

Abstract
Methods for site-specific modification of proteins are in high demand. Reactions that yield bioconjugates should be quantitative, site-specific, and versatile with respect to nature and size of the biological/chemical targets involved, require minimal modification of the target, display acceptable kinetics under physiological conditions, and be orthogonal to other labeling methods. Sortase-mediated transpeptidation reactions meet these criteria. Here we describe the expression and purification conditions for two orthogonal sortase A enzymes and provide the protocol that allows functionalization of any given protein at its C-terminus or for select proteins at an internal site. Sortase-mediated reactions take only a few minutes, but reaction times can be extended to increase yields.
INTRODUCTION
One of the goals of protein engineering is the installation of desirable features, template-encoded or otherwise, on proteins that naturally lack them. The ability to confer different functionalities onto a protein of interest enables a broad array of applications. Attachment of a fluorophore to a protein allows its use in live-cell microscopy, while generation of a fusion between an antibody and a payload of interest, such as a toxin or an antigen, can find use in therapeutics and vaccine development, respectively.
Several strategies based on genetic, chemical, enzymatic, or chemo-enzymatic methods equip proteins with functional groups. Genetic engineering is the method of choice when modification at a precise site is required. However, the effect of the genetically appended sequence on expression, folding, and function of the final product is difficult to predict. Some proteins are simply refractory to the construction of functional fusions by standard genetic means 1. The range of modifications that can be applied to a protein as a fusion product is limited in the first instance to those that are template-encoded. Chemical modifications of proteins are more versatile but lack precision, as they usually target exposed cysteine or lysine residues. Moreover, because the reactions often call for non-physiological reaction conditions (pH, reducing conditions, ionic composition), chemical damage of the target protein can occur. While enzymatic methods can overcome some of drawbacks and afford site-specific protein modification, they often require genetic installation of sizable catalytically active protein domains (such as the O6-alkylguanine-DNA alkyltransferases (SNAP or CLIP-base technology 2,3), haloalkane dehalogenases (halo-tag technology, 20–40 kDa, 4) onto the protein substrate; installation of the 15-amino acid BirA acceptor peptide 5, the use of which is limited to biotin and its synthetically demanding chemical derivatives; engineering of a 13-amino acid acceptor peptide for lipoic acid ligase 6, with the limitation that it primarily accepts lipid substrates and therefore mutant screens are required to incorporate new functionalities; the use of the formylglycine-generating enzyme that converts a cysteine residue within the context of a LCTPSR sequence (aldehyde tag) into formylglycine that can be used in oxime ligations 7,8; or exploiting the enzyme phosphopantetheinyl transferase to conjugate CoA-derived molecules to a specific 11-amino acid sequence 9. Sortase-mediated transpeptidation reactions are a versatile complement to these protein modification strategies 10,11,12,13 and predominantly rely on the use of modified peptides, readily accessible by solid phase peptide synthesis using commercially available building blocks. Using sortases, we achieve labeling with similar precision afforded by genetic fusions, and moreover provide ready access to protein derivatives structures that are unattainable genetically.
Sortases and mechanism of action
Gram-positive bacteria display proteins at their surface to enable them to acquire nutrients, evade host immunity, or adhere to sites of infection 14. Sortases comprise a family of membrane-associated transpeptidases that anchor those proteins to the cell wall 11,15. The different members of the sortase family can be divided into four subfamilies, based on their distinct primary sequence and substrates 16. While sortases A accept a large number of protein substrates, the sortases of B-D type have more specialized functions and therefore fewer substrates. In Staphylococcus aureus, proteins targeted to the bacterial surface display a conserved sortase recognition motif: Leu-Pro-Xxx-Thr-Gly (LPXTG, where X is any amino acid and Gly cannot be a free carboxylate), at or near their C-terminus. Upon recognition, sortase A cleaves between the threonine and glycine residues to form an acyl-enzyme intermediate. The active site cysteine of sortase A bonds with the carbonyl of the threonine residue of the target protein. This intermediate is then resolved by nucleophilic attack by the free amino group of the cell wall precursor lipid II. This lipid II-linked protein conjugate is incorporated during cell wall synthesis and consequently the protein is displayed at the surface 17 (Fig. 1).
Figure 1.

Schematic representation of how proteins are anchored to the cell wall via sortase in Gram-positive bacteria. See text for details. Adapted from 17.
Diversity of applications
Sortase-mediated reactions are applicable to any protein of interest, provided it comprises a LPXTG motif as the sortase target, or a suitably exposed Gly residue to serve as the incoming nucleophile. Both modifications (LPXTG, Gly) can be introduced using standard molecular cloning protocols. Also, sortases A are easily expressed in soluble recombinant form and in excellent yield in E.coli (see “Expression and production of sortases ”). The natural nucleophile, lipid II, can be replaced by any peptide with an oligoglycine (Gly1-5) at the N-terminus. In turn, the peptides can be decorated with any molecule accessible through chemical synthesis (e.g., fluorophores, biotin, cross-linkers, lipids, carbohydrates, nucleic acids) 1,10,18,19,20,21,22,23 provided a free N-terminal Gly remains available on the peptide used as incoming nucleophile. Thus, incubation of sortase, LPXTG-containing protein, and nucleophile leads to the covalent attachment of that nucleophile to the protein of interest, in a site-specific manner. Because the oligoglycine peptide that serves as the nucleophile is functionalized beforehand, the chemical reaction conditions used to incorporate the functional group inflict damage on neither sortase nor the protein substrate, as long as the modified oligoglycine peptide remains in solution once added to the sortase reaction. Proteins can also serve as nucleophiles, provided they display a suitably exposed (stretch of) glycine(s) at their N-terminus (Gly1-5). Such modification allows the proteins to be N-terminally labeled with functionalized peptides 24,25 or to form protein-protein adducts 1,26,27.
Relying on a common mechanistic principle, sortagging affords ready access to a wealth of site-specific modifications: C-terminal 10,19,20, internal loop regions 1,28, N-terminal 24,25, and formation of cyclized (poly)peptides 29,30,31. We have mainly used sortases A derived from Staphylococcus aureus and Streptococcus pyogenes. Versions of Staphylococcus aureus with improved Kcat values 32, as well as mutant versions that do not require Ca++ ions 33 have been reported and further extend the range of reaction conditions and applications. Streptococcus pyogenes sortase A accepts dialanine (poly)peptides as nucleophiles 34. The possibility of using two orthogonal sortases increases the versatility of these labeling reactions, as one can attach two different labels to one and the same molecule of choice 24.
Limitations
More than 50 different substrates including peptides 21,30, soluble proteins 10,20,28,29, membrane proteins displayed at the cell surface 10,35, M13 bacteriophage 36, budding influenza virus 37, antibodies 22,26,27, bacterial toxins 1,24, pre-assembled complexes 1,24 have yielded to sortase labeling. We have yet to encounter a protein that could not be labeled using sortase. One initial limitation of using sortase A from S. aureus was its obligate Ca2+ dependent activity 10. The presence of calcium in the reaction buffer precludes the use of phosphate-based buffers. Sortase A from S. pyogenes 34 or a mutant form of S. aureus (E105K/E108A33) are Ca2+ independent and thus circumvent this limitation.
Not every lab is equipped to perform peptide synthesis, and commercial vendors provide such services. To assist those interested in synthesizing their own peptides, we have included protocols that describe the synthesis of probes of general utility (biotin and fluorophores). It requires minimum specialized equipment (reaction vessels for peptide synthesis) and involves reactions readily executed in a laboratory outfitted for biochemical work (fume hoods, appropriate organic waste disposal, lyophilizer).
Advantages of the method
Site-specific.
Quantitative.
Versatile with respect to the moieties to be attached to the protein of interest (biotin, fluorophores, reporter peptides, sugars, lipids, etc).
Versatile regarding the labeling position on a protein: N-, C-, both N- and C-terminus, solvent-accessible loops.
Enable cyclization of proteins or peptides, if N- and C-termini are in close proximity.
Carried out under physiological conditions.
Require minimal modification of the target: 5 amino acid recognition sequence.
Orthogonal sortases are available.
Reaction can be performed using sortase A in solution or sortase A immobilized on a solid-phase support (described elsewhere in this issue of Nature Protocols [ref]).
Experimental design
The components of any sortagging reaction are: sortase, substrate, and nucleophile. Thus, we have divided this protocol in five sections: “Engineering substrates for sortagging”, “Expression and production of sortases”, “Peptide synthesis”, and “C-terminal sortagging reactions ”.
ENGINEERING SUBSTRATES FOR SORTAGGING
A) C-terminal labeling
Substrates equipped with the LPXTG recognition motif for S. aureus, or LPXTA for S. pyogenes can be engineered using standard molecular cloning protocols. Although any amino acid can precede the Thr residue, a glutamic acid is often used because it is commonly found in the natural sortase A substrates 38. The sortase recognition sequence is normally engineered at the C-terminus of the protein to be modified, with the G or A residue in amide linkage, followed by an affinity purification handle (e.g., His6) that is lost upon reaction (Fig. 2). The efficiency of the sortase-mediated reaction depends on the flexibility and accessibility of the region comprising the sortase A-recognition motif. Thus, if the C-terminus of the protein is known to be hidden (in the absence of a known structure, a failure to yield to sortase labeling would be a clear indication of lack of accessibility), we recommend engineering a flexible linker composed of (Gly4Ser)n preceding the LPETG/A sequence. The length of such linkers needs to be tested empirically for each protein of interest. Critical step. Identify the first amino acid of the protein substrate. Gly or Ala residues may cause cyclization as a competing side-reaction during labeling 29.
Figure 2. C-terminal labeling of proteins.
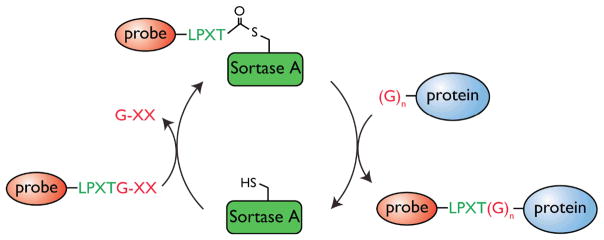
A protein modified at its C-terminus with the LPXTG sortase recognition motif followed by a handle (usually His6) is incubated with S. aureus Sortase A. Sortase cleaves the Thr-Gly bond and via its active site Cys residue forms an acyl intermediate with Thr in the protein. Addition of a peptide probe comprising of a series of N-terminal glycine residues and a functional moiety of choice resolves the intermediate, thus regenerating the active site cysteine on sortase and ligating the peptide probe to the C-terminus of the protein.
(B) Internal loop structure labeling
Site-specific modification of an internal solvent-exposed region in the protein of interest is a particular case of C-terminal labeling. As long as the LPXTG/A motif is introduced in an unstructured segment of the substrate protein, sortase can recognize it 28. An LPXTG motif that is highly structured is usually a poor sortase substrate 10,28. Flexibility can be ensured through installation of a specific protease cleavage site, immediately downstream of the sortase motif 1. Upon cleavage, the newly exposed C-terminus including the LPETG/A motif is likely to be unstructured. Accessibility to protease cleavage is a useful indicator for successful execution of a subsequent sortase reaction. Depending on the sequence of the protein, we rely on established site-directed mutagenesis strategies to engineer or to insert a LPXTG/A motif at the intended site. Since sortase cleaves the protein at the site of recognition, it is likely that the two halves of the protein will separate upon sortagging unless otherwise stabilized, for example through a disulfide bond 1 or for topological reasons 28. Thus, selecting an internal location for a sortase site in a loop region constrained by a disulfide bond might minimize the risk of such disintegration (Fig. 3). Critical step. Identifying the adequate protease to use has to be tested empirically to ensure that the protease does not cleave elsewhere within the protein. Trypsin and Factor Xa are examples of proteases used for this purpose 1.
Figure 3. Labeling of an internal loop of a protein.
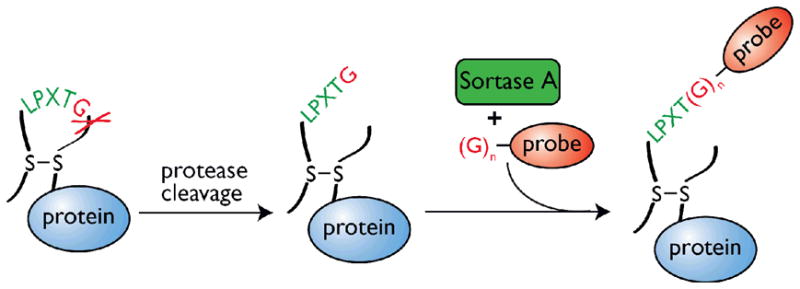
A protein comprising a loop formed by the establishment of a disulfide bond is modified to contain the sortase recognition motif (LPXTG) followed by a specific protease cleavage site. To increase flexibility of the LPXTG-containing region, the loop is nicked with the protease of choice and the sortase-mediated reaction follows as described for C-terminal labeling. Note however that as long as the region containing the LPXTG motif is flexible and accessible, a proteolytic event might not even be required. The presence of a disulfide bridge is also not critical for the reaction. While such a bond is convenient to ensure that the protein will not disintegrate upon labeling, at times the topology and conformation of the protein of interest naturally ensures integrity.
EXPRESSION AND PRODUCTION OF SORTASES
We commonly use three different sortases: the Ca2+ dependent sortase A from S. aureus, its mutant version [E105K/E108A 33] and sortase A from S. pyogenes; the latter two are Ca2+ independent. Because sortase is a membrane protein in Gram-positive bacteria, we use versions where the transmembrane domain has been eliminated and replaced with a hexahistidine purification tag. Two soluble versions exist for S. aureus wild-type sortase A, with either a N-terminal deletion of 25 amino acids 17 or 59 amino acids. The enzymatic activity of both versions is identical 39, but the molecular weight is different. This is a useful trait to explore in those cases where the molecular weights of the protein to be labeled and of the sortase to be used are similar. In addition, we equipped the Δ59 truncated version with a thrombin cleavage site that releases the His tag upon digestion. This not only facilitates further downstream purification but also increases the mobility of sortase in SDS-PAGE gels, allowing a clear-cut distinction between sortase and substrate if so required.
The following standard procedure for protein expression and purification yields approximately 40 mg L−1 S. aureus sortase A and 10 mg L−1 S. pyogenes sortase A.
MATERIALS
REAGENTS
pQE30 sortase A S. aureus (Δ25), available upon request 17
pET28a sortase A S. aureus (Δ59), available upon request
pET28a sortase A S. aureus (Δ59, E105K, E108A, Ca2+ independent), available upon request
pET28a sortase A S. pyogenes (Δ81), available upon request
E. coli BL21(DE3) (Invitrogen C6000-03)
Kanamycin (Fisher Scientific, cat. no. BP906-5)
Ampicillin (Sigma, cat. no. A0166)
Luria-Broth (LB) medium (Gentaur, cat. no. SD7002(S518)) and LB plates (BD, cat. no. 244510) (ampicillin 100 μg ml−1 for the pQE30 vector or kanamycin 30 μg ml−1 for the pET28-derived constructs)
Isopropyl-β-D-thiogalactopyranoside (Chem-impex international, cat. no. 00194)
Nickel-nitrilotriacetic acid (Ni-NTA)-agarose resin (Qiagen, cat. no. 1018240)
Lysozyme (Sigma, cat. no. L6876)
DNAse I (Roche, cat no. 10 104 159 001)
Imidazole (Alfa Aesar, cat. no. A10221)
Common reagents for SDS-PAGE analysis
Brilliant blue R (Sigma-Aldrich, cat. no. B7920)
Methanol, MeOH (EMD, cat. no. MX0488-1)
Acetic acid, AcOH (VWR, cat. no. BDH3094)
Hydrochloric acid, HCl (EMD, cat. no. HX0603-4) ! Caution
Ethanol (Pharmco-AAPER, cat. no. 111000190)
BCA-protein reagent assay kit (Pierce cat. no. 23227)
EQUIPMENT
Water bath at 42°C
Bacterial shaker
Floor centrifuge capable of spinning 1 L bacterial cultures at 6,000 g and spinning 50-ml tubes at 20,000 g
Motorized mechanical pipette
French Press for lysis of bacterial cells
Poly-prep chromatography column (Bio-Rad)
FPLC (Äkta system or similar; GE Healthcare)
HiLoad 16/60 Superdex 75 prep grade size exclusion chromatography column
Amicon ultra concentrators 10 kDa NMWL (Millipore)
50 mL polypropylene conical tubes (Corning or similar)
REAGENT SETUP
LB media. Autoclave. Store at 4°C. Add the desired antibiotic just before using the media.
LB media agar plates. Prepare according to instructions. Autoclave. Let the media cool-down and add the desired antibiotic. Pour into plates. Store at 4°C.
Kanamycin, 30 mg mL−1 in water and filter sterilize. CRITICAL: store at −20 °C (1,000× stock).
Ampicillin, sodium salt 100 mg mL−1 in water and filter sterilize. CRITICAL: store at −20 °C (1,000× stock).
Isopropyl-β-D-thiogalactopyranoside, 0.5 M in water. CRITICAL: store at −20 °C (1,000× stock).
1 M Imidazole in nickel-binding buffer, adjusted with HCl to pH 7.8. Dilute the imidazole stock solution in nickel-binding buffer to make the lysis, wash, and elution buffer as indicated. Store at 4 °C.
Lysozyme. CRITICAL: Store powder at −20 °C and dissolve at 10 mg mL−1 in water just before use (1,000× stock).
DNAse. CRITICAL: Store powder at 4 °C and dissolve at 10 mg mL−1 in water just before use (1,000× stock).
Nickel-binding buffer: 50 mM Tris·Cl, pH 7.5, 150 mM NaCl. Store at 4 °C.
Lysis Buffer: 50 mM Tris-Cl, pH 7.5, 150 mM NaCl, 5 mM MgCl2, 10 mM imidazole, 10% glycerol, 1 mg mL−1 DNase, 1 mg mL−1 lysozyme. Store at 4°C.
Wash Buffer: 50 mM Tris-Cl, pH 7.5, 150 mM NaCl, 5 mM MgCl2, 10 mM imidazole, 10% glycerol. Store at 4°C.
Elution Buffer: 50 mM Tris-Cl, pH 7.5, 150 mM NaCl, 500 mM imidazole, 10% glycerol. Store at 4 °C.
Coomassie blue staining: Dissolve 1.25 g Brilliant Blue R in a mixture of methanol (200 mL), water (250 mL) and acetic acid (50 mL). Store in a dark container at RT.
Destaining solution: Mix water, ethanol and acetic acid in a ratio of 6:3:1. Store at RT.
PROCEDURE
(A) Transform plasmid, amplify bacteria, and harvest bacteria TIMING 3 d
Transform competent BL21(DE3) E.coli with pET28a S. aureus sortase A (Δ59), pET28a Ca2+ independent S. aureus sortase A (Δ59), or pET28a S. pyogenes sortase A onto LB-kanamycin plates or E.coli with pQE30 S. aureus sortase A (Δ25)onto LB-ampicillin plates and incubate at 37 °C overnight.
Start overnight culture: inoculate an individual colony from the plate into 100 mL LB-ampicillin or LB-kanamycin media (depending on the construct) and shake at 220 rpm at 37 °C overnight.
Start expression culture: dilute 10 mL of the overnight culture into 1 L LB media with the corresponding antibiotic. Shake at 37°C until A600 is 0.4–0.6 (~3 h). Reserve a 100 μL sample of the culture as the preinduction control for testing sortase expression.
Induce protein expression: add Isopropyl-β-D-thiogalactopyranoside to a final concentration of 0.5 mM and shake at 25°C for 16 h.
-
Harvest bacteria: Reserve a 100 μL sample of the culture as the postinduction control for testing sortase expression. Centrifuge the culture at 6,000 g for 15 min at 4°C. Decant supernatant. Resuspend the pellet in 50 mL nickel-binding buffer, transfer to a 50 mL centrifuge tube using a motorized pipette and centrifuge again.
PAUSE POINT: The pellet can be frozen at −80°C and processed when convenient. We advise analyzing the pre- and post-induction samples by SDS-PAGE before proceeding to the purification step.
(B) Preparation of sortases TIMING 1–2d
Lyse the bacteria in 25 mL ice-cold lysis buffer and transfer to the chamber of a pre-chilled French press. Lyse the cells under a pressure of 1000 psi. Allow the bacterial lysate to cool on ice for 2 min and repeat the process. Clarify the cell lysate at 20,000 g for 30 min at 4 °C. Keep the supernatant on ice.
Pack a Poly-prep column with 1–1.5 mL (bed volume) nickel-nitrilotriacetic acid (Ni-NTA) agarose resin and wash with 10 column volumes of wash buffer (by gravity flow). CRITICAL STEP: Do not allow the column to run dry.
Load the bacterial supernatant onto the column by gravity flow.
Wash the column with 50 column volumes of wash buffer to remove nonspecifically bound proteins.
Elute with 3 column volumes of ice-cold elution (high imidazole) buffer.
Analyze a sample of the eluate by SDS-PAGE.
The sortase preparation is best further purified by gel filtration. This affords removal of remaining contaminants and of the imidazole contained in the elution buffer. Pre-equilibrate a Hi-Load Superdex 75 16/60 column with 40 mL nickel-binding buffer (no imidazole), at a rate of 1 mL min−1.
Inject the eluted protein sample and maintain a flow rate of 1 mL min−1. Collect 1.3 mL fractions.
Based on the UV spectra, identify and collect the protein-containing fractions and analyze by SDS-PAGE. Pool the fractions that contain pure sortase and concentrate to 25 mg mL−1 using an Amicon ultra concentrator. The protein concentration is determined by BCA assay. An aliquot of the protein is analyzed by SDS-PAGE to check for purity.
-
Add 10% glycerol to the preparation and aliquot the enzyme. Snap-freeze and store at −80°C, where it is stable for a long time (months). Once thawed, sortase A can be stored at 4 °C for 1 month with no noticable loss of activity.
Note: We often detect the presence of contaminants in the preparation of the S. pyogenes sortase A. These contaminants do not affect the activity of the enzyme, but the enzyme preparation can be further purified by anion-exchange chromatography (Mono Q) using a linear gradient 0–40% NaCl over 75 mL in a Mono Q column (GE Healthcare) at a flow rate of 0.5 mL min−1. Under these conditions, the enzyme elutes around 15–20% NaCl. Mono Q Buffer A: 50 mM Tris·Cl, pH 8.0. Mono Q Buffer B: 50 mM Tris·Cl, pH 8.0, 1 M NaCl.
PEPTIDE SYNTHESIS
We here describe the manual synthesis of several peptides that can be used in reactions mediated by sortase A from S. aureus, and therefore contain glycine residues at the N-terminus. For reactions using sortase A from S. pyogenes an alanine based peptide is used as a nucleophile. Their synthesis follows the same protocol except for the use of Fmoc-Ala-OH in place of Fmoc-Gly-OH. Perform two or three repeated couplings of Fmoc-Ala-OH to obtain the di and tri-alanine sequence, respectively. Also, Fmoc-Lys(biotin)-OH, Fmoc-Lys(5-TAMRA)-OH, and other conjugates can be purchased as premade building blocks. These building blocks are more expensive, but reduce the time required for synthesis and ease purification of the desired final product.
MATERIALS
REAGENTS
N,N-Dimethylformamide (DMF; Applied Biosystems, cat. no. GEN002007) ! Caution Flammable/toxic
Acetonitrile (JT Baker Analytical, cat. no. 9017-03) ! Caution Flammable/toxic
N-methyl-2-pyrrolidone (NMP; Sigma Aldrich, cat. no. 328634-2L) ! Caution Flammable/irritant/toxic
Diisopropylethylamine (DIPEA; Fisher BioReagent, cat. no. BP592500) ! Caution Highly flammable/corrosive
Dichloromethane (DCM; VWR, cat. no. JT9305-3) ! Caution Carcinogen
Dimethylsulfoxide (DMSO; EMD Chemicals Inc, cat. no. MX1458-6) ! Caution Irritant/flammable
Piperidine (Sigma Aldrich, cat. no. 104094) ! Caution Flammable/corrosive
Pyridine (Sigma Aldrich, cat. no. 360570) ! Caution Highly flammable/toxic
Diethyl ether (EMD Chemicals Inc, cat. no. EX0185-8)! Caution Highly flammable
Trifluoroacetic acid (TFA; Sigma Aldrich, cat. no. T6508) ! Caution Strongly corrosive/toxic
Triisopropylsilane (TIS; Sigma Aldrich, cat. no. 233781) ! Caution Flammable
Ethanedithiol (Sigma Aldrich, cat. no. w348406) ! Caution Flammable and toxic
5(6)-carboxy-tetramethylrhodamine (TAMRA; Novabiochem, cat. no, 815030)
Biotin (Sigma Aldrich, cat. no. B4501)
Fmoc-Lysine(Mtt)-OH (EMD biosciences, cat. no. 04-12-1137)
Fmoc-Cys(Trt)-OH (Novabiochem, cat. no. 852008)
Fmoc-Gly3-OH (Chem-Impex international, cat. no. 08072)
Fmoc-Gly-OH (Novabiochem, cat. no. 852001)
Fmoc-Ala-OH (Novabiochem, cat. no. 852003)
Fmoc-Lys(biotin)-OH (Novabiochem, cat. no. 852097)
Fmoc-Lys(5-TAMRA)-OH (AAT Bioquest, cat. no. 5045)
2-(1H-Benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HBTU; Novabiochem, cat. no. 851006)! Caution Irritant/harmful
benzotriazol-1-yl-oxytripyrrolidinophosphonium hexafluorophosphate (PyBOP; Novabiochem, cat. no. 851009) ! Caution Irritant/harmful
Ninhydrin (Eastman, cat. no. 2495) ! Caution Harmful
Potassium cyanide (Sigma-Aldrich, cat. no. 31252) ! Caution Highly toxic
Phenol (J.T. Baker, cat. no.2858-04) ! Caution Toxic/corrosive
Ethanol (Pharmco-AAPER, cat. no. 111000190)
Rink amide resin SS, 100–200 mesh, 1% DVB (Advanced Chemtech, cat. no. SA5030)
tert-Butanol (Aldrich, cat. no. 36,053-8)
EQUIPMENT
3 mL syringe and filter frit (New England Peptide, AC0-003)
Screw cap glass peptide column with a frit filter bottom
Wrist Action shaker (St. John Associate Inc.)
Swing bucket centrifuge (Beckman)
HPLC system (Agilent 1100 series)
Reverse phase protein and peptide C18 column (Vydac, cat. no. 218TP1010)
Liquid Chromatography/Mass Spectrometry (LC/MS)
Nuclear Magnetic Resonance (NMR) spectrometer
Vacuum line
Lyophilizer
Microfuge tubes
Test tube racks
Glass pipettes and Pasteur pipets
Syringes
Graduated cylinders
Pipet bulbs
Heating block
Rotary evaporator (Buchi)
50 mL polypropylene conical tubes (Corning or equivalent)
REAGENT SETUP
Kaiser test solution A dissolve 500 mg of ninhydrin in 10 mL ethanol.
Kaiser test solution B dissolve 80 g of phenol in 20 mL of ethanol.
Kaiser test solution C dissolve 1.3 mg potassium cyanide (20 μmol) in 20 mL of water. Add 2 mL of the potassium cyanide solution to 100 mL of pyridine.
20% piperidine in NMP mix 20 mL piperidine and 80 mL NMP.
Kaiser Test TIMING 5 min
Monitor peptide couplings by performing a Kaiser test 40.
Mix 2 μL of solution A, 2 μL of solution B and 4 μL of solution C in a microcentrifuge tube.
Add 5–10 beads of the dried resin (obtained after the final wash step after the coupling reaction) to the mixture.
-
Heat the tube to 95°C for 3 min. A dark blue color indicates incomplete coupling.
Note: This test works on primary amines and does not work for testing the attachment of an amino acid to a Pro residue. Alternative methods such as the acetaldehyde/p-chloroanil test or microcleavage can be used to monitor these reactions 41,42.
Microcleavage Test TIMING 45 min
Prepare a 30 μL solution of 95% TFA and 5% H2O.
Add 5–10 beads to the solution and cleave for 20 min at room temperature.
Take 5 μL of the supernatant and dilute in 30 μL of H2O and analyze by mass spectrometry.
A) GGGK-Biotin and GGGK-TAMRA peptides
Resin Preparation TIMING 15 min
-
1
Add 100 μmol of Rink amide resin (167 mg) into a capped glass column with a fritted glass bottom, solvate the resin in DCM (7 mL) by shaking for 15 min in a wrist-action shaker at room temperature and remove the DCM by vacuum filtration.
Deprotection TIMING 30 min
-
2
Add 20% piperidine solution in NMP (7 mL) and shake for 15 min at room temperature to remove the resin’s Fmoc protecting groups.
-
3
Remove the piperidine solution by vacuum filtration and wash the resin three times with NMP (7ml), three times with DCM (7ml) and an additional time with NMP (7ml).
Coupling Reaction TIMING 2–3 h per coupling until pause point, 3.5 h per coupling cycle
-
4
Dissolve Fmoc-Lys(Mtt)-OH (188 mg, 300 μmol), HBTU (114 mg, 300 μmol), and DIPEA (104 μL, 600 μmol) in NMP (7 mL) and add to the resin. Shake the suspension for 2 h at room temperature.
-
5
Remove the reaction solution by vacuum filtration and wash the resin three times with NMP (7ml) and three times with DCM (7ml). Confirm the coupling reaction by performing a Kaiser test.
Note: If the reaction is incomplete, repeat steps 4–5 with half the amount of reagents used for a standard coupling and shake for 1 h at room temperature.
PAUSE POINT: Dry the resin under vacuum and store at 4 °C. CRITICAL STEP: Only store protected peptides.
-
6
Selectively remove the 4-methyl trityl (Mtt) protecting group with a solution of 97% DCM, 2% TIS and 1% TFA. Shake the peptide resin with the cleavage solution (5 mL) for 30 min. Remove the solution by vacuum filtration; add an additional 5 mL of cleavage solution and shake for 30 min to ensure complete removal of the Mtt. The side chain deprotected peptide remains attached to the resin.
Note: The cleaved Mtt produces an orange color. Eventually the color will fade from reaction with the TIS. If the color remains after the second 30 min reaction, perform a third 30 min deprotection.
-
7
Wash the resin three times with NMP (7ml), three times with DCM (7ml), and one additional brief wash with NMP (7ml) containing DIPEA (500 μmol, 5 equiv).
Note: This final wash step neutralizes the resin and improves the coupling efficiency in the next step. This wash step should be brief. Prolonged exposure to alkaline conditions may result in partial removal of the Fmoc-protecting group.
-
8
To synthesize the biotin-containing probe, make a solution of biotin (74 mg, 300 μmol), HBTU (114 mg, 300 μmol) and DIPEA (104 μL, 600 μmol) in NMP (7 mL). Add the solution to the resin and shake for 2 h at room temperature. The reaction product consists of the G3K-biotin peptide.
To synthesize the TAMRA-containing probe, prepare a solution of 5(6)-TAMRA (52 mg, 120 μmol), PyBOP (63 mg, 120 μmol), and DIPEA (42 μL, 240μmol) in NMP (7 mL). Add the solution to the resin and shake overnight at room temperature. To prevent photobleaching, avoid exposure to light where possible and wrap the column in aluminum foil.
-
9
Repeat step 5 to monitor biotin/TAMRA coupling.
-
10
Repeat steps 2–5, except use Fmoc-Gly3-OH (123 mg, 300 μmol) instead of the Lys residue.
Note: Three repeated couplings of Fmoc-Gly-OH can be used instead of Fmoc-Gly3-OH.
-
11
Remove the terminal Fmoc group as indicated in step 2–3.
Cleavage from Resin TIMING 3 h
-
12
Wash the residue an additional two times with DCM. Then suspend the resin in a solution consisting of 95% TFA, 2.5% H2O, and 2.5% TIS (5 mL) for 2 h at room temperature.
-
13
Elute the cleavage solution into 90 mL of ice cold (-20 °C) diethyl ether and rinse the resin with an additional 3 mL of the cleavage solution into the ether.
-
14
Store the ether solution at −20 °C for 20 min to precipitate the peptide. Centrifuge the suspension at 3,500 rpm for 15 min at 4 °C, decant the supernatant and gently evaporate the remaining ether under reduced pressure.
PAUSE POINT. The crude peptide can be stored as a solid at −20 °C.
HPLC purification
-
15
Dissolve the dried peptide in water (2 mL) and centrifuge at 14,000 rpm for 10 min.
Note: Up to 50% of tert-butanol may be added to peptides that do not readily dissolve in pure water.
-
16
Purify the centrifuged supernatant by reverse-phase HPLC on a C18 column using a 10–90% water-acetonitrile gradient with 0.1% TFA.
-
17
Analyze the fractions for the presence of the desired product by LC/MS and lyophilize the desired fractions to dryness.
Critical step. Verify the identity and purity by LC/MS analysis (linear gradient 5–45% acetonitrile in 10 min) and NMR spectroscopy. If LC/MS shows the crude peptide is of sufficient purity, this HPLC purification may be omitted and the peptide may be used directly in sortase reactions.
Note: TAMRA-containing probes consist of mixture of regio-isomers, which are easily separated by reverse phase HPLC.
PAUSE POINT. The lyophilized peptide can be stored at −20 °C indefinitely.
B) GGGK NHS Ester Probes
In-solution coupling of NHS esters to (partially) protected peptides provides a route to couple base/acid labile and/or more costly dyes (such as Alexa fluor 647) or other suitable precursors to a GGG-containing peptide.
Peptide synthesis
-
1
Follow steps 2–5 of the GGGK-biotin/TAMRA probe synthesis, but using Fmoc-Lys(Boc)-OH (141 mg, 300 μmol) instead of the Mtt protected Lys.
-
2
Follow steps 2–5 from the GGGK-biotin/TAMRA, substituting Fmoc-Gly3-OH (123 mg, 300 μmol) for the Lys residue.
-
3
Cleave and precipitate the peptide from the resin with the Fmoc group still on the terminal Gly as described in steps 12–15 for the GGGK-biotin/TAMRA probes.
Critical step. It is crucial to leave the Fmoc-protecting group on the peptide, as it prevents unwanted side reactions during the NHS ester coupling.
Critical step. Hydrophobic peptides may precipitate poorly. Dry the supernatant of the ether precipitation by rotary evaporation to increase the yield.
PAUSE POINT. Store the crude peptide at −20 °C.
-
4
Purify the peptide by reverse phase HPLC as indicated in steps 15–17 for the GGGK-biotin/TAMRA probes.
Note: If LC/MS shows the crude peptide is of sufficient purity, this HPLC purification may be omitted.
Note: tert-Butanol may be added to peptides that do not dissolve in pure H2O prior to HPLC purification.
NHS ester coupling
-
5
Dissolve the peptide in DMSO.
-
6
Add three to five equivalents of the Fmoc-GGGK peptide in DMSO to the NHS ester and add 5 equivalents of DIPEA.
-
7
Incubate the reaction for 16 h at room temperature.
Note: to prevent photobleaching or photoconversions, exclude the peptide from light by covering its containers in aluminum foil.
Critical step. It is important to leave the reaction for 16 h. Although the coupling of the NHS ester may be faster, the added DIPEA also facilitates (slow) removal of the Fmoc-protecting group.
-
8
Acidify the reaction mixture with 0.1% aqueous TFA and purify the peptide by reverse phase HPLC as described in steps 15–17 for the GGGK probes synthesis.
C) GGGC Maleimide-containing Probes
An alternative for the installation of chemical groups of interest onto a peptide is to use a cysteine-maleimide reaction. The procedure is similar to that of the GGGK NHS ester peptide, except the lysine is replaced by a cysteine and a maleimide-derived dye/probe is substituted for the NHS ester.
Note: Under the appropriate conditions, the maleimide will react with the cysteine exclusively and therefore fully deprotected peptides can be used.
Note: Prolonged storage of the probe in aqueous solution can result in hydrolysis of the maleimide. The resulting ring-opened product is still an excellent probe for the sortase reaction, but may cause difficulty during ion-exchange purification of the sortagged proteins. It is therefore crucial that probes containing maleimide-coupled functional groups are stored as lyophilized powders.
Peptide synthesis
-
1
Repeat steps 1–5 as described for the GGGK probes with Fmoc-Cys(Trt)-OH to load the resin.
-
2
Repeat steps 2–5 as described for the GGGK probes using Fmoc-Gly-Gly-Gly-OH.
-
3
Repeat steps 2–3 from the GGGK probes to remove the N-terminal Fmoc-protecting group.
-
4
Cleave the peptide from the resin by incubating the resin twice with 1 mL of a cleavage cocktail containing 93% trifluoracetic acid, 2.5% triisopropylsilane, 2% ethanedithiol and 2.5% water for 2 h.
Critical step. Addition of ethanedithiol prevents formation of disulfide bridges.
-
5
Precipitate and collect the peptide as described in steps 13–14 for the GGGK probes.
-
6
Remove the supernatant and dissolve the pellet in 2 mL of methanol.
-
7
Precipitate and collect the peptide as described in steps 13–15 for the GGGK probes. Critical step. Removal of residual of ethanedithiol is critical since even trace amounts can interfere in the maleimide-thiol reaction.
-
8
Analyze, purify (if needed) and lyophilize the crude H2N-GGGC-CONH2 as described in step 17 for GGGK probes.
PAUSE POINT. Store the lyophilized peptide at −20 °C until needed.
Maleimide-thiol coupling
-
9
Dissolve the peptide (2 equiv.) in PBS (0.25 mL) and add maleimide (1 equiv.) in DMF or DMSO (0.25 mL).
-
10
Incubate overnight at room temperature and subsequently quench the reaction with 0.1% aqueous TFA (2 mL).
-
11
Purify, analyze and lyophilize the product as described in steps 16–17 for GGGK probes.
Critical step. Verify the identity and purity by LC/MS analysis (linear gradient 5–45% acetonitrile in 10 min) and NMR spectroscopy.
PAUSE POINT. Store the purified product at −20 °C.
C-TERMINAL SORTAGGING REACTIONS
MATERIALS
REAGENTS
Purified target protein in buffer (no phosphate-based buffer if a Ca2+-dependent sortase A is used)
Purified sortase A in buffer (no phosphate-based buffer if a Ca2+-dependent sortase A is used)
Oligoglycine peptide (if using S. aureus sortase A) or dialanine or trialanine peptide (if using S. pyogenes sortase A) stock solution: 10mM in DMSO or water (10x stock). If an oligoglycine protein is used as the nucleophile, then dissolve it in buffer (no phosphate-based buffer if a Ca2+-dependent sortase A is used)
Nickel-nitrilotriacetic acid (Ni-NTA)-agarose resin (Qiagen, cat. no. 1018240)
Imidazole (Alfa Aesar, cat. no. A10221)
4x loading LDS-buffer (Invitrogen NP0008)
Common reagents for SDS-PAGE and for Coomassie staining (see “Expression and production of sortases” section)
EQUIPMENT
Micropipettes (5–1000 μL)
1.5 mL centrifuge tubes
Centrifuge for 1.5 mL centrifuge tubes
37 °C water bath
End-over-end shaker
Heating block
Desalting PD-10 columns (GE Healthcare, cat. no. 17-0851-01)
REAGENT SETUP
Sortase buffer: 500 mM Tris-HCl pH 7.5, 1.5 M NaCl, 100 mM CaCl2 (not required if using a Ca2+-independent sortase) (10x stock).
Nickel-binding buffer: 50 mM Tris-HCl pH 7.5, 0.5 M NaCl, 10 mM imidazole. Store at 4°C.
1 M Imidazole in nickel-binding buffer, adjusted with HCl to pH 7.8. Dilute the imidazole stock solution in nickel-binding buffer to make the lysis, wash, and elution buffer as indicated. Store at 4°C.
PROCEDURE
A) Setting up the reaction conditions
Mix a solution containing 10–50 μM target protein, 20–150 μM sortase, 1–2 mM oligoglycine probe in 1x sortase buffer (final concentrations). The controls to be included are: target protein only, sortase only, oligoglycine probe only, target protein and sortase, target protein and oligoglycine probe, sortase and oligoglycine probe.
-
Incubate the reactions at RT or at 37°C. Take 1 μL aliquots after 2, 4, 6, 8 and 16 h. Add 1x LDS-gel loading buffer to the aliquots to stop the reaction and boil 2 min.
PAUSE POINT: The aliquots can be frozen at −20 °C until further analysis.
Analyze the aliquots by SDS-PAGE followed by Coomassie staining.
Anticipated result
A successful sortase reaction often results in the formation of the acyl-enzyme intermediate if the oligoglycine probe is omitted. Small quantities of hydrolysis product (loss of (His)6 or epitope tag may occur upon cleavage of the sortase motif in the absence of added nucleophile. The acyl-enzyme intermediate usually survives reducing SDS-PAGE in detectable amounts. A full sortase reaction often yields a reaction product of mobility distinct from that of the input substrate and the hydrolysis product. The ability to distinguish the various intermediates critically depends on the molecular weight of the anticipated products and the gel systems used to analyze them.
(B) Purification
Overview: Because the protein substrate is constructed with a His6 handle downstream of the LPXTG/A motif (see section “Engineering substrates for sortagging”), the protein substrate molecules that were not labeled will retain the His6 tag upon reaction. The sortases are themselves tagged with His6. Thus, a convenient strategy to separate the labeled product from sortase and unreacted substrate is the use of affinity-chromatography (Ni-NTA beads), followed by HLPC or by a desalting column to remove the unreacted peptide nucleophile. Critical step. Check compatibility of the incorporated functionality with Ni-NTA purification. We have noted that some functional groups, including acylhydrazones, bind to the resin.
-
Pipette 500 μL of the Ni-NTA resin (50% slurry) into a microcentrifuge tube. Centrifuge at 280 g, 5 min, 4 °C. Discard the supernatant. Wash the resin three times using 1 mL of nickel-binding buffer.
CAUTION: Do not let the beads dry.
Dilute the sortase reaction with nickel-binding buffer to a final volume of 500 μL (We recommend taking an aliquot of 5 μL from the diluted reaction to later determine the efficiency of purification). Add to the Ni-NTA resin. Agitate on an end-over-end shaker for 30 min, 4°C.
Centrifuge at 280 g, 5 min, 4°C. Keep the supernatant.
Add 500 μL of nickel-binding buffer to the resin, agitate on an end-over-end shaker for 10 min, 4°C.
Centrifuge at 280 g, 5 min, 4°C. Keep the supernatant.
Combine the supernatants obtained in steps 3 and 5. Take an aliquot and analyze together with the aliquot taken in step 2 by SDS-PAGE.
If further purification is required to remove free peptide nucleophile, we recommend using a desalting column equilibrated in a buffer of choice, depending on the use of and compatibility with the labeled protein. Purification by gel filtration is an alternative. We use the same protocol described in the section “Expression and production of sortases”.
Anticipated results
The exact reaction conditions for protein labeling need to be determined empirically for each protein substrate. The range provided has yielded acceptable results in the majority of the cases. To achieve maximal levels of labeling, it is helpful to titrate the sortase, substrate, and probe concentrations relative to one another, to vary the time of labeling, and the temperature of reaction.
Troubleshooting
| Problem | Possible cause | Solution |
|---|---|---|
| No sortase expression | Wrong antibiotic selection | Confirm you used the right antibiotic |
| No induction | Make sure you are using BL21(DE3) E.coli bacteria Prepare a fresh solution of IPTG |
|
| No protein labeling | No acyl-intermediate is being formed | (see below) |
| Not enough nucleophile added to the reaction | Reaction conditions have to be determined ad-hoc. Increase the amount of nucleophile to 10 mM and/or decrease the amount of substrate | |
| pH of the reaction buffer not compatible with sortase activity | Ensure that the pH of the reaction buffer is neutral. Check the pH of the stock solutions. Probe solutions have the tendency to have a low pH due to residual traces of TFA. Multiple rounds of lyophilization and/or neutralization with buffer or aq. NaHCO3 solves this issue | |
| Proteolysis of sortase | Verify the integrity of sortase upon reaction by coomassie staining or anti-His blot. The amount of sortase before and after reaction should be equal. If not, consider the presence of a contaminating protease, which most probably co-purified with the protein of interest. We recommend further purification of the protein substrate. | |
| No acyl-intermediate is observed in the substrate-sortase control | The substrate is not correctly engineered | Confirm that the LPXTG/A motif is in frame and that no stop codons exist upstream of this sequence |
| Ensure that one or a few amino acids are present downstream of the Gly/Ala residues (Gly/Ala in LPXTG/A must be in amide linkage) | ||
| The LPXTG/A motif is not well exposed or it is designed in a conformationally constrained region of the protein | Extend the C-terminus of the protein introducing a linker (Gly4Ser)2 immediately upstream of the sortase motif | |
| Sortase is inactive | Test the preparation of sortase using GFP.LPETG/A.His6 as the substrate | |
| Not enough sortase added to the reaction | Sortase concentration has to be titrated for each substrate to be labeled. Increase the substrate concentration and/or decrease the amount of protein to be labeled | |
| Detection of a circular form of the protein when attempting labeling the C-terminus. The circular versions of such sortase substrates often show more rapid migration on SDS-PAGE than their linear counterparts. | The N- and C-termini of the protein are in close proximity and the N-terminal amino acid of the protein (Gly or Ala) acts as a nucleophile | Confirm that the first amino acid in the protein of interest is not a Gly or Ala. If it is, mutate it to Ser for example. If the first amino acid has to be a glycine or an alanine, design a thrombin cleavage site (LVPR) immediately upstream the first amino acid and proceed to digestion with thrombin upon sortase labeling, to expose the Gly or Ala residues. It is possible that a Lys residue close to the N-terminus can serve as the incoming nucleophile for circularization. Consider this possibility if replacement of N-terminal Gly or Ala fails to suppress circularization. |
| Detection of a white fluffy precipitate during the sortase-labeling reaction | Ca2+ precipitate | Do not use phosphate buffers |
| Protein is precipitating during the labeling reaction | High concentration of protein, specially when attempting protein-protein fusions | Optimize reaction temperature and incubation time so less protein may be used to achieve the same reaction yield without precipitation |
| The protein of interest precipitates at high temperatures | Perform the labeling reaction at RT and extend the reaction time | |
| No labeling when attempting modification at an internal site even when the site is nicked by a protease | The region is not flexible or accessible. | Extend the sequence of the protein by a few amino acids. The number of amino acids required must be determined empirically. |
References
- 1.Guimaraes CP, et al. Identification of host cell factors required for intoxication through use of modified cholera toxin. J Cell Biol. 2011;195:751–764. doi: 10.1083/jcb.201108103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Keppler A, et al. A general method for the covalent labeling of fusion proteins with small molecules in vivo. Nat Biotechnol. 2003;21:86–89. doi: 10.1038/nbt765. [DOI] [PubMed] [Google Scholar]
- 3.Gautier A, et al. An engineered protein tag for multiprotein labeling in living cells. Chem Biol. 2008;15:128–136. doi: 10.1016/j.chembiol.2008.01.007. [DOI] [PubMed] [Google Scholar]
- 4.Los GV, et al. HaloTag: a novel protein labeling technology for cell imaging and protein analysis. ACS Chem Biol. 2008;3:373–382. doi: 10.1021/cb800025k. [DOI] [PubMed] [Google Scholar]
- 5.Howarth M, Ting AY. Imaging proteins in live mammalian cells with biotin ligase and monovalent streptavidin. Nat Protoc. 2008;3:534–545. doi: 10.1038/nprot.2008.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cohen JD, Zou P, Ting AY. Site-specific protein modification using lipoic acid ligase and bis-aryl hydrazone formation. Chembiochem. 2012;13:888–894. doi: 10.1002/cbic.201100764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Carrico IS, Carlson BL, Bertozzi CR. Introducing genetically encoded aldehydes into proteins. Nat Chem Biol. 2007;3:321–322. doi: 10.1038/nchembio878. [DOI] [PubMed] [Google Scholar]
- 8.Rabuka D, Rush JS, deHart GW, Wu P, Bertozzi CR. Site-specific chemical protein conjugation using genetically encoded aldehyde tags. Nat Protoc. 2012;7:1052–1067. doi: 10.1038/nprot.2012.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yin J, Lin AJ, Golan DE, Walsh CT. Site-specific protein labeling by Sfp phosphopantetheinyl transferase. Nat Protoc. 2006;1:280–285. doi: 10.1038/nprot.2006.43. [DOI] [PubMed] [Google Scholar]
- 10.Popp MW, Antos JM, Grotenbreg GM, Spooner E, Ploegh HL. Sortagging: a versatile method for protein labeling. Nat Chem Biol. 2007;3:707–708. doi: 10.1038/nchembio.2007.31. [DOI] [PubMed] [Google Scholar]
- 11.Popp MW, Ploegh HL. Making and breaking peptide bonds: protein engineering using sortase. Angew Chem Int Ed Engl. 2011;50:5024–5032. doi: 10.1002/anie.201008267. [DOI] [PubMed] [Google Scholar]
- 12.Parthasarathy R, Subramanian S, Boder ET. Sortase A as a novel molecular “stapler” for sequence-specific protein conjugation. Bioconjug Chem. 2007;18:469–476. doi: 10.1021/bc060339w. [DOI] [PubMed] [Google Scholar]
- 13.Ton-That H, Mazmanian SK, Faull KF, Schneewind O. Anchoring of surface proteins to the cell wall of Staphylococcus aureus. Sortase catalyzed in vitro transpeptidation reaction using LPXTG peptide and NH(2)-Gly(3) substrates. J Biol Chem. 2000;275:9876–9881. doi: 10.1074/jbc.275.13.9876. [DOI] [PubMed] [Google Scholar]
- 14.Marraffini LA, Dedent AC, Schneewind O. Sortases and the art of anchoring proteins to the envelopes of gram-positive bacteria. Microbiol Mol Biol Rev. 2006;70:192–221. doi: 10.1128/MMBR.70.1.192-221.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mazmanian SK, Liu G, Ton-That H, Schneewind O. Staphylococcus aureus sortase, an enzyme that anchors surface proteins to the cell wall. Science. 1999;285:760–763. doi: 10.1126/science.285.5428.760. [DOI] [PubMed] [Google Scholar]
- 16.Spirig T, Weiner EM, Clubb RT. Sortase enzymes in Gram-positive bacteria. Mol Microbiol. 2011;82:1044–1059. doi: 10.1111/j.1365-2958.2011.07887.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ton-That H, Liu G, Mazmanian SK, Faull KF, Schneewind O. Purification and characterization of sortase, the transpeptidase that cleaves surface proteins of Staphylococcus aureus at the LPXTG motif. Proc Natl Acad Sci U S A. 1999;96:12424–12429. doi: 10.1073/pnas.96.22.12424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pritz S, et al. Synthesis of biologically active peptide nucleic acid-peptide conjugates by sortase-mediated ligation. J Org Chem. 2007;72:3909–3912. doi: 10.1021/jo062331l. [DOI] [PubMed] [Google Scholar]
- 19.Antos JM, Miller GM, Grotenbreg GM, Ploegh HL. Lipid modification of proteins through sortase-catalyzed transpeptidation. J Am Chem Soc. 2008;130:16338–16343. doi: 10.1021/ja806779e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Popp MW, Dougan SK, Chuang TY, Spooner E, Ploegh HL. Sortase-catalyzed transformations that improve the properties of cytokines. Proc Natl Acad Sci U S A. 2011;108:3169–3174. doi: 10.1073/pnas.1016863108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sinisi A, et al. Development of an Influenza virus Protein Array Using Sortagging Technology. Bioconjug Chem. 2012 doi: 10.1021/bc200577u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Witte MD, et al. Preparation of unnatural N-to-N and C-to-C protein fusions. Proc Natl Acad Sci U S A. 2012;109:11993–11998. doi: 10.1073/pnas.1205427109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Samantaray S, Marathe U, Dasgupta S, Nandicoori VK, Roy RP. Peptide-sugar ligation catalyzed by transpeptidase sortase: a facile approach to neoglycoconjugate synthesis. J Am Chem Soc. 2008;130:2132–2133. doi: 10.1021/ja077358g. [DOI] [PubMed] [Google Scholar]
- 24.Antos JM, et al. Site-specific N- and C-terminal labeling of a single polypeptide using sortases of different specificity. J Am Chem Soc. 2009;131:10800–10801. doi: 10.1021/ja902681k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Williamson DJ, Fascione MA, Webb ME, Turnbull WB. Efficient N-terminal labeling of proteins by use of sortase. Angew Chem Int Ed Engl. 2012;51:9377–9380. doi: 10.1002/anie.201204538. [DOI] [PubMed] [Google Scholar]
- 26.Levary DA, Parthasarathy R, Boder ET, Ackerman ME. Protein-protein fusion catalyzed by sortase A. PLoS One. 2011;6:e18342. doi: 10.1371/journal.pone.0018342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Swee LK, et al. Sortase-mediated modification of alphaDEC205 affords optimization of antigen presentation and immunization against a set of viral epitopes. Proc Natl Acad Sci U S A. 2013;110:1428–1433. doi: 10.1073/pnas.1214994110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Popp MW, Artavanis-Tsakonas K, Ploegh HL. Substrate filtering by the active site crossover loop in UCHL3 revealed by sortagging and gain-of-function mutations. J Biol Chem. 2009;284:3593–3602. doi: 10.1074/jbc.M807172200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Antos JM, et al. A straight path to circular proteins. J Biol Chem. 2009;284:16028–16036. doi: 10.1074/jbc.M901752200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bolscher JG, et al. Sortase A as a tool for high-yield histatin cyclization. FASEB J. 2011;25:2650–2658. doi: 10.1096/fj.11-182212. [DOI] [PubMed] [Google Scholar]
- 31.Wu Z, Guo X, Guo Z. Sortase A-catalyzed peptide cyclization for the synthesis of macrocyclic peptides and glycopeptides. Chem Commun (Camb) 2011;47:9218–9220. doi: 10.1039/c1cc13322e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen I, Dorr BM, Liu DR. A general strategy for the evolution of bond-forming enzymes using yeast display. Proc Natl Acad Sci U S A. 2011;108:11399–11404. doi: 10.1073/pnas.1101046108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hirakawa H, Ishikawa S, Nagamune T. Design of Ca2+-independent Staphylococcus aureus sortase A mutants. Biotechnol Bioeng. 2012;109:2955–2961. doi: 10.1002/bit.24585. [DOI] [PubMed] [Google Scholar]
- 34.Race PR, et al. Crystal structure of Streptococcus pyogenes sortase A: implications for sortase mechanism. J Biol Chem. 2009;284:6924–6933. doi: 10.1074/jbc.M805406200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Esteban A, et al. Fungal recognition is mediated by the association of dectin-1 and galectin-3 in macrophages. Proc Natl Acad Sci U S A. 2011;108:14270–14275. doi: 10.1073/pnas.1111415108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hess GT, et al. M13 bacteriophage display framework that allows sortase-mediated modification of surface-accessible phage proteins. Bioconjug Chem. 2012;23:1478–1487. doi: 10.1021/bc300130z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Popp MW, Karssemeijer RA, Ploegh HL. Chemoenzymatic site-specific labeling of influenza glycoproteins as a tool to observe virus budding in real time. PLoS Pathog. 2012;8 doi: 10.1371/journal.ppat.1002604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Boekhorst J, de Been MW, Kleerebezem M, Siezen RJ. Genome-wide detection and analysis of cell wall-bound proteins with LPxTG-like sorting motifs. J Bacteriol. 2005;187:4928–4934. doi: 10.1128/JB.187.14.4928-4934.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ilangovan U, Ton-That H, Iwahara J, Schneewind O, Clubb RT. Structure of sortase, the transpeptidase that anchors proteins to the cell wall of Staphylococcus aureus. Proc Natl Acad Sci U S A. 2001;98:6056–6061. doi: 10.1073/pnas.101064198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kaiser E, Colescott RL, Bossinger CD, Cook PI. Color test for detection of free terminal amino groups in the solid-phase synthesis of peptides. Anal Biochem. 1970;34:595–598. doi: 10.1016/0003-2697(70)90146-6. [DOI] [PubMed] [Google Scholar]
- 41.Vojkovsky T. Detection of secondary amines on solid phase. Pept Res. 1995;8:236–237. [PubMed] [Google Scholar]
- 42.Gisin BF. The monitoring of reactions in solid-phase peptide synthesis with picric acid. Anal Chim Acta. 1972;58:248–249. doi: 10.1016/S0003-2670(00)86882-8. [DOI] [PubMed] [Google Scholar]