

Abstract
DNA double strand breaks (DSBs) are deleterious lesions that can lead to chromosomal anomalies, genomic instability and cancer. The histone protein H2AX plays an important role in the DNA damage response (DDR) and the presence of phospho-H2AX (γH2AX) nuclear foci is the hallmark of DSBs. We hypothesize that ongoing DNA damage provides a mechanism by which chromosomal abnormalities and intratumor heterogeneity are acquired in malignant plasma cells (PCs) in patients with multiple myeloma (MM). Therefore, we assessed PCs from patients with the premalignant condition, monoclonal gammopathy of undetermined significance (MGUS) and MM, as well as human MM cell lines (HMCLs) for evidence of DSBs. γH2AX foci were detected in 2/5 MGUS samples, 37/40 MM samples and 6/6 HMCLs. Notably, the DSB response protein 53BP1 colocalized with γH2AX in both MM patient samples and HMCLs. Treatment with wortmannin decreased phosphorylation of H2AX and suggests phosphoinositide (PI) 3-kinases and/or PI3-kinase like family members underlie the presence of γH2AX foci in MM cells. Taken together, these data imply that ongoing DNA damage intensifies across the disease spectrum of MGUS to MM and may provide a mechanism whereby clonal evolution occurs in the monoclonal gammopathies.
Keywords: Myeloma, H2AX, PI3-kinase, ATR, DNA damage
INTRODUCTION
MM is an aggressive and typically fatal hematological malignancy that is characterized by the clonal expansion of malignant PCs in the bone marrow (BM).1 It is now known that MM is preceded by a stable precursor condition termed MGUS2,3 and the progression rate of MGUS to MM is ~1% per year.4 Unfortunately, the precise mechanism(s) underlying progression has yet to be determined; however, acquisition of additional chromosomal abnormalities and/or mutations is believed to play a role.
Importantly, abnormal PCs in both MGUS and MM possess numerous chromosomal abnormalities, each of which alone or in combination may underlie initial abnormal PC clonal expansion. It is also generally believed that additional PC intrinsic genetic changes are required for the progression from MGUS to MM.5 A specific example of a commonly observed chromosomal abnormality in both MGUS and MM are translocations involving the immunoglobulin heavy chain (IgH) locus. Indeed, IgH translocations are detected in over half of patients with MGUS and MM and this high frequency is believed to reflect errors associated with repair of DNA DSBs that are naturally introduced during the process of IgH class switch.6 Other natural processes involving the generation of DSBs within the Ig locus include VDJ recombination and somatic hypermutation.6 Although each of these naturally occurring genetic events is believed to be tightly regulated, it remains possible that they could also lead to oncogenic off-target mutations.7 The activation of oncogenes is known to promote unscheduled replication of premalignant and malignant cells, which can result in replicative stress and additional DSBs.8 Consequently, we hypothesize that inadvertent DSBs and genomic instability may underlie both transformation and clonal evolution in the monoclonal gammopathies.
Because DSBs can clearly lead to chromosomal abnormalities,9 the integrity of cellular DNA is closely monitored and an intricate repair program is immediately activated following detection of DNA damage.10 In this regard, H2AX, a member of the H2A family of histones, is one of the key components involved in the initial DDR. Within minutes of DSB formation, one of the PI3-like kinases (ataxia telangiectasia mutated (ATM), DNA-dependent protein kinase (DNA-PK) and/or Ataxia-telangiectasia-and-Rad3 related (ATR))11 becomes activated and phosphorylates H2AX on a carboxyl serine residue (Ser 139) to generate γH2AX. Previous findings suggest that multiple H2AX molecules within a 2-Mbp region around each DSB become phosphorylated to create a γH2AX focus.12,13 The function of γH2AX foci remains unclear but it is believed that they may aid in the recruitment and accumulation of DNA damage repair proteins such as 53BP1, Mre11, Rad50 and Nsb1.14,15 Given that each γH2AX focus is believed to represent a single DSB, immunohistochemical detection of γH2AX foci is a sensitive method for detection of DNA damage.16
Recently, several reports have shown that a variety of primary malignancies8,17–19 possess γH2AX foci in the absence of DNA inducing agents. To our knowledge, whether primary patient MM cells exhibit γH2AX foci has yet to be reported, however, several reports have demonstrated that γH2AX foci can be experimentally induced by various compounds in MM cells.20–22 There is also evidence for phosphorylated H2AX in freshly plated human MM cell lines (HMCLs), however, analysis of constitutive γH2AX foci was not the focus of this study.23 Given that MGUS and MM are both known to possess significant chromosomal abnormalities, the primary goal of our study was to assess primary MM patient samples and HMCLs for evidence of DSBs.
MATERIALS and METHODS
Patient material
MGUS and MM patient samples were collected as part of routine clinical examination. Written informed consent to participate in this study was provided by all subjects in accordance with the Declaration of Helsinki and the Mayo Clinic Institutional Review Board. MGUS patients were asymptomatic and had less than 10% BM clonal PCs while patients diagnosed with MM had 10% or greater clonal BM PCs, as defined.24 Normal control PCs were isolated from the BM of patients undergoing spine surgeries without coincident B lineage malignancies. MM patients and normal control subjects were on average 65.2 ± 12.3 and 63.1 ± 14.1 years old, respectively. Malignant and control BM aspirates were enriched for PCs by magnetic cell separation using a human CD138 positive selection kit (STEMCELL Technologies, Vancouver, Canada) and a Robosep Cell Separator. Control B cells were purified from normal donor blood using a human B cell enrichment kit (STEMCELL Technologies) and a RoboSep.
Cell lines, culture medium, and reagents
The HMCLs ANBL-6, DP-6, and KAS-6/1 have been previously described.25,26 VP-6 was established from primary patient MM cells but has not yet been described. Sister HMCLs, ALMC-1 and ALMC-2 were derived from a patient diagnosed with primary amyloidosis, who ultimately relapsed with MM.27 HL-60 cells were purchased from ATCC. The ANBL-6, ALMC-1, ALMC-2, KAS-6/1 and VP-6 HMCLs were maintained in IMDM (Invitrogen, Carlsbad, CA) while the DP-6 HMCL and HL-60 cells were maintained in RPMI 1640 (Invitrogen). Media was supplemented with 10% fetal calf serum, 100 U/ml penicillin G, 50 μg/ml gentamicin, 100 μg/ml streptomycin, 2 mmol/l glutamine and 1 ng/ml IL-6 (Novartis Pharma, Basel, Switzerland). HMCL experiments were performed at least 30–36 hrs following plating. Wortmannin was purchased from Sigma-Aldrich (St. Louis, MO).
Immunofluorescence (IF) analysis
γH2AX foci analysis was performed using cytospin preparations of cells mounted on glass slides using a Thermo Shandon cytospin 2. For γH2AX foci analysis of primary patient and normal control cells, slides were prepared as soon as PC or B cell sorting was completed, which was typically within 4 hours following collection of BM or blood. Cells were fixed with 4% paraformaldehyde (PFA), permeabilized with 0.2% Triton-X in PBS for 10 minutes and then incubated with pre-cleared 5% milk in PBS for 1 hour at RT to reduce non-specific binding. Cells were then incubated with an anti-γH2AX-FITC antibody (Ab) (Biolegend, San Diego, CA) (1 μg/ml) for 1 hour at RT in the dark. Subsequently, cells were incubated with an anti-Ig (H+L)-Texas-Red Ab (Southern Biotech, Birmingham, AL)(1 μg/ml) for 30 min at RT in the dark. To assess PCs in the S phase of the cell cycle, cells were pulsed with 10 μM 5-bromo-2′-deoxyuridine (BrdU; Sigma-Aldrich) for 1 hr before cytospin preparation. Prior to γH2AX staining, cells were incubated with the BrdU-specific monoclonal antibody, BU-1 (prepared internally at Mayo Clinic), for 1 hr at 37°C and then incubated with a rhodamine conjugated anti-mouse Ig secondary Ab for 30 min at 37°C.28 Following all staining protocols, cells were mounted with Vectashield containing DAPI (Vector Laboratories Inc., Burlingame, CA) and viewed using an Olympus AX 70 fluorescence microscope (Olympus Imaging America Inc., Center Valley, PA). Images were acquired using an Olympus DP71 microscope digital camera equipped with Olympus DP Manager Software. In order to calculate percentage of γH2AX expressing cells, all cells within a field were analyzed until at least one hundred cells were scored. In addition, the numbers of foci within all γH2AX positive cells were counted in order to determine the mean number of foci ± SD. If no γH2AX foci were observed following this protocol, the entire slide, containing ~10,000 cells, was scanned.
Plasma cell labeling index (PCLI), karyotyping, and FISH studies
PCLI, metaphase karotype and FISH analyses are clinical tests routinely performed for patients with monoclonal gammopathies and were completed by the Mayo Clinic Hematopathology Cell Kinetics and Cytogenetics Laboratories, respectively.
Flow cytometry
On the day of analysis, cells were pulsed with 10 μM BrdU for 2 h. BrdU incorporation and phosphorylated H2AX was determined according to the BrdU flow kit protocol (BD Biosciences, San Diego, CA). Cells were analyzed for BrdU incorporation and γH2AX content using a FACSCAN flow cytometer. Data were analyzed using FlowJo analytical software (TreeStar, Ashland, OR).
Immunoblotting
Western blotting and detection of immunoreactive proteins was performed as previously described.29 γH2AX, p53 and β-actin were detected using an anti-γH2AX Ab (Biolegend), an anti-p53 Ab (Santa Cruz Biotechnology, Inc.), and an anti-β-actin Ab (Novus Biologicals, Littleton, CO). The DNA damage proteins ATM, ATR, Chk1, and Chk2 were detected using primary Abs from a DNA damage Ab sampler kit (Cell Signaling Technology, Inc., Danvers, MA).
p53 mutational analysis
Total RNA was isolated from the HMCLs using the Trizol method (Invitrogen). Two μg of total RNA was converted to cDNA using the GE Healthcare First Strand cDNA Synthesis kit with random hexamer primers. Amplification was carried out using the QIAGEN HotStarTaq MasterMix kit with Q solution (QIAGEN Inc., Valencia, CA). Beta (β)-actin served as an internal control as previously described.30 p53 primers used to amplify p53 exons 5 to 11 were: forward 5′ CATTCTGGGACAGCCAAGTC 3′ and reverse 5′ ATGGCAGGGGAGGGAGAGAT 3′. PCR cycling conditions were as follows: 15 minutes at 94°C, 5 cycles of [30″@94°C, 30″@65°C, 1′@72°C]; 5 cycles of [30″@94°C, 30″@60°C, 1′@72°C]; 35 cycles of [30″@94°C, 30″@55°C, 1′@72°C]; and 10 minutes at 72°C. Amplified products were electrophoresed on a 1.5% agarose-TAE gel and visualized with ethidium bromide (200 ng/ml). Visible bands were cut out and purified with the Invitrogen Purelink gel extraction kit and sequenced directly with the primers used for amplification in both the forward and reverse directions.
RESULTS
γH2AX is localized to nuclear foci in primary MM plasma cells
In a study by Yang et al. (2009)23 phosphorylated H2AX was detected in freshly plated HMCLs by immunoblot; primary patient MM cells were not assessed in this study. Although immunoblotting is an excellent method of detecting phosphorylated H2AX, detection of γH2AX foci via IF is a more sensitive assay and allows for distinction between discrete γH2AX foci and pan-nuclear staining.10 Distinct nuclear γH2AX foci have also been well established as indicators of DSBs, and thus, we began our study by performing IF staining for γH2AX in freshly isolated primary MM patient samples. 37/40 MM samples were found to possess cells exhibiting γH2AX foci. Notably, the numbers of foci were generally found to vary from cell to cell, however, cells were considered to be γH2AX positive if they possessed one or more γH2AX foci. This scoring system was employed based on the knowledge and importance of the fact that even a single DSB is so deleterious, that if unrepaired, can be lethal.31 Importantly, the majority of γH2AX positive cells possessed more than one γH2AX foci. Figure 1A illustrates representative results from 5 different MM patient samples with various levels of γH2AX foci. Table 1 displays the results from all analyzed MM patients arranged, in ascending order, according to the percentage of cells possessing one or more γH2AX foci. Of note, samples MM 20 and MM 36 represent samples taken from the same patient, eleven months apart. In addition, we also examined MGUS patient samples and detected a limited number of γH2AX positive cells in 2 of 5 patients. An image of PCs from one of the MGUS patients exhibiting γH2AX foci is shown in Figure 1A.
Figure 1. Detection of γH2AX nuclear foci in MM patient samples and HMCLs.
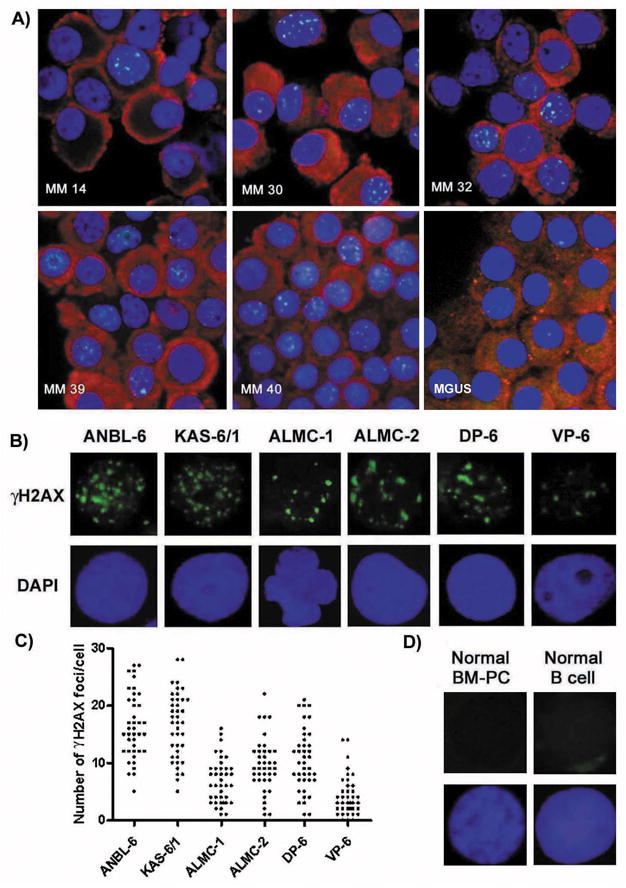
A) IF detection of γH2AX nuclear foci in freshly isolated CD138+ cells of 5 MM patients and 1 MGUS patient using an antiγH2AX-FITC Ab (green), anti-Ig (H + L) Ab (red) and DAPI (blue). B) Representative IF detection of nuclear γH2AX foci in HMCLs using an anti-γH2AX-FITC Ab (green) and DAPI (blue). C) The number of γH2AX foci/cell present in 50 individual cells for each HMCL. D) Representative IF detection of nuclear γH2AX foci in normal BM-PCs and normal B cells using an anti-γH2AX-FITC Ab (green) and DAPI (blue).
Table 1.
Clinical Characteristics of MM Patients. Metaphase karotype analysisa//FISHb.
| Pt | Disease Status | G | Age | PC % | H2AX % | Mean H2AX Foci ± SD per nucleus | PCLI | Chromosomal Aberrationsa//FISHb |
|---|---|---|---|---|---|---|---|---|
| MM1 | Treated | F | 67 | 62 | 0 | 0.0 ± 0.0 | 3.7 | 46,XX[20]//t(11;14) |
| MM2 | Never Treated | F | 73 | 42 | 0 | 0.0 ± 0.0 | 1 | 46,XX[20]//+7, +9, +15, −13 |
| MM3 | Relapse | F | 60 | 52 | 0 | 0.0 ± 0.0 | 1.7 | 54,X,−X,−2,−2,+add(3)(q25),−4,+add(6)(q21)x2,del(6)(q13q23), dic(7;?)(q36;?), −8,del(8)(q21.2q22),+9,add(10)(q24)+add(15)(p11.2),add(17)(p11.2),+19,+add(21)(p11.2), +6mar[5]/108,idemx2[2]/46,XX[13]//+3, +7, +9, +15 |
| MM4 | Relapse | F | 63 | 47 | 10 | 3.2 ± 2.7 | 2.2 | 84,XX,−X,−X,add(1)(p13)x2,−6,−6,−13,−13,−14,−14, add(14)(q32)x2[1]/46,XX[19]//−13, t(6;14) |
| MM5 | Treated | M | 52 | 20 | 13 | 2.6 ± 1.1 | 1 | 46,XY[20]//+3, +7, +9, +15, −13, −14, −17p |
| MM6 | Never Treated | M | 77 | 38 | 16 | 3.3 ± 2.5 | 1.3 | 46,XY[20]//−13, t(11;14) |
| MM7 | Treated | M | 65 | 33 | 17 | 6.2 ± 5.4 | 0 | 46,XY[30]//+3, +7, +9, +11, +15 |
| MM8 | Post PBMSCT | M | 71 | 25 | 24 | 2.0 ± 1.2 | 0.2 | 46,XY[20]//FISH not done |
| MM9 | SMM | M | 74 | 61 | 29 | 2.5 ± 1.6 | 0 | 46,XY[20]//t(11;14) |
| MM10 | Never Treated | M | 77 | 57 | 31 | 3.4 ± 2.3 | 4.6 | 43–45,XY,+1,der(1;16)(q10;p10)x2,t(2;16)(p11.2;q24), del(8)(q24.1)[5],der(8;21)(q10;q10)[5],add(9)(p22)[8],−13[8],add(14)(q32)[9],+16[7],−20[4],+21[5],−22[8], +1-2mar[5][cp10]/46,XY[10]//−13, IGH/? |
| MM11 | Pre PBSCT | F | 50 | 43 | 34 | 3.9 ± 2.8 | 3.6 | 46–47,X,−X,add(1)(p13)[2],add(6)(q13)[2],+7[2], der(7)t(1;7)(q12;p22)x2[2],t(11;14)(q13;q32),+15[2], add(22)(q11.2)[2][cp4]/46,XX[16]//+7, +15, t(11;14) |
| MM12 | Treated | M | 49 | 21 | 35 | 2.7 ± 1.7 | 0 | 46,XY[20]//+3, +9, +11, +15 |
| MM13 | Relapse | M | 70 | 34 | 36 | 4.3 ± 2.2 | 0.4 | 46,XY[20]//+3, +9, +11, +15 |
| MM14 | Never Treated | M | 63 | 65 | 38 | 4.1 ± 3.1 | 0 | 47,XY,add(8)(p11.2),+add(11)(q13),t(11;14)(q13;q32),−13, +mar[1]/92,idemx2,+4mar[1]/46,XY,add(5)(q13)[1]/46,XY[27]//−13, t(11;14) |
| MM15 | Never Treated | M | 61 | 94 | 42 | 6.4 ± 4.2 | 1 | karyotype not done//13q-, t(14;16) |
| MM16 | Never Treated | F | 62 | 26 | 42 | 4.9 ± 2.8 | 0.2 | 53,XX,+3,+5,−8,+9,+11,add(13)(p11.2),+15,+21,+2mar[4]/46, XX[16]//+3, +7, +9, +15 |
| MM17 | Never Treated | M | 57 | 45 | 43 | 3.1 ± 2.2 | 5.6 | 46,XY[20]//−13, −17 |
| MM18 | Post PBSCT | M | 62 | 22 | 43 | 5.0 ± 2.6 | 0 | 61,XY,+add(1)(p13)x3,+2,+3,+3,+5,+6,+add(7)(q22)x2,−8,+9, +11,+13,+15,+17,+20[3]/46,XY[17]//FISH not done |
| MM19 | Relapse | F | 70 | 98 | 44 | 7.9 ± 4.1 | 4.5 | 41–43,X,−X,+1,+add(1)(q12)[5], der(1;6)t(1;6)(p12;q27)ins(6;?)(q27;?)[15], dic(1;6)(p13;q27)[2],dic(1;16)(p13;p13.3)[5],−4,add(6)(q13),er(8)t(4;8)(q12;p21),−10[11],add(10)(p11.2)[6], add(12)(p13),−13,add(13(p11.2),−14,add(16)(p11.2)[7], dd(16)(p13.1)[15],add(17)(q25),−22,+1-2mar[cp17]/84–86,XX, G27-X,−X,+1,+1,der(1;6)t(1;6)(p12;q27)ins(6;?)(q27;?)[2], dic(1;6)(p13;q27)x2[1],−4,−4,add(6)(q13)x2, der(8)t(4;8)(q12;p21)x2,add(10), (p11.2)x2,add(12)(p13)x2,−13, −13,add(13)(p11.2)x2,−14,−14,add(16)(p11.2)[2], add(16)(p13.1)[3],add(16)(p13.1)[2],add(17)(q25)x2,−22,−22,+2-5mar[cp3]//−13, t(4;14) |
| MM20 | Relapse | F | 75 | 23 | 44 | 7.0 ± 3.6 | 4.4 | 46,XX,t(1;19)(q23;q13.1)[2]/48,X,−X,−1,+3,add(4)(q35), add(6)(q12),−8,del(8)(p21),+9,+11,−12,−14,add(14)(q32), +15, +19, +2mar[1]/46,XX[17]//+3, +9, +15, −17p, IGH/? |
| MM21 | Relapse | M | 67 | 61 | 46 | 3.2 ± 2.0 | 5.8 | 53–55,XY,+Y,+add(1)(p13),der(1)del(1)(p32p34)del(1)(q12),+3, der(4)t(3;4)(q21;p16),+7,−8,+9,+11,+15,add(17)(p11.2),+19,+21, +1-2mar[cp4]/46,XY[16]//+3, +7, +9, +11, +15, −17p |
| MM22 | Relapse | M | 28 | 99 | 50 | 2.9 ± 2.4 | 3.4 | 56,XY,+X,+add(1)(p13),add(1)(p36.1),+3,+5,+5,+add(6)(q21), +7,+9,+10, +15, add(16)(q13),+add(19)(q13.3), −22[11]/46,XY[9]//+3, +7, +9, +15, −13 |
| MM23 | Pre PBSCT | M | 62 | 61 | 52 | 4.0 ± 4.3 | 1.2 | 44,psu dic(X;9)(q24;p13),−Y,i(1)(q10),der(7)t(3;7)(p21;p22), del(13)(q12q14), −14,add(16)(q12.1), −22, +mar[5]/46,XY[15]//−13q, −14 |
| MM24 | Treated | M | 81 | 55 | 54 | 4.0 ± 2.9 | 0 | 46,XY[20]//+9, +11,+15 |
| MM25 | Never Treated | F | 92 | 41 | 55 | 4.3 ± 2.6 | 2.4 | 45,X,−X,add(3)(q27),+5,psu dic(5;18)(q33;p11.2),add(9)(p13),+add(9)(p24),−12,−14,+15,−20,add(22)(q13), +der(?)t(?;14)(?;q11.2)[1]/46, XX[29]//+9, +15 |
| MM26 | Never Treated | M | 72 | 74 | 56 | 5.4 ± 2.7 | 7.7 | 46,XY,+1,dic(1;22)(p13;p11.2),add(2)(p11.2), t(11;14)(q13;q32)[2]/46,XY[18]//t(11;14) |
| MM27 | Relapse | F | 72 | 93 | 61 | 4.0 ± 2.8 | 9 | 42–50,−X,−X[3],add(X)(q13)[16], trc(1;16;20)(p34.1;q22p11.2;q13.3)del(1)(p13p22)[18],add(1)(q21)[15], add(2)(q31),−4,+5,−6,add(6)(q21),add(7)(q32)[16],i(7)(p10)[2],−8,+9, −10[12],−10[3],der(10) add(10)(p11.2)add(10)(q24)[14], +add(11)(p11.2),−12,−14[3], add(14)(q32)[15],+15[16],add(15)(q24)[15], add(15)(p11.2)[14],−17[18],−18[6], −19[4],inv(19)(p13.3q13.3)[15],+add(21)(p11.2)[10], +del(22)(q13)×2, +3-8mar[cp19]/46,XX[1]//+9, +11, +15, −17p |
| MM28 | Never Treated | F | 58 | 43 | 63 | 5.1 ± 2.8 | 1.7 | 46,XX[20]//+3, +7, +9, +15 |
| MM29 | Relapse | F | 66 | 43 | 64 | 6.3 ± 3.5 | 1.4 | 54–55,XY,+Y,+add(1)(p11.2),add(1)(p13),+3,+5,−8,+9,+11,−13, add(14)(q32), der(14)t(1;14)(p22;q32),+15,+15, +19,+21,+0-1mar[cp2]/46, XY[18]//+3, +9, +15, −13, IGH/? |
| MM30 | Relapse | M | 65 | 42 | 67 | 5.5 ± 3.8 | 1.5 | 46,XY[20]//−13, −17p, t(4;14) |
| MM31 | Never Treated | M | 84 | 21 | 67 | 5.9 ± 2.5 | 0.2 | 46,XY,t(11;14)(q13;q32)[3]/46,XY[17]//t(11;14) |
| MM32 | Relapse | F | 55 | 94 | 68 | 4.9 ± 3.6 | 19 | 50–52,XX,der(4)t(3;4)(q12;p14),add(4)(q21),add(5)(p13), add(6)(q25),+7, add(8)(p21),+9, add(16)(q12),+15,+16, add(17)(q11.2), −18,+der(19)t(11;19)(q13;p13.1),+21,+add(22)(p11.2), der(14;16)(q32;p11.2), +1-3mar[cp18]/46,XX[2]//+3, +7, +9, +11, +15, −17p |
| MM33 | Never Treated | M | 77 | 20 | 68 | 3.0 ± 1.9 | 0 | 51,XY,der(1;16)(q10;p10),add(1)(q32),−2,+3,−4,+5,+7,−8,+9, add(11)(q25),+19,+4mar[1]/46,XY[19]//+3, +7, +9, +11, +15, IGH/? |
| MM34 | Treated | M | 65 | 75 | 68 | 5.7 ± 3.0 | 3.8 | 50,XY,der(1)del(1)(p13p22)add(1)(q21),+del(3)(p13),add(4)(p14),add(4)(q21), +add(5)(q11.2), der(5;10)(p10;q10), add(8)(p11.2),add(9)(q22),+der(9;12)(q10;q10),add(10)(p11.2),+del(11)(q11), add(12)(q13),−14,+15, der(17)t(11;17)(q13;p11.2),+19,add(22)(p11.2)[20]//+3, +9, +11, −17p |
| MM35 | Post PBSCT | M | 67 | 38 | 75 | 4.8 ± 2.8 | 10.6 | 51,XY,del(1)(p13p22),+7,+add(8)(p11.2),+11,+15,+19[8]/46,XY[12]//FISH not done |
| MM36 | Post PBSCT | F | 75 | 76 | 78 | 2.5 ± 1.7 | 1.8 | 52–53,X,−X,+add(1)(p13),der(1;12)(p10;p10),+3[3], add(4)(q35),+5,add(6)(q12), +add(6)[2],del(8)(p21),+9, +11, add(14)(q32),+15,+15,−17[2],+19,+0-2mar[cp5]/46,XX,t(1;19)(q23;q13.1)[1]/46,XX[14]//+3, +9, +11, +15, −17p, IGH/? |
| MM37 | Pre PBSCT | M | 55 | 41 | 78 | 7.5 ± 3.9 | 4.8 | 46,XY,add(1)(p13),t(1;5)(p22;q22)[2],add(6)(q13), add(8)(p21)[2], der(14)t(11;14)(q13;q32), +mar[2]cp[4]/46,XY[16]//−13, t(11;14) |
| MM38 | Pre PBSCT | F | 71 | 82 | 80 | 7.7 ± 5.4 | 1.7 | 86–90,XXXX,+add(1)(p13)×2,add(3)(q21)×2,add(6)(q13)×2,−8, add(9)(p13)×2,add(11)(q23)×2,−13,−13, t(14:16)(q32;q23)×2, −16,−16,+0-2mar[cp3]/46,XX[17]//−13, t(14;16) |
| MM39 | Never Treated | M | 32 | 35 | 90 | 6.7 ± 4.1 | ND | 46,XY[20]//13q-, −16, IGH/? |
| MM40 | Never Treated | M | 67 | 60 | 90 | 4.6 ± 2.1 | 1.3 | 46,XY,add(2)(p13),del(13)(q12q14),add(14)(q22), der(14)t(11;14)(q13;q32),−15,+mar[1]/46,XY[29]//−17p, t(11;14) |
HMCLs also exhibit constitutive γH2AX foci
The data described above clearly demonstrate that a large proportion of primary MM patient PCs exhibit γH2AX foci. Because numbers of primary MM cells are typically limited and are thereby prohibitive to mechanistic studies, we next wished to assess whether HMCLs also exhibit γH2AX foci. As shown in Figure 1B, all six of the HMCLs tested were found to exhibit constitutive γH2AX foci. The number of γH2AX positive cells was found to vary considerably between the HMCLs and was also found to vary from cell to cell within a given cell line (Figure 1C).
Normal BM-PCs and normal B cells do not show evidence of γH2AX foci
In order to ascertain whether γH2AX foci were specific to MGUS and MM cells, we examined freshly isolated BM-PCs from 15 normal donors and freshly isolated normal blood B cells from 7 normal donors for the presence of γH2AX foci. Figure 1D shows representative IF findings from these studies. In each case, all cells on the slide were scanned and we failed to observe even a single normal B cell or normal BM-PC exhibiting γH2AX foci.
The presence of constitutive nuclear γH2AX foci does not correlate with phase of the cell cycle
The extent of H2AX phosphorylation has previously been shown to be associated with the phase of the cell cycle.32 In general, S and G2/M phase cells tend to demonstrate higher levels of H2AX phosphorylation compared to G1 cells. Thus, it remained possible that the γH2AX positive cells that we observed in the MM patient samples simply reflected cells in the S phase of the cell cycle. To address this possibility, we labeled HMCLs with BrdU and measured the levels of γH2AX in the G1, S and G2/M phases of the cell cycle. Under normal culture conditions the majority of γH2AX was found to be present in the G1 and S phases of the cell cycle (data not shown). IF analysis revealed both BrdU positive (S/G2M phase) and BrdU negative (G1) cells possessing γH2AX foci (Figure 2A). Finally, we examined whether there was a correlation between the number of cells possessing γH2AX foci in primary MM patient samples and their respective PCLI. As illustrated in Figure 2B, we did not observe an obvious correlation. Statistical analysis using the Spearman’s rank correlation test resulted in a Spearman coefficient r = 0.29 and a p-value of 0.07 and thus, confirmed a lack of correlation between % of γH2AX foci and PCLI. These data suggest that constitutive γH2AX in MM patient samples may not be primarily attributed to cycling cells and could indeed be reflective of DSBs induced in a cell cycle-independent manner.
Figure 2. γH2AX foci do not correlate with phase of the cell cycle and γH2AX colocalizes with 53BP1.

A) IF analysis of BrdU labeled ANBL-6 cells revealed both BrdU positive (S/G2M phase) and BrdU negative (G1) cells possessing γH2AX foci. B) No correlation is observed between the % of cells possessing γH2AX foci in primary MM patient samples and their respective PCLI. C) HMCLs and primary MM patient samples were double stained with an anti-γH2AX-FITC Ab (green) and an anti-53BP1/Alexa 594 Ab (red). The resulting images were then overlaid in order to determine colocalization. Representative results are shown using one MM patient sample as well as the HMCL ANBL-6.
53BP1 colocalizes with γH2AX nuclear foci
H2AX knockout cells have been shown to have impaired recruitment of the DNA repair proteins NBS1, BRCA1 and 53BP1.33 Recruitment of DNA repair proteins to γH2AX foci is generally thought to provide further evidence that γH2AX foci reflect genuine sites of DSB.34 To investigate whether the γH2AX foci in MM cells show evidence of additional DNA repair proteins, we costained primary MM patient cells and HMCLs for γH2AX and 53BP1. Clear co-localization of γH2AX and 53BP1 was observed in all 4 of the primary MM samples tested and in all 6 HMCLs (representative results are displayed in Figure 2C). These findings suggest that the γH2AX foci in MM cells reflect genuine sites of DSBs and provide evidence of ongoing DNA damage in BM resident MM cells.
Activation of DNA damage signaling molecules in HMCLs
H2AX is a substrate of the PI3-kinase like kinases ATM, ATR, and DNA-PK.10 Upon cellular sensing of DSBs, one or more of these kinases becomes activated and in turn phosphorylates H2AX and several other downstream signaling molecules.35 We next sought to determine which DNA damage signaling molecules, in addition to γH2AX, were activated in the HMCLs in the absence of DNA damage inducing agents. As shown in Figure 3A, all 6 of the HMCLs showed robust phosphorylation of ATR and varying levels of phosphorylation of the down-stream ATR signaling target Chk1. ANBL-6 was the only HMCL to show constitutive phosphorylation of Chk2. Of note, phosphorylation of ATR, Chk1, and H2AX was not observed in normal B cells, normal T cells, or normal PBMCs (Figure 3A, right panel). Although robust phosphorylation of ATM was not detected in the HMCLs (data not shown), exposure to ionizing radiation was able to induce phosphorylation of ATM in the HMCLs (ANBL-6 is shown as a representative HMCL in Figure 3B). These results suggest that the DNA damage pathway that is activated in MM cells may be largely ATR-dependent.
Figure 3. Activation of DNA damage signaling molecules in HMCLs.
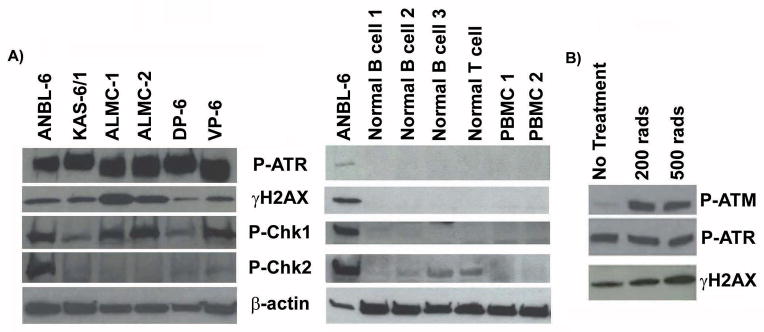
A) Immunoblotting demonstrates that HMCLs show evidence of constitutively activated DNA damage signaling molecules while normal control cells lack constitutive activation. Cell lines were maintained under normal culture conditions. B) Irradiation induced activation of P-ATM in the ANBL-6 HMCL. Irradiated cells were allowed to recover for 30 minutes.
PI3-kinase inhibition decreases γH2AX
Wortmannin is a PI3-kinase inhibitor that is able to inhibit the DNA damage signaling proteins ATM and DNA-PK at concentrations around 20μM while inhibition of ATR requires concentrations closer to 200μM.36 Because ATR is thought to be responsible for preventing replication of damaged DNA and generally signals upstream of γH2AX in the DNA damage signaling pathway10, we next treated cells with wortmannin in an effort to determine if either concentration of drug had an impact on levels of phosphorylated H2AX. As shown in Figure 4A, treatment of the HMCLs with both 20μM and 200μM wortmannin resulted in decreased phosphorylation of H2AX (Figure 4A). Of note, complete inhibition of ATR and H2AX phosphorylation was only observed when the cells were treated with the higher concentration of wortmannin. These data suggest that ATR may be playing a dominant role in the phosphorylation of H2AX in MM cells.
Figure 4. PI3-kinase inhibition decreases γH2AX.
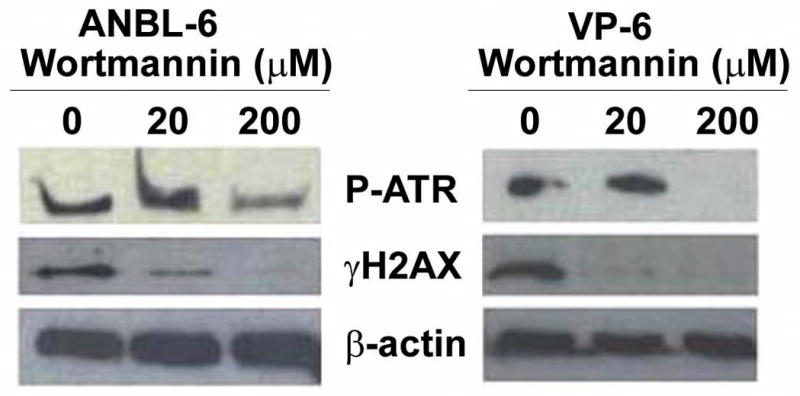
HMCLs were either untreated or treated for 1 hr with 20 μM or 200 μM wortmannin and analyzed via immunoblot.
HMCLs display evidence of p53 mutations
Activation of the DDR is believed to initially function as a barrier against tumor progression.8,17,18 However, continuous activation of this pathway in malignant cells is believed to select for those cells containing p53 mutations or deletion of p53.37 Reexamination of the clinical characteristics of our primary MM patients (Table 1) showed that patients possessing a higher % of cells with γH2AX foci tended to be positive for loss of 17p. Thus, we next examined our HMCLs for expression of p53 and p53 mutations. Compared to HL-60 cells, which are known to be p53 negative38, all HMCLs, with the exception of DP-6, expressed relatively high protein levels of p53 (Figure 5A). High protein expression or accumulation of p53 is often indicative of p53 mutations.38 Indeed, p53 mutation analysis revealed that all HMCLs tested possessed p53 mutations (Figure 5B). Using the IRAC TP53 database (http://www-p53.iarc.fr/), we were able to determine that each of these specific p53 mutations have previously been detected and reported in various malignancies. These data suggest that constitutive activation of the DDR may provide selective pressure for p53 mutations or loss of p53 in MM cells.
Figure 5. HMCLs express high levels of p53 and possess p53 mutations.
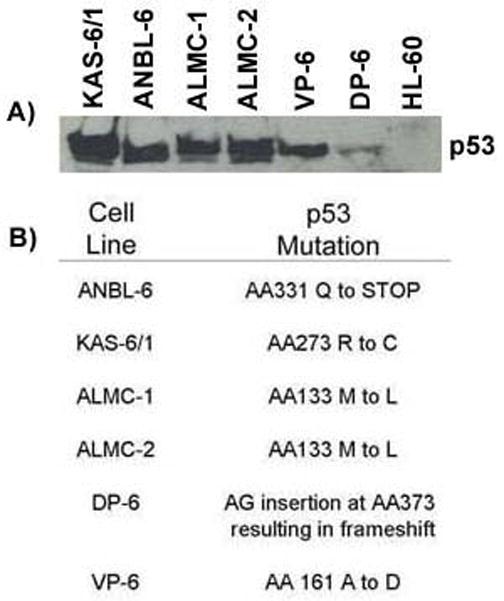
A) Immunoblotting demonstrates that HMCLs generally show evidence of high levels of expression of p53. Cell lines were maintained under normal culture conditions. B) Specific p53 mutations harbored by each HCML.
DISCUSSION
The goal of the current study was to examine primary malignant PCs from patients with MM for evidence of genetic instability as revealed by the presence of phosphorylated H2AX, a sensitive marker of DSBs. Indeed, IF analysis revealed the presence of distinct nuclear γH2AX foci in a high percentage of freshly isolated MM patient samples, and all HMCLs tested. A limited analysis of MGUS PCs provocatively revealed that 2 out of 5 MGUS patients studied included PCs with evidence of γH2AX foci. Importantly, γH2AX foci were not observed in any of the normal PC or B cell samples examined. Our study also demonstrates that MM cells display high levels of constitutive ATR activation, which may be indicative of replicative stress. Finally, we show that the PI3-kinase inhibitor, wortmannin, inhibits H2AX phosphorylation suggesting a critical role for this family of kinases in the DDR exhibited by MM cells. To our knowledge this is the first evidence demonstrating constitutive γH2AX foci and activation of a DDR in primary MM cells.
The presence of γH2AX foci, as well as variability in the number of γH2AX foci, has previously been shown in a variety of primary tumors and cell lines. In fact, Yu et al.39 observed γH2AX foci in 17 different tumor cell lines and demonstrated that foci numbers varied from cell to cell within the cell lines and that cell lines possessing overall higher numbers of γH2AX foci tended to have more structural chromosomal rearrangements and/or possessed mutant p53. Examination of the clinical characteristics of our MM patients revealed that, in general, patients possessing higher levels of γH2AX foci also tended to exhibit greater chromosomal abnormalities as revealed by metaphase karyotyping and FISH analysis. Because the cohort of patients studied was heterogeneous and included untreated and treated patients, we lack the statistical power to correlate γH2AX foci numbers with clinical outcome data. Of interest, however, within our patient cohort we had one instance in which we had obtained two samples from the same patient, eleven months apart. Notably, the latter sample displayed an increased percentage of γH2AX positive PCs, which was taken following stem cell transplant relapse. This very preliminary result suggests there may be clinical utility in measuring levels of γH2AX foci as a tool to identify patients undergoing disease progression. In this regard, larger studies of γH2AX foci in serial MM patient samples obtained over time are clearly warranted and are currently in progress. Overall, these results suggest that a large proportion of MM patient samples exhibit γH2AX foci and that these foci tend to be associated with greater numbers of chromosomal abnormalities, which may signify chromatin instability.
Because the presence of γH2AX foci may have utility in predicting disease progression, we also studied a very small series of patients with the premalignant condition, MGUS. We provocatively observed that 2 out of 5 MGUS patients displayed γH2AX positive PCs. It is interesting to note that upon re-review of the two γH2AX positive MGUS patients, one MGUS patient displayed a monoclonal immunoglobulin (M protein) level (0.5 g/dL) consistent with a diagnosis of MGUS, yet 15% BM-PCs, a level more consistent with a diagnosis of smoldering MM. This patient died from MM approximately one year after diagnosis of MGUS. Although our MGUS results are anecdotal due to the size of our cohort, they do suggest that analysis of a larger cohort of MGUS patients for H2AX positive PCs and disease progression may be warranted. It is also important to acknowledge that normal PC contamination is a known limitation of CD138 sorting of MGUS and MM patient samples. Because MGUS samples are much more likely to contain normal PCs, it was possible that the three MGUS patients (and three MM patients) that were negative for H2AX positive PCs were false negatives due to our scoring system which typically evaluated 100 cells. To overcome this limitation, all the cells on these slides were scanned for H2AX foci and all were found to be negative. Therefore, we consider it unlikely that normal BM-PCs are obscuring detection of H2AX positive abnormal PCs in these patient samples. Concerning patient samples that did display H2AX positive PCs, because normal BM-PCs are completely negative for H2AX foci, we believe that the actual percentage of γH2AX positive cells in the individual samples may actually be higher than reported due to this factor.
Although a variety of factors can induce the appearance of nuclear γH2AX foci, a number of recent studies have suggested that γH2AX foci in tumor cells arise as a result of replication stress.8,17 Replication stress or a block in replication arises when replication fork barriers such as secondary DNA structures or DNA damage are encountered during the S phase of the cell cycle40 resulting in activation of the DDR pathway in order to repair the DNA and faithfully restore DNA integrity. However, if the DNA damage is irreparable, cells undergo apoptosis in order to avoid the accumulation of deleterious chromosomal anomalies.18 In early cancer development it is believed that cells experience replication stress due to the unscheduled proliferation induced by the activation of oncogenes.18 Consequently, cells activate the DDR pathway and γH2AX becomes phosphorylated. Based on the fact that oncogenes are constitutively active in malignant cells, constitutive phosphorylation of H2AX in these cells is believed to be attributed to constitutive activation of the DDR. Indeed, Bartkova et al.17 recently demonstrated that in both low and high grade gliomas the DDR is constitutively activated and that several markers of ongoing DNA-replication stress could be detected. Thus, the authors concluded that activation of the DDR may be an early event in glioma tumorigenesis that may initially limit cell proliferation resulting in the selection of cells possessing mutations that facilitate checkpoint escape, and ultimately leading to tumor progression.17 Interestingly, prior to the development of MM, MGUS clonal PCs clearly increase significantly in number allowing detection of the abnormal PC’s secreted M protein. However, following this initial expansion, many MGUS patients display a remarkably stable M protein, suggesting the MGUS PC clone has reached a plateau. The biological reasons for the latter remain unclear, however, current dogma holds that progression to MM occurs following acquisition of additional critical mutations. Despite the fact that MGUS samples were limited in the current study, there appears to be an overall increase in γH2AX foci over the disease spectrum of MGUS to MM patient to HMCL. These data suggest that activation of the DDR may also occur early in MM disease development and may result in reduced or limited replication. The selection of cells possessing mutations able to overcome the DDR-induced checkpoint could then potentially result in disease progression to MM.
Our observations that ATR was constitutively activated in MM cells suggested the possibility that replication stress might underlie the induction of γH2AX foci. Unfortunately, there is no biochemical inhibitor that is absolutely specific for ATR, however, others have used the PI3-kinase inhibitor, wortmannin, to draw conclusions about the relative activity of ATM vs. ATR. Thus, Sarkaria et al.36 were the first to demonstrate that low wortmannin concentrations inhibit ATM (20 μM) while higher concentrations of wortmannin inhibit ATR (200 μM). This work was later confirmed in independent studies similarly showing ATR inhibition at the higher concentration.41–43 Although the higher concentration of wortmannin was needed in our studies to completely inhibit phosphorylation of H2AX and is therefore suggestive of a primary role for ATR activation, we acknowledge that the lack of specificity of this inhibitor does not permit a definitive conclusion. In additional experimentation not shown, we have used ATR-specific siRNA as a more specific method of inhibiting ATR. Although we were able to achieve nearly complete expression of ATR using this approach, the effects of ATR silencing on levels of phosphorylated H2AX were modest, suggesting that other PI3-kinases may also play a role. Of note, prior work by Chanoux et al.44 demonstrated that although ATR plays an important role in H2AX phosphorylation in embryonic fibroblasts under replication stress, ATM and DNA-PK play a role in H2AX phosphorylation in ATR-deficient cells. Thus, it is remains possible that ATR does indeed play a primary role in replication stress related H2AX phosphorylation in MM cells.
p53 is a tumor suppressor that plays a critical role in regulation of the cell cycle.45 In general, when a cell encounters stress (e.g., DNA damage, replication stress, environmental stress) p53 becomes activated and induces cell-cycle arrest. Recently, several reports have demonstrated that primary tumor samples and cell lines which demonstrate constitutive activation of the DDR and/or replication stress also possess mutations and/or loss of p53.8,17,18 Constitutive activation of the DDR is hypothesized to result in selection of cells which possess either loss or mutation of p53, as such cells could then overcome cell cycle arrest. Overcoming cell cycle arrest prior to repair of the aberrations which initiated it may then lead to the accumulation of additional mutations that stimulate proliferation.37 Of note, loss or mutation of p53 is believed to be a late occurring event in MM.5,46 Analysis of the clinical characteristics of our MM patients demonstrated that in general patients with a higher percentage of γH2AX and additional chromosomal abnormalities also exhibited loss of 17p. Moreover, the PCLI of these patients also tended to be generally higher. Taken together, we hypothesize that MM patients that exhibit constitutive γH2AX foci also possess constitutive activation of the DDR, which leads to the selection of those cells that have acquired p53 mutations and/or loss of 17p. As a result, these cells accumulate additional chromosomal aberrations, which may contribute to disease progression.
Overall, our results suggest that constitutive activation of the PI3-kinase-dependent DNA damage pathway underlies constitutive phosphorylation of H2AX in MM patient samples and cell lines. Additionally, constitutive activation of this DDR pathway may be responsible for selection of cells with inactivated or deleted p53, which allows cells to overcome cell-cycle arrest and/or apoptosis and may ultimately lead to tumor and/or disease progression.
Acknowledgments
This work was supported by a grant from the National Institutes of Health (CA062242).
Footnotes
Authorship Contributions DKW designed and performed research, analyzed data, and wrote the manuscript. RCT, BKA, and KJH performed research and analyzed data. XW designed research and interpreted data. PMH and AD designed research and enrolled patients. DFJ designed research, wrote and approved the manuscript.
Disclosure of Conflicts of Interest The authors declare no conflicts of interest.
References
- 1.Raab MS, Podar K, Breitkreutz I, Richardson PG, Anderson KC. Multiple myeloma. Lancet. 2009;374:324–339. doi: 10.1016/S0140-6736(09)60221-X. [DOI] [PubMed] [Google Scholar]
- 2.Landgren O, Kyle RA, Pfeiffer RM, Katzmann JA, Caporaso NE, Hayes RB, et al. Monoclonal gammopathy of undetermined significance (MGUS) consistently precedes multiple myeloma: a prospective study. Blood. 2009;113:5412–5417. doi: 10.1182/blood-2008-12-194241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Weiss BM, Abadie J, Verma P, Howard RS, Kuehl WM. A monoclonal gammopathy precedes multiple myeloma in most patients. Blood. 2009;113:5418–5422. doi: 10.1182/blood-2008-12-195008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kyle RA, Rajkumar SV. Monoclonal gammopathies of undetermined significance. Rev Clin Exp Hematol. 2002;6:225–252. doi: 10.1046/j.1468-0734.2002.00076.x. [DOI] [PubMed] [Google Scholar]
- 5.Bergsagel PL, Kuehl WM. Molecular pathogenesis and a consequent classification of multiple myeloma. J Clin Oncol. 2005;23:6333–6338. doi: 10.1200/JCO.2005.05.021. [DOI] [PubMed] [Google Scholar]
- 6.Pasqualucci L, Neumeister P, Goossens T, Nanjangud G, Chaganti RS, Kuppers R, et al. Hypermutation of multiple proto-oncogenes in B-cell diffuse large-cell lymphomas. Nature. 2001;412:341–346. doi: 10.1038/35085588. [DOI] [PubMed] [Google Scholar]
- 7.Gabrea A, Leif Bergsagel P, Michael Kuehl W. Distinguishing primary and secondary translocations in multiple myeloma. DNA Repair (Amst) 2006;5:1225–1233. doi: 10.1016/j.dnarep.2006.05.012. [DOI] [PubMed] [Google Scholar]
- 8.Gorgoulis VG, Vassiliou LV, Karakaidos P, Zacharatos P, Kotsinas A, Liloglou T, et al. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature. 2005;434:907–913. doi: 10.1038/nature03485. [DOI] [PubMed] [Google Scholar]
- 9.McKinnon PJ, Caldecott KW. DNA strand break repair and human genetic disease. Annu Rev Genomics Hum Genet. 2007;8:37–55. doi: 10.1146/annurev.genom.7.080505.115648. [DOI] [PubMed] [Google Scholar]
- 10.Bonner WM, Redon CE, Dickey JS, Nakamara AJ, Sedelnikova OA, Solier S, et al. GammaH2AX and cancer. Nat Rev Cancer. 2008;8:957–967. doi: 10.1038/nrc2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kinner A, Wu W, Staudt C, Iliakis G. Gamma-H2AX in recognition and signaling of DNA double-strand breaks in the context of chromatin. Nucleic Acids Res. 2008;36:5678–5694. doi: 10.1093/nar/gkn550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rogakou EP, Boon C, Redon C, Bonner WM. Megabase chromatin domains involved in DNA double-strand breaks in vivo. J Cell Biol. 1999;146:905–916. doi: 10.1083/jcb.146.5.905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Muslimovic A, Ismail IH, Gao Y, Hammarsten O. An optimized method for measurement of gamma-H2AX in blood mononuclear and cultured cells. Nat Protoc. 2008;3:1187–1193. doi: 10.1038/nprot.2008.93. [DOI] [PubMed] [Google Scholar]
- 14.Kruhlak MJ, Celeste A, Dellaire G, Fernandez-Capetillo O, Muller WG, McNally JG, et al. Changes in chromatin structure and mobility in living cells at sites of DNA double-strand breaks. J Cell Biol. 2006;172:823–834. doi: 10.1083/jcb.200510015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Paull TT, Rogakou EP, Yamazaki V, Kirchgessner CU, Gellert M, Bonner WM. A critical role for histone H2AX in recruitment of repair factors to nuclear foci after DNA damage. Curr Biol. 2000;10:886–895. doi: 10.1016/s0960-9822(00)00610-2. [DOI] [PubMed] [Google Scholar]
- 16.Pilch DR, Sedelnikova OA, Redon C, Celeste A, Nussenzweig A, Bonner WM. Characteristics of gamma-H2AX foci at DNA double-strand breaks sites. Biochem Cell Biol. 2003;81:123–129. doi: 10.1139/o03-042. [DOI] [PubMed] [Google Scholar]
- 17.Bartkova J, Horejsi Z, Koed K, Kramer A, Tort F, Zieger K, et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature. 2005;434:864–870. doi: 10.1038/nature03482. [DOI] [PubMed] [Google Scholar]
- 18.Bartkova J, Hamerlik P, Stockhausen MT, Ehrmann J, Hlobilkova A, Laursen H, et al. Replication stress and oxidative damage contribute to aberrant constitutive activation of DNA damage signalling in human gliomas. Oncogene. 2010;29:5095–5102. doi: 10.1038/onc.2010.249. [DOI] [PubMed] [Google Scholar]
- 19.Warters RL, Adamson PJ, Pond CD, Leachman SA. Melanoma cells express elevated levels of phosphorylated histone H2AX foci. J Invest Dermatol. 2005;124:807–817. doi: 10.1111/j.0022-202X.2005.23674.x. [DOI] [PubMed] [Google Scholar]
- 20.Dai Y, Chen S, Pei XY, Almenara JA, Kramer LB, Venditti Ca, et al. Interruption of the Ras/MEK/ERK signaling cascade enhances Chk1 inhibitor-induced DNA damage in vitro and in vivo in human multiple myeloma cells. Blood. 2008;112:2439–2449. doi: 10.1182/blood-2008-05-159392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kiziltepe T, Hideshima T, Ishitsuka K, Ocio EM, Raje N, Catley L, et al. JS-K, a GST-activated nitric oxide generator, induces DNA double-strand breaks, activates DNA damage response pathways, and induces apoptosis in vitro and in vivo in human multiple myeloma cells. Blood. 2007;110:709–718. doi: 10.1182/blood-2006-10-052845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ocio EM, Maiso P, Chen X, Garayoa M, Alverez-Fernandez S, San Segundo L, et al. Zalypsis: a novel marine-derived compound with potent antimyeloma activity that reveals high sensitivity of malignant plasma cells to DNA double-strand breaks. Blood. 2009;113:3781–3791. doi: 10.1182/blood-2008-09-177774. [DOI] [PubMed] [Google Scholar]
- 23.Yang C, Betti C, Singh S, Toor A, Vaughan A. Impaired NHEJ function in multiple myeloma. Mutat Res. 2009;660:66–73. doi: 10.1016/j.mrfmmm.2008.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kyle RA, Rajkumar SV. Criteria for diagnosis, staging, risk stratification and response assessment of multiple myeloma. Leukemia. 2009;23:3–9. doi: 10.1038/leu.2008.291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Westendorf JJ, Ahmann GJ, Greipp PR, Witzig TE, Lust JA, Jelinek DF. Establishment and characterization of three myeloma cell lines that demonstrate variable cytokine responses and abilities to produce autocrine interleukin-6. Leukemia. 1996;10:866–876. [PubMed] [Google Scholar]
- 26.Jelinek DF, Ahmann GJ, Greipp PR, Jalal SM, Westendorf JJ, Katzmann JA, et al. Coexistence of aneuploid subclones within a myeloma cell line that exhibits clonal immunoglobulin gene rearrangement: clinical implications. Cancer Res. 1993;53:5320–5327. [PubMed] [Google Scholar]
- 27.Arendt BK, Ramirez-Alvarado M, Sikkink LA, Keats JJ, Ahmann GJ, Dispenzieri A, et al. Biologic and genetic characterization of the novel amyloidogenic lambda light chain-secreting human cell lines, ALMC-1 and ALMC-2. Blood. 2008;112:1931–1941. doi: 10.1182/blood-2008-03-143040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Greipp PR, Kumar S. Plasma cell labeling index. Methods Mol Med. 2005;113:25–35. doi: 10.1385/1-59259-916-8:25. [DOI] [PubMed] [Google Scholar]
- 29.Walters DK, French JD, Arendt BK, Jelinek DF. Atypical expression of ErbB3 in myeloma cells: cross-talk between ErbB3 and the interferon-alpha signaling complex. Oncogene. 2003;22:3598–3607. doi: 10.1038/sj.onc.1206512. [DOI] [PubMed] [Google Scholar]
- 30.Arora T, Jelinek DF. Differential myeloma cell responsiveness to interferon-alpha correlates with differential induction of p19(INK4d) and cyclin D2 expression. J Biol Chem. 1998;273:11799–11805. doi: 10.1074/jbc.273.19.11799. [DOI] [PubMed] [Google Scholar]
- 31.Resnick MA, Martin P. The repair of double-strand breaks in the nuclear DNA of Saccharomyces cerevisiae and its genetic control. Mol Gen Genet. 1976;143:119–129. doi: 10.1007/BF00266917. [DOI] [PubMed] [Google Scholar]
- 32.MacPhail SH, Banath JP, Yu Y, Chu E, Olive PL. Cell cycle-dependent expression of phosphorylated histone H2AX: reduced expression in unirradiated but not X-irradiated G1-phase cells. Radiat Res. 2003;159:759–767. doi: 10.1667/rr3003. [DOI] [PubMed] [Google Scholar]
- 33.Celeste A, Petersen S, Romanienko PJ, Fernandez-Capetillo O, Chen HT, Sedelnikova OA, et al. Genomic instability in mice lacking histone H2AX. Science. 2002;296:922–927. doi: 10.1126/science.1069398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.DiTullio RA, Jr, Mochan TA, Venere M, Bartkova J, Sehested M, Bartek J, et al. 53BP1 functions in an ATM-dependent checkpoint pathway that is constitutively activated in human cancer. Nat Cell Biol. 2002;4:998–1002. doi: 10.1038/ncb892. [DOI] [PubMed] [Google Scholar]
- 35.Shiotani B, Zou L. Single-stranded DNA orchestrates an ATM-to-ATR switch at DNA breaks. Mol Cell. 2009;33:547–558. doi: 10.1016/j.molcel.2009.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sarkaria JN, Tibbetts RS, Busby EC, Kennedy AP, Hill DE, Abraham RT. Inhibition of phosphoinositide 3-kinase related kinases by the radiosensitizing agent wortmannin. Cancer Res. 1998;58:4375–4382. [PubMed] [Google Scholar]
- 37.Marusyk A, DeGregori J. Replicational stress selects for p53 mutation. Cell Cycle. 2007;6:2148–2151. doi: 10.4161/cc.6.17.4732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Collins SJ. The HL-60 promyelocytic leukemia cell line: proliferation, differentiation, and cellular oncogene expression. Blood. 1987;70:1233–1244. [PubMed] [Google Scholar]
- 39.Yu T, MacPhail SH, Banath JP, Klokov D, Olive PL. Endogenous expression of phosphorylated histone H2AX in tumors in relation to DNA double-strand breaks and genomic instability. DNA Repair (Amst) 2006;5:935–946. doi: 10.1016/j.dnarep.2006.05.040. [DOI] [PubMed] [Google Scholar]
- 40.Osborn AJ, Elledge SJ, Zou L. Checking on the fork: the DNA-replication stress-response pathway. Trends Cell Biol. 2002;12:509–516. doi: 10.1016/s0962-8924(02)02380-2. [DOI] [PubMed] [Google Scholar]
- 41.Andreassen PR, D’Andrea AD, Taniguchi T. ATR couples FANCD2 monoubiquitination to the DNA-damage response. Genes Dev. 2004;18:1958–1963. doi: 10.1101/gad.1196104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ward IM, Chen J. Histone H2AX is phosphorylated in an ATR-dependent manner in response to replicational stress. J Biol Chem. 2001;276:47759–47762. doi: 10.1074/jbc.C100569200. [DOI] [PubMed] [Google Scholar]
- 43.Jirmanova L, Bulavin DV, Fornace AJ., Jr Inhibition of the ATR/Chk1 pathway induces a p38-dependent S-phase delay in mouse embryonic stem cells. Cell Cycle. 2005;4:1428–1434. doi: 10.4161/cc.4.10.2055. [DOI] [PubMed] [Google Scholar]
- 44.Chanoux RA, Yin B, Urtishak KA, Asare A, Bassing CH, Brown EJ. ATR and H2AX cooperate in maintaining genome stability under replication stress. J Biol Chem. 2009;284:5994–6003. doi: 10.1074/jbc.M806739200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Efeyan A, Serrano M. p53: guardian of the genome and policeman of the oncogenes. Cell Cycle. 2007;6:1006–1010. doi: 10.4161/cc.6.9.4211. [DOI] [PubMed] [Google Scholar]
- 46.Neri A, Baldini L, Trecca D, Cro L, Polli E, Maiolo AT. p53 gene mutations in multiple myeloma are associated with advanced forms of malignancy. Blood. 1993;81:128–135. [PubMed] [Google Scholar]