

Abstract
NPM-ALK chimeric oncogene is aberrantly expressed in an aggressive subset of T-cell lymphoma that frequently occurs in children and young adults. The mechanisms underlying the oncogenic effects of NPM-ALK are not completely elucidated. Inducible nitric oxide synthase (iNOS) promotes the survival and maintains the malignant phenotype of cancer cells by generating NO, a highly active free radical. We tested the hypothesis that iNOS is deregulated in NPM-ALK+ T-cell lymphoma and promotes the survival of this lymphoma. In line with this possibility, an iNOS inhibitor and NO scavenger decreased the viability, adhesion and migration of NPM-ALK+ T-cell lymphoma cells, and an NO donor reversed these effects. Moreover, the NO donor salvaged the viability of lymphoma cells treated with ALK inhibitors. In further support of an important role of iNOS, we found iNOS protein to be highly expressed in NPM-ALK+ T-cell lymphoma cell lines and in 79% of primary tumors but not in human T lymphocytes. Although expression of iNOS mRNA was identified in NPM-ALK+ T-cell lymphoma cell lines and tumors, iNOS mRNA was remarkably elevated in T lymphocytes, suggesting posttranscriptional regulation. Consistently, we found that miR-26a contains potential binding sites and interacts with the 3′-UTR of iNOS. In addition, miR-26a was significantly decreased in NPM-ALK+ T-cell lymphoma cell lines and tumors compared with T lymphocytes and reactive lymph nodes. Restoration of miR-26a in lymphoma cells abrogated iNOS protein expression and decreased NO production and cell viability, adhesion, and migration. Importantly, the effects of miR-26a were substantially attenuated when the NO donor was simultaneously used to treat lymphoma cells. Our investigation of the mechanisms underlying the decrease in miR-26a in this lymphoma revealed novel evidence that STAT3, a major downstream substrate of NPM-ALK tyrosine kinase activity, suppresses MIR26A1 gene expression.
Keywords: NPM-ALK, iNOS, microRNA-26a, STAT3, T-cell lymphoma, CTDSPL
Introduction
NPM-ALK (nucleophosmin-anaplastic lymphoma kinase) is a chimeric oncogene that molecularly characterizes an aggressive subset of T-cell lymphoma known as anaplastic large-cell lymphoma (ALCL). NPM-ALK-expressing (NPM-ALK+) T-cell lymphoma commonly affects children and young adults [1]. The generation of NPM-ALK results from the fusion of ALK on chromosome 2 to NPM on chromosome 5, which results in the t(2;5)(p23;q35) [2]. This translocation encodes the aberrant expression of NPM-ALK, a constitutively activated tyrosine kinase [3]. Previous studies have demonstrated that NPM-ALK induces cellular transformation and initiates lymphomagenesis [4–8]. To induce its effects, NPM-ALK acts as the centerpiece within a comprehensive molecular network that promotes cellular survival. In this network, NPM-ALK interacts with and phosphorylates the adapter proteins Grb2, Shc, IRS-1 and SNT, the enzymes phospholipase C-γ, PI3K/Akt, MAP kinases, IGF-IR, and the transcription factor STAT3 [5, 9–15]. Most likely, there are other unidentified molecular pathways that interact with NPM-ALK to complement its effects.
Nitric oxide (NO) is a highly active free radical that produces many reactive intermediates. NO plays important roles in numerous physiological and pathological cellular events including those occurring in cancerous cells [16, 17]. These roles include the regulation of immune defense, vasomotor activity, neurotransmission and platelet aggregation [18]. NO also acts as an intracellular messenger that inhibits apoptosis [19–21]. NO is generated from L-arginine by three distinct isoforms of NADPH-dependent NO synthases (NOSs): neuronal (nNOS), endothelial (eNOS) and inducible (iNOS). nNOS and eNOS are expressed constitutively and are transiently activated because their activation is dependent on the Ca2+-activated calmodulin. Owing to the transient nature of elevated Ca2+ levels, the activity of NO produced by nNOS and eNOS is short-lived. In contrast, activation of iNOS is initiated by inflammatory mediators and cytokines and is Ca2+ independent because calmodulin is tightly bound to iNOS even at basal Ca2+ levels [22]. Therefore, iNOS is distinguished from the constitutive isoforms by the production of much larger amounts of NO, which is associated with significant mutagenic effects that can occur through DNA damage, transition or transactivation of nucleic acid bases and/or inactivation of DNA-repair proteins [23]. Indeed, increased iNOS expression is observed in several types of solid tumors, including lung, prostate, breast, and colon cancers. Conversely, downregulation of iNOS attenuates the activity of these tumors [24–26]. For these reasons, iNOS is considered a potential therapeutic target for the treatment of malignant diseases [27]. Although the expression of iNOS has been described in some types of leukemia and lymphoma [28–34], the status of its expression and its potential role in NPM-ALK+ T-cell lymphoma are not known. In this study, we tested the hypothesis that iNOS is deregulated in NPM-ALK+ T-cell lymphoma and that this deregulation contributes to the pathogenesis of this lymphoma.
Materials and methods
Cell lines and cell culture
The cell lines Karpas 299, DEL, SR-786, SU-DHL-1 and SUP-M2 (DSMZ, Braunschweig, Germany) [35]; FE-PD (from Dr. Karen Pulford, John Radcliffe Hospital, Oxford, UK) [35]; Mac-2A (from Dr. George Rassidakis, MD Anderson Cancer Center, Houston, Texas, USA) [35]; 293T, MCF7 and U-937 (ATCC, Manassas, VA); and BAEC (Lonza, Allendale, NJ) were used. Human CD3+ pan-T lymphocytes were purchased (Stemcell Technologies, Vancouver, BC, Canada). Cell lines were maintained in RPMI 1640 (NPM-ALK+ and U-937), MEM (MCF7) or DMEM (293T and BAEC) supplemented with 10% FBS (Sigma, St. Louis, MO). Additional cell lines and reagents used in the study are included in Supporting Information.
Human tissues and immunohistochemical (IHC) staining
Patients had granted informed consent in accordance with the Declaration of Helsinki. After approval of the institutional ethical research committee, 19 archival ALK+ lymphoma tumors, 12 ALK-negative tumors and 5 reactive lymph nodes were probed by IHC for the expression of iNOS and nitrotyrosine as previously described [14]. IHC was also performed on formalin-fixed, paraffin-embedded cellblock sections prepared from the cell lines and T lymphocytes. Antibody dilution was 1:100 for iNOS and nitrotyrosine.
Transfection of siRNA and microRNA
Knockdown of ALK and STAT3 was achieved by transient transfection of specific targeting SMARTpool-designed siRNA (Dharmacon, Lafayette, CO) using the Nucleofector “V” solution (Lonza) and A-030 program. The siCONTROL non-targeting siRNA was used as a negative control. Cells were also transfected by electroporation with hsa-miR-26a or the negative control cel-miR-67 (no sequence identity with human microRNA; Dharmacon).
MicroRNA expression array
Total RNA isolation was performed using the Trizol reagent (Invitrogen, Grand Island, NY). RNA labeling and hybridization on microRNA array chips were performed as previously described [36]. Briefly, 5 μg of total RNA from each sample was biotin- labeled by reverse transcription using 5′ biotin end-labeled random octomer oligo primer. Hybridization of biotin-labeled cDNA was carried out on a microRNA array chip (MD Anderson Cancer Center, Version 5.0), which contains 678 human microRNA genes in duplicate. Hybridization signals were detected by biotin binding of a streptavidin–Alexa647 conjugate b using an Axon GenePix 4000B scanner and the images were quantified by GenePix 6.0 software (Axon Instruments, Union City, CA).
Potential targets of microRNA
Analysis of microRNA-predicted targets was performed using 3 different Web-based algorithms including: TargetScan (http://www.targetscan.org), miRanda (http://www.microrna.org/microrna/home.do), and PicTar (http://pictar.mdc-berlin.de/).
Chromatin immunoprecipitation (ChIP) assay
Using Genomatix and the Web-based software Matinspector (http://www.genomatix.de), we identified 2 sequences (S1 and S2) within the promoter of the CTDSPL gene (3p21.3), the host gene of MIR26A1 (http://www.mirbase.org/), which contain putative STAT3 binding sites. ChIP was performed using a commercially available kit (Upstate Biotechnology, Lake Placid, NY) (detailed in Supporting Information).
Plasmids, cloning, and luciferase assay
After screening by ChIP, S1 and S2 sequences (Supporting Information) were cloned into a luciferase vector (pGL4.17, Promega, Madison, WI) and then amplified by PCR using STAT3 binding sites. The sequences and the luciferase vector were double- digested with kpn1 and Bg1II restriction endonucleases. The vectors and PCR products were purified and ligated overnight at 16°C using T4 DNA ligase. Clones positive for S1-pGL4.17 or S2-pGL4.17 were confirmed by PCR. Transfection of S1-pGL4.17 and S2-pGL4.17 with/out STAT3 siRNA was performed using Lipofectamin (Invitrogen). As a transfection efficiency control, the Renilla luciferase reporter pRL-TK (Promega) was co-transfected. After 24 h, cells were harvested and subjected to luciferase assay using the DualGlo luciferase assay kit (Promega). Mean luciferase activity was derived from three independent experiments.
The miR-26a and iNOS 3′-UTR reporter clones were purchased from OriGene (Rockville, MD). Mutated miR-26a was generated using site-directed mutagenesis (Agilent, Santa Clara, CA) (Supporting Information). The 293T cells were transfected using Lipofectamin with iNOS 3′-UTR and reporter and the Renilla luciferase reporter construct pRL-TK combined with empty vector (pCMV, OriGene) or wild-type (WT) or mutated miR-26a reporter for 24 h. Experiments were performed in quadruplicate. WT iNOS 3′-UTR plasmid was purchased (OriGene). Site directed mutagenesis (Agilent) was used to generate iNOS that does not include 3′-UTR (Supporting Information). WT iNOS 3′-UTR expression plasmid as a template was used to incorporate the mutation. 293T cells were transfected with EV, WT or mutated iNOS along with either control miR or miR-26a for 48 h using the 4D electroporation system and program DG-130 (Lonza).
Quantitative real-time PCR (qPCR)
The High Pure FFPE RNA Micro kit (Roche Applied Science, Indianapolis, IN) was used to measure microRNA levels in formalin-fixed, paraffin-embedded tissue sections prepared from cell line pellets and tumor samples. Also, qPCR was used to measure iNOS, CTDSPL, and CTDSP2 mRNA levels. Details are included in Supporting Information.
Western blotting, migration, adhesion, viability, NO production
These assays were performed using standard techniques as described previously [14, 37] (see Supporting Information for details of methods and reagents).
Statistical analysis
For statistical analysis of microRNA array data, an individual “p” value was not considered meaningful because of multiple comparisons. The distribution of “p” values, under the null hypothesis that microRNA levels were not statistically different, was considered to be uniform. When differences between some microRNA levels were significant, we considered that there was an overabundance of the “p” values. This situation was captured by modeling the distribution of “p” values as a β-uniform mixture. To identify differences in microRNA levels that were statistically significant, a cutoff for the single “p” value was selected by controlling the false recovery rate defined as the percentage of the microRNA results that were found to be significant and were expected to be false positive. For other experiments, Student’s t-test was used and a “p” value of < 0.05 was considered significant.
Results
Expression of iNOS and nitrotyrosine in NPM-ALK+ T-cell lymphoma
Western blotting showed expression of iNOS and nitrotyrosine proteins in NPM-ALK+ T-cell lymphoma cell lines Karpas 299, DEL and SR-786 (Fig. 1A); SU-DHL-1 and SUP-M2 (not shown) – but not in T-cells (Fig. 1A). Immunostaining also revealed iNOS and nitrotyrosine in paraffin- embedded cell line pellets but not in T-cells (Fig. 1B). The iNOS protein was detected in 15 of 19 (79%) of the ALK+ lymphomas (Fig. 1C). Notably, iNOS and nitrotyrosine were simultaneously detected in 8 of 10 ALK+ tumors probed for the expression of nitrotyrosine (Fig. 1C). iNOS mRNA was significantly higher in T-cells (Fig. 1D) and reactive lymph nodes (Fig. 1E) than in NPM-ALK+ lymphoma cell lines and ALK+ lymphoma tumors. In ALK-negative ALCL lines, iNOS protein was very weakly expressed in FE-PD and totally absent in Mac-2A cells (Supporting Fig. 1A). Furthermore, iNOS protein was detected in only 7 of 12 (58%) ALK-negative ALCL tumors. Higher levels of iNOS mRNA were detected in FE-PD cells compared with Mac-2A cells (Supporting Fig. 1B). Furthermore, significant differences were not detected between iNOS mRNA levels in ALK-negative ALCL tumors and reactive lymph nodes (Supporting Fig. 1C). In cell lines of other hematological malignancies (Supporting Fig. 1A), western blotting showed that iNOS was expressed in mantle cell lymphoma (MCL) cell lines SP-53, JeKo-1 and Mino, and in the Jurkat T lymphoblastic leukemia/lymphoma cell line. In contrast, iNOS protein was not detected in chronic myelogenous leukemia (CML) cell lines KBM-5, MEG-01 and BV173. Expression of iNOS mRNA was remarkably variable with SP-53, JeKo-1 and MEG-01 demonstrating the lowest levels and KBM-5 and BV173 showing the highest levels (Supporting Fig. 1B).
Figure 1. Expression of iNOS and nitrotyrosine proteins and iNOS mRNA in NPM-ALK+ T- cell lymphoma.
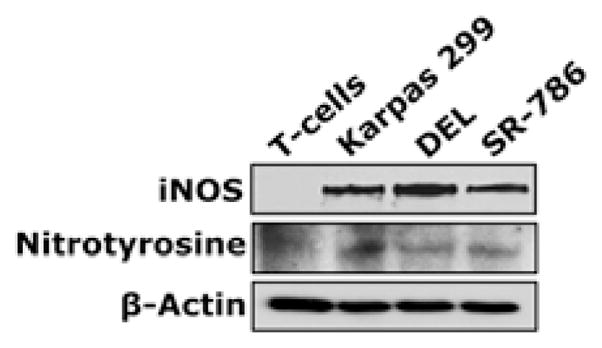

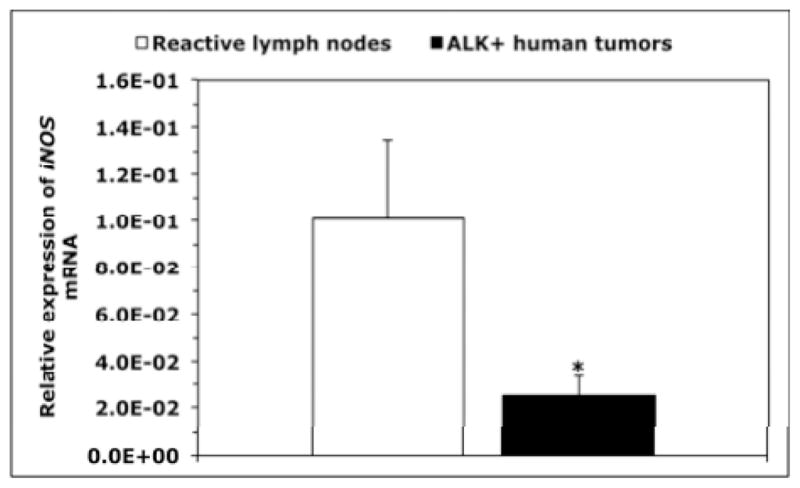
(A) Western blotting shows iNOS and nitrotyrosine in NPM-ALK+ T-cell lymphoma cell lines and lack of expression in T lymphocytes. (B) Immunohistochemistry demonstrates iNOS and nitrotyrosine in the lymphoma cell lines, but not in T lymphocytes. The 293T and BAEC cell lines were used as negative and positive controls, respectively, for iNOS; U-937 and MCF7 were used as negative and positive controls, respectively, for nitrotyrosine (original magnification, ×1000 for T lymphocytes and ×400 for the cell lines); (C) Immunohistochemistry of iNOS and nitrotyrosine in representative examples of ALK+ T-cell lymphomas. Lymphoma tissues from patients #2 and #3 were positive for both iNOS and nitrotyrosine and the specimen from Patient #1 was negative for both. Expression of iNOS was not detected in the lymphoid cells in reactive lymph nodes; however, iNOS demonstrated intense expression in stromal cells and scattered plasma cells. Nitrotyrosine was seen in scattered plasma cells and weak staining in reactive germinal centers, which became much less pronounced in the mantle zones and mostly absent in inter-follicular areas (original magnification, ×400); (D) qPCR demonstrates that the expression of iNOS mRNA was remarkably higher in T lymphocytes than in the lymphoma cell lines (*: p < 0.01, **: p < 0.001 vs. T-cells); (E) Similarly, iNOS mRNA was significantly higher in reactive lymph nodes compared with ALK+ T-cell lymphoma tumors (*: p < 0.01 vs. reactive lymph nodes). Data shown in D and E represent the means ± SE of 3 independent experiments.
Effects of iNOS inhibitors, NO scavengers, and NO donors on NPM-ALK+ T-cell lymphoma
To test whether NO/iNOS signaling contributes to the survival of NPM-ALK+ T-cell lymphoma, we used 1400W (iNOS inhibitor) or C-PTIO (NO scavenger). Treatment of T lymphocytes with 1400W or C-PTIO did not decrease the viability of these cells (Fig. 2A). Nonetheless, treatment of Karpas 299 and DEL cells with 1400W or C-PTIO was associated with a significant decrease in their viability (Fig. 2B), adhesion to endothelial cells (Fig. 2C), and IGF-I-induced migration (Fig. 2D). Importantly, the NO donor SNAP significantly diminished the effects of 1400W and C-PTIO. Furthermore, SNAP reversed the decrease in cell viability induced by ALK inhibitors TAE684 and PF-2341066 (Fig. 2E and Fig. 2F), which suggests that the survival-promoting effects of NPM-ALK are mediated, at least partially, through iNOS/NO signaling.
Figure 2. Inhibition of iNOS/NO signaling decreases the viability, adhesion, and migration of NPM-ALK+ T-cell lymphoma cells.
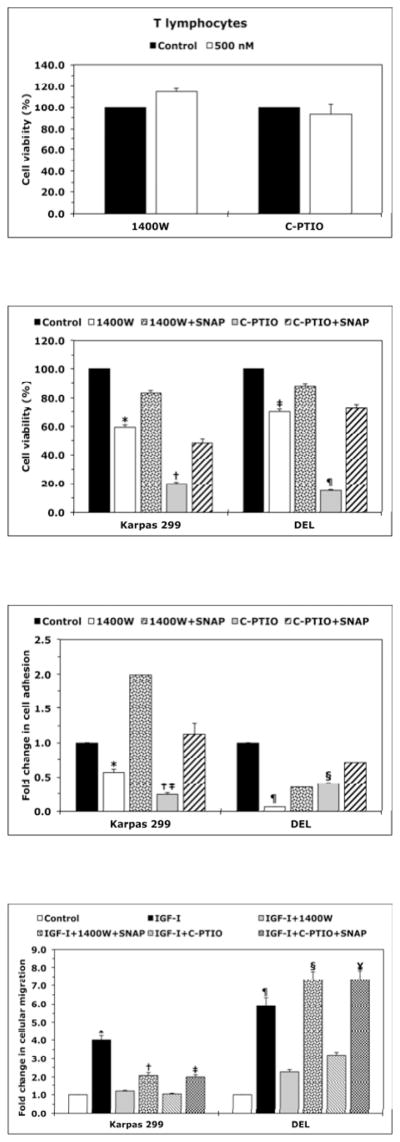

(A) The iNOS inhibitor 1400W (500 nM) and the NO scavenger C-PTIO (500 nM) did not change viability of T lymphocytes at 48 h after treatment; (B) In contrast, 1400W and C-PTIO decreased the viability of Karpas 299 and DEL cells at 24 h after treatment, and the NO donor SNAP (500 nM) attenuated these effects (*: p < 0.001 vs. control and 1400W+SNAP, †: p < 0.001 vs. control and C- PTIO+SNAP; ‡: p < 0.0001 vs. control and 1400W+SNAP; ¶: p < 0.0001 vs. control and C- PTIO+SNAP); (C) Inhibition of iNOS/NO signaling reduced the adhesion of Karpas 299 and DEL cells to endothelial cells and SNAP reversed these effects (*: p < 0.001 vs. control and 1400W+SNAP, †: p < 0.0001 vs. control, p < 0.01 vs. C-PTIO+SNAP, ‡: p < 0.00001 vs. control and 1400W+SNAP, ¶: p < 0.00001 vs. control and C-PTIO+SNAP); (D) IGF-I stimulated the migration of Karpas 299 and DEL cells, and 1400W and C-PTIO inhibited the effects of IGF-I. Additional treatment with SNAP reversed the effects of 1400W and C-PTIO (*: p < 0.001 vs. control, IGF-I+1400W, and IGF-I+C-PTIO, †: p < 0.01 vs. control and IGF-I+1400W, ‡: p < 0.001vs. control and IGF-I+C-PTIO, ¶: p < 0.001 vs. control and IGF-I+1400W and p < 0.01 vs. IGF-I+C- PTIO, §: p: < 0.001 vs. control and IGF-I+1400W, ¥: p < 0.001 vs. control and p < 0.01 vs. IGF-I+C- PTIO); The ALK inhibitors TAE684 or PF-2341066 induced a concentration-dependent decrease in the viability of Karpas 299 (E) and DEL (F) cells at 24 h. These effects were significantly diminished when cells were simultaneously treated with the ALK inhibitor and SNAP (*: p < 0.001, **: p < 0.0001 for cells treated with the ALK inhibitors and SNAP vs. cells treated with ALK inhibitors alone). Data shown are the means ± SE of 3 independent experiments.
MiR-26a is a posttranscriptional suppressor of iNOS expression in NPM-ALK+ T-cell lymphoma
Because T-cells lacked the expression of iNOS protein and expressed high levels of iNOS mRNA, we questioned whether posttranscriptional expression of iNOS is physiologically suppressed by microRNA. Three Web-based algorithms were used to analyze array data generated in our laboratory to compare the microRNA expression between human T lymphocytes and NPM-ALK+ lymphoma cell lines, and showed that miR-26a potentially binds to the 3′-UTR of iNOS (Fig. 3A). These data demonstrated that the expression of miR-26a was significantly decreased in the lymphoma cell lines compared with T-cells, which was confirmed by qPCR, not only in the NPM-ALK+ lymphoma cell lines and T-cells but also in 5 ALK+ T-cell lymphomas compared with 3 reactive lymph nodes (Fig. 3B). Notably, abundant levels of miR-26a were detected in ALK-negative ALCL tumors (Fig. 3B). We also found that WT miR-26a decreased the luciferase activity of iNOS 3′-UTR transfected into 293T cells, whereas miR-26a mutated at its binding site with iNOS 3′-UTR failed to induce this effect (Fig. 3C). Furthermore, miR-26a decreased WT iNOS protein transfected into 293T cells, but not iNOS construct that lacked 3′-UTR (Fig. 3D). These results support functional interactions between miR-26a and iNOS 3′-UTR. Importantly, transfection of miR-26a in Karpas 299 and DEL cells was associated with a dramatic downregulation of endogenous iNOS protein (Fig. 3E), but not iNOS mRNA (Fig. 3F), suggesting that the suppressive effect of miR-26a on iNOS occurs posttranscriptionally.
Figure 3. miR-26a is markedly decreased in NPM-ALK+ T-cell lymphoma, and its restoration decreases iNOS protein expression.




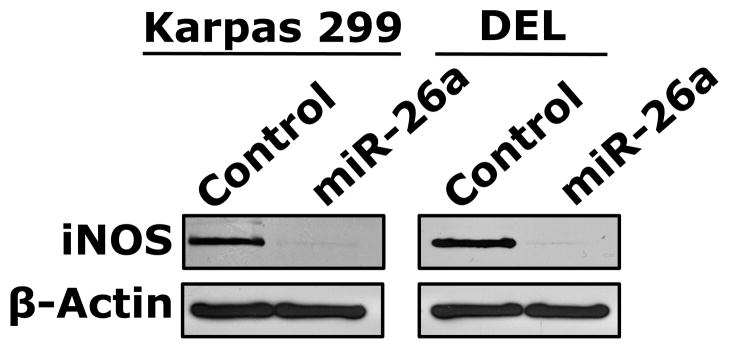

(A) In the upper panel, microRNA array heat map illustrates the expression of microRNA in the NPM-ALK+ T-cell lymphoma cell lines and T lymphocytes (yellow/orange/red = upregulated; blue/green = downregulated). The red arrow marks miR-26a that ranks 12th top microRNA downregulated in the NPM-ALK+ T-cell lymphoma cell lines versus T-cells (p = 0.000003). The lower panel shows potential binding sites between miR-26a and iNOS 3′-UTR; (B) qPCR shows miR-26a is decreased in the NPM-ALK+ T-cell lymphoma cell lines and ALK+ T-cell lymphomas compared with T lymphocytes, reactive lymph nodes and ALK-negative ALCL tumors (*: p < 0.05, **: p < 0.01 vs. T lymphocytes, reactive lymph nodes, and ALK-negative tumors, †: p < 0.05 vs. reactive lymph nodes); (C) WT miR-26a significantly decreased the luciferase activity of iNOS 3′-UTR, and MT miR-26a that lacked the binding sites with iNOS 3′-UTR failed to induce a similar effect (*: p < 0.0001); (D) In the upper panel, miR-26a remarkably decreased the levels of WT iNOS transfected into 293T cells and failed to induce a similar effect on MT iNOS that lacked 3′-UTR. Vertical lines indicate repositioned gel lanes. In the lower panel, densitometry shows a marked decrease in the iNOS:β-actin ratio when miR-26a was transfected with WT iNOS, but not with MT iNOS, into 293T cell line; (E) WB shows that transfection of miR-26a decreased endogenous iNOS protein in Karpas 299 and DEL cells; (F) but did not change iNOS mRNA in these cells. For results presented in D, E, and F, cel-miR-67 was used as a negative control. The results shown in B, C, and F represent the means ± SE of 3 different experiments performed in quadruplet.
Downregulation of iNOS expression by miR-26a decreases NO production and cellular viability, adhesion and migration of NPM-ALK+ T-cell lymphoma
Transfection of miR-26a induced a significant decrease in NO released by Karpas 299 and DEL cells (Fig. 4A). Further analysis showed that miR-26a caused a pronounced decrease in the viability of Karpas 299 and DEL cells (Fig. 4B) as well as in their adhesion to endothelial cells (Fig. 4C) and migration in response to IGF-I (Fig. 4D). Importantly, the effects of miR-26a were markedly attenuated when the NO donor SNAP was simultaneously used to treat the cells.
Figure 4. miR-26a inhibits NO production and decreases the viability, adhesion, and migration of NPM-ALK+ T-cell lymphoma.


(A) miR-26a decreased NO production by Karpas 299 and DEL cells (*: p < 0.01 vs. control; RFU = Relative Fluorescence Units); (B) miR-26a decreased viability and this effect was diminished by the NO donor SNAP (*: p < 0.0001 vs. control and p < 0.001 vs. miR-26a+SNAP, †: p < 0.00001 vs. control and miR-26a+SNAP); (C) miR-26a decreased adhesion of lymphoma cell lines to endothelial cells and SNAP effectively reversed the effects of miR-26a (*: p < 0.00001 vs. control and p < 0.01 vs. miR-26a+SNAP, †: p < 0.000001 vs. control and miR-26a+SNAP); (D) miR-26a abrogated the stimulatory effects of IGF-I on migration of Karpas 299 and DEL cells, and this effect was inhibited by SNAP (*: p < 0.01 vs. untreated cells and IGF-I+miR-26a; †: p < 0.01 vs. untreated cells and p < 0.05 vs. IGF-I+miR-26a). cel-miR-67 was used as the negative control miR. Results represent 3 independent experiments and are shown as means ± SE.
Deregulation of miR-26a/iNOS expression is mediated through NPM-ALK/STAT3 signaling
Next, we set out to analyze the mechanisms underlying the preferential expression of iNOS protein in NPM-ALK+ T-cell lymphoma compared with T-cells. Since our results suggested that miR-26a negatively regulates the expression of iNOS protein and that miR-26a is markedly reduced in NPM- ALK+ T-cell lymphoma compared with T-cells, we decided to investigate whether NPM-ALK contributes to this phenomenon. Also, because we identified putative binding sites between STAT3, a downstream target of NPM-ALK, and CTDSPL gene promoter, the host gene of MIR26A1, we wanted to determine whether STAT3 is involved in the aberrant expression of miR-26a/iNOS. Transfection of Karpas 299 and DEL cells with ALK siRNA caused a substantial decrease in NPM- ALK, pSTAT3 and iNOS proteins (Fig. 5A). Similarly, direct targeting of STAT3 by siRNA decreased iNOS protein (Fig. 5B). Downregulation of NPM-ALK or STAT3 by siRNA increased miR-26a levels (Fig. 5C). Neither ALK nor STAT3 siRNA induced significant effects on iNOS mRNA (Fig. 5D), suggesting that NPM-ALK and STAT3 are not directly involved in the regulation of iNOS gene transcription.
Figure 5. Downregulation of miR-26a and upregulation of iNOS protein expression in NPM- ALK+ T-cell lymphoma is mediated through an NPM-ALK/STAT3 signaling axis.

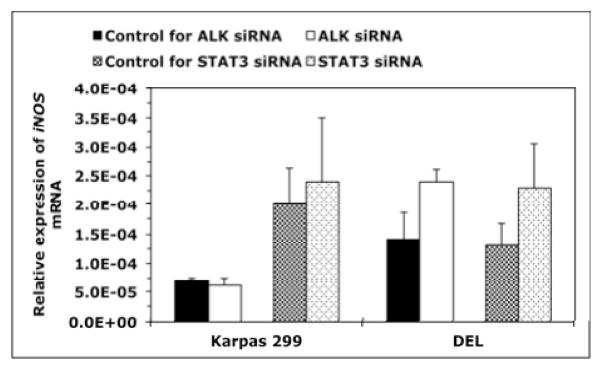
(A) Western blot shows that downregulation of ALK by siRNA in Karpas 299 and DEL cell lines was associated with a marked decrease in pSTAT3 and iNOS proteins; (B) Transfection of STAT3 siRNA decreased iNOS protein levels; (C) qPCR analysis shows that transfection of Karpas 299 and DEL cell lines with ALK or STAT3 siRNA increased miR-26a levels (*: p < 0.05, **: p < 0.00001); (D) ALK and STAT3 siRNA did not cause significant changes in iNOS mRNA levels by qPCR analysis. In all experiments, control cells were transfected with scrambled siRNA. Experiments were repeated 3 times and the data represent means ± SE.
Inhibition of NPM-ALK/STAT3 signaling upregulates CTDSPL gene expression and STAT3 suppresses CTDSPL gene promoter activity
We analyzed the effects of downregulation of NPM-ALK or STAT3 on the expression of CTDSPL gene, the host gene of MIR26A1 gene. Downregulation of NPM-ALK by ALK siRNA induced substantial increase in CTDSPL mRNA in Karpas 299 (Fig. 6A) and DEL (Fig. 6B) cells. Similar effects were noted when downregulation of STAT3 was achieved (Fig. 6C). In contrast, targeting NPM-ALK did not induce significant changes in the expression levels of CTDSP2, the host gene of MIR26A2 (Fig. 6D). These results suggest that the inhibition of miR-26a expression by the NPM- ALK/STAT3 signaling axis is mediated through suppression of MIR26A1 and not MIR26A2. Last, we further examined the interactions between STAT3 and the CTDSPL gene. ChIP assay in Karpas 299 and DEL cells demonstrated the binding between STAT3 and S1 and S2, two sequences within CTDSPL gene promoter identified by Genomatix that can potentially bind with STAT3 (Fig. 6E, left and right panels, respectively). Moreover, downregulation of STAT3 by siRNA in Karpas 299 and DEL cells increased luciferase activity of the CTDSPL gene promoter (Fig. 6F).
Figure 6. Specific targeting of NPM-ALK and STAT3 upregulates expression of CTDSPL, the host gene of MIR26A1, and STAT3 suppresses CTDSPL gene promoter activity.


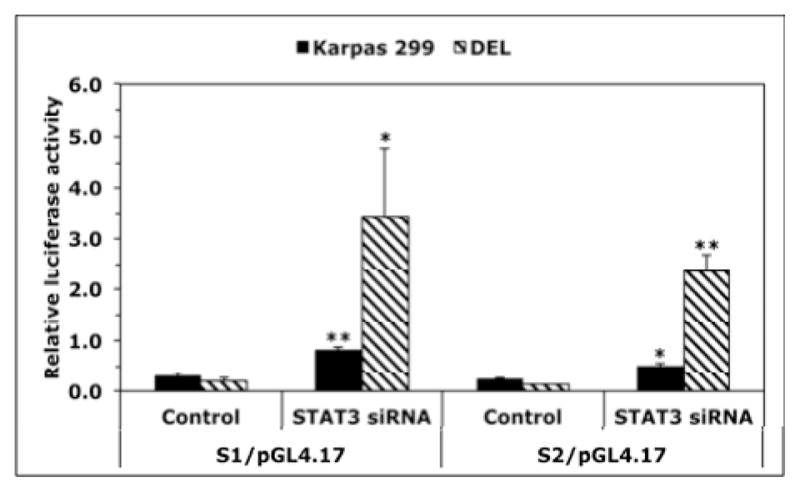
Downregulation of NPM-ALK in Karpas 299 (A) and DEL (B) cells increased CTDSPL mRNA levels (*: p < 0.001 for Karpas 299 and < 0.05 for DEL); (C) Similar results were obtained when STAT3 was downregulated, although the increase in CTDSPL levels was statistically significant only in Karpas 299 cells (p < 0.05) and not in DEL cells because of marked variability; (D) Specific targeting of NPM-ALK by ALK siRNA did not affect the expression levels of CTDSP2 mRNA, the host gene of MIR26A2; (E) ChIP analysis showed binding (black arrows) between STAT3 and S1 (left panel) and S2 (right panel) sequences of the CTDSPL gene. Colours have been inverted to facilitate visualization of the bands; (F) Targeting STAT3 in Karpas 299 and DEL cells was associated with a significant increase in the luciferase activity of the CTDSPL gene promoter (*: p < 0.05, **: p < 0.01 vs. control).
Discussion
In this study, we show that iNOS protein is highly expressed in NPM-ALK+ T-cell lymphoma cell lines and in 79% of ALK+ lymphomas. Notably, nitrotyrosine is also expressed in the cell lines and tumor tissues that simultaneously express iNOS suggesting that iNOS is activated in this lymphoma. In addition, iNOS appears to maintain the survival of this lymphoma because selective inhibitors of iNOS signaling decreased the viability, adhesion and migration of the lymphoma cells, and an NO donor significantly diminished the effects of these inhibitors. Using the NO donor salvaged the NPM-ALK+ T-cell lymphoma cells from the lethal effects of ALK inhibitors, which indicates that the survival-promoting effects of NPM-ALK might be mediated through iNOS/NO signaling.
When the mechanisms underlying the aberrant expression of iNOS in NPM-ALK+ T-cell lymphoma were investigated, we found novel evidence to support a working model (Fig. 7). Under physiological conditions, miR-26a is highly expressed in T-cells and suppresses iNOS expression posttranscriptionally. In support of this idea, significant levels of iNOS mRNA and miR-26a were detected in T-cells and reactive lymph nodes, but iNOS protein was absent in T-cells and small lymphoid cells residing in reactive lymph nodes. Whereas negligible levels of miR-26a were detected in NPM-ALK+ T-cell lymphoma cell lines and ALK+ tumors, the expression of iNOS protein was pronounced. Importantly, miR-26a contains binding sites and interacts with iNOS 3′-UTR. Restoration of miR-26a was associated with a marked decrease in iNOS protein expression, but not iNOS mRNA. The decrease in iNOS protein levels caused by miR-26a was associated with a significant reduction in NO released by NPM-ALK+ lymphoma cells and a marked decrease in the viability, adhesion to endothelial cells and IGF-I-induced migration of these cells. Our model also demonstrates that the decrease in miR-26a and resultant increase in iNOS are dependent on NPM-ALK. Downregulation of NPM-ALK by siRNA was associated with a significant increase in miR-26a and a marked decrease in iNOS protein. Two distinct genes encode the expression of the mature form of miR-26a. The first is the MIR26A1 gene located on the intron of CTDSPL (CTD [carboxy- terminal domain, RNA polymerase II, polypeptide A] small phosphatase-like) gene (3p21.3), and the second is the MIR26A2 gene on the intron of CTDSP2 (12q14.1). Downregulation of NPM-ALK in Karpas 299 and DEL cells was associated with a marked increase in CTDSPL mRNA levels and no significant changes in CTDSP2 mRNA levels. When we examined the contribution of STAT3, we found that the decrease in miR-26a occurs because STAT3 suppresses the expression and promoter activity of CTDSPL. Notably, STAT3 is an oncogenic transcription factor that contributes significantly to the survival of this lymphoma after being phosphorylated by NPM-ALK [15, 38, 39]. It has been previously shown that iNOS promotes the survival of some types of leukemia and lymphoma including chronic lymphocytic leukemia (CLL) [28–34]. In contrast to B-cells that lack iNOS expression, CLL lymphocytes express iNOS mRNA and protein [33]. It has also been shown that iNOS protects CLL lymphocytes from apoptosis [32]. Several mechanisms have been proposed for the aberrant expression of iNOS in CLL. The presence of iNOS spliced variants suggests that posttranscriptional regulation of iNOS expression occurs in CLL [34]. The expression of iNOS in CLL was also shown to be induced posttranscriptionally by interleukin-4 and transcriptionally and posttranscriptionally by interferon-γ [33]. In addition, stimulation of toll-like receptor 7 or CD23 was reported to induce iNOS expression in CLL [29–32].
Figure 7. Proposed model for the mechanisms underlying upregulation of iNOS protein expression in NPM-ALK+ T-cell lymphoma.

Under physiological conditions in T lymphocytes (purple), miR-26a is transcribed, processed, and then functions to inhibit the translation of iNOS mRNA to iNOS protein. In NPM-ALK+ T-cell lymphoma, NPM-ALK activates STAT3 by phosphorylation. Thereafter, pSTAT3 translocates to the nucleus where it suppresses transcription of MIR26A1. Decreased miR-26a levels in the cytoplasm allow translation of iNOS mRNA to iNOS protein. Dotted arrows indicate translocation to the nucleus or the cytoplasm. The lower panel details that pSTAT3 binds to the CTDSPL gene promoter at two sites, S1 and S2, to inhibit transcription. In addition, pSTAT3 inhibits transcription of MIR26A1, which is located in the intron of the CTDSPL gene.
Herein, we also analyzed the expression of iNOS in ALK-negative ALCL. iNOS protein was absent in Mac-2A cells, weakly expressed in FE-PD cells, and present in only 58% of the ALK- negative tumors. In contrast to NPM-ALK+ lymphoma, significant differences were not present between the levels of iNOS mRNA in reactive lymph nodes vs. ALK-negative ALCL tumors. Another important difference was that abundant levels of miR-26a were detected in the ALK- negative tumors. Although the contribution of iNOS signaling to the survival of ALK-negative ALCL cannot be ruled out, the regulatory mechanisms for iNOS expression in this lymphoma remain to be identified. When iNOS expression was analysed in other types of lymphoma/leukemia cell lines, we noticed significant variability. Similar to CLL, iNOS protein was detected in another B-cell lymphoma/leukemia, i.e., MCL. The expression of iNOS was also seen in the T lymphoblastic leukemia/lymphoma cell line Jurkat. In contrast, iNOS protein was not detected in CML cell lines. These results are similar to previous studies that showed that iNOS protein is absent or very weakly expressed in CML cell lines and primary neoplastic cells [40]. Interestingly, our data show that iNOS protein is more abundant in Karpas 299 cells compared with other types of leukemia/lymphoma cell lines that express iNOS.
Previous reports have proposed that STAT3 plays a role in the regulation of iNOS expression. Interestingly, the conclusions of these reports varied and were cell type- and experimental conditions-dependent. In a recent study, downregulation of STAT3 in murine macrophages was associated with decreased iNOS expression and iNOS gene promoter activity [41]. However, the binding of STAT3 to iNOS promoter was not documented in that study [41]. Therefore, it is difficult to exclude that the effect of STAT3 was mediated through intermediate molecules. In line with this concept, STAT3 alone was found to possess minimal effect on iNOS gene promoter activity in breast cancer cells, but STAT3/EGFR complex was capable of binding to iNOS promoter and enhanced its transcription [42]. In contrast, studies in murine mesangial cells showed that STAT3 suppresses iNOS expression through interactions with NF-κB [43, 44]. Herein, we could not support a direct effect of STAT3 on the transcription of iNOS because downregulation of STAT3 did not cause significant changes in iNOS mRNA levels.
Posttranscriptional mechanisms are also involved in the regulation of iNOS expression and, similar to its transcriptional regulation, these mechanisms vary based on the cell type and experimental conditions. Proteins such as JNK, HuR, and tristetraprolin were found to enhance the stability of iNOS mRNA, whereas KH-type splicing regulatory protein attenuates this stability [45, 46]. It has also been shown that ubiquitinated iNOS can be regulated by proteasome degradation [47]. Few studies have investigated the posttranscriptional regulation of iNOS expression by microRNA. Recently, it was shown that miR-939 downregulates iNOS protein expression in cytokine-stimulated iNOS protein in lipopolysaccharide-stimulated murine splenic lymphocytes and murine renal cell carcinoma [49, 50]. Nonetheless, these two studies did not document the binding and interactions between miR-146a and iNOS 3′-UTR. Furthermore, we could not identify iNOS 3′-UTR as a potential target of miR-146a by searching several Web-based algorithms. Defining the role of aberrantly expressed microRNA in the pathogenesis of NPM-ALK+ T-cell lymphoma has become a focus of research [51–54]. To our knowledge, our study is the first to document direct regulation of iNOS expression by a microRNA, i.e., miR-26a, in any type of cancer cells. MiR-26a was originally described as a tumor suppressor gene that is negatively regulated by Myc in B-cell lymphoma [55]. More recent studies have proposed that miR-26a has a dual effect: depending on the cancer cell type, it can function as an oncogene or a tumor suppressor. For example, as an oncogene, miR-26a targets PTEN and GSK-3β and contributes to the survival of glioma and cholangiocarcinoma, respectively [56, 57]. As a tumor suppressor gene, miR-26a targets several oncogenic proteins, including EZH2 [58]. Nonetheless, other investigators could not support a suppressive effect of miR-26a on EZH2 protein expression [59]. Although we cannot completely exclude that the negative impact of miR-26a on NPM-ALK+ T-cell lymphoma cells is partially mediated through its interactions with other known or unknown targets, the marked attenuation of the effects of miR-26a by the NO donor supports that the effects of miR-26a were mediated through targeting iNOS signaling.
In conclusion, we describe a new role of iNOS in cancer, namely in the pathogenesis of NPM-ALK+ T-cell lymphoma. We also found that miR-26a can suppress the expression of iNOS protein at the posttranscriptional level. Our results proposed novel oncogenic contributions of NPM-ALK and STAT3 to NPM-ALK+ T-cell lymphoma through downregulation of miR-26a and upregulation of iNOS protein. These findings expand current understanding of the pathobiology of NPM-ALK+ T-cell lymphoma, which could improve the therapeutic approaches used for the treatment of this aggressive lymphoma.
Supplementary Material
Acknowledgments
The authors are grateful for Dawn Chalaire for outstanding assistance with the preparation of this manuscript. The authors also thank Chang-Gong Liu, Ph.D. and Yong Li, Ph.D. for their help with the microRNA array methods and experiments and Jing Wang, Ph.D. for his assistance with the statistical analysis of the microRNA array data. This study was supported by grant R01 CA151533 from the National Cancer Institute and the Physician Scientist Program Award and Bridge Funding Grant from MD Anderson Cancer Center to H.M.A., and was also supported in part by MD Anderson Cancer Center support grant CA016672 from the National Cancer Institute. The contents of this paper are solely the responsibility of the authors and do not necessarily represent the official views of the National Cancer Institute or the National Institutes of Health.
Footnotes
Conflict of interest statement
The authors declare no competing financial interests
Statement of author contributions
HZ and DV designed and performed research, analyzed data and contributed to writing the paper; CVC provided experimental tools; RM, LD and AK performed research and analyzed data; HMA designed and performed research, analyzed data, provided experimental tools and wrote the paper. All of the authors read and approved the manuscript.
References
- 1.Amin HM, Lai R. Pathobiology of ALK+ anaplastic large-cell lymphoma. Blood. 2007;110:2259–2267. doi: 10.1182/blood-2007-04-060715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Morris SW, Kirstein MN, Valentine MB, et al. Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin’s lymphoma. Science. 1994;263:1281–1284. doi: 10.1126/science.8122112. [DOI] [PubMed] [Google Scholar]
- 3.Morris SW, Naeve C, Mathew P, et al. ALK, the chromosome 2 gene locus altered by the t(2;5) in non-Hodgkin’s lymphoma, encodes a novel neural receptor tyrosine kinase that is highly related to leukocyte tyrosine kinase (LTK) Oncogene. 1997;14:2175–2188. doi: 10.1038/sj.onc.1201062. [DOI] [PubMed] [Google Scholar]
- 4.Chiarle R, Voena C, Ambrogio C, et al. The anaplastic lymphoma kinase in the pathogenesis of cancer. Nat Rev Cancer. 2008;8:11–23. doi: 10.1038/nrc2291. [DOI] [PubMed] [Google Scholar]
- 5.Fujimoto J, Shiota M, Iwahara T, et al. Characterization of the transforming activity of p80, a hyperphosphorylated protein in a Ki-1 lymphoma cell line with chromosomal translocation t(2;5) Proc Natl Acad Sci USA. 1996;93:4181–4186. doi: 10.1073/pnas.93.9.4181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wellmann A, Doseeva V, Butscher W, et al. The activated anaplastic lymphoma kinase increases cellular proliferation and oncogene up-regulation in rat 1a fibroblasts. FASEB J. 1997;11:965–972. doi: 10.1096/fasebj.11.12.9337149. [DOI] [PubMed] [Google Scholar]
- 7.Kuefer MU, Look AT, Pulford K, et al. Retrovirus-mediated gene transfer of NPM-ALK causes lymphoid malignancy in mice. Blood. 1997;90:2901–2910. [PubMed] [Google Scholar]
- 8.Chiarle R, Gong JZ, Guasparri I, et al. NPM-ALK transgenic mice spontaneously develop T-cell lymphomas and plasma cell tumors. Blood. 2003;101:1919–1927. doi: 10.1182/blood-2002-05-1343. [DOI] [PubMed] [Google Scholar]
- 9.Riera L, Lasorsa E, Ambrogio C, et al. Involvement of Grb2 adaptor protein in nucleophosmin- anaplastic lymphoma kinase (NPM–ALK)-mediated signaling and anaplastic large cell lymphoma growth. J Biol Chem. 2010;285:26441–26450. doi: 10.1074/jbc.M110.116327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chikamori M, Fujiomto J, Tokai–Nishizumi N, et al. Identification of multiple SNT-binding sites on NPM-ALK oncoprotein and their involvement in cell transformation. Oncogene. 2007;26:2950–2954. doi: 10.1038/sj.onc.1210095. [DOI] [PubMed] [Google Scholar]
- 11.Bai RY, Dieter P, Peschel C, et al. Nucleophosmin-anaplastic lymphoma kinase of large-cell anaplastic lymphoma is a constitutively active tyrosine kinase that utilizes phospholipase C-gamma to mediate its mitogenicity. Mol Cell Biol. 1998;18:6951–6961. doi: 10.1128/mcb.18.12.6951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bai RY, Ouyang T, Miething C, et al. Nucleophosmin-anaplastic lymphoma kinase associated with anaplastic large-cell lymphoma activates the phosphatidylinositol 3-kinase/Akt antiapoptotic pathway. Blood. 2000;96:4319–4327. [PubMed] [Google Scholar]
- 13.Staber PB, Vesely P, Haq N, et al. The oncoprotein NPM-ALK of anaplastic large-cell lymphoma induces JUNB transcription via ERK1/2 and JunB translation via mTOR signaling. Blood. 2007;110:3374–3383. doi: 10.1182/blood-2007-02-071258. [DOI] [PubMed] [Google Scholar]
- 14.Shi P, Lai R, Lin Q, et al. IGF-IR tyrosine kinas interacts with NPM-ALK oncogene to induce survival of T-cell ALK+ anaplastic large-cell lymphoma. Blood. 2009;114:360–370. doi: 10.1182/blood-2007-11-125658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chiarle R, Simmons WJ, Cai H, et al. Stat3 is required for ALK-mediated lymphomagenesis and provides a possible therapeutic target. Nat Med. 2005;11:623–629. doi: 10.1038/nm1249. [DOI] [PubMed] [Google Scholar]
- 16.Lala PK, Chakraborty C. Role of nitric oxide in carcinogenesis and tumor progression. Lancet Oncol. 2001;2:149–156. doi: 10.1016/S1470-2045(00)00256-4. [DOI] [PubMed] [Google Scholar]
- 17.Fukumura D, Kashiwagi S, Jain RK. The role of nitric oxide in tumor progression. Nat Rev Cancer. 2006;6:521–534. doi: 10.1038/nrc1910. [DOI] [PubMed] [Google Scholar]
- 18.Palmer RM, Ferrige AG, Moncada S. Nitric oxide release accounts for the biological activity of endothelium-derived relaxing factor. Nature. 1987;327:524–526. doi: 10.1038/327524a0. [DOI] [PubMed] [Google Scholar]
- 19.Lander HM, Sehajpal PK, Novogrodsky A. Nitric oxide signaling: a possible role for G proteins. J Immunol. 1993;151:7182–7187. [PubMed] [Google Scholar]
- 20.Genaro AM, Hortelano S, Alvarez A, et al. Splenic B lymphocytes programmed cell death is prevented by nitric oxide release through mechanisms involving sustained Bcl-2 levels. J Clin Invest. 1995;95:1884–1890. doi: 10.1172/JCI117869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Beltrán B, Mathur A, Duchen MR, et al. The effect of nitric oxide on cell respiration: a key to understanding its role in cell survival or death. Proc Natl Acad Sci USA. 2000;97:14602–14607. doi: 10.1073/pnas.97.26.14602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cho HJ, Xie Q-W, Calaycay J, et al. Calmodulin is a subunit of nitric oxide synthase from macrophages. J Exp Med. 1992;176:599–604. doi: 10.1084/jem.176.2.599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wink DA, Kasprzak KS, Maragos CM, et al. DNA deaminating ability and genotoxicity of nitric oxide and its progenitors. Science. 1991;254:1001–1003. doi: 10.1126/science.1948068. [DOI] [PubMed] [Google Scholar]
- 24.Alatoma SH, Lipponen PK, Kosam VM. Inducible nitric oxide synthase (iNOS) expression and its prognostic value in prostate cancer. Anticancer Res. 2001;21:3101–3106. [PubMed] [Google Scholar]
- 25.Vakkala M, Kahlos K, Lakari E, et al. Inducible nitric oxide synthase expression, apoptosis, and angiogenesis in in situ and invasive breast carcinomas. Clin Cancer Res. 2000;6:2408–2416. [PubMed] [Google Scholar]
- 26.Land SC, Rae C. iNOS initiates and sustains metabolic arrest in hypoxic lung adenocarcinoma cells: mechanism of cell survival in solid tumor core. Am J Physiol Cell Physiol. 2005;289:C918–C933. doi: 10.1152/ajpcell.00476.2004. [DOI] [PubMed] [Google Scholar]
- 27.Fitzpatrick B, Mehiblel M, Cowen RL, et al. iNOS as a therapeutic target for treatment of human tumors. Nitric Oxide. 2008;19:217–224. doi: 10.1016/j.niox.2008.05.001. [DOI] [PubMed] [Google Scholar]
- 28.Billard C, Menasria F, Quiney C, et al. Flavopiridol-induced iNOS downregulation during apoptosis of chronic lymphocytic leukemia cells is caspase-dependent. Leuk Res. 2008;32:755–760. doi: 10.1016/j.leukres.2007.09.016. [DOI] [PubMed] [Google Scholar]
- 29.Hammadi A, Billard C, Faussat A-M, et al. Stimulation of iNOS expression and apoptosis resistance in B-cell chronic lymphocytic leukemia (B-CLL) cells through engagement of Toll-like receptor 7 (TLR-7) and NF-κB activation. Nitric Oxide. 2008;19:138–145. doi: 10.1016/j.niox.2008.04.017. [DOI] [PubMed] [Google Scholar]
- 30.Barreiro Arcos ML, Gorelik G, Klecha A, et al. Inducible nitric oxide synthase-mediated proliferation of a T lymphoma cell line. Nitric Oxide. 2003;8:111–118. doi: 10.1016/s1089-8603(02)00181-7. [DOI] [PubMed] [Google Scholar]
- 31.Li H-L, Sun B-Z, Ma F-C. Expression of COX-2, iNOS, p53 and Ki-67 in gastric mucosa-associated lymphoid tissue lymphoma. World J Gastroenterol. 2004;10:1862–1866. doi: 10.3748/wjg.v10.i13.1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhao H, Dugas N, Mathiot C, et al. B-cell chronic lymphocytic leukemia cells express a functional inducible nitric oxide synthase displaying anti-apoptotic activity. Blood. 1998;92:1031–1043. [PubMed] [Google Scholar]
- 33.Levesque MC, Misukonis MA, O’Loughlin CW, et al. IL-4 and interferon gamma regulate expression of inducible nitric oxide synthase in chronic lymphocytic leukemia cells. Leukemia. 2003;17:442–450. doi: 10.1038/sj.leu.2402783. [DOI] [PubMed] [Google Scholar]
- 34.Tiscornia AC, Cayota A, Landoni AI, et al. Post-transcriptional regulation of inducible nitric oxide synthase in chronic lymphocytic leukemia B cells in pro- and antiapoptotic culture conditions. Leukemia. 2004;18:48–56. doi: 10.1038/sj.leu.2403169. [DOI] [PubMed] [Google Scholar]
- 35.Drexler HG. Guide to leukemia-lymphoma cell lines. 2. Braunschweig; Germany: 2010. [Google Scholar]
- 36.Liu C-G, Calin GA, Volinia S, et al. MicroRNA expression profiling using microarrays. Nat Protoc. 2008;3:563–578. doi: 10.1038/nprot.2008.14. [DOI] [PubMed] [Google Scholar]
- 37.Stone WL, Yang H, Qui M. Assays for nitric oxide expression. Methods Mol Biol. 2006;315:245–256. doi: 10.1385/1-59259-967-2:245. [DOI] [PubMed] [Google Scholar]
- 38.Zamo A, Chiarle R, Piva R, et al. Anaplastic lymphoma kinase (ALK) activates STAT3 and protects hematopoietic cells from cell death. Oncogene. 2002;21:1038–1047. doi: 10.1038/sj.onc.1205152. [DOI] [PubMed] [Google Scholar]
- 39.Amin HM, McDonnell TJ, Ma Y, et al. Selective inhibition of STAT3 induces apoptosis and G1 cell cycle arrest in ALK-positive anaplastic large cell lymphoma. Oncogene. 2004;23:5426–5434. doi: 10.1038/sj.onc.1207703. [DOI] [PubMed] [Google Scholar]
- 40.Selleri C, Maciejewski JP, Montuori N, et al. Involvement of nitric oxide in farnesyltransferase inhibitor-mediated apoptosis in chronic myeloid leukemia cells. Blood. 2003;102:1490–1498. doi: 10.1182/blood-2003-01-0178. [DOI] [PubMed] [Google Scholar]
- 41.Park S-Y, Baik YH, Cho JH, et al. Inhibition of lipopolysaccharide-induced nitric oxide synthase through S6K1-p42/44 MAPK pathway and STAT3 (Ser 727) phosphorylation in Raw 262. 7 cells. Cytokine. 2008;44:126–134. doi: 10.1016/j.cyto.2008.07.006. [DOI] [PubMed] [Google Scholar]
- 42.Lo HW, Hsu SC, Ali-Seyed M, et al. Nuclear interaction of EGFR and STAT3 in the activation of the iNOS/NO pathway. Cancer Cell. 2005;7:575–589. doi: 10.1016/j.ccr.2005.05.007. [DOI] [PubMed] [Google Scholar]
- 43.Yu Z, Zhang W, Kone BC. Signal transducers and activators of transcription 3 (STAT3) inhibits transcription of the inducible nitric oxide synthase gene by interacting with nuclear factor κB. Biochem J. 2002;367:97–105. doi: 10.1042/BJ20020588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yu Z, Kone BC. The STAT3 DNA-binding domain mediates interaction with NF-κB p65 and inducible nitric oxide synthase transrepression in mesangial cells. J Am Soc Nephrol. 2004;15:585–591. doi: 10.1097/01.asn.0000114556.19556.f9. [DOI] [PubMed] [Google Scholar]
- 45.Linker K, Pautz A, Fechir M, et al. Involvement of KSRP in the post–transcriptional regulation of human iNOS expression–complex interplay of KSRP and TTP and HuR. Nucleic Acid Res. 2005;33:4813–4827. doi: 10.1093/nar/gki797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fechir M, Linker K, Pautz A, et al. Tristetraprolin regulates the expression of human inducible nitric–oxide synthase gene. Mol Pharmacol. 2005;67:2148–2161. doi: 10.1124/mol.104.008763. [DOI] [PubMed] [Google Scholar]
- 47.Kolodziejski PJ, Musial A, Koo J–S, et al. Ubiquitination of inducible nitric oxide synthase is required for its degradation. Proc Natl Acad Sci USA. 2002;99:12315–12320. doi: 10.1073/pnas.192345199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Guo Z, Shao L, Zheng L, et al. miRNA-939 regulates human inducible nitric oxide synthase posttranscriptional gene expression in human hepatocytes. Proc Natl Acad Sci USA. 2012;109:5826–5831. doi: 10.1073/pnas.1118118109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dai R, Phillips RA, Zhang Y, et al. Suppression of LPS-induced interferon–γ and nitric oxide in splenic lymphocytes by select estrogen-regulated microRNAs: a novel mechanism of immune modulation. Blood. 2008;112:4591–4597. doi: 10.1182/blood-2008-04-152488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Perske C, Lahat N, Levin SS, et al. Loss of inducible nitric oxide synthase expression in the mouse renal cell carcinoma cell line RENCA is mediated by microRNA miR-146a. Am J Pathol. 2010;177:2046–2054. doi: 10.2353/ajpath.2010.091111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Desjobert C, Renalier M-H, Bergalet J, et al. MiR-29a down-regulation in ALK-positive anaplastic large cell lymphomas contributes to apoptosis blockade through MCL-1 overexpression. Blood. 2011;117:6627–6637. doi: 10.1182/blood-2010-09-301994. [DOI] [PubMed] [Google Scholar]
- 52.Matsuyama H, Suzuki HI, Nishimori H, et al. miR-135b mediates NPM-ALK-driven oncogenicity and renders IL-17–producing immunophenotype to anaplastic large cell lymphoma. Blood. 2011;118:6881–6892. doi: 10.1182/blood-2011-05-354654. [DOI] [PubMed] [Google Scholar]
- 53.Dejean E, Renalier MH, Fiosseau M, et al. Hypoxia-microRNA-16 downregulation induces VEGF expression in anaplastic lymphoma kinase (ALK)-positive anaplastic large-cell lymphoma. Leukemia. 2011;25:1882–1890. doi: 10.1038/leu.2011.168. [DOI] [PubMed] [Google Scholar]
- 54.Vishwamitra D, Li Y, Wilson D, et al. MicroRNA 96 is a post-transcriptional suppressor of anaplastic lymphoma kinase expression. Am J Pathol. 2012;180:1772–1780. doi: 10.1016/j.ajpath.2012.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chang T-C, Yu D, Lee Y-S, et al. Widespread microRNA repression by Myc contributes to tumorigenesis. Nat Genet. 2007;40:43–50. doi: 10.1038/ng.2007.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Huse JT, Brennan C, Hambardzumyan D, et al. The PTEN-regulating microRNA miR-26a is amplified in high–grade glioma and facilitates gliomagenesis in vivo. Genes Dev. 2009;23:1327–1337. doi: 10.1101/gad.1777409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhang J, Han C, Wu T. MicroRNA-26a promotes cholangiocarcinoma growth by activating β–catenin. Gastroenterology. 2012;143:246–256. doi: 10.1053/j.gastro.2012.03.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sander S, Bullinger L, Klapproth K, et al. MYC stimulates EZH2 expression by repression of its negative regulator miR-26a. Blood. 2008;112:4202–4212. doi: 10.1182/blood-2008-03-147645. [DOI] [PubMed] [Google Scholar]
- 59.Varambally S, Cao Q, Mani RS, et al. Genomic loss of microRNA-101 leads to overexpression of methyltransferase EZH2 in cancer. Science. 2008;322:1695–1699. doi: 10.1126/science.1165395. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.