

Abstract
Targeted gene disruption studies have established that the c-Jun NH2-terminal kinase (JNK) is required for the stress-induced release of mitochondrial cytochrome c and apoptosis, and that the Bax subfamily of Bcl-2-related proteins is essential for JNK-dependent apoptosis. However, the mechanism by which JNK regulates Bax has remained unsolved. Here we demonstrate that activated JNK promotes Bax translocation to mitochondria through phosphorylation of 14-3-3, a cytoplasmic anchor of Bax. Phosphorylation of 14-3-3 led to dissociation of Bax from this protein. Expression of phosphorylation-defective mutants of 14-3-3 blocked JNK-induced Bax translocation to mitochondria, cytochrome c release and apoptosis. Collectively, these results have revealed a key mechanism of Bax regulation in stress-induced apoptosis.
Keywords: apoptosis, Bax, JNK, phosphorylation, 14-3-3
Introduction
Apoptosis is essential for normal development and maintenance of tissue homeostasis. Disruption of the equilibrium between pro- and anti-apoptotic factors results in pathological conditions such as cancer, autoimmune disease and neurodegenerative disorders (Krammer, 2000; Yuan and Yankner, 2000). The loss of mitochondrial membrane integrity and the consequent release of cytochrome c into the cytosol are important events during apoptosis and are regulated by the Bcl-2 family of proteins (Wang, 2001). As the members of the Bcl-2 family exert their actions mostly at the level of mitochondria and reside upstream of the onset of irreversible cellular damage, they play a pivotal role in determining whether a cell will live or die (Gross et al, 1999; Tsujimoto and Shimizu, 2000). All Bcl-2 family members possess at least one of four conserved motifs known as Bcl-2 homology domains (BH1–BH4). Most anti-apoptotic members, including Bcl-2 and Bcl-xL, contain all the four domains. Pro-apoptotic members such as Bax and Bak lack the BH4 domain, whereas other pro-apoptotic members, the so-called BH3 domain-only proteins that include Bid, Bim and Bad, contain only the BH3 domain (Gross et al, 1999; Tsujimoto and Shimizu, 2000).
Recently, it has been demonstrated that Bax plays an essential role in inducing apoptosis in response to stress stimuli, as revealed by gene disruption of Bax and of Bax and Bak (Knudson et al, 1995; Lindsten et al, 2000; Wei et al, 2001; Zong et al, 2001). Bax is localized mostly in the cytoplasm, but redistributes to mitochondria in response to stress stimuli (Hsu et al, 1997; Wolter et al, 1997). After translocation to mitochondria, Bax induces cytochrome c release either by forming a pore by oligomerization in the outer mitochondrial membrane, or by opening other channels (Shimizu et al, 1999; Saito et al, 2000; Kuwana et al, 2002). The mechanisms underlying Bax translocation, however, are not fully understood.
Recent studies have shown that several proteins including 14-3-3 proteins prevent apoptosis through sequestration of Bax (Samuel et al, 2001; Guo et al, 2003; Nomura et al, 2003; Sawada et al, 2003). 14-3-3 proteins, which include seven isoforms in humans, are also implicated in antagonizing apoptotic signals through association with other pro-apoptotic proteins such as Bad, FKHRL1, ASK1 and Nur77 (Zha et al, 1996; Datta et al, 1997; Brunet et al, 1999; Zhang et al, 1999; Masuyama et al, 2001; van Hemert et al, 2001; Yaffe, 2002). Most 14-3-3 target proteins require phosphorylation to interact with 14-3-3, and consensus phosphoserine containing 14-3-3 binding motifs (RSXpSXP and RXXXpSXP) have been defined (Muslin et al, 1996; Yaffe et al, 1997). On the other hand, there are several examples of proteins containing dramatic variations from these motifs, including some that do not even require phosphorylation for binding, such as exoenzyme S, the platelet glycoprotein IB-IX-V complex, the plasma membrane H+-ATPase AHA2 and Drosophila PAR-1 (Du et al, 1996; Masters et al, 1999; Svennelid et al, 1999; Benton et al, 2002). Bax also interacts with 14-3-3 proteins in a phosphorylation-independent manner (Nomura et al, 2003). A substantial proportion of Bax molecules is bound to 14-3-3 proteins in the cytosol of healthy cells; in response to stress stimuli, however, Bax dissociates from 14-3-3 and redistributes to mitochondria (Nomura et al, 2003). Moreover, caspases activated by stress stimuli cleave 14-3-3θ within its COOH-terminal region and promote its dissociation from Bax (Nomura et al, 2003). These observations are consistent with previous results showing that disruption of the 14-3-3σ gene promotes Bax translocation to mitochondria in response to cellular stresses (Samuel et al, 2001). However, the translocation of Bax to mitochondria occurs independently of caspase activation in a number of systems (Putcha et al, 1999; Gilmore et al, 2000; Tsuruta et al, 2002), suggesting the existence of another mechanism responsible for the dissociation of Bax from 14-3-3 proteins.
The c-Jun N-terminal kinase (JNK) represents a group of mitogen-activated protein kinases (MAPKs), which is activated when cells are exposed to environmental stresses (Davis, 2000). In all, 10 members of the JNK family are generated by the alternative splicing of transcripts derived from the JNK1, JNK2 and JNK3 genes. The studies of JNK gene disruption in mice have confirmed that JNK contributes to stress responses (Davis, 2000). JNK3 is essential for apoptosis of hippocampal neurons following exposure to excitotoxic stresses (Yang et al, 1997). JNK1 and JNK2 are required for apoptosis of thymocytes in response to ligation of the T-cell receptor and of neurons in the developing hindbrain (Kuan et al, 1999; Sabapathy et al, 2001). Furthermore, JNK1 and JNK2 double-knockout cells are resistant to apoptosis induced by UV, anisomycin or DNA damage (Tournier et al, 2000). Moreover, recent studies have shown that Bax and Bak are required for JNK-induced apoptosis, and that Bax remains inactive upon exposure of JNK-deficient fibroblasts to environmental stress (Lei et al, 2002). How then does JNK activate Bax?
In this study, we show that 14-3-3ζ and 14-3-3σ are direct targets of JNK and that phosphorylation of 14-3-3 proteins by JNK results in dissociation of Bax from 14-3-3 proteins, leading to apoptosis. This novel function of JNK may provide the missing link between the stress-activated kinase cascade and Bax translocation to mitochondria, a critical step in the regulation of apoptosis.
Results
Active JNK promotes Bax translocation to the mitochondria
We first examined whether JNK activation is sufficient to promote Bax translocation to the mitochondria in COS-1 cells, since Bax has been reported to be required for JNK-induced apoptosis in mouse embryonic fibroblasts (Lei et al, 2002). To this end, we utilized a green fluorescent protein (GFP)–Bax fusion construct to monitor Bax localization in real time (Tsuruta et al, 2002). In all the experiments with GFP–Bax, p35, a pan-caspase inhibitor, was co-transfected to block caspase activation induced by overexpression of GFP–Bax. As a constitutively active form of JNK, we used a fusion protein (MKK7–JNK) in which MKK7α1 and JNK1β1 are connected by the Flag epitope tag (Figure 1A); in this construct, which is similar to the one used by Lei et al (2002), MKK7 phosphorylates and activates JNK1 intramolecularly (K Yoshioka, unpublished data). We found that expression of MKK7–JNK (wild type, WT) promoted GFP–Bax translocation to the mitochondria (Figure 1B and C). In contrast, the fusion construct of MKK7α1 and a kinase negative JNK (MKK7–JNK (KN)) had no effect on GFP–Bax localization (Figure 1B and C), suggesting that JNK promotes Bax translocation by phosphorylation of some target(s). To determine if JNK activity triggers translocation of the endogenous Bax protein, the distribution of endogenous Bax was assessed by subcellular fractionation after transfection of COS-1 cells with the MKK7–JNK constructs. The amount of endogenous Bax detected in the mitochondrial fraction was increased, and that detected in the cytosolic fraction was decreased, by expression of MKK7–JNK(WT) but not by that of MKK7–JNK(KN). On the other hand, the abundance of the mitochondrial marker F0F1-ATPase subunit α (or that of the cytosolic marker α-tubulin) in the corresponding fraction was unaffected by the expression of either construct (Figure 1D). Although expression of MKK7–JNK(WT) resulted in the activation of caspase-3 in COS-1 cells (Supplementary Figure 1) (Lei et al, 2002), coexpression of p35, which prevented caspase-3 activation by MKK7–JNK(WT) (Supplementary Figure 1), failed to inhibit Bax translocation to the mitochondrial fraction (Figure 1D), suggesting that MKK7–JNK(WT) promoted the translocation of endogenous Bax to mitochondria independently of caspase activation.
Figure 1.
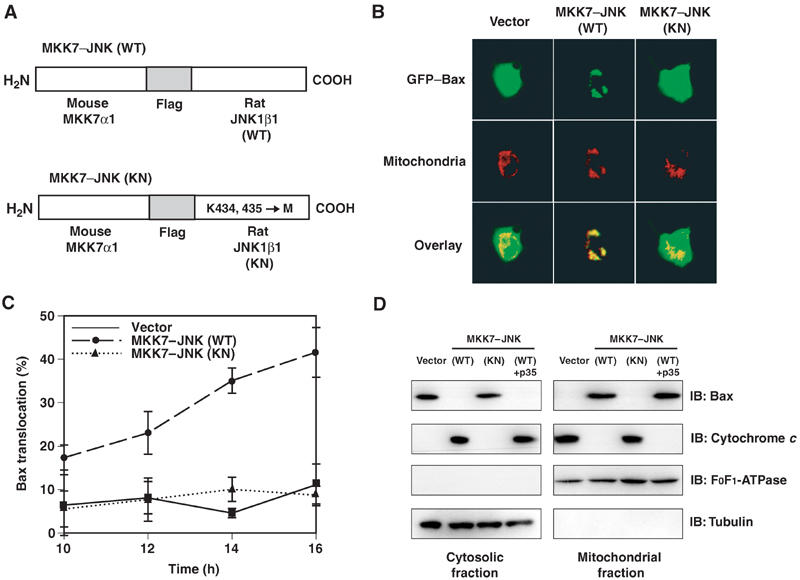
Expression of active JNK promotes Bax translocation to the mitochondria. (A) Schematic representation of the structure of the MKK7–JNK fusion proteins (WT or KN), which comprise mouse MKK7α1 linked to rat JNK1β1 by the Flag epitope sequence. The kinase-negative mutant of JNK1 was generated by replacement of Lys434 and Lys435 with methionines. (B) COS-1 cells were transfected for 13 h with expression vectors for GFP–Bax, the mitochondrial marker DsRed-Mito and the caspase inhibitor p35, together with a vector for MKK7-JNK (WT or KN) or the corresponding empty vector, as indicated. They were then examined for the distribution of GFP–Bax (green) and mitochondria (red) by fluorescence microscopy; the two separate images for each representative cell are also shown superimposed (overlay). (C) COS-1 cells were transfected for the indicated times as in (B), and the percentage of cells exhibiting GFP–Bax localization to mitochondria was determined. Data are means±s.d. of values obtained from five fields of 30–150 cells in each of three independent experiments. (D) COS-1 cells were transfected for 20 h with expression vectors for GFP and MKK7–JNK(WT or KN) in the absence or presence of a vector for p35, as indicated. The transfection efficiency was ∼75%, as determined by monitoring the expression of GFP. The cells were then subjected to subcellular fractionation, and the amount of endogenous Bax and that of cytochrome c in the mitochondrial and cytosolic fraction were assessed by immunoblot analysis (IB) with antibodies specific for these proteins. The amounts of the mitochondrial marker F0F1-ATPase and cytosolic marker α-tubulin were similarly assessed as an internal standard.
To determine whether JNK activity is required for stress-induced translocation of Bax to the mitochondria, we examined the effect of SP600125, a JNK inhibitor, on GFP–Bax redistribution induced by the protein synthesis inhibitor anisomycin. Inhibition of protein synthesis by anisomycin triggers apoptosis in a broad variety of cells. Treatment of COS-1 cells with anisomycin resulted in phosphorylation of the JNK substrate c-Jun, and pretreatment with SP600125 blocked this effect of anisomycin (Figure 2A). Anisomycin also induced a gradual redistribution of GFP–Bax to mitochondria, and this effect of anisomycin was inhibited by SP600125 (Figure 2B). We also measured Bax translocation to the mitochondria using subcellular fractionation and found that SP600125 inhibited the translocation of endogenous Bax to mitochondria in response to treatment with anisomycin (Supplementary Figure 2). The concentration of SP600125 required for the inhibition of Bax translocation was the same as that required for the inhibition of c-Jun phosphorylation (data not shown). To verify that the effect of SP600125 was specific for JNK, we also used the JNK-binding domain peptide (JBD) (Dickens et al, 1997) and a dominant-negative (DN) JNK to block JNK activity. The JBD blocked anisomycin-induced GFP–Bax translocation (Figure 2C) and expression of DN form of JNK blocked GFP–Bax translocation induced by staurosporine (Supplementary Figure 3), another agent known to induce apoptosis. Taken together, these results suggest that JNK activity is required for Bax translocation to the mitochondria induced by anisomycin or by staurosporine.
Figure 2.
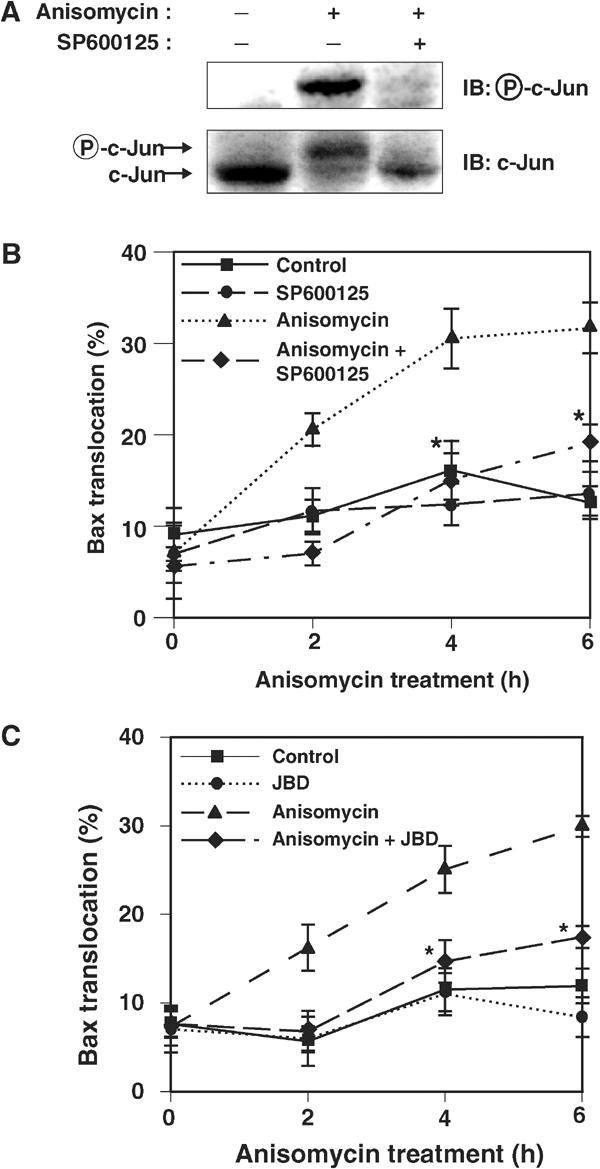
JNK is required for stress-induced translocation of Bax to the mitochondria. (A) COS-1 cells were incubated first for 30 min with or without 20 μM SP600125 and then for 1 h in the presence or absence of anisomycin (10 μg/ml). Cell lysates were then subjected to immunoblot analysis with antibodies to phosphorylated c-Jun or to c-Jun. The anisomycin-induced shift in the electrophoretic mobility of the band detected by the antibody to c-Jun reflects phosphorylation of c-Jun. (B) COS-1 cells were transfected for 11 h with expression vectors for GFP–Bax and p35, were pretreated with or without 20 μM SP600125 for 30 min, and were then incubated for the indicated times in the presence or absence of anisomycin (10 μg/ml). The percentage of cells in which GFP–Bax was localized to mitochondria was then determined. Data are means±s.d. of values obtained from five fields of 30–150 cells in each of three independent experiments (*P<0.0005 as compared with the anisomycin group). (C) COS-1 cells were transfected for 11 h with expression vectors for GFP–Bax and p35, together with a vector for JBD or the corresponding empty vector, as indicated. The cells were then incubated for the indicated times in the presence or absence of 10 μg/ml anisomycin, after which the percentage of cells exhibiting GFP–Bax localization to mitochondria was determined. Data are means±s.d. of values obtained from five fields of 30–150 cells in each of two independent experiments (*P<0.0005 as compared with the anisomycin group).
JNK can regulate Bax localization independently of c-Jun, Akt and Bim
We first tested the ability of JNK to phosphorylate Bax and found that Bax is not phosphorylated by JNK in an in vitro kinase assay (Supplementary Figure 4). We therefore hypothesized that there is another JNK target that regulates Bax localization in response to stress stimuli. Apoptosis induced by neurotrophic factor deprivation in sympathetic neurons is mediated by the JNK-catalyzed phosphorylation of c-Jun (Harris and Johnson, 2001; Putcha et al, 2001; Whitfield et al, 2001). We examined whether c-Jun phosphorylation is required for Bax translocation to the mitochondria. MKK7–JNK(WT) phosphorylated c-Jun and induced activation of c-Jun-dependent transcription, as measured by a luciferase reporter gene controlled by an AP-1-dependent promoter (which monitors the activity of the Jun–Fos complex). Activation of c-Jun-dependent transcription was blocked by coexpression of a DN form of c-Jun (Figure 3A). However, expression of DN c-Jun did not inhibit the translocation of GFP–Bax to mitochondria induced by MKK7–JNK(WT) (Figure 3B). The MKK7–JNK(WT)-induced increase in the amount of endogenous Bax in the mitochondrial fraction was also unaffected by DN c-Jun (Supplementary Figure 5). These results therefore suggest that JNK induces Bax translocation to mitochondria independently of the transcriptional activity of c-Jun.
Figure 3.
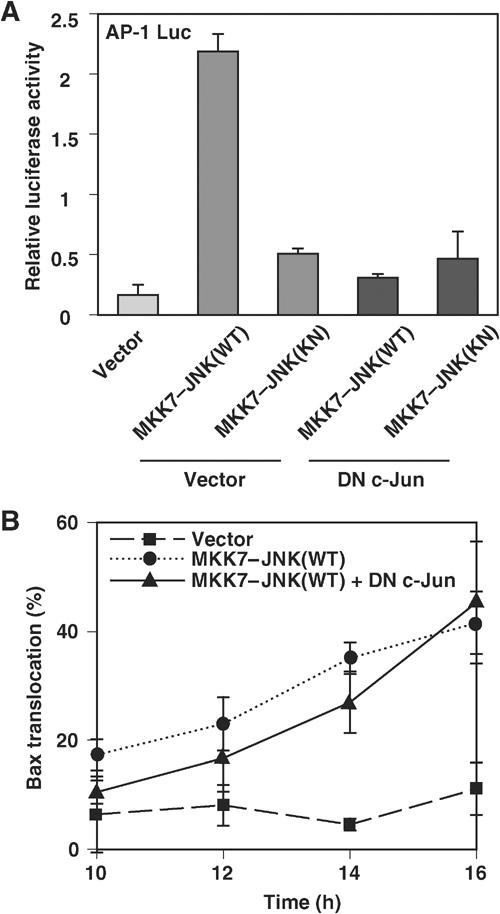
JNK induces Bax translocation to the mitochondria independently of c-Jun. (A) COS-1 cells were transfected for 1 day with an AP-1-luciferase reporter plasmid and expression vectors for MKK7–JNK(WT or KN) and a DN form of c-Jun or the corresponding empty vectors, as indicated. The normalized luciferase activity of cell lysates was then determined. Data represent the means±s.d. of triplicate determinations from three independent experiments. (B) COS-1 cells were transfected for the indicated times with expression vectors for GFP–Bax and p35 together with vectors for MKK7–JNK(WT or KN) and DN c-Jun, as indicated. The percentage of cells exhibiting GFP–Bax localization to mitochondria was then determined. Data are means±s.d. of values obtained from five fields of 30–150 cells in each of two independent experiments.
Since previous reports have shown that Bax translocation to the mitochondria in response to apoptotic stimuli is suppressed by the PI3K–Akt pathway (Yamaguchi and Wang, 2001; Tsuruta et al, 2002; Molton et al, 2003), we examined the involvement of the PI3K–Akt pathway in JNK-mediated Bax translocation. We found that expression of MKK7–JNK(WT) or of MKK7–JNK(KN) had no effect on the EGF-induced activation and phosphorylation of Akt at Ser-473 in COS-1 cells (Supplementary Figure 6). Moreover, expression of constitutively active or DN Akt constructs did not affect the level of JNK phosphorylation (Tsuruta et al, 2002) or the JNK-induced GFP–Bax translocation to the mitochondria (Supplementary Figure 7). These results suggest that JNK does not induce Bax translocation by inhibiting the PI3K–Akt pathway.
The Bcl-2 family member Bim can regulate Bax translocation and has been reported to be a target of JNK (Lei and Davis, 2003; Putcha et al, 2003). We therefore examined whether Bim is phosphorylated by JNK under the conditions used in this study and whether Bim phosphorylation correlates with Bax translocation to mitochondria. Bim phosphorylation was monitored by a shift in its electrophoretical mobility on SDS–PAGE as described (Lei and Davis, 2003; Putcha et al, 2003). Immunoblot analysis showed a mobility shift of Bim in response to anisomycin treatment. The JNK inhibitor SP600125, however, had little effect on the anisomycin-induced mobility shift of Bim, whereas it effectively blocked phosphorylation of c-Jun (Supplementary Figure 8). Importantly, expression of MKK7–JNK(WT) promoted Bax translocation to mitochondria, but did not induce the mobility shift of Bim (Supplementary Figure 8). These data suggest that a kinase or kinases other than JNK are activated by anisomycin and phosphorylate Bim, and that JNK induces Bax translocation independently of Bim phosphorylation at least in this system.
JNK phosphorylates 14-3-3ζ at Ser-184 and 14-3-3σ at Ser-186 in vitro
Under resting conditions, 14-3-3 proteins function as cytoplasmic anchors of Bax and prevent Bax from translocating to the mitochondria (Nomura et al, 2003). Consistent with this observation, targeted disruption of the 14-3-3σ gene was shown to accelerate Bax translocation to the mitochondria in human HCT116 cells (Samuel et al, 2001). These findings prompted us to investigate the possibility that JNK promotes Bax translocation to mitochondria by regulating the interaction between Bax and 14-3-3 proteins. Previous reports have shown that several isoforms of 14-3-3 are phosphorylated in vivo at a serine residue in the region between α-helices 7 and 8 (Ser-186 of 14-3-3β and Ser-184 of 14-3-3ζ), although the kinase or kinases responsible for this phosphorylation were not identified (Aitken et al, 1995). Interestingly, this residue conforms to the consensus sequence for JNK phosphorylation (Ser-Pro), and resides within the putative Bax-binding region (Figure 4A) (Nomura et al, 2003). We thus first examined whether JNK is capable of phosphorylating 14-3-3 proteins. Active JNK immunoprecipitated from anisomycin-treated COS-1 cell extracts efficiently phosphorylated recombinant His-tagged 14-3-3β, 14-3-3ζ and 14-3-3σ in an in vitro kinase assay (Figure 4B). JNK immunoprecipitates prepared from untreated cells did not phosphorylate recombinant 14-3-3 proteins, confirming that the kinase activity of JNK was responsible for this phosphorylation. Mutant proteins in which Ser-184 of 14-3-3ζ or Ser-186 of 14-3-3β or 14-3-3σ was replaced with alanine were not phosphorylated by the active JNK immunoprecipitated from anisomycin-treated cells (Figure 4B), indicating that these serine residues are the JNK-mediated phosphorylation sites in vitro.
Figure 4.
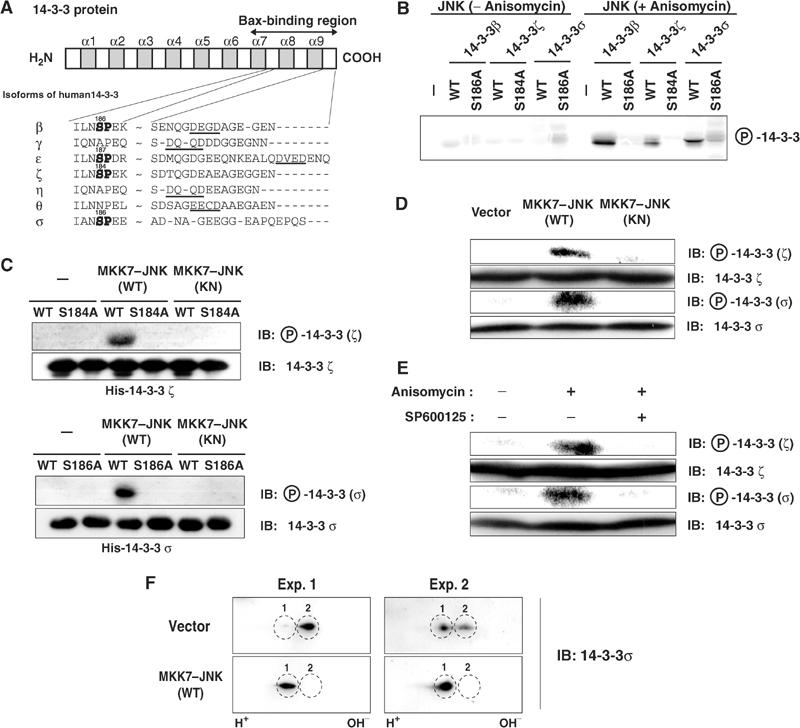
JNK phosphorylates 14-3-3ζ and 14-3-3σ both in vitro and in vivo. (A) Schematic representation of the structure of human 14-3-3 proteins and the amino-acid sequences surrounding putative JNK phosphorylation sites (SP, shown in bold) and caspase-3 cleavage sites ((D/E)XXD, underlined) in the Bax-binding region of the indicated isoforms. The numbers indicate the position of putative phosphorylation sites. (B) COS-1 cells were transfected for 24 h with an expression vector for Flag-tagged JNK and then incubated for 1 h in the absence or presence of anisomycin (10 μg/ml). Flag–JNK was immunoprecipitated from cell lysates with antibodies to Flag and incubated with the indicated recombinant His6-tagged 14-3-3 proteins (WT or Ser → Ala mutants) in the presence of [γ-32P]ATP. The amount of JNK immunoprecipitated from anisomycin-treated cell extracts was comparable to that from untreated cell extracts under the conditions used in this in vitro kinase assay (data not shown). Phosphorylation of 14-3-3 proteins was detected by electrophoresis and autoradiography. (C) Recombinant His6-tagged 14-3-3 proteins (WT or mutant) were incubated with or without recombinant MKK7–JNK(WT or KN) in the presence of ATP and were then subjected to immunoblot analysis with antibodies specific for 14-3-3ζ or 14-3-3ζ phosphorylated on Ser184 (upper panels) or for 14-3-3σ or 14-3-3σ phosphorylated on Ser186 (lower panels). (D) HCT116 cells were transfected for 1 day with an expression vector for MKK7–JNK(WT or KN) or the corresponding empty vector, after which cell lysates were subjected to immunoblot analysis with the antibodies described in (C). (E) HCT116 cells were incubated first for 30 min with or without 20 μM SP600125 and then for 3 h in the presence or absence of anisomycin (10 μg/ml). Cell lysates were then subjected to immunoblot analysis as described in (D). (F) HCT116 cells were transfected for 1 day with an expression vector for MKK7–JNK(WT) or the corresponding empty vector, after which cell lysates were subjected to two-dimensional gel electrophoresis and immunoblot analysis with anti-14-3-3σ antibody. Two typical results (Exp. 1 and Exp. 2) are shown. The ratio between Spot 1 and Spot 2 in control cells varied among experiments, but expression of MKK7–JNK (WT) repeatedly increased Spot 1 and abolished Spot 2.
To further indicate that Ser-184 of 14-3-3ζ and Ser-186 of 14-3-3σ are phosphorylated by JNK, we prepared antibodies to phosphopeptides corresponding to these phosphorylation sites. The anti-phospho-Ser184-14-3-3ζ and anti-phospho-Ser186-14-3-3σ antibodies recognized recombinant 14-3-3 proteins (14-3-3ζ and σ) that had been phosphorylated by recombinant MKK7–JNK(WT), but not those incubated with MKK7–JNK(KN) (Figure 4C). Moreover, these antibodies did not react with the Ser → Ala (SA) mutants of the corresponding 14-3-3 proteins, confirming that they specifically recognize phosphorylated 14-3-3 proteins at these serine residues.
JNK phosphorylates 14-3-3ζ at Ser-184 and 14-3-3σ at Ser-186 in vivo
To determine whether JNK also phosphorylates 14-3-3 in vivo, we examined the phosphorylation status of 14-3-3 proteins with the anti-phospho-14-3-3 antibodies. We used HCT116 cells in which ζ and σ isoforms of 14-3-3 are expressed endogenously. Expression of MKK7–JNK(WT), but not MKK7–JNK(KN), in HCT116 cells increased the bands reactive to the antibodies raised against phosphopeptides for 14-3-3ζ at Ser-184 and 14-3-3σ at Ser-186 (Figure 4D), suggesting that endogenous 14-3-3 proteins became phosphorylated upon JNK activation. We then addressed whether the phosphorylation of 14-3-3 is induced by cellular stresses. Exposure of HCT116 cells to anisomycin induced the phosphorylation of 14-3-3 proteins, revealed by the antibodies for 14-3-3ζ at Ser-184 and 14-3-3σ at Ser-186 (Figure 4E). The concentration of anisomycin required for phosphorylation of 14-3-3 was similar to that required for JNK activation (data not shown). Furthermore, anisomycin-induced phosphorylation of 14-3-3 proteins (14-3-3ζ and 14-3-3σ) was inhibited by pretreatment of cells with the JNK inhibitor SP600125 (Figure 4E), suggesting that JNK activation is required for anisomycin-induced 14-3-3 phosphorylation in vivo.
To further examine phosphorylation of 14-3-3 upon JNK activation in vivo, we carried out two-dimensional gel electrophoresis to separate hyper- and hypophosphorylated forms of 14-3-3. We detected two spots of 14-3-3σ in control (vector-transfected) HCT116 cells. Expression of MKK7–JNK(WT) (Figure 4F) or treatment with anisomycin (data not shown) resulted in a marked increase in the spot with greater negative charge (Spot 1) and a corresponding decrease in the other spot (Spot 2). We confirmed that Spot 1 is a phosphorylated form of Spot 2, as alkaline phosphatase treatment of MKK7–JNK(WT)-expressing cell extracts reduced Spot 1 and increased Spot 2 (data not shown). These results suggest that a high proportion of 14-3-3σ becomes phosphorylated upon JNK activation.
JNK promotes dissociation of Bax from 14-3-3 proteins
We next asked whether JNK-mediated phosphorylation of 14-3-3 affects the interaction between Bax and 14-3-3. Immunoprecipitation of 14-3-3σ from HCT116 cell lysates resulted in the co-precipitation of endogenous Bax (Figure 5A). Expression of MKK7–JNK(WT), but not that of MKK7–JNK(KN), resulted in a marked decrease in the amount of Bax that co-precipitated with 14-3-3σ. This effect of MKK7–JNK(WT) was not affected by coexpression of p35, suggesting that JNK promotes the dissociation of Bax from 14-3-3 independently of caspase activation.
Figure 5.
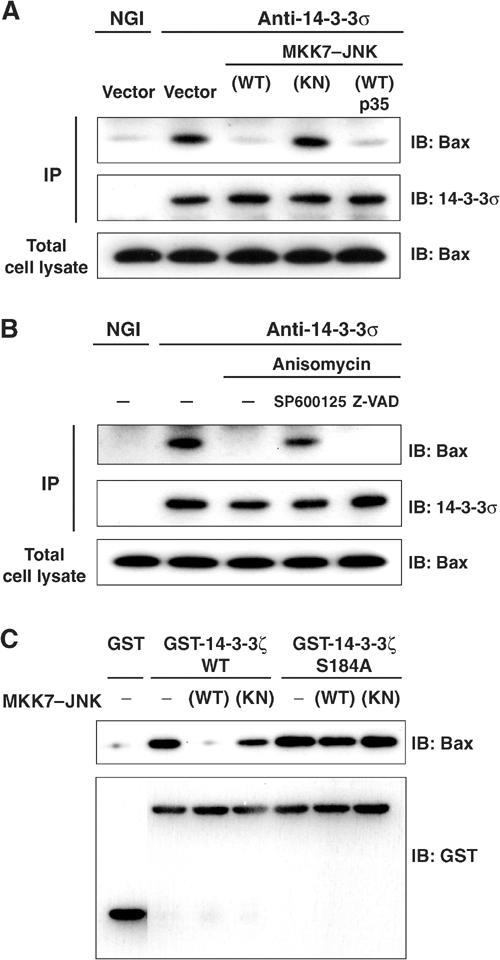
JNK promotes dissociation of Bax from 14-3-3 proteins. (A) HCT116 cells were transfected for 24 h with expression vectors for GFP, MKK7–JNK(WT or KN) and p35, as indicated. Cell lysates were then subjected either to immunoblot analysis with antibodies to Bax or to immunoprecipitation (IP) with antibodies to 14-3-3σ (or to normal goat IgG (NGI) precipitation); the resulting precipitates were subjected to immunoblot analysis with antibodies to 14-3-3σ and to Bax. (B) HCT116 cells were incubated first for 30 min with or without 20 μM SP600125 or 100 μM Z-VAD-CH2DCB and then for 3 h in the presence or absence of anisomycin (10 μg/ml). Cell lysates were then subjected to immunoprecipitation and immunoblot analysis as described in (A). (C) Equal amounts of recombinant GST–14-3-3ζ (WT or Ser184 → Ala mutant) or GST alone were incubated with or without recombinant MKK7–JNK(WT or KN) in the presence of ATP for 30 min, and the GST proteins were precipitated with glutathione-sepharose beads. The beads were then incubated with HeLa cell lysates, and subjected to immunoblot analysis with antibodies to Bax or to GST.
Exposure of HCT116 cells to anisomycin also reduced the amount of Bax that co-immunoprecipitated with 14-3-3σ (Figure 5B). Pretreatment of cells with the caspase inhibitor Z-VAD-CH2DCB had no effect on the anisomycin-induced dissociation of Bax from 14-3-3, whereas pretreatment with the JNK inhibitor SP600125 blocked this effect of anisomycin. These results thus suggest that JNK mediates the anisomycin-induced dissociation of Bax from 14-3-3.
To examine further whether JNK-induced dissociation of Bax from 14-3-3 is dependent on the phosphorylation status of 14-3-3, we performed glutathione S-transferase (GST) pull-down assays with a GST–14-3-3ζ fusion protein (Figure 5C). Incubation of GST–14-3-3ζ, but not that of GST alone, with HeLa cell lysates resulted in the co-precipitation of Bax by glutathione-Sepharose. However, prior phosphorylation of GST-14-3-3ζ by MKK7–JNK(WT) led to a marked decrease (approximately 90% reduction) in the amount of Bax that co-precipitated with GST-14-3-3ζ. The S184A mutant of GST-14-3-3ζ precipitated similar amounts of Bax, regardless of whether or not it had been preincubated with MKK7–JNK(WT). These results are thus consistent with the notion that JNK-mediated phosphorylation of 14-3-3ζ at Ser184 reduces its affinity for Bax.
14-3-3 mutants inhibit Bax translocation, cytochrome c release and cell death
If 14-3-3 is a major target for JNK in induction of Bax translocation, one might expect that expression of a phosphorylation site mutant of 14-3-3 would block JNK-induced Bax translocation. Indeed, expression of either 14-3-3ζ S184A or 14-3-3σ S186A mutant inhibited MKK7–JNK- and anisomycin-induced GFP–Bax translocation to mitochondria (Figure 6A and B and Supplementary Figure 9). In addition, when distribution of endogenous Bax was assessed by subcellular fractionation, expression of 14-3-3ζ S184A mutant inhibited the increase of Bax protein in the mitochondrial fraction induced by MKK7–JNK (Supplementary Figure 10). Importantly, in both assays, 14-3-3 WT was less effective than its SA mutant in suppression of JNK-induced Bax translocation (Figure 6A and B and Supplementary Figure 10), suggesting strongly that phosphorylation of 14-3-3 at this Ser residue is critical for Bax translocation to mitochondria upon JNK activation.
Figure 6.
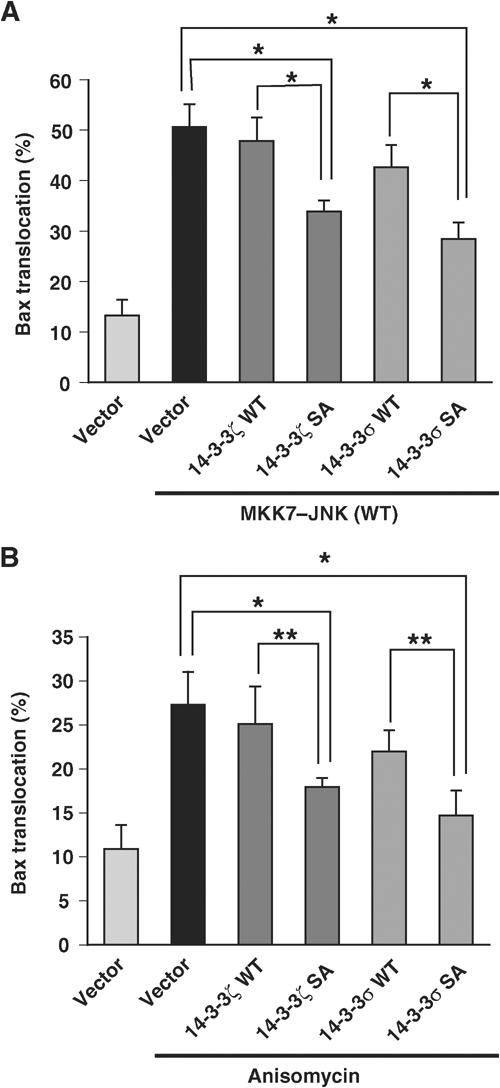
14-3-3 mutants inhibit JNK-induced Bax translocation to mitochondria. (A) COS-1 cells were transfected for 16 h with expression vectors for GFP–Bax and p35 together with those for MKK7–JNK(WT) and either 14-3-3ζ or 14-3-3σ. The percentage of cells exhibiting GFP–Bax localization to the mitochondria was then determined. Data are means±s.d. of values obtained from five fields of 30–150 cells in each of three independent experiments (*P<0.0005). (B) COS-1 cells were transfected for 11.5 h with expression vectors for GFP–Bax, p35 and either 14-3-3ζ or 14-3-3σ, and were incubated for 6 h in the presence or absence of anisomycin (10 μg/ml). The percentage of cells in which GFP–Bax was localized to the mitochondria was then determined and shown as in (A) (*P<0.0005 and **P<0.005).
We next examined whether 14-3-3 mutants inhibited JNK-induced cytochrome c release and cell death. The amount of endogenous cytochrome c detected in the cytosolic fraction was increased when MKK7–JNK(WT), but not MKK7–JNK(KN), was expressed in COS-1 cells (Figure 7A and data not shown). In contrast, co-expression of 14-3-3ζ S184A mutant, and that of the corresponding 14-3-3 WT to a lesser extent, inhibited MKK7–JNK (WT)-induced cytochrome c release, whereas the amount of the cytosolic marker α-tubulin in the cytosolic fraction was unaffected by MKK7–JNK or the 14-3-3 mutants (Figure 7A). Expression of MKK7–JNK (WT) in COS-1 cells induced chromatin condensation, an indication of cell death (Figure 7B). However, expression of 14-3-3σ SA mutant rendered the cells resistant to this effect of MKK7–JNK (WT) (Figure 7B). Moreover, 14-3-3σ WT was less effective in protecting cells from MKK7–JNK (WT)-induced cell death, compared to the 14-3-3σ SA mutant (Figure 7B). We next asked whether the 14-3-3σ mutant can inhibit anisomycin-induced cell death. Treatment of HeLa cells with anisomycin resulted in cell death, and pretreatment with SP600125 or coexpression of JBD or DN JNK partially blocked this effect (Figure 7C and D and Supplementary Figure 11), suggesting that JNK is required for anisomycin-induced cell death in HeLa cells. We found that expression of 14-3-3σ SA mutant blocked anisomycin-induced cell death, and 14-3-3σ WT was less effective compared to the 14-3-3σ SA mutant (Figure 7E), supporting the notion that phosphorylation of 14-3-3 by JNK plays an important role in JNK-mediated cell death.
Figure 7.
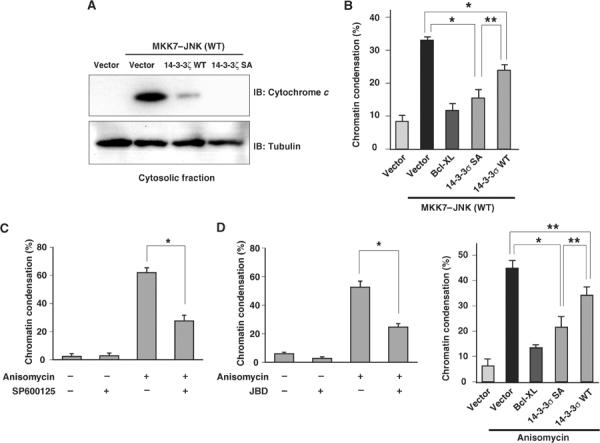
14-3-3 mutants inhibit JNK-induced cell death. (A) COS-1 cells were transfected for 20 h with expression vectors for GFP, MKK7–JNK(WT), and either 14-3-3ζ WT or 14-3-3ζ S184A, as indicated, and were then subjected to subcellular fractionation. The amounts of endogenous cytochrome c and α-tubulin (internal control) in the cytosolic fraction were determined by immunoblot analysis with specific antibodies. (B) COS-1 cells were transfected for 20 h with expression vectors for GFP, MKK7–JNK(WT) and either Bcl-XL, 14-3-3σ S186A or the corresponding WT proteins, as indicated. They were then stained with Hoechst 33342 (6.7 μg/ml) for 10 min, and the percentage of GFP-positive cells with pyknotic nuclei was determined. Data are means±s.d. of values obtained from three fields of 200–300 cells in each of three independent experiments (*P<0.0005 and **P<0.005). (C) HeLa cells were incubated first for 30 min with or without 20 μM SP600125 and then for 3 h in the presence or absence of anisomycin (10 μg/ml). They were then stained with Hoechst 33342 (6.7 μg/ml) for 10 min, and the percentage of cells with pyknotic nuclei was determined. Data are means±s.d. of values obtained from three fields of 100–200 cells in each of two independent experiments (*P<0.0005). (D) HeLa cells were transfected with expression vectors for GFP and JBD and then for 3 h in the presence or absence of anisomycin (10 μg/ml). They were then stained with Hoechst 33342 (6.7 μg/ml) for 10 min, and the percentage of GFP-positive cells with pyknotic nuclei was determined. Data are means±s.d. of values obtained from three fields of 100–150 cells (*P<0.0005). (E) HeLa cells were transfected with expression vectors for GFP and either Bcl-XL, 14-3-3σ S186A or the corresponding WT protein, and then for 3 h in the presence or absence of anisomycin (10 μg/ml). They were then stained with Hoechst 33342 (6.7 μg/ml) for 10 min, and the percentage of GFP-positive cells with pyknotic nuclei was determined and shown as in (C) (*P<0.0005 and **P<0.005).
Discussion
Recent studies have demonstrated that JNK plays a pivotal role in activation of the intrinsic apoptotic pathway that is mediated by mitochondria in response to cellular stress (Davis, 2000). Although Bax has been shown to be necessary for this action of JNK, the nature of the functional relation between these two proteins has been unclear. We now provide several lines of evidence that demonstrate that JNK-mediated phosphorylation of 14-3-3 induces the release of Bax from 14-3-3 and triggers its translocation to the mitochondria: (1) expression of an active form of JNK promoted Bax translocation to mitochondria; (2) inhibition of JNK (either by a chemical inhibitor or by a DN mutant) reduced the extent of Bax translocation to mitochondria in response to cellular stress; (3) JNK phosphorylated 14-3-3ζ at Ser-184 and 14-3-3σ at Ser-186 both in vitro and in vivo, and such phosphorylation reduced the affinity of 14-3-3 proteins for Bax; (4) expression of active JNK or treatment of cells with anisomycin induced the dissociation of Bax from 14-3-3 proteins; and (5) expression of phosphorylation site mutants of 14-3-3 proteins inhibited Bax translocation to mitochondria. These results strongly indicate that JNK regulates the activity of Bax by phosphorylating 14-3-3 proteins.
The initial studies of the role of JNK in apoptotic signals were performed by investigating neuronal cell death in response to neurotrophic factor withdrawal (Xia et al, 1995). The role for JNK in stress-induced neuronal cell death was confirmed by the studies of mice with a targeted disruption of the neuronal gene JNK3 (Yang et al, 1997). Although the JNK3 knockout mice are developmentally normal, they are resistant to excitotoxins. A similar resistance was observed in mice with a germ-line mutation in the c-Jun gene that replaced the JNK phosphorylation sites with alanine (Behrens et al, 1999). These data suggested that JNK mediates transcription-dependent apoptotic signals in neurons. In contrast, in non-neuronal cells, the JNK-mediated cytochrome c release and apoptosis induced by UV do not require de novo gene expression (Tournier et al, 2000). It is thus likely that relevant targets of JNK required for cytochrome c release and apoptosis are already present in these cells. Potential targets of JNK that may regulate cytochrome c release and apoptosis include members of the Bcl-2 family. The anti-apoptotic members Bcl-2, Bcl-xL and Mcl-1, and the pro-apoptotic members Bad, Bim and Bmf are phosphorylated by JNK, although the significance of these phosphorylation events remains unclear (Ito et al, 1997; Maundrell et al, 1997; Kharbanda et al, 2000; Donovan et al, 2002; Inoshita et al, 2002; Lei and Davis, 2003; Putcha et al, 2003). We have now shown that expression of phosphorylation site mutants of 14-3-3, and 14-3-3 WT to a lesser extent, suppressed JNK-induced Bax translocation, mitochondrial cytochrome c release and subsequent cell death. We confirmed that the 14-3-3 mutant did not hamper JNK-mediated phosphorylation of other targets such as c-Jun (F Tsuruta and Y Gotoh, unpublished results). These results strongly suggest that 14-3-3 protein is a major target of JNK in induction of these apoptotic events.
The JNK-mediated phosphorylation of 14-3-3β and 14-3-3ζ was found to occur at the previously identified phosphorylation sites (Aitken et al, 1995); 14-3-3β and 14-3-3ζ phosphorylated at these sites were named 14-3-3α and 14-3-3δ, respectively. It remains to be determined how such phosphorylation leads to the dissociation of Bax from 14-3-3. However, since the JNK phosphorylation sites reside within the domain responsible for the interaction of 14-3-3 with Bax (Nomura et al, 2003), it is likely that phosphorylation impairs the interaction at the binding interface. Alternatively, although the overall structure of 14-3-3 proteins is thought to be relatively rigid (Liu et al, 1995; Xiao et al, 1995), it is possible that phosphorylation might change the global conformation of 14-3-3 and thereby induce the dissociation of Bax.
On the Bax side, the 14-3-3-interacting domains are in the NH2- and COOH-terminal regions (Nomura et al, 2003). Interestingly, previous studies have proposed that these regions are masked when Bax is in the inactive form and are exposed upon activation. Indeed, antibodies against certain epitopes at the NH2-terminal region (cf. 6A7 monoclonal antibody) can react only with active Bax (Nechushtan et al, 1999; Lei et al, 2002). It is thus possible that 14-3-3 proteins physically conceal the NH2- and COOH-terminal domains of Bax and thereby maintain it in its inactive form, and that dissociation of Bax from 14-3-3 may be essential for its activation in addition to the conformational change of Bax. According to this scenario, our observation that phosphorylation of 14-3-3 by JNK releases Bax from 14-3-3 may explain how JNK activates Bax. Consistent with this notion, targeted disruption of JNK genes has been shown to prevent the activation of Bax in response to cellular stresses, as judged by the lack of reactivity toward the 6A7 antibody (Lei et al, 2002).
Another proposed mechanism of Bax activation is a conformational change due to acidification or alkalization of the cytoplasmic pH (Khaled et al, 1999; Nomura et al, 2003). However, we could not detect a significant change in the signal level of a pH indicator incorporated into the cytoplasm in the presence or absence of activated JNK in our preliminary experiments (F Tsuruta and Y Gotoh, unpublished results), suggesting that it is unlikely that JNK promotes Bax activation indirectly through regulation of cytoplasmic pH.
Whereas we have shown that three 14-3-3 isoforms (β, ζ and σ) serve as JNK targets, some isoforms (γ, η and θ) do not possess a corresponding JNK phosphorylation site. One of these isoforms (θ) has been reported to be cleaved by caspase, resulting in dissociation of Bax (Nomura et al, 2003). Our results indicate that caspase activity is dispensable for the JNK-dependent dissociation of Bax from 14-3-3, indicating the existence of two independent mechanisms to regulate their dissociation. Since caspase is activated downstream of JNK, it is possible that phosphorylation-dependent release of Bax from 14-3-3 takes place first, and the caspase-dependent release of Bax subsequently amplifies the apoptotic signal through positive feedback.
Recently, it has been reported that Ku70 and Humanin also prevent apoptosis through sequestration of Bax (Guo et al, 2003; Sawada et al, 2003). However, Sawada et al (2003) have shown that the absence of Ku70 is not sufficient for inducing the apoptotic level of Bax translocation to mitochondria in the absence of apoptotic stimuli. Guo et al (2003) have also demonstrated that elimination of endogenous Humanin does not induce Bax translocation to mitochondria and apoptosis in healthy cells. It is thus likely that several cytoplasmic anchors for Bax work in parallel to sequester Bax from mitochondria, and dissociation of Bax from all or some of these cytoplasmic anchors may be required for initiation of Bax translocation to mitochondria.
In conclusion, we have demonstrated that JNK promotes Bax translocation to mitochondria through phosphorylation of 14-3-3 proteins. This finding may account at least in part for the apoptosis-inducing activity of the JNK and shed light on the mechanism of stress-induced apoptosis.
Materials and methods
The Materials and methods used in this study are described in Supplementary data.
Supplementary Material
Supplemental data
Acknowledgments
We thank J Inagawa and K Ueda for technical supports with the two-dimensional gel electrophoresis, Drs R Dolmetsch and X Wang for critical reading of the manuscript, Drs M Miura, M Yaffe, H Fu, B Vogelstein and R Davis for providing reagents, and members of the Gotoh laboratory for helpful discussions and technical support.
References
- Aitken A, Howell S, Jones D, Madrazo J, Patel Y (1995) 14-3-3 alpha and delta are the phosphorylated forms of raf-activating 14-3-3 beta and zeta. In vivo stoichiometric phosphorylation in brain at a Ser-Pro-Glu-Lys MOTIF. J Biol Chem 270: 5706–5709 [DOI] [PubMed] [Google Scholar]
- Behrens A, Sibilia M, Wagner EF (1999) Amino-terminal phosphorylation of c-Jun regulates stress-induced apoptosis and cellular proliferation. Nat Genet 21: 326–329 [DOI] [PubMed] [Google Scholar]
- Benton R, Palacios IM, Johnston DS (2002) Drosophila 14-3-3/PAR-5 is an essential mediator of PAR-1 function in axis formation. Dev Cell 3: 659–671 [DOI] [PubMed] [Google Scholar]
- Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, Anderson MJ, Arden KC, Blenis J, Greenberg ME (1999) Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 96: 857–868 [DOI] [PubMed] [Google Scholar]
- Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, Greenberg ME (1997) Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell 91: 231–241 [DOI] [PubMed] [Google Scholar]
- Davis RJ (2000) Signal transduction by the JNK group of MAP kinases. Cell 103: 239–252 [DOI] [PubMed] [Google Scholar]
- Dickens M, Rogers JS, Cavanagh J, Raitano A, Xia Z, Halpern JR, Greenberg ME, Sawyers CL, Davis RJ (1997) A cytoplasmic inhibitor of the JNK signal transduction pathway. Science 277: 693–696 [DOI] [PubMed] [Google Scholar]
- Donovan N, Becker EB, Konishi Y, Bonni A (2002) JNK phosphorylation and activation of BAD couples the stress-activated signaling pathway to the cell death machinery. J Biol Chem 277: 40944–40949 [DOI] [PubMed] [Google Scholar]
- Du X, Fox JE, Pei S (1996) Identification of a binding sequence for the 14-3-3 protein within the cytoplasmic domain of the adhesion receptor, platelet glycoprotein Ib alpha. J Biol Chem 271: 7362–7367 [DOI] [PubMed] [Google Scholar]
- Gilmore AP, Metcalfe AD, Romer LH, Streuli CH (2000) Integrin-mediated survival signals regulate the apoptotic function of Bax through its conformation and subcellular localization. J Cell Biol 149: 431–446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross A, McDonnell JM, Korsmeyer SJ (1999) BCL-2 family members and the mitochondria in apoptosis. Genes Dev 13: 1899–1911 [DOI] [PubMed] [Google Scholar]
- Guo B, Zhai D, Cabezas E, Welsh K, Nouraini S, Satterthwait AC, Reed JC (2003) Humanin peptide suppresses apoptosis by interfering with Bax activation. Nature 423: 456–461 [DOI] [PubMed] [Google Scholar]
- Harris CA, Johnson EM Jr (2001) BH3-only Bcl-2 family members are coordinately regulated by the JNK pathway and require Bax to induce apoptosis in neurons. J Biol Chem 276: 37754–37760 [DOI] [PubMed] [Google Scholar]
- Hsu YT, Wolter KG, Youle RJ (1997) Cytosol-to-membrane redistribution of Bax and Bcl-X(L) during apoptosis. Proc Natl Acad Sci USA 94: 3668–3672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoshita S, Takeda K, Hatai T, Terada Y, Sano M, Hata J, Umezawa A, Ichijo H (2002) Phosphorylation and inactivation of myeloid cell leukemia 1 by JNK in response to oxidative stress. J Biol Chem 277: 43730–43734 [DOI] [PubMed] [Google Scholar]
- Ito T, Deng X, Carr B, May WS (1997) Bcl-2 phosphorylation required for anti-apoptosis function. J Biol Chem 272: 11671–11673 [DOI] [PubMed] [Google Scholar]
- Khaled AR, Kim K, Hofmeister R, Muegge K, Durum SK (1999) Withdrawal of IL-7 induces Bax translocation from cytosol to mitochondria through a rise in intracellular pH. Proc Natl Acad Sci USA 96: 14476–14481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kharbanda S, Saxena S, Yoshida K, Pandey P, Kaneki M, Wang Q, Cheng K, Chen YN, Campbell A, Sudha T, Yuan ZM, Narula J, Weichselbaum R, Nalin C, Kufe D (2000) Translocation of SAPK/JNK to mitochondria and interaction with Bcl-x(L) in response to DNA damage. J Biol Chem 275: 322–327 [DOI] [PubMed] [Google Scholar]
- Knudson CM, Tung KS, Tourtellotte WG, Brown GA, Korsmeyer SJ (1995) Bax-deficient mice with lymphoid hyperplasia and male germ cell death. Science 270: 96–99 [DOI] [PubMed] [Google Scholar]
- Krammer PH (2000) CD95's deadly mission in the immune system. Nature 407: 789–795 [DOI] [PubMed] [Google Scholar]
- Kuan CY, Yang DD, Samanta Roy DR, Davis RJ, Rakic P, Flavell RA (1999) The Jnk1 and Jnk2 protein kinases are required for regional specific apoptosis during early brain development. Neuron 22: 667–676 [DOI] [PubMed] [Google Scholar]
- Kuwana T, Mackey MR, Perkins G, Ellisman MH, Latterich M, Schneiter R, Green DR, Newmeyer DD (2002) Bid, Bax, and lipids cooperate to form supramolecular openings in the outer mitochondrial membrane. Cell 111: 331–342 [DOI] [PubMed] [Google Scholar]
- Lei K, Davis RJ (2003) JNK phosphorylation of Bim-related members of the Bcl2 family induces Bax-dependent apoptosis. Proc Natl Acad Sci USA 100: 2432–2437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei K, Nimnual A, Zong WX, Kennedy NJ, Flavell RA, Thompson CB, Bar-Sagi D, Davis RJ (2002) The Bax subfamily of Bcl2-related proteins is essential for apoptotic signal transduction by c-Jun NH(2)-terminal kinase. Mol Cell Biol 22: 4929–4942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsten T, Ross AJ, King A, Zong WX, Rathmell JC, Shiels HA, Ulrich E, Waymire KG, Mahar P, Frauwirth K, Chen Y, Wei M, Eng VM, Adelman DM, Simon MC, Ma A, Golden JA, Evan G, Korsmeyer SJ, MacGregor GR, Thompson CB (2000) The combined functions of proapoptotic Bcl-2 family members bak and bax are essential for normal development of multiple tissues. Mol Cell 6: 1389–1399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D, Bienkowska J, Petosa C, Collier RJ, Fu H, Liddington R (1995) Crystal structure of the zeta isoform of the 14-3-3 protein. Nature 376: 191–194 [DOI] [PubMed] [Google Scholar]
- Masters SC, Pederson KJ, Zhang L, Barbieri JT, Fu H (1999) Interaction of 14-3-3 with a nonphosphorylated protein ligand, exoenzyme S of Pseudomonas aeruginosa. Biochemistry 38: 5216–5221 [DOI] [PubMed] [Google Scholar]
- Masuyama N, Oishi K, Mori Y, Ueno T, Takahama Y, Gotoh Y (2001) Akt inhibits the orphan nuclear receptor Nur77 and T-cell apoptosis. J Biol Chem 276: 32799–32805 [DOI] [PubMed] [Google Scholar]
- Maundrell K, Antonsson B, Magnenat E, Camps M, Muda M, Chabert C, Gillieron C, Boschert U, Vial-Knecht E, Martinou JC, Arkinstall S (1997) Bcl-2 undergoes phosphorylation by c-Jun N-terminal kinase/stress-activated protein kinases in the presence of the constitutively active GTP-binding protein Rac1. J Biol Chem 272: 25238–25242 [DOI] [PubMed] [Google Scholar]
- Molton SA, Todd DE, Cook SJ (2003) Selective activation of the c-Jun N-terminal kinase (JNK) pathway fails to elicit Bax activation or apoptosis unless the phosphoinositide 3′-kinase (PI3K) pathway is inhibited. Oncogene 22: 4690–4701 [DOI] [PubMed] [Google Scholar]
- Muslin AJ, Tanner JW, Allen PM, Shaw AS (1996) Interaction of 14-3-3 with signaling proteins is mediated by the recognition of phosphoserine. Cell 84: 889–897 [DOI] [PubMed] [Google Scholar]
- Nechushtan A, Smith CL, Hsu YT, Youle RJ (1999) Conformation of the Bax C-terminus regulates subcellular location and cell death. EMBO J 18: 2330–2341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nomura M, Shimizu S, Sugiyama T, Narita M, Ito T, Matsuda H, Tsujimoto Y (2003) 14-3-3 interacts directly with and negatively regulates pro-apoptotic Bax. J Biol Chem 278: 2058–2065 [DOI] [PubMed] [Google Scholar]
- Putcha GV, Deshmukh M, Johnson EM Jr (1999) BAX translocation is a critical event in neuronal apoptosis: regulation by neuroprotectants, BCL-2, and caspases. J Neurosci 19: 7476–7485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putcha GV, Le S, Frank S, Besirli CG, Clark K, Chu B, Alix S, Youle RJ, LaMarche A, Maroney AC, Johnson EM Jr (2003) JNK-mediated BIM phosphorylation potentiates BAX-dependent apoptosis. Neuron 38: 899–914 [DOI] [PubMed] [Google Scholar]
- Putcha GV, Moulder KL, Golden JP, Bouillet P, Adams JA, Strasser A, Johnson EM (2001) Induction of BIM, a proapoptotic BH3-only BCL-2 family member, is critical for neuronal apoptosis. Neuron 29: 615–628 [DOI] [PubMed] [Google Scholar]
- Sabapathy K, Kallunki T, David JP, Graef I, Karin M, Wagner EF (2001) c-Jun NH2-terminal kinase (JNK)1 and JNK2 have similar and stage-dependent roles in regulating T cell apoptosis and proliferation. J Exp Med 193: 317–328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito M, Korsmeyer SJ, Schlesinger PH (2000) BAX-dependent transport of cytochrome c reconstituted in pure liposomes. Nat Cell Biol 2: 553–555 [DOI] [PubMed] [Google Scholar]
- Samuel T, Weber HO, Rauch P, Verdoodt B, Eppel JT, McShea A, Hermeking H, Funk JO (2001) The G2/M regulator 14-3-3sigma prevents apoptosis through sequestration of Bax. J Biol Chem 276: 45201–45206 [DOI] [PubMed] [Google Scholar]
- Sawada M, Sun W, Hayes P, Leskov K, Boothman DA, Matsuyama S (2003) Ku70 suppresses the apoptotic translocation of Bax to mitochondria. Nat Cell Biol 5: 320–329 [DOI] [PubMed] [Google Scholar]
- Shimizu S, Narita M, Tsujimoto Y (1999) Bcl-2 family proteins regulate the release of apoptogenic cytochrome c by the mitochondrial channel VDAC. Nature 399: 483–487 [DOI] [PubMed] [Google Scholar]
- Svennelid F, Olsson A, Piotrowski M, Rosenquist M, Ottman C, Larsson C, Oecking C, Sommarin M (1999) Phosphorylation of Thr-948 at the C terminus of the plasma membrane H(+)-ATPase creates a binding site for the regulatory 14-3-3 protein. Plant Cell 11: 2379–2391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tournier C, Hess P, Yang DD, Xu J, Turner TK, Nimnual A, Bar-Sagi D, Jones SN, Flavell RA, Davis RJ (2000) Requirement of JNK for stress-induced activation of the cytochrome c-mediated death pathway. Science 288: 870–874 [DOI] [PubMed] [Google Scholar]
- Tsujimoto Y, Shimizu S (2000) Bcl-2 family: life-or-death switch. FEBS Lett 466: 6–10 [DOI] [PubMed] [Google Scholar]
- Tsuruta F, Masuyama N, Gotoh Y (2002) The phosphatidylinositol 3-kinase (PI3K)-Akt pathway suppresses Bax translocation to mitochondria. J Biol Chem 277: 14040–14047 [DOI] [PubMed] [Google Scholar]
- van Hemert MJ, Steensma HY, van Heusden GP (2001) 14-3-3 proteins: key regulators of cell division, signalling and apoptosis. BioEssays 23: 936–946 [DOI] [PubMed] [Google Scholar]
- Wang X (2001) The expanding role of mitochondria in apoptosis. Genes Dev 15: 2922–2933 [PubMed] [Google Scholar]
- Wei MC, Zong WX, Cheng EH, Lindsten T, Panoutsakopoulou V, Ross AJ, Roth KA, MacGregor GR, Thompson CB, Korsmeyer SJ (2001) Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science 292: 727–730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitfield J, Neame SJ, Paquet L, Bernard O, Ham J (2001) Dominant-negative c-Jun promotes neuronal survival by reducing BIM expression and inhibiting mitochondrial cytochrome c release. Neuron 29: 629–643 [DOI] [PubMed] [Google Scholar]
- Wolter KG, Hsu YT, Smith CL, Nechushtan A, Xi XG, Youle RJ (1997) Movement of Bax from the cytosol to mitochondria during apoptosis. J Cell Biol 139: 1281–1292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia Z, Dickens M, Raingeaud J, Davis RJ, Greenberg ME (1995) Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science 270: 1326–1331 [DOI] [PubMed] [Google Scholar]
- Xiao B, Smerdon SJ, Jones DH, Dodson GG, Soneji Y, Aitken A, Gamblin SJ (1995) Structure of a 14-3-3 protein and implications for coordination of multiple signalling pathways. Nature 376: 188–191 [DOI] [PubMed] [Google Scholar]
- Yaffe MB (2002) How do 14-3-3 proteins work? Gatekeeper phosphorylation and the molecular anvil hypothesis. FEBS Lett 513: 53–57 [DOI] [PubMed] [Google Scholar]
- Yaffe MB, Rittinger K, Volinia S, Caron PR, Aitken A, Leffers H, Gamblin SJ, Smerdon SJ, Cantley LC (1997) The structural basis for 14-3-3:phosphopeptide binding specificity. Cell 91: 961–971 [DOI] [PubMed] [Google Scholar]
- Yamaguchi H, Wang HG (2001) The protein kinase PKB/Akt regulates cell survival and apoptosis by inhibiting Bax conformational change. Oncogene 20: 7779–7786 [DOI] [PubMed] [Google Scholar]
- Yang DD, Kuan CY, Whitmarsh AJ, Rincon M, Zheng TS, Davis RJ, Rakic P, Flavell RA (1997) Absence of excitotoxicity-induced apoptosis in the hippocampus of mice lacking the Jnk3 gene. Nature 389: 865–870 [DOI] [PubMed] [Google Scholar]
- Yuan J, Yankner BA (2000) Apoptosis in the nervous system. Nature 407: 802–809 [DOI] [PubMed] [Google Scholar]
- Zha J, Harada H, Yang E, Jockel J, Korsmeyer SJ (1996) Serine phosphorylation of death agonist BAD in response to survival factor results in binding to 14-3-3 not BCL-X(L). Cell 87: 619–628 [DOI] [PubMed] [Google Scholar]
- Zhang L, Chen J, Fu H (1999) Suppression of apoptosis signal-regulating kinase 1-induced cell death by 14-3-3 proteins. Proc Natl Acad Sci USA 96: 8511–8515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zong WX, Lindsten T, Ross AJ, MacGregor GR, Thompson CB (2001) BH3-only proteins that bind pro-survival Bcl-2 family members fail to induce apoptosis in the absence of Bax and Bak. Genes Dev 15: 1481–1486 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental data