has no effect on lipogenesis or VLDL secretion.
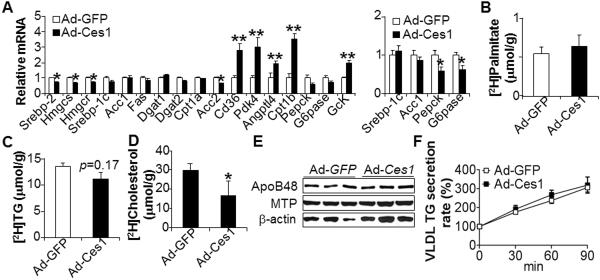
(A) Hepatic mRNA levels in wild-type (left panel) or ob/ob mice (right panel) were determined by qRT-PCR. (B–D) De novo lipogenesis was determined in mice after injection of 2H2O (n=5 mice per group). The levels of newly synthesized [2H]palmitate (B), [2H]TG (C) or [2H]cholesterol (D) in the liver were quantified. (E) Hepatic protein level was assessed by Western blot assays. (F) VLDL secretion rate was determined (n=6 mice per group).
The data of Figure 2A also suggest that over-expression of CES1 may have little effect on lipogenic pathways. We then investigated de novo lipogenesis by injection of mice with 2H2O. Consistent with the gene expression data (Figure 2A), over-expression of hepatic Ces1 had no effect on hepatic de novo biosynthesis of palmitate (Figure 2B) or TG (Figure 2C), but reduced hepatic cholesterol biosynthesis (Figure 2D). Finally, we also determined the effect of hepatic Ces1 expression on VLDL secretion. Over-expression of hepatic Ces1 had no effect on the protein levels of microsomal triglyceride transfer protein (MTP) or ApoB (Figure 2E), or VLDL secretion rate (Figure 2F). Thus, lipogenesis or VLDL secretion does not contribute to the reduced hepatic TG levels following hepatic CES1 expression.
Over-expression of hepatic CES1 increases hepatic TG hydrolysis and stimulates fatty acid oxidation
The data of Figure 2A suggest that CES1 over-expression may induce PPARα activity. To test this hypothesis, we co-transfected a Ces1-expressing plasmid and a luciferase-reporter plasmid containing 3 copies of PPARα responsive element (PPRE). Over-expression of CES1 highly induced PPARα activity (Figure 3A, top panel). Free fatty acids (FFAs), which may derive from TG hydrolysis, have been well documented as endogenous ligands for PPARα (39). Thus, we hypothesized that CES1 had TGH activity (Figure 3A, bottom panel). Over-expression of Ces1 significantly increased the release of [3H]FFAs from [3H]triolein in both COS-7 cells (Figure 3B) and the liver (Figure 3C), indicating that CES1 has TGH activity.
Figure 3. Hepatic expression of CES1 increases triglyceride hydrolase activity and activates PPARα.
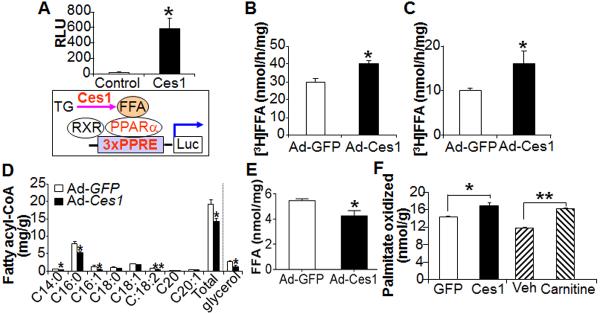
(A) HepG2 cells were transfected with a control plasmid or a Ces1-expressing plasmid together with a 3xPPRE-Luc reporter plasmid. Luciferase activity was determined (top panel). In the bottom panel, the diagram shows that CES1 hydrolyzes TG and releases FFAs, which bind to PPARα/RXR complex and then induce PPARα activity. (B, C) TGH activity was assessed using lysates from COS-7 cells (B) or the liver (C). (D, E) Hepatic fatty acid profile was determined by GC-mass spectrometry (D) and hepatic FFA levels were quantified (n=8 mice per group) (E). (F) FAO was performed in the liver cell line AML12 cells that were infected with Ad-GFP or Ad-CES1 for 48 h, or treated with either vehicle or carnitine (1 mM) (n=3–5 per group). Carnitine treatment serves as a positive control. Veh, vehicle.
Although the expression of CES1 increased hepatic TGH activity (Figure 3C), hepatic total fatty acids and a number of long-chain fatty acids, such as C14:0, C16:0, C16:1, and C18:2, were significantly reduced (Figure 3D). Hepatic FFA levels were also significantly reduced (Figure 3E). The repression of Acc2 and induction of Cd36, Pdk4, Angptl4 and Cpt1b (Figure 2A), all of which are involved in FAO, suggest that CES1 may regulate FAO. As predicted, over-expression of CES1 increased FAO (Figure 2F). In addition, hepatic expression of CES1 also tended to increase plasma levels of β-hydroxybutyrate (p=0.07) (Figure S3). Thus, the reduced hepatic FFA levels in Ces1-over-expressing mice may be a net effect of increased TGH activity and increased FAO.
Knockdown of hepatic CES1 causes hepatic steatosis and elevated plasma cholesterol levels
To determine whether hepatic CES1 is required for maintaining normal lipid and glucose homeostasis, we generated three adenoviruses expressing shRNA specifically against murine Ces1 (Ad-shCes1). One of the three Ad-shCes1 adenenoviruses was particularly efficient in knocking down endogenous Ces1 expression in cultured cells (Figure S4), and was used for subsequent studies. Ad-shLacZ (control) or Ad-shCes1 was i.v. injected into C57BL/6 mice. Remarkably, hepatic expression of Ces1 shRNA reduced hepatic levels of Ces1 mRNA (Figure 4A) and protein (Figure 4B) by ~ 95%, increased plasma total cholesterol (TC) levels by ~ 2 fold (Figure 3C), but had no effect on plasma TG (Figure 4C) or glucose levels (data not shown). Analysis of plasma by fast protein liquid chromatography (FPLC) indicated that hepatic Ces1 deficiency markedly increased plasma non-HDL-C levels (Fig. 4D), but had no much effect on plasma TG lipoprotein profile (data not shown). Hepatic Ces1 deficiency also caused development of fatty liver (Fig. 4E), which was associated with more than 2-fold increase in hepatic TG levels but unchanged hepatic TC levels (Fig. 4F). Thus, hepatic CES1 is indispensable for maintaining normal hepatic TG homeostasis and plasma cholesterol levels.
Figure 4. Loss of hepatic CES1 causes fatty liver and increased plasma cholesterol level.
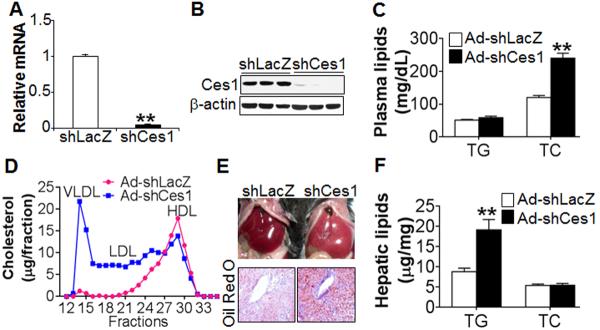
(A–F) C57BL/6 mice were i.v. injected with either Ad-shLacZ or AdshCes1 (n=8 mice per group). After a 6-h fast, mice were euthanized. Hepatic mRNA (A) and protein (B) levels were determined. Plasma TG and total cholesterol (TC) levels were quantified (C). Plasma cholesterol lipoprotein profile was determined by FPLC (D). Representative liver images (top panel) and oil red O staining of liver sections (lower panel) are shown (E). Hepatic TG and TC levels were quantified (F).
Hepatic CES1 deficiency results in increased lipogenesis
The data of Figure 4 show that the effect of hepatic CES1 deficiency on lipid metabolism is quite striking. Analysis of hepatic gene expression indicated that many SREBP- or cholesterol/LXR (liver X receptor)-regulated genes, including Hmgcs, Acc-1, Dgat1, Dgat2, ATP citrate lyase (Acl) (40, 41), Abca1, and Cd36 (42), were significantly induced (Figure 5A). The induction of these genes was selective as many other genes were unchanged (Figure S5). Consistent with the induction of numerous lipogenic genes, hepatic de novo lipogenesis of palmitate (Figure 5B), TG (Figure 5C), and cholesterol (Figure 5D), were induced by 2.1, 2.9 and 2.1 fold, respectively, indicating that loss of hepatic CES1 has profound effects on lipogenesis. Investigations of hepatic mature/nuclear SREBPs (nSREBPs) showed that loss of hepatic CES1 resulted in an increase in the levels of hepatic nSREBP-1 and nSREBP-2 by 2 fold and 2.4 fold, respectively (Figure 5E). The latter data suggest that elevated mature SREBP levels are responsible for increased lipogenesis.
Figure 5. Loss of hepatic CES1 induces de novo lipogenesis.
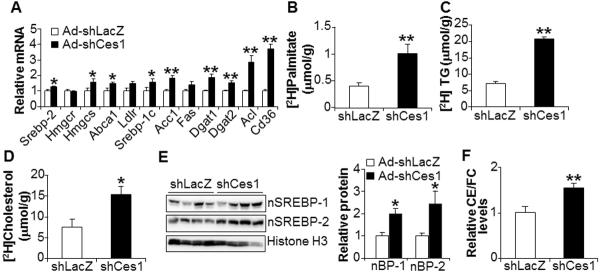
(A) Hepatic mRNA levels were determined by qRT-PCR (n=8 per group). (B–D) De novo lipogenesis was determined in mice after injection of 2H2O (n=5 per group). The levels of newly synthesized [2H]palmitate (B), [2H]TG (C), or [2H]cholesterol (D) in the liver were quantified. (E) Hepatic protein levels were determined by Western blot assays (E, left panel) and then quantified by using ImageJ software (E, right panel). nBP-1, nuclear form SREBP-1. nBP-2, nuclear form SREBP-2. (F) Hepatic ratio of CE to FC was determined (n=8 mice per group). The CE/FC ratio in shLacZ-treated mice was set at 1.
SREBP processing is known to be regulated by the change in intracellular sterol levels; when cellular sterol levels are low, mature SREBPs are increased by inducing SREBP processing (43). Consistent with previous data showing that CES1 has CEH activity (6, 44), loss of hepatic Ces1 signficantly reduced hepatic free cholesterol (FC) level (Figure S6A) whereas hepatic FFA level remained unchanged (Figure S6B). Ces1-deficient mice also had significantly higher levels of cholesterol esters (CE) to FC (Figure 5F). Thus, the reduced hepatic FC levels may account, at least in part, for the increased levels of hepatic nSREBP-1 and nSREBP-2.
Finally, we investigated the effect of hepatic Ces1 deficiency on VLDL secretion. Loss of hepatic Ces1 had no effect on the protein levels of MTP, ApoB100 or ApoB48 (Figure S6C), but tended to increase hepatic VLDL secretion rate (Figure S6D). Thus, loss of hepatic CES1 causes hepatic steatosis and hyperlipidemia (Figure 4) likely resulting from increased lipogenesis.
Activation of FXR induces hepatic CES1 expression
FXR is highly expressed in the liver and plays an important role in regulating lipid and carbohydrate metabolism. Activation of FXR by the specific agonist GW4064 reduced hepatic TG level by ~ 40% but did not alter hepatic cholesterol levels (Figure 6A). Analysis of hepatic gene expression showed that GW4064 treatment induced hepatic mRNA levels of Shp and Ces1, but had no effect on Ces2 or Ces3 mRNA levels (Figure 6B). GW4064 treatment also highly induced hepatic CES1 protein levels, but did not alter hepatic ApoB100 or MTP protein levels (Figure 6C), suggesting that GW4064 does not affect VLDL secretion. In addition to GW4064, hepatic Ces1 mRNA was also induced by treatment with obeticholic acid (OCA, INT-747) (Figure 6D), another potent and selective FXR agonist (32), or cholic acid, the endogenous FXR ligand (Figure 6E). In contrast, hepatic Ces1 mRNA level was reduced in Fxr−/− mice (Figure 6F). Thus, hepatic CES1 expression is regulated by FXR.
Figure 6. Hepatic CES1 is regulated by FXR.
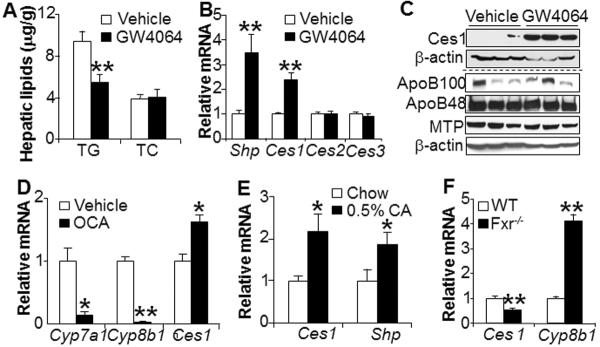
(A–C) C57BL/6 mice were gavaged with vehicle (0.5% CMC (carboxymethyl cellulose)) or GW4064 (30 mg/kg, twice a day) for 7 days (n=8 mice per group). Hepatic TG and TC levels were determined (A). Hepatic mRNA levels were quantified by qRT-PCR (B) and hepatic protein levels were determined by Western blot assays (C). (D) C57BL/6 mice were gavaged with either 0.5% CMC (vehicle) or INT-747 (OCA, 30 mg/kg/d) for 7 days (n=5 mice per group). Hepatic mRNA levels were determined. Cyp7a1 and Cyp8b1 serve as positive controls. (E) C57BL/6 mice were fed a chow diet or 0.5% cholic acid (CA) for 7 days. Hepatic mRNA levels were quantified. (F) Hepatic mRNA levels were quantified by qRT-PCR in wild-type or Fxr−/− mice (n=8 mice per group).
CES1 is a direct FXR target gene
To investigate how FXR regulates CES1 expression, we first determined the FXR response element(s) (FXRE) in the Ces1 gene promoter. Analysis of the promoter activity by using a serial of 5'-deletion constructs (Figure 7A) and mutagenesis (Figure 7B) uncovered an FXRE located between 300 bp and 150 bp upstream of transcription start site. Electrophoretic mobility shift assays showed that the FXR/RXR complex bound to this FXRE in vitro and this binding was competed away by cold wild-type oligonucleotides but not by mutant oligonucleotides (Figure 7C). Finally, chromatin immunoprecipitation assay showed that FXR bound to the promoter of Akr1b7, a known FXR target gene (45, 46), as well as the Ces1 promoter in the liver (Figure 7D). Collectively, the data of Figure 7 demonstrate that Ces1 is a direct FXR target gene, and that CES1 is induced by FXR through an FXRE located between 150 bp and 300 bp upstream of the transcription start site.
Figure 7. CES1 is a direct FXR target gene.
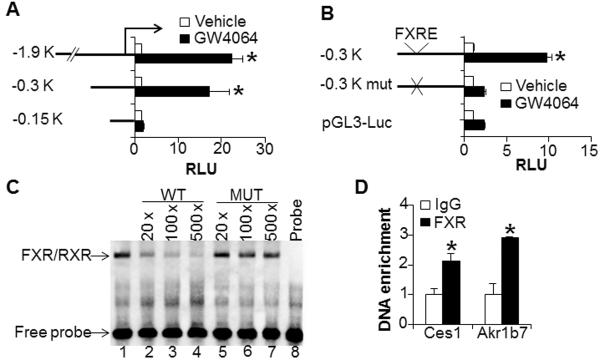
(A, B) Transient transfection assays were performed using promoter-luciferase constructs containing a serial of 5'-deletions (A) or mutations (B). (C) EMSA assays were performed using in vitro transcribed/translated proteins. Wild-type (WT) and mutant (MUT) oligos were used in the competition assays. (D) Chromatin immunoprecipitation (ChIP) assays were performed using liver lysates (n=3 per group). Akr1b7 serves as a positive control.
Hepatic CES1 is essential for activated FXR to improve lipid homeostasis
Activation of FXR has beneficial effects on lipid homeostasis. The findings that hepatic CES1 is important for maintaining lipid homeostasis and is regulated by FXR prompted us to study the role of hepatic CES1 in FXR-controlled lipid homeostasis. C57BL/6 mice were injected i.v. with either Ad-shLacZ or Ad-shCes1, followed by treatment with either vehicle or OCA for 7 days. As expected, knockdown of hepatic Ces1 increased plasma cholesterol levels by ~ 4 fold (Figure 8A). Interestingly, OCA treatment reduced plasma cholesterol levels in the control mice but not in Ces1-deficient mice (Figure 8A). Similarly, OCA treatment also reduced plasma TG level in the control mice but not in the Ces1-deficient mice (Figure 8B). In contrast, OCA lowered plasma glucose levels in both the control mice and Ces1-deficient mice (Figure S7A). Analysis of plasma by FPLC showed that loss of hepatic Ces1 markedly increased plasma non-HDL-C levels (Figure S7B) and also slightly increased LDL triglyceride levels (Figure S7C), and these changes were exacerbated following OCA treatment (Figure S7B and S7C). Thus, hepatic CES1 is critical for an FXR agonist to lower plasma TG and cholesterol levels.
Figure 8. Essential roles of hepatic CES1 in FXR-regulated lipid homeostasis.
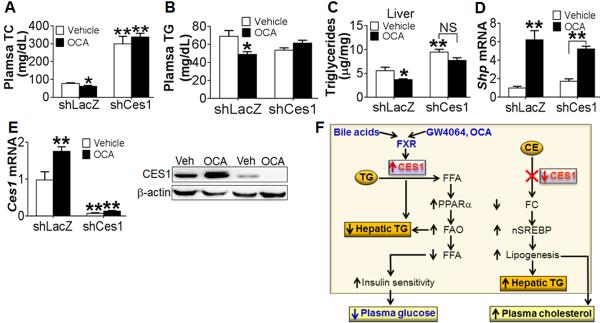
(A–E) C57BL/6 mice were i.v. injected with Ad-shLacZ or Ad-shCes1. The next day, these mice were gavaged with either vehicle (0.5% CMC) or INT-747 (OCA, 30 mg/kg/d) for 7 days (n=8–10 mice per group). After a 5-h fast, mice were euthanized. Plasma total cholesterol (TC) (A) and TG (B) levels as well as hepatic TG levels (C) were determined. Hepatic mRNA levels of Shp (D) and Ces1 (E, left panel) were determined by qRT-PCR. Western blot assays were also performed (E, right panel). NS, not significant. In (C), treatment of the control group versus the Ces1-deficient group by OCA was significantly different (p<0.01). (F) Central roles of hepatic CES1 in lipid and carbohydrate metabolism and in FXR signaling. Expression of hepatic CES1 lowers hepatic TG level by increasing TG hydrolysis and FAO. As a result, hepatic FFA level is reduced, which in turn causes an increase in hepatic insulin sensitivity and a reduction in plasma glucose level. When hepatic CES1 is deficient, hepatic free cholesterol (FC) levels are reduced, resulting in an increase in hepatic SREBP processing. The increased nSREBP1 and nSERBP2 levels promote lipogenesis, resulting in increased levels of hepatic TG and plasma cholesterol. Hepatic CES1 can be induced FXR and bile acids. The induction of hepatic CES1 is critical for activated FXR to improve lipid homeostasis.
In the liver, OCA treatment had no effect on hepatic cholesterol levels in control mice but they were increased in Ces1-deficient mice (Figure S7D). As expected, OCA treatment reduced hepatic TG levels in control mice (Figure 8C). Interestingly, OCA treatment had no significant effect on hepatic TG levels in Ces1-deficient mice (Figure 8C). Thus, hepatic CES1 is important for activated FXR to lower hepatic TG levels.
Analysis of hepatic gene expression showed that OCA induced the FXR target gene Shp in both control mice and Ces1-deficient mice (Figure 8D). OCA also induced hepatic Ces1 expression in control mice but this induction was blunted in Ces1-deficient mice (Figure 8E).
We also treated ob/ob mice with OCA. Similar to what we observed in C57BL/6 mice, we found that OCA treatment lowered plasma cholesterol and TG levels as well as hepatic TG levels in a CES1-dependent manner (Figure S8A, B, D), but had no effect on hepatic cholesterol levels (Figure S8C). Interestingly, loss of hepatic CES1 did not further increase hepatic TG levels in ob/ob mice that were deficient in hepatic Ces1 (Figure S8D). One possibility is that hepatic TG accumulation in ob/ob mice had reached the maximum level. Analysis of hepatic gene expression showed that OCA treatment induced hepatic mRNA levels of Ces1 in the control mice (Figure S8E) and Shp in both the control and Ces1-deficient mice (Figure S8F). Together, the data of Figure 8 and Figure S8 demonstrate that hepatic CES1 is indispensable for activated FXR to regulate both plasma lipid and hepatic TG levels.
Discussion
CES1 is highly expressed in the liver, but the role of hepatic CES1 in lipid and carbohydrate metabolism has not been investigated before. Herein, we have used both gain- and loss-of-function approaches to demonstrate that hepatic CES1 plays a critical role in regulating both lipid and carbohydrate metabolism. In addition, we also demonstrate that FXR regulates hepatic CES1 expression and that the induction of hepatic CES1 is indispensable for activated FXR to improve lipid homeostasis (Figure 8F). Numerous studies have demonstrated that FXR is a therapeutic target for treatment of metabolic disease (9, 24), particularly for treatment of NAFLD (47). Our data suggest that hepatic CES1 is a promising therapeutic target for treatment of NAFLD. Consequently, the findings from the current study may have significant therapeutic implications.
One key finding in our study is the demonstration that hepatic CES1 has TGH activity and is essential for maintaining hepatic TG homeostasis. Hepatic TG mobilization has been poorly understood. Previous data suggest that ATGL may play an important role in mobilizing hepatic TG (2, 3). However, ATGL expression is low in hepatocytes (4, 48). Since CES1 is highly expressed in the liver and plays an essential role is controlling hepatic TG levels, CES1 may represent one of the major TG hydrolases in the liver.
Previous data have shown that CES1 also has CEH activity (6, 44). Thus, the effect of hepatic CES1 on lipid and carbohydrate metabolism may depend on its TGH activity and/or CEH activity (Figure 8F). Expression of hepatic CES1 lowers hepatic TG levels and improves glucose homeostasis likely through increased TG hydrolysis and subsequent increase in PPARα activity and FAO, as neither lipogenesis nor VLDL secretion is changed (Figures 2 and 3). In contrast, loss of hepatic CES1 increases hepatic TG levels and plasma cholesterol levels likely through increased lipogenesis, which may result from loss of CEH activity and subsequent increase in SREBP processing (Figures 6 and 8F).
Very recently and after this study was completed, Quiroga et al. reported that global Ces1−/− mice develop hepatic steatosis, increased obesity, and hyperlipidemia (49). Given that CES1 is expressed in multiple tissues, including liver, intestine and other metabolic tissues (50) (data not shown), one cannot conclude which tissue(s) play a role in the observed metabolic changes. Moreover, intestinal CES1 is also reported to regulate chylomicron secretion (50), which can contribute significantly to both hepatic TG and plasma lipid metabolism. Thus, it is critical to study the relative roles of hepatic vs. intestinal CES1 in lipid and carbohydrate metabolism. Our study provides the first evidence demonstrating that hepatic CES1 plays a critical role in regulating both lipid and glucose homeostasis.
The finding that expression of hepatic CES1 improves glucose homeostasis is intriguing (Figure 1). Our data show that expression of hepatic CES1 enhances hepatic insulin signaling, as hepatic glucokinase and p-AKT levels are increased whereas hepatic gluconeogenic genes are repressed (Figures 1 and 2). The increased hepatic insulin signaling may result from reduced hepatic FFA level (Figure 3E).
FXR has been recognized as an interesting target for treatment of fatty liver disease (24, 47). However, the mechanism underlying FXR-mediated alleviation of hepatic steatosis remains undetermined. Our study provides compelling evidence demonstrating that CES1 is a direct downstream target gene of FXR (Figures 6 and 7) and is necessary for activated FXR to lower plasma TG and cholesterol levels (Figure 8). We also demonstrate that activation of FXR lowers hepatic TG level, at least in part, through induction of hepatic CES1 (Figure 8). Thus, hepatic CES1 plays a crucial role in FXR-controlled lipid homeostasis. Interestingly, CES1 over-expression is not sufficient to lower plasma TG or cholesterol levels (Figure S2). One possibility is that CES1 is just one of the downstream components of FXR signaling that are responsible for FXR-controlled lipid homeostasis.
Both human CES1 and mouse CES1 are highly expressed in the liver and share 74% amino acid homology. However, none has been known about the role of human CES1 in triglyceride or glucose metabolism. One of our future directions will be to investigate whether human CES1 and mouse CES1 have similar effects on lipid and glucose homeostasis.
In summary, we have shown that hepatic expression of CES1 has beneficial effects on lipid and carbohydrate metabolism whereas loss of hepatic CES1 causes fatty liver and pro-atherogenic lipid profile. Such effects of CES1 may result from its TGH and/or CEH activity and subsequent change in FAO and/or lipogenesis. In addition, activation of FXR induces hepatic CES1, which is critical for FXR-mediated improvement of lipid homeostasis. Together, our data indicate that hepatic CES1 is an important component of FXR signaling in metabolic control, and also suggest that hepatic CES1 may represent a therapeutic target for treatment of NAFLD.
Supplementary Material
Acknowledgements
This work was supported by NIH grants R15DK088733, R01HL103227, and R01DK095895 to Y.Z.
References
- 1.Brunt EM. Pathology of nonalcoholic fatty liver disease. Nat Rev Gastroenterol Hepatol. 2010;7:195–203. doi: 10.1038/nrgastro.2010.21. [DOI] [PubMed] [Google Scholar]
- 2.Wu JW, Wang SP, Alvarez F, Casavant S, Gauthier N, Abed L, Soni KG, et al. Deficiency of liver adipose triglyceride lipase in mice causes progressive hepatic steatosis. Hepatology. 2011;54:122–132. doi: 10.1002/hep.24338. [DOI] [PubMed] [Google Scholar]
- 3.Ong KT, Mashek MT, Bu SY, Greenberg AS, Mashek DG. Adipose triglyceride lipase is a major hepatic lipase that regulates triacylglycerol turnover and fatty acid signaling and partitioning. Hepatology. 2011;53:116–126. doi: 10.1002/hep.24006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zimmermann R, Strauss JG, Haemmerle G, Schoiswohl G, Birner-Gruenberger R, Riederer M, Lass A, et al. Fat mobilization in adipose tissue is promoted by adipose triglyceride lipase. Science. 2004;306:1383–1386. doi: 10.1126/science.1100747. [DOI] [PubMed] [Google Scholar]
- 5.Ellinghaus P, Seedorf U, Assmann G. Cloning and sequencing of a novel murine liver carboxylesterase cDNA. Biochim Biophys Acta. 1998;1397:175–179. doi: 10.1016/s0167-4781(98)00023-2. [DOI] [PubMed] [Google Scholar]
- 6.Zhao B, Natarajan R, Ghosh S. Human liver cholesteryl ester hydrolase: cloning, molecular characterization, and role in cellular cholesterol homeostasis. Physiol Genomics. 2005;23:304–310. doi: 10.1152/physiolgenomics.00187.2005. [DOI] [PubMed] [Google Scholar]
- 7.Zhao B, Song J, Chow WN, St Clair RW, Rudel LL, Ghosh S. Macrophage-specific transgenic expression of cholesteryl ester hydrolase significantly reduces atherosclerosis and lesion necrosis in Ldlr mice. J Clin Invest. 2007;117:2983–2992. doi: 10.1172/JCI30485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ko KW, Erickson B, Lehner R. Es-x/Ces1 prevents triacylglycerol accumulation in McArdle-RH7777 hepatocytes. Biochim Biophys Acta. 2009;1791:1133–1143. doi: 10.1016/j.bbalip.2009.07.006. [DOI] [PubMed] [Google Scholar]
- 9.Zhang Y, Edwards PA. FXR signaling in metabolic disease. FEBS Lett. 2008;582:10–18. doi: 10.1016/j.febslet.2007.11.015. [DOI] [PubMed] [Google Scholar]
- 10.Bilz S, Samuel V, Morino K, Savage D, Choi CS, Shulman GI. Activation of the farnesoid X receptor improves lipid metabolism in combined hyperlipidemic hamsters. Am J Physiol Endocrinol Metab. 2006;290:E716–722. doi: 10.1152/ajpendo.00355.2005. [DOI] [PubMed] [Google Scholar]
- 11.Ma K, Saha PK, Chan L, Moore DD. Farnesoid X receptor is essential for normal glucose homeostasis. J Clin Invest. 2006;116:1102–1109. doi: 10.1172/JCI25604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang Y, Lee FY, Barrera G, Lee H, Vales C, Gonzalez FJ, Willson TM, et al. Activation of the nuclear receptor FXR improves hyperglycemia and hyperlipidemia in diabetic mice. Proc Natl Acad Sci U S A. 2006;103:1006–1011. doi: 10.1073/pnas.0506982103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cariou B, van Harmelen K, Duran-Sandoval D, van Dijk TH, Grefhorst A, Abdelkarim M, Caron S, et al. The farnesoid X receptor modulates adiposity and peripheral insulin sensitivity in mice. J Biol Chem. 2006;281:11039–11049. doi: 10.1074/jbc.M510258200. [DOI] [PubMed] [Google Scholar]
- 14.Cipriani S, Mencarelli A, Palladino G, Fiorucci S. FXR activation reverses insulin resistance and lipid abnormalities and protects against liver steatosis in Zucker (fa/fa) obese rats. J Lipid Res. 2010;51:771–784. doi: 10.1194/jlr.M001602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nozawa H. Xanthohumol, the chalcone from beer hops (Humulus lupulus L.), is the ligand for farnesoid X receptor and ameliorates lipid and glucose metabolism in KK-A(y) mice. Biochem Biophys Res Commun. 2005;336:754–761. doi: 10.1016/j.bbrc.2005.08.159. [DOI] [PubMed] [Google Scholar]
- 16.Zhang Y, Yin L, Anderson J, Ma H, Gonzalez FJ, Willson TM, Edwards PA. Identification of novel pathways that control farnesoid X receptor-mediated hypocholesterolemia. J Biol Chem. 2010;285:3035–3043. doi: 10.1074/jbc.M109.083899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hartman HB, Gardell SJ, Petucci CJ, Wang S, Krueger JA, Evans MJ. Activation of farnesoid X receptor prevents atherosclerotic lesion formation in LDLR−/− and apoE−/− mice. J Lipid Res. 2009;50:1090–1100. doi: 10.1194/jlr.M800619-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mencarelli A, Renga B, Distrutti E, Fiorucci S. Antiatherosclerotic effect of farnesoid X receptor. Am J Physiol Heart Circ Physiol. 2009;296:H272–281. doi: 10.1152/ajpheart.01075.2008. [DOI] [PubMed] [Google Scholar]
- 19.Watanabe M, Houten SM, Wang L, Moschetta A, Mangelsdorf DJ, Heyman RA, Moore DD, et al. Bile acids lower triglyceride levels via a pathway involving FXR, SHP, and SREBP-1c. J Clin Invest. 2004;113:1408–1418. doi: 10.1172/JCI21025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cipriani S, Mencarelli A, Palladino G, Fiorucci S. FXR activation reverses insulin resistance and lipid abnormalities and protects against liver steatosis in Zucker (fa/fa) obese rats. J Lipid Res. 2009 doi: 10.1194/jlr.M001602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang S, Wang J, Liu Q, Harnish DC. Farnesoid X receptor agonist WAY-362450 attenuates liver inflammation and fibrosis in murine model of non-alcoholic steatohepatitis. J Hepatol. 2009;51:380–388. doi: 10.1016/j.jhep.2009.03.025. [DOI] [PubMed] [Google Scholar]
- 22.Fiorucci S, Rizzo G, Antonelli E, Renga B, Mencarelli A, Riccardi L, Orlandi S, et al. A farnesoid x receptor-small heterodimer partner regulatory cascade modulates tissue metalloproteinase inhibitor-1 and matrix metalloprotease expression in hepatic stellate cells and promotes resolution of liver fibrosis. J Pharmacol Exp Ther. 2005;314:584–595. doi: 10.1124/jpet.105.084905. [DOI] [PubMed] [Google Scholar]
- 23.Cariou B. The farnesoid X receptor (FXR) as a new target in non-alcoholic steatohepatitis. Diabetes Metab. 2008;34:685–691. doi: 10.1016/S1262-3636(08)74605-6. [DOI] [PubMed] [Google Scholar]
- 24.Adorini L, Pruzanski M, Shapiro D. Farnesoid X receptor targeting to treat nonalcoholic steatohepatitis. Drug Discov Today. 2012;17:988–997. doi: 10.1016/j.drudis.2012.05.012. [DOI] [PubMed] [Google Scholar]
- 25.Goodwin B, Jones SA, Price RR, Watson MA, McKee DD, Moore LB, Galardi C, et al. A Regulatory Cascade of the Nuclear Receptors FXR, SHP-1, and LRH-1 Represses Bile Acid Biosynthesis. Molecular Cell. 2000;6:517–526. doi: 10.1016/s1097-2765(00)00051-4. [DOI] [PubMed] [Google Scholar]
- 26.Lu TT, Makishima M, Repa JJ, Schoonjans K, Kerr TA, Auwerx J, Mangelsdorf DJ. Molecular basis for feedback regulation of bile acid synthesis by nuclear receptors. Molecular Cell. 2000;6:507–515. doi: 10.1016/s1097-2765(00)00050-2. [DOI] [PubMed] [Google Scholar]
- 27.Boulias K, Katrakili N, Bamberg K, Underhill P, Greenfield A, Talianidis I. Regulation of hepatic metabolic pathways by the orphan nuclear receptor SHP. EMBO J. 2005;24:2624–2633. doi: 10.1038/sj.emboj.7600728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang L, Liu J, Saha P, Huang J, Chan L, Spiegelman B, Moore DD. The orphan nuclear receptor SHP regulates PGC-1alpha expression and energy production in brown adipocytes. Cell Metab. 2005;2:227–238. doi: 10.1016/j.cmet.2005.08.010. [DOI] [PubMed] [Google Scholar]
- 29.Huang J, Iqbal J, Saha PK, Liu J, Chan L, Hussain MM, Moore DD, et al. Molecular characterization of the role of orphan receptor small heterodimer partner in development of fatty liver. Hepatology. 2007;46:147–157. doi: 10.1002/hep.21632. [DOI] [PubMed] [Google Scholar]
- 30.Matsukuma KE, Bennett MK, Huang J, Wang L, Gil G, Osborne TF. Coordinated control of bile acids and lipogenesis through FXR-dependent regulation of fatty acid synthase. J Lipid Res. 2006;47:2754–2761. doi: 10.1194/jlr.M600342-JLR200. [DOI] [PubMed] [Google Scholar]
- 31.Maloney PR, Parks DJ, Haffner CD, Fivush AM, Chandra G, Plunket KD, Creech KL, et al. Identification of a chemical tool for the orphan nuclear receptor FXR. Journal of Medicinal Chemistry. 2000;43:2971–2974. doi: 10.1021/jm0002127. [DOI] [PubMed] [Google Scholar]
- 32.Pellicciari R, Fiorucci S, Camaioni E, Clerici C, Costantino G, Maloney PR, Morelli A, et al. 6alpha-ethyl-chenodeoxycholic acid (6-ECDCA), a potent and selective FXR agonist endowed with anticholestatic activity. J Med Chem. 2002;45:3569–3572. doi: 10.1021/jm025529g. [DOI] [PubMed] [Google Scholar]
- 33.Bligh EG, Dyer WJ. A rapid method of total lipid extraction and purification. Can J Biochem Physiol. 1959;37:911–917. doi: 10.1139/o59-099. [DOI] [PubMed] [Google Scholar]
- 34.Rakhshandehroo M, Hooiveld G, Muller M, Kersten S. Comparative analysis of gene regulation by the transcription factor PPARalpha between mouse and human. PLoS One. 2009;4:e6796. doi: 10.1371/journal.pone.0006796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wu P, Peters JM, Harris RA. Adaptive increase in pyruvate dehydrogenase kinase 4 during starvation is mediated by peroxisome proliferator-activated receptor alpha. Biochem Biophys Res Commun. 2001;287:391–396. doi: 10.1006/bbrc.2001.5608. [DOI] [PubMed] [Google Scholar]
- 36.Huang B, Wu P, Bowker-Kinley MM, Harris RA. Regulation of pyruvate dehydrogenase kinase expression by peroxisome proliferator-activated receptor-alpha ligands, glucocorticoids, and insulin. Diabetes. 2002;51:276–283. doi: 10.2337/diabetes.51.2.276. [DOI] [PubMed] [Google Scholar]
- 37.Kersten S, Mandard S, Tan NS, Escher P, Metzger D, Chambon P, Gonzalez FJ, et al. Characterization of the fasting-induced adipose factor FIAF, a novel peroxisome proliferator-activated receptor target gene. J Biol Chem. 2000;275:28488–28493. doi: 10.1074/jbc.M004029200. [DOI] [PubMed] [Google Scholar]
- 38.Iynedjian PB, Jotterand D, Nouspikel T, Asfari M, Pilot PR. Transcriptional induction of glucokinase gene by insulin in cultured liver cells and its repression by the glucagon-cAMP system. J Biol Chem. 1989;264:21824–21829. [PubMed] [Google Scholar]
- 39.Sapiro JM, Mashek MT, Greenberg AS, Mashek DG. Hepatic triacylglycerol hydrolysis regulates peroxisome proliferator-activated receptor alpha activity. J Lipid Res. 2009;50:1621–1629. doi: 10.1194/jlr.M800614-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Moon YA, Lee JJ, Park SW, Ahn YH, Kim KS. The roles of sterol regulatory element-binding proteins in the transactivation of the rat ATP citrate-lyase promoter. J Biol Chem. 2000;275:30280–30286. doi: 10.1074/jbc.M001066200. [DOI] [PubMed] [Google Scholar]
- 41.Sato R, Okamoto A, Inoue J, Miyamoto W, Sakai Y, Emoto N, Shimano H, et al. Transcriptional regulation of the ATP citrate-lyase gene by sterol regulatory element-binding proteins. J Biol Chem. 2000;275:12497–12502. doi: 10.1074/jbc.275.17.12497. [DOI] [PubMed] [Google Scholar]
- 42.Zhou J, Febbraio M, Wada T, Zhai Y, Kuruba R, He J, Lee JH, et al. Hepatic fatty acid transporter Cd36 is a common target of LXR, PXR, and PPARgamma in promoting steatosis. Gastroenterology. 2008;134:556–567. doi: 10.1053/j.gastro.2007.11.037. [DOI] [PubMed] [Google Scholar]
- 43.Brown MS, Goldstein JL. A proteolytic pathway that controls the cholesterol content of membranes, cells, and blood. Proc Natl Acad Sci U S A. 1999;96:11041–11048. doi: 10.1073/pnas.96.20.11041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shimada A, Tamai T, Oida K, Takahashi S, Suzuki J, Nakai T, Miyabo S. Increase in neutral cholesteryl ester hydrolase activity produced by extralysosomal hydrolysis of high-density lipoprotein cholesteryl esters in rat hepatoma cells (H-35) Biochim Biophys Acta. 1994;1215:126–132. doi: 10.1016/0005-2760(94)90101-5. [DOI] [PubMed] [Google Scholar]
- 45.Ge X, Yin L, Ma H, Li T, Chiang JY, Zhang Y. Aldo-keto reductase 1B7 is a target gene of FXR and regulates lipid and glucose homeostasis. J Lipid Res. 2011;52:1561–1568. doi: 10.1194/jlr.M015859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schmidt DR, Schmidt S, Holmstrom SR, Makishima M, Yu RT, Cummins CL, Mangelsdorf DJ, et al. AKR1B7 is induced by the farnesoid X receptor and metabolizes bile acids. J Biol Chem. 2011;286:2425–2432. doi: 10.1074/jbc.M110.181230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fuchs M. Non-alcoholic Fatty liver disease: the bile Acid-activated farnesoid x receptor as an emerging treatment target. J Lipids. 2012;2012:934396. doi: 10.1155/2012/934396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Reid BN, Ables GP, Otlivanchik OA, Schoiswohl G, Zechner R, Blaner WS, Goldberg IJ, et al. Hepatic overexpression of hormone-sensitive lipase and adipose triglyceride lipase promotes fatty acid oxidation, stimulates direct release of free fatty acids, and ameliorates steatosis. J Biol Chem. 2008;283:13087–13099. doi: 10.1074/jbc.M800533200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Quiroga AD, Li L, Trotzmuller M, Nelson R, Proctor SD, Kofeler H, Lehner R. Deficiency of carboxylesterase 1/esterase-x results in obesity, hepatic steatosis, and hyperlipidemia. Hepatology. 56:2188–2198. doi: 10.1002/hep.25961. [DOI] [PubMed] [Google Scholar]
- 50.Quiroga AD, Lian J, Lehner R. Carboxylesterase1/Esterase-x regulates chylomicron production in mice. PLoS One. 2012;7:e49515. doi: 10.1371/journal.pone.0049515. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.