

Abstract
Most dsDNA viruses, including bacteriophages and herpesviruses, rely on a staged assembly process of capsid formation. A viral protease is required for many of them to disconnect scaffolding domains/proteins from the capsid shell, therefore priming the maturation process. We used the bacteriophage HK97 as a model system to decipher the molecular mechanisms underlying the recruitment of the maturation protease by the assembling procapsid and the influence exerted onto the latter. Comparisons of the procapsid with and without protease using single-particle electron cryomicroscopy reconstructions, hydrogen/deuterium exchange coupled to mass spectrometry and native mass spectrometry demonstrated that the protease interacts with the scaffolding domains within the procapsid interior and stabilizes them as well as the whole particle. The results suggest that the thermodynamic consequences of protease packaging are to shift the equilibrium between isolated coat subunit capsomers and procapsid in favor of the latter by stabilizing the assembled particle before making the process irreversible through proteolysis of the scaffolding domains.
Virus maturation corresponds to a transition from an initial non-infectious, often fragile assembly product to an infectious and robust virion. Initial subunit interactions occur under conditions where the assembling entities have an association energy that favors assembly over disassembly, but that is near equilibrium to allow “self-correction” of misassembled subunits through annealing (Caspar, 1980; Katen and Zlotnick, 2009). As viruses require sturdy stability to survive in the extra-cellular environment, they undergo a staged assembly process relying on a mechano-chemical reorganization program encoded in the capsid structure that governs events underlying maturation.
Assembly and maturation of dsDNA phage capsids are tightly regulated processes, both at the genetic and biochemical levels, exhibiting conserved features in all Caudovirales and in some eukaryotic viruses such as herpesviruses (Veesler and Johnson, 2012). Moreover, the conservation of the coat subunit fold observed in all tailed phages and herpesviruses as well as some archeal viruses, suggests that their capsids share a common evolutionary origin, putatively through the existence of a common viral ancestor that preceded the divergence of Eukarya, Bacteria and Archaea (Veesler and Cambillau, 2011; Veesler and Johnson, 2012). The lambdoid dsDNA phage HK97 constitutes an accessible model system for studying maturation of such viruses due to its well-characterized genetics and ease of handling. Its capsid maturation pathway involves discrete intermediate particle forms (Supplementary Fig. 1), comparable to transition states in protein folding, which can be isolated using a combination of molecular biology and biochemical techniques.
The HK97 capsid precursor protein is a fusion of the scaffolding protein (δ-domain, residues 2-103) and of the coat subunit (residues 104-385) that forms a mixture of hexameric and pentameric capsomers upon expression. In vivo, 415 coat subunits (60 hexamers and 11 pentamers) assemble with a dodecameric portal and an undefined number of copies of the viral protease to form the first icosahedral particle termed Prohead-1 (Supplementary Fig. 1). Activation of the viral protease, that occurs with completion of particle assembly, results in digestion of the scaffolding domains and auto-digestion of the protease to produce small peptide fragments (Duda et al., 2013) that diffuse out of the particle to yield Prohead-2. The two Prohead intermediates (1 and 2) exhibit distorted coat subunit tertiary structures readily recognized by the bent spine helix and the twisted P-domain β-sheet (Gertsman et al., 2009; Huang et al., 2011). Their quaternary structures are also characterized by departure from canonical symmetry as the hexameric capsomers are skewed, displaying only 2-fold symmetry. These structural distortions are believed to be induced by interactions among scaffolding domains during capsomer formation and later stabilized by quaternary interactions following δ-domain proteolysis (Johnson, 2010). Prohead-2 is thus a metastable intermediate, trapped in a local free-energy minimum, that is primed to transition to a lower-energy conformation in response to small perturbations (Lee et al., 2005). Genome packaging triggers Prohead-2 expansion, resulting in the formation of successive Expansion Intermediates characterized by an increase of capsid diameter, a reduction of the shell thickness and a “curing” of the hexon asymmetry. The first Expansion Intermediate (EI-1) features 6-fold symmetric hexons with subunit tertiary structures still displaying the distortions observed in the Proheads (Veesler et al., 2012). The large conformational changes occurring during EI-1 formation makes it competent for the autocatalytic formation of isopeptide bonds between residues Lys169, on the E-loop of one coat subunit, and Asn356, on the P-domain of an adjacent subunit in a neighboring capsomer (Duda, 1998; Wikoff et al., 2000). Crosslinking promotes formation of the subsequent Expansion Intermediates and has been proposed to modulate the capsid structural reorganization by biasing thermal motions via a Brownian ratchet mechanism (Lee et al., 2008; Ross et al., 2005). The mature capsid is termed Head-2 and harbors 415 crosslinks with a chainmail topology that exert a critical stabilizing effect required to harbor the viral genome at near liquid-crystalline density (Wikoff et al., 2000). All studies in which maturation intermediates and mature capsids were isolated and studied by crystallography or electron microscopy employed an E. coli expression system in which only the protease and capsid protein genes are co-expressed. The expression system leads to spontaneous, in vivo, particle assembly and recapitulates capsid intermediates and mature capsid identified during the virus infection, but they lack the portal and dsDNA and are therefore icosahedrally symmetric (e.g. Head II contains no DNA and 420 crosslinks).
In the present study, we characterized the interactions underlying the co-assembly of the viral maturation protease with the phage HK97 procapsid. Employing a mutant protease (described below) in the expression system, without enzymatic activity but otherwise normal, we obtained subnanometer electron cryomicroscopy reconstructions of the protease-free as well as the protease-loaded Prohead-1 and used these as a basis to analyze the influence of the protease on the assembly. The viral protease is anchored to the scaffolding domains and exerts an overall stabilizing effect on the procapsid. Hydrogen/Deuterium exchange studies coupled to mass spectrometry (HDXMS) further confirmed these results and allowed us to accurately map the influence of the protease on the coat subunit structure. Finally, we provide an estimate of the number of protease molecules that are packaged into the procapsid based on native mass spectrometry and quantitative proteomics analysis of the two Prohead-1 particle forms used to perform three-dimensional reconstructions. This study constitutes the most detailed analysis available to date of the interactions between a dsDNA viral capsid and the maturation protease and provides a basis to further our understanding of the first steps of virion assembly in Caudovirales phages and herpesviruses.
RESULTS
CryoEM reconstruction of protease-free Prohead-1
We computed an icosahedrally-averaged reconstruction of Prohead-1 without the viral protease at 7.8 Å resolution using 5680 particle images (Supplementary Fig. 2). This procapsid, which does not naturally occur in the virus life cycle, is a ~540 Å, round particle with a rough surface, due to the radial orientation of subunits relative to the shell, and harboring skewed hexameric coat subunit capsomers with approximate 2-fold symmetry (Fig 1 A). The subunits are distorted at the level of the spine helix and P-domain β sheet (compared to the tertiary structure of the subunits in the mature particle), as observed in the Prohead-1 (containing inactive protease) crystal structure previously reported (Huang et al., 2011) (Fig 1 B). The T=7 capsid lattice displays three different classes of P-loop interactions: (i) class I - 20 icosahedral 3-fold contacts involving subunits D (Fig 1 A and C); (ii) class II - 60 quasi-3-fold contacts involving subunits B, C and E (Fig 1 A and D); (iii) class III - 60 quasi-3-fold contacts involving subunits A, F and G (Fig 1 A and E). Limited interactions of the coat subunit P-loops are observed at the class II contacts resulting in holes in the capsid shell that connect the capsid interior with the extracellular milieu. The unsealed interactions arise from the different degree of distortion of subunits involved in such contacts in Prohead-1 compared to Prohead-2 (Supplementary Fig 3) (Gertsman et al., 2009; Huang et al., 2011). In Prohead-1, these subunits position their P-loops away from each other whereas they are tightly interacting in Prohead-2, emphasizing the correlation between the magnitude of quasi-equivalent distortion and the strength of capsomer interactions.
Figure 1. Subnanometer CryoEM reconstruction of protease-free HK97 Prohead-1.
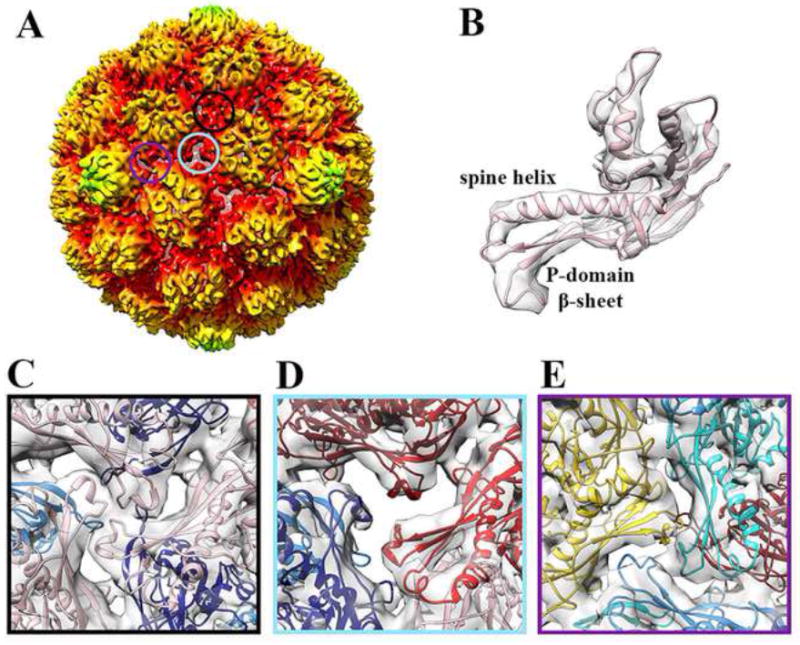
(A) Surface rendering of the procapsid reconstruction low-pass filtered at 7.8 Å resolution, sharpened with a B factor of -700 Å2 and radially colored as in Figure 1. The three circles overlaid onto the particle correspond to the three classes of 3-fold contacts and their respective color point to the corresponding frame in panels (C), (D) and (E). The reconstruction is radially colored according to the color key used in Figure S1. (B) Fit of a Prohead-1 coat subunit (PDB ID 3QPR) into the corresponding region of the reconstruction shown in (A). The seven subunits of the icosahedral asymmetric unit are conformationally distorted at the level of the spine helix and P-domain β-sheet. (C) Class I icosahedral 3-fold contacts involving subunits D. (D) Class II quasi-3-fold contacts involving subunits B, C and E. The coat subunit N-arms have been removed for clarity on this panel. (E) Class III quasi-3-fold contacts involving subunits A, F and G. The coat subunit coordinates are colored as follows: A (cyan), B (dark red), C (red), D (pink), E (navy blue), F (dodger blue) and G (gold). Panels C-E represent views from the virion interior.
The virion interior exhibits extended densities intruding nearly radially relative to the capsid shell with a high-degree of apparent disorder (Fig. 2A), as judged by the significantly lower resolution of the reconstruction in this region. These rod-shaped densities are appended to the periphery of the crevice defined by each capsomer (hexons and pentons) on its internal face and correspond to the scaffolding domains fused to the coat subunit N-termini. They appear to be arranged as dimers of trimers (in agreement with previous observations (Conway et al., 2007; Ross et al., 2006)), reminiscent of the skewing of the coat subunit cores, but the identity of quasi-equivalent subunits belonging to each cluster is different at the level of the capsid shell and the scaffolding domains. Indeed, subunits B, C, D and E, F, A form the two trimers present in each hexameric capsomer (Fig. 2B) whereas subunits C, D, E and F, A, B form the two scaffolding protein trimeric moieties (Fig. 2C). The coat subunit N-arm (104-119) was modeled as well as an elongated helical segment corresponding to the scaffolding domain C-terminal region (Fig. 2B-E). The modeling was based on the secondary-structure predictions and biophysical analyses (Benevides et al., 2004; Němeček et al., 2009) as the limited resolution of the reconstruction within the capsid interior precludes distinguishing individual α-helices. A marked deviation from quasi-equivalence was observed in the organization of the scaffolding domains with helices belonging to domains A/D and C/F being respectively the best and worst defined in the density. As a result, we modeled only a short portion of the scaffolding domains belonging to subunits C and F and their assignment to either of the above-mentioned clusters is more ambiguous than for the other four subunits of the hexon. The reconstruction suggests that the N-termini could extend further toward the capsid center, which corresponds to the 80 residues missing from the model and predicted to be in an α-helical conformation.
Figure 2. Organization of the scaffolding domains.
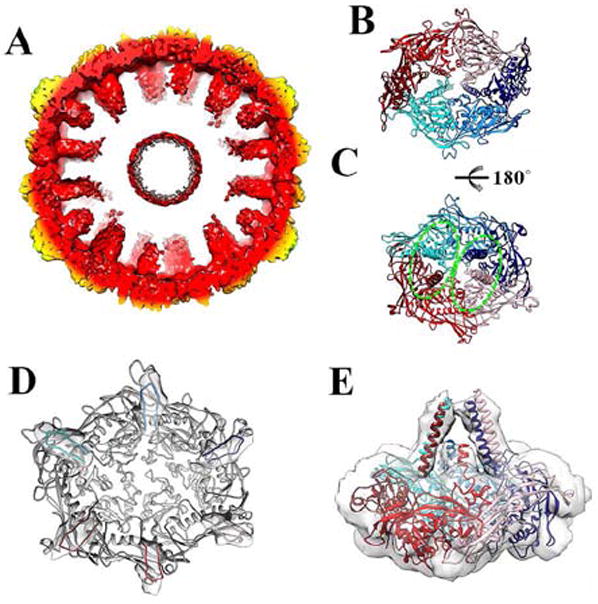
(A) Cross-section of the protease-free Prohead-1 reconstruction revealing the scaffolding domains extending toward the procapsid center. The reconstruction is radially colored as in Figure 1. (B) Each Prohead-1 hexon (viewed from the capsid exterior) can be divided into two-halves according to the position of the coat subunit cores (delimited by the two shades of red and blue). (C) The scaffolding domains belonging to each Prohead-1 hexon (viewed from the capsid interior) can also be divided into two groups (delimited by the dashed ellipsoids) that differ from the ones formed by the coat subunit cores. (D) The coat subunit N-arms (residues 104-130) lie on the internal face of each capsomer and connect to the scaffolding domains (viewed from the capsid interior). The N-arms are colored by subunit identity and depicted within the corresponding region of the reconstruction while the rest of the subunits are colored grey. (E) Lateral view of a Prohead-1 hexon fitted into the reconstruction to show the approximate organization of the scaffolding domain C-terminal moieties (residues 80-104) relative to the coat subunits. The coat subunit coordinates are colored as in Figure 2.
CryoEM reconstruction of protease-containing Prohead-1
To overcome the transient nature of Prohead-1 in the presence of the viral protease, we engineered an inactivating point mutant of the catalytic histidine (H65A) allowing the production of stalled protease-containing Prohead-1 particles (Duda et al., 2013; Huang et al., 2011). We computed an icosahedrally-averaged reconstruction of this maturation intermediate at 8.3 Å resolution (Fig. 3A-B & Supplementary Fig. 2) to compare with the protease-free reconstruction. The features observed in the region of the capsid shell are similar in the two reconstructions and the coat subunits exhibit identical conformations at this resolution in the two maps. Although this reconstruction also exhibits a dampened resolution in the capsid interior, the presence of the protease clearly reinforces the density corresponding to the scaffolding domains as compared to the protease-free map. A difference map computed by subtracting the density of this latter to the protease-containing Prohead-1 reconstruction allows a better appreciation of the structural differences between the two particles (Fig. 3C). Addition of the protease strengthens the scaffolding domain region that extends further toward the capsid center while retaining the same quasi-equivalent organization. The dynamic nature of the scaffolding domains is preserved and prevents modeling of their N-termini as well as of the protease due to blurring of the map in this region. The effect of the viral protease on the reconstruction suggests that it is incorporated into the procapsid shell in one of the following ways: (i) the protease molecules may be interacting with the scaffolding domains in a transient manner through moderate affinity contacts; (ii) or they remain anchored to the scaffolding domains (high-affinity) but are not organized with icosahedral symmetry (similarly to what was proposed for herpes viruses (Zhou et al., 1998)) as a result of the scaffolding domain disorder and/or due to the non stoichiometric ratio of encapsidated protease to scaffold. The absence of discernable density attributable to the protease in our reconstruction is in agreement with either of these two hypotheses but doesn’t allow their discrimination.
Figure 3. Subnanometer CryoEM reconstruction of protease-containing HK97 Prohead-1.
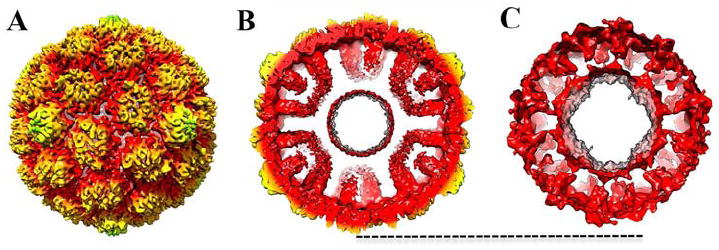
(A) Surface rendering of the procapsid reconstruction low-pass filtered at 8.3 Å resolution and radially colored as in Figure 1. (B) Cross-section of the reconstruction showing the scaffolding domains extending toward the procapsid center. (C) Difference map (low-pass filtered to 20 Å resolution) resulting from the subtraction of the protease-free Prohead-1 to the protease-containing one after scaling of the two maps. The dashed line indicates the outer most surface of the capsid shell.
Interactions between the viral protease and the coat subunits
We implemented a comparative HDXMS approach to map solvent accessibility as a function of time in the two Prohead-1 forms and to characterize the contacts existing between the protease and the procapsid shell. We obtained a peptide coverage corresponding to 93% of the coat subunit sequence and the results are presented as mirror and difference plots comparing the two procapsid forms, i.e. protease-free minus protease-containing Prohead-1 (Fig. 4 A-B and Supplementary Fig. 4). The mirror plot displays the relative deuterium exchange across all pepsin-generated peptides listed from N to C-terminus. In both states, the fragment peptides spanning the N-terminal 50 residues (scaffolding domain) overall showed higher deuterium exchange compared to the rest of the protein. This is indicative of the dynamic character of this region. The derived difference plot shows the magnitude of the difference in deuterium exchange across all the peptides between the two states. Positive values represent regions that are protected from deuterium exchange in presence of the protease. Strikingly, viral protease interaction conferred protection to the entire scaffolding domain across all time points examined. We observed that the magnitude of protection was at a maximum after 30 s deuterium labeling and reduced with increasing labeling time. These results are in agreement with the cryoEM data and support the hypothesis that the protease directly interacts with the scaffolding domains. The coat subunit also exhibited pronounced solvent protection in the presence of the protease at the level of the N-arm, the C-terminal region of the spine helix and the A-loop that occupies the center of the capsomers (Fig. 4 C-D). We can easily rationalize the susceptibility of the N-arm to protease interactions as it is fused and is directly adjacent to the scaffolding domain. The same holds for the C-terminal region of the spine helix that is tightly interacting with the N-arm, through formation of salt bridges and hydrogen bonds (observed in the Prohead-2 crystal structure (Gertsman et al., 2009)), explaining the similar behavior of these two protein segments. It is less apparent why solvent protection was observed for the A-loop as it is at the external surface of the capsomer (far from the scaffolding domains). It may be the result of subtle, induced conformational changes propagated through a network of interactions involving the entire coat subunit A domain and that are not discernable at the moderate resolution of the cryoEM structures (Engen et al., 1999; Mayne et al., 1992). The regions outside the scaffolding domain showed larger shifts in deuterium exchange at longer deuterium labeling times. Hydrogen/Deuterium exchange is highly sensitive, allows mapping the effects of ligand interactions and has been documented to report both direct and indirect binding effects on proteins (Mayne et al., 1992). The indirect effects have been attributed to decreased protein motions or dynamics (Engen et al., 1999). It has also been proposed that direct protein-protein interaction sites can be separately identified from indirect conformational changes by focusing solely on the fast exchanging amides (Mandell et al., 1998). In an extension, we predict that direct binding can be discerned from allosteric changes by comparing the timescales of deuterium exchange protection at distinct locations of a protein. When allostery operates at timescales detectably slower than direct binding interactions, the latter will be detected as maximal protection in the deuterium exchange profile at earlier time points than the former. In the case of Prohead-1, the scaffolding domain featured the most pronounced deuterium exchange protection at the earliest time point (Fig. 4 A). In contrast, the other coat subunit regions exhibited the largest protection at later time points (Fig. 4 A). These observations suggest that the protease directly interacts with the scaffolding domain and induces conformational changes in the regions described above. Such action at a distance was described in the bacteriophage P22 capsid protein where mutations at position 170 (within the homologous HK97 A-domain) affects its ability to interact with scaffolding proteins, inhibiting proper procapsid assembly (Suhanovsky et al., 2010). The protease molecules significantly modulate solvent accessibility of more than 35% of the coat subunit aminoacid residues, likely through a combination of direct interactions and allosteric effects. Overall these data demonstrate the direct interaction between the viral protease and the scaffolding domains in dsDNA phages.
Figure 4. Interactions between the viral protease and the coat subunits studied by Hydrogen/Deuterium exchange coupled to mass-spectrometry.
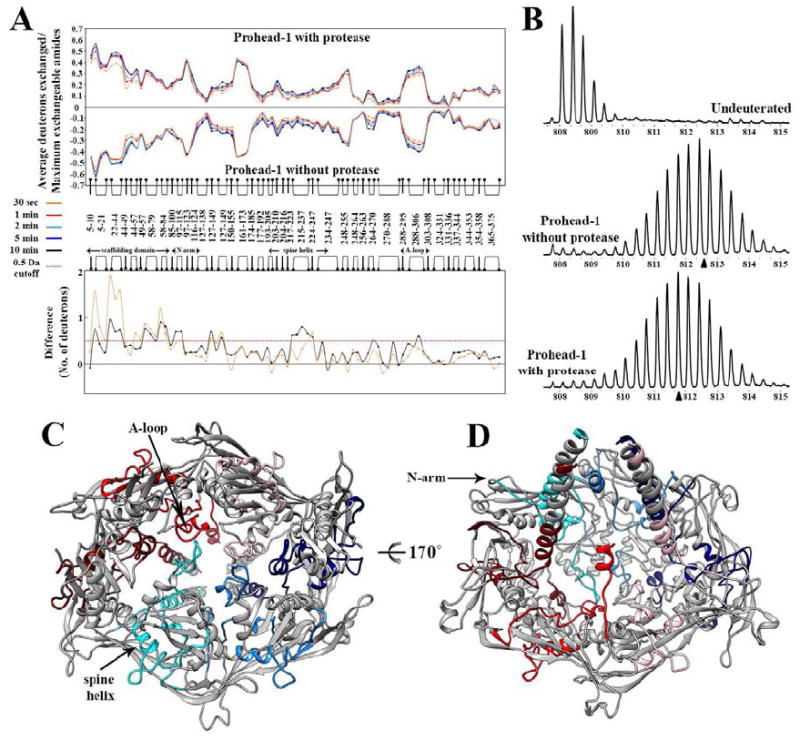
(A) Top panel: Mirror plot of the relative deuterium exchange across pepsin digest fragments from N to C-terminus. Each plot represents a time point of deuterium labeling (t=30s, 1min, 2min, 5min, 10min), color coded as indicated. Bottom panel: Difference plot showing the average mass difference resulting from deuterium exchange between the two Prohead-1 forms, i.e. protease-free minus protease-containing procapsids. Positive differences represent regions that are protected from deuterium exchange in presence of the protease. (B) Illustration of the mass spectra obtained for the peptide 22-43. (Top) Undeuterated Prohead-1 without protease, (Middle) Prohead-1 without protease after 2 min of exchange, (Bottom) Prohead-1 with protease after 2 min of exchange. The triangles indicate the centroid of each spectrum obtained after 2 min of exchange. (C) View of the coat subunit hexon from the procapsid exterior. The regions featuring the highest solvent protection in presence of the protease are colored according to the scheme defined in Figure 1 whereas the other parts of the coat subunits are colored gray. (D) View from the procapsid interior of the coat subunit hexon colored as in (C). In panels A and B, peptides are depicted as linked bullet points and correspond to the composite residues in each location analyzed generated by overlapping pepsin digest fragments.
MS-based quantification of protease packaging and cleavage efficiency
The stoichiometry defining the number of protease molecules incorporated into the procapsid of dsDNA viruses has been elusive. Densitometry analysis of SDS protein gels of HK97 Prohead I with a non functional protease indicated ~50 protease molecules per particle (Hendrix and Duda, 1998). We analyzed the two Prohead-1 forms described in this study by native mass-spectrometry to get better estimates. A well-defined spectrum for the protease-free procapsid allowed resolution of individual charge states and yielded a mass estimate of 17.9 +/- 0.004 MDa (Fig. 5, top), in excellent agreement with the theoretical mass (17.7MDa) (Snijder et al., 2013). Analysis of the protease-containing particle resulted in an unresolved spectrum at a higher m/z than the empty Prohead-1 (Figure 5, middle). In this case, a mass estimate of the particle requires assumptions about the charging behavior of the procapsid particles in electrospray ionization. A plot of mass in function of charge obtained in our laboratory shows a power-law function with strong correlation (R2=0.9965, Supplementary Figure 5). The average residual from the obtained fit was 2 ± 3 charges, with no clear mass dependence, indicating that we can reasonably estimate z within approximately 5 units. The m/z position of the particle is determined at 53190 ± 180 (average ± standard deviation, n=5). We can estimate the mass as: m=[m/z]*z, with an error that is estimated as the sum of the relative errors on [m/z] and z. We thereby estimated the mass of the protease-filled Prohead-1 particle at approximately 21.4 ± 0.34 MDa. This corresponds to 144 ± 14 copies of the protease. It should be noted that this estimate only holds under the assumption of normal charging. For instance, as charge is considered to be distributed on the exterior of a droplet in the final stages of electrospray ionization, the internal surface of the capsid might not be equally accessible to charge.
Figure 5. Native mass-spectrometry quantification of protease packaging and cleavage efficiency.
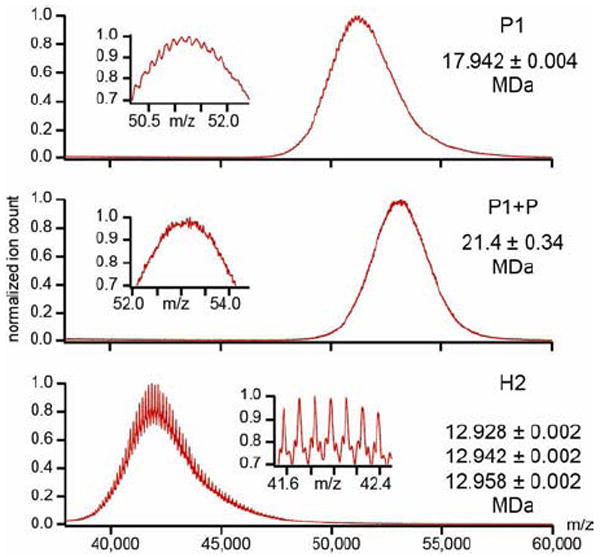
(top) The mass spectrum obtained for the protease-free procapsid indicates a mass of 17.9 +/- 0.004 MDa while the theoretical one is 17.7MDa (mass accuracy of 1%). (middle) The mass spectrum obtained for the protease-containing procapsid is noisier and yields a mass estimate of 21.4 ± 0.34 MDa. (bottom) Mass spectrum of Head-2 reveals three distributions with a mass of approximately 12.9 MDa. The masses are consistent with a highly efficient maturation cleavage but clearly reveal some residual material in the Head-2 particles. (insets) Zoom-in of the indicated m/z region to reveal the fine structures on the signal.
To confirm this estimate of the number of packaged protease molecules, we also performed a label-free quantitative proteomics experiment, in which the protease-containing particle was digested and analyzed by LC-MS/MS. We used the number of peptide-spectrum-matches (PSM) as a quantitative measure of protein abundance. Some issues may arise in PSM based quantitation, especially of low abundant proteins that are identified with a low number of peptides in complex background matrix, which results in high stochastic fluctuations in the obtained PSM. However, the Prohead-1 with protease sample is a very simple mixture, generating a total of ~70 identified peptides. Therefore, no issue is expected due to sample complexity. In addition, the total number of PSM is based on 50 and 20 unique peptides for gp5 and gp4, respectively. Both proteins generate a comparable set of tryptic peptides, given the distribution of cleavable sites in the sequence, such that little bias in observable peptides is expected. Based on replicate injections from the same digest we obtained an estimate of 104 ± 18 copies (average ± stdev), indicating that instrumental reproducibility was ~20%. Out of six independent digest using two independent purification batches, we reached an estimate of 116 ± 23 copies (average ± standard deviation), which agrees reasonably well with the native MS estimate.
It should be noted that the lack of resolution in the native MS experiment is a clear indication of heterogeneity in the number of packaged protease molecules and supports the hypothesis put forward based on the cryoEM results. We therefore presumed that the varying amount of protease in the particles could lead to a heterogeneous population of mature capsids following proteolysis. We analyzed the mature Head-2 particle by native MS and observed a large shift in mass to 12.9 MDa (Figure 5, bottom). Closer inspection of the signal revealed that at least three overlapping distributions could be distinguished. Their mass deviates by approximately 10, 24 and 39 kDa from a theoretically fully cleaved Head-2 particle, indicating that at least 99.5% of all delta-domains were cleaved. Although maturation proceeds with extraordinarily high efficiency in HK97, native MS revealed a small amount of residual material in the Head-2 particle probably not detectable by conventional methods. The residual masses may correspond to one copy of the protease molecule that could not exit the particle (24kDa), or perhaps to partly cleaved fragments of the delta-domain (10kDa).
DISCUSSION
We described the comparative analysis of the first maturation intermediate of the bacteriophage HK97 assembly pathway, in the absence and presence of the maturation protease. The combination of single-particle EM and mass-spectrometry provided a unique opportunity to characterize the influence of the protease on the procapsid structure. We addressed largely unanswered questions such as: What is the structural organization of the scaffolding domains? How do the scaffolding domains induce distortions within the coat subunit tertiary structure? How do the protease molecules interact with the coat subunits in terms of regions involved and stoichiometry? What is the thermodynamic influence of the presence of the protease within the procapsid? The subnanometer Prohead-1 reconstructions described here show that the presence or absence of protease has no effect on the quaternary structure, with unsealed quasi-3-fold contacts present at the interface between coat subunits B, C and E (class II) while the other two classes of (quasi)-3-fold contacts are sealed with the formation of tight P-loop interactions. In Prohead-2, the class II contacts are sealed, due to the different degree of distortion of the corresponding subunits, and remain as such throughout the entire maturation process (Gertsman et al., 2010a; 2010b; 2009; Huang et al., 2011). These structural variations correlate with the requirement of each particle form to fulfill different tasks (Veesler and Johnson, 2012). Prohead-1 is an organizational intermediate bearing nearly half of its (quasi)-3-fold inter-capsomer contacts partially unformed. Such modest interactions are required at this stage to permit the associating subunits to self-correct through annealing as well as to putatively provide an exit pathway for the fragments of the scaffolding domain and protease, released upon proteolysis. In contrast, Prohead-2 exhibits sealed P-loop interactions at all trimer contacts as these regions serve as invariant pivot points for the dramatic rearrangements that follow (Gertsman et al., 2010a; 2009; Wikoff et al., 2000).
Incorporation of the maturation protease within the HK97 procapsid is mediated via direct interactions with the scaffolding domains, as demonstrated by comparing protease-free and protease-loaded Prohead-1 particles using electron cryo-microscopy reconstructions and HDXMS. Beyond the stabilizing effect exerted on the scaffolding domains, probably by reducing their positional fluctuations, the presence of the protease results in an overall stabilization of the procapsid. Indeed, the intrinsic variance (Wang et al., 2013) of the capsid shell region (corresponding to the coat subunit core only) proves to be 10% higher when the protease is absent indicating an increase degree of destabilization. These results are in agreement with the previous observations that Prohead-1 disassembly occurs at a higher temperature when the protease is packaged (82.3°C) as compared to when it is absent (79.3°C), and that diffraction quality crystals could only be obtained with protease (Huang et al., 2011). The weak protease association with the scaffolding domain provides an effective environment for efficient proteolysis since the substrate is immediately accessible and with the opportunity for dynamic and broad availability of target sequences. From an assembly standpoint, it is probable that the presence of the protease shifts the equilibrium between isolated capsomers and Prohead-1 in favor of the latter by contributing to its stabilization before making the process irreversible by exerting its proteolytic action on the scaffolding domains to yield Prohead-2.
The quasi-equivalent organization of the scaffolding domains revealed by electron microscopy reconstructions provides initial insights into a putative mechanism of induction of coat subunit distortion during assembly. The subunit mismatch existing within the dimers of trimers formed at the level of each hexon by the coat subunit cores and the scaffolding domains might be responsible for generating the observed conformational strain of the former. However, such a hypothesis does not provide a satisfactory explanation for the distortion of the coat subunit monomers involved in the formation of pentons as their scaffolding domains are 5-fold symmetric. The dynamic nature of the scaffolding domains attenuates the resolution of the reconstructions in the capsid interior and limits the analysis of their influence onto the capsid shell conformation. This characteristic seems to be a conserved feature shared by many phage procapsids (Chen et al., 2011) but the reconstructions presented here constitute a significant step forward in understanding the interplay between scaffolding proteins and procapsid shells. Further studies are required to understand the molecular basis for generating capsid subunit conformational strain and to determine the mechanism of initiation of protease activity upon completion of prohead I assembly.
EXPERIMENTAL PROCEDURES
HK97 Prohead-1 with inactive protease or protease-free particles were produced using a pT7-Hd2.9:4 vector encoding for the gp4 viral protease carrying the H65A mutation along with the wild-type gp5 coat subunit or only this latter, respectively. Detailed procedures for the single-particle analysis, HDXMS, native mass-spectrometry and quantitative proteomics analysis can be found in the Supplementary information.
Supplementary Material
Acknowledgments
We acknowledge funding from the NIH (R01 AI040101) to J.E.J and from a FP7 Marie-Curie IOF fellowship (273427) awarded to D.V. We also acknowledge funding from Ministry of Education (MOE2012-T3-1-008), Singapore and Mechanobiology Institute, NUS, Singapore to G.S.A. Part of this work was carried out at the National Resource for Automated Molecular Microscopy which is supported by the National Institutes of Health though the National Center for Research Resources (2P41RR017573-11) and the National Institute of General Medical Sciences (9 P41 GM103310-11). J.S and A.J.R.H. are supported by the Netherlands Proteomics Centre. Molecular graphics and analyses were performed with the UCSF Chimera package. Chimera is developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco, with support from the National Institutes of Health (National Center for Research Resources grant 2P41RR001081, National Institute of General Medical Sciences grant 9P41GM103311).
Footnotes
ACCESSION NUMBERS
The Prohead-1 cryoEM maps have been deposited to the EMDB with accession numbers and for Prohead-1 without and with protease, respectively.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Benevides JM, Bondre P, Duda RL, Hendrix RW. Domain structures and roles in bacteriophage HK97 capsid assembly and maturation. Biochemistry. 2004 doi: 10.1021/bi0302494. [DOI] [PubMed] [Google Scholar]
- Caspar DL. Movement and self-control in protein assemblies. Quasi-equivalence revisited. Biophys J. 1980;32:103–138. doi: 10.1016/S0006-3495(80)84929-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen DH, Baker ML, Hryc CF, DiMaio F. Structural basis for scaffolding-mediated assembly and maturation of a dsDNA virus. 2011 doi: 10.1073/pnas.1015739108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conway JF, Cheng N, Ross PD, Hendrix RW. A thermally induced phase transition in a viral capsid transforms the hexamers, leaving the pentamers unchanged. J Struct Biol. 2007;158:224–232. doi: 10.1016/j.jsb.2006.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duda RL. Protein chainmail: catenated protein in viral capsids. Cell. 1998;94:55–60. doi: 10.1016/s0092-8674(00)81221-0. [DOI] [PubMed] [Google Scholar]
- Duda RL, Oh B, Hendrix RW. Functional domains of the HK97 capsid maturation protease and the mechanisms of protein encapsidation. J Mol Biol. 2013 doi: 10.1016/j.jmb.2013.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engen JR, Gmeiner WH, Smithgall TE, Smith DL. Hydrogen exchange shows peptide binding stabilizes motions in Hck SH2. Biochemistry. 1999;38:8926–8935. doi: 10.1021/bi982611y. [DOI] [PubMed] [Google Scholar]
- Gertsman I, Fu CY, Huang R, Komives EA, Johnson JE. Critical Salt Bridges Guide Capsid Assembly, Stability, and Maturation Behavior in Bacteriophage HK97. Mol Cell Proteomics. 2010a;9:1752–1763. doi: 10.1074/mcp.M000039-MCP201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gertsman I, Komives EA, Johnson JE. HK97 Maturation Studied by Crystallography and H/ 2 H Exchange Reveals the Structural Basis for Exothermic Particle Transitions. J Mol Biol. 2010b doi: 10.1016/j.jmb.2010.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gertsman I, Gan L, Guttman M, Lee K, Speir JA, Duda RL, Hendrix RW, Komives EA, Johnson JE. An unexpected twist in viral capsid maturation. Nature. 2009;458:646–650. doi: 10.1038/nature07686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendrix RW, Duda RL. Bacteriophage HK97 head assembly: a protein ballet. Advances in Virus Research. 1998 doi: 10.1016/s0065-3527(08)60810-6. [DOI] [PubMed] [Google Scholar]
- Huang RK, Khayat R, Lee KK, Gertsman I, Duda RL, Hendrix RW, Johnson JE. The Prohead-I Structure of Bacteriophage HK97: Implications for Scaffold-Mediated Control of Particle Assembly and Maturation. J Mol Biol. 2011;408:541–554. doi: 10.1016/j.jmb.2011.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson JE. Virus particle maturation: insights into elegantly programmed nanomachines. Curr Opin Struct Biol. 2010;20:210–216. doi: 10.1016/j.sbi.2010.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katen S, Zlotnick A. The thermodynamics of virus capsid assembly. Meth Enzymol. 2009;455:395–417. doi: 10.1016/S0076-6879(08)04214-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KK, Tsuruta H, Hendrix RW, Duda RL. Cooperative reorganization of a 420 subunit virus capsid. J Mol Biol. 2005;352:723–735. doi: 10.1016/j.jmb.2005.07.024. [DOI] [PubMed] [Google Scholar]
- Lee KK, Gan L, Tsuruta H, Moyer C, Conway JF, Duda RL, Hendrix RW, Steven AC, Johnson JE. Virus capsid expansion driven by the capture of mobile surface loops. Structure (London, England : 1993) 2008;16:1491–1502. doi: 10.1016/j.str.2008.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandell JG, Falick AM, Komives EA. Identification of protein-protein interfaces by decreased amide proton solvent accessibility. Proc Natl Acad Sci USa. 1998;95:14705–14710. doi: 10.1073/pnas.95.25.14705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayne L, Paterson Y, Cerasoli D, Englander SW. Effect of antibody binding on protein motions studied by hydrogen-exchange labeling and two-dimensional NMR. Biochemistry. 1992;31:10678–10685. doi: 10.1021/bi00159a006. [DOI] [PubMed] [Google Scholar]
- Němeček D, Overman SA, Hendrix RW. Unfolding thermodynamics of the Δ-domain in the prohead I subunit of phage HK97: determination by factor analysis of Raman spectra. Journal of Molecular …. 2009 doi: 10.1016/j.jmb.2008.10.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross PD, Cheng N, Conway JF, Firek BA, Hendrix RW, Duda RL, Steven AC. Crosslinking renders bacteriophage HK97 capsid maturation irreversible and effects an essential stabilization. Embo J. 2005;24:1352–1363. doi: 10.1038/sj.emboj.7600613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross PD, Conway JF, Cheng N, Dierkes L, Firek BA, Hendrix RW, Steven AC, Duda RL. A free energy cascade with locks drives assembly and maturation of bacteriophage HK97 capsid. J Mol Biol. 2006;364:512–525. doi: 10.1016/j.jmb.2006.08.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snijder J, Rose RJ, Veesler D. Studying 18 MDa virus assemblies with native mass spectrometry. Angew Chem Int Ed Engl. 2013;52:4020–4023. doi: 10.1002/anie.201210197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suhanovsky MM, Parent KN, Dunn SE, Baker TS, Teschke CM. Determinants of bacteriophage P22 polyhead formation: the role of coat protein flexibility in conformational switching. Mol Microbiol. 2010;77:1568–1582. doi: 10.1111/j.1365-2958.2010.07311.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veesler D, Cambillau C. A common evolutionary origin for tailed-bacteriophage functional modules and bacterial machineries. Microbiol Mol Biol Rev. 2011;75:423–33. doi: 10.1128/MMBR.00014-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veesler D, Johnson JE. Virus maturation. Annu Rev Biophys. 2012;41:473–496. doi: 10.1146/annurev-biophys-042910-155407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veesler D, Quispe J, Grigorieff N, Potter CS, Carragher B, Johnson JE. Maturation in Action: CryoEM Study of a Viral Capsid Caught during Expansion. Structure (London, England : 1993) 2012;20:1384–1390. doi: 10.1016/j.str.2012.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Matsui T, Domitrovic T, Zheng Y, Doerschuk PC, Johnson JE. Dynamics in cryo EM reconstructions visualized with maximum-likelihood derived variance maps. J Struct Biol. 2013;181:195–206. doi: 10.1016/j.jsb.2012.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wikoff WR, Liljas L, Duda RL, Tsuruta H, Hendrix RW. Topologically linked protein rings in the bacteriophage HK97 capsid. Science. 2000 doi: 10.1126/science.289.5487.2129. [DOI] [PubMed] [Google Scholar]
- Zhou ZH, Macnab SJ, Jakana J, Scott LR, Chiu W, Rixon FJ. Identification of the sites of interaction between the scaffold and outer shell in herpes simplex virus-1 capsids by difference electron imaging. Proc Natl Acad Sci USa. 1998;95:2778–2783. doi: 10.1073/pnas.95.6.2778. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.