

Abstract
Understanding the patterns and causes of phenotypic divergence is a central goal in evolutionary biology. Much work has shown that mRNA abundance is highly variable between closely related species. However, the extent and mechanisms of post-transcriptional gene regulatory evolution are largely unknown. Here we used ribosome profiling to compare transcript abundance and translation efficiency in two closely related yeast species (S. cerevisiae and S. paradoxus). By comparing translation regulatory divergence to interspecies differences in mRNA sequence features, we show that differences in transcript leaders and codon bias substantially contribute to divergent translation. Globally, we find that translation regulatory divergence often buffers species differences in mRNA abundance, such that ribosome occupancy is more conserved than transcript abundance. We used allele-specific ribosome profiling in interspecies hybrids to compare the relative contributions of cis- and trans-regulatory divergence to species differences in mRNA abundance and translation efficiency. The mode of gene regulatory divergence differs for these processes, as trans-regulatory changes play a greater role in divergent mRNA abundance than in divergent translation efficiency. Strikingly, most genes with aberrant transcript abundance in F1 hybrids (either over- or underexpressed compared to both parent species) did not exhibit aberrant ribosome occupancy. Our results show that interspecies differences in translation contribute substantially to the evolution of gene expression. Compensatory differences in transcript abundance and translation efficiency may increase the robustness of gene regulation.
The relationship between genotype and phenotype is a long-standing biological puzzle. Genetic mutations can affect phenotypes both by changing the coding sequence of proteins and by changing the regulation of gene expression. As much as 40 years ago, it became clear that differences in protein-coding sequences are too infrequent to explain differences in phenotype (King and Wilson 1975). Consequently, evolution of gene regulation has been suggested as a major source of phenotypic diversity (Carroll 2005; Rockman and Kruglyak 2006).
With the advent of DNA microarrays in the 1990s, many researchers began examining gene expression differences between species. mRNA abundance levels were found to vary greatly both among and between biological species, including yeast (Brem et al. 2002; Bullard et al. 2010; Tirosh et al. 2011), fruit flies (Ranz et al. 2003; Rifkin et al. 2003), mice (Schadt et al. 2003), and humans (Pickrell et al. 2010). In general, these studies report that polymorphic mRNA abundance is present in at least 25% of genes in a species, and mRNA levels are even more divergent between closely related species. However, a few recent studies suggest that protein abundance is more conserved than mRNA abundance across all domains of life, including E. coli, S. cerevisiae, and humans (Schrimpf et al. 2009; Laurent et al. 2010). Because these studies depended upon orthologous genes identifiable across vast evolutionary distance, their results could be biased toward highly abundant housekeeping genes. Nonetheless, these works suggest that post-transcriptional processes, including mRNA translation and protein turnover, could act to reduce the effects of variation in mRNA abundance and offset divergent gene expression.
Gene expression requires numerous steps, including transcription, mRNA translation, and protein turnover. Each of these processes is regulated through complex networks of cis-regulatory sequence elements and trans-acting factors. Cis-regulatory elements (e.g., transcription factor binding sites and miRNA target sites) are localized to the alleles they regulate and thus affect gene expression in an allele-specific manner. In contrast, trans-acting factors (e.g., transcription factors, RNA binding proteins, and miRNAs) can affect the expression of both alleles in a diploid cell. Consequently, the roles of these network components can be dissected by comparing allele-specific gene expression in F1 hybrids. In F1 hybrids, two parental alleles are subjected to identical trans-regulatory environments. Thus, differences in gene expression from the two alleles reflect divergent activity of their cis-regulatory sequences. Trans-regulatory divergence can then be inferred by comparing interspecific expression differences with allele-specific expression differences in F1 hybrids (Wittkopp et al. 2004; Tirosh et al. 2009). This hybrid approach has revealed that most intraspecies variation in mRNA abundance results from trans-regulatory differences, but cis-regulatory differences accumulate over evolutionary time (Lemos et al. 2008; Wittkopp et al. 2008a; Emerson et al. 2010).
Divergence of gene regulatory networks has also been implicated in inheritance of gene expression levels. Like any phenotype, mRNA abundance can be inherited either additively or nonadditively. One extreme form of nonadditive inheritance includes cases in which genes are misexpressed in F1 hybrids compared to parent species. This misexpression includes both over- and underdominant inheritance, in which total mRNA abundance in F1 hybrids is either much higher or lower than both parent species, respectively. Misexpression of mRNA abundance can affect many genes (Gibson et al. 2004; Ranz et al. 2004), and is associated with antagonistic cis- and trans-regulatory changes (Landry 2005; McManus et al. 2010; Schaefke et al. 2013).
Although the evolution of mRNA abundance has been studied extensively, divergent regulation of mRNA translation has received much less attention. We used ribosome profiling to compare mRNA abundance and translation efficiency in two species of yeast, S. cerevisiae and S. paradoxus. Interspecies differences in mRNA abundance and translation efficiency affected similar proportions of the transcriptome. Importantly, differences in translation efficiency tend to buffer differences in mRNA abundance. We further compared the relative contributions of cis- and trans-regulatory differences in mRNA abundance and translation efficiency using F1 hybrids. Differences in both mRNA levels and translation efficiency are largely attributed to trans-regulatory divergence. Finally, we investigated inheritance patterns of mRNA abundance and ribosome occupancy in F1 hybrids. Strikingly, ribosome occupancy was much less likely to exhibit misexpression, especially for genes involved in cell cycle regulation. These results reveal the importance of translation regulation in evolution, provide insights into the genetic and molecular mechanisms of translation regulatory divergence, and indicate that translation regulatory networks often act as a buffer to gene regulatory divergence in yeast.
Results
Measuring translation efficiency with ribosome profiling
To investigate translation regulatory divergence, we used ribosome profiling in diploid S. cerevisiae, S. paradoxus, and their F1 hybrid (Fig. 1A). This technique provides estimates of the relative number of ribosomes translating mRNA from each gene (ribosome occupancy). Compared with RNA-seq or microarray measurements of mRNA abundance, ribosome occupancy is a better proxy of protein abundance in yeast (Ingolia et al. 2009). This is consistent with the common model that yeast protein synthesis is largely regulated at the translation initiation step (Lackner et al. 2007; Sonenberg and Hinnebusch 2009; Shah et al. 2013). Ribosome-bound mRNA was extracted from cycloheximide-treated cells and digested with RNase I to produce short (28–30 nucleotides) ribosome protected mRNA fragments (RPFs). RPFs were then purified and used to generate strand-specific libraries for Illumina high-throughput sequencing (Ingolia et al. 2011). Strand-specific polyA-positive mRNA-seq libraries were made in parallel. Together, this approach measures both ribosome occupancy (RPF) and mRNA abundance (mRNA) that can be compared to estimate relative translation efficiency, defined here as the ratio of ribosome footprints to mRNA fragments (RPF/mRNA) (Ingolia et al. 2009).
Figure 1.

Overview of allele-specific ribosome profiling to measure divergence in ribosome occupancy, mRNA abundance, and translational efficiency. (A) Ribosome profiling was performed on log-phase cultures of S. cerevisiae, S. paradoxus, and their F1 hybrid. Ribosome protected fragments (RPFs) were purified and cloned into Illumina high-throughput sequencing libraries (left). Poly-adenylated mRNA sequencing libraries were prepared in parallel (right). (B) Sequence reads from each sample were aligned to both species genomes. Allele-specific reads were identified by comparing genomic alignments and mapped to corresponding regions of orthologous ORFs. (C) Comparison of separate and allele-specific coverage in ribosome profiling experiments. IGV browser tracks showing normalized coverage of RPF and mRNA sequence reads from S. cerevisiae (blue) and S. paradoxus (magenta) over the COX6 gene (YHR051W). Measurements of interspecies differences in read coverage are equivalent for separate species (upper) and allele-specific alignments (“mock hybrid,” lower) (see also Supplemental Fig. S1).
We developed an analysis pipeline to compare translation efficiency between species and between alleles in F1 hybrids that leverages the high-quality genome sequences and gene annotations from each species (Scannell et al. 2011). To improve the annotations of introns and translation start sites, we sequenced the transcriptome of S. paradoxus (Scannell et al. 2011). Our reannotation of the S. paradoxus genome resulted in a working set of 5474 orthologous protein-coding genes. Sequence reads from each species were aligned to their respective genomes. Reads from F1 hybrid samples were aligned to each species genome separately, and alignments were compared to identify reads that map preferentially to one allele (Fig. 1B; McManus et al. 2010). This analysis resulted in an average of 7.4 million RPF and 6.9 million mRNA reads covering ORFs in each replicate (Supplemental Table S1).
In some cases, allele-specific alignment can be biased toward higher quality genomes (Degner et al. 2009). However, these effects appear to be largely absent when using two high-quality genomes, as was the design of this study (McManus et al. 2010; Graze et al. 2012; Stevenson et al. 2013). To investigate the effectiveness of our allele-specific mapping pipeline, we combined sequence data from individual species to create a “mock hybrid” and compared divergence estimates from allele-specific and single-species alignment procedures. Estimates of gene expression differences from allele-specific and single-species alignment procedures were highly consistent for ribosome occupancy (RPFs; Pearson's R2 > 0.97), mRNA abundance (R2 > 0.98) and translation efficiency (RPF/mRNA; R2 > 0.96) (Fig. 1C; Supplemental Fig. S1), suggesting that the allele-specific analysis pipeline generates accurate estimates of regulatory divergence. We therefore used our allele-specific pipeline for both interspecies and F1 hybrid allelic comparisons.
Interspecies differences in translation efficiency and mRNA abundance
We compared interspecies differences in mRNA abundance and ribosome occupancy using a custom analysis pipeline (see Methods). Count-based tests were applied to identify statistically significant differences using 5% false discovery rate (FDR) and 1.5-fold minimum thresholds in both replicates. Our biological replicates were well correlated for both mRNA and RPF libraries (ρ > 0.97) (Supplemental Fig. S2). By requiring at least 20 total allele-specific mapped reads (S. cerevisiae reads + S. paradoxus reads > 20) for both RPF and mRNA in each replicate, we identified 1308 genes (26.9%) with differences in ribosome occupancy (RPFs), 1739 genes (35.8%) with differences in mRNA abundance, and 1345 genes (27.7%) with differences in translational efficiency (RPF/ mRNA) (Fig. 2A–C). Similar results were obtained using minimum cutoffs of 10, 50, and 100 reads (Supplemental Table S2). These results suggest that translation regulation may play as significant a role as mRNA abundance in the evolution of gene expression.
Figure 2.
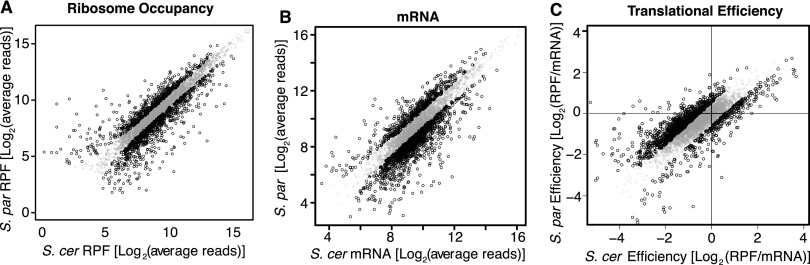
Comparing regulatory divergence of ribosome occupancy, mRNA abundance, and translation efficiency. (A–C) Scatter plots compare the normalized average number of sequence reads for S. cerevisiae (x-axis) and S. paradoxus (y-axis). Genes with statistically significant differences in read counts (FDR < 5%, minimum 1.5-fold difference) are plotted as open circles with black edges. Translation efficiency is defined here as the number of ribosome protected fragment reads (RPF) divided by the number of mRNA-seq reads covering an ORF.
Many features of mRNA transcripts influence translation efficiency. Long transcript leaders (5′ UTRs), frequent upstream AUGs (uAUGs), and uORF activity have all been associated with reduced translation efficiency (Ingolia et al. 2009; Brar et al. 2011; Rojas-Duran and Gilbert 2012; Waern and Snyder 2013). We used our RNA-seq data to annotate transcript leaders for 3014 genes in each species. Species differences in transcript leader length were negatively correlated with divergent translation efficiency such that the species with the longer transcript leader tended to have lower efficiency of translation (ρ = −0.27; P = 1.9 × 10−13). Differences in the number of uAUGs were also negatively correlated with divergent translation efficiency, suggesting that orthologs with more uAUGs tend to be less efficiently translated (ρ = −0.27; P = 6.3 × 10−8). Partial correlation analysis of transcript leader length, controlling for uAUG frequency and vice versa, show that they contribute equally to divergent translation efficiency (ρ = −0.126; P = 0.00054; and ρ = −0.129; P = 0.00038). In comparison, the frequency of upstream stop codons was not correlated with divergent translation efficiency (P > 0.3). Thus, divergent translation regulation is due in part to divergent transcript leader length and uAUG frequency.
The importance of uAUGs suggests that upstream open reading frames (uORFs) may also contribute to divergent translation efficiency. Once thought to be rare, recent studies have identified thousands of uORFs in S. cerevisiae, many of which appear to initiate translation at noncanonical “NTG” start codons (Ingolia et al. 2009; Brar et al. 2011). We searched for putative uORFs starting with NTG and ending with an in-frame stop codon in transcript leader regions from each species. RPF reads were mapped to uORF regions and the ratios of uORF to main ORF reads from each species were compared. Species differences in ribosome occupancy over uORFs were also negatively correlated with divergent translation efficiency (ρ = −0.25; P = 3.6 × 10−8), such that orthologs with higher uORF occupancy showed generally lower efficiency of main ORF translation (Supplemental Fig. S3). These results indicate that translation regulatory divergence can be attributed in part to interspecies differences in uORF presence and utilization.
Due to redundancy in the genetic code, codon usage bias can also contribute to regulation of translation efficiency (Plotkin and Kudla 2011). A previous comparison of nine yeast species revealed divergent codon usage bias for hundreds of orthologous ORFs (Man and Pilpel 2007). We calculated codon bias using the tRNA Adaptation Index (tAI) (dos Reis et al. 2004) to compare species differences in codon bias to translation regulatory differences. Codon bias was positively correlated to mRNA abundance, ribosome occupancy, and translation efficiency (ρ > 0.52; P < 2 × 10−16), such that highly expressed genes are biased toward using more abundant tRNA in both species. In contrast, interspecies differences in codon bias were weakly correlated with divergent translation efficiency (ρ = 0.11; P = 4.706 × 10−15).
Translation regulatory divergence buffers species differences in mRNA abundance
Genes with interspecies differences in mRNA abundance were more than twice as likely to have divergent translation efficiency compared to genes with conserved mRNA abundance (Fisher's exact test [FET], P < 10−16). The co-occurrence of divergence in translation efficiency could work in the same (amplifying) or opposite (buffering) direction (Fig. 3A). Strikingly, we found that buffering effects were ∼5.5 times more common (N = 560) than amplifying effects (N = 101; FET, P < 10−16). Indeed there was a significant negative correlation between mRNA abundance and translation efficiency genome-wide (ρ = −0.413, P < 10−16) (Fig. 3B,C). This tendency toward buffering would reduce divergent expression overall at the protein level.
Figure 3.

Translation regulatory divergence buffers interspecies differences in mRNA abundance. (A) Cartoon depicting buffering (left) and amplification (right). mRNA are shown as blue lines, and ribosomes are shown as black circles. Buffered genes have divergent mRNA abundance, with less divergent ribosome occupancy such that protein production is more conserved. In contrast, amplified genes have divergent mRNA abundance and even more divergent ribosome occupancy. (B) Scatterplot comparing divergent translation efficiency (y-axis, RPF/mRNA) with divergent mRNA abundance (x-axis). Buffered and amplified genes are plotted in red and blue, respectively. The negative correlation between mRNA abundance and translation efficiency suggests a genome-wide trend toward buffering. (C) Example of RPF and RNA-seq coverage over FPK1, a buffered gene. IGV browser tracks showing normalized coverage for S. cerevisiae (blue) and S. paradoxus (red).
We investigated several features of buffered and amplified genes. Highly expressed genes often have housekeeping functions and therefore might be buffered more frequently than genes with low mRNA abundance. However the mRNA abundance of buffered genes was either average (in S. cerevisiae) or slightly lower than average (S. paradoxus) (Supplemental Fig. S4). We also compared the biological functions of buffered and amplified genes. Gene ontology (GO) analysis (Supplemental Table S3) revealed that buffered genes were enriched in processes involved in cellular communication and catabolism (P < 0.01), whereas amplified genes, were enriched in functions related to more specific metabolic processes, including amino acid metabolism, sulfur assimilation (P < 0.01), iron import, and redox reactions (P < 0.004). The enrichment of these categories is consistent with species/strain differences in metabolic strategies, as S. paradoxus metabolism uses aerobic respiration more intensely, whereas S. cerevisiae has more auxotrophies to amino acid biosynthesis.
Cis- and trans-contributions to translation regulatory divergence
To gain further insight into the molecular and genetic changes underlying translation regulatory divergence, we performed allele-specific ribosome profiling on an F1 hybrid strain made by crossing our strains of S. cerevisiae and S. paradoxus. In F1 hybrids, differences in allele-specific translation efficiency reflect divergence of their cis-regulatory sequences. The effects of trans-regulatory divergence can then be inferred by comparing interspecies expression differences with F1 hybrid allele-specific expression (Wittkopp et al. 2004; Tirosh et al. 2009; Bullard et al. 2010; McManus et al. 2010). This approach is feasible with our strains, as S. cerevisiae and S. paradoxus are sufficiently divergent to allow proper allele assignment for 71% of sequence reads (Supplemental Table S1). Figure 1C shows an example comparison of sequence coverage with separate species and allele-specific read assignments.
At each level of gene regulatory control—transcript abundance, ribosome occupancy, and translational efficiency—more genes were affected by differences in the activity of trans-regulatory factors than by cis-regulatory differences (Fig. 4A). This is consistent with previous studies of regulatory divergence and shows that the mode of regulatory evolution is similar for mRNA abundance and translation efficiency. However, processes revealed that cis-regulatory divergence contributes more to differences in translation efficiency (36.7% cis) than to differences in mRNA abundance (30.2% cis; Wilcoxon rank-sum test [WRT], P = 3.9 × 10−14) (Fig. 4B).
Figure 4.

Contributions of cis- and trans-regulatory divergence in mRNA abundance, ribosome occupancy, and translation efficiency. (A) Bar plot shows the number of genes affected by significant regulatory divergence in cis-acting sequences (C) and trans-acting factors (T). Trans-acting factors contribute most to mRNA abundance. (B) Box plot showing the fraction of regulatory divergence attributable to differences in cis-regulatory elements (%cis). Ribosome occupancy and translation efficiency both have higher %cis than mRNA abundance. (C) Venn diagram showing the overlap of genes with cis-regulatory divergence in mRNA abundance, ribosome occupancy, and translation efficiency. (D) Box plot depicting %cis for genes with buffering (Buf) and amplifying (Amp) regulatory divergence of mRNA abundance (left) and translation efficiency (right). Asterisks indicate results of Wilcoxon rank-sum test comparisons: (*) P < 0.001; (**) P < 0.0005.
We investigated the overlap of genes with cis-regulatory divergence in mRNA abundance, translation efficiency, and ribosome occupancy (Fig. 4C). Cis-regulatory divergence in translation efficiency was found three times more often in genes with cis-regulatory divergence in mRNA levels than compared to those without (FET, P < 2.2 × 10−16). Coupled cis-regulatory evolution of mRNA abundance and translation efficiency was biased more than threefold toward buffering allele-specific ribosome occupancy. As a result, 345 genes (55%) with cis-regulatory differences in mRNA levels do not exhibit cis-regulatory divergence in ribosome occupancy. We refer to these genes as “cis-mRNA buffered,” whereas genes with concurrent cis-regulatory differences in mRNA abundance and ribosome occupancy are “cis-mRNA penetrant.” The median magnitude of cis-regulatory divergence for cis-mRNA buffered genes is significantly lower than that of cis-mRNA penetrant genes (1.84-fold versus 2.31-fold; WRT, P < 2.2 × 10−16). This suggests that small magnitude cis-regulatory differences in mRNA levels are less likely to penetrate into the phenotype of protein production.
We next examined how cis- and trans-regulatory divergence in mRNA abundance and translation efficiency contribute to buffered and amplified interspecies expression differences. Compared to buffered genes, amplified genes were more likely to exhibit cis-regulatory divergence of both mRNA abundance (FET, 1.6-fold, P = 0.018) and translation efficiency (FET, 2.3-fold, P = 5.3 × 10−5). Amplified genes also had a higher percentage of cis-regulatory divergence than buffered genes, both for mRNA abundance (31.1% versus 25.5%; WRT, P = 0.07) and translation efficiency (60.7% versus 31.7%; WRT, P = 0.0003342) (Fig. 4). Thus, cis-regulatory divergence contributes more to amplified genes than to buffered genes.
Buffering of misexpression in F1 hybrids
Previous studies have revealed aberrant mRNA abundance in interspecies hybrids—either lower (underexpressed) or higher (overexpressed) than both parental species. This misexpression may be related to interspecies network incompatibilities and, potentially, to speciation (Ranz et al. 2004; Landry 2005; Moehring et al. 2007). However, a recent proteomics study estimated that only 3% of 398 analyzed genes exhibit misexpression of protein abundance in F1 hybrids of S. cerevisiae and S. bayanus (Khan et al. 2012). We analyzed the inheritance of divergent expression in both mRNA abundance and ribosome occupancy to test the hypothesis that mRNA misexpression could be translationally buffered. Compared to mRNA abundance, ribosome occupancy showed strikingly fewer genes with nonadditive inheritance modes (Fig. 5), with 6.3-fold fewer underexpressed genes (FET, P < 10−16) and 1.9-fold fewer overexpressed genes (FET, P = 0.0003194). This result indicates that divergent translation regulation also buffers misexpression of mRNA abundance, such that total protein synthesis in F1 hybrids is more consistent with that of parent species.
Figure 5.

Inheritance of gene expression in F1 hybrids of S. cerevisiae and S. paradoxus. (A) Hypothetical patterns of gene expression in S. paradoxus (red), S. cerevisiae (blue), and F1 hybrid yeast (purple), depicting six modes of gene expression inheritance. (B) Scatterplots showing the difference between mRNA expression levels (left) in the F1 hybrid and S. cerevisiae (x-axis) and S. paradoxus (y-axis). The difference between ribosome occupancy levels (RPF) in the F1 hybrid and parental species is shown on the right. Bar plots show the number of genes in each inheritance category from ribosome occupancy data. Genes with overdominant (overexpressed) and underdominant (underexpressed) inheritance occur much less frequently when considering ribosome occupancy, as compared to misexpression in mRNA abundance.
Buffering of misexpression could reduce hybrid incompatibilities. To investigate the effects of misexpression and buffering, we compared enrichment of GO functions in genes misexpressed at the mRNA level and at the level of ribosome occupancy. Genes underexpressed at the mRNA level were functionally enriched in cell cycle regulation and cellular metabolic processes, whereas genes with overexpressed mRNA largely functioned in purine biosynthesis pathways (P < 0.01) (Supplemental Table S4). Consistent with this, genes with aberrantly high ribosome occupancy in the F1 hybrid strain were enriched for functions in inosine monophosphate biosynthesis, the precursor to purine synthesis. In contrast, no GO functional categories were enriched in the 34 genes underexpressed at the translation level. In conclusion, compensatory changes in translation efficiency appear to correct aberrant mRNA abundance in F1 hybrids of S. paradoxus and S. cerevisiae, possibly restoring the expression of cell cycle control genes.
Discussion
The evolution of translation efficiency
Although undoubtedly important, evolutionary changes in mRNA abundance may not always reflect interspecies differences in gene expression. Indeed, prior work has highlighted a disconnect between variation in mRNA and protein abundance within yeast (Foss et al. 2007), mice (Ghazalpour et al. 2011), and humans (Wu et al. 2013). Together, these studies suggest that variation in mRNA translation and protein turnover contribute to polymorphic gene expression. Our results indicate that translation regulation plays a substantial role in the evolution of gene expression. Roughly one-quarter of all expressed genes exhibited divergent translation efficiency, showing that divergence in translation regulation and mRNA abundance affect similar numbers of genes.
Changes in codon usage have been previously implicated in yeast gene regulatory evolution (Man and Pilpel 2007). More recent studies have highlighted important translation regulatory roles of transcript leaders in S. cerevisiae (Ingolia et al. 2009; Brar et al. 2011; Rojas-Duran and Gilbert 2012; Waern and Snyder 2013). Our results indicate that interspecies differences in transcript leaders appear to play a larger role than codon bias. One explanation for this observation is that changes in codon bias are more likely to be pleiotropic, as changes in codon usage can alter translation elongation and disrupt cotranslational protein folding (Angov 2011; Plotkin and Kudla 2011). In contrast, changes in transcript leader length and sequence composition are likely to affect only translation initiation. Much like transcription promoter regions, transcript leaders provide important opportunities for evolutionary changes in gene expression without affecting protein-coding sequences.
Differences in modes of regulatory evolution for mRNA abundance and translation efficiency
We used allele-specific ribosome profiling to compare the contributions of cis- and trans-regulatory changes toward divergence of mRNA abundance and translation efficiency. We found that cis-regulatory differences appear to contribute more to divergence in translation efficiency than to interspecies differences in mRNA abundance. This is inconsistent with other work suggesting that fewer cis-acting loci contribute to polymorphic regulation of mRNA abundance than of protein abundance among S. cerevisiae strains (Foss et al. 2011). This may be due in part to a smaller sample size (354 genes) and less power of mass spectrometry-based studies for QTL mapping. In fact, recent work from the same group has shown that local pQTL are more common than previously appreciated (FW Albert, S Treusch, AH Shockley, JS Bloom, L Kruglyak, in prep.). Differences in divergence time in the Foss study (between strains) and ours (between species) could also contribute to their differing conclusions, as it has been shown that cis-regulatory divergence accumulates over evolutionary time (Lemos et al. 2008; Wittkopp et al. 2008b; Emerson et al. 2010).
Buffering of divergent mRNA abundance and translation efficiency
Our results indicate that regulatory interactions between mRNA abundance and translation efficiency more often reduce than amplify interspecies differences in gene expression. Buffering of gene expression may help explain recent work showing that protein abundance is more conserved than mRNA abundance across organisms (Schrimpf et al. 2009; Laurent et al. 2010). Changes in translation efficiency may compensate for changes in mRNA abundance by balancing the numbers of translating ribosomes.
There are two potential, nonexclusive mechanisms for buffering divergent gene regulation. First, regulatory networks controlling mRNA abundance and translation efficiency could have diverged through compensatory genetic mutations (“genetic compensation”), such that mutations in one pathway counteract the effects of mutations in a different pathway. Secondly, the gene regulatory networks may be inherently robust, such that changes in mRNA abundance are automatically buffered by feedback loops and regulatory circuitry (“network robustness”). The contributions of cis- and trans-regulatory changes in buffered genes may help differentiate between these mechanisms of buffering. For example, the genetic compensation model predicts that buffered genes would more frequently exhibit counteracting cis-regulatory changes in mRNA abundance and translation efficiency. In contrast, counteracting trans-acting differences are more consistent with network robustness. Of the 560 buffered genes in our data set, 75 (13%) exhibit counteracting cis-regulatory changes, whereas 254 (45%) have counteracting trans-acting changes. Consequently, we propose that network robustness is likely to be more common than genetic compensation.
Compared to buffered gene expression levels, amplified gene expression levels have fewer concordant trans-regulatory changes in both mRNA levels and translation efficiency. Of the 101 amplified genes, 23 (23%) have amplifying cis-regulatory changes in both mRNA abundance and translation efficiency, whereas 24 (24%) have amplifying trans-regulatory changes. Amplifying genes also had a higher percentage of cis-regulatory divergence, showing that cis-regulatory changes contribute more to divergent regulation of amplified genes, compared to buffered genes. Importantly, cis-regulatory divergence is thought to be more frequently mediated by positive selection than trans-regulatory divergence (Emerson et al. 2010; Graze et al. 2012; Schaefke et al. 2013). Thus amplifying divergence of mRNA abundance and translation efficiency may reflect positive selection for gene expression, as has been suggested for combinations of cis-regulatory mutations affecting mRNA abundance alone in Drosophila (Graze et al. 2012).
Many biological systems, including gene regulatory networks, are known to exhibit robustness to genetic and environmental perturbation (Stark et al. 2005; Levy and Siegal 2008; Masel and Siegal 2009; MacNeil and Walhout 2011; Denby et al. 2012). By increasing the robustness of protein production, translational buffering could reduce the phenotypic impacts of variation in mRNA abundance. This in turn would allow increased variation in mRNA abundance which, if unmasked by disruption of buffering, could give organisms more variation upon which selection might act. This would create a situation identical to canalization in development (Waddington 1942; Rutherford and Lindquist 1998), in which capacitor genes allow the buildup of latent variation, which is only observed under extraordinary conditions. Our results suggest that translational buffering serves a similar purpose by allowing variation in mRNA abundance to accumulate through genetic drift without substantially changing protein levels.
Earlier studies on gene regulatory evolution have documented widespread misexpression of mRNA abundance in interspecies hybrids (Gibson et al. 2004; Ranz et al. 2004), often associated with counteracting cis- and trans-regulatory divergence of mRNA abundance (Landry 2005; McManus et al. 2010; Schaefke et al. 2013). Surprisingly, our analysis revealed translational buffering of misexpressed genes. Hundreds of genes had aberrant mRNA levels in hybrid cells, yet few showed aberrant ribosome occupancy patterns. As with interspecies differences in mRNA abundance, over- and underexpressed genes could be buffered through genetic compensation or network robustness. It has been shown that protein–protein interactions are functional in F1 hybrids of S. cerevisiae and S. kudriavzevii (Leducq et al. 2012), suggesting that many biochemical features of regulatory networks are robust in interspecies hybrids. Thus, network robustness is an attractive answer. By reducing aberrant gene expression patterns, translational buffering of misexpression could contribute to the considerable success of yeast hybrids (Morales and Dujon 2012).
Thus far, we have investigated translation regulatory evolution only in the context of optimal growth conditions. Because many stress responses involve changes in mRNA translation, repeating these experiments under different conditions could shed light on the role of translation in species-specific responses to stress. It will also be important to identify the genes required to maintain translational buffering, as they could act as capacitors for variation in mRNA abundance. Although there is much yet to learn, our results underscore the importance of translation in the evolution of gene expression, help define the molecular and genetic causes of divergence in translation efficiency, and show that interspecies differences in translation efficiency often act as a buffer to gene regulatory evolution.
Methods
Yeast strains
Saccharomyces strains: S. cerevisiae (GSY83; S288C), S. paradoxus (GSY82; CBS432), and their F1 hybrid (GSY88) were kind gifts from Gavin Sherlock (Kao et al. 2010). The original GSY83 is a haploid strain, whereas GSY82 and GSY88 are diploid strains. For this study, a diploid version of GSY83 was produced by galactose-induced transient expression of the HO gene from plasmid YCP50-Gal∷HO, a gift from John Woolford. Diploid GSY83 were identified via PCR screening.
Ribosome profiling library preparation
Yeast polysome extracts were prepared for ribosome profiling as previously described (Ingolia 2010) with slight modifications (see Supplemental Methods). RNA for matched RNA-seq was extracted from 50 μL of cell lysate using TRIzol (Life Technologies) and vortexing with glass beads. The sample was then enriched for mRNA using magnetic oligo-DT DynaBeads (Life Technologies) according to the manufacturer's instructions. The resulting RNA was then fragmented by incubating in alkaline fragmentation buffer (50 mM NaCO3, pH 9.2; 1 mM EDTA) for 40 min at 95°C, precipitated with one volume of isopropanol and resuspended in 20 μL of nuclease-free water.
High-throughput sequencing library preparation was performed as previously described (Ingolia et al. 2011) with slight modifications (see Supplemental Methods). RPF and mRNA library cDNA templates were amplified by 12 cycles of PCR using Phusion-polymerase (New England Biolabs), with primers incorporating barcoded Illumina TruSeq library sequences. The resulting PCR products were then purified using PCR purification columns (Qiagen) according to the manufacturer's instructions. The quality and size of the PCR products were assessed using an Agilent Tapestation. Libraries were sequenced on an Illumina HiSeq 2000 at the University of Southern California Epigenome Center.
Sequence data alignment
Raw sequences were trimmed of 3′ adapters using custom Perl scripts. For RPF data, reads with lengths of 27–33 nucleotides were retained for further analysis as this size is most likely to represent footprinted fragments. For mRNA, reads with lengths of 27–40 nucleotides were retained. Adapter trimmed reads were first aligned to genomes using Bowtie (Langmead et al. 2009) (version 0.12.8) with parameters –best –chunkmbs 500. Reads that failed to map to genomic regions were recovered and aligned to splice-junction databases requiring a minimum overlap of 6 nt. Splice-junction read alignments were converted to SAM formatted genomic coordinates using a custom Perl script.
Allele-specific read assignment was performed as previously described (McManus et al. 2010) with slight modifications. Briefly, adapter trimmed reads from separate species were concatenated into single “mixed parent” files. Mixed parent and hybrid sequence files were aligned to both species genome and junction databases as described above, and alignments were compared with a custom Perl script to identify reads that map preferentially to one genome (Fig. 1B). The library preparation procedure used here frequently adds untemplated nucleotides during reverse transcription (Brar et al. 2012). Considering this, we ignored mismatches at the first two bases of the 5′ end of sequence reads when comparing alignments between two species. Reads were assigned to the allele to which they map with the fewest mismatches. Estimates of interspecies differences in gene expression made through our allele-specific pipeline were highly correlated with those made by single-species alignment used in traditional RNA-seq and ribosome profiling (ρ > 0.96 for RPF, mRNA, and translation efficiency) (Supplemental Fig. S1).
Gene expression analyses
Allele-specific reads were mapped to ORFs and uORFs and normalized by down-sampling all genes from the species or allele with more mapped reads (McManus et al. 2010). We also accounted for species differences in ORF length by down-sampling reads from the longer ortholog in proportion to the ratio of ortholog lengths (i.e., if one species' ORF was twice as long, we would divide the number of assigned reads by two). Tests of statistical significance were performed essentially as previously described (McManus et al. 2010) using R (version 2.15.2). For analyses of expression differences, raw P-values were corrected using the Benjamini-Hochberg method (Benjamini and Hochberg 1995), and significant differences were identified using 5% FDR and 1.5-fold magnitude thresholds. Significant differences in mRNA abundance and ribosome occupancy (RPF) were identified using binomial exact tests (BET), requiring both replicates to meet significance thresholds. Differences in translation efficiency (RPF/mRNA) were identified using Cochran Mantel-Haenszel tests (CMH), which directly incorporate experimental replicates. CMH tests were used to evaluate trans-regulatory divergence by comparing the ratio of expression differences in parents and hybrids for mRNA abundance and ribosome occupancy. The method of Altman and Bland (2003) was used to test for trans-regulatory divergence in translation efficiency. Supplemental Table S5 contains all read-count data, tAI calculations, expression ratios, and results of significance test for divergent expression. For full expression analysis details, see the Supplemental Methods.
Genes were classified as “buffered” or “amplified” using the following criteria. Both buffered and amplified genes have significant differences in mRNA abundance and translational efficiency. In buffered genes, the interspecies difference in ribosome occupancy (RPF) is at least 1.5-fold lower than the interspecies difference in mRNA abundance. In contrast, amplified genes have at least 1.5-fold higher differences in ribosome occupancy compared to differences in mRNA abundance.
Categories of gene expression inheritance were determined using a custom R program as previously described (McManus et al. 2010). Total expression in F1 hybrid cells was normalized to that of parental samples and log-transformed. Genes with total expression more than 1.25-fold different from that of either parent species were considered to have nonconserved inheritance and were classified as having additive, dominant, underdominant, or overdominant inheritance modes based on the magnitude of differences between hybrid and parental expression.
Additional analyses
Gene ontology enrichment analysis was performed using the generic gene ontology term finder (http://go.princeton.edu), and GO terms were further evaluated using revigo (http://revigo.irb.hr) (Supek et al. 2011). The tRNA adaptation index (tAI) was calculated using the codonR package (http://people.cryst.bbk.ac.uk/∼fdosr01/tAI/index.html) (dos Reis et al. 2004). Genome Browser tracks (bedGraph) were produced for visual analysis using genomeCoverageBed from BEDTools. Images for all figures were produced using the Integrative Genome Browser (Robinson et al. 2011).
Data access
High-throughput sequencing data have been submitted to the NCBI Sequence Read Archive (SRA; http://www.ncbi.nlm.nih.gov/sra) under accession number SRP028552. A read-count table for mRNA and RPFs, genome-browser tracks (bedGraph), and gene annotations (bed) have been submitted to the NCBI Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE52119.
Supplementary Material
Acknowledgments
We are grateful to Gavin Sherlock for sharing strains, and John Woolford and Adam Lindstedt for use of equipment. We also thank Zia Khan, Christine Vogel, and members of the McManus laboratory for comments and suggestions on the manuscript. This work was supported by laboratory start-up funding provided by the Department of Biological Sciences at Carnegie Mellon University.
Author contributions: C.J.M. conceived of the experiments and designed the study. C.J.M., G.E.M., and P.S. performed the experiments. C.J.M. and A.S. analyzed the data. C.J.M. wrote the manuscript with input from all authors.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.164996.113.
Freely available online through the Genome Research Open Access option.
References
- Altman DG, Bland JM 2003. Interaction revisited: The difference between two estimates. BMJ 326: 219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angov E 2011. Codon usage: Nature's roadmap to expression and folding of proteins. Biotechnol J 6: 650–659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y 1995. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J R Stat Soc Ser B Methodol 57: 289–300 [Google Scholar]
- Brar GA, Yassour M, Friedman N, Regev A, Ingolia NT, Weissman JS 2011. High-resolution view of the yeast meiotic program revealed by ribosome profiling. Science 335: 552–557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brar GA, Rouskin S, McGeachy AM, Ingolia NT, Weissman JS 2012. The ribosome profiling strategy for monitoring translation in vivo by deep sequencing of ribosome-protected mRNA fragments. Nat Protoc 7: 1534–1550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brem RB, Yvert G, Clinton R, Kruglyak L 2002. Genetic dissection of transcriptional regulation in budding yeast. Science 296: 752–755 [DOI] [PubMed] [Google Scholar]
- Bullard JH, Mostovoy Y, Dudoit S, Brem RB 2010. Polygenic and directional regulatory evolution across pathways in Saccharomyces. Proc Natl Acad Sci 107: 5058–5063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll SB 2005. Evolution at two levels: On genes and form. PLoS Biol 3: e245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degner JF, Marioni JC, Pai AA, Pickrell JK, Nkadori E, Gilad Y, Pritchard JK 2009. Effect of read-mapping biases on detecting allele-specific expression from RNA-sequencing data. Bioinformatics 25: 3207–3212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denby CM, Im JH, Yu RC, Pesce CG, Brem RB 2012. Negative feedback confers mutational robustness in yeast transcription factor regulation. Proc Natl Acad Sci 109: 3874–3878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- dos Reis M, Savva R, Wernisch L 2004. Solving the riddle of codon usage preferences: A test for translational selection. Nucleic Acids Res 32: 5036–5044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emerson JJ, Hsieh LC, Sung HM, Wang TY, Huang CJ, Lu HHS, Lu MYJ, Wu SH, Li WH 2010. Natural selection on cis and trans regulation in yeasts. Genome Res 20: 826–836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foss EJ, Radulovic D, Shaffer SA, Ruderfer DM, Bedalov A, Goodlett DR, Kruglyak L 2007. Genetic basis of proteome variation in yeast. Nat Genet 39: 1369–1375 [DOI] [PubMed] [Google Scholar]
- Foss EJ, Radulovic D, Shaffer SA, Goodlett DR, Kruglyak L, Bedalov A 2011. Genetic variation shapes protein networks mainly through non-transcriptional mechanisms. PLoS Biol 9: e1001144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghazalpour A, Bennett B, Petyuk VA, Orozco L, Hagopian R, Mungrue IN, Farber CR, Sinsheimer J, Kang HM, Furlotte N, et al. 2011. Comparative analysis of proteome and transcriptome variation in mouse. PLoS Genet 7: e1001393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson G, Riley-Berger R, Harshman L, Kopp A, Vacha S, Nuzhdin S, Wayne M 2004. Extensive sex-specific nonadditivity of gene expression in Drosophila melanogaster. Genetics 167: 1791–1799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graze RM, Novelo LL, Amin V, Fear JM, Casella G, Nuzhdin SV, Mcintyre LM 2012. Allelic imbalance in Drosophila hybrid heads: Exons, isoforms, and evolution. Mol Biol Evol 29: 1521–1532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingolia NT 2010. Genome-wide translational profiling by ribosome footprinting. Methods Enzymol 470: 119–142 [DOI] [PubMed] [Google Scholar]
- Ingolia NT, Ghaemmaghami S, Newman JRS, Weissman JS 2009. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science 324: 218–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingolia NT, Lareau LF, Weissman JS 2011. Ribosome profiling of mouse embryonic stem cells reveals the complexity and dynamics of mammalian proteomes. Cell 147: 789–802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao KC, Schwartz K, Sherlock G 2010. A genome-wide analysis reveals no nuclear Dobzhansky-Muller pairs of determinants of speciation between S. cerevisiae and S. paradoxus, but suggests more complex incompatibilities. PLoS Genet 6: e1001038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan Z, Bloom JS, Amini S, Singh M, Perlman DH, Caudy AA, Kruglyak L 2012. Quantitative measurement of allele-specific protein expression in a diploid yeast hybrid by LC-MS. Mol Syst Biol 8: 1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- King MC, Wilson AC 1975. Evolution at two levels in humans and chimpanzees. Science 188: 107–116 [DOI] [PubMed] [Google Scholar]
- Lackner DH, Beilharz TH, Marguerat S, Mata J, Watt S, Schubert F, Preiss T, Bähler J 2007. A network of multiple regulatory layers shapes gene expression in fission yeast. Mol Cell 26: 145–155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landry CR 2005. Compensatory cis-trans evolution and the dysregulation of gene expression in interspecific hybrids of Drosophila. Genetics 171: 1813–1822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg S 2009. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10: R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurent JM, Vogel C, Kwon T, Craig SA, Boutz DR, Huse HK, Nozue K, Walia H, Whiteley M, Ronald PC, et al. 2010. Protein abundances are more conserved than mRNA abundances across diverse taxa. Proteomics 10: 4209–4212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leducq J-B, Charron G, Diss G, Gagnon-Arsenault I, Dubé AK, Landry CR 2012. Evidence for the robustness of protein complexes to inter-species hybridization. PLoS Genet 8: e1003161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemos B, Araripe LO, Fontanillas P, Hartl DL 2008. Dominance and the evolutionary accumulation of cis- and trans-effects on gene expression. Proc Natl Acad Sci 105: 14471–14476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy SF, Siegal ML 2008. Network hubs buffer environmental variation in Saccharomyces cerevisiae. PLoS Biol 6: e264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacNeil LT, Walhout AJM 2011. Gene regulatory networks and the role of robustness and stochasticity in the control of gene expression. Genome Res 21: 645–657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Man O, Pilpel Y 2007. Differential translation efficiency of orthologous genes is involved in phenotypic divergence of yeast species. Nat Genet 39: 415–421 [DOI] [PubMed] [Google Scholar]
- Masel J, Siegal ML 2009. Robustness: Mechanisms and consequences. Trends Genet 25: 395–403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McManus CJ, Coolon JD, Duff MO, Eipper-Mains J, Graveley BR, Wittkopp PJ 2010. Regulatory divergence in Drosophila revealed by mRNA-seq. Genome Res 20: 816–825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moehring AJ, Teeter KC, Noor MAF 2007. Genome-wide patterns of expression in Drosophila pure species and hybrid males. II. Examination of multiple-species hybridizations, platforms, and life cycle stages. Mol Biol Evol 24: 137–145 [DOI] [PubMed] [Google Scholar]
- Morales L, Dujon B 2012. Evolutionary role of interspecies hybridization and genetic exchanges in yeasts. Microbiol Mol Biol Rev 76: 721–739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickrell JK, Marioni JC, Pai AA, Degner JF, Engelhardt BE, Nkadori E, Veyrieras J-B, Stephens M, Gilad Y, Pritchard JK 2010. Understanding mechanisms underlying human gene expression variation with RNA sequencing. Nature 464: 768–772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plotkin JB, Kudla G 2011. Synonymous but not the same: The causes and consequences of codon bias. Nat Rev Genet 12: 32–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranz JM, Castillo-Davis CI, Meiklejohn CD, Hartl DL 2003. Sex-dependent gene expression and evolution of the Drosophila transcriptome. Science 300: 1742–1745 [DOI] [PubMed] [Google Scholar]
- Ranz JM, Namgyal K, Gibson G, Hartl DL 2004. Anomalies in the expression profile of interspecific hybrids of Drosophila melanogaster and Drosophila simulans. Genome Res 14: 373–379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rifkin SA, Kim J, White KP 2003. Evolution of gene expression in the Drosophila melanogaster subgroup. Nat Genet 33: 138–144 [DOI] [PubMed] [Google Scholar]
- Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP 2011. Integrative genomics viewer. Nat Biotechnol 29: 24–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rockman MV, Kruglyak L 2006. Genetics of global gene expression. Nat Rev Genet 7: 862–872 [DOI] [PubMed] [Google Scholar]
- Rojas-Duran MF, Gilbert WV 2012. Alternative transcription start site selection leads to large differences in translation activity in yeast. RNA 18: 2299–2305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutherford SL, Lindquist S 1998. Hsp90 as a capacitor for morphological evolution. Nature 396: 336–342 [DOI] [PubMed] [Google Scholar]
- Scannell DR, Zill OA, Rokas A, Payen C, Dunham MJ, Eisen MB, Rine J, Johnston M, Hittinger CT 2011. The awesome power of yeast evolutionary genetics: New genome sequences and strain resources for the Saccharomyces sensu stricto genus. G3 (Bethesda) 1: 11–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schadt EE, Monks SA, Drake TA, Lusis AJ, Che N, Colinayo V, Ruff TG, Milligan SB, Lamb JR, Cavet G, et al. 2003. Genetics of gene expression surveyed in maize, mouse and man. Nature 422: 297–302 [DOI] [PubMed] [Google Scholar]
- Schaefke B, Emerson JJ, Wang T-Y, Lu MYJ, Hsieh LC, Li W-H 2013. Inheritance of gene expression level and selective constraints on trans- and cis-regulatory changes in yeast. Mol Biol Evol 30: 2121–2133 [DOI] [PubMed] [Google Scholar]
- Schrimpf SP, Weiss M, Reiter L, Ahrens CH, Jovanovic M, Malmström J, Brunner E, Mohanty S, Lercher MJ, Hunziker PE, et al. 2009. Comparative functional analysis of the Caenorhabditis elegans and Drosophila melanogaster proteomes. PLoS Biol 7: e48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah P, Ding Y, Niemczyk M, Kudla G, Plotkin JB 2013. Rate-limiting steps in yeast protein translation. Cell 153: 1589–1601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonenberg N, Hinnebusch AG 2009. Regulation of translation initiation in eukaryotes: Mechanisms and biological targets. Cell 136: 731–745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark A, Brennecke J, Bushati N, Russell RB, Cohen SM 2005. Animal microRNAs confer robustness to gene expression and have a significant impact on 3′UTR evolution. Cell 123: 1133–1146 [DOI] [PubMed] [Google Scholar]
- Stevenson KR, Coolon JD, Wittkopp PJ 2013. Sources of bias in measures of allele-specific expression derived from RNA-seq data aligned to a single reference genome. BMC Genomics 14: 536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Supek F, Bošnjak M, Škunca N, Šmuc T 2011. REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS ONE 6: e21800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirosh I, Reikhav S, Levy AA, Barkai N 2009. A yeast hybrid provides insight into the evolution of gene expression regulation. Science 324: 659–662 [DOI] [PubMed] [Google Scholar]
- Tirosh I, Wong KH, Barkai N, Struhl K 2011. Extensive divergence of yeast stress responses through transitions between induced and constitutive activation. Proc Natl Acad Sci 108: 16693–16698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waddington C 1942. Canalization of development and the inheritance of acquired characters. Nature 150: 563–565 [DOI] [PubMed] [Google Scholar]
- Waern K, Snyder M 2013. Extensive transcript diversity and novel upstream open reading frame regulation in yeast. G3 (Bethesda) 3: 343–352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittkopp PJ, Haerum BK, Clark AG 2004. Evolutionary changes in cis and trans gene regulation. Nature 430: 85–88 [DOI] [PubMed] [Google Scholar]
- Wittkopp PJ, Haerum BK, Clark AG 2008a. Independent effects of cis- and trans-regulatory variation on gene expression in Drosophila melanogaster. Genetics 178: 1831–1835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittkopp PJ, Haerum BK, Clark AG 2008b. Regulatory changes underlying expression differences within and between Drosophila species. Nat Genet 40: 346–350 [DOI] [PubMed] [Google Scholar]
- Wu L, Candille SI, Choi Y, Xie D, Jiang L, Li-Pook-Than J, Tang H, Snyder M 2013. Variation and genetic control of protein abundance in humans. Nature 499: 79–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.