

Abstract
The transcription factor serum response factor (SRF) controls the expression of genes involved in cellular proliferation and differentiation. Interestingly, SRF also promotes cell survival by regulating the expression of antiapoptotic genes. In in vitro differentiating murine embryonic stem (ES) cells, SRF deficiency leads to increased apoptosis. Loss of SRF correlates with impaired expression of the antiapoptotic Bcl-2 and Bcl-xl genes. SRF binds the Bcl-2 promoter in vivo and activates Bcl-2 transcription. Reconstituting Bcl-2 in Srf(−/−) ES cells rescues these cells from apoptosis, demonstrating that SRF-dependent Bcl-2 expression is critical for ES cell survival. At the multicellular level, SRF deficiency leads to impaired cavitation and reduced Bcl-2 expression in embryoid bodies (EBs) and inappropriate apoptosis in both EBs and pregastrulation mouse embryos. Thus, our data from genetic and cellular studies uncover SRF-regulated Bcl-2 expression as a novel mechanism that is important for cell survival during early murine embryogenesis.
Keywords: apoptosis, Bcl-2, cavitation, ES cells, SRF
Introduction
Serum response factor (SRF) regulates genes encoding immediate-early transcription factors, components of the actin cytoskeleton, and neuron- or muscle-specific proteins (Treisman, 1995; Herdegen and Leah, 1998; Miano, 2003). SRF regulates transcription by binding to CArG box sequences in target promoters (Treisman and Ammerer, 1992; Johansen and Prywes, 1995). SRF recruits ternary complex factors (TCFs) to ets binding sites adjacent to CArG boxes (Shaw et al, 1989; Treisman, 1994). TCFs are regulated by phosphorylation upon MAPK signaling (Gille et al, 1992; Janknecht et al, 1993a; Marais et al, 1993; Whitmarsh et al, 1995). SRF also regulates genes independently of TCFs, for example, cytoskeletal- and muscle-specific genes, as well as the Srf gene itself (Wei et al, 1998; Sotiropoulos et al, 1999). On TCF-independent promoters, SRF can be activated by other coactivators, for example, of the myocardin family (Wang et al, 2001, 2002; Miralles et al, 2003).
Mouse embryos homozygous for an Srf null allele die before gastrulation and do not form detectable mesoderm (Arsenian et al, 1998). The exact molecular mechanism leading to this early embryonic lethality is unknown, since Srf(−/−) embryonic stem (ES) cells are able to differentiate into mesoderm (Weinhold et al, 2000).
In early mouse embryogenesis, formation of the proamniotic cavity restructures the embryo at the transition from the blastocyst to the egg cylinder stage and prepares it for gastrulation. Later, cavitation is involved in kidney and lung development, as well as gland formation (Jacobson et al, 1997; Vaux and Korsmeyer, 1999). Cavity formation requires the coordinate regulation of both apoptosis of inner cells and survival of cells lining the cavity. Embryoid body (EB) formation has been utilized as an in vitro system to study cavitation (Martin and Evans, 1975). Cavitation requires two antagonistic signals: (i) a death signal from outer endoderm cells inducing apoptosis in inner ectodermal cells, and (ii) a survival signal for the columnar epithelial epiblast (CEE) cells bordering the cavity (Coucouvanis and Martin, 1995). Only a few regulators of cavitation have been described, including Bmp2/4 and laminin (Coucouvanis and Martin, 1999; Murray and Edgar, 2000).
Apoptosis is mainly executed by proteases of the caspase family, whereby members of the Bcl-2 family, for example, Bcl-2, Bcl-xl, or Mcl-1, inhibit caspase activation (Adams and Cory, 1998; Gross et al, 1999). Phosphorylation of Bcl-2 proteins in response to PI-3-kinase/Akt signaling favors formation of antiapoptotic Bcl-2 dimers (Datta et al, 1997; Franke and Cantley, 1997). Bcl-2 is also regulated transcriptionally. Transcription factors that can activate Bcl-2 include CREB (Wilson et al, 1996; Pugazhenthi et al, 1999, 2000), NF-κB (Kurland et al, 2001), Wilms-tumour (WT) protein (Mayo et al, 1999), brain-specific Brn-3a (Smith et al, 1998), and Aiolos (Romero et al, 1999).
We show that SRF is required for the survival of differentiating murine ES cells in vitro and mouse epiblast cells in vivo. In SRF-deficient EBs, enhanced apoptosis causes defective cavitation. We identify SRF as a direct transcriptional regulator of the antiapoptotic Bcl-2 gene in differentiating ES cells. This suggests a novel developmental function of SRF, that is, regulating the balance between cell survival and apoptosis during embryogenesis.
Results
SRF deficiency causes increased apoptosis in differentiating ES cells in vitro
The lethal pregastrulation defect of SRF-deficient embryos (Arsenian et al, 1998) suggested that SRF might affect the survival of differentiating ES cells. Therefore, TUNEL staining of cultured ES cells of different Srf genotype was used to monitor apoptosis in vitro (Figure 1A–H). After 4 days of differentiation upon LIF (leukemia inhibitory factor) withdrawal, a substantial fraction of Srf(−/−) ES cells had undergone apoptosis (Figure 1B and C). In contrast, in SRF-containing cultures, apoptotic cells occurred less frequently and were specifically localized at the boundaries of cavity-like regions (asterisks in Figure 1A and D).
Figure 1.
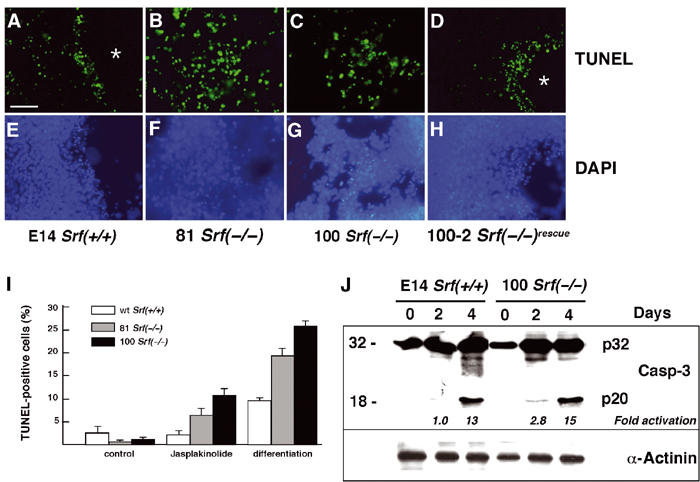
Srf(−/−) ES cells display increased apoptosis when differentiated under monolayer conditions. (A–H) ES cells of the indicated Srf genotype were grown on coverslips and differentiated under monolayer conditions for 4 days. Apoptotic cells were visualized by TUNEL staining (A–D). The same slides were counterstained with DAPI (E–H). Asterisks indicate cavity-like regions that formed exclusively in SRF-containing cells after 4 days of differentiation. Bar=50 μm. (I) ES cells of the indicated Srf genotype were grown for 48 h in the presence of LIF, followed by the addition for 24 h of either solvent (MeOH; control) or 10 nM jasplakinolide. Alternatively, cells were differentiated upon LIF removal under monolayer conditions for 4 days. Apoptotic cells were quantified by TUNEL staining followed by FACS analysis. Values represent the mean of two (differentiation) or three (jasplakinolide) independent experiments ±s.d. (J) E14 Srf(+/+) and 100 Srf(−/−) ES cells were differentiated as monolayer cultures for up to 4 days, and protein extracts were prepared after 0, 2, and 4 days. Western blotting was performed with an antiserum recognizing both the inactive caspase-3 precursor (CPP32; p32) and the active cleavage product (p20). Fold activation of caspase-3 activity, as indicated on the bottom of the blot, was calculated by dividing the normalized intensity of the respective p20 band by the normalized intensity of the p20 band at day 0. Normalized intensities were first derived by dividing the p20 signal by the respective signal from the loading control (α-actinin).
We next quantified apoptosis in our ES cell system, as induced by either jasplakinolide treatment or cell differentiation. The F-actin-stabilizing drug jasplakinolide induces apoptosis in various cell types (Posey and Bierer, 1999; Odaka et al, 2000). FACS analysis revealed that both jasplakinolide treatment and differentiation by LIF withdrawal for 4 days yielded significantly more apoptosis in the two Srf(−/−) ES cell lines than in wild-type (wt) cells (Figure 1I). Enhanced apoptosis of differentiating Srf(−/−) ES cells was accompanied by a slight elevation in caspase-3 activity, as judged by cleavage of the inactive caspase-3 precursor CPP32 (p32) into the p20 derivative at days 2 and 4 of differentiation (Figure 1J). Together, our data suggest that SRF deficiency results in increased apoptosis in an in vitro system of ES cell differentiation.
Constitutively active SRF counteracts ES cell apoptosis upon jasplakinolide treatment
We next tested whether expression of wt SRF (wtSRF), or a constitutively active SRF variant (SRF-VP16), was sufficient to rescue Srf(−/−) ES cells from apoptosis. In contrast to nonactivated wt SRF, SRF-VP16 is a strong activator of SRF target genes in Srf(−/−) ES cells (Schratt et al, 2002). Srf(−/−) ES cells were transfected with expression constructs for either (i) the unrelated red fluorescent protein (RFP), (ii) wt SRF, or (iii) the constitutively active SRF-VP16 variant. Cells were then treated with jasplakinolide and cell death was assayed by TUNEL. Expression of the nonrelated RFP protein did not protect Srf(−/−) ES cells from jasplakinolide-induced apoptosis (Figure 2A and B). Transient expression of wt SRF in Srf(−/−) ES cells led to a significant reduction in the total number of apoptotic cells appearing upon jasplakinolide treatment (Figure 2C and D), as quantified by FACS analysis (Figure 2G). However, some apoptotic cells expressing wt SRF were still observed (arrows in Figure 2D). In contrast, overexpression of the constitutively active SRF-VP16 protein (Figure 2E and F) eliminated all jasplakinolide-induced cell death. These results indicate that SRF-mediated transcriptional activation contributes to ES cell survival.
Figure 2.
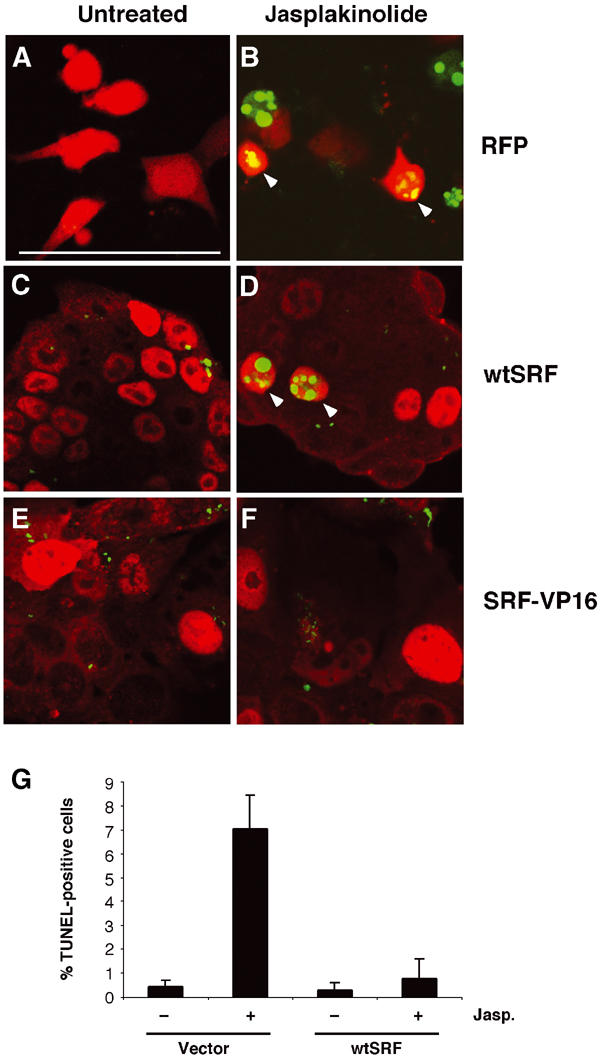
Activated SRF is sufficient to rescue survival of ES cells after jasplakinolide treatment. 100 Srf(−/−) ES cells were transiently transfected with expression vectors encoding red fluorescent protein (RFP; A, B), wt SRF (wtSRF; C, D), or constitutively active SRF-VP16 (E, F). At 48 h after transfection, cells were treated for 24 h either with solvent control (A, C, E; untreated) or with 10 nM jasplakinolide (B, D, F). Transfected cells were identified by their red fluorescence. RFP was detected directly (A, B) and SRF- and SRF-VP16-expressing cells were identified by immunofluorescent staining (red) using antibodies against SRF (C, D) or VP16 (E, F), respectively. Apoptotic cells were visualized by TUNEL staining (green). Note the presence of double positive cells in (B) and, at very low frequency, in (D) (arrowheads), as well as the absence of such cells in (F). Bar=50 μm. (G) Apoptotic Srf(−/−) ES cells were quantified by FACS of untreated or jasplakinolide-treated cultures transfected with either empty vector or wtSRF. Values represent the mean of two independent experiments ±s.d.
SRF is required for differentiation-induced expression of Bcl-2 family proteins
We next assessed expression levels of Bcl-2, Bcl-xl, and Mcl-1 mRNA during in vitro ES cell differentiation using quantitative RT–PCR (Figure 3A). Bcl-2 mRNA levels were increased at 2 days of wt ES cell differentiation, whereas they remained at low levels in SRF-deficient cells. Bcl-xl mRNA levels stayed fairly constant in wt ES cells. In Srf(−/−) cells, Bcl-xl levels, although gradually increasing during differentiation, were significantly lower after the onset (day 2) of differentiation as compared to wt cells. The observed differences between wt and Srf(−/−) cells were more pronounced for Bcl-2 than for Bcl-xl mRNA levels. Impaired mRNA expression was not a general defect of differentiating Srf(−/−) ES cells, since Mcl-1 mRNA levels did not appear affected by SRF deficiency. Western blotting revealed that Bcl-2 protein levels dramatically increased upon wt ES cell differentiation, reaching stable maximum levels at day 4 (Figure 3B). In contrast, no significant induction of Bcl-2 protein was found in SRF-deficient cells within 6 days of differentiation. The levels of the related Bcl-xl protein initially increased in both wt and Srf(−/−) ES cells. Subsequently, however, Bcl-xl levels increased further in wt cells, whereas a decrease was observed in Srf(−/−) cells. SRF protein levels also increased gradually during the differentiation period, thereby paralleling Bcl-2 expression. Taken together, our data reveal that Bcl-2, Bcl-xl, and SRF are developmentally regulated during ES cell differentiation and that the expression of Bcl-2, and to a lesser extent Bcl-xl, is compromised in the absence of SRF.
Figure 3.
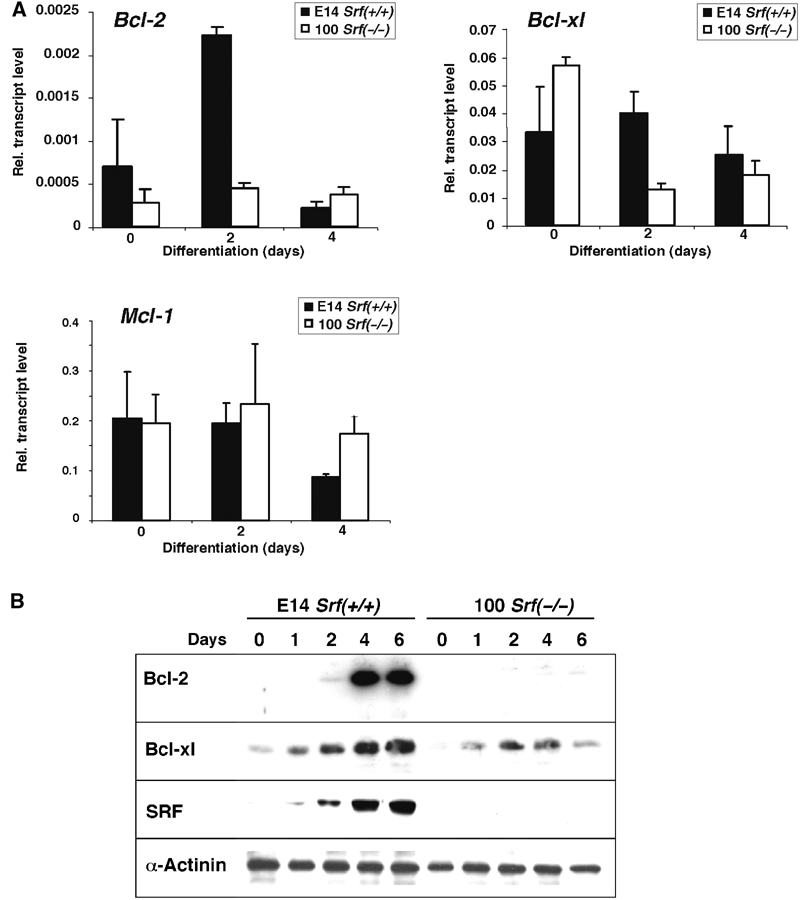
Expression of antiapoptotic Bcl-2 family members in differentiating ES cells. (A) mRNA expression. Quantitative RT–PCR analysis was performed with RNA from E14 Srf(+/+) and 100 Srf(−/−) ES cells differentiated for 0, 2, and 4 days under monolayer conditions. Values represent the mean of three independent experiments ±s.d. (B) Expression of antiapoptotic Bcl-2 proteins. ES cell differentiation and extract preparation was carried out as described in Figure 1. Western blotting was performed using hamster anti-Bcl-2, mouse anti-Bcl-xl, and rabbit anti-SRF, as well as anti-α-actinin antibody for loading control.
Constitutively active SRF induces endogenous Bcl-2 expression in Srf(−/−) ES cells
We next investigated whether SRF was sufficient to induce expression of the endogenous Bcl-2 gene. Constitutively active SRF-VP16, wt SRF, or a control construct that lacked the SRF DNA-binding domain (SRFΔM-VP16) were introduced into undifferentiated SRF-deficient ES cells and the expression of Bcl-2 family members was monitored. At the RNA level, SRF-VP16 expression induced Bcl-2 and Bcl-xl mRNA 3- to 6-fold in Srf(−/−) cells, as judged by quantitative RT–PCR (Figure 4A). This induction was dependent on the SRF DNA-binding domain, since SRFΔM-VP16 failed to induce Bcl-2 and Bcl-xl mRNA levels. wt SRF was unable to activate Bcl-2 and Bcl-xl expression, indicating that SRF-activating signals, which are not present in undifferentiated ES cells, are necessary for SRF-mediated increase in Bcl-2/Bcl-xl mRNA. SRF-VP16 did not induce endogenous Mcl-1 mRNA levels. Western blotting revealed a strong induction of endogenous Bcl-2 protein by SRF-VP16 in Srf(−/−) ES cells (Figure 4B). SRF-VP16 induced Bcl-xl protein levels more weakly. These effects were specific, as α-actinin protein levels were not affected by SRF-VP16 expression. Together, reintroduction of SRF-VP16 into Srf(−/−) ES cells is sufficient to induce endogenous Bcl-2 and Bcl-xl expression, even in the absence of differentiation-inducing cues.
Figure 4.

Constitutively active SRF-VP16 induces ectopic Bcl-2 expression, at both the mRNA and protein levels, in undifferentiated ES cells. (A) 81 Srf(−/−), 100 Srf(−/−), 99 Srf(−/+), and E14 wt ES cells were transfected with either a control vector, or vectors encoding SRFΔM-VP16, wt SRF, or SRF-VP16. At 72 h after transfection, total cell extracts were prepared and mRNA levels within the samples were determined by quantitative RT–PCR analysis using primers specific for Bcl-2, Bcl-xl, and Mcl-1. Fold activation was calculated by dividing relative mRNA levels of SRF- or SRF-VP16-transfected cells by the respective mRNA levels of control (SRFΔM-VP16)-transfected cells. The graph represents the mean of three independent RNA preparations, including error bars. (B) Transfection was as described in (A), except that only SRFΔM-VP16 (lanes 1 and 3) and SRF-VP16 (lanes 2 and 4) were transfected. Western blotting of whole-cell extracts used either hamster anti-Bcl-2, mouse anti-Bcl-xl, or mouse anti-α-actinin antibody.
SRF binds and activates the Bcl-2 promoter
Does SRF regulate the expression of antiapoptotic Bcl-2 genes directly by binding to the respective promoters? Transcription of both the mouse and human Bcl-2 genes is mainly driven by two promoters, P1 and P2. Genome sequence inspection identified two putative SRF-binding sites within the P2 promoter of the murine Bcl-2 gene (Figure 5A), and one putative SRF-binding site in the P2 promoter of the human homolog (not shown). No consensus SRF-binding site was found in the Bcl-x gene promoter. Using EMSA, we observed SRF-containing DNA–protein complexes when extracts from SRF-encoding ES cells were incubated with either Bcl-2 CArG1 or CArG2 oligonucleotides (Figure 5B). No such complexes were seen with extracts from SRF-deficient cells. This suggested SRF to be capable of binding specifically to both CArG box sequences of the murine Bcl-2 P2 promoter in vitro.
Figure 5.
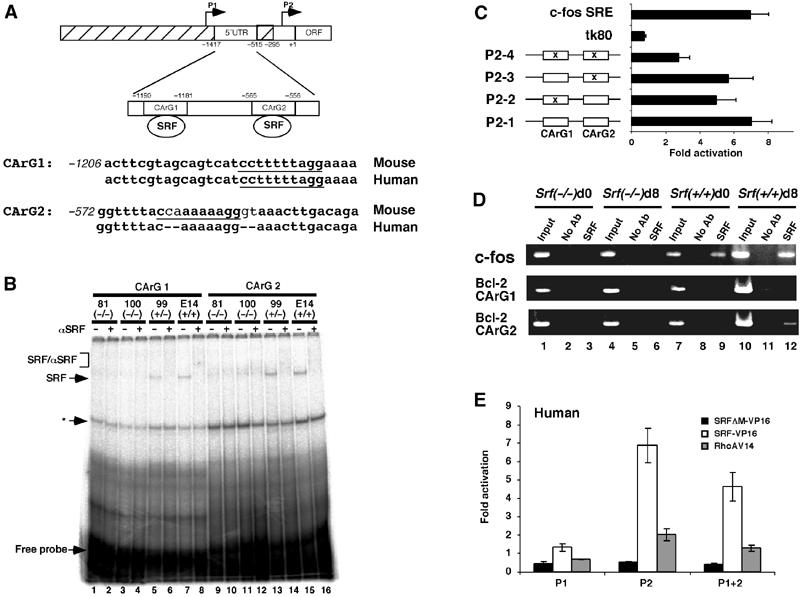
SRF binds directly to the murine Bcl-2 regulatory region in vitro and in vivo and activates Bcl-2 P2 promoter-driven reporter gene transcription. (A) Schematic of the murine Bcl-2 gene regulatory region. Transcriptional start sites under the control of the P1 and P2 regions are indicated by arrows. Nucleotide coordinates are oriented with regard to the beginning of the murine Bcl-2 ORF (+1). The two conserved CArG box sequences lie within a 5′-untranslated exon. Intronic sequences are hatched. Murine and human genomic sequence comparison of the CArG1- and CArG2-containing DNA segments is given. (B) EMSA analysis of Bcl-2 CArG box binding activity in ES cells of different Srf genotype. In all, 15 μg of total ES cell extract was incubated with CArG 1 (lanes 1–8) or CArG 2 (lanes 9–16) oligonucleotides in the presence or absence of anti-SRF antibody (α-SRF). SRF: SRF/DNA complex; SRF/αSRF: SRF/α-SRF/DNA complex. Asterisk (*) denotes an unidentified complex that is not supershifted by an anti-SRF antibody. (C) SRF-VP16 activates the murine Bcl-2 P2 promoter in a CArG box-dependent manner. P2-1 to -4, tk80-luc, and c-fos SRE-luc were transfected into NIH3T3 along with an SRF-VP16 expression vector. Conserved CArG box motives are indicated by open boxes, and CArG box mutations are marked by x. Fold activation over empty vector is given, representing the mean of three independent experiments ±s.d. (D) ChIP analysis of SRF binding to the genomic c-fos and Bcl-2 CArG elements. PCR was performed on immunoprecipitated chromatin fragments of 100 Srf(−/−) (lanes 1–6) or wt (lanes 7–12) ES cells, either undifferentiated (d0; lanes 1–3 and 7–9) or in vitro differentiated for 8 days (d8; lanes 4–6 and 10–12). Lanes 1, 4, 7, and 10 show amplification of total input DNA (1:100 dilution). Lanes 2, 5, 8, and 11 show amplification of ChIP reactions with no antibody. Lanes 3, 6, 9, and 12 show amplification of target sequences in ChIP reactions with anti-SRF antibody. (E) NIH3T3 cells were transfected with the luciferase reporter gene constructs driven by human Bcl-2 promoters (P1-luc, P2-luc, P1+2-luc) along with either empty vector or expression vectors encoding for SRF-VP16, SRFΔM-VP16, or constitutively active RhoA-V14. Normalized luciferase activity and fold activation was derived as in (C). Values represent the mean of three independent experiments ±s.d.
To test the functional significance of the CArG sequences for Bcl-2 transcription, we performed luciferase reporter assays in NIH3T3 fibroblasts. We cloned a 1.8 kb fragment of the murine Bcl-2 gene, which contains the two putative SRF-binding sites (Figure 5A). Site-specific mutagenesis of the CArG box sequences of P2-1 resulted in constructs P2-2 to P2-4 (Figure 5C). P2-1 was efficiently activated by cotransfected SRF-VP16 (Figure 5C). However, P2-2 and P2-3, which each lack one of the two CArG box sequences, gave a 30–40% lowered induction. Mutation of both CArG boxes in P2-4 further reduced activation by SRF-VP16 to a residual 2- to 3-fold activation.
To address whether endogenous SRF binds the genomic Bcl-2 CArG elements inside murine ES cells, we employed chromatin immunoprecipitation (ChIP) assays (Figure 5D). The SRF target c-fos promoter was enriched in SRF immunoprecipitates from both undifferentiated (d0, lane 9) and differentiated (d8, lane 12) wt ES cells. This effect was specific, as no signal was observed using Srf(−/−) ES cells. With respect to the Bcl-2 promoter, the anti-SRF antiserum specifically enriched the Bcl-2 CArG box 2 element in differentiated (d8), but not undifferentiated, wt ES cells (lane 12). Enhanced occupancy of the c-fos and Bcl-2 CArG boxes in differentiated ES cells correlated with increasing SRF expression upon ES cell differentiation (Figure 3). No enrichment of the Bcl-2 CArG1 element could be detected under our assay conditions. Taken together, the ChIP experiments indicate that at least the Bcl-2 CArG box 2 is bound by SRF in differentiated ES cells in vivo.
We also transfected fibroblasts with three previously characterized human Bcl-2 reporter constructs (P1-luc, P2-luc, and P1+P2-luc; see Figure 5A) (Aillet et al, 1998). Coexpressed SRF-VP16 induced luciferase gene expression from P2-luc and P1+P2-luc, but not P1-luc (Figure 5E). This finding indicates that sequences in the P2 promoter, most likely the identified CArG sequences, are responsible for the observed induction of Bcl-2 transcription by SRF-VP16. Consistent with these observations, the constitutively active SRF activator RhoA-V14 stimulated luciferase expression only with P2 promoter-containing constructs (Figure 5E).
Overexpression of Bcl-2 rescues jasplakinolide-induced apoptosis in Srf(−/−) ES cells
Having shown that Bcl-2 is a transcriptional target of SRF, we next tested whether reintroduction of the Bcl-2 protein alone was sufficient to rescue Srf(−/−) ES cells from apoptosis. Srf(−/−) ES cells were transfected either with empty vector (Figure 6A and B) or with HA-tagged Bcl-2 expression vector (Figure 6C and D). In agreement with the previous results, jasplakinolide treatment induced apoptosis in mock-transfected cells in two independent experiments (21±3% apoptotic cells, n=100) (Figure 6A and B). However, Srf(−/−) ES cells transiently overexpressing an HA-Bcl-2 fusion protein (Figure 6C and D) were completely protected from jasplakinolide-induced apoptosis (0% apoptotic cells, n=100) (arrowheads in Figure 6D). To obtain further evidence that the SRF-regulated expression of Bcl-2 family members is important for ES cell survival, we performed RNAi-mediated knockdown of Bcl-2 and Bcl-xl expression in differentiating wild-type ES cells (Supplementary Figure S1). Simultaneous knockdown of Bcl-2 and Bcl-xl led to a significant increase in the apoptotic frequency compared to wt control cells. We conclude that SRF-regulated Bcl-2 expression contributes to the survival of differentiating ES cells.
Figure 6.
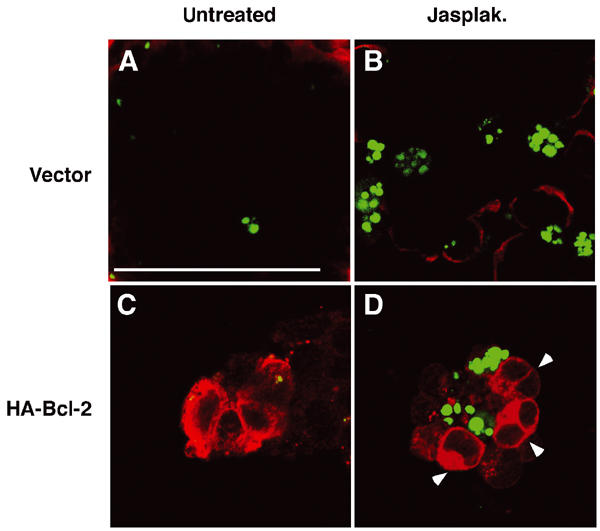
Jasplakinolide-induced apoptosis in Srf(−/−) ES cells is rescued by overexpression of Bcl-2. 100 Srf(−/−) ES cells were transiently transfected with empty vector (A, B) or with an expression vector encoding HA-tagged Bcl-2 (C, D). At 48 h after transfection, cells were treated for 24 h either with solvent control (A, C) or 10 nM jasplakinolide (B, D). HA-Bcl-2-expressing cells (red) were identified by immunocytochemistry using an antibody against hemagglutinin (C, D). Apoptotic cells (green) were simultaneously visualized by TUNEL staining. Note that HA-Bcl-2-positive cells (arrowheads in (D)) were always TUNEL negative. Bar=50 μm.
EBs derived from Srf(−/−) ES cells display increased apoptosis and impaired cavitation
Apoptosis occurs first in the early vertebrate embryo during formation of the proamniotic cavity. Embryoid bodies (EBs) recapitulate this cavitation process, and therefore represent a suitable system to study the regulation of apoptosis. We first monitored apoptosis, using TUNEL staining, in day-5 EBs generated from ES cells of different Srf genotype (Figure 7A–D). In Srf(+/+), Srf(−/+), and Srf(−/−)rescue EBs, apoptotic cells were primarily found near developing cavities (Figure 7A and B, and data not shown). In contrast, inside EBs derived from two independent Srf(−/−) ES cell lines, a much larger number of cells, distributed across the entire EB, had already initiated an apoptotic program (Figure 7C and D, and data not shown). The cell survival defect of Srf(−/−) ES cells is cell autonomous, since wt ES cells fail to rescue Srf(−/−) ES cells from apoptosis in chimeric EBs, which were grown from a mixture of Srf(−/−) and Srf(+/+) ES cells (data not shown). To investigate potential effects of SRF deficiency on EB cavitation at later stages, histological sections of day-8 EBs were prepared and cavitation monitored in Srf(+/+) and Srf(−/−) EBs (Figure 7E–H). At day 8, all Srf(+/+) EBs investigated had formed distinct cavities of variable sizes, which were bordered by cells that displayed the typical morphology of CEE cells (Figure 7E and F). In contrast, Srf(−/−) EBs were greatly disorganized, and no CEE-bordered cavities were observed inside Srf(−/−) EBs (Figure 7G and H). In addition, a high number of cells inside Srf(−/−) EBs displayed condensed chromatin, a hallmark of apoptotic cell death. These data suggest that SRF is required for cell survival during EB cavitation.
Figure 7.
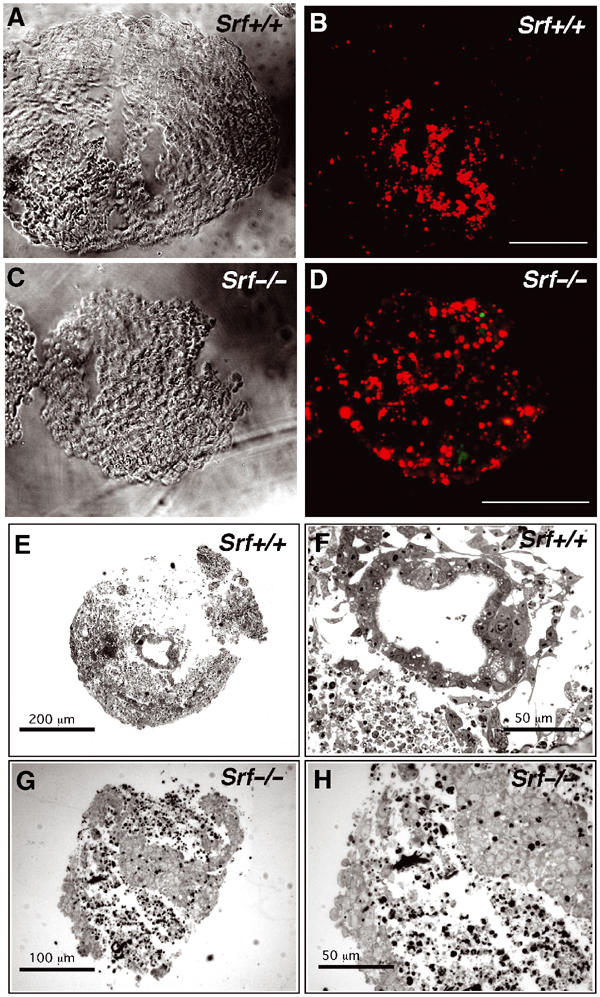
EBs derived from Srf(−/−) ES cells display increased apoptosis and impaired cavity formation. (A–D) Increased, widespread apoptosis in day-5 Srf(−/−) EBs revealed by TUNEL staining (bar=100 μm). Note the size difference between Srf(+/+) and Srf(−/−) EBs. (E–H) Light microscopy on cryosections (10 μm) of day-8 EBs derived from either Srf(+/+) (E, F) or Srf(−/−) (G, H) ES cells. The results show representative examples of multiple EBs examined. Srf(+/+) EBs form cavities, which are lined by polarized columnar epithelial epiblast (CEE) cells (F). Srf(−/−) EBs display large areas of loosely arranged, fragmented cells, without forming distinct cavities (G). Note that a large fraction of cells inside Srf(−/−) EBs display condensed chromatin, a hallmark of apoptotic cell death (H).
Increased apoptosis in Srf(−/−) EBs is accompanied by the inability to induce Bcl-2 expression
Our results from differentiating Srf(−/−) ES cells (Figure 3) prompted us to assess the expression of Bcl-2 genes in EBs using quantitative RT–PCR. In SRF-containing EBs, Bcl-2 and Bcl-xl mRNAs gradually increased over 8 days of differentiation (Figure 8A). In contrast, Bcl-2 mRNA was almost undetectable in day-8 Srf(−/−) EBs. Bcl-xl mRNA gradually increased with differentiation in Srf(−/−) EBs, but only reached about 60% of wt levels at day 8. For Mcl-1, no significant differences were observed between the EBs of differing Srf genotype (not shown). EBs from an independent Srf(−/−) ES cell line gave similar results (not shown). The spatial distribution of Bcl-2 protein expression in day-8 EBs was investigated by immunohistochemistry (Figure 8B). Bcl-2 expression in wt EBs was highly restricted to cells that surrounded a large cavity (CA), most likely representing CEE cells (Figure 8B, upper panel). Apoptotic, TUNEL-positive cells were found inside the developing cavity and lacked Bcl-2 protein. In contrast, no Bcl-2 protein expression was detectable in day-8 Srf(−/−) EBs (Figure 8B, lower panel), and apoptotic cells were scattered throughout these EBs. These results suggest an important role for SRF-driven Bcl-2 expression in the regulation of cell survival during EB cavitation.
Figure 8.
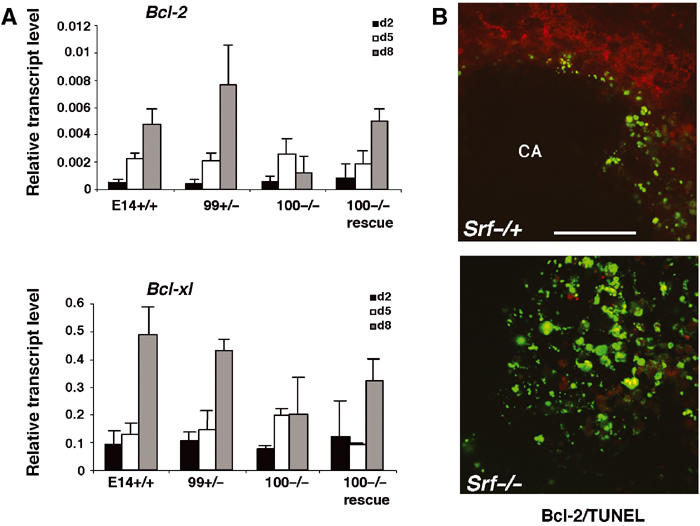
Increased apoptosis in Srf(−/−) EBs is accompanied by impaired Bcl-2 expression. (A) Expression of Bcl-2 family members during EB differentiation. Quantitative RT–PCR analysis on extracts from day-2, -5, and -8 EBs of the indicated Srf genotype. Expression of Bcl-2, Bcl-xl, and Mcl-1 mRNA is displayed relative to the endogenous Hprt gene. The graph represents the mean of three independent RNA preparations ±s.d. (B) Anti-Bcl-2 indirect immunofluorescence demonstrates the presence of Bcl-2-expressing cells (red) inside day-8 Srf(−/+) EBs (B, upper) and their almost complete absence in Srf(−/−) EBs (B, lower). Of note, Bcl-2 is expressed in the cytoplasm of Srf(−/+) cells that are found near cavities (CA). Simultaneous TUNEL staining (green) reveals apoptotic cells at the inner margin of the cavity of Srf(−/+) EBs and their widespread existence in Srf(−/−) EBs (B; bar=50 μm).
Srf(−/−) pregastrulation embryos display enhanced apoptosis
Finally, we investigated whether loss of SRF expression also affected cell survival in vivo. E6.5 embryos of different Srf genotype were prepared, fixed, and processed for whole-mount TUNEL staining. Interestingly, the fraction of apoptotic cells in homozygous Srf mutant embryos was about four-fold higher than in Srf(+/+) or Srf(−/+) control embryos (Figure 9A and B). Elevated apoptosis was accompanied by a significant size reduction of the SRF-deficient embryos as compared to SRF-containing embryos. These results demonstrate that SRF is required in vivo for cell survival in pregastrulation mouse embryos.
Figure 9.
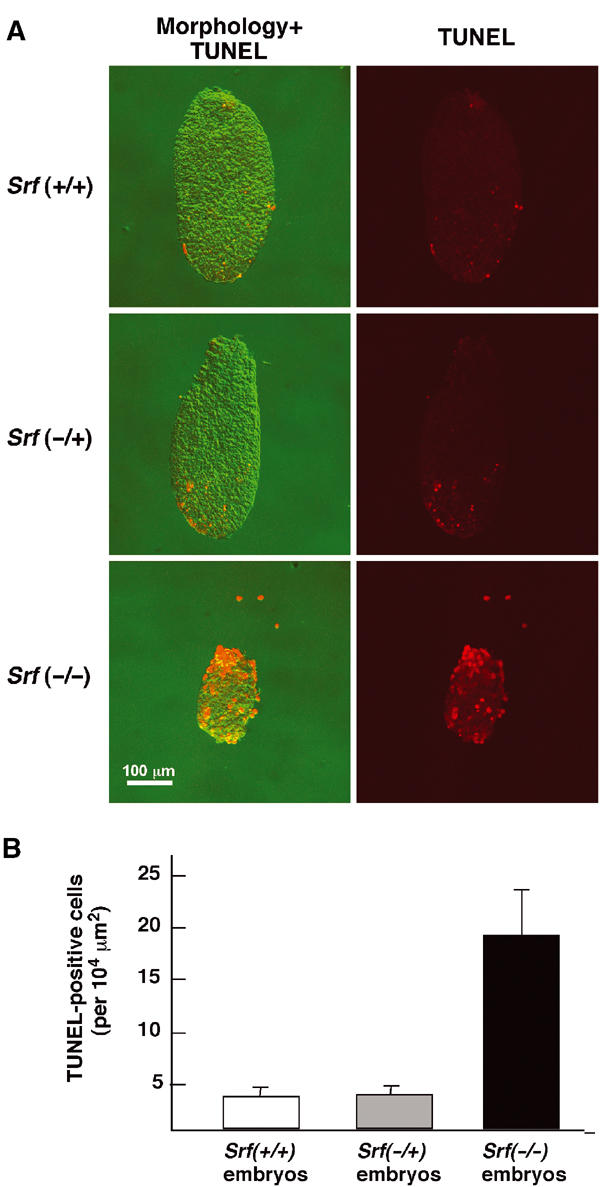
Increased apoptosis in Srf(−/−) pregastrulation embryos. (A) Whole-mount TUNEL staining on E6.5 mouse embryos of the indicated Srf genotypes. (B) Quantitation of apoptosis in wt and Srf mutant E6.5 embryos. Srf(+/+): n=13; Srf(−/+): n=15; Srf(−/−): n=10.
Discussion
SRF was previously shown to regulate specific target genes, including immediate-early genes, neuronal genes, and muscle genes (Johansen and Prywes, 1995; Treisman, 1995; Herdegen and Leah, 1998; Miano, 2003). In this study, we uncovered a new role for SRF in the regulation of cell survival during embryogenesis. We show that SRF-dependent Bcl-2 expression is critical for the survival of differentiating ES cells. First, SRF-deficient ES cells display increased apoptosis upon in vitro differentiation by LIF withdrawal and fail to express the antiapoptotic Bcl-2 protein. Second, constitutively active SRF induces ectopic Bcl-2 expression in undifferentiated ES cells and reintroduction into Srf null cells of either SRF or Bcl-2 rescues these cells from apoptosis. Third, during EB differentiation, enhanced cell death, defective cavitation, and impaired Bcl-2 expression are detected in the absence of SRF. Finally, Srf(−/−) pregastrulation embryos are reduced in size and display enhanced apoptosis. Together, our results describe a novel SRF–Bcl-2 signaling pathway that is operative during embryonic development. Defective expression of antiapoptotic Bcl-2 family members therefore may provide, at least in part, a molecular explanation for the early embryonic lethality of Srf knockout mice.
We note that in our initial characterization of Srf knockout mice, the presence of pyknotic cells in the epiblast of these embryos was emphasized (Arsenian et al, 1998). However, in this previous study, the aspects of cell death were not explored any further. In Arsenian et al (1998), reduced size of SRF-deficient embryos was seen at E7.5, rather than at E6.5, as reported here. We interpret this time shift to reflect differences in genetic backgrounds of the animals used since the studies were performed with mice of mixed genetic background. Current breedings with Srf(flex1/lx) mice (Srf-flex1 representing a floxed Srf allele and Srf-lx its Cre-derived null allele), in a C57BL/6N congenic background, also display size reduction of E6.5 SRF-deficient embryos. Therefore, enhanced apoptosis of Srf(−/−) pregastrulation embryos is displayed in an inbred mouse line (S Raimundo, S Alberti, and A Nordheim, unpublished), permitting the search for potential modifier genes that might impinge on cell survival during embryogenesis.
Other studies indicated SRF to play a role in the regulation of cell survival. SRF-dependent expression of the antiapoptotic Bcl-2 family member Mcl-1 was found to correlate with survival during TPA-induced differentiation of myelomonocytic cells (Townsend et al, 1999). More recently, caspase-dependent SRF protein cleavage has been described to occur at the onset of Fas-induced apoptosis in B cells, and a noncleavable SRF mutant was able to prevent apoptosis in this system (Drewett et al, 2001). We provide here the first genetic evidence that SRF is required for cell survival during early embryonic development in vivo. It will be interesting to test whether SRF-regulated expression of Bcl-2 family members is also important in other developmental settings that require a tight regulation between cell survival and death. We will be able to address this issue using a recently generated conditional Srf null allele (Wiebel et al, 2002).
Our data identify SRF as a direct regulator of Bcl-2 transcription. Binding of SRF to the Bcl-2 promoter occurs specifically in differentiating ES cells, which correlates well with increasing SRF protein levels and concomitant Bcl-2 expression during ES cell differentiation. Of the two CArG box motives present in the Bcl-2 promoter, only CArG box 2 appears to be bound by SRF in ES cells in vivo. These findings contrast with results obtained in fibroblasts, where both CArG box motives are necessary for activation of Bcl-2 transcription by SRF. This discrepancy may be explained by cell-type- and developmental-stage-specific differences in the accessibility of the Bcl-2 CArG box motives. Further experiments are required to determine the exact contribution of CArG box motives to Bcl-2 transcription in different tissues and organisms.
Although we found that SRF deficiency almost completely prevents Bcl-2 expression in differentiating ES cells, additional transcription factors, for example, CREB, might contribute to Bcl-2 regulation (Wilson et al, 1996; Pugazhenthi et al, 1999, 2000; Riccio et al, 1999).
We report that Bcl-2 is expressed in differentiating ES cells at the cavitation stage. Consistent with a role for Bcl-2 in early development, this protein is widely expressed in embryonic tissues (LeBrun et al, 1993). Nonetheless, Bcl-2 knockout mice gastrulate normally and die postnatally due to kidney failure (Veis et al, 1993), arguing against an exclusive role for Bcl-2 in early embryonic development. The viability of Bcl-2(−/−) embryos may be explained by expression of Bcl-2-related molecules providing redundancy in survival functions, for example, Bcl-xl. Since we could not locate CArG consensus elements within 3.5 kb upstream of the Bcl-xl coding region, the observed SRF effect on Bcl-xl transcription may be indirect. In support of this notion, apoptosis correlates with decreased expression of c-fos (Bertolotto et al, 2000), encoding a component of the AP-1 transcription complex. AP-1 activates Bcl-xl expression by directly binding to the Bcl-xl promoter region (Sevilla et al, 1999).
Mechanisms regulating SRF activity during embryonic cavitation require elucidation. In the surviving inner cells that line the embryonic cavity, increased SRF levels might be responsible for SRF activation. In support, we observed increased SRF protein levels upon ES cell differentiation in vitro. Also, pre-existing SRF molecules might be activated by extracellular cues. For example, attachment of inner cells to a basement membrane, which has been shown to rescue these cells from apoptosis (Coucouvanis and Martin, 1995), could trigger signaling cascades that lead to SRF activation. PI-3-kinase and RhoA signaling were implicated in the regulation of Bcl-2 expression upon cell adhesion (Matter and Ruoslahti, 2001), indicating that PI-3-kinase and/or RhoA might act upstream of SRF to activate Bcl-2 expression. In support of this hypothesis, we found that RhoA is able to induce the expression of a Bcl-2 reporter construct containing SRF-binding sites.
Since SRF promotes cell survival during cavitation, we postulate that SRF has to be inactivated in inner cells that undergo apoptosis during the cavitation process. It is intriguing to speculate that SRF is cleaved in a caspase-dependent manner, as previously observed at the onset of apoptosis in B cells (Drewett et al, 2001). The nature of the pathways that mediate death signaling in differentiating ES cells is not completely understood. Since we observe only slightly enhanced caspase-3 activation in SRF-deficient cells, other caspase-dependent or -independent pathways likely contribute to the enhanced apoptosis of SRF-deficient ES cells (Hakem et al, 1998; Joza et al, 2001).
In summary, our results indicate that the SRF–Bcl-2 pathway contributes in an important way to the survival of differentiating ES cells. However, we anticipate that other SRF-regulated genes are involved in this process. By genome-wide expression profiling using SRF mutant and wt ES cells, it should be possible to identify novel SRF target genes that modulate cell death and survival.
Materials and methods
Mouse embryos
E6.5 embryos of different Srf genotype were prepared from matings of heterozygous Srf(−/+) mice (mixed genetic background) (Arsenian et al, 1998). Embryos were dissected free of extraembryonic membranes, fixed in 4% paraformaldehyde, permeabilized with proteinase K, and refixed with 4% paraformaldehyde/0.1% glutaraldehyde. Subsequent whole-mount TUNEL staining was performed with the 10-fold diluted In Situ Cell Death Detection Kit (TMR red, Roche, Basel). Apoptotic cells were detected by fluorescence with a laser scanning microscope and quantitated upon normalization to overall embryo surface area (Image master software; Pharmacia). Genotyping of embryos required protease K digestion followed by PCR with specific primers (forward-A: 5′-TGCCACTCCCACTGTCCTTTCCTAATAA; forward-B: 5′-GAGATGCTGCGAACGTCCGGAATC; reverse: 5′-CTTCGCGCACACCAGGACACAGAGGAT).
Antibodies. Rabbit anti-Bcl-2 (N-19), rabbit anti-HA (Y-11), rabbit anti-SRF and mouse anti-VP16 were all obtained from Santa Cruz Biotechnology, Inc. Mouse anti-Bcl-2 was from Transduction Labs. Hamster anti-Bcl-2, mouse anti-Bcl-xl, and mouse anti-caspase-3 were from Pharmingen. Mouse anti-α-actinin was from Sigma.
Plamids. Human Bcl-2 promoter constructs P1-luc, P2-luc, and P1+P2-luc were described previously (Aillet et al, 1998). A corresponding fragment of the mouse regulatory Bcl-2 region (−1296 to −279 relative to the start of the open reading frame (ORF)) was amplified by genomic PCR with the following primers: Bcl-2prom fw: 5′-GAGGAGAAAGGGTGCGCAG-3′; Bcl-2 prom bw: 5′-TACTTCCTCCGCATGCTGA-3′.
The obtained fragment was cloned into tk-80 luc. Mutations of the CArG sequences were performed with the QuickChange Site-directed Mutagenesis Kit (Stratagene) using the following mutagenesis primers:
Bcl-2CArG1mut fw: 5′-GGGGACTTCGTAGCAGTCATCGCGGATCCGA AAAAGAGGGGGGC-3′
Bcl-2CArG1mut bw: 5′-GCCCCCCCTCTTTTTCGGATCCGCGATGACTGCTACGAAG TCCCC-3′
Bcl-2CArG2mut fw: 5′-TTTGTTTGGGTTTTACCAGAGCTCGTAAACT TGACAGAAGATCATGCCG-3′
Bcl-2CArG2mut bw: 5′-CGGCATGATCTTCTGTCAAGTTTACGAGCTCTGGTAAAAC CCAAACAAA-3′
Mutated CArG box sequences are underlined. Mutations were verified by restriction digestion and sequencing. Plasmids encoding SRF-VP16, SRFΔM-VP16, wtSRF, and RhoA-V14 (Schratt et al, 2002), reporter gene constructs tk80-luc and SRE2-luc (Janknecht et al, 1993b), and expression vectors for constitutively active MEKK, BXB, Erk-1, and Elk-1 (Janknecht and Nordheim, 1996) were described. HA-Bcl-2 expression plasmid was provided by S Kuegler (Homburg/Saar).
Cell culture, transfection, and apoptosis induction
The ES cell lines E14.1 Srf(+/+), 99 Srf(−/+), 81 Srf(−/−), 100 Srf(−/−), and 100 Srf(−/−)rescue, their culture conditions, and the differentiation protocol for EBs were described previously (Weinhold et al, 2000). For the differentiation of ES cells as monolayer cultures, subconfluent cells were washed twice with ES media lacking LIF, and further cultured in the absence of LIF for up to 8 days. The differentiation medium was replaced every 2 days. NIH3T3 mouse fibroblasts were maintained in DMEM containing 10% FCS (DMEM) and passaged every 3–4 days. ES cells were transfected with LipofectAmine (Gibco BRL) (Schratt et al, 2002).
For reporter gene assays, NIH3T3 cells were seeded the day before transfection at 2 × 105 cells per six wells. Transfection with LipofectAmine in serum-free DMEM was according to the manufacturer's protocol. Total DNA amount was maintained at 1.5 μg, and a 10-fold excess of LipofectAmine (Gibco BRL) reagent was used. In all, 100 ng expression vector and 300 ng reporter plasmid were used per six wells. A measure of 100 ng of the lacZ expression vector pEQ176 was cotransfected to monitor transfection efficiency (Janknecht et al, 1993a). At 5 h after transfection, serum was added to the samples to a final concentration of 10%. After 48 h, cells were lysed and both luciferase and lacZ activities were measured.
For induction of apoptosis, ES cells were grown in the presence of LIF for 48 h and then treated with 10 nM jasplakinolide (Molecular Probes) for 24 h.
Indirect immunofluorescence and TUNEL staining
Cells grown on gelatin-coated coverslips were fixed in 4% formaldehyde and permeabilized in 0.2% Triton X-100. Nonspecific binding was blocked by incubation for 1 h in 1% BSA in PBS at 37°C, before staining with primary antibody for 1 h at 37°C. Incubation with fluorescence-conjugated secondary antibodies (Molecular Probes, 1:200 in 0.2% BSA) was performed for 30 min at 37°C. Cells were TUNEL-stained with the In Situ Cell Death Detection Kit Fluorescein (Roche) according to the manufacturer's protocol and counterstained with 4,6-diamidino-2-phenylindole (DAPI). Coverslips were then washed four times with PBS, once in water, air-dried, and mounted in Moviol. Image acquisition was performed with LSM 510 (Zeiss).
EBs were embedded in tissue-freezing media (Jung) for serial cryosectioning (10 μm). Sections were postfixed for 1 h with 4% paraformaldehyde in PBS. Staining procedures and image acquisition were as described above for ES cells.
FACS analysis
ES cells were collected 24 h after induction of apoptosis, fixed in 4% formaldehyde for 10 min, permeabilized with 0.2% Triton X-100 for 5 min, and blocked in 1% BSA for 1 h. After three washes in PBS, the cells were incubated for 1 h in TUNEL reaction mixture (Roche, Basel). For reference, incubation was performed in the absence of any fluorescent dye. After washing, cells were subjected to FACScan analysis (Becton Dickinson). A total of 104 cells were counted for each sample. Data analysis was performed with CellQuest software (Becton Dickinson).
Quantitative RT–PCR
Preparation of total RNA, cDNA synthesis, semiquantitative PCR using the SYBR green technology (Perkin Elmer), as well as primer sequences for the housekeeping gene Hprt have been described previously (Weinhold et al, 2000). In addition, the following primers were used:
Tm-bcl2 fw: 5′-TGGCATCTTCTCCTTCCAGC-3′
Tm-bcl2 bw: 5′-ACGTCCTGGCAGCCATCTC-3′
Tm-mcl1 fw: 5′-TGGAGTTCTTCCACGTACAGGA-3′
Tm-mcl1 bw: 5′-AGCAACACCCGCAAAAGC-3′
Tm-bclxl fw: 5′- TCTACGGGAACAATGCAGCA-3′
Tm-bclxl bw: 5′- AGGAACCAGCGGTTCAAGC-3′
Tm-srf fw: 5′-TGTGCAGGCCATTCATGTG-3′
Tm-srf bw: 5′-ACAGACGACGTCATGATGGTG-3′
Specificity of the PCRs was confirmed by melting point analysis and agarose gel electrophoresis.
Western blotting and EMSA
Preparation of total cell extracts, Western blotting, and EMSA were carried out as described (Weinhold et al, 2000). The following oligonucleotides were used in EMSA:
Bcl-2 CArG1 fw: 5′-TAGCAGTCATCCTTTTTAGGAAAAAGA-3′
Bcl-2 CArG1 bw: 5′-TAGCCCCTCTTTTTCCTAAAAAGGATGACT-3′
Bcl-2 CArG2 fw: 5′-TAGCTTTTACCAAAAAAGGGTAAACTTGACAGAA-3′
Bcl-2 CArG2bw: 5′-GATCTTCTGTCAAGTTTACCCTTTTTTGGTAAAA-3′
Bcl-2 CArG1mut fw: 5′-TAGCAGTCATCGCGGATCCGAAAAAGAGGG-3′
Bcl-2 CArG1mut bw: 5′-TAGCCCCTCTTTTTCGGATCCGCGATGACT-3′
Bcl-2-CArG2mut fw: 5′-AAAATGGTTTTTTCCCATTGAACTGTCTTCTA-3′
Bcl-2 CArG2mut bw: 5′-GATCTTCTGTCAAGTTTACGAGCTCTGGTAAAA-3′
Complementary oligonucleotides were annealed and labeled with 32P-dCTP (Hartmann/Braunschweig).
Chromatin immunoprecipitation
ChIP assays were performed with a ChIP assay kit (Upstate Biotechnology Inc.) as described (Manabe and Owens, 2001). Samples were derived from 2 × 106 cells of either undifferentiated ES cells (d0), or of those differentiated upon LIF removal for 8 days under monolayer conditions (d8). IP was performed with polyclonal rabbit anti-SRF antiserum (Santa Cruz). PCR primers used are as follows: Bcl-2 CArG1: 5′-gggacttcgtagcagtcatcctttttagg-3′, 5′-aaaaagcagcttgctaaatgcaggc-3′; Bcl-2 CArG2: 5′-agaccccaactcccgattcattg-3′, 5′-aaggacggcatgatcttctgtcaagt-3′; c-fos: 5′-cggttccccccctgcgctgcaccctcagag-3′, 5′-agaacaacagggaccggccgtggaaacctg-3′. For PCR, 32 (c-fos, Bcl-2 CArG1) or 35 (Bcl-2 CArG2) cycles were used. Samples from 100 Srf(−/−) ES cells and no-Ab controls showed low levels of background amplification.
Supplementary Material
Supplementary Figure S1
Acknowledgments
We thank S Kuegler (Homburg) and N Israel (Paris) for plasmids and ME Greenberg for comments on the manuscript. This work was sponsored by the DFG (SFB446/B7) and the Fonds der Chemischen Industrie. UP received a Boehringer Ingelheim Fonds fellowship.
References
- Adams JM, Cory S (1998) The Bcl-2 protein family: arbiters of cell survival. Science 281: 1322–1326 [DOI] [PubMed] [Google Scholar]
- Aillet F, Masutani H, Elbim C, Raoul H, Chene L, Nugeyre MT, Paya C, Barre-Sinoussi F, Gougerot-Pocidalo MA, Israel N (1998) Human immunodeficiency virus induces a dual regulation of Bcl-2, resulting in persistent infection of CD4(+) T- or monocytic cell lines. J Virol 72: 9698–9705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arsenian S, Weinhold B, Oelgeschlager M, Ruther U, Nordheim A (1998) Serum response factor is essential for mesoderm formation during mouse embryogenesis. EMBO J 17: 6289–6299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertolotto C, Ricci JE, Luciano F, Mari B, Chambard JC, Auberger P (2000) Cleavage of the serum response factor during death receptor-induced apoptosis results in an inhibition of the c-FOS promoter transcriptional activity. J Biol Chem 275: 12941–12947 [DOI] [PubMed] [Google Scholar]
- Coucouvanis E, Martin GR (1995) Signals for death and survival: a two-step mechanism for cavitation in the vertebrate embryo. Cell 83: 279–287 [DOI] [PubMed] [Google Scholar]
- Coucouvanis E, Martin GR (1999) BMP signaling plays a role in visceral endoderm differentiation and cavitation in the early mouse embryo. Development 126: 535–546 [DOI] [PubMed] [Google Scholar]
- Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, Greenberg ME (1997) Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell 91: 231–241 [DOI] [PubMed] [Google Scholar]
- Drewett V, Devitt A, Saxton J, Portman N, Greaney P, Cheong NE, Alnemri TF, Alnemri E, Shaw PE (2001) Serum response factor cleavage by caspases 3 and 7 linked to apoptosis in human BJAB cells. J Biol Chem 276: 33444–33451 [DOI] [PubMed] [Google Scholar]
- Franke TF, Cantley LC (1997) Apoptosis. A Bad kinase makes good. Nature 390: 116–117 [DOI] [PubMed] [Google Scholar]
- Gille H, Sharrocks AD, Shaw PE (1992) Phosphorylation of transcription factor p62TCF by MAP kinase stimulates ternary complex formation at c-fos promoter. Nature 358: 414–417 [DOI] [PubMed] [Google Scholar]
- Gross A, McDonnell JM, Korsmeyer SJ (1999) BCL-2 family members and the mitochondria in apoptosis. Genes Dev 13: 1899–1911 [DOI] [PubMed] [Google Scholar]
- Hakem R, Hakem A, Duncan GS, Henderson JT, Woo M, Soengas MS, Elia A, de la Pompa JL, Kagi D, Khoo W, Potter J, Yoshida R, Kaufman SA, Lowe SW, Penninger JM, Mak TW (1998) Differential requirement for caspase 9 in apoptotic pathways in vivo. Cell 94: 339–352 [DOI] [PubMed] [Google Scholar]
- Herdegen T, Leah JD (1998) Inducible and constitutive transcription factors in the mammalian nervous system: control of gene expression by Jun, Fos and Krox, and CREB/ATF proteins. Brain Res Brain Res Rev 28: 370–490 [DOI] [PubMed] [Google Scholar]
- Jacobson MD, Weil M, Raff MC (1997) Programmed cell death in animal development. Cell 88: 347–354 [DOI] [PubMed] [Google Scholar]
- Janknecht R, Ernst WH, Houthaeve T, Nordheim A (1993a) C-terminal phosphorylation of the serum-response factor. Eur J Biochem 216: 469–475 [DOI] [PubMed] [Google Scholar]
- Janknecht R, Ernst WH, Pingoud V, Nordheim A (1993b) Activation of ternary complex factor Elk-1 by MAP kinases. EMBO J 12: 5097–5104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janknecht R, Nordheim A (1996) Regulation of the c-fos promoter by the ternary complex factor Sap-1a and its coactivator CBP. Oncogene 12: 1961–1969 [PubMed] [Google Scholar]
- Johansen FE, Prywes R (1995) Serum response factor: transcriptional regulation of genes induced by growth factors and differentiation. Biochim Biophys Acta 1242: 1–10 [DOI] [PubMed] [Google Scholar]
- Joza N, Susin SA, Daugas E, Stanford WL, Cho SK, Li CY, Sasaki T, Elia AJ, Cheng HY, Ravagnan L, Ferri KF, Zamzami N, Wakeham A, Hakem R, Yoshida H, Kong YY, Mak TW, Zuniga-Pflucker JC, Kroemer G, Penninger JM (2001) Essential role of the mitochondrial apoptosis-inducing factor in programmed cell death. Nature 410: 549–554 [DOI] [PubMed] [Google Scholar]
- Kurland JF, Kodym R, Story MD, Spurgers KB, McDonnell TJ, Meyn RE (2001) NF-kappaB1 (p50) homodimers contribute to transcription of the bcl-2 oncogene. J Biol Chem 276: 45380–45386 [DOI] [PubMed] [Google Scholar]
- LeBrun DP, Warnke RA, Cleary ML (1993) Expression of bcl-2 in fetal tissues suggests a role in morphogenesis. Am J Pathol 142: 743–753 [PMC free article] [PubMed] [Google Scholar]
- Manabe I, Owens GK (2001) CArG elements control smooth muscle subtype-specific expression of smooth muscle myosin in vivo. J Clin Invest 107: 823–834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marais R, Wynne J, Treisman R (1993) The SRF accessory protein Elk-1 contains a growth factor-regulated transcriptional activation domain. Cell 73: 381–393 [DOI] [PubMed] [Google Scholar]
- Martin GR, Evans MJ (1975) Differentiation of clonal lines of teratocarcinoma cells: formation of embryoid bodies in vitro. Proc Natl Acad Sci USA 72: 1441–1445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matter ML, Ruoslahti E (2001) A signaling pathway from the alpha5beta1 and alpha(v)beta3 integrins that elevates bcl-2 transcription. J Biol Chem 276: 27757–27763 [DOI] [PubMed] [Google Scholar]
- Mayo MW, Wang CY, Drouin SS, Madrid LV, Marshall AF, Reed JC, Weissman BE, Baldwin AS (1999) WT1 modulates apoptosis by transcriptionally upregulating the bcl-2 proto-oncogene. EMBO J 18: 3990–4003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miano JM (2003) Serum response factor: toggling between disparate programs of gene expression. J Mol Cell Cardiol 35: 577–593 [DOI] [PubMed] [Google Scholar]
- Miralles F, Posern G, Zaromytidou AI, Treisman R (2003) Actin dynamics control SRF activity by regulation of its coactivator MAL. Cell 113: 329–342 [DOI] [PubMed] [Google Scholar]
- Murray P, Edgar D (2000) Regulation of programmed cell death by basement membranes in embryonic development. J Cell Biol 150: 1215–1221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odaka C, Sanders ML, Crews P (2000) Jasplakinolide induces apoptosis in various transformed cell lines by a caspase-3-like protease-dependent pathway. Clin Diagn Lab Immunol 7: 947–952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posey SC, Bierer BE (1999) Actin stabilization by jasplakinolide enhances apoptosis induced by cytokine deprivation. J Biol Chem 274: 4259–4265 [DOI] [PubMed] [Google Scholar]
- Pugazhenthi S, Miller E, Sable C, Young P, Heidenreich KA, Boxer LM, Reusch JE (1999) Insulin-like growth factor-I induces bcl-2 promoter through the transcription factor cAMP-response element-binding protein. J Biol Chem 274: 27529–27535 [DOI] [PubMed] [Google Scholar]
- Pugazhenthi S, Nesterova A, Sable C, Heidenreich KA, Boxer LM, Heasley LE, Reusch JE (2000) Akt/protein kinase B up-regulates Bcl-2 expression through cAMP-response element-binding protein. J Biol Chem 275: 10761–10766 [DOI] [PubMed] [Google Scholar]
- Riccio A, Ahn S, Davenport CM, Blendy JA, Ginty DD (1999) Mediation by a CREB family transcription factor of NGF-dependent survival of sympathetic neurons. Science 286: 2358–2361 [DOI] [PubMed] [Google Scholar]
- Romero F, Martinez AC, Camonis J, Rebollo A (1999) Aiolos transcription factor controls cell death in T cells by regulating Bcl-2 expression and its cellular localization. EMBO J 18: 3419–3430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schratt G, Philippar U, Berger J, Schwarz H, Heidenreich O, Nordheim A (2002) Serum response factor is crucial for actin cytoskeletal organization and focal adhesion assembly in embryonic stem cells. J Cell Biol 156: 737–750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sevilla L, Aperlo C, Dulic V, Chambard JC, Boutonnet C, Pasquier O, Pognonec P, Boulukos KE (1999) The Ets2 transcription factor inhibits apoptosis induced by colony-stimulating factor 1 deprivation of macrophages through a Bcl-xL-dependent mechanism. Mol Cell Biol 19: 2624–2634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw PE, Schroter H, Nordheim A (1989) The ability of a ternary complex to form over the serum response element correlates with serum inducibility of the human c-fos promoter. Cell 56: 563–572 [DOI] [PubMed] [Google Scholar]
- Smith MD, Ensor EA, Coffin RS, Boxer LM, Latchman DS (1998) Bcl-2 transcription from the proximal P2 promoter is activated in neuronal cells by the Brn-3a POU family transcription factor. J Biol Chem 273: 16715–16722 [DOI] [PubMed] [Google Scholar]
- Sotiropoulos A, Gineitis D, Copeland J, Treisman R (1999) Signal-regulated activation of serum response factor is mediated by changes in actin dynamics. Cell 98: 159–169 [DOI] [PubMed] [Google Scholar]
- Townsend KJ, Zhou P, Qian L, Bieszczad CK, Lowrey CH, Yen A, Craig RW (1999) Regulation of MCL1 through a serum response factor/Elk-1-mediated mechanism links expression of a viability-promoting member of the BCL2 family to the induction of hematopoietic cell differentiation. J Biol Chem 274: 1801–1813 [DOI] [PubMed] [Google Scholar]
- Treisman R (1994) Ternary complex factors: growth factor regulated transcriptional activators. Curr Opin Genet Dev 4: 96–101 [DOI] [PubMed] [Google Scholar]
- Treisman R (1995) Journey to the surface of the cell: Fos regulation and the SRE. EMBO J 14: 4905–4913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treisman R, Ammerer G (1992) The SRF and MCM1 transcription factors. Curr Opin Genet Dev 2: 221–226 [DOI] [PubMed] [Google Scholar]
- Vaux DL, Korsmeyer SJ (1999) Cell death in development. Cell 96: 245–254 [DOI] [PubMed] [Google Scholar]
- Veis DJ, Sorenson CM, Shutter JR, Korsmeyer SJ (1993) Bcl-2-deficient mice demonstrate fulminant lymphoid apoptosis, polycystic kidneys, and hypopigmented hair. Cell 75: 229–240 [DOI] [PubMed] [Google Scholar]
- Wang D, Chang PS, Wang Z, Sutherland L, Richardson JA, Small E, Krieg PA, Olson EN (2001) Activation of cardiac gene expression by myocardin, a transcriptional cofactor for serum response factor. Cell 105: 851–862 [DOI] [PubMed] [Google Scholar]
- Wang DZ, Li S, Hockemeyer D, Sutherland L, Wang Z, Schratt G, Richardson JA, Nordheim A, Olson EN (2002) Potentiation of serum response factor activity by a family of myocardin-related transcription factors. Proc Natl Acad Sci USA 99: 14855–14860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei L, Zhou W, Croissant JD, Johansen FE, Prywes R, Balasubramanyam A, Schwartz RJ (1998) RhoA signaling via serum response factor plays an obligatory role in myogenic differentiation. J Biol Chem 273: 30287–30294 [DOI] [PubMed] [Google Scholar]
- Weinhold B, Schratt G, Arsenian S, Berger J, Kamino K, Schwarz H, Rüther U, Nordheim A (2000) Srf(−/−) ES cells display non-cell autonomous impairment in mesodermal differentiation. EMBO J 19: 5835–5844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitmarsh AJ, Shore P, Sharrocks AD, Davis RJ (1995) Integration of MAP kinase signal transduction pathways at the serum response element. Science 269: 403–407 [DOI] [PubMed] [Google Scholar]
- Wiebel FF, Rennekampff V, Vintersten K, Nordheim A (2002) Generation of mice carrying conditional knockout alleles for the transcription factor SRF. Genesis 32: 124–126 [DOI] [PubMed] [Google Scholar]
- Wilson BE, Mochon E, Boxer LM (1996) Induction of bcl-2 expression by phosphorylated CREB proteins during B-cell activation and rescue from apoptosis. Mol Cell Biol 16: 5546–5556 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1