

Abstract
Comparative analysis of methanogen compositions in the feces of horse and pony was carried out by constructing the α-subunit of methyl coenzyme-M reductase (mcrA) gene and 16S ribosomal RNA gene (16S rRNA) clone libraries. The mcrA clone library analysis indicated that Methanomicrobiales was predominant in both horse and pony. Furthermore, most of the clones of the 16S rRNA gene library showed that Methanomicrobiales was also predominant in horse and pony, but the LIBSHUFF analysis showed that the horse and pony libraries were significantly different (P < 0.05). Most of operational taxonomic units (OTUs) showed low similarity to the identified methanogens in both the mcrA and the 16S rRNA clone libraries. The results suggest that horse and pony harbor unidentified and novel methanogens in their hindgut. The methanogen population was higher in horse than in pony; however, the anaerobic fungal population was similar in horse and pony. The methanogen diversity was different between two breeds of Equus caballus.
1. Introduction
Members of the Equidae family, such as horse and pony, possess an anatomically specialized hindgut that accommodates a microbial ecosystem consisting of bacteria, protozoa, and anaerobic rumen fungi that are capable of degrading and fermenting structural polysaccharides of the plant cell walls [1]. Furthermore, methanogens that reduce CO2 with H2 to form methane are also common inhabitants of the hindgut of Equidae [2].
To date, many studies have been published on the microbial diversity in the hindgut of horse and pony. These studies include the diversity of bacteria [2–9], protozoa [3, 9], and anaerobic rumen fungi [3]. However, only limited information is available on the methanogen population in the hindgut of horses [10]. Many studies have been conducted to analyze the composition and population size of methanogens from the rumen of ruminants and other types of herbivores [11–24]. These studies have shown that methanogens that are affiliated to Methanobacteriales, Methanomicrobiales, and Methanoplasmatales [25] are major constituents of the rumen of ruminants. Information about the methanogen density and diversity in the hindgut of horse and pony are important for fully understanding the microbial ecosystem in their hindgut.
The diversity in the microbial communities of the rumen is influenced by many factors, such as location, environment, feed composition, feeding frequency, supplements, animal species, and genetic background of individual animals within a species [26]. The composition of the microbiota in the hindgut of equine could depend on the hindgut capacity as a reflection of body size or even horse breed [9].
This study aimed to comparatively analyze the archaeal 16S rRNA gene and the mcrA gene in thoroughbred horses and Japanese local ponies kept under the same management.
2. Materials and Methods
2.1. Animals, Diets, and Sample Collection
Six mature thoroughbred horses and 3 mature Japanese local ponies were used for this study. Sex and age of each animal are as follows: horse 1 (gelding, 17), horse 2 (female, 12), horse 3 (gelding, 17), horse 4 (gelding, 12), horse 5 (gelding, 14), horse 6 (female, 14), pony 1 (gelding, 4), pony 2 (gelding, 10), and pony 3 (gelding, 7). Animals were fed regularly twice per day with Timothy hay, barley, and rice bran at a ratio of 2.0 : 1.5 : 1.0 kg for horses and 1.5 : 1.0 : 0.5 kg for ponies. Salt and calcium were used as supplements and water was available ad libitum. The animals are used for equestrian art and perform exercises every morning under the same management. To determine the microbial population and methanogen diversity, freshly voided fecal samples were collected and kept at 4°C during delivery to the laboratory and then stored at −25°C.
2.2. pH and Real-Time PCR Analysis of Total Bacteria, Methanogen, and Anaerobic Rumen Fungi Populations
Five grams of feces was mixed with 20 mL of distilled water and homogenized, and its pH was measured using a glass electrode [27]. The remaining feces was used for DNA analysis and kept at −80°C. The DNA was extracted using the QIAamp DNA stool kit (QIAGEN, Inc., Valencia, CA, USA) according to the manufacturer's instructions. The genomic DNA concentration was adjusted to 10 ng μL−1 and stored at −25°C until analysis.
Real-time PCR analysis was performed as literature instruction, and the PCR primers that were used in this study are shown in Table 1. Real-time PCR amplification and detection were performed using an ABI prism 7000 sequence detection system (Applied Biosystems, Foster City, CA, USA). PCR was carried out in a reaction mixture with a final volume of 25 μL containing 10 ng of template DNA, 12.5 μL of SYBR Green master mix (Applied Biosystem, Foster City, CA, USA), and 4.5 μL each of forward and reverse primers (0.5 μL of forward and reverse primers for methanogen). Amplification consisted of 1 cycle of polymerase activation at 95°C for 10 min followed by 40 cycles of denaturing at 95°C for 15 s and 60°C for 30 s. The product size was confirmed by agarose gel electrophoresis after each determination. Standard DNA was prepared from the 16S rDNA fragment of Escherichia coli cloned into pCR 2.1 vector. Sample-derived standards for methanogen were prepared from the treatment pool set of community DNA [17]. The gene fragment encoding mcrA from cow rumen was cloned into pCR 2.1 vector. Standard DNA for fungi was prepared from a pure culture of ITS1 fragment of Neocallimastix sp. strain SR 6 cloned into pCR 2.1 vector as described in Lwin et al. [28]. PCR products from these cloned DNA were used as standards. Amplified standard DNAs were purified using a QIAquick PCR purification kit (QIAGEN, Inc., Valencia, CA, USA) and quantified with spectrophotometry (at 260 nm). The standards were serially diluted by 10-fold and were prepared just prior to real-time PCR. All measurements were performed in triplicate.
Table 1.
PCR primers used in real-time PCR assays and in clone library analysis.
| Application | Target | Name (direction) | Sequence (5′→3′) | Annealing temperature (°C) | Number of cycles | Product size (bp) | Reference |
|---|---|---|---|---|---|---|---|
| Quantitative real-time PCR | Anaerobic fungi | q-pcr (f) | GAGGAAGTAAAAGTCGTAACAAGGTTTC | 60 | 40 | 120 | [29] |
| q-pcr (r) | CAAATTCACAAAGGGTAGGATGATT | ||||||
| Methanogen | q-mcra (f) | TTCGGTGGATCDCARAGRGC | 60 | 40 | 141 | [23] | |
| q-mcra (r) | GBARGTCGWAWCCGTAGAATCC | ||||||
| Total bacteria | 1114 (f) | CGGCAACGAGCGCAACCC | 60 | 40 | 130 | [29] | |
| 1275 (r) | CCATTGTAGCAGGTG | ||||||
|
| |||||||
| 16S rRNA library | Methanogen | Met 86 (f) | GCTCAGTAACACGTGG | 53 | 30 | 1254 | [30] |
| Met 1340 (r) | CGGTGTGTGCAAGGAG | ||||||
|
| |||||||
| mcrA library | Methanogen | mcrA(f) | GGTGGTGTMGGATTCACACARTAYGCWACAGC | 58 | 30 | 480 | [31] |
| mcrA (r) | TTCATTGCRTAGTTWGGRTAGTT | ||||||
f: forward; r: reverse.
2.3. Amplification of Archaeal 16S rRNA and mcrA Genes
DNA samples from each individual animal were pooled into 1 portion for each animal breed. The mcrA-specific primers described in Luton et al. [31] and the archaeal 16S rRNA gene primers [30] (Table 1) were used for DNA amplification. The PCR reaction mixture (25 μL) contained 1.0 μL of template, 0.5 μM of each primer, 200 μM of a dNTP mixture, 1× Ex Taq buffer, 0.5 mg/mL BSA, and 0.625 units of Ex Taq polymerase. PCR was carried out on a thermal cycler (Dice TP 600; TaKaRa, Otsu, Japan) with the following conditions: initial denaturation at 95°C for 3 min for the archaeal 16S rRNA gene and 94°C for 2 min for the mcrA gene, denaturation at 94°C for 30 s, elongation at 72°C for 90 s for the 16S rRNA gene and 30 s for the mcrA gene, and a final extension at 72°C for 10 min. The annealing temperatures and the number of cycles are shown in Table 1. Following electrophoresis on 1.0% agarose gels in Tris-acetate EDTA buffer, PCR products were visualized by ethidium bromide staining.
2.4. Cloning and Sequencing
The PCR products were then cloned into the pCR 2.1 vector using the TA Cloning Kit (Invitrogen, Carlsbad, CA, USA). Positive transformants were randomly picked, and the cloned DNA fragments were sequenced, as described by Matsui et al. [32]. A homology search of archaeal 16S rRNA gene sequences and deduced amino acid sequences of mcrA was performed with the Blast N and Blast X programs [33]. Chimeric artifacts of PCR were checked with the CHECK_CHIMERA online program of the Ribosomal Database Project (RDP-II) and omitted from analysis [34].
2.5. Phylogenetic Analysis
The archaeal 16S rRNA genes and deduced amino acid sequences of mcrA gene were aligned with Clustal X ver. 2.0 [35], and phylogenetic trees were constructed using the neighbor-joining method [36]. The stability of the branches was analyzed with 1000 bootstrap replications. Operational taxonomic units (OTUs), richness observations (Chao 1), and Shannon-Wiener index (H′) were calculated using the DOTUR program [37]. A 98% sequence similarity criterion was employed for OTU of 16S rRNA gene sequence. A criterion for OTU of mcrA was calculated from correlation between mcrA sequence distance and 16S rRNA gene sequences distance obtained from following methanogens of 23 species from 7 orders including Methanoplasmatales proposed by Paul et al. [25]; accession numbers of mcrA gene and 16S rDNA are shown in the parenthesis after species name; order Methanobacteriales: Methanobrevibacter gottschalkii (EU919431/U55239), Methanobrevibacter millerae (EU919430/NR_042785), Methanobrevibacter ruminantium (AF414046/NR_042784), Methanobrevibacter smihii (NC_009515/AY196669), and Methanobrevibacter woosei (EU919432/NR_044788), order Methanomicrobiales: Methanocorpusculum bavaricum (AF414049/NR_042787), Methanocorpusculum labreanum (AAP20896/NR_074173), Methanocorpusculum parvum (AY260445/AY260435), Methanoculleus bourgensis (NC_018227/AY196674), Methanofollis liminatans (AF414041/Y16429), Methanogenium thermophilum (AB300783/M59129), Methanomicrobium mobile (AF414044/M59142), Methanospirillum hungatei (YP_503573/M60880), order Methanosarcinales: Methanosarcina barkeri (Y00158/NR_074253), Methanosaeta concilii (YP_004383383/NR_102903), order Methanococcales: Methanocaldococcus jannaschii (L77117/NR_074233), Methanococcus vannielii (P07961/NR_074175), and Methanospaera stadtmanae (YP_447374/JQ346752), order Methanocellales: Methanocella arvoryzae (AM114193/NR_074232), Methanocella conradii (YP_005380187/JN048683), order Methanopyrales: Methanopyrus kandleri (NP_613940/NR_074539), order Methanoplasmatales: Methanogenic archaeon CRM1 (GQ339872/GQ339875) and Methanogenic archaeon DCM1 (GQ339873/GQ339876). The correlation of the mcrA distance data to the 16S rRNA distance data gave an equation Y = 2.1944X (R 2 = 0.6196) when the line was forced through the origin. When 0.02 (criterion for 16S rRNA OTU) was plugged into X, Y = 0.0439. Therefore, the criterion of OTU of amino acid sequence of mcrA was determined as 95% similarity. Distances of protein and DNA were calculated with protodist and dnadist of Phylip package (ver. 3.68), respectively. The coverage was calculated from the following formula: coverage (%) = [1 − (n/N)] × 100, where N is the total number of clones and n is an OTU that consists of only 1 clone. The evenness (E) was calculated from the H′ using the following formula: E = H′/H max [38], where H max = 1n(S). LIBSHUFF analysis was used to calculate the statistical significance of the differences between the 2 libraries using the mothur program [39].
2.6. Statistical Analysis
The statistical analysis of pH and microbial population in the feces was performed using the Student's t-test. The significance was set at P < 0.05.
2.7. Nucleotide Sequence Accession Numbers
All nucleic acid sequences obtained in this study were deposited in the DNA Data Bank of Japan (DDBJ), European Molecular Biology Laboratory (EMBL), and GenBank databases, under accession numbers AB739303–AB739402 for 16S rRNA gene sequences and AB739403–AB739502 for mcrA gene sequences.
3. Results
3.1. Fecal pH and Microbial Population Densities
pH and microbial population density are shown in Table 2. The average pH of the fecal samples from horse was higher than that from pony (P > 0.05). The total bacterial population density was similar in horse and pony. Densities of anaerobic rumen fungi and methanogen populations were higher in horse than in pony (P > 0.05).
Table 2.
pH and population densities (copy numbers per gram wet weight of feces) of anaerobic rumen fungi, methanogen, and bacteria in feces of the horse and pony.
| Item | Horse | Pony |
|---|---|---|
| pH | 7.19 ± 0.38 | 6.79 ± 0.04 |
| Total bacteria (×1010 copy/gram) | 2.72 ± 0.81 | 2.31 ± 0.99 |
| Methanogens (×106 copy/gram) | 11.30 ± 17.88 | 4.38 ± 1.48 |
| Anaerobic rumen fungi (×105 copy/gram) |
2.31 ± 2.23 | 1.52 ± 0.80 |
3.2. Phylogenetic Analysis of the mcrA Gene and the Archaeal 16S rRNA Gene
No chimeric sequence was found in the present study. A total of 50 clones were analyzed from the mcrA gene clone libraries of both horse and pony. The deduced amino acid sequences of the mcrA gene clones from horse and pony libraries were classified into 9 OTUs (Figure 1 and Table 3). The phylogenetic analysis showed that the OTUs were classified into 3 clades—Methanobacteriales, Methanomicrobiales, and Methanoplasmatales in both horse and pony (Figure 1). Most of the clones (4 OTUs and 46 clones, 92% in horse; 3 OTUs and 47 clones, 94% in pony) affiliated with Methanomicrobiales. Only 1 OTU was affiliated to genus Methanobrevibacter in both horse (OTU6) and pony (OTU5). OTU6 from the horse library showed 100% similarity to Methanobrevibacter gottschalkii. OTU5 showed 95% similarity to uncultured methanogens detected in the foregut of the tammar wallaby [40] (data not shown). The remaining 3 OTUs (OTU7, 8, and 9) in horse (6% of clones) and 1 OTU (OTU7) in pony (2% of clones) were classified as Methanoplasmatales (Table 3 and Figure 1). OTU9 showed a high similarity (96%) to the mcrA sequence of unidentified gut methanogenic archaeon DCM1 (published only in the database). Most of OTUs showed low similarity (less than 95%) to the identified methanogens (Table 3). OTU1, 2, 4, and 7 were commonly detected in both horse and pony library.
Figure 1.
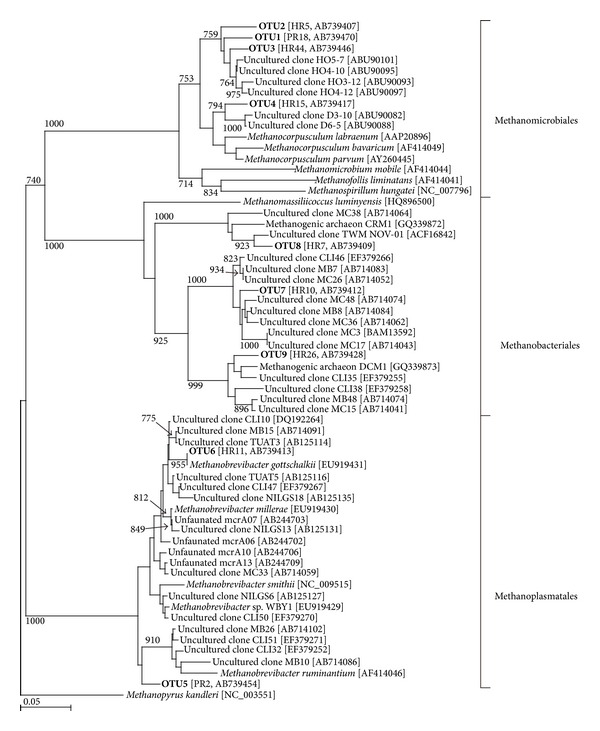
A phylogenetic tree showing the relationship between mcrA deduced amino acid sequences in the horse and pony. The tree was constructed using neighbor-joining analysis. The scale bar represents a 5% sequence divergence of amino acid sequence. Reference sequences were retrieved from the GenBank database, and their accession numbers are in brackets. OTU names from this study are labeled in bold. Representative clone name and its accession number are shown in brackets after OTU name. The Methanopyrus kandleri sequence was used as an outgroup to root the tree.
Table 3.
The number of clones and similarity of the deduced amino acid sequences of the mcrA gene of each operational taxonomic unit (OTU) to cultured methanogens in horse and pony.
| OTUs | Nearest known methanogen* | Number of clones | ||
|---|---|---|---|---|
| Horse | Pony | Total | ||
| Methanomicrobiales | ||||
| OTU1 | Methanocorpusculum labreanum [NC_008942] (93) | 29 | 33 | 62 |
| OTU2 | Methanocorpusculum labreanum [NC_008942] (90) | 10 | 13 | 23 |
| OTU3 | Methanocorpusculum labreanum [NC_008942] (93) | 6 | 0 | 6 |
| OTU4 | Methanocorpusculum labreanum [NC_008942] (94) | 1 | 1 | 2 |
|
| ||||
| Methanobacteriales | ||||
| OTU5 | Methanobrevibacter smithii [DQ251046] (94) | 0 | 2 | 2 |
| OTU6 | Methanobrevibacter gottschalkii [ACK56066] (100) | 1 | 0 | 1 |
|
| ||||
| Methanoplasmatales | ||||
| OTU7 | Candidatus Methanomethylophilus alvus [KC412011] (80) | 1 | 1 | 2 |
| OTU8 | Candidatus Methanomethylophilus alvus [KC412011] (92) | 1 | 0 | 1 |
| OTU9 | Candidatus Methanomethylophilus alvus [KC412011] (83) | 1 | 0 | 1 |
*Number in brackets and in parenthesis is accession number and similarity value (%), respectively.
Fifty clones of the archaeal 16S rRNA gene from the horse and pony libraries were analyzed. The cloned sequences from horse and pony libraries were classified into 10 OTUs (Figure 2 and Table 4). Phylogenetic analysis showed that the OTUs were classified into 4 clades (Figure 2). Similar to mcrA clone libraries, the majority of clones (32 clones, 64% in horse; 37 clones, 74% in pony) were affiliated with Methanomicrobiales (Table 4 and Figure 2). OTU2 did belong to Methanosarcinalesthat was not observed in mcrA clone library. OTU4, 5, and 6 showed a high similarity (97%–99%) to Methanobrevibacter ruminantium. These OTUs consisted of 18% and 14% of the total number of clones from the horse and the pony library, respectively. OTU8, 9 and 10 were classified into Methanoplasmatales clade (Figure 2). The clones from the horse library in these OTUs consisted of 10% of the total clone number, and 1 OTU from the pony library consisted of 6% of the total clone number (Table 3). OTU9 and OTU10 showed a high similarity (97% and 96%) to unidentified gut methanogenic archaeon DCM1 (GQ339876) and CRM1 (GQ339875) (published only in the database), respectively. OTU1, 2, 4, 6, and 8 were commonly detected in both horse and pony library.
Figure 2.
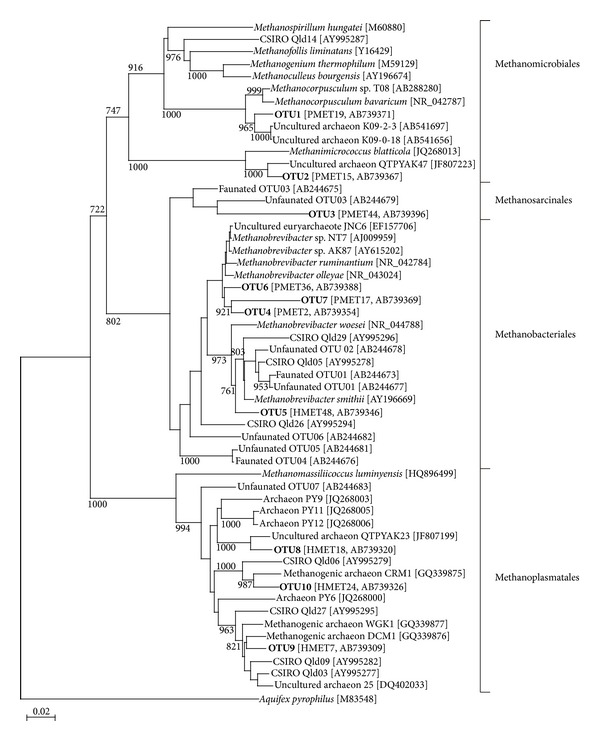
A phylogenetic tree showing the relationship between archaeal 16S rRNA sequences in the horse and pony. The tree was constructed using neighbor-joining analysis. The scale bar represents a 2% sequence divergence of DNA sequence. Reference sequences were retrieved from the GenBank database, and their accession numbers are in brackets. OTU names from this study are labeled in bold. Representative clone name and its accession number are shown in brackets after OTU name. The Aquifex pyrophilus sequence was used as an outgroup to root the tree.
Table 4.
The number of clones and similarity of the archaeal 16S rRNA gene sequences of each operational taxonomic unit (OTU) to cultured methanogens in horse and pony.
| OTUs | Nearest known methanogen* | Number of clones | ||
|---|---|---|---|---|
| Horse | Pony | Total | ||
| Methanomicrobiales | ||||
| OTU1 | Methanocorpusculum labreanum [NR_074173] (96) | 32 | 37 | 69 |
|
| ||||
| Methanosarcinales | ||||
| OTU2 | Methanomicrococcus blatticola [AJ238002] (94) | 4 | 1 | 5 |
|
| ||||
| Methanobacteriales | ||||
| OTU3 | Methanocorpusculum sinense [FR749947] (90) | 0 | 1 | 1 |
| OTU4 | Methanobrevibacter ruminantium [CP001719] (97) | 1 | 5 | 6 |
| OTU5 | Methanobrevibacter smithii [NR_074235] (97) | 6 | 0 | 6 |
| OTU6 | Methanobrevibacter ruminantium [CP001719] (98) | 2 | 2 | 4 |
| OTU7 | Methanobrevibacter ruminantium [CP001719] (94) | 0 | 1 | 1 |
|
| ||||
| Methanoplasmatales | ||||
| OTU8 | Candidatus Methanomethylophilus alvus [KC412010] (91) | 1 | 3 | 4 |
| OTU9 | Candidatus Methanomethylophilus alvus [KC412010] (93) | 2 | 0 | 2 |
| OTU10 | Candidatus Methanomethylophilus alvus [KC412010] (97) | 2 | 0 | 2 |
*Number in brackets and in parenthesis is accession number and similarity value (%), respectively.
In the analysis of the mcrA gene, the Shannon-Wiener index (H′), evenness, and Chao-1 species richness were higher in the horse library than in the pony library; however, LIBSHUFF analysis revealed that there was no significant difference in the diversity of mcrA genes in horse and pony (Table 5). Similar trends in H′ and evenness were observed in the analysis of the 16S rRNA gene. However, the LIBSHUFF analysis of the 16S rRNA gene library showed that there was a significant difference between the 2 libraries (P < 0.05). Lower coverages and higher Chao-1 species richness observed for the 16S rRNA gene clone libraries were higher than those observed for the mcrA gene clone libraries.
Table 5.
General information and diversity indices of the mcrA gene and archaeal 16S rRNA gene clone libraries recovered from microbial populations in the fecal contents of horse and pony.
| Item | mcrA gene | 16S rRNA gene | ||
|---|---|---|---|---|
| Horse | Pony | Horse | Pony | |
| Number of clones | 50 | 50 | 50 | 50 |
| Number of OTUs | 8 | 5 | 8 | 7 |
| Coverage (%) | 37.5 | 86 | 75 | 57 |
| Shannon-Wiener index (H′) | 1.28 | 0.91 | 1.29 | 0.99 |
| Evenness | 0.33 | 0.23 | 0.33 | 0.25 |
| Chao-1 species richness | 18 | 7 | 12 | 13 |
| LIBSHUFF analysis | Ns | P < 0.05 | ||
Ns: not significant.
4. Discussion
A greater understanding of the microbial diversity of the hindgut is essential for improving the digestive process. The diversity of methanogens in the gastrointestinal tract of equine is also important for understanding the mitigation of methane emission. In the hindgut of nonruminants, methanogens use H2 and CO2 to produce methane [41]; however, methane production by monogastric animals is lower than methane production by ruminants. Additionally, among the monogastric animals, large herbivorous animals such as horses, mules, and asses produce a large amount of methane [42]. There is little information about the microbial ecosystem and methanogen diversity in equines. This study was conducted to establish further information about the methanogen diversity in horse and pony.
Morvan et al. [43] showed that the methanogen population was 104 to 106 cells per gram wet weight of cecal contents in horse. This study showed a similar density as that described by Morvan et al. [43]; however, there was no significant difference between horse and pony (Table 2).
Methanogens of the Methanomicrobiales are the most prevalent in the rumen of sheep (approximately 54%) and cattle (21–54%) [15, 16], and they were also dominant in Korean native cattle [12] and Murrah buffalo [44]. This study of a thoroughbred horse and Japanese native pony showed a similar tendency as that observed with the ruminal methanogen composition of sheep, cattle, and buffalo in previous studies.
Criterion for OTU assignment in the mcrA clone library was determined at 95% from correlation between mcrA distance and 16S rRNA gene distance. The criterion at 95% of mcrA corresponds to 98% of 16S rRNA gene. The analysis of mcrA revealed that most of the clones showed similarity less than 95% to the identified methanogen (Table 3). Furthermore, most of the clones from 16S rRNA gene library showed similarity less than 98% to the identified methanogen (Table 4). Therefore, these results suggest that most of the clones were derived from unidentified and novel species of methanogens.
The Methanoplasmatales were the second most dominant clade for both the mcrA and 16S rRNA gene analysis from the thoroughbred horse. The Methanoplasmatales clade represents a novel group of Archaea in the rumen of cattle [15], uncultured methanogens in the rumen of buffalo [44], uncultured Archaea in the rumen of cattle [13], or putative “new taxa” in the rumen of cattle [14]. Methanoplasmatales clade in horse also belongs to those new groups, and the OTUs in Methanoplasmatales clade showed 80%–92% similarity to amino acid sequence of mcrA genes and 91–97% similarity to 16S rRNA genes against identified or candidatus methanogen species in this study (Tables 3 and 4). Although OTU3 of 16S rRNA library showed 90% similarity to Methanocorpusculum sinense (FR749947), which is a member of Methanomicrobiales(Table 4), phylogenetic placement of the OTU was within the Methanobacteriales clade (Figure 2).
About half of the OTUs were commonly found in both breeds in the present study (Tables 3 and 4). Remaining OTUs were specifically found in each breed.
King et al. [13] showed that significant differences in 16S rRNA gene methanogen diversity were observed in different breeds of cows (Jersey versus Holstein) that were kept under the same dietary regimen. Similar to this report, a significant difference was found between horse and pony in the analysis of the 16S rRNA gene clone libraries by LIBSHUFF analysis (P < 0.05) (Table 5). Furthermore, the hindgut of horse was more diverse than that of pony. Phylogenetic analyses using 2 different gene clone libraries resulted in similar tendencies. However, the details of these distributions were different. The mcrA gene can be used for detecting more Methanomicrobialesspecies, while the 16S rRNA gene can be used to detect more Methanobrevibacter species. OTU that belongs to Methanosarcinales was detected only in the 16S rRNA clone library but not in the mcrA clone library. Thus using two different marker genes provides better resolution for the analysis of methanogen diversity.
5. Conclusions
The present study is the first report on the molecular diversities of methanogens in the hindgut of horse and pony based on mcrA and 16S rRNA gene clone library analysis. Although both animals harbored diverse group of methanogens, the composition was different (P < 0.05). The phylum Methanomicrobiales were the most abundant group in their hindgut. The clones affiliated to the phylum Methanoplasmatales which is recently proposed new phylum were also detected in the both libraries. Most of the clones obtained in this study were originated from unidentified methanogens, showing that the ecosystem is still unexplored environment. Isolation and characterization of the unidentified methanogens from hindgut of horse and pony should be done to clarify their function in the hindgut.
Acknowledgments
The authors would like to thank Mr. Kawakita Hirone, Takada High School (Tsu, Mie Prefecture, Japan), for providing horse and pony feces. This study was financially supported by Grants-in-Aid for Scientific Research, Japan Society for the Promotion of Science (22580308). Nucleotide sequencing was performed at the Life Science Research Center (Center for Molecular Biology and Genetics), Mie University (Tsu, Mie Prefecture, Japan).
Conflict of Interests
The authors declare that there is no conflict of interests regarding the publication of this paper.
References
- 1.Stewart CS. Microorganisms in hindgut fermentors. In: Mackie I, White BA, Issacson RE, editors. GastroIntestInal Microbiology. Vol. 2. New York, NY, USA: Chapman and Hall; 1996. pp. 162–171. [Google Scholar]
- 2.Daly K, Stewart CS, Flint HJ, Shirazi-Beechey SP. Bacterial diversity within the equine large intestine as revealed by molecular analysis of cloned 16S rRNA genes. FEMS Microbiology Ecology. 2001;38(2-3):141–151. [Google Scholar]
- 3.Moore BE, Dehority BA. Effects of diet and hindgut defaunation on diet digestibility and microbial concentrations in the cecum and colon of the horse. Journal of Animal Science. 1993;71(12):3350–3358. doi: 10.2527/1993.71123350x. [DOI] [PubMed] [Google Scholar]
- 4.Lin C, Stahl DA. Taxon-specific probes for the cellulolytic genus Fibrobacter reveal abundant and novel equine-associated populations. Applied and Environmental Microbiology. 1995;61(4):1348–1351. doi: 10.1128/aem.61.4.1348-1351.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yamano H, Koike S, Kobayashi Y, Hata H. Phylogenetic analysis of hindgut microbiota in Hokkaido native horses compared to light horses. Animal Science Journal. 2008;79(2):234–242. [Google Scholar]
- 6.Hastie PM, Mitchell K, Murray J-AMD. Semi-quantitative analysis of Ruminococcus flavefaciens, Fibrobacter succinogenes and Streptococcus bovis in the equine large intestine using real-time polymerase chain reaction. British Journal of Nutrition. 2008;100(3):561–568. doi: 10.1017/S0007114508968227. [DOI] [PubMed] [Google Scholar]
- 7.Koike S, Shingu Y, Inaba H, et al. Fecal bacteria in Hokkaido native horses as characterized by microscopic enumeration and competitive polymerase chain reaction assays. Journal of Equine Science. 2000;11(2):45–50. [Google Scholar]
- 8.Julliand V, de Vaux A, Millet L, Fonty G. Identification of Ruminococcus flavefaciens as the predominant cellulolytic bacterial species of the equine cecum. Applied and Environmental Microbiology. 1999;65(8):3738–3741. doi: 10.1128/aem.65.8.3738-3741.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kobayashi Y, Koike S, Miyaji M, Hata H, Tanaka K. Hindgut microbes, fermentation and their seasonal variations in Hokkaido native horses compared to light horses. Ecological Research. 2006;21(2):285–291. [Google Scholar]
- 10.Lin C, Miller TL. Phylogenetic analysis of Methanobrevibacter isolated from feces of humans and other animals. Archives of Microbiology. 1998;169(5):397–403. doi: 10.1007/s002030050589. [DOI] [PubMed] [Google Scholar]
- 11.Wright A-DG, Williams AJ, Winder B, Christophersen CT, Rodgers SL, Smith KD. Molecular diversity of rumen methanogens from sheep in Western Australia. Applied and Environmental Microbiology. 2004;70(3):1263–1270. doi: 10.1128/AEM.70.3.1263-1270.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shin EC, Choi BR, Lim WJ, et al. Phylogenetic analysis of archaea in three fractions of cow rumen based on the 16S rDNA sequence. Anaerobe. 2004;10(6):313–319. doi: 10.1016/j.anaerobe.2004.08.002. [DOI] [PubMed] [Google Scholar]
- 13.King EE, Smith RP, St-Pierre B, Wright A-DG. Differences in the rumen methanogen populations of lactating Jersey and holstein dairy cows under the same diet regimen. Applied and Environmental Microbiology. 2011;77(16):5682–5687. doi: 10.1128/AEM.05130-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tan HY, Sieo CC, Lee CM, Abdullah N, Liang JB, Ho YW. Diversity of bovine rumen methanogens in vitro in the presence of condensed tannins, as determined by sequence analysis of 16S rRNA gene library. Journal of Microbiology. 2011;49(3):492–498. doi: 10.1007/s12275-011-0319-7. [DOI] [PubMed] [Google Scholar]
- 15.Tajima K, Nagamine T, Matsui H, Nakamura M, Aminov RI. Phylogenetic analysis of archaeal 16S rRNA libraries from the rumen suggests the existence of a novel group of archaea not associated with known methanogens. FEMS Microbiology Letters. 2001;200(1):67–72. doi: 10.1111/j.1574-6968.2001.tb10694.x. [DOI] [PubMed] [Google Scholar]
- 16.Yanagita K, Kamagata Y, Kawaharasaki M, Suzuki T, Nakamura Y, Minato H. Phylogenetic analysis of methanogens in sheep rumen ecosystem and detection of Methanomicrobium mobile by fluorescence in situ hybridization. Bioscience, Biotechnology and Biochemistry. 2000;64(8):1737–1742. doi: 10.1271/bbb.64.1737. [DOI] [PubMed] [Google Scholar]
- 17.Lwin KO, Kondo M, Ban-Tokuda T, et al. Ruminal fermentation and microbial ecology of buffaloes and cattle fed the same diet. Animal Science Journal. 2012;83(12):767–776. doi: 10.1111/j.1740-0929.2012.01031.x. [DOI] [PubMed] [Google Scholar]
- 18.Skillman LC, Toovey AF, Williams AJ, Wright A-DG. Development and validation of a real-time PCR method to quantify rumen protozoa and examination of variability between Entodinium populations in sheep offered a hay-based diet. Applied and Environmental Microbiology. 2006;72(1):200–206. doi: 10.1128/AEM.72.1.200-206.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Regensbogenova M, McEwan NR, Javorsky P, et al. A re-appraisal of the diversity of the methanogens associated with the rumen ciliates. FEMS Microbiology Letters. 2004;238(2):307–313. doi: 10.1016/j.femsle.2004.07.049. [DOI] [PubMed] [Google Scholar]
- 20.Tokura M, Chagan I, Ushida K, Kojima Y. Phylogenetic study of methanogens associated with rumen ciliates. Current Microbiology. 1999;39(3):123–128. doi: 10.1007/s002849900432. [DOI] [PubMed] [Google Scholar]
- 21.Whitford MF, Teather RM, Forster RJ. Phylogenetic analysis of methanogens from the bovine rumen. BMC Microbiology. 2001;1, article 5 doi: 10.1186/1471-2180-1-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sharp R, Ziemer CJ, Stern MD, Stahl DA. Taxon-specific associations between protozoal and methanogen populations in the rumen and a model rumen system. FEMS Microbiology Ecology. 1998;26(1):71–78. [Google Scholar]
- 23.Denman SE, Tomkins NW, McSweeney CS. Quantitation and diversity analysis of ruminal methanogenic populations in response to the antimethanogenic compound bromochloromethane. FEMS Microbiology Ecology. 2007;62(3):313–322. doi: 10.1111/j.1574-6941.2007.00394.x. [DOI] [PubMed] [Google Scholar]
- 24.Ozutsumi Y, Tajima K, Takenaka A, Itabashi H. The mcrA gene and 16S rRNA gene in the phylogenetic analysis of methanogens in the rumen of faunated and unfaunated cattle. Animal Science Journal. 2012;83(11):727–734. doi: 10.1111/j.1740-0929.2012.01023.x. [DOI] [PubMed] [Google Scholar]
- 25.Paul K, Nonoh JO, Mikulski L, Brune A. Methanoplasmatales, thermoplasmatales-related archaea in termite guts and other environments, are the seventh oder of methanogens. Applied and Environmental Microbiology. 2012;78(23):8245–8253. doi: 10.1128/AEM.02193-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dehority BA, Orpin CG. Development of, and natural fluctuations in, rumen microbial populations. In: Hobson PN, editor. The Rumen Microbial Ecosystem. London, UK: Elsevier Applied Science; 1988. pp. 151–183. [Google Scholar]
- 27.Sato H, Nakajima J. Fecal ammonia, urea, volatile fatty acid and lactate levels in dairy cows and their pathophysiological significance during diarrhea. Animal Science Journal. 2005;76(6):595–599. [Google Scholar]
- 28.Lwin KO, Hayakawa M, Ban-Tokuda T, Matsui H. Real-time PCR assays for monitoring anaerobic fungal biomass and population size in the rumen. Current Microbiology. 2011;62(4):1147–1151. doi: 10.1007/s00284-010-9843-7. [DOI] [PubMed] [Google Scholar]
- 29.Denman SE, McSweeney CS. Development of a real-time PCR assay for monitoring anaerobic fungal and cellulolytic bacterial populations within the rumen. FEMS Microbiology Ecology. 2006;58(3):572–582. doi: 10.1111/j.1574-6941.2006.00190.x. [DOI] [PubMed] [Google Scholar]
- 30.Wright A-DG, Pimm C. Improved strategy for presumptive identification of methanogens using 16S riboprinting. Journal of Microbiological Methods. 2003;55(2):337–349. doi: 10.1016/s0167-7012(03)00169-6. [DOI] [PubMed] [Google Scholar]
- 31.Luton PE, Wayne JM, Sharp RJ, Riley PW. The mcrA gene as an alternative to 16S rRNA in the phylogenetic analysis of methanogen populations in landfill. Microbiology. 2002;148(11):3521–3530. doi: 10.1099/00221287-148-11-3521. [DOI] [PubMed] [Google Scholar]
- 32.Matsui H, Ban-Tokuda T, Wakita M. Detection of fiber-digesting bacteria in the ceca of ostrich using specific primer sets. Current Microbiology. 2010;60(2):112–116. doi: 10.1007/s00284-009-9513-9. [DOI] [PubMed] [Google Scholar]
- 33.Altschul SF, Madden TL, Schäffer AA, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Research. 1997;25(17):3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cole JR, Chai B, Marsh TL, et al. The Ribosomal Database Project (RDP-II): previewing a new autoaligner that allows regular updates and the new prokaryotic taxonomy. Nucleic Acids Research. 2003;31(1):442–443. doi: 10.1093/nar/gkg039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Larkin MA, Blackshields G, Brown NP, et al. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23(21):2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 36.Saitou N, Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Molecular Biology and Evolution. 1987;4(4):406–425. doi: 10.1093/oxfordjournals.molbev.a040454. [DOI] [PubMed] [Google Scholar]
- 37.Schloss PD, Handelsman J. Introducing DOTUR, a computer program for defining operational taxonomic units and estimating species richness. Applied and Environmental Microbiology. 2005;71(3):1501–1506. doi: 10.1128/AEM.71.3.1501-1506.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Magurran AE. Ecological Diversity and Its Measurement. Princeton, NJ, USA: Princeton University Press; 1988. [Google Scholar]
- 39.Schloss PD, Westcott SL, Ryabin T, et al. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Applied and Environmental Microbiology. 2009;75(23):7537–7541. doi: 10.1128/AEM.01541-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Evans PN, Hinds LA, Sly LI, McSweeney CS, Morrison M, Wright A-DG. Community composition and density of Methanogens in the foregut of the Tammar wallaby (Macropus eugenii) Applied and Environmental Microbiology. 2009;75(8):2598–2602. doi: 10.1128/AEM.02436-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Miller TL, Wolin MJ. Methanogens in human and animal intestinal tracts. Systematic and Applied Microbiology. 1986;7(2-3):223–229. [Google Scholar]
- 42.Jensen BB. Methanogenesis in monogastric animals. Environmental Monitoring and Assessment. 1996;42(1-2):99–112. doi: 10.1007/BF00394044. [DOI] [PubMed] [Google Scholar]
- 43.Morvan B, Bonnemoy F, Fonty G, Gouet P. Quantitative determination of H2-utilizing acetogenic and sulfate-reducing bacteria and methanogenic archaea from digestive tract of different mammals. Current Microbiology. 1996;32(3):129–133. doi: 10.1007/s002849900023. [DOI] [PubMed] [Google Scholar]
- 44.Chaudhary PP, Sirohi SK, Saxena J. Diversity analysis of methanogens in rumen of Bubalus bubalis by 16S riboprinting and sequence analysis. Gene. 2012;493(1):13–17. doi: 10.1016/j.gene.2011.11.041. [DOI] [PubMed] [Google Scholar]