

Abstract
Red blood cells (RBCs) based drug carrier appears to be the most appealing for protein drugs due to their unmatched biocompatability, biodegradability, and long lifespan in the circulation. Numerous methods for encapsulating protein drugs into RBCs were developed, however, most of them induce partial disruption of the cell membrane, resulting in irreversible alterations in both physical and chemical properties of RBCs. Herein, we introduce a novel method for encapsulating proteins into intact RBCs, which was meditated by a cell penetrating peptide (CPP) developed in our lab—low molecular weight protamine (LMWP). L-asparaginase, one of the primary drugs used in treatment of acute lymphoblastic leukemia (ALL), was chosen as a model protein to illustrate the encapsulation into erythrocytes mediated by CPPs. In addition current treatment of ALL using different L-asparaginase delivery and encapsulation methods as well as their associated problems were also reviewed.
Keywords: Red blood cells, erythrocytes, cell penetrating peptide, acute lymphoblastic leukemia, encapsulatation
1. Introduction
Proteins including hormones[1, 2], monoclonal antibodies[3-5] and vaccines[6-9] are being widely used as drugs for therapeutic purposes, and many of them display powerful and selective therapeutic activities [10]. However, clinical applications of these proteins are limited by their potential immunogenicity[11], degradation by circulating serum proteases, glomerular filtration, and processing by the immune system such as uptake by the reticuloendothelial system (RES), leading to a short plasma half-life, poor bioavailability, and reduced in vivo activities [12, 13]. These problems demand either frequent injection or the use of large doses to achieve the required therapeutic efficacy, which in a way further increases the risk of inducing severe allergic responses [14]. Prolonging the in vivo half-lives of proteins, therefore, becomes one of the essential elements in improving their pharmacokinetic and pharmacodynamic properties [14, 15].
Possessing a relatively long half-life should be a key aspect in the clinical application of protein drugs, and various strategies, including chemical modification [16-20], fusion with other proteins or peptides [21-23], glycosylation [24-26], incorporation of protease resistant variants [27], and encapsulation into micro- or nano-carriers [28-30] etc., were therefore developed during the past decades [31]. Of all the methods established to-date, PEGylation and micro-/nano-encapsulation technologies have been proven effective and applied in clinics [19, 20]. Indeed, the first PEGylated protein drug on the market PEG-adenosine deaminase was approved by FDA in 1991. Nevertheless, PEGylation has its own drawbacks, such as the modification process is non-specific, incomplete and with side reactions, resulting in impairment of the therapeutic functions of the proteins drugs [32].
Micro-/nano-encapsulation technology provides a possible strategy to overcome limitations in applying protein drugs [12, 33, 34]. The pharmacokinetic profiles such as half-life and stability of therapeutic proteins can be vastly improved via their entrapment into polymer-based carriers or living cells [35]. Poly(D,L-lactic-coglycolic-acid) (PLGA) have been extensively exploited as the carrier for protein drugs [36], due to its acceptance by FDA as a biocompatible material. For instance Lupron Depot® (leuprolide acetate), the first marketed injectable PLGA microspheres in 1989, has been shown to provide continuous and sustained release of leuprolide for at least 1 month [37]. However, the degradation of PLGA often leads to accumulation of lactic and glycolic acids within the carrier device, causing a low-pH microenvironment that subsequently induces denaturation and activity loss of the encapsulated protein drug [31, 36]. Among all the drug carriers, red blood cells (RBCs) appears to be most appealing, simply because they are completely biocompatible and biodegradable, and also possess a long life-span of (120 days) that can’t be matched by any other existing carriers [38-41].
A variety of techniques have been successfully developed to entrap protein drugs into RBCs. However, these methods all require disruption of the RBC membranes with chemical (e.g. drug-induced endocytosis [41]), mechanical (hypotonic osmosis/dialysis) [42], or electrical force (electroporation [43, 44]) to create large pores for proteins drugs to diffuse in. Unfortunately, these forces cause membrane deterioration and, as a consequence, result in a loss of structural integrity and cellular components of the erythrocytes, rendering them prone to destruction or recognition by the host immune system. It should be specifically pointed out that in order to inherit the benefits of RBC as a natural and long-circulating drug carrier, it is absolutely essential to retain complete structural and functional integrity of the erythrocytes. Unfortunately most of the existing RBC encapsulation methods to date fail to meet this critical point. In an effort to overcome these obstacles, application of the extraordinarily potent cell-penetrating peptides (CPPs) has been attempted [11], and was later revealed that CPP was able to ferry covalently attached macromolecular species, including proteins and drug carriers, across cell membranes of all organ types including the brain. This discovery could make CPP an elegant solution for delivery of proteins into cells including RBCs [45-47].
Herein, we use L-asparaginase, one of the primary drugs used in treatment of acute lymphoblastic leukemia (ALL), as an example to illustrate the process of RBC encapsulation mediated by CPP. Moreover, current treatment of ALL using different L-asparaginase delivery and encapsulation methods as well as their problems were also reviewed and compared with our CPP mediated encapsulation method.
2. L-asparaginase in Treatment of Acute lymphoblastic leukemia (ALL)
ALL is a type of cancer of the white blood cells in which the bone marrow makes too many immature white blood cells called lymphocytes that are unable to help the body fight infections. As the number of lymphocytes increases in the blood and bone marrow, there is less room for healthy white blood cells, red blood cells, and platelets[48, 49]. As a consequence, ALL patients often suffer infections, anemia, and easy bleeding. Almost 4,000 cases of ALL are diagnosed annually in the United States alone, approximately two thirds of which are in adolescent children, making ALL the most common cancer in this age group[49, 50]. Indeed, although cancer in children and adolescents is rare, ALL represents 23% of the cancers diagnosed among children younger than 15 years of age, occurring at an annual rate of 30 to 40 per million [51]. While a cure rate of ~80% was estimated for childhood ALL, the experience with adult ALL was far less rewarding, as the reported cure rate seldom exceeded 40% [49].
2.1. Reaction Mechanism of L-asparaginase (ASNase) in the Treatment of ALL
One of the primary drugs used in the treatment of ALL is ASNase, which has been in clinical practice since 1967 [14, 52-56]. ASNase is an enzyme which hydrolyzes amino acid L-asparagine (ASN) into L-aspartic acid and ammonia; as shown in Figure 1A. The presence of the enzyme leads to reduction in L-asparagine level and, when given at appropriate times, can lead to persistent depletion of L-asparagine which is non-essential under physiological conditions.
Fig. 1.

A: The reaction mechanism of L-asparaginase. L-asparaginase is hydrolyzed into L-aspartic acid and ammonia[57]; B: Mechanism of action of L-asparaginase. Figure from Müller and Boos [55].
The mechanism of action of the anti-leukemic effect of L-asparaginase is associated with a specific characteristic of malignant lymphoblastic cells related to the metabolic deficiency. Unlike most healthy cells, certain neoplastic tissues, including ALL cells, exist a significantly reduced expression of the enzyme asparagines synthetase (AS), and are therefore unable to synthesize sufficient amounts of L-asparagine. As a consequence, these cells have to rely solely on an extracellular source of L-asparagine to maintain protein synthesis [58]. With an interruption of L-asparagine supply, induced by the administration of L-asparaginase, the malignant blasts no longer have resource of L-asparagine. Systemic depletion of ASN by ASNase would therefore lead to impairment of protein biosynthesis and, subsequently, arrest the cell cycle in leukemic cells, causing their deaths through cellular dysfunction [59]. On the other hand, most healthy cells are able to convert L-aspartic acid to L-asparagine by the action of asparagines synthetase and thus conventions L-glutamine as the amine acid donor. (See Fig. 1B)
2.2. Pitfalls of L-asparaginase (ASNase) Treatment
ASNase formulations currently in clinical use are originated from two bacterial sources, Escherichia coli and Erwinia chrysanthemi. This enzyme is a tetramer with each monomer containing an active site, and has an overall molecular weight of 133-140kDa. The specific activity of purified ASNase ranges between 300- ole of substrate/min/mg of protein. The isoelectric point lies between pH 4.5-5.5 for the E. coli enzyme and 8.6 for the Erwinia chrysanthemi enzyme [14]. The Km is approximately 1×10−5 M [60]. ASNase is not adsorbed from the GI track, and thus in clinical practice, it is normally administered via the intravenous or intramuscular route.
Like most protein drugs, clinical application of ASNase faces two major obstacles. First, ASNase is a non-human protein, and its clinical use is therefore associated with a high incidence of hypersensitive reactions. When a patient has a reaction, the route of administration, form, or source of the drug will be changed. The reaction rate with intravenous (iv) administration of the free form of ASNase is very high. For this reason, the free enzyme is almost always given via the intramuscular (im) or subcutaneous (sc) route. Specifically, with its bacterial origin, ASNase is capable of triggering significant immunological consequences including activation of B lymphocytes and production of antibodies, causing severe anaphylactic and, in certain cases, even fatal reactions [61] . Most allergic reactions occur within one to several hours after drug administration, and include signs and symptoms typically seen in anaphylaxis.
Secondly, like most protein drugs, ASNase is susceptible to degradation by serum proteases, leads to its elimination by the reticuloendothelial system (RES). The plasma half-life of ASNase, which is not related to dose or organ (e.g. liver, kidney, etc.) function, is estimated to be in the range of 8-30 hours [62]. This rapid body clearance demands frequent injection of large doses of ASNase, further elevating the chance of inducing severe immunological responses.
Aside from those problems mentioned above, the use of L-asparaginase can also result in liver dysfunction. The elevated transaminase activity, abnormal bilirubin and alkaline phosphatase levels, and depression in albumine and lipoprotein levels were observed [56]. Other toxicities of L-asparaginase include coagulation abnormalities, pancreatitis, cerebral dysfunction, parotitis, and immune suppression.
3. Red Blood Cells As a Promising Carrier for Protein Drugs
Among all carriers employed for ASNase encapsulation, the use of erythrocytes (red blood cells; RBCs) appears to be most appealing [15, 43, 63-68]. The benefits of utilizing erythrocytes as the drug carrier can be listed as follows. First, RBCs are completely biodegradable without generation of toxic bi-products, and their breakdown products are completely recycled. Second, the erythrocytes protect the loaded proteins from inactivation by endogenous factors and proteolytic degradation. While keeping the drug “safe” from the physiological environment, the erythrocytes can also help avoiding any undesired immune response torwards the encapsulated drug. In addition, erythrocytes are the most abundant cells in the human body (5.4×106 and 4.8×106 RBCs/mL in healthy men and women, respectively), thereby providing an affordable source of supply for use in drug encapsulation. Moreover, RBCs are biconcave in shape and have a mean diameter of 7 to 8 μm and a thickness of approximately 2 μm, which endows them with the largest surface-to-volume ratio (1.9×104 cm/g) usable for drug encapsulation; when comparing to all other cell types [69]. RBCs are also very elastic and able to pass the smallest capillaries to reach the diseased organ/tissue. Altogether, RBCs are well suited to be used as drug carriers, particularly for the unstable protein drugs.
Most importantly, the carrier erythrocytes possess a remarkably longer life-span in circulation when comparing to any of the presently existing drug carriers. On average, the natural erythrocytes have a life-span of 120 days [39, 64, 68]. This means if physical and biological properties of erythrocyte can be fully maintained, the drug encapsulated erythrocytes will be able to circulate much longer than any of the synthetic carriers. This also means that a considerable increase in drug dosing intervals can be managed with drug concentration being maintained at a safe yet effective level for an extremely long time.
The pharmacokinetic and pharmacodynamic parameters of the drug will be changed following modification. For instance, half-life of the pegylated asparginase is approximately 15 days [54, 70] whereas the free enzyme has half-life of only 26h [71]. As being discussed later, when asparginase was encapsulated in erythrocytes, a half-life of up to 29 days was observed. In principle, encapsulation of asparginase within a completely intact erythrocytes can potentially provide the drug with a systemic lifespan similar to that of the erythrocyte.
With the quick development of the RBC carriers, numerous human and animal studies have been conducted to examine drug encapsulation in RBC [72-75]. From these studies, a number of significant challenges and limitations have been identified. Indeed, ex vivo modification (e.g., using donor or autologous blood), leading to the unintentional reduction of the RBC biocompatibility, thereby limits treatment options to the hemo-transfusion setting and restricts a widespread use of this technology [76].
In the case of L-asparaginase, previous study by Ataullakhanov et al indicated that L-asparagine was able to penetrate into human erythrocytes from external medium[77]. To this regard, L-asparaginase encapsulated erythrocytes could function as bioreactor, converting incoming L-asparagine to aspartic acid. If, however, the drug of interest is leached out from the erythrocyte carrier, the amount of the released drug in the circulation would still be less than the level if free drug were being administered. This means that the drug-associated resulting side effects will still be significantly reduced [78].
4. Methods to Entrap Protein Drugs into RBCs
4.1.Existing Methods to Entrap Protein drugs into RBCs and their Disadvantages
A variety of methods have already been established to entrap protein drugs into RBCs. The most adapted techniques thus far include drug (e.g. primaquine, hydrocortisone, etc.)-induced endocytosis [41], electroporation [69], and hypo-osmotic-based pre-swelling [79], rupture/resealing[15, 65], or dialysis [38]. Using these methods to create sufficiently large pores or perturbations on the cell membrane, a number of the impermeable protein drugs including L-asparaginase, erythropoietin, acetaldehyde dehydrogenase, and alcohol dehydrogenase have been successfully loaded into RBCs. It is important to point out that in order to inherit the benefits of RBC as a natural and long-lasting drug carrier, retaining both the physical and chemical attributes of native RBCs is absolutely essential. In view of this requirement, all of the above methods are still beset by a host of shortcomings, despite their reported success.
The most crucial drawbacks in RBC protein encapsulation by existing methods come from two aspects. First, all of the currently available techniques require the application of either a chemical (e.g. drug-induced endocytosis), mechanical (e.g. osmotic dialysis seen in Fig. 2A), or electrical (e.g. electroporation seen in Fig. 2B) force to disrupt the RBC membrane thus creating sufficiently large pores for the protein drug to diffuse through.
Fig. 2.

Schematic illustration of methods for loading protein drug into erythrocyte. (A) Osmotic dialysis, (B) electroporation, and (C) CPP-mediation method.
Such a disruption of the cell membrane, however, often leads to a partial but irreversible deterioration of the structural integrity and morphology of RBCs [64, 76]. As displayed in Fig. 3, a significant alteration of the erythrocyte morphology from the native discocyte form (i.e. normal erythrocyte with a small area of central pallor biconcave disc shape; right panels of both pictures) to stomatocyte (i.e. abnormal erythrocyte with oval or rectangular area of central pallor; left panels of both pictures) following treatment by hypo-osmosis (top column) or electroporation (bottom column) was clearly observed. Yet, the structural integrity of RBC membrane is the primary basis for ensuring the physical integrity and physiologic functions of the RBC. Any morphological or physical change in RBC could result in the recognition of the processed RBCs as foreign entities by the phagocytic systems, Consequently, these processed RBCs will be recognized by the phagocytic system as foreign entities, rendering their rapid destruction and clearance by the host immune system.
Fig. 3.
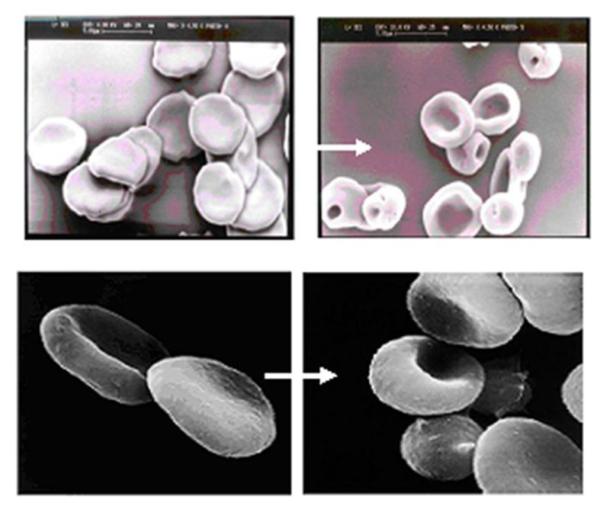
RBC processed by Hypo-osmosis (Top); and Electroporation (Bottom) methods. Left panels: native RBCs with the normal discocyte form; Right panels: processed RBCs with the abnormal stomatocyte form. [80]
Second, erythrocytes processed by the above methods would inevitably result in a loss of important cellular constituents, such as hemoglobin and cytoskeleton, from the cells. This is because all of these methods rely on a pore-opening and a resealing mechanism [42, 80], and while they allow the protein drug to diffuse in, they also allow the cell constituents to diffuse out. Indeed, loss of hemoglobin was clearly observed in erythrocytes treated with the hypo-osmosis dialysis method, as evidenced by a decrease in MCH and presence of a pinkish color after resealing [38, 80]. A loss of hemoglobin would not only impair the oxygen transport function of RBCs, but also affect their ability to manage oxidative stress. Similarly, a loss of cytoskeleton from the erythrocyte would compromise it with a much weakened the structural and functional integrity of the cell is absolutely structural integrity, rendering it prone to destruction or recognition by the phagocytic system.
4.2.Proposed RBC Encapsulation Technology by Utilizing Cell Penetrating Peptides
4.2.1. The Concept of the Proposed RBC Encapsulation Technology
The concept of the proposed encapsulation technology was fostered primarily from non-selective cell internalization phenomena mediated from the aforementioned cell-penetrating peptides (CPPs; sometimes termed as the protein transduction domain, PTD). Based on this nature, in principle and practice, all cell types including erythrocytes should be transducible. In addition, since it was also demonstrated that CPP-mediated cell internalization did not induce any perturbation or alteration of the cell membrane, these CPP peptides could potentially be applied as a powerful tool to achieve non-invasive encapsulation of biologically active protein therapeutics into intact and fully functional erythrocytes. These hypotheses provide the framework of our proposed innovative method for encapsulation of ASNase into RBCs.
As displayed in Fig. 4., the new encapsulation method called for covalent conjugation of the protein with a CPP via a disulfide linkage. Because of the universal and potent membrane-penetrating activity of CPP, even without the involvement of any invasive membrane-disrupting procedures, the CPP-ASNase conjugate is still able to internalize into erythrocytes without altering RBC’s structural and/or functional attributes. Within the erythrocyte, CPP would be, by design, dissociated from its protein counterpart via degradation of the disulfide linkage, due to the presence of a high level of cytoplasmic glutathione and reductase activities. This bond dissociation would enable the cell-impermeable ASNase to remain permanently entrapped in the erythrocytes, ensuring protection of the enzyme drug from detection by the host immune system and clearance by RES and other endogenous factors.
Fig. 4.
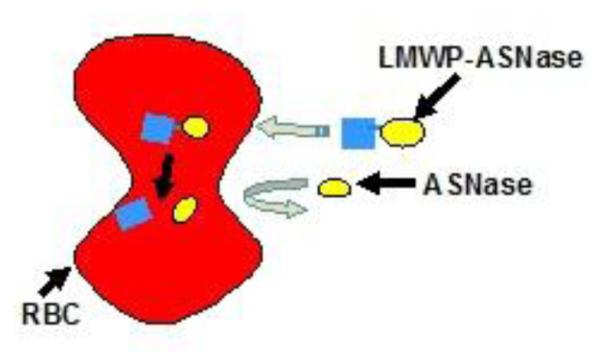
Schematic illustration of the proposed RBC encapsulation technology
Ataullakhanov and co-workers demonstrated that the substrate asparagine was able to permeate into human erythrocytes from external medium [77]. Hence, ASNase-encapsulated erythrocytes would function as a living bioreactor, depleting ASN in the circulation and depriving leukemic cells of their essential nutrient and, consequently, leading to death of these cells (see Fig. 5).
Fig. 5.
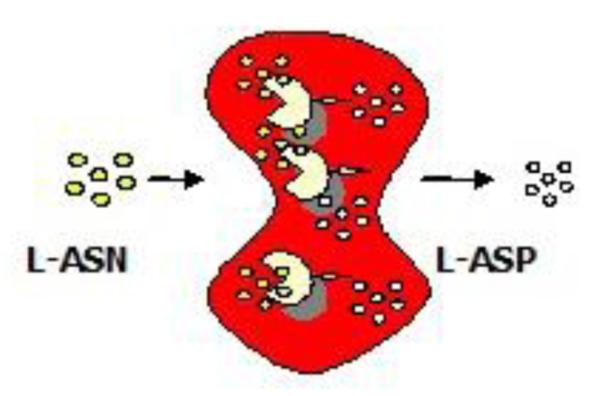
Conversion of asparagine (ASN) to aspartic acid (ASP) by RBC-encapsulated asparaginase (ASNase)
4.2.2. LMWP Functions as a Potent CPP
To inherit the benefits of RBCs as a natural and long-lasting drug carrier, it is absolutely essential to retain both the structural and functional integrity of erythrocyte. As described previously, recent discovery of a family of small but extraordinarily potent CPPs, provides an innovative means to encapsulate therapeutic proteins into fully functional erythrocytes. In general, currently existing CPPs are short peptides containing (3 - 30 amino acid residues) that are capable of transporting molecules across the cell membrane [81-85]. CPPs found to date include TAT28 [86, 87], ANTP29 [42], VP2230 [88], poly (arginine) peptides [63], and the non-toxic, naturally occurring low molecular weight protamine (LMWP) developed in our own laboratory [89, 90]. These CPPs have been attempted to enhance extracellular and intracellular internalization of various biomolecules including peptides, proteins (MW > 150 kDa; more than 60 different proteins have already been tested), plasmid DNAs, siRNAs, oligonucleotides, peptide-nucleic acids (PNA), nano-carriers, and liposomes [45, 89-99].
Low molecular weight protamine (LMWP) (MW: ~1.8 kDa, VSRRRRRRGGRRRR) is an arginine-rich peptide previously developed in our lab by enzymatic digestion of native protamine with thermolysin [90, 100]. This peptide was previously shown to retain a nearly complete heparin-neutralizing ability yet with a significantly less toxicity in vivo. Extensive animal studies have been conducted to demonstrate that LMWP is neither immunogenic nor antigenic [90]. In addition, unlike most of the commonly encountered highly cationic peptides, administration of LMWP into dogs did not elicit acute hypotensive responses or other toxicity such as complement activation [91, 100]. Recently, it was demonstrated that since LMWP shared certain sequence similarity with TAT, the most established CPP to date, it also similarly possesses the potent cell-penetrating activity [93, 94, 96]. This finding was of great significance, simply because LMWP possesses several major advantages over all of the existing CPPs. First, unlike other CPPs that rely solely on chemical synthesis for their production, LMWP can be manufactured in mass quantities using enzymatic hydrolysis and an established single step purification system. Secondly, unlike most other CPPs which are derived from viral resources and thus present health concerns, LMWP is obtained from digestion of native protamine; a FDA approved clinical drug. Thirdly, unlike all existing CPPs, the toxicology profile of LMWP has already been thoroughly established; as previously presented. Last but not least, since LMWP possesses only one single -NH2 group at the N-terminus, its conjugation to a protein drug can be precisely regulated and easily carried out using the standard and well-established N-succinimidyl-3-(2-pyridyldithio) propionate (SPDP) activation method [98, 101].
4.3.RBC Encapsulation of Protein Drugs by LMWP Mediated Encapsulation
4.3.1. Ovalbumin as the Testing Protein
To examine the general feasibility of utilizing LMWP to translocate proteins into erythrocytes, we first adopted Alexa Fluor 488 (a fluorescent dye)-labeled ovalbumin as the protein model. Briefly, LMWP was introduced with a thiol (-SH) functional group at its N-terminus by using the bifunctional cross-linker 3-(2-pyridyldithio)propionic acid N-hydroxysuccinimide (SPDP) activating agent, and was then coupled with a similarly SPDP-activated ovalbumin molecule. As shown in the confocal microscopic images in Fig. 6, RBCs incubated with the dye-labeled ovalbumin displayed only vague auto-fluorescence from hemoglobin excitation on the cell surface, with no observable uptake of the labeled protein within the interior of the cells (Fig. 6 second row); a phenomenon that was almost identical to that of the untreated control RBCs (Fig. 6 first row). On the contrary, after conjugation with LMWP, significant intracellular fluorescence was detected within the RBC carriers, confirming the occurrence of LMWP-mediated cell internalization of the otherwise impermeable ovalbumin (Fig. 6 third row).
Fig. 6.
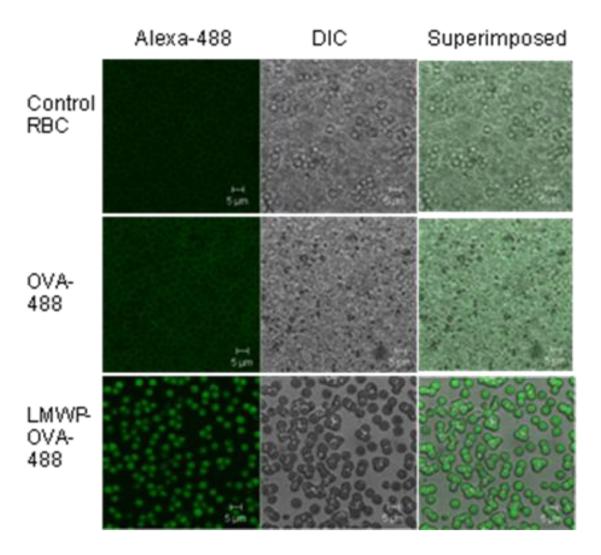
Confocol microscopic images of untreated RBC (Control RBC; first row); RBC incubated with Alex a Fluor 488-labeled ovalbumin (OVA-488, second row); and LMWP-ovalbumin (LMWP-OVA-488; third row). First column: fluorescence mode; second column: DIC mode; and third column: superimposition. [101]
4.3.2. RBC Encapsulation of L-Asparaginase
For encapsulation, RBCs were incubated with the LMWP-ASNase conjugates at a total ASNase concentration of 100 IU/mL. A loading efficiency of 4% was observed, with a loading capacity of 8 IU ASNase per packed volume of 100 μL of packed RBCs. During the loading process, no hemolysis or loss of hemoglobin was detected. In addition, no leaching of ASNase from the loaded RBCs was detected during the first 14 days of incubation in isotonic buffer at 4°C. After this period, both control and ASNase-loaded RBCs began to show signs of disintegration in vitro. When the loaded RBCs were lysed after 14 days of incubation, 70% of ASNase activity could be recovered. Since the RBCs were treated with trypsin and washed with Alsever’s solution after ASNase loading, the recovered enzymatic activity was clearly from the intracellularly entrapped ASNase. By utilizing an optimized encapsulation protocol, our results showed that reasonable ASNase loading efficiency (~4%) and RBC loading capacity (8 IU of ASNase per 100 L of packed RBCs) were consistently achieved. It should be noted that clinically, the dose regimen for ASNase as a sole induction agent in the treatment of ALL is about 200 IU/kg body weight. Hence, even based on our currently established loading protocol, this clinical dosing regimen, which can be translated into a dose of 3 mL of ASNase-loaded RBCs per kg of body weight, is obviously quite achievable.
It was speculated that the detachment of LMWP from ASNase via degradation of the disulfide bonds by the elevated glutathione activity in the cytosol caused the membrane-impermeable ASNase to be trapped inside. On the other hand, so far there has been no report to implicate that the CPP-mediated cell entry is a reversible process. Hence, the permanent entrapment of the protein drug inside RBCs could also result from this irreversible translocation process. Albeit controversial, most literature reports suggested that the final destiny of the CPP-mediate event was the nucleus. Since RBCs are non-dividing and non-nucleated cells, it is without any doubt that the entrapped ASNase stay in the cytosol of the RBC. Overall, the absence of leaching and activity decay of the entrapped ASNase indeed fulfills one of the essential prerequisites for the ASNase-loaded RBCs to be considered as a real clinical remedy (see Fig. 7).
Fig. 7.

Morphological and structural integrity of RBCs after Encapsulation with LMWP-ASNase. Scanning electron microscopic images of (A) Untreated normal RBCs (i.e. Control); and LMWP-ASNase-.encapsulated RBCs; Hematological Parameters of loaded RBCs (B); Osmotic fragility curves for normal (red line) and ASNase-loaded (green line) RBCs (C); oxygen-carrying
Scanning electron microscopic (SEM) observations revealed that RBCs, after incubation with the LMWP-ASNase at 37 °C for 1 hour, maintained a complete morphological integrity (Fig. 7A) [101]. Examination of the key hematological parameters, including the mean corpuscular volume (MCV), mean hemoglobin content (MHC), and mean corpuscular hemoglobin concentrations (MCHC) (Fig. 7B), and osmotic fragility (Fig. 7C) confirmed that RBCs virtually maintained a complete structural integrity. All above results provide support to our hypothesis, i.e. CPP-mediated protein encapsulation is a non-invasive process that does not compromise the RBC membrane with any weakened structural behavior.
5. In Vivo application of RBC-Encapsulated ASNase in the Treatment of Acute Lymphoblastic Lenkemia
To evaluate the feasibility of this novel RBC encapsulation method, L-asparaginase (ASNase), a clinically approved protein drug used commonly in the treatment of acute lymphoblastic leukemia (ALL), was selected as the model protein therapeutics and conjugated to the cell-penetrating low molecular weight protamine (LMWP) peptide via a disulfide bond [101]. As reported, the LMWP peptide developed in our laboratory was proven to be a potent yet non-toxic CPP [90].
The true proof of the benefits of the CPP-mediated cell encapsulation method was stemmed from the in vivo pharmacokinetic investigation of the plasma half-life of ASNase-loaded erythrocytes [101]. After intravenous injection of ASNase-loaded RBCs into DBA/2 mice, the circulating half-life based on enzymatic activity was observed to follow the linear pattern of the elimination phase. For comparison, RBCs loaded with ASNase via standard hypotonic methods were used as the control. As shown in Fig. 8A, a half-life of approximately 5.9 days was observed for the ASNase-loaded erythrocytes processed with the hypotonic methods. In a sharp contrast, ASNase-loaded erythrocytes processed with our novel LMWP-mediated encapsulation method exhibited a significant prolonged plasma half-life of 9.2 days; a period that almost doubled the value obtained for the control, hypotonic-treated erythrocytes. In addition, preliminary animal studies concerning the therapeutic functions of ASNase-loaded erythrocytes against DBA/2 mice harboring the L5178Y lymphoma tumor cells were conducted. As displayed in Fig. 8B, administration of 100μL of the LMWP-ASNase-loaded RBCs (8 IU/mouse) was already capable of significantly increasing the survival time of the mice (14.4 ±2.3 days), when compared to that (10.0±1.4 days) of the control, saline-injected animal group; a dramatic enhancement of the survival time by 44%. Consistent with findings reported by other investigators (102), our in vivo experiments by intravenous injection to the L5178Y tumor-bearing mice of a total dose of 8 IU of free ASNase showed a maximum enhancement of 16.7% in the survival time over the control group administered with saline only. In sharp contrast, a nearly 3-fold increase over their results (i.e. 44% versus 16.7%) in the enhancement of life span was observed for our RBC-ASNase treated mice, particularly considering the fact that merely a volume as small as 100μL of the ASNase-encapsulated RBCs were infused into these tumor-bearing mice. Further studies to more conclusively demonstrate the therapeutic benefits of the proposed system by infusion a larger volume of the ASNase-encapsulated RBC is currently under way. All together, the asparaginase-entrapped RBCs prepared by using our novel encapsulation method not only yielded a long circulating half-life similar to that of untreated RBCs, but also produced a much enhanced therapeutic efficacy of the entrapped protein drug, presumably via protection of the drug by RBCs from possible proteolytic degradation and phagocytic clearance.
Fig. 8.
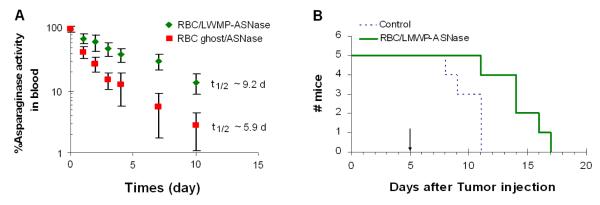
Pharmacokinetic profiles of ASNase activity in blood in DBA2 mice (A) and Kaplan-Meier survival curve for DBA/2 mice bearing L5178Y lymphoma cells (B). [101]
6. Discussion and Prospectives
Presented herein is a proof-of-concept demonstration of the feasibility of encapsulating protein drugs into intact and fully functional erythrocytes. As displayed in Fig. 1C, the new encapsulation method calls for covalent conjugation of the protein with a CPP via a disulfide linkage. Because of the universal and potent membrane-penetrating activity of CPP, even without the involvement of any invasive membrane-disrupting procedures, the CPP-protein conjugate is still able to internalize erythrocytes without altering the structural and/or functional attributes of RBCs. Within the erythrocyte, CPP would be, by design, automatically dissociated from its protein counterpart via degradation of the disulfide linkage, due to the presence of a high level of cytoplasmic glutathione and reductase activities. This detachement of CPP would enable the cell-impermeable protein drug to remain permanently entrapped inside the erythrocytes, thus shielding it from detection by the host immune system and clearance by RES and other circulating endogenous factors.
Based on our preliminary findings, the proposed novel RBC encapsulation method could be applied to encapsulating liposomes, nanoparticles, or micelles into intact RBCs without any structural or functional damage. Nevertheless, there are still many limitations that could hinder the use of this method in clinics. One of the major hurdles lies in the lack of appropriate expertise and “know-how” in the clinical setting to handle the RBC-loading procedure. A typical hospital pharmacy, although it may have the stock of the cell-permeable LMWP-protein conjugates, lacks the required expertise to manage RBC encapsulation. On the other hand, while professional blood banks are capable of handling blood products, the ex vivo loading of a drug into the RBC may still be beyond the scope of their routine work. Most critically, even all the problems associated with RBC encapsulation have been resolved, the erythrocytes thus prepared may still not be suitable for clinical application, simply because they may not be compatible with the patient’s blood type and thus would cause immune resistance and rejection. To overcome these hurdles, an engineering approach that could solve these problems is currently being explored and under development in our own laboratories.
Acknowledgments
This work was supported in part by NIH R01 Grant CA114612, School of Pharmacy, Fudan University & The Open Project Program of Key Lab of Smart Drug Delivery (Fudan University), Ministry of Education, China(SDD2013-04). This research was also partially sponsored by National Natural Science Foundation of China (NSFC, 81361140344), Tianjin Municipal Science and Technology Commission (12JCZDJC34000) and National Key Basic Research Program of China(2011CB933100)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Vandenberg LN, Colborn T, Hayes TB, Heindel JJ, Jacobs DR, Jr., Lee DH, Shioda T, Soto AM, vom Saal FS, Welshons WV, Zoeller RT, Myers JP. Hormones and endocrine-disrupting chemicals: low-dose effects and nonmonotonic dose responses. Endocr. Rev. 2012;33:378–455. doi: 10.1210/er.2011-1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Zhang J, Lazar MA. The mechanism of action of thyroid hormones. Annu. Rev. Physiol. 2000;62:439–466. doi: 10.1146/annurev.physiol.62.1.439. [DOI] [PubMed] [Google Scholar]
- [3].Debbio C.B. Del, Tonon LM, Secoli SR. Monoclonal antibody therapy: a literature review. Rev. Gaucha. Enferm. 2007;28:133–142. [PubMed] [Google Scholar]
- [4].Leavy O. Therapeutic antibodies: past, present and future. Nat. Rev. Immunol. 2010;10:297. doi: 10.1038/nri2763. [DOI] [PubMed] [Google Scholar]
- [5].Scott AM, Wolchok JD, Old LJ. Antibody therapy of cancer. Nat. Rev. Cancer. 2012;12:278–287. doi: 10.1038/nrc3236. [DOI] [PubMed] [Google Scholar]
- [6].Cheever MA, Higano CS. PROVENGE (Sipuleucel-T) in prostate cancer: the first FDA-approved therapeutic cancer vaccine. Clin. Cancer Res. 2011;17:3520–3526. doi: 10.1158/1078-0432.CCR-10-3126. [DOI] [PubMed] [Google Scholar]
- [7].Gulley JL, Madan RA, Heery CR. Therapeutic vaccines and immunotherapy in castration-resistant prostate cancer. Am. Soc. Clin. Oncol. Educ Book/ASCO. Am. Soc. Clin. Oncol. Meeting. 2012;2013:166–170. doi: 10.1200/EdBook_AM.2013.33.e166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Lubaroff DM. Prostate cancer vaccines in clinical trials. Expert Rev. Vaccines. 2012;11:857–868. doi: 10.1586/erv.12.54. [DOI] [PubMed] [Google Scholar]
- [9].Ogi C, Aruga A. Clinical evaluation of therapeutic cancer vaccines. Hum. Vaccin Immunother. 2013;9:1049–1057. doi: 10.4161/hv.23917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Carter PJ. Introduction to current and future protein therapeutics: a protein engineering perspective. Exp. Cell Res. 2011;317:1261–1269. doi: 10.1016/j.yexcr.2011.02.013. [DOI] [PubMed] [Google Scholar]
- [11].Huang Y, Park YS, Wang J, Moon C, Kwon YM, Chung HS, Park YJ, Yang VC. ATTEMPTS system: a macromolecular prodrug strategy for cancer drug delivery. Curr. Pharm. Des. 2010;16:2369–2376. doi: 10.2174/138161210791920441. [DOI] [PubMed] [Google Scholar]
- [12].Haag R, Kratz F. Polymer therapeutics: concepts and applications. Angew Chem. Int. Ed. Engl. 2006;45:1198–1215. doi: 10.1002/anie.200502113. [DOI] [PubMed] [Google Scholar]
- [13].Tang L, Persky AM, Hochhaus G, Meibohm B. Pharmacokinetic aspects of biotechnology products. J. Pharm. Sci. 2004;93:2184–2204. doi: 10.1002/jps.20125. [DOI] [PubMed] [Google Scholar]
- [14].Graham ML. Pegaspargase: a review of clinical studies. Adv. Drug Deliv. Rev. 2003;55:1293–1302. doi: 10.1016/s0169-409x(03)00110-8. [DOI] [PubMed] [Google Scholar]
- [15].Updike SJ, Wakamiya RT, Lightfoot EN., Jr. Asparaginase entrapped in red blood cells: action and survival. Science. 1976;193:681–683. doi: 10.1126/science.821145. [DOI] [PubMed] [Google Scholar]
- [16].Katre NV, Knauf MJ, Laird WJ. Chemical modification of recombinant interleukin 2 by polyethylene glycol increases its potency in the murine Meth A sarcoma model. Proc. Natl. Acad. Sci. 1987;84:1487–1491. doi: 10.1073/pnas.84.6.1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Tsutsumi Y, Onda M, Nagata S, Lee B, Kreitman RJ, Pastan I. Site-specific chemical modification with polyethylene glycol of recombinant immunotoxin anti-Tac(Fv)-PE38 (LMB-2) improves antitumor activity and reduces animal toxicity and immunogenicity. Proc. Natl. Acad. Sci. 2000;97:8548–8553. doi: 10.1073/pnas.140210597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Bailon P, Palleroni A, Schaffer CA, Spence CL, Fung WJ, Porter JE, Ehrlich GK, Pan W, Xu ZX, Modi MW, Farid A, Berthold W, Graves M. Rational design of a potent, long-lasting form of interferon: a 40 kDa branched polyethylene glycol-conjugated interferon alpha-2a for the treatment of hepatitis C. Bioconjug. Chem. 2001;12:195–202. doi: 10.1021/bc000082g. [DOI] [PubMed] [Google Scholar]
- [19].Veronese FM, Pasut G. PEGylation, successful approach to drug delivery. Drug Discov. Today. 2005;10:1451–1458. doi: 10.1016/S1359-6446(05)03575-0. [DOI] [PubMed] [Google Scholar]
- [20].Greenwald RB, Choe YH, McGuire J, Conover CD. Effective drug delivery by PEGylated drug conjugates. Adv. Drug Deliv. Rev. 2003;55:217–250. doi: 10.1016/s0169-409x(02)00180-1. [DOI] [PubMed] [Google Scholar]
- [21].Buscaglia CA, Alfonso J, Campetella O, Frasch AC. Tandem amino acid repeats from Trypanosoma cruzi shed antigens increase the half-life of proteins in blood. Blood. 1999;93:2025–2032. [PubMed] [Google Scholar]
- [22].Dennis MS, Zhang M, Meng YG, Kadkhodayan M, Kirchhofer D, Combs D, Damico LA. Albumin binding as a general strategy for improving the pharmacokinetics of proteins. J. Biol. Chem. 2002;277:35035–35043. doi: 10.1074/jbc.M205854200. [DOI] [PubMed] [Google Scholar]
- [23].Kontermann RE. Strategies to extend plasma half-lives of recombinant antibodies. BioDrugs. 2009;23:93–109. doi: 10.2165/00063030-200923020-00003. [DOI] [PubMed] [Google Scholar]
- [24].Jefferis R. Glycosylation as a strategy to improve antibody-based therapeutics. Nat. Rev. Drug Discov. 2009;8:226–234. doi: 10.1038/nrd2804. [DOI] [PubMed] [Google Scholar]
- [25].Sinclair AM, Elliott S. Glycoengineering: the effect of glycosylation on the properties of therapeutic proteins. J. Pharm. Sci. 2005;94:1626–1635. doi: 10.1002/jps.20319. [DOI] [PubMed] [Google Scholar]
- [26].Baynes JW, Wold F. Effect of glycosylation on the in vivo circulating half-life of ribonuclease. J. Biol. Chem. 1976;251:6016–6024. [PubMed] [Google Scholar]
- [27].Marshall SA, Lazar GA, Chirino AJ, Desjarlais JR. Rational design and engineering of therapeutic proteins. Drug Discov. Today. 2003;8:212–221. doi: 10.1016/s1359-6446(03)02610-2. [DOI] [PubMed] [Google Scholar]
- [28].Yavlovich A, Smith B, Gupta K, Blumenthal R, Puri A. Light-sensitive lipid-based nanoparticles for drug delivery: design principles and future considerations for biological applications. Mol. Membr. Biol. 2010;27:364–381. doi: 10.3109/09687688.2010.507788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Thierry B. Drug nanocarriers and functional nanoparticles: applications in cancer therapy. Curr. Drug Deliv. 2009;6:391–403. doi: 10.2174/156720109789000474. [DOI] [PubMed] [Google Scholar]
- [30].Sultana S, Khan MR, Kumar M, Kumar S, Ali M. Nanoparticles-mediated drug delivery approaches for cancer targeting: a review. J. Drug Target. 2013;21:107–125. doi: 10.3109/1061186X.2012.712130. [DOI] [PubMed] [Google Scholar]
- [31].Pisal DS, Kosloski MP, Balu-Iyer SV. Delivery of therapeutic proteins. J. Pharm Sci. 2010;99:2557–2575. doi: 10.1002/jps.22054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Veronese FM. Peptide and protein PEGylation: a review of problems and solutions. Biomaterials. 2001;22:405–417. doi: 10.1016/s0142-9612(00)00193-9. [DOI] [PubMed] [Google Scholar]
- [33].Baran ET, Ozer N, Hasirci V. In vivo half life of nanoencapsulated L-asparaginase. J. Mater. Sci. Mater. Med. 2002;13:1113–1121. doi: 10.1023/a:1021125617828. [DOI] [PubMed] [Google Scholar]
- [34].DeLoach JR. Encapsulation of exogenous agents in erythrocytes and the circulating survival of carrier erythrocytes. J. Appl. Biochem. 1983;5:149–157. [PubMed] [Google Scholar]
- [35].Bilati U, Allemann E, Doelker E. Strategic approaches for overcoming peptide and protein instability within biodegradable nano- and microparticles. Eur. J. Pharm. Biopharm. 2005;59:375–388. doi: 10.1016/j.ejpb.2004.10.006. [DOI] [PubMed] [Google Scholar]
- [36].Zolnik BS, Burgess DJ. Effect of acidic pH on PLGA microsphere degradation and release. J. Control Release. 2007;122:338–344. doi: 10.1016/j.jconrel.2007.05.034. [DOI] [PubMed] [Google Scholar]
- [37].Okada H, Inoue Y, Heya T, Ueno H, Ogawa Y, Toguchi H. Pharmacokinetics of once-a-month injectable microspheres of leuprolide acetate. Pharm. Res. 1991;8:787–791. doi: 10.1023/a:1015818504906. [DOI] [PubMed] [Google Scholar]
- [38].Garin MI, Lopez RM, Sanz S, Pinilla M, Luque J. Erythrocytes as carriers for recombinant human erythropoietin. Pharm. Res. 1996;13:869–874. doi: 10.1023/a:1016049027661. [DOI] [PubMed] [Google Scholar]
- [39].Magnani M, Rossi L, D’Ascenzo M, Panzani I, Bigi L, Zanella A. Erythrocyte engineering for drug delivery and targeting. Biotechnol. Appl. Biochem. 1998;28(Pt 1):1–6. [PubMed] [Google Scholar]
- [40].Cremel M, Guerin N, Horand F, Banz A, Godfrin Y. Red blood cells as innovative antigen carrier to induce specific immune tolerance. Int. J. Pharm. 2013;443:39–49. doi: 10.1016/j.ijpharm.2012.12.044. [DOI] [PubMed] [Google Scholar]
- [41].Matovcik LM, Junga IG, Schrier SL. Drug-induced endocytosis of neonatal erythrocytes. Blood. 1985;65:1056–1063. [PubMed] [Google Scholar]
- [42].Joliot A, Pernelle C, Deagostini-Bazin H, Prochiantz A. Antennapedia homeobox peptide regulates neural morphogenesis. Proc. Natl. Acad. Sci. 1991;88:1864–1868. doi: 10.1073/pnas.88.5.1864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Rossi L, Serafini S, Pierige F, Antonelli A, Cerasi A, Fraternale A, Chiarantini L, Magnani M. Erythrocyte-based drug delivery. Expert Opin. Drug Deliv. 2005;2:311–322. doi: 10.1517/17425247.2.2.311. [DOI] [PubMed] [Google Scholar]
- [44].Millan CG, Marinero ML, Castaneda AZ, Lanao JM. Drug, enzyme and peptide delivery using erythrocytes as carriers. J. Control Release. 2004;95:27–49. doi: 10.1016/j.jconrel.2003.11.018. [DOI] [PubMed] [Google Scholar]
- [45].Lee TY, Park YS, Garcia GA, Sunahara RK, Woods JH, Yang VC. Cell permeable cocaine esterases constructed by chemical conjugation and genetic recombination. Mol. Pharm. 2012;9:1361–1373. doi: 10.1021/mp200623w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Gu G, Xia H, Hu Q, Liu Z, Jiang M, Kang T, Miao D, Tu Y, Pang Z, Song Q, Yao L, Chen H, Gao X, Chen J. PEG-co-PCL nanoparticles modified with MMP-2/9 activatable low molecular weight protamine for enhanced targeted glioblastoma therapy. Biomaterials. 2013;34:196–208. doi: 10.1016/j.biomaterials.2012.09.044. [DOI] [PubMed] [Google Scholar]
- [47].David AE, Gong J, Chertok B, Domszy RC, Moon C, Park YS, Wang NS, Yang AJ, Yang VC. Immobilized thermolysin for highly efficient production of low-molecular-weight protamine--an attractive cell-penetrating peptide for macromolecular drug delivery applications. J. Biomed. Mater. Res. A. 2012;100:211–219. doi: 10.1002/jbm.a.33244. [DOI] [PubMed] [Google Scholar]
- [48].Apostolidou E, Swords R, Alvarado Y, Giles FJ. Treatment of acute lymphoblastic leukaemia : a new era. Drugs. 2007;67:2153–2171. doi: 10.2165/00003495-200767150-00004. [DOI] [PubMed] [Google Scholar]
- [49].Pui CH, Evans WE. Treatment of acute lymphoblastic leukemia. N. Engl. J. Med. 2006;354:166–178. doi: 10.1056/NEJMra052603. [DOI] [PubMed] [Google Scholar]
- [50].Langebrake C, Reinhardt D, Ritter J. Minimising the long-term adverse effects of childhood leukaemia therapy. Drug Saf. 2002;25:1057–1077. doi: 10.2165/00002018-200225150-00002. [DOI] [PubMed] [Google Scholar]
- [51]. http://www.cancer.gov/
- [52].Hill JM, Roberts J, Loeb E, Khan A, MacLellan A, Hill RW. L-asparaginase therapy for leukemia and other malignant neoplasms. Remission in human leukemia. JAMA. 1967;202:882–888. [PubMed] [Google Scholar]
- [53].Becker FF, Broome JD. L-asparaginase: inhibition of endogenous RNA polymerase activity in regenerating liver. Arch. Biochem. Biophys. 1969;130:332–336. doi: 10.1016/0003-9861(69)90041-1. [DOI] [PubMed] [Google Scholar]
- [54].Ho DH, Brown NS, Yen A, Holmes R, Keating M, Abuchowski A, Newman RA, Krakoff IH. Clinical pharmacology of polyethylene glycol-L-asparaginase. Drug Metab. Dispos. 1986;14:349–352. [PubMed] [Google Scholar]
- [55].Muller HJ, Boos J. Use of L-asparaginase in childhood ALL. Crit. Rev. Oncol. Hematol. 1998;28:97–113. doi: 10.1016/s1040-8428(98)00015-8. [DOI] [PubMed] [Google Scholar]
- [56].Oettgen HF, Tallal L, Tan CC, Murphy ML, Clarkson BD, Golbey RD, Krakoff IH, Karnofsky DA, Burchenal HJ. Clinical experience with L-asparaginase. Recent Results Cancer Res. 1970;33:219–235. doi: 10.1007/978-3-642-99984-0_27. [DOI] [PubMed] [Google Scholar]
- [57]. http://www.man.poznan.pl/CBB/POSTER1/poster.html.
- [58].Cooney DA, Handschumacher RE. L-asparaginase and L-asparagine metabolism. Annu. Rev. Pharmacol. 1970;10:421–440. doi: 10.1146/annurev.pa.10.040170.002225. [DOI] [PubMed] [Google Scholar]
- [59].Becker FF, Broome JD. L-asparaginase: inhibition of early mitosis in regenerating rat liver. Science. 1967;156:1602–1603. doi: 10.1126/science.156.3782.1602. [DOI] [PubMed] [Google Scholar]
- [60].Wriston JC, Jr., Yellin TO. L-asparaginase: a review. Adv. Enzymol. Relat. Areas Mol. Biol. 1973;39:185–248. doi: 10.1002/9780470122846.ch3. [DOI] [PubMed] [Google Scholar]
- [61].Shepherd GM. Hypersensitivity reactions to chemotherapeutic drugs. Clin. Rev. Allergy. Immunol. 2003;24:253–262. doi: 10.1385/CRIAI:24:3:253. [DOI] [PubMed] [Google Scholar]
- [62].Information AD. Asparaginase. American Society of Health System Pharmacists. 2001:872–875. [Google Scholar]
- [63].Hamidi M, Tajerzadeh H. Carrier erythrocytes: an overview. Drug Deliv. 2003;10:9–20. doi: 10.1080/713840329. [DOI] [PubMed] [Google Scholar]
- [64].Biagiotti S, Paoletti MF, Fraternale A, Rossi L, Magnani M. Drug delivery by red blood cells. IUBMB Life. 2011;63:621–631. doi: 10.1002/iub.478. [DOI] [PubMed] [Google Scholar]
- [65].Ihler GM, Glew RH, Schnure FW. Enzyme loading of erythrocytes. Proc. Natl. Acad. Sci. U S A. 1973;70:2663–2666. doi: 10.1073/pnas.70.9.2663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Luo R, Mutukumaraswamy S, Venkatraman SS, Neu B. Engineering of erythrocyte-based drug carriers: control of protein release and bioactivity. J. Mater. Sci. Mater. Med. 2012;23:63–71. doi: 10.1007/s10856-011-4485-2. [DOI] [PubMed] [Google Scholar]
- [67].Pierige F, Serafini S, Rossi L, Magnani M. Cell-based drug delivery. Adv. Drug Deliv. Rev. 2008;60:286–295. doi: 10.1016/j.addr.2007.08.029. [DOI] [PubMed] [Google Scholar]
- [68].Zolla L, Lupidi G, Marcheggiani M, Falcioni G, Brunori M. Red blood cells as carriers for delivering of proteins. Ann. Ist. Super Sanita. 1991;27:97–103. [PubMed] [Google Scholar]
- [69].Lizano C, Sanz S, Luque J, Pinilla M. In vitro study of alcohol dehydrogenase and acetaldehyde dehydrogenase encapsulated into human erythrocytes by an electroporation procedure. Biochim. Biophys. Acta. 1998;1425:328–336. doi: 10.1016/s0304-4165(98)00085-3. [DOI] [PubMed] [Google Scholar]
- [70].Fu CH, Sakamoto KM. PEG-asparaginase. Expert Opin. Pharmacother. 2007;8:1977–1984. doi: 10.1517/14656566.8.12.1977. [DOI] [PubMed] [Google Scholar]
- [71].Avramis VI, Sencer S, Periclou AP, Sather H, Bostrom BC, Cohen LJ, Ettinger AG, Ettinger LJ, Franklin J, Gaynon PS, Hilden JM, Lange B, Majlessipour F, Mathew P, Needle M, Neglia J, Reaman G, Holcenberg JS, Stork L. A randomized comparison of native Escherichia coli asparaginase and polyethylene glycol conjugated asparaginase for treatment of children with newly diagnosed standard-risk acute lymphoblastic leukemia: a Children’s Cancer Group study. Blood. 2002;99:1986–1994. doi: 10.1182/blood.v99.6.1986. [DOI] [PubMed] [Google Scholar]
- [72].Alanazi FK, Gel D. Harisa, Maqboul A, Abdel-Hamid M, Neau SH, Alsarra IA. Biochemically altered human erythrocytes as a carrier for targeted delivery of primaquine: an in vitro study. Arch Pharm. Res. 2011;34:563–571. doi: 10.1007/s12272-011-0406-7. [DOI] [PubMed] [Google Scholar]
- [73].Foroozesh M, Hamidi M, Zarrin A, Mohammadi-Samani S, Montaseri H. Preparation and in-vitro characterization of tramadol-loaded carrier erythrocytes for long-term intravenous delivery. J. Pharm. Pharmacol. 2011;63:322–332. doi: 10.1111/j.2042-7158.2010.01207.x. [DOI] [PubMed] [Google Scholar]
- [74].Lizano C, Perez MT, Pinilla M. Mouse erythrocytes as carriers for coencapsulated alcohol and aldehyde dehydrogenase obtained by electroporation in vivo survival rate in circulation, organ distribution and ethanol degradation. Life Sci. 2001;68:2001–2016. doi: 10.1016/s0024-3205(01)00991-2. [DOI] [PubMed] [Google Scholar]
- [75].Lizano C, Weissig V, Torchilin VP, Sancho P, Garcia-Perez AI, Pinilla M. In vivo biodistribution of erythrocytes and polyethyleneglycol-phosphatidylethanolamine micelles carrying the antitumour agent dequalinium. Eur. J. Pharm. Biopharm. 2003;56:153–157. doi: 10.1016/s0939-6411(03)00089-4. [DOI] [PubMed] [Google Scholar]
- [76].Muzykantov VR. Drug delivery by red blood cells: vascular carriers designed by mother nature. Expert Opin. Drug Deliv. 2010;7:403–427. doi: 10.1517/17425241003610633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Ataullakhanov FI, Vitvitskii VM, Zhabotinskii AM, Pichugin AV. Permeability of human erythrocytes to asparagine. Biokhimiia. 1985;50:1733–1737. [PubMed] [Google Scholar]
- [78].Avramis VI, Panosyan EH. Pharmacokinetic/pharmacodynamic relationships of asparaginase formulations: the past, the present and recommendations for the future. Clin. Pharmacokinet. 2005;44:367–393. doi: 10.2165/00003088-200544040-00003. [DOI] [PubMed] [Google Scholar]
- [79].Hamidi M, Zarei N, Zarrin A, Mohammadi-Samani S. Preparation and validation of carrier human erythrocytes loaded by bovine serum albumin as a model antigen/protein. Drug Deliv. 2007;14:295–300. doi: 10.1080/10717540701203000. [DOI] [PubMed] [Google Scholar]
- [80].Chalmers RA. Comparison and potential of hypo-osmotic and iso-osmotic erythrocyte ghosts and carrier erythrocytes as drug and enzyme carriers. Bibl. Haematol. 1985:15–24. doi: 10.1159/000410223. [DOI] [PubMed] [Google Scholar]
- [81].Bachran C, Heisler I, Fuchs H, Sutherland M. Influence of protein transduction domains on target-specific chimeric proteins. Biochem. Biophys. Res. Commun. 2005;337:602–609. doi: 10.1016/j.bbrc.2005.09.095. [DOI] [PubMed] [Google Scholar]
- [82].Dietz GP, Bahr M. Delivery of bioactive molecules into the cell: the Trojan horse approach. Mol. Cell Neurosci. 2004;27:85–131. doi: 10.1016/j.mcn.2004.03.005. [DOI] [PubMed] [Google Scholar]
- [83].Suzuki T, Futaki S, Niwa M, Tanaka S, Ueda K, Sugiura Y. Possible existence of common internalization mechanisms among arginine-rich peptides. J. Biol. Chem. 2002;277:2437–2443. doi: 10.1074/jbc.M110017200. [DOI] [PubMed] [Google Scholar]
- [84].Schmidt N, Mishra A, Lai GH, Wong GC. Arginine-rich cell-penetrating peptides. FEBS Lett. 2010;584:1806–1813. doi: 10.1016/j.febslet.2009.11.046. [DOI] [PubMed] [Google Scholar]
- [85].Coupade C. De, Fittipaldi A, Chagnas V, Michel M, Carlier S, Tasciotti E, Darmon A, Ravel D, Kearsey J, Giacca M, Cailler F. Novel human-derived cell-penetrating peptides for specific subcellular delivery of therapeutic biomolecules. Biochem. J. 2005;390:407–418. doi: 10.1042/BJ20050401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Schwarze SR, Ho A, Vocero-Akbani A, Dowdy SF. In vivo protein transduction: delivery of a biologically active protein into the mouse. Science. 1999;285:1569–1572. doi: 10.1126/science.285.5433.1569. [DOI] [PubMed] [Google Scholar]
- [87].Cao L, Si J, Wang W, Zhao X, Yuan X, Zhu H, Wu X, Zhu J, Shen G. Intracellular localization and sustained prodrug cell killing activity of TAT-HSVTK fusion protein in hepatocelullar carcinoma cells. Mol. Cells. 2006;21:104–111. [PubMed] [Google Scholar]
- [88].Elliott G, O’Hare P. Intercellular trafficking and protein delivery by a herpesvirus structural protein. Cell. 1997;88:223–233. doi: 10.1016/s0092-8674(00)81843-7. [DOI] [PubMed] [Google Scholar]
- [89].Chang LC, Lee HF, Yang Z, Yang VC. Low molecular weight protamine (LMWP) as nontoxic heparin/low molecular weight heparin antidote (I): preparation and characterization. AAPS Pharm. Sci. 2001;3:E17. doi: 10.1208/ps030317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Park YJ, Chang LC, Liang JF, Moon C, Chung CP, Yang VC. Nontoxic membrane translocation peptide from protamine, low molecular weight protamine (LMWP), for enhanced intracellular protein delivery: in vitro and in vivo study. FASEB J. 2005;19:1555–1557. doi: 10.1096/fj.04-2322fje. [DOI] [PubMed] [Google Scholar]
- [91].Byun Y, Singh VK, Yang VC. Low molecular weight protamine: a potential nontoxic heparin antagonist. Thromb Res. 1999;94:53–61. doi: 10.1016/s0049-3848(98)00201-1. [DOI] [PubMed] [Google Scholar]
- [92].Chang LC, Liang JF, Lee HF, Lee LM, Yang VC. Low molecular weight protamine (LMWP) as nontoxic heparin/low molecular weight heparin antidote (II): in vitro evaluation of efficacy and toxicity. AAPS Pharm. Sci. 2001;3:E18. doi: 10.1208/ps030318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Kharidia R, Friedman KA, Liang JF. Improved gene expression using low molecular weight peptides produced from protamine sulfate. Biochemistry (Mosc) 2008;73:1162–1168. doi: 10.1134/s0006297908100143. [DOI] [PubMed] [Google Scholar]
- [94].Choi YS, Lee JY, Suh JS, Kwon YM, Lee SJ, Chung JK, Lee DS, Yang VC, Chung CP, Park YJ. The systemic delivery of siRNAs by a cell penetrating peptide, low molecular weight protamine. Biomaterials. 2010;31:1429–1443. doi: 10.1016/j.biomaterials.2009.11.001. [DOI] [PubMed] [Google Scholar]
- [95].Lee LM, Chang LC, Wrobleski S, Wakefield TW, Yang VC. Low molecular weight protamine as nontoxic heparin/low molecular weight heparin antidote (III): preliminary in vivo evaluation of efficacy and toxicity using a canine model. AAPS Pharm. Sci. 2001;3:E19. doi: 10.1208/ps030319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Park YS, David AE, Huang Y, Park JB, He H, Byun Y, Yang VC. In vivo delivery of cell-permeable antisense hypoxia-inducible factor 1alpha oligonucleotide to adipose tissue reduces adiposity in obese mice. J. Control Release. 2012;161:1–9. doi: 10.1016/j.jconrel.2012.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Xia H, Gao X, Gu G, Liu Z, Zeng N, Hu Q, Song Q, Yao L, Pang Z, Jiang X, Chen J, Chen H. Low molecular weight protamine-functionalized nanoparticles for drug delivery to the brain after intranasal administration. Biomaterials. 2011;32:9888–9898. doi: 10.1016/j.biomaterials.2011.09.004. [DOI] [PubMed] [Google Scholar]
- [98].He H, Sheng J, David AE, Kwon YM, Zhang J, Huang Y, Wang J, Yang VC. The use of low molecular weight protamine chemical chimera to enhance monomeric insulin intestinal absorption. Biomaterials. 2013;34:7733–7743. doi: 10.1016/j.biomaterials.2013.06.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Choi JK, Jang JH, Jang WH, Kim J, Bae IH, Bae J, Park YH, Kim BJ, Lim KM, Park JW. The effect of epidermal growth factor (EGF) conjugated with low-molecular-weight protamine (LMWP) on wound healing of the skin. Biomaterials. 2012;33:8579–8590. doi: 10.1016/j.biomaterials.2012.07.061. [DOI] [PubMed] [Google Scholar]
- [100].Park YJ, Liang JF, Ko KS, Kim SW, Yang VC. Low molecular weight protamine as an efficient and nontoxic gene carrier: in vitro study. J. Gene Med. 2003;5:700–711. doi: 10.1002/jgm.402. [DOI] [PubMed] [Google Scholar]
- [101].Kwon YM, Chung HS, Moon C, Yockman J, Park YJ, Gitlin SD, David AE, Yang VC. L-Asparaginase encapsulated intact erythrocytes for treatment of acute lymphoblastic leukemia (ALL) J. Control Release. 2009;139:182–189. doi: 10.1016/j.jconrel.2009.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Kodera Y, Sekine T, Yasukohchi T, Kiriu Y, Hiroto M, Matsushima A, Inada Y. Stabilization of L-asparaginase modified with comb-shaped poly(ethylene glycol) derivatives, in vivo and in vitro. Bioconjug. Chem. 1994;5:283–286. doi: 10.1021/bc00028a001. [DOI] [PubMed] [Google Scholar]