

Abstract
The concept of the heart as a terminally differentiated organ incapable of replacing damaged myocytes has been at the center of cardiovascular research and therapeutic development for the last fifty years. The progressive decline in myocyte number with aging and the formation of scarred tissue following myocardial infarction have been interpreted as irrefutable proofs of the post-mitotic characteristics of the adult heart. However, emerging evidence supports a more dynamic view of the myocardium in which cell death and cell restoration are vital components of the remodeling process that governs organ homeostasis, aging and disease. The identification of dividing myocytes throughout the life span of the organisms and the recognition that undifferentiated primitive cells regulate myocyte turnover and tissue regeneration indicate that the heart is a self-renewing organ controlled by a compartment of resident stem cells. Moreover, exogenous progenitors of bone marrow origin transdifferentiate and acquire the cardiomyocyte and vascular lineages. This new reality constitutes the foundation of the numerous cell-based clinical trials that have been conducted in the last decade for the treatment of ischemic and non-ischemic cardiomyopathies.
Introduction
The possible application of autologous cell products in the management of human heart failure requires the acquisition of basic knowledge on the growth and differentiation of ckit-positive cardiac stem cells (CSCs) [1] and the inevitable comparison with the currently used cardiospheres [2], bone marrow mononuclear cells [3], and bone marrow-derived mesenchymal stromal cells [4]. But the most challenging task for all of us is to establish whether the therapeutic efficacy of resident CSCs is superior, equal, or inferior to c-kit-positive hematopoietic stem cells (HSCs). The entire field of regenerative cardiology was triggered by observations supporting the notion that HSCs transdifferentiate and acquire the cardiomyocyte and vascular lineage restoring the infarcted heart experimentally [5]. Surprisingly, c-kit-positive HSCs have never been tested clinically, a deficiency that has to be overcome to actually define the more powerful primitive cell for myocardial regeneration. Although this is a critical issue for the proponents of cell therapy in patients with acute and chronic heart failure (HF), a strong debate has been initiated by the adversaries of cardiomyocyte renewal via stem cell activation. The same establishment that violently attacked the concept of myocyte replication now uses this argument against the fundamental role that CSCs have in heart homeostasis and tissue repair. In this commentary, we will discuss these viewpoints and emphasize what has to be done to resolve the confusion that permeates the new field of regenerative cardiology to-date.
Deciphering CSC function is fundamental for the implementation of this cell class in the daily treatment of the decompensated human heart. The recognition that in small and large animals and humans the heart is a constantly renewing organ where the capacity to replace dying cells depends on the persistence of a stem cell compartment has dramatically changed our understanding of myocardial biology. Slowly replicating CSCs give rise to proliferating, lineage-restricted progenitor-precursor cells, which then become highly dividing amplifying cells that eventually reach terminal differentiation and growth arrest [6]. Stem cells have a high propensity for cell division and this property is maintained throughout the lifespan of the organ and organism. In contrast, transient amplifying cells represent a group of cells which have a limited proliferation capacity. Amplifying cells divide and concurrently differentiate [7], and when differentiation is completed, the ability to reenter the cell cycle is permanently lost. A new paradigm of the heart has emerged: multipotent resident CSCs are implicated in the constant turnover of myocytes, endothelial cells (ECs), smooth muscle cells (SMCs) and fibroblasts. The recognition that activated CSCs translocate to areas of need where they grow and differentiate makes the possibility of myocardial regeneration a feasible reality. In a manner comparable to HSCs that repopulate and completely reconstitute the ablated bone marrow [8], CSCs may rebuild the damaged myocardium and convert a severely diseased heart into a physiologically functional heart. Whether HSCs released from the bone marrow into the systemic circulation participate in the homeostatic control of the myocardium and in tissue reconstitution following injury is an important question that has only been partially considered thus far.
To impact on the late stages of severe ventricular dysfunction, we have to regenerate large quantities of cardiac muscle, create coronary vessels, reverse the process of negative remodeling and ultimately rebuild the entire heart (Figure 1). The chronically dilated failing heart has to be restructured into a smaller, less spherical, properly functioning organ. These dramatic changes in cardiac size and shape can only be accomplished by replacing injured, poorly contracting myocardium with new cardiomyocytes integrated with a newly regenerated coronary vasculature. Four major characteristics of cardiac pathophysiology have to be corrected in HF: 1. the segmental and focal areas of myocardial scarring have to be restored; 2. the damaged large and intermediate-sized coronary arteries have to be replaced; 3. the rarefaction of resistance coronary arterioles and capillaries has to be corrected; and 4. the hypertrophied mechanically inefficient cardiomyocytes have to be replaced by smaller better functioning cells. Although the enormous effort made in the last 3 decades has been successful in developing new drugs that delay the progression of HF [9], reversal of the process remains to be obtained. Would cell therapy accomplish this goal? This is a critical question that has no plausible answer at present. However, this novel experimental treatment has to be tested since HF has reached endemic proportion in the Western world and there is nothing as promising as stem cells in the clinical arena.
Fig. 1.
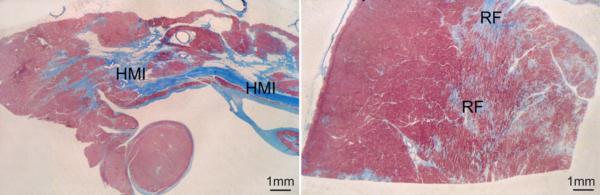
Photomicrographs of sections of paraffin-embedded left ventricular tissue showing a healed myocardial infarct with thinning of the wall (left) and multiple sites of replacement fibrosis in the noninfarcted viable left ventricular tissue (right). Trichrome staining. Bar corresponds to 2 mm in A and B. HMI, human myocardial infarction; RF, replacement fibrosis. Figure adapted from Beltrami CA et al. Circulation 1994;89:151-63.
Myocardial Biology Has Changed
For several decades, the human heart has been considered a post-mitotic organ formed of a predetermined number of myocytes, which is established at birth and is preserved throughout life [10]. Based on this premise, the age of myocytes corresponds to the age of the organ and organism, i.e., cellular, organ and organism age coincide. Myocytes must age at the same rate and, at any given time, the heart is composed of a homogeneous population of cells of identical age. Because of this static view, aging has been construed as a time-dependent process that interacts with ischemic injury, hypertension, diabetes and other disorders, which together define the senescent cardiac phenotype. Similarly, primitive cardiac pathology has been interpreted as a process exclusively dictated by excessive, maladaptive hypertrophy of pre-existing myocytes, a concept that persists in part of the scientific community [11]. Despite the appreciation that myocyte death occurs physiologically and increases dramatically with myocardial aging and disease processes [12, 13], the possibility that myocyte formation is an important compensatory determinant of ventricular performance has been dismissed easily.
The recognition that human CSCs (hCSCs) live in the heart and generate cardiac cell lineages has imposed a reevaluation of the current view of cardiac homeostasis, aging and pathology. A novel conceptual framework of the heart has emerged; the heart is a self-renewing organ characterized by resident hCSCs stored in niches [14]. The first documentation of resident c-kit-positive CSCs was obtained in rodents 10 years ago [15]. This study adhered to the basic principles required for the recognition of adult stem cells: c-kit-positive CSCs are lineage-negative clonogenic cells that differentiate in vitro into cardiomyocytes, and vascular SMCs and ECs. In vivo, CSCs create myocytes and coronary vessels, forming de novo myocardium. The newly generated myocytes possess the mechanical and electric properties of functionally-competent cells, which improve ventricular performance [15].
Importantly, the human atrial and ventricular myocardium contains a pool of c-kit-positive hCSCs which are stored in small microdomains with the characteristics of stem cell niches [1]. The niches control the physiological turnover of cardiac cells mediated by migration and commitment of hCSCs that leave the niche structure to replace old, dying cells within the myocardium. Myocyte and fibroblasts are structurally and functionally connected to hCSCs and operate as supporting cells within the niches; hCSCs divide symmetrically and asymmetrically and are able to form new stem cells and cells destined to acquire specialized functions [1, 16]. The preferential localization of these microdomains in the atria and apex is consistent with the lower level of hemodynamic stress in these anatomical regions of the heart. Whether CSC niches are more abundant in the epimyocardium than in the endomyocardium reflecting the distribution of stress across the ventricular wall is currently unknown.
The self-renewing, clonogenicity and multipotentiality of hCSCs has been demonstrated in vitro and in vivo by various protocols including genetic marking (Figure 2A) [17], which is considered the gold standard assay for HSCs [18]. The fundamental principle of hCSC self-renewal in vivo was established by serial transplantation. This strategy has been used in the last 40 years to test the functional properties of HSCs [18]. The lethally irradiated recipient of a bone marrow transplant is a successful donor for a subsequent serial transplant only if the HSCs from the original donor undergo substantial self-renewal within the primary recipient. This approach has been applied to single cell derived clonal hCSCs. Two weeks after infarction in immunosuppressed rats and implantation of EGFP-labeled hCSCs, the left ventricle of the primary recipient was enzymatically dissociated and cells were double sorted for c-kit and EGFP. Nearly 10,000 hCSCs were recovered from each heart [16]. These cells were delivered immediately to the infarcted region of subsequent recipients. Fifteen days later, human myocytes and coronary vessels were identified, replacing large areas of the infarcted myocardium. Undifferentiated hCSCs were detected in the secondary recipient, providing further evidence in support of the self-renewal and long-term proliferation in vivo of hCSCs (Figure 2B).
Fig. 2.

(A) Various clones were detected in distinct cardiac cell populations from the regenerated myocardium of one treated rat. Sites of integration in isolated cell populations. Some clones were common to different cell classes (same color arrowheads). Key DNA sequences are ≈80 bp shorter than the corresponding clonal bands. (B) In a serially transplanted infarcted heart, EGFP-positive (insets) c-kit-positive (white) human CSCs are present (arrows). Panel adapted from reference [17]; panel B adapted from reference [16].
After the discovery of c-kit-positive CSCs, different classes of progenitor cells have been characterized in the adult heart [6]. A variety of surface antigens, transcription factors, and functional assays have been used to define these primitive cells, which include ISL1 progenitors, epicardial progenitors, side population progenitors, Sca1 progenitors, and progenitors generating cardiospheres. An interesting class of cardiac progenitors has been described in the mouse heart; these primitive cells have the ability to expel toxic compounds and dyes through an ATP-binding cassette transporter [19]. This property defines a pool of putative cardiac progenitors that form colonies in semisolid media and differentiate into cardiomyocytes. However, a depletion of side population cells occurs after infarction in mice overexpressing a dominant-negative MEF2C, documenting that side population cells are committed to the myocyte lineage. This work introduced the concept of a myocardial stem cell that participates in the response of the heart to ischemic injury [20]. With the exception of cardiospheres, these various progenitors have yet to be introduced clinically and have been discussed in detail elsewhere [6].
Although several laboratories now concur that the heart contains a compartment of primitive cells with the characteristics of stem cells, the identification of the actual long-term repopulating hCSC, equivalent to the HSC in the bone marrow, remains uncertain. Stem cells are relatively rare; the frequency of hCSCs in the adult organ, one every 30,000–40,000 myocardial cells [1], is consistent with that of HSCs in the bone marrow, one every 10,000–100,000 [21]. But whether a subset of hCSCs with enhanced growth reserve and differentiation potential is present in the myocardium throughout the organ lifespan is currently unknown. This CSC category would be ideal for the recovery in structure and function of the failing heart.
Myocyte Death and Renewal in the Human Heart
In the last decade, a new scientific field aiming by various approaches at the restoration of the damaged myocardium has emerged. However, the controversy on the growth reserve of the adult human heart has not been resolved and the extent of myocyte renewal claimed by different laboratories varies dramatically. A recent study, based on retrospective 14C birth dating of cells, has claimed that ~1% and ~0.45% replacement of myocytes occurs annually in the human heart at 25 and 75 years of age, respectively [22]. These findings indicate that only 50% of myocytes are renewed once during the entire life of the human heart, from birth to death, while an equal number lives as long as the organ and organism, up to 100 years of age and longer. However, the magnitude of the process is in contrast with the level of myocyte apoptosis present in the adult human heart and the progressive increase in myocyte loss that occurs with aging in humans [12, 13].
Importantly, this crucial issue has been addressed in a rather comprehensive manner by determining the interaction of myocyte regeneration, cellular senescence, growth inhibition and apoptosis in normal female and male human hearts, collected from patients, 19 to 104 years of age, who died from causes other than cardiovascular diseases [23]. A large number of hearts (n = 72: women: n = 32 and men: n = 42) was included to obtain a common denominator of the processes that regulate replication and death of ventricular myocytes. In the human heart, apoptosis is invariably coupled with the expression of the aging-associated protein p16INK4a that is a marker of replicative senescence and irreversible growth arrest of progenitor cells in various organs including hCSCs [24]. Myocytes positive for p16INK4a were found at all ages but the frequency of senescent cells was higher in men than in women throughout life. In the oldest man, 104 years of age, 80% of myocytes expressed this nuclear protein. This was not the case in the oldest woman, 102 years of age, where a relevant proportion of myocytes, 45%, did not reach the senescent phenotype. From 19 to 104 years of age, the time-dependent increase in old myocytes was 0.68% per year in women and 0.89% per year in men; the 31% higher rate of accumulation of senescent myocytes in the aging male heart was significant (Figure 3A). In comparison with cellular senescence, myocyte apoptosis was relatively low but it occurred only in p16INK4a-positive cells. Cell death was higher in men than in women at 19 to 49, 50 to 69 and 72 to 104 years of age (Figure 3B). However, the rate of increase in myocyte apoptosis with age did not differ with gender: 131 cells/108 per year in men and 123 cells/108 per year in women (Figure 3C).
Fig. 3.
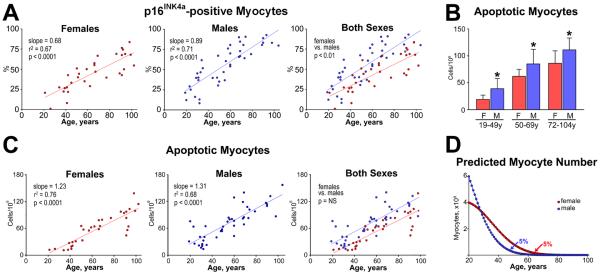
(A) The rate of increase in the fraction of p16INK4a-positive myocytes with age is lower in the female than in the male left ventricle (LV). (B) The fraction of apoptotic myocytes in the LV is lower in women than men. F indicates female; M, male. *P<0.05 vs. female. (C) The rate of increase of apoptotic myocytes with age is similar in the female and male LV. (D) At 20 years of age, the female (red) and male (blue) LV contains 4 and 6×109 cardiomyocytes, respectively. Curves are derived by combining the yearly levels of apoptosis shown in panel C and the prediction that cell death lasts 4 hours. In the absence of cell regeneration, 5% of LV myocytes (200×106 in the female LV and 300×106 in the male LV) would be left at 63 and 48 years of age in the female and male LV, respectively. Figure adapted from reference [23].
Cell apoptosis is completed in all organs in less than 4 hours [25], suggesting that these rates of cell death result in a massive loss of myocytes with aging. If we consider the number of cells present in the left ventricle in women and men at 20 years of age (men: 6 × 106; women: 4 × 106) and the rate of myocyte regeneration claimed in the early report [22], the myocardium would essentially disappear with time. In the absence of myocyte formation but in the presence of cell apoptosis, only 5% of cardiomyocytes would persist at 63 and 48 years of age in women and men, respectively (Figure 3D). This value does not include cell necrosis, which has been documented by the detection of cardiac troponin in the circulation of apparently healthy individuals [26], suggesting that a significant high degree of myocyte regeneration has to occur to preserve cardiac mass and function in humans. Moreover, myocyte regeneration has to increase with age in view of the remarkable increase in cell apoptosis and necrosis in the old human heart [12, 13]. This concept has been shown to be correct in a rather definitive manner by studies measuring the fraction of human cardiomyocytes labeled by the thymidine analog iododeoxyuridine (IdU) [27], and retrospective 14C birth dating of cardiomyocytes in the aging and failing heart [28]. Myocyte turnover in humans occurs at a rate of ~15–20% per year and is dramatically enhanced in the presence of end-stage heart failure.
Controversy on Myocyte Regeneration in the Human Heart
Three cellular processes control the maturation, steady state, aging and failure of the human heart: cell generation, hypertrophy and death. The balance between cell growth and cell death characterizes organ homeostasis in which cell loss is compensated by a corresponding rate of cell formation. Myocyte hypertrophy typically occurs postnatally and in response to a hemodynamic challenge in post-mitotic myocytes which can increase only in size being unable to reenter the cell cycle. Although there is no controversy on the role of myocyte hypertrophy in the expansion of cardiac mass postnatally and following an overload in the adult heart, a debate exists on the extent of myocyte turnover physiologically and on the degree of myocyte regeneration pathologically. Similarly, the origin and contribution of new myocytes under these conditions is matter of disagreement. For the scientific community that is not directly involved in this area of research it might be relevant to discuss the strategies that have been applied to address these issues of human myocardial biology and the potential sources of the discrepancy that exists among several laboratories.
Retrospective 14C Birth Dating of Cells
14C birth dating of cells has been introduced a few years ago in the analysis of the average age of human cardiomyocytes and their turnover rates as a function of organ and organism age [22]. This protocol is based on the increase in 14C in the atmosphere during aboveground nuclear testing in the 1950s and the dramatic drop in 14C after the agreement on nuclear testing was signed in 1963 [29]. As a result of this treaty, the atmospheric 14C has decreased exponentially at a rate of ~50% per 11 years. 14C is not directly assimilated by humans but it is absorbed by plants and through daily ingestion is integrated in replicating cells in S phase [29]. Changes in the level of 14C in the DNA of human cells can be used to define their birth date since the decay of 14C in the atmosphere continues to be closely monitored. Therefore, measurements of 14C in the DNA can be traced back to the year in which a corresponding level of atmospheric 14C was detected. 14C birth dating has been applied to teeth, brain, fat tissue and the heart [22, 28, 30–35]. 14C birth dating provides information similar to that generated by the incorporation of thymidine analogs in animal models, an analysis which has only been rarely possible in human beings [27, 32, 36]. As all methodologies, 14C birth dating is a powerful technique, but limitations exist and several parameters have to be accurately measured to obtain reliable information. These variables are critical for a valid implementation of this procedure.
Variations in the approach introduced in the evaluation of 14C birth dating of cardiomyocytes may explain the striking differences in the results reported by independent laboratories [22, 28]. First, the purity of each myocyte cell preparation has to be measured and corrected for the contribution of ECs and fibroblasts; both these cardiac cell classes may profoundly influence the computed results. Similarly, the proportion of mononucleated, binucleated, and multinucleated cells has to be determined for a correct analysis of [14C] in myocytes, ECs, and fibroblasts; in fact, binucleation would yield degrees of cell formation higher than those actually present as DNA replication and karyokinesis would occur in the absence of cytokinesis. At 2, 3 and 7 years of organ age myocytes are almost exclusively mononucleated; binucleated myocytes become apparent at 16 and 20 years of age and later in life [28]. Trinucleated myocytes have only been occasionally seen and tetranucleated myocytes have not been found. Importantly, from shortly after birth to nearly 80 years of age, mononucleated myocytes decrease and binucleated myocytes increase up to ~25 years of age and remain constant thereafter. An average 77% mononucleated and 23% binucleated myocytes are present in the adult human heart and these values are not affected by myocardial aging [28]. This possibility does not involve ECs and fibroblasts since these cell classes are all mononucleated.
Importantly, the interpretation of [14C] in the DNA involves the analysis of polyploidy since they would mimic cell replication, resulting in an overestimation of cell turnover. Polyploidy is characterized by an exponential increase in DNA content dictated by the number of doublings of the entire genome from 2n to 4n to 8n and 16n. This exponential increase in the amount of DNA is reflected by a significant increase in nuclear volume. Thus far, there is little or no information on this critical process of DNA endoreplication in the human heart, and frequently cited results [22, 37] were collected nearly 4 decades ago [38]. These old studies were based on the measurement of DNA content per nucleus by Feulgen staining and cytophotometry. Labeling of nuclear DNA by the Feulgen reaction was developed in 1924 and was combined with cytophotometry in 1964 [39]. Unfortunately, this protocol continues to be used, despite significant limitations, which include type and duration of fixation [40], extent of DNA hydrolysis required for the Schiff reaction [41], and the chromatin structure. The latter is crucial for the accessibility of the DNA by this technique [42]. By this outdated methodology, the number of polyploid myocyte nuclei has been argued to increase dramatically after birth, comprising 50% of cardiomyocytes at 10 years of age [43]. Values up to nearly 100% of polyploid myocytes have been alleged as a result of aging and cardiac hypertrophy [43]. These findings are inconsistent with reality; this engrained absurd view of myocyte biology must be corrected. The inherent problems present in the Feulgen staining of the nuclear DNA can be avoided by the use of flow cytometry that has become the gold standard for measurements of nuclear ploidy in various organs including liver, brain, intestine, skin, kidney, and cancer cells.
An attempt has also been made to evaluate nuclear ploidy by confocal microscopy of myocardial sections 30 μm in thickness [44]. By necessity, the intensity of labeling of nuclei varied across the thickness of the section, becoming progressively weaker with the depth of the section. To compensate for this artifact, the signal intensity of the labeled DNA was amplified to levels saturating the photodetection system. This is apparent in the images shown in this report [44]. As a result, all nuclei had comparable brightness and the major variable was represented by the size of the nucleus, rather than by the actual DNA content. This makes the measurement of ploidy by this protocol invalid.
By flow cytometry, the majority of myocyte, EC, and fibroblast nuclei show a 2n diploid DNA content. Tetraploid and octaploid myocyte nuclei are rare and hexadecaploid myocyte nuclei are not detected. Myocyte, EC, and fibroblast nuclei with DNA content higher than 2n and lower than 4n reflect dividing cells, positive for Ki67. Tetraploid myocyte nuclei are in part Ki67-positive, while octaploid myocyte nuclei are Ki67-negative, excluding that they represent cycling cells in G2 [28].
During postnatal maturation, adulthood and aging, diploid myocyte nuclei decrease and tetraploid and octaploid myocyte nuclei increase up to ~25 years of age and remain constant thereafter. An average 88% diploid and 12% tetraploid and octaploid myocyte nuclei combined are found in the adult human heart and these values are not affected by myocardial aging. Similarly, diploid EC and fibroblast nuclei constitute the large majority of cells with aging, accounting for 99% of the population. Although the level of ploidy is small, the [14C] in each cell class of each heart has to be corrected for the contribution of ploidy to avoid introducing confounding variables in these determinations. Thus, the human heart is mostly composed of diploid cells, a fundamental factor for the determination of myocyte turnover by 14C birth dating of cells. Cell number is required for the measurement of retrospective 14C birth dating of cells but, most importantly, for the evaluation of cell turnover. In fact, an increase in number of cells per unit volume of myocardium would indicate a higher turnover rate, despite an identical cellular age [28]. Cell volume can be easily obtained in isolated myocyte preparations by optical section reconstruction and confocal microscopy; and the volume fraction of myocytes in the myocardium can be determined morphometrically in tissue samples collected from the same hearts for histological analysis. Based on these two parameters, the number of myocytes per 10 cm3 of myocardium can be computed in each heart. And an identical protocol can be employed for ECs and fibroblasts [28].
A few comments have to be made regarding the potential confounding variables present in the evaluation of [14C] in myocyte nuclei; they include DNA repair of damaged or mutated DNA, turnover of mitochondrial DNA, and nucleotide salvage pathways triggered by cell death. The first two possibilities would yield younger myocytes and higher turnover rates, while the third would lead to opposite effects; however, these processes do not significantly alter the measurements of [14C] in the nuclear DNA by Accelerator Mass Spectrometry (AMS).
Nearly 2,000 to 10,000 nucleotide bases are replaced per day in the human genome as a result of DNA damage [45]. Diploid human cells contain 6 × 109 base pairs, i.e., 12 × 109 nucleotide bases [46]. When the highest value of 10,000 bases being repaired per day is considered, it would require 1.2 × 106 days or 3,288 years to restore the entire genomic DNA. Over 80 years of life, only 2.4% of 14C in the genomic DNA would be derived by this mechanism. The mitochondrial genome contains 1.6 × 104 base pairs, i.e., 3.2 × 104 nucleotide bases [47]. Even if we assume that 3,000 mitochondria are present in adult human cardiomyocytes, aggregate mitochondrial DNA would be composed of 9.6 × 106 nucleotide bases. This value is equivalent to 0.1% of the DNA, making its contribution to the measurement of 14C in the genomic DNA essentially negligible. Finally, incorporation of 14C-labeled nucleotides released from dying myocytes during DNA degradation would be minimal. In a model of irradiated cells in vitro it was demonstrated that less than 0.05% metabolites originating from the degraded DNA is re-used for new DNA synthesis [48]. This value is significantly below the level of accuracy of AMS. Importantly, 14C was found to be undetectable in human neurons of individuals born prior to the rise in 14C in the atmosphere resulting from above ground nuclear testing [30], further questioning the relevance of these biological events in the evaluation of average myocyte age and turnover rate by this methodology.
Therefore, three fundamental parameters required for a correct interpretation of the [14C] in the nuclear DNA have to be measured: binucleation, polyploidization, and number of cell per unit volume of myocardium. These cellular determinants are critical for the evaluation of 14C birth dating of myocytes and their renewal. Conversely, DNA repair, turnover of mitochondrial DNA, and nucleotide salvage pathways associated with cell death have no effects on the evaluation of myocyte age and myocyte turnover rate.
Contrasting Results on Retrospective 14C Birth Dating of Human Cardiomyocytes
Thus far, there are only two studies that have analyzed by AMS the [14C] in the nuclear DNA of cardiomyocytes as a function of organ and organism age [22, 28]. However, striking differences have been reported. Data from our laboratory indicate that the human heart is a dynamic organ characterized by a high turnover of myocytes. In adulthood and with physiological aging, from ~20 to ~80 years, cardiomyocytes are replaced approximately 8 times [28].
A certain degree of skepticism may exist concerning these observations, since previous results utilizing retrospective 14C birth dating have suggested that myocyte turnover in the human heart is minimal and decreases with age [22]. In contrast to this early work, several confounding factors were considered here in the analysis of 14C incorporation into the nuclear DNA of cardiomyocytes [28]; they included unbiased collection of cardiomyocytes, purity of the cell preparation, proportion of mononucleated and multinucleated cells, fraction of polyploid nuclei, and number of cardiomyocytes. As discussed above, these variables have profound consequences on the quantitative evaluation of cardiomyocyte renewal in the aging heart by 14C birth dating of cells. Our data are consistent with the necessary balance between myocyte death and myocyte regeneration present during an individual's lifespan. Myocyte apoptosis and necrosis occur physiologically, and cell death has to be accompanied by cell formation for the heart to continue to exist. The simple concept of a requisite equilibrium between myocyte death and renewal has often been ignored. Myocyte death increases with age so that a significantly higher level of myocyte regeneration than that purported in the previous study on 14C birth dating of cells [22] has to occur to preserve cardiac mass and function in humans. Although the human heart is a highly dynamic organ that retains a significant degree of plasticity throughout life, the ability to regenerate cardiomyocytes cannot prevent the manifestations of myocardial aging or oppose the negative effects of ischemic and idiopathic dilated cardiomyopathy and end-stage heart failure.
First, an inherent limitation of the 14C birth dating study [22] was related to the need to introduce mathematical models with assumptions that affected the computed values. The scenario chosen [22] presupposes that the number of myocytes in the heart is constant and that cells turn over at a constant rate. This form of invariant growth [49] defines parenchyma in a steady state in which cell death is compensated by cell regeneration in young healthy individuals. However, this scenario can hardly be applied to pathologic states or to the biology of aging in which from 17 to 90 years of age an average of 38 × 106 myocytes are lost per year [50]; 3 of the 12 patients were 62, 67 and 73 years old, 1 had hypertension, 2 had myocardial infarction, hypertrophy was present in 5 and coronary disease in 4 [22]. Also, myocyte number increases postnatally and cell loss occurs with age and cardiac diseases. Importantly, the analysis of 14C was restricted to nuclei expressing the contractile protein troponin I (TnI) which is present exclusively in senescent myocyte nuclei [27]. It is unrealistic to define the physiological mechanisms of myocardial aging with a sample size of 12 individuals of both genders, some of which affected by severe cardiac pathologies [22].
Second, another major problem involves the way in which the data on atmospheric 14C were utilized. It is not possible from the data obtained from patients 50 to 73 years of age to indicate the age of the cells, since the measured 14C level may reflect its incorporation during the rising or decaying part of the 14C curve. The difference in the rate of myocyte renewal between young and old individuals may simply reflect an artifact in calculation due to assumptions of the model imposed and not a real biological phenomenon. Thus, the claim that myocyte renewal decreases with age has no basis.
Third, the right side of the published 14C curve was employed to compute myocyte age in young hearts while the left side of the 14C curve was used to calculate myocyte age in old hearts. Without basis 14C levels were calculated and interpreted differently in young (19 to 42 years) and old (50 to 73 years) hearts to arbitrarily draw an exponential curve that indicated a progressive decline in cell generation as a function of age. There is no gradual decrease in the rate of myocyte turnover but rather two distinct sets of data, highlighted by us by adding a red line to the original graph. In this regard, the runs test showed a significant deviation (p<0.0001) from the exponential decay model used further questioning the accuracy of these results. In summary, it is highly risky to study aging employing a sample of 12 hearts only, divide them in two age groups and calculate myocyte turnover on arbitrary decisions.
Conversely, the analysis of a significant larger number of human hearts indicates that the adult and aging myocardium is a dynamic organ characterized by high turnover of myocytes that increases with chronic HF [28]. This conclusion was reached by applying a protocol that: a) is independent from a potential bias, i.e., 14C birth dating ; b) excludes the human factor in the collection of the data, i.e., AMS; c) uses a non-modified, unperturbed biological system, i.e., physiological myocardial aging; d) introduces an established condition, chronic HF, known to affect the myocardium structurally and functionally; and e) is redundant, from the point of view of information theory, reducing the amount of noise in the data, i.e., misinformation, as defined by Shannon's law [51].
In fact, the 14C data were complemented with independent determinations of the degree of myocyte regeneration at organ death; three markers of the cell cycle were employed: Ki67 [52], phospho-H3 [53], and aurora B kinase [54]. Ki67 is expressed in late G1, S, G2 and early mitosis (Figure 4A) and is not implicated in DNA repair or ploidy formation [52]. Phospho-H3 is upregulated in late G2 and mitosis (Figure 4B); it is highly phosphorylated at Ser10 during chromatin condensation and remains phosphorylated up to the end of telophase [53]. Aurora B kinase ensures accurate segregation of the duplicated chromosomes and controls proper cytoplasmic division (Figure 4C), preventing polyploidization [55]. Thus, the degree of myocyte replication was measured by the expression of Ki67, the myocyte mitotic index was evaluated by the presence of phospho-H3 during karyokinesis, and myocyte cytokinesis was identified by the localization of aurora B kinase at the cleavage furrow of dividing myocytes.
Fig. 4.
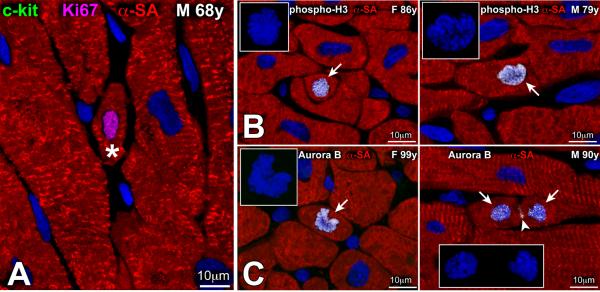
(A) Amplifying myocytes (asterisk) are small cycling cells, positive for Ki67 (magenta) and lack c-kit. (B, C) Small amplifying myocytes are positive for phospho-H3 (B, arrow) (B) and aurora B kinase (C, arrow). In the right panel in C, aurora B kinase is located in the 2 sets of telophase chromosomes and at the cleavage furrow of the dividing myocyte (arrowhead). Insets in B and C illustrate the organization of chromosomes in the dividing cells. Figure adapted from reference [23].
In summary, the human heart is a highly dynamic organ that retains a significant degree of plasticity throughout life and in the presence of HF. However, the ability to regenerate cardiomyocytes cannot prevent the manifestations of myocardial aging or oppose the negative effects of ischemic and idiopathic dilated cardiomyopathy. Recently, the clinical implementation of autologous CSCs and cardiosphere-derived cells in patients with HF has provided encouraging results [56–58], suggesting that cell therapy may interfere with the devastating consequences of this myocardial disease.
Contrasting Results on Cardiomyocyte Turnover in the Adult Mouse Heart
The identification and characterization of resident CSCs in small and large animals [15, 19, 59–65] has not resolved the controversy regarding the turnover rate of cardiomyocytes in the adult heart which continues inexorably. Several laboratories have repeatedly supported the view that the heart has near none regenerative capacity, whether intrinsic to the organ, via activation of CSCs, or extrinsic to the myocardium, by delivery and transdifferentiation of HSCs. A recent publication [66] concerning the level of myocyte renewal in the mouse heart, and the origin of the irrelevant number of newly-formed parenchymal cells is another example of this position that affects negatively the field of adult stem cell biology and the critical role that these cells may have in the management of the human disease. In this report, the claim is made that myocyte renewal in the adult mouse heart comprises 0.76% of cells per year, a rate that declines with age. Since the mouse lifespan is ~2.5 years, only ~2.0% of myocytes would be replaced during this time. Additionally, resident CSCs may exist but are inconsequential for the regulation of organ homeostasis and tissue repair [66].
The function of the rodent heart is dictated by the number and mechanical properties of cardiomyocytes, which both decrease as a function of age. A loss of 50% of cardiomyocytes leads to ventricular failure that is incompatible with life in this model [67]. A balance between cell death and cell regeneration has to be present to preserve cardiac mass and ventricular performance throughout the organ lifespan. The magnitude of myocyte renewal purported in this study [66] does not reflect the actual biological reality of the aging mouse heart. Myocyte death increases dramatically with age [68], pointing to the unrealistic significance of the claim made in this study. Moreover, the identification of cardiac troponin in the circulation of healthy mice provides additional evidence of a chronic loss of cardiomyocytes by the necrotic pathway [69], further emphasizing the unreliability of this report, and the impossibility for the heart to exist and perform its function without significant cardiomyocyte regeneration. Although the argument has been made that post-mitotic cardiomyocytes regulate cell renewal, the marker of cytokinesis, aurora B kinase was not identified [66]. Conversely, aurora B kinase labeling of duplicated chromosomes and cleavage furrow of dividing cardiomyocytes has been detected in the mouse heart [70]. Small replicating myocytes correspond to amplifying cells derived from commitment and differentiation of CSCs [6].
Some comments on the technique employed are in order [71]. There is no need to use a complex but morphologically incorrect approach simply because it seems sophisticated neglecting its inherent errors. Multi-isotope imaging mass spectrometry (MIMS) is a powerful analytical tool [71], but the speed of data acquisition is slow, making the necessary sampling totally inadequate; this problem is magnified when rare events are measured, as the number of cycling myocytes. Moreover, the unparalleled high sensitivity of this instrument can be easily lost when an insufficient delivery of thymidine is used; 15N-thymidine was given at ~1 mg/kg body weight per hour. The half-life time of thymidine in the circulation is ~15 minutes [72], so that its concentration in the organism was 250 μg/kg body weight, a value at least two orders of magnitude smaller than that typically used in studies of cell kinetics.
Importantly, MIMS identifies cells in the myocardium by the background 14N signal, being unable to discriminate the identity of cardiac cells; 14N is present in all cell types making it impossible to recognize small developing myocytes. In most cases, antibodies against specific contractile proteins and high resolution microcopy are required to define the early stages of CSC commitment and differentiation into amplifying cardiomyocytes. Finally, the myocyte-restricted EGFP expression in these mice is dependent on the cell-specific induction of the α-myosin heavy chain (α-MHC) promoter, which is operative in activated CSCs, as shown previously [73], challenging the conclusion of the authors that CSCs are not involved in the process. These deficiencies are easily overcome by the injection of correct quantities of thymidine analogs and analysis of the myocardium by immunolabeling and confocal microscopy [24, 27, 68, 74, 75].
Translation of Basic Discoveries to Clinical Applications
Despite the controversy concerning the magnitude of myocyte turnover, the origin of the newly formed cardiomyocytes, the existence of CSCs, and the ability of bone marrow cells (BMCs) to form new parenchymal structures in the injured heart, several cell-based clinical trials have been conducted in the United States and Europe. Shortly after the experimental evidence that HSCs induce myocardial regeneration after infarction, unfractionated mononuclear BMCs and CD34 positive cells have been administered to patients affected by acute and chronic myocardial infarction, dilated cardiomyopathy, and refractory angina. Although the individual outcomes have been inconsistent and variability exists among trials, meta-analyses of pooled data indicate that BMC therapy results in all cases in a 3–4% increase in ejection fraction [76]. Reductions in infarct scar size and left ventricular end-systolic volume have been observed, together with small decreases in left ventricular end-diastolic volume. In spite of differences in the type of cells injected, choice of clinical end-points, methods for the evaluation of cardiac function, interval between the onset of the cardiac disease and BMC infusion, the positive consequences of BMCs have consistently been documented. Allogeneic and autologous mesenchymal stromal cells (MSCs) have also been employed in small clinical trials with encouraging results [77, 78]. Although the benefits may seem modest, these initial data have favored the conduct of larger randomized trials designed to critically evaluate the long-term effects of BMC therapy on a broader patient population [79].
However, the mechanisms involved in the positive impact of BMC therapy on human beings remains to be identified. The impossibility to permanently label the cells to be delivered and the difficulty to obtain cardiac biopsies to assess parameters consistent with myocardial regeneration leaves uncertain our understanding of the cellular processes mediating partial myocardial recovery. Measurements of coronary flow suggest that vasculogenesis may be operative while the contribution of de novo myocyte formation is uncertain. Reductions in infarct scar size speak in favor of myocardial regeneration, although unequivocal data have not been obtained yet. The most popular hypotheses include development of coronary vessels and enhanced blood supply to areas of hibernating myocardium, vasculogenesis and cardiomyogenesis, and the activation of growth and differentiation of resident CSCs via a paracrine effect, mediated by the release of a multiplicity of cytokines by the administered BMCs. Importantly, the recent identification of CSCs has shifted the attention to endogenous cell mechanisms as a novel target of cell therapy for the failing heart [6, 13, 79].
Human beings up to 104 years of age possess a significant number of hCSCs with long telomeres and remarkable growth reserve, which, however, are in a quiescent state [23]. Conversely, the large majority of activated hCSCs in the senescent myocardium have short telomeres that are inherited by the specialized progeny. In the young healthy heart, the asymmetric kinetics of stem cell growth efficiently preserves organ homeostasis. The structural and functional decline of the old and diseased heart may be coupled with defects in the hierarchical growth of the organ, suggesting that quantitative and qualitative alterations occur in resident hCSCs and/or in the pool of transit amplifying cardiomyocytes. Both possibilities have been documented in stem cell-regulated organs.
The environmental and/or cell-autonomous mechanisms that maintain “young” hCSCs in a state of long-term quiescence remains to be identified. However, the existence of a pool of hCSCs with intact telomeres, 8 to 12 kbp, in the senescent female and male heart and in explanted hearts [23, 80] is of great clinical relevance. This category of hCSCs with high growth reserve is expected to generate a young myocyte progeny within the failing and senescent heart. Because each division of hCSCs results in the loss of ~130 bp of telomeric DNA [1], an extremely large number of cardiomyocytes can be formed by these cells, before critical telomeric shortening and growth arrest occur. From a clinical perspective, the recognition that a subset of telomerase-competent hCSCs with long telomeres persists at all ages and with HF has raised the possibility that autologous cell-based therapy may be feasible in patients with severe ventricular dysfunction. Recently, a methodology has been developed to isolate this compartment of functionally-competent hCSCs from endomyocardial biopsies of patients undergoing cardiac transplantation or left ventricular assist device implantation [81]. After in vitro amplification, a clinically relevant number of hCSCs with high myogenic and vasculogenic potential was obtained. Importantly, expanded hCSCs possess a significant growth reserve as documented by the short population doubling time, the high telomerase activity, and the relatively long telomeres.
The Phase 1 trial SCIPIO (Stem Cell Infusion in Patients with Ischemic cardiOmyopathy) involves the delivery of autologous c-kit-positive lineage-negative hCSCs for the treatment of severe chronic heart failure of ischemic origin [56, 58]. Patients with EF lower than 40% at 4 months after coronary artery bypass grafting were enrolled in the treatment and control groups. Treated patients received a single intracoronary infusion of 1 million autologous hCSCs. The primary endpoint was short-term safety of treatment and the secondary endpoint was efficacy. Importantly, no hCSC-related adverse effects were reported. In 14 CSC-treated patients who were analyzed, EF increased from 30% to 38% at 4 months after infusion. In contrast, 7 control patients, during the corresponding time interval, did not show any change in this functional parameter. The beneficial effects of CSCs were even more pronounced at 1 year. In 7 treated patients, infarct size decreased 24% and 30% at 4 and 12 months, respectively [56]. These initial results are encouraging and warrant further studies.
Prior to infusion in patients enrolled in SCIPIO trial, c-kit-positive CSCs were extensively characterized by immunolabeling and confocal microscopy, and FACS analysis. In each of the 20 treated patients, the fraction of c-kit-positive cells varied from 75% to 98%; early markers of commitment to the myocyte, SMC, and EC lineages were found only in 0.1–2.7% of the entire cell population. Mean telomere length was 7.5 kbp, varying from 6.8 to 8.1 kbp, and telomerase activity was high in all CSC samples, indicating that, after passaging in culture, CSCs retained a significant growth reserve. The characterization of the telomere-telomerase axis should be introduced as standard quality control assay for the evaluation of the functional properties of cells to be administered to patients.
In addition to c-kit-positive CSCs, another class of cardiac derived cells has been administered to patients. Cell culture in serum-free media has been utilized for the isolation and expansion of cardiospheres. These aggregates contain a core of cells positive for the stem cell antigen c-kit and an outer layer composed of cells positive for CD105, a membrane glycoprotein commonly expressed in bone marrow mesenchymal stromal cells [2]. Cardiosphere-derived cells undergo spontaneous maturation toward the myocyte lineage, and this process of commitment can be coaxed by co-culture with neonatal ventricular myocytes. Connexin 43 is expressed between highly dividing cells within the cardiospheres and in the expanded differentiating cardiosphere-derived cells (CDCs). Clinically, CDCs may represent the ideal combination of primitive and early committed cells for the treatment of cardiac diseases. However, the delivery of a partially defined heterogeneous cell preparation may result in a vaster array of unpredictable, undesired effects than a uniform population of identical cells with well-established biological characteristics.
Cardiospheres may recapitulate the microenvironment of the stem cell niche. Mesenchymal-like and committed cells of the external layer may act as supporting cells for the internally distributed c-kit-positive CSCs. It has been suggested that direct implantation of niche-like cardiospheres in the damaged myocardium may be associated with a homing advantage with respect to monolayer-cultured CDCs. The presence of supporting cells may transiently protect the adjacent primitive cells, enhancing their survival in the hostile environment of the damaged myocardium. However, a direct contact between the delivered and recipient cardiac cells is required for actual engraftment in the host tissue and acquisition of the cardiogenic fate. Donor stem cells have to integrate structurally within the surrounding myocardium by forming junctional and adhesion complexes with adjacent myocytes and fibroblasts. An important difference may exist between the mechanisms involved in the therapeutic effects of CDCs and c-kit-positive CSCs. The former seems to operate predominantly via a paracrine mechanism while the latter exerts its function primarily though differentiation into integrated cardiomyocytes and coronary vessels. But this view is based on experimental findings leaving unanswered the question whether a similar difference persists in human beings.
In the prospective, randomised CArdiosphere-Derived aUtologous stem CElls to reverse ventricUlar dySfunction (CADUCEUS) trial, patients 2–4 weeks after myocardial infarction with EF of 25–45% were enrolled [58]. Autologous cells were isolated from endomyocardial biopsies, expanded in vitro, and infused in the infarct-related artery 1.5–3 months after myocardial infarction. The primary endpoint of the clinical study was the proportion of patients at 6 months who died due to ventricular tachycardia, ventricular fibrillation, or sudden unexpected death, or had myocardial infarction after cell infusion, new cardiac tumor formation, or a major adverse cardiac event (MACE; composite of death and hospital admission for heart failure or non-fatal recurrent myocardial infarction). Four treated patients, 24% in the CDC group, had serious adverse events compared with one control. At 6 months, MRI analysis of patients treated with CDCs showed reductions in scar mass and increases in viable heart mass, regional contractility, and regional systolic wall thickening. However, EF did not differ between control and treated groups [58].
Conclusions
The human heart is a highly dynamic organ regulated by a pool of resident hCSCs that modulate cardiac homeostasis and condition organ aging. Hopefully, recent findings will resolve the long debate that has divided the scientific community in strong opponents and passionate supporters of the regenerative potential of the human heart, offering a more biologically valid understanding of cardiac growth and repair. A common ground can now be found to translate this different perspective of cardiac biology into the development of novel strategies for the management of the human disease.
Non-Standard Abbreviations
- α-MHC
α-myosin heavy chain
- AMS
Accelerator mass spectrometry
- BMCs
Bone marrow cells
- CDCs
Cardiosphere-derived cells
- CSCs
Cardiac stem cells
- ECs
Endothelial cells
- EGFP
Enhanced green fluorescent protein
- hCSCs
Human CSCs
- HF
Heart failure
- HSCs
Hematopoietic stem cells
- MIMS
Multi-isotope imaging mass spectrometry
- SMCs
Smooth muscle cells
- MSCs
Mesenchymal stromal cells
- TnI
Troponin I
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bearzi C, Rota M, Hosoda T, Tillmanns J, Nascimbene A, De Angelis A, et al. Human cardiac stem cells. Proc Natl Acad Sci U S A. 2007;104:14068–73. doi: 10.1073/pnas.0706760104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Smith RR, Barile L, Cho HC, Leppo MK, Hare JM, Messina E, et al. Regenerative potential of cardiosphere-derived cells expanded from percutaneous endomyocardial biopsy specimens. Circulation. 2007;115:896–908. doi: 10.1161/CIRCULATIONAHA.106.655209. [DOI] [PubMed] [Google Scholar]
- 3.Yoon CH, Koyanagi M, Iekushi K, Seeger F, Urbich C, Zeiher AM, et al. Mechanism of improved cardiac function after bone marrow mononuclear cell therapy: role of cardiovascular lineage commitment. Circulation. 2010;121:2001–11. doi: 10.1161/CIRCULATIONAHA.109.909291. [DOI] [PubMed] [Google Scholar]
- 4.Amado LC, Saliaris AP, Schuleri KH, St John M, Xie JS, Cattaneo S, et al. Cardiac repair with intramyocardial injection of allogeneic mesenchymal stem cells after myocardial infarction. Proc Natl Acad Sci U S A. 2005;102:11474–9. doi: 10.1073/pnas.0504388102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Orlic D, Kajstura J, Chimenti S, Jakoniuk I, Anderson SM, Li B, et al. Bone marrow cells regenerate infarcted myocardium. Nature. 2001;410:701–5. doi: 10.1038/35070587. [DOI] [PubMed] [Google Scholar]
- 6.Anversa P, Kajstura J, Rota M, Leri A. Regenerating new heart with stem cells. J Clin Invest. 2013;123:62–70. doi: 10.1172/JCI63068. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 7.Hsu YC, Fuchs E. A family business: stem cell progeny join the niche to regulate homeostasis. Nat Rev Mol Cell Biol. 2012;13:103–14. doi: 10.1038/nrm3272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Logan AC, Weissman IL, Shizuru JA. The road to purified hematopoietic stem cell transplants is paved with antibodies. Curr Opin Immunol. 2012;24:640–8. doi: 10.1016/j.coi.2012.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Go AS, Mozaffarian D, Roger VL, Benjamin EJ, Berry JD, Borden WB, et al. Heart disease and stroke statistics--2013 update: a report from the American Heart Association. Circulation. 2013;127:e6–e245. doi: 10.1161/CIR.0b013e31828124ad. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Laflamme MA, Murry CE. Heart regeneration. Nature. 2011;473:326–35. doi: 10.1038/nature10147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mahmoud AI, Porrello ER. Turning back the cardiac regenerative clock: lessons from the neonate. Trends Cardiovasc Med. 2012;22:128–33. doi: 10.1016/j.tcm.2012.07.008. [DOI] [PubMed] [Google Scholar]
- 12.Shih H, Lee B, Lee RJ, Boyle AJ. The aging heart and post-infarction left ventricular remodeling. J Am Coll Cardiol. 2011;57:9–17. doi: 10.1016/j.jacc.2010.08.623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Anversa P, Leri A. Innate regeneration in the aging heart: healing from within. Mayo Clin Proc. 2013;88:871–83. doi: 10.1016/j.mayocp.2013.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Urbanek K, Cesselli D, Rota M, Nascimbene A, De Angelis A, Hosoda T, et al. Stem cell niches in the adult mouse heart. Proc Natl Acad Sci U S A. 2006;103:9226–31. doi: 10.1073/pnas.0600635103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Beltrami AP, Barlucchi L, Torella D, Baker M, Limana F, Chimenti S, et al. Adult cardiac stem cells are multipotent and support myocardial regeneration. Cell. 2003;114:763–76. doi: 10.1016/s0092-8674(03)00687-1. [DOI] [PubMed] [Google Scholar]
- 16.Kajstura J, Bai Y, Cappetta D, Kim J, Arranto C, Sanada F, et al. Tracking chromatid segregation to identify human cardiac stem cells that regenerate extensively the infarcted myocardium. Circ Res. 2012;111:894–906. doi: 10.1161/CIRCRESAHA.112.273649. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 17.Hosoda T, D'Amario D, Cabral-Da-Silva MC, Zheng H, Padin-Iruegas ME, Ogorek B, et al. Clonality of mouse and human cardiomyogenesis in vivo. Proc Natl Acad Sci U S A. 2009;106:17169–74. doi: 10.1073/pnas.0903089106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mazurier F, Gan OI, McKenzie JL, Doedens M, Dick JE. Lentivector-mediated clonal tracking reveals intrinsic heterogeneity in the human hematopoietic stem cell compartment and culture-induced stem cell impairment. Blood. 2004;103:545–52. doi: 10.1182/blood-2003-05-1558. [DOI] [PubMed] [Google Scholar]
- 19.Unno K, Jain M, Liao R. Cardiac side population cells: moving toward the center stage in cardiac regeneration. Circ Res. 2012;110:1355–63. doi: 10.1161/CIRCRESAHA.111.243014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hierlihy AM, Seale P, Lobe CG, Rudnicki MA, Megeney LA. The post-natal heart contains a myocardial stem cell population. FEBS Lett. 2002;530:239–43. doi: 10.1016/s0014-5793(02)03477-4. [DOI] [PubMed] [Google Scholar]
- 21.Shepherd BE, Kiem HP, Lansdorp PM, Dunbar CE, Aubert G, LaRochelle A, et al. Hematopoietic stem-cell behavior in nonhuman primates. Blood. 2007;110:1806–13. doi: 10.1182/blood-2007-02-075382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bergmann O, Bhardwaj RD, Bernard S, Zdunek S, Barnabé-Heider F, Walsh S, et al. Evidence for cardiomyocyte renewal in humans. Science. 2009;324:98–102. doi: 10.1126/science.1164680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kajstura J, Gurusamy N, Ogórek B, Goichberg P, Clavo-Rondon C, Hosoda T, et al. Myocyte turnover in the aging human heart. Circ Res. 2010;107:1374–86. doi: 10.1161/CIRCRESAHA.110.231498. [DOI] [PubMed] [Google Scholar]
- 24.Beausejour CM, Campisi J. Ageing: balancing regeneration and cancer. Nature. 2006;443:404–5. doi: 10.1038/nature05221. [DOI] [PubMed] [Google Scholar]
- 25.De Saint-Hubert M, Prinsen K, Mortelmans L, Verbruggen A, Mottaghy FM. Molecular imaging of cell death. Methods. 2009;48:178–87. doi: 10.1016/j.ymeth.2009.03.022. [DOI] [PubMed] [Google Scholar]
- 26.Mahajan VS, Jarolim P. How to interpret elevated cardiac troponin levels. Circulation. 2011;124:2350–4. doi: 10.1161/CIRCULATIONAHA.111.023697. [DOI] [PubMed] [Google Scholar]
- 27.Kajstura J, Urbanek K, Perl S, Hosoda T, Zheng H, Ogórek B, et al. Cardiomyogenesis in the adult human heart. Circ Res. 2010;107:305–15. doi: 10.1161/CIRCRESAHA.110.223024. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 28.Kajstura J, Rota M, Cappetta D, Ogórek B, Arranto C, Bai Y, et al. Cardiomyogenesis in the aging and failing human heart. Circulation. 2012;126:1869–81. doi: 10.1161/CIRCULATIONAHA.112.118380. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 29.Arlotta P, Macklis JD. Archeo-cell biology: carbon dating is not just for pots and dinosaurs. Cell. 2005;122:4–6. doi: 10.1016/j.cell.2005.06.037. [DOI] [PubMed] [Google Scholar]
- 30.Spalding KL, Bhardwaj RD, Buchholz BA, Druid H, Frisén J. Retrospective birth dating of cells in humans. Cell. 2005;122:133–43. doi: 10.1016/j.cell.2005.04.028. [DOI] [PubMed] [Google Scholar]
- 31.Spalding KL, Buchholz BA, Bergman LE, Druid H, Frisén J. Forensics: age written in teeth by nuclear tests. Nature. 2005;437:333–4. doi: 10.1038/437333a. [DOI] [PubMed] [Google Scholar]
- 32.Bhardwaj RD, Curtis MA, Spalding KL, Buchholz BA, Fink D, Björk-Eriksson T, et al. Neocortical neurogenesis in humans is restricted to development. Proc Natl Acad Sci U S A. 2006;103:12564–8. doi: 10.1073/pnas.0605177103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Spalding KL, Arner E, Westermark PO, Bernard S, Buchholz BA, Bergmann O, et al. Dynamics of fat cell turnover in humans. Nature. 2008;453:783–7. doi: 10.1038/nature06902. [DOI] [PubMed] [Google Scholar]
- 34.Bergmann O, Liebl J, Bernard S, Alkass K, Yeung MS, Steier P, et al. The age of olfactory bulb neurons in humans. Neuron. 2012;74:634–9. doi: 10.1016/j.neuron.2012.03.030. [DOI] [PubMed] [Google Scholar]
- 35.Spalding KL, Bergmann O, Alkass K, Bernard S, Salehpour M, Huttner HB, et al. Dynamics of hippocampal neurogenesis in adult humans. Cell. 2013;153:1219–27. doi: 10.1016/j.cell.2013.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Eriksson PS, Perfilieva E, Björk-Eriksson T, Alborn AM, Nordborg C, Peterson DA, et al. Neurogenesis in the adult human hippocampus. Nat Med. 1998;4:1313–7. doi: 10.1038/3305. [DOI] [PubMed] [Google Scholar]
- 37.Laflamme MA, Murry CE. Heart regeneration. Nature. 2011;473:326–35. doi: 10.1038/nature10147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Adler CP. Relationship between deoxyribonucleic acid content and nucleoli in human heart muscle cells and estimation of cell number during cardiac growth and hyperfunction. Recent Adv Stud Cardiac Struct Metab. 1975;8:373–86. [PubMed] [Google Scholar]
- 39.Sandritter W, Scomazzoni G. Deoxyribonucleic acid content (feulgen photometry) and dry weight (interference microscopy) of normal and hypertrophoc heart muscle fibers. Nature. 1964;202:100–1. doi: 10.1038/202100a0. [DOI] [PubMed] [Google Scholar]
- 40.Kiss R, Salmon I, Camby I, Gras S, Pasteels JL. Characterization of factors in routine laboratory protocols that significantly influence the Feulgen reaction. J Histochem Cytochem. 1993;41:935–45. doi: 10.1177/41.6.8315284. [DOI] [PubMed] [Google Scholar]
- 41.Dolezel J, Greilhuber J, Suda J. Estimation of nuclear DNA content in plants using flow cytometry. Nat Protoc. 2007;2:2233–44. doi: 10.1038/nprot.2007.310. [DOI] [PubMed] [Google Scholar]
- 42.Darzynkiewicz Z, Traganos F, Kapuscinski J, Staiano-Coico L, Melamed MR. Accessibility of DNA in situ to various fluorochromes: relationship to chromatin changes during erythroid differentiation of Friend leukemia cells. Cytometry. 1984;5:355–63. doi: 10.1002/cyto.990050411. [DOI] [PubMed] [Google Scholar]
- 43.Oberpriller JO, Oberpriller JC, Mauro A. The Development and Regenerative Potential of Cardiac Muscle. Hardwood Academic Publishers; New York, NY: 1991. pp. 227–252. [Google Scholar]
- 44.Bergmann O, Zdunek S, Alkass K, Druid H, Bernard S, Frisén J. Identification of cardiomyocyte nuclei and assessment of ploidy for the analysis of cell turnover. Exp Cell Res. 2011;317:188–94. doi: 10.1016/j.yexcr.2010.08.017. [DOI] [PubMed] [Google Scholar]
- 45.Lindahl T. Instability and decay of the primary structure of DNA. Nature. 1993;362:709–15. doi: 10.1038/362709a0. [DOI] [PubMed] [Google Scholar]
- 46.International Human Genome Sequencing Consortium Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- 47.Tsutsui H, Kinugawa S, Matsushima S. Mitochondrial oxidative stress and dysfunction in myocardial remodelling. Cardiovasc Res. 2009;81:449–56. doi: 10.1093/cvr/cvn280. [DOI] [PubMed] [Google Scholar]
- 48.Cleaver JE, Trosko JE. DNA-degradation products from mammalian cells irradiated with ultra-violet light. Int J Radiat Biol Relat Stud Phys Chem Med. 1969;15:411–24. doi: 10.1080/09553006914550701. [DOI] [PubMed] [Google Scholar]
- 49.Watt FM, Hogan BL. Out of Eden: stem cells and their niches. Science. 2000;287:1427–30. doi: 10.1126/science.287.5457.1427. [DOI] [PubMed] [Google Scholar]
- 50.Olivetti G, Giordano G, Corradi D, Melissari M, Lagrasta C, Gambert SR, et al. Gender differences and aging: effects on the human heart. J Am Coll Cardiol. 1995;26:1068–79. doi: 10.1016/0735-1097(95)00282-8. [DOI] [PubMed] [Google Scholar]
- 51.McLellan MR, Ryan MD, Breneman CM. Rank order entropy: why one metric is not enough. J Chem Inf Model. 2011;51:2302–19. doi: 10.1021/ci200170k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Endl E, Gerdes J. The Ki-67 protein: fascinating forms and an unknown function. Exp Cell Res. 2000;257:231–7. doi: 10.1006/excr.2000.4888. [DOI] [PubMed] [Google Scholar]
- 53.Jacobberger JW, Frisa PS, Sramkoski RM, Stefan T, Shults KE, Soni DV. A new biomarker for mitotic cells. Cytometry A. 2008;73:5–15. doi: 10.1002/cyto.a.20501. [DOI] [PubMed] [Google Scholar]
- 54.Bersell K, Arab S, Haring B, Kühn B. Neuregulin1/ErbB4 signaling induces cardiomyocyte proliferation and repair of heart injury. Cell. 2009;138:257–70. doi: 10.1016/j.cell.2009.04.060. [DOI] [PubMed] [Google Scholar]
- 55.Losada A, Hirano M, Hirano T. Cohesin release is required for sister chromatid resolution, but not for condensin-mediated compaction, at the onset of mitosis. Genes Dev. 2002;16:3004–16. doi: 10.1101/gad.249202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bolli R, Chugh AR, D'Amario D, Loughran JH, Stoddard MF, Ikram S, et al. Cardiac stem cells in patients with ischaemic cardiomyopathy (SCIPIO): initial results of a randomised phase 1 trial. Lancet. 2011;378:1847–57. doi: 10.1016/S0140-6736(11)61590-0. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 57.Makkar RR, Smith RR, Cheng K, Malliaras K, Thomson LE, Berman D, et al. Intracoronary cardiosphere-derived cells for heart regeneration after myocardial infarction (CADUCEUS): a prospective, randomised phase 1 trial. Lancet. 2012;379:895–904. doi: 10.1016/S0140-6736(12)60195-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chugh AR, Beache GM, Loughran JH, Mewton N, Elmore JB, Kajstura J, et al. Administration of cardiac stem cells in patients with ischemic cardiomyopathy: the SCIPIO trial: surgical aspects and interim analysis of myocardial function and viability by magnetic resonance. Circulation. 2012;126:S54–64. doi: 10.1161/CIRCULATIONAHA.112.092627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hatzistergos KE, Quevedo H, Oskouei BN, Hu Q, Feigenbaum GS, Margitich IS, et al. Bone marrow mesenchymal stem cells stimulate cardiac stem cell proliferation and differentiation. Circ Res. 2010;107:913–22. doi: 10.1161/CIRCRESAHA.110.222703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Linke A, Müller P, Nurzynska D, Casarsa C, Torella D, Nascimbene A, et al. Stem cells in the dog heart are self-renewing, clonogenic, and multipotent and regenerate infarcted myocardium, improving cardiac function. Proc Natl Acad Sci U S A. 2005;102:8966–71. doi: 10.1073/pnas.0502678102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cottage CT, Bailey B, Fischer KM, Avitable D, Collins B, Tuck S, et al. Cardiac progenitor cell cycling stimulated by pim-1 kinase. Circ Res. 2010;106:891–901. doi: 10.1161/CIRCRESAHA.109.208629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bolli R, Tang XL, Sanganalmath SK, Rimoldi O, Mosna F, Abdel-Latif A, et al. Intracoronary delivery of autologous cardiac stem cells improves cardiac function in a porcine model of chronic ischemic cardiomyopathy. Circulation. 2013;128:122–31. doi: 10.1161/CIRCULATIONAHA.112.001075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Angert D, Berretta RM, Kubo H, Zhang H, Chen X, Wang W, et al. Repair of the injured adult heart involves new myocytes potentially derived from resident cardiac stem cells. Circ Res. 2011;108:1226–37. doi: 10.1161/CIRCRESAHA.110.239046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lee ST, White AJ, Matsushita S, Malliaras K, Steenbergen C, Zhang Y, et al. Intramyocardial injection of autologous cardiospheres or cardiosphere-derived cells preserves function and minimizes adverse ventricular remodeling in pigs with heart failure post-myocardial infarction. J Am Coll Cardiol. 2011;57:455–65. doi: 10.1016/j.jacc.2010.07.049. [DOI] [PubMed] [Google Scholar]
- 65.Mohsin S, Khan M, Toko H, Bailey B, Cottage CT, Wallach K, et al. Human cardiac progenitor cells engineered with Pim-1 kinase enhance myocardial repair. J Am Coll Cardiol. 2012;60:1278–87. doi: 10.1016/j.jacc.2012.04.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Senyo SE, Steinhauser ML, Pizzimenti CL, Yang VK, Cai L, Wang M, et al. Mammalian heart renewal by pre-existing cardiomyocytes. Nature. 2013;493:433–6. doi: 10.1038/nature11682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Urbanek K, Rota M, Cascapera S, Bearzi C, Nascimbene A, De Angelis A, et al. Cardiac stem cells possess growth factor-receptor systems that after activation regenerate the infarcted myocardium, improving ventricular function and long-term survival. Circ Res. 2005;97:663–73. doi: 10.1161/01.RES.0000183733.53101.11. [DOI] [PubMed] [Google Scholar]
- 68.Torella D, Rota M, Nurzynska D, Musso E, Monsen A, Shiraishi I, et al. Cardiac stem cell and myocyte aging, heart failure, and insulin-like growth factor-1 overexpression. Circ Res. 2004;94:514–24. doi: 10.1161/01.RES.0000117306.10142.50. [DOI] [PubMed] [Google Scholar]
- 69.Torzewski M, Wenzel P, Kleinert H, Becker C, El-Masri J, Wiese E, et al. Chronic inflammatory cardiomyopathy of interferon γ-overexpressing transgenic mice is mediated by tumor necrosis factor-α. Am J Pathol. 2012;180:73–81. doi: 10.1016/j.ajpath.2011.09.006. [DOI] [PubMed] [Google Scholar]
- 70.Urbanek K, Cabral-da-Silva MC, Ide-Iwata N, Maestroni S, Delucchi F, Zheng H, et al. Inhibition of notch1-dependent cardiomyogenesis leads to a dilated myopathy in the neonatal heart. Circ Res. 2010;107:429–41. doi: 10.1161/CIRCRESAHA.110.218487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Steinhauser ML, Bailey AP, Senyo SE, Guillermier C, Perlstein TS, Gould AP, et al. Multi-isotope imaging mass spectrometry quantifies stem cell division and metabolism. Nature. 2012;481:516–9. doi: 10.1038/nature10734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kriss JP, Revesz L. The distribution and fate of bromodeoxyuridine and bromodeoxycytidine in the mouse and rat. Cancer Res. 1962;22:254–65. [PubMed] [Google Scholar]
- 73.Bailey B, Izarra A, Alvarez R, Fischer KM, Cottage CT, Quijada P, et al. Cardiac stem cell genetic engineering using the alphaMHC promoter. Regen Med. 2009;4:823–33. doi: 10.2217/rme.09.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Limana F, Urbanek K, Chimenti S, Quaini F, Leri A, Kajstura J, et al. bcl-2 overexpression promotes myocyte proliferation. Proc Natl Acad Sci U S A. 2002;99:6257–62. doi: 10.1073/pnas.092672899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gonzalez A, Rota M, Nurzynska D, Misao Y, Tillmanns J, Ojaimi C, et al. Activation of cardiac progenitor cells reverses the failing heart senescent phenotype and prolongs lifespan. Circ Res. 2008;102:597–606. doi: 10.1161/CIRCRESAHA.107.165464. [DOI] [PubMed] [Google Scholar]
- 76.Jeevanantham V, Butler M, Saad A, Abdel-Latif A, Zuba-Surma EK, Dawn B. Adult bone marrow cell therapy improves survival and induces long-term improvement in cardiac parameters: a systematic review and meta-analysis. Circulation. 2012;126:551–68. doi: 10.1161/CIRCULATIONAHA.111.086074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Trachtenberg B, Velazquez DL, Williams AR, McNiece I, Fishman J, Nguyen K, et al. Rationale and design of the Transendocardial Injection of Autologous Human Cells (bone marrow or mesenchymal) in Chronic Ischemic Left Ventricular Dysfunction and Heart Failure Secondary to Myocardial Infarction (TAC-HFT) trial: A randomized, double-blind, placebo-controlled study of safety and efficacy. Am Heart J. 2011;161:487–93. doi: 10.1016/j.ahj.2010.11.024. [DOI] [PubMed] [Google Scholar]
- 78.Hare JM, Fishman JE, Gerstenblith G, DiFede Velazquez DL, Zambrano JP, Suncion VY, et al. Comparison of allogeneic vs autologous bone marrow–derived mesenchymal stem cells delivered by transendocardial injection in patients with ischemic cardiomyopathy: the POSEIDON randomized trial. JAMA. 2012;308:2369–79. doi: 10.1001/jama.2012.25321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Leri A, Anversa P. Stem cells: bone-marrow-derived cells and heart failure--the debate goes on. Nat Rev Cardiol. 2013;10:372–3. doi: 10.1038/nrcardio.2013.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Cesselli D, Beltrami AP, D'Aurizio F, Marcon P, Bergamin N, Toffoletto B, et al. Effects of age and heart failure on human cardiac stem cell function. Am J Pathol. 2011;179:349–66. doi: 10.1016/j.ajpath.2011.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.D'Amario D, Fiorini C, Campbell PM, Goichberg P, Sanada F, Zheng H, et al. Functionally competent cardiac stem cells can be isolated from endomyocardial biopsies of patients with advanced cardiomyopathies. Circ Res. 2011;108:857–61. doi: 10.1161/CIRCRESAHA.111.241380. [DOI] [PMC free article] [PubMed] [Google Scholar]