

Abstract
Intensive scientific research devoted in the recent years to understand the molecular mechanisms or neurodegeneration in spinocerebellar ataxias (SCAs) are identifying new pathways and targets providing new insights and a better understanding of the molecular pathogenesis in these diseases. In this consensus manuscript, the authors discuss their current views on the identified molecular processes causing or modulating the neurodegenerative phenotype in spinocerebellar ataxias with the common opinion of translating the new knowledge acquired into candidate targets for therapy. The following topics are discussed: transcription dysregulation, protein aggregation, autophagy, ion channels, the role of mitochondria, RNA toxicity, modulators of neurodegeneration and current therapeutic approaches. Overall point of consensus includes the common vision of neurodegeneration in SCAs as a multifactorial, progressive and reversible process, at least in early stages. Specific points of consensus include the role of the dysregulation of protein folding, transcription, bioenergetics, calcium handling and eventual cell death with apoptotic features of neurons during SCA disease progression. Unresolved questions include how the dysregulation of these pathways triggers the onset of symptoms and mediates disease progression since this understanding may allow effective treatments of SCAs within the window of reversibility to prevent early neuronal damage. Common opinions also include the need for clinical detection of early neuronal dysfunction, for more basic research to decipher the early neurodegenerative process in SCAs in order to give rise to new concepts for treatment strategies and for the translation of the results to preclinical studies and, thereafter, in clinical practice.
Keywords: Aggregation, Ataxia, Autophagy, Calcium, Cerebellum, Mitochondria, Neurodegeneration, Polyglutamine, Purkinje cell, Therapy, Transcription dysregulation, Neuronal death
Introduction
Some 20 years ago, the first genetic defect associated with a specific spinocerebellar ataxia (SCA) subtype, hence denoted as SCA1 (SCA type 1), was identified [1]. In addition to enabling unequivocal molecular diagnosis of the disease, this finding opened up multiple lines of basic scientific research aimed at understanding the biological functions of the gene products, the identification of the cellular and molecular pathways implicated, and the disease mechanisms of pathogenesis with the ultimate objective of identifying, developing and implementing effective treatments. These efforts have identified major etiological roles of transcriptional dysregulation, protein aggregation and clearance, autophagy, alterations of calcium homeostasis, mitochondria defects, toxic RNA gain-of-function mechanisms and activation of pro-apoptotic routes, amongst others, all leading to early synaptic neurotransmission deficits, progressive spinocerebellar dysfunction and, ultimately, neuronal and specifically cerebellar Purkinje cell demise (Fig. 1 and Table 1). The identification of the molecular targets influencing the critical pathways in the pathogenesis and, in particular, those triggering the onset of symptoms open the way for effective treatments by reversing the neurotoxic process during the early stages of neurodegeneration. Whilst the underlying mechanisms of pathogenesis are not yet well understood, the authors discuss their current interests and consensus views on the different basic mechanisms triggering neurodegeneration in SCAs and the ongoing and upcoming efforts to prevent them.
Fig. 1.
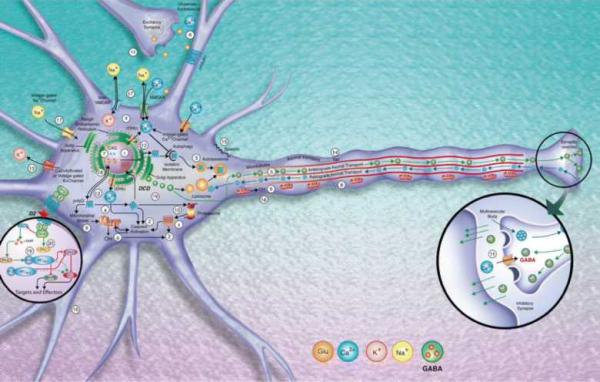
Most common molecular pathways identified triggering neurodegeneration in the SCAs. DCD dark cell degeneration (SCA2, SCA3, SCA7, SCA28). 1 Aggregation (SCA1, SCA2, SCA3, SCA6, SCA7, SCA8, SCA17, SCA35, DRPLA). 2 Caspase activation (SCA1, SCA2, SCA3. SCA6, SCA7, SCA8, SCA12, SCA17, DRPLA). 3 Autophagy (SCA1, SCA2, SCA3, SCA6, SCA7, SCA17, DRPLA). 4 Ca2+ homeostasis/signaling alterations (SCA1, SCA2, SCA6, SCA14, SCA15, SCA16). 5 Disruption of axonal transport and vesicle trafficking (SCA5, SCA11, SCA27). 6 Glutamate excitotoxicity (SCA1, SCA2, SCA3, SCA6, SCA12). 7 Interference with transcription (SCA1, SCA2, SCA3, SCA6, SCA7, SCA8, SCA12, SCA17, DRPLA). 8 Mitochondrial impairment (SCA1, SCA2, SCA3, SCA6, SCA17, SCA8, SCA17, SCA28, DRPLA). 9 Oxidative stress (SCA2, SCA3, SCA7, SCA12). 10 Alterations of proteasome degradation (SCA1, SCA3, SCA5, SCA7, SCA14). 11 Early synaptic neurotransmission deficits (SCA1, SCA2, SCA3, SCA5, SCA6, SCA7, SCA8, SCA14, SCA17, SCA31, DRPLA). 12 Unfolded protein response (UPR) (SCA1, SCA2, SCA3, SCA6, SCA7, SCA8, SCA17, DRPLA). 13 Potassium channel dysfunction (SCA13, SCA19/22). 14 Tau phosphorylation dysregulation (SCA11). 15 Neuronal membrane skeleton defects (SCA5). 16 Neurite alterations (SCA1, SCA10, SCA14). 17 Voltage-gated Na+ channel dysregulation (SCA27). 18 Proteostatic disruption (SCA26). 19 Protein phosphatase 2A (PP2A) activity dysregulation (SCA1, SCA2, SCA12). 20 Protein kinase C (PKC) activity deficits (SCA1, SCA14).
Table 1.
Genetic heterogeneity and molecular pathways underlying spinocerebellar ataxias
| SCA subtype |
Genomic location |
Gene | Protein | Function | DNA mutation |
References |
|---|---|---|---|---|---|---|
| SCA1 | 6p22.3 | ATXN1 | Ataxin-1 | Transcription regulation |
(CAG)n | [1, 34] |
| SCA2 | 12q24.12 | ATXN2 | Ataxin-2 | RNA metabolism | (CAG)n | [384–386] |
| SCA3/MJD | 14q32.12 | ATXN3 | Ataxin-3 | De-ubiquitination, transcription regulation |
(CAG)n | [387] |
| SCA4 | 16q24-ter | SCA4 | U | U | U | [388] |
| SCA5 | 11q13.2 | SPTBN2 | β-spectrin, non- erythrocytic 2 |
Neuronal membrane skeleton |
ID, MM | [389] |
| SCA6 | 19p13.2 | CACNA1 A |
CACNA1A | Ca2+ signalling/homeosta sis |
(CAG)n | [187] |
| SCA7 | 3p14.1 | ATXN7 | Ataxin-7 | Transcription regulation |
(CAG)n | [390] |
| SCA8 | 13q21 | ATXN8O S/ATXN8 |
Ataxin-8 | U | (CUG/CAG)n | [266, 271] |
| SCA9 | Reserved | U | U | U | U | [391] |
| SCA10 | 22q13.31 | ATXN10 | Ataxin-10 | Neuritogenesis | Intronic (ATTCT)n |
[232] |
| SCA11 | 15q15.2 | TTBK2 | Tau tubulin kinase 2 | Implicated in tau phosphorylation |
FM, MM | [112] |
| SCA12 | 5q32 | PPP2R2B | PPP2R2B | Regulation of PP2 activity, transcription regulation |
5’-UTR (CAG)n |
[392] |
| SCA13 | 19q13.33 | KCNC3 | KCNC3 | K+ signalling | MM | [203, 393] |
| SCA14 | 19q13.42 | PRKCG | PRKCG | Phosphorylation | ID, MM | [394] |
| SCA15a | 3p26.1 | ITPR1 | Inositol 1,4,5- triphosphate receptor |
Inositol 1,4,5- triphosphate calcium signalling |
D,MM | [217, 219] |
| SCA16 | 8q23- q24.1 |
ITPR1 | Inositol 1,4,5- triphosphate receptor |
Inositol 1,4,5- triphosphate calcium signalling |
D,MM | [218] |
| SCA17/HDL 4 |
6q27 | TBP | TBP | General transcription (TFIID complex) |
(CAG)n | [77] |
| SCA18 | 7q22-q32b | U | U | U | U | [395, 396] |
| SCA19 | 1p21-q21b | KCND3 | Potassium Voltage- Gated Channel, Shal-Related Subfamily, Member 3 |
K+ signalling | D, MM | [216] |
| SCA20 | 11q12.2- 11q12.3 |
U | U | Chromosomal duplication |
U | [397] |
| SCA21 | 7p21.3- p15.1b |
U | U | U | U | [398, 399] |
| SCA22 | 1p21-q23b | KCND3 | Potassium voltage- gated channel, Shal-related subfamily, member 3 |
K+ signalling | D, MM | [215] |
| SCA23 | 20p13 | PDYN | Prodynorphin | Synaptic transmission |
MM | [400] |
| SCA24 | U | U | U | U | U | – c |
| SCA25 | 2p21-p15b | U | U | U | U | [393] |
| SCA26 | 19p13.3b | EEF2 | Eukaryotic translation elongation factor 2 |
RNA metabolism translation elongation proteostatic disruption |
MM | [401] |
| SCA27 | 13q33.1 | FGF14 | FGF14 | Signal transduction, regulation NaV channels |
FM, MM | [402] |
| SCA28 | 18p11.21 | AFG3L2 | ATPase family gene 3-like 2 |
ATP-dependent protease activity |
MM | [13, 403] |
| SCA29a | 3p26 | U | U | U | U | [404] |
| SCA30 | 4q34.3- q35.1b |
U | U | U | U | [405] |
| SCA31 | 16q21- q22 |
BEAN | BEAN | U | Intronic (TGGAA)n |
[233] |
| SCA32 | 7q32-q33 | U | U | U | U | [406] |
| SCA33 | U | U | U | U | U | - |
| SCA34 | 6p12.3- q16.2 |
U | U | U | U | [407] |
| SCA35 | 20p13 | TGM6 | Transglutaminase 6 | Cross-linking of proteins and the conjugation of polyamines to proteins |
MM | [408, 409] |
| SCA36 | 20p13 | NOP56 | NOP56 ribonucleoprotein |
Involved in the early to middle stages of 60S ribosomal subunit biogenesis |
Intronic (GGCCTG) |
[83] |
| SCA37 | 1p32 | U | U | U | U | [410] |
| DRPLA | 12p13.31 | ATN1 | Atrophin-1 | Transcription repression (nuclear receptor co- repressor) |
(CAG)n | [411–413] |
| 16q-ADCA | 16q22.1 | PLEKHG4 | Puratrophin-1 | Intracellular signalling, cytoskeleton dynamics |
5’-UTR SNP | [414, 415] |
Genes noted in genomic location according to Ensembl
SCA29 maps to the same genomic location than SCA15
Genes noted according to HGNC
Previously noted as autosomal recessive spinocerebellar ataxia with saccadic intrusions mapping to 1p36 [416]
Cell Death in Spinocerebellar ataxias (Ivelisse Sánchez and Antoni Matilla-Dueñas)
Cerebellar cell degeneration and loss is a major neuropathological feature in spinocerebellar ataxias [2-4]. In fact, by the time the patients demonstrate ataxia, the most prominent motor symptoms of SCA, brain atrophy is already detected in most cases [3]. As disease progresses, substantial loss of the Purkinje cell layer and all four deep cerebellar nuclei is evident and the remaining neurons are atrophied and/or misplaced (heterotopy) within the cerebellum, as shown by neuroimaging and histological analyses [5-7]. However, the specific mechanisms leading to neuronal dysfunction and their eventual death in SCAs are not well understood. With this in mind, many laboratories are currently focusing on identifying early preclinical measurable markers or clinical signs with prognostic value. This would allow effective treatment strategies to be applied prior to cell loss or the irreversible disruption of the neuronal circuitry. In addition to gaining an understanding of the possible mechanisms responsible for the disease symptoms from early biomarkers and clinical signs, the morphological features indicative of a specific type of cell death are also shedding light on the possible signalling routes involved. Therefore, these findings, together with functional studies, will reveal potential targets for treatment. The general morphological features based on histological and ultrastructural observations include cell shrinkage, nuclear condensation or DNA fragmentation, vacuolation, abnormal mitochondria and neurite morphology, and changes in neurite branching complexity or synaptic connections. Of these, changes in neurite morphology have been shown to be reversible to some extent in cell and animal models of several ataxias.
Few articles have been published describing the morphological features of Purkinje cell death process during disease progression in spinocerebellar ataxias, and for obvious reasons, most of the work has been done in animal models. The common morphological features described in several genetic mouse models resemble those of “dark cell degeneration” (DCD). During this form of cell death, neurons exhibit both apoptotic and necrotic features, including caspase activation, shrunken cell bodies, protein inclusions and initially swollen mitochondria and endoplasmic reticulum. However, the cardinal morphological feature of this form of cell death is the highly electron-dense cytoplasm. Interestingly, a morphologically similar cell death is detected in linker cells—specific gonad cells present in the male reproductive system of Caenorhabditis elegans—during male reproductive development [8, 9]. This cell death type was found to be independent of two essential players in apoptotic cell death, ced-3 or ced-4. The human homologs of these two C. elegans proteins are the caspases and the apoptosis scaffold protein, apoptotic protease activating factor 1 (APAF-1), respectively [8, 9]. Similar to the DCD described in several polyglutamine (from here denoted polyQ) SCA models, dying linker cells do not show nuclear condensation and contain large numbers of single-membrane cytoplasmic vesicles [10-13]. These cytoplasmic vesicles may be responsible for the high electron density of the cytoplasm described in DCD. Likewise, caspase-3 or caspase-9 knockout mouse embryos have also shown the DCD form of cell death instead of the typical apoptosis, suggesting that DCD may be a default program in itself at least during development, or that in mammals it may exist as an alternative pathway [14]. It remains to be determined whether lack of the other caspases such as caspases 8 or 10 would elicit the same effect. Interestingly, a polyQ protein, pqn-41, was found essential for linker cell death in C. elegans [15]. Although it is not yet clear whether the dark cell death observed in polyQ SCAs or other SCAs are genetically and functionally similar to that of linker cells in C. elegans, these data suggest that polyQ SCA proteins may be in fact triggering a specialized cell death program [16, 17].
Interestingly, in addition to Purkinje cells from SCAs 2, 3, 7 and 28 mice, the ataxic Purkinje cell degeneration (Pcd) mutant mouse containing a mutation on the Zn carboxypeptidase Nna1 gene also showed similar cell death morphology [18]. Therefore, DCD may be a mechanism of cell death to which Purkinje cells are particularly sensitive. In fact, similar morphological features have been described in the cerebellum of models of AMPA-induced delayed excitotoxicity and of hypoxia [19, 20]. Purkinje neurons are highly innervated by excitatory input from granular cells in an environment containing Bergmann glia and other supporting cells which can serve to buffer glutamate levels at the synapses. It is then understandable that changes in the Ca2+ buffering capacity upon glutaminergic input due to extracellular or intracellular changes may cause or further enhance Purkinje cell dysfunction. It has been proposed that alterations in cellular functions directly or indirectly leading to Ca2+ signalling dysregulation are eventually responsible for DCD mitochondria-mediated cell death [21]. Interestingly, in both SCA2 and SCA3, ataxins 2 and 3 respectively have been shown to interact with inositol 1,4,5-triphosphate receptor type 1 (IP3R1), inducing abnormal Ca2+ release to the cytoplasm and potentially inducing Ca2+ buffering defects by the mitochondria. In fact, suppression of inositol 1,4,5-triphosphate receptor-mediated calcium signalling by inositol 1,4,5-phosphatase (Inpp5a) enzyme (5PP) overexpression or treatment with dantrolene appeared to prevent DCD in SCA2 and SCA3 mouse models [10, 11, 22]. Similarly, mutant spinocerebellar ataxia type 7 (SCA7) expression in Bergmann glia cells was shown sufficient to interfere with their glutamate uptake functions, resulting in DCD of Purkinje cells [12]. On the other hand, the mutation underlying SCA28 is caused by haploinsufficiency of Afg3l2 encoding for the inner mitochondria membrane m-AAA protease, directly resulting in respiratory chain dysfunction, increased oxidative stress and calcium buffering dysregulation [11]. It is then possible that the different mutated proteins in SCAs may ultimately trigger the same mode of cell death by exerting their effects at different levels in the signalling route.
Do these lines of evidence suggest that DCD is a common cell death mechanism in polyQ SCAs? This appears to be the case, but more scientific data are yet needed to support it. Furthermore, the temporal pattern of these morphological changes and the relationship of spinocerebellar ataxia causing mutations to the specific alterations in cellular functions leading to this specific form of cell death are not well understood. It is clear that DCD morphology has been detected in many SCAs and other polyglutamine disorders including the activation of caspases, which are key apoptotic players. However, the molecular mechanisms involved in the DCD of C. elegans and how these may relate to cell death mechanisms in mammals are just now being deciphered. Whilst it is clear that the time for therapeutic intervention should precede the late stages of the cell death pathway, we propose that dissecting the mechanisms leading to neuronal death in SCAs will yield important insights on shared signalling routes and potential early targets. Many questions still remain on what causes the defective excitatory input levels or its mismanagement as well as the Ca2+ and other signalling alterations in each of the spinocerebellar ataxias. Most importantly, understanding the critical early triggers to Purkinje cell dysfunction by each of the SCA genes should serve to propose rational therapy strategies. The most recent investigations pursuing all of these questions are herein addressed and discussed.
Dysregulation of Gene Transcription as a Trigger of Ataxia Pathogenesis (Antoni Matilla-Dueñas and Ivelisse Sánchez)
Transcriptional dysregulation in the brain has been implicated as a common generic molecular mechanism underlying pathogenesis in a myriad of spinocerebellar ataxias [23, 24]. Many transcription factors, such as the cAMP response element-binding protein TATA-binding protein (TBP) and Sp1, amongst others, contain polyglutamine regions within their primary sequence. These polyglutamine regions or domains appear necessary for their association with the chromatin in vivo and facilitate or prevent transcription, thereby acting as activators or repressors, respectively, depending on which promoter area they bind and the co-regulators with which they interact. There is already some experimental evidence suggesting that the function of polyQ could be to modulate protein–protein interactions. For example, the polyQ sequence in TBP modulates its interaction with TFIIB [25]. Further evidence has shown that mutations affecting polyglutamine expansions within ataxins in spinocerebellar ataxias interfere with transcription through different mechanisms, the most common being protein–protein or protein–DNA interactions, acetylation, phosphorylation and RNA interference, which would provide variable effects on gene expression. Among the 24 proteins directly responsible for spinocerebellar neurodegeneration in the SCAs identified to date (Table 1), most of them participate in the regulation of gene expression either at the transcriptional or posttranscriptional levels or in the regulation of cellular and molecular events that somehow target transcription [26-31]. This is the reason why, at least in the case of polyQ ataxias, they are increasingly denoted as transcriptionopathies, in which neuronal toxicity would first arise from large-scale alterations of transcription. In these ataxias, dysregulation of gene expression occurs as a consequence of the direct effects of the respective mutations on transcription or RNA expression and/or by the indirect actions exerted by aberrant interactions participated by the mutated SCA gene products. The tissue-specific neurodegenerative nature of polyglutamine expansion spinocerebellar ataxias implies that their associated polyglutamine-expanded proteins affect a discreet set of regulatory factors and specific subsets of genes that are particularly vital for normal function in the affected neuronal cells and tissues. Most of the studies aiming to identify these genes and pathways dysregulated by transcription alterations in SCAs have been assessed by global transcriptome analyses either in cellular or animal models.
Sufficient evidence has been set forth to support a link between altered histone acetylation and altered gene expression in several polyglutamine expansion disorders [32]. As a result, histone deacetylase (HDAC) inhibitors have been tested as a therapeutic approach for the treatment of these disorders in laboratory models. Treatment with these drugs has been shown to reduce the expression of proteins involved in DNA synthesis, upregulate the expression of pro-apoptotic factors, promote cell cycle arrest and differentiation and, more generally, modulate the expression of a broad range of genes, which are amongst the main reasons why the wide application of these drugs to treat neurodegeneration has been limited to date (reviewed in [33]). However, focused research exploring specific regulators of transcription at different levels may provide new therapeutic approaches to treat neurodegeneration in spinocerebellar ataxias. This is thoroughly addressed in later sections in this manuscript.
Spinocerebellar Ataxia Type 1
SCA1 was the first ataxia with the gene identified and the associated protein characterized, and therefore there is a considerable amount of knowledge acquired relative to its pathogenic mechanisms (reviewed in [34]). The first suspicion linking ATXN1, the spinocerebellar ataxia type 1 protein, to transcription was provided by the fact that Atxn1 includes an AXH sequence identified first in the high-mobility group transcription activator HMG1 [35, 36]. The AXH domain has been shown to directly regulate transcription by interacting with at least one trans-activator, SP1 [26]. Determination of the AXH structure in Atxn1 has shown that it forms stable homodimers and contains an OB fold [37, 38], a structural motif found in many oligonucleotide-binding proteins, supporting the proposed role of Atxn1 in RNA binding [39]. Furthermore, the AXH module contains a cluster of charged surface residues that provide a surface for protein–protein interactions; many of them are crucial for the regulation of transcription [40]. Among the most characterised interactions regulating transcription include both co-activators and co-repressors such as ANP32A/LANP [41], ATXN1L [42], CBF1 [43], the Capicua homolog CIC [44], GFI-1 [45], HDAC3 [46], polyQ-binding protein 1 [47], RORα-Tip60 [48], SMRTER [46] and SP1 [26]. ATXN1 has also been shown to interact with at least two RNA-splicing factors: RBM17 [49] and U2AF65 [50]. All these interactions point to the relevant nuclear functions of ATXN1 as a co-regulator of transcription-related processes. Remarkably, some of these factors have been shown to modify the pathogenesis of SCA1 in mouse and fly models [44, 45, 48, 51-53]. For instance, Rorα haploinsufficiency results in enhanced pathogenesis [48] and partial Tip60 loss delays cerebellar degeneration during mid-stage disease progression in SCA1 transgenic mice [53]. Even though much of the genetic evidence suggests that SCA1 is mainly caused by a gain-of-function mechanism, additional data from studies with knockout and transgenic mice have revealed that both share common transcriptional and molecular phenotypes. This highlights the contribution of the biological functions of ataxin-1 in SCA1 neurodegeneration, indicating the role of loss of ataxin-1 function in pathogenesis. Global microarray analysis of both knockout and transgenic mice revealed that ataxin-1 regulates genetic programs involved in cerebellar motor functions in mice through intracellular signalling [26, 27, 54]. Of these, the evolutionary conserved Wnt signalling pathway interacts with RA signalling to regulate multiple cell differentiation processes during embryonic and adult life, including neuron cell differentiation, neurite development, central nervous system (CNS) plasticity and nervous system development [55, 56]. Through the direct interaction with the transcription factor CBF1, Atxn1 acts as an integral component of the Notch signalling pathway [43]. Thus, more functions regulated by transcription are being assigned to ATXN1, which are highly relevant to understand SCA1 pathogenesis. Remarkably, modulating the transcriptional functions of ataxin-1 has been proven beneficial in transgenic mice, thereby opening new strategies to therapy [57]. Very recently, a previously undescribed function of ataxin-1 in the modulation of protein phosphatase 2a activity and the regulation of its holoenzyme composition has been described and found altered in the SCA1 mouse cerebellum [31]. Taken together, the evidence highlights that dysregulation of the biological functions of ataxin-1 underlies the early events in SCA1, further supporting the combination of both gain and loss of functions of ataxin-1 in SCA1 pathogenesis. This in turn is very relevant to the identification of therapeutic targets.
Spinocerebellar Ataxia Type 2
In SCA2, ataxin-2 has been linked to cellular RNA metabolism and endocytosis processes [58, 59] (reviewed in [60]). A role in transcription and translation regulation by ataxin-2 has been proposed through its interaction with poly(A)-binding protein 1, and the assemblage into polyribosomes suggests a role for ATXN2 in RNA metabolism [58, 61]. More recently, ATXN2 has been shown to interact with Krüppel-associated box-containing zinc finger repressor proteins [62]. Members of this family are involved in the transcriptional repression of RNA polymerase I, II and III promoters as well as in the binding and splicing of RNA, which result in crucial functions regarding the maintenance of the nucleolus, cell differentiation, and proliferation and apoptosis [63]. Thus, ATXN2 may as well act as a transcription co-regulator, together forming a multi-protein complex with ZBRK1.
Spinocerebellar Ataxia Type 3
Ataxin-3 (ATXN3), the disease protein in SCA3, interacts with several transcriptional components such as the TATA-binding protein-associated factor TAFII130 [64, 65], the cAMP response element-binding protein CBP [66, 67], the DNA repair protein RAD23 [68], the nuclear co-repressor receptor NCoR, and the histone deacetylases HDAC3 and HDAC6 [69] (reviewed in [70]). Normal ATXN3 is involved in the expression of genes implicated in stress response and extracellular matrix modelling, whereas mutant ATXN3 is associated with the upregulation of genes mainly involved in inflammatory reactions [71]. Studies have shown that ATXN3 modulates gene transcription through chromatin binding and recruitment of histone deacetylating complexes to target gene promoters [69]. In screenings for transcription factors specifically interacting with ATXN3, the forkhead box class O (FOXO) transcription factor 4 (FOXO4) was identified [72]. FOXO family transcription factors are involved in various cellular processes including regulation of cell cycle arrest, differentiation, cell death and resistance to cellular oxidative stress. ATXN3 interacts with and stabilizes the FOXO transcription factor FOXO4, and upon oxidative stress, they both translocate to the nucleus and activate manganese superoxide dismutase (SOD2) transcription, which in turn protects cells from oxidative damage. Thus, mutant ATXN3 associates with a significantly reduced capability to counteract oxidative stress, contributing to neuronal cell death in SCA3.
Spinocerebellar Ataxia Type 7
The SCA7 gene product, ataxin-7 (ATXN7), localises to the nucleus and has been shown to function as a component of the TATA-binding protein-free TAF-containing SPT3-TAF9-GCN5 acetyltransferase transcription complex [73]. ATXN7 is highly homologous to the yeast protein, Sgf73, which acts as a subunit of the SAGA chromatin remodelling and transcription complex which is made up of 21 proteins highly conserved from yeast to man. The SAGA modular complex includes, amongst others Spt, Ada, and Gcn5 acetylase and acts as a histone acetyltransferase complex like cAMP response element-binding protein (CBP) and also harbours histone deubiquitination activity (reviewed in [74]). In human and mammals, SAGA complex has multiple homologues: TATA-binding protein-free TAF complex, SPT3/TAF9/GCN5 acetyltransferase complex and the PCAF/GCN5 complex. These co-activator complexes are recruited by specific transcription factors to enhancer/promoter regions in target genes and induce the unwinding of DNA from nucleosomes via histone acetylation to upregulate transcription. In a polyQ–Atxn7 knock-in mouse model, transcription downregulation was demonstrated to be an early event leading to photoreceptor dysfunction, retinal degeneration and visual impairment [75]. SCA7 neuropathology has been linked to the functions of ATXN7-interacting proteins within the multicomponent complexes [75]. Thus, the presence of polyQ in ATXN7 would diminish the transcriptional co-activator functions of SAGA.
Spinocerebellar Ataxia Type 17
Of the nine polyglutamine diseases causing ataxia, spinocerebellar ataxia type 17 (SCA17) is caused by polyglutamine expansion (>43 glutamines) within the TATA box-binding protein (TBP) [76, 77], which is an essential transcription factor for the expression of most genes. The identification of the mutation in TBP as the direct responsible causative defect in SCA17 pointed to alterations in transcription as the main primary trigger of SCA pathogenesis. Like other polyglutamine diseases, SCA17 shows late-onset neurodegeneration that is particularly prominent in the cerebellum. Of all the identified polyglutamine proteins, TBP is the smallest and has been well characterised for its function. Thus, SCA17 has become an ideal disease model for understanding how polyglutamine expansion alters transcription and causes selective neuropathology. Although TBP associates with a myriad of transcription factors to regulate the expression of the vast majority of genes, global transcriptomics studies with transgenic SCA17 mice revealed no overt abnormalities of transcriptional profiling in the brain of these mutant mice [25], suggesting that polyglutamine expansion may selectively affect the function of specific transcription factors and their targets. In these studies, proposed negative effects of the mutant TBP on chaperone expression were uncovered. Chaperones are known to be protective against neuronal damage under cellular stress [78] and acute and chronic stresses which occur to neuronal cells in the brain over the course of ageing. Reduced levels of chaperones in the brain are expected to decrease the cellular capacity to refold polyglutamine proteins and to protect against oxidative stress during ageing, which can promote ageing-related neuropathological changes. Although improving chaperone function in neuronal cells has been proposed to reduce the toxicity of misfolded proteins [79-81], the complexity of chaperones has made it difficult to pinpoint specific targets for effective treatment. This highlights the need of deciphering the transcriptionally dysregulated pathways in SCAs for the identification of putative therapeutic targets.
Spinocerebellar Ataxia Type 36
Aberrant GGCCTG expansions are found in intron 1 of the NOP56 gene in SCA36, but these expansions are not shown to alter the levels of NOP56 RNA or protein expression. The NOP56 protein is a core component of the box C+D small nuclear ribonucleoprotein, which is required for ribosome biogenesis [82]. NOP56 and SRSF2 co-localise with RNA foci, and the transcription of MIR1292 decreases as the repeat was expanded, suggesting that a toxic RNA gain-of-function mechanism is responsible for SCA36 [83].
Dentatorubral-Pallidoluysian Atrophy
Dentatorubral–pallidoluysian atrophy (DRPLA) is a human polyQ ataxia caused by the expansion of a CAG stretch in the atrophin-1 gene. The protein atrophin-1 (ATN1) takes part in several cellular processes and functions as a bimodal transcriptional cofactor that is recruited to regulatory elements by a number of transcription factors [84-86]. Several studies provide a role of ATN1 as a nuclear co-repressor (reviewed in [87]). Atrophin-1 was shown to interact with the transcriptional repressor ETO/MTG8 and to repress transcription in tissue culture cells [88]. Drosophila atrophin mutants show defects in multiple developmental processes and derepression of several genes [84]. Both human atrophin-1 and Drosophila atrophin repress transcription in vivo when tethered to DNA, and polyQ expansion in atrophin-1 reduces repression. A recent genome-wide transcriptional profiling focusing on primary events preceding neurodegeneration in flies led to prove that mutant polyQ-expanded atrophin represses the transcription of the fat tumour suppressor gene, the function of which in this system protects from degeneration and atrophin toxicity [89]. Remarkably, in fat mutants, neurons undergo progressive degeneration with autophagic hallmarks. These data uncover specific mechanisms of toxicity by examining the dysregulated transcription profiles exerted by the expanded mutation.
Transcription Regulation by Calcium Signalling
Calcium homeostasis is critical in neurons for establishing and maintaining the synaptic transmission properties, including long-term potentiation and depression, and intracellular signalling. Ca2+ is able to specify distinct genomic responses by the differential activation of transcriptional regulators that decipher the information contained in Ca2+ signals. It triggers electrical activity-dependent gene expression, and one of the earliest genomic consequences is the induction of immediate early genes, many of them have been identified, including transcription factors, protein kinases, synaptic vesicle proteins and neurotrophins. Many immediate early genes encode transcription factors such as c-fos, fosB, c-jun and zif268, which in turn regulate the expression of late response genes the products of which contribute to the structural and functional changes underlying neuronal plasticity. Others, such as the neurotrophin brain-derived neurotrophic factor (BDNF), act directly. BDNF promotes neuronal survival, acts as a neurotransmitter eliciting postsynaptic action potentials and modulates synaptic transmission by enhancing neurotransmitter release.
A few SCAs are caused by the dysregulation of neuronal calcium homeostasis. Mutations causing insufficiency in the smooth endoplasmic reticulum calcium channel IP3R1 in humans are directly responsible for ataxia in SCAs 15 and 16. However, abnormal neuronal Ca2+ signalling may also play an important role in the pathogenesis of other ataxia subtypes, such as in SCAs l, 2, 5, 6 and 14. There is evidence supporting this. Microarray analyses of the SCA1 transgenic mouse models have revealed early-altered calcium signalling impairment [90]. These mice express significantly reduced levels of the calcium buffers calbindin and parvalbumin, ITPR1, type 1 inositol phosphate 5-phosphatase, endoplasmic reticulum (ER) calcium transporter SERCA, glutamate transporter EAAT4, EAAT4 stabilizer β-spectrin III, T-type voltage-gated calcium channels and transient receptor potential type 3 calcium channels [54, 90]. A calcium-mediated pathway that influences transcription involves the mitogen-activated protein kinase (MAPK/ERK) cascade, which transduces the information from the site of calcium signal generation at the plasma membrane to the nucleus [91]. Nuclear signalling of the MAPK cascades catalyses the phosphorylation of transcription factors, but also regulates gene expression more globally at the level of chromatin remodelling. These data point to the convergence of calcium intrasignalling pathways in the regulation of transcription.
MicroRNAs in Spinocerebellar Ataxias
Bilen et al. [92] provided the first evidence that microRNAs (miRNAs), including the anti-apoptotic miRNA bantam, limit the severity of polyglutamine repeat-induced neurodegeneration. These findings were further supported by other studies where a few microRNAs were identified on the basis of their central role in tuning the fine expression of polyQ disease-causing proteins in mammals, such as ataxins 1 and 3, and atrophin-1 and their modulatory roles of miRNA-mediated mechanisms in toxicity [93-95]. These studies extend the types of biological processes regulated by miRNAs to the long-term survival of neurons and the response and handling of toxic disease proteins. Identifying the miRNAs involved should address the nature and type of targets and the extent of the biological regulation of the occurring pathways.
The Role of Aggregation in the Neuropathogenesis of SCAs (Thorsten Schmidt, Jana Schmidt and Olaf Riess)
The accumulation of proteins is the hallmark of most neurodegenerative disorders. However, the localisation of these accumulations differs; that is, the Lewy bodies in Parkinson’s disease (PD) or dementia with Lewy bodies can be found in the cytoplasm whereas plaques and tangles formed in Alzheimer’s disease (AD) are cytoplasmic or even extracellular, and the aggregates in most spinocerebellar ataxias caused by polyglutamine expansions or Huntington’s disease (HD) are found inside the nucleus and are therefore termed neuronal intranuclear inclusion bodies (NII) [77, 96-102]. However, there are contradictory reports in SCA2 describing the existence or the lack of nuclear aggregates [103, 104], whereas in spinocerebellar ataxia type 6 (SCA6), cytoplasmic but no intranuclear aggregates have been described so far [105]. Having in most cases the intracellular localisation of the aggregated protein in common (in the nucleus) whilst the normal localisation of the affected protein differs between polyQ diseases indicates that also the pathways leading to the nuclear import and thereby to the protein aggregation may differ.
Recent studies demonstrated that major misfolded proteins associated with neurodegenerative diseases like AD, PD, HD and amyotrophic lateral sclerosis (ALS) share seeded aggregation properties of prions, whereas in all these cases self-propagation is not clear so far (reviewed in [106]). Although a prion-like neuron-to-neuron transmission of aggregates [107] has not been shown for polyglutamine diseases yet, it was demonstrated that cultured cells are able to take up aggregated polyglutamines [108], which then may seed further aggregation and recruitment of proteins [109].
In SCAs, not only the altered proteins with expanded polyglutamine repeats but also repeat-containing RNA may accumulate. In SCA8 for instance, the occurrence of CUG-positive ribonuclear inclusions both in human SCA8 patients and in transgenic mice was described [110]. Comparable RNA foci were also found in SCAs 31 and 36, respectively [111]. On the protein level, widespread tau accumulations were described in SCA11 [112]. Due to the limited postmortem neuropathological data about non-polyglutamine SCAs [111], we focus next on those SCAs caused by polyglutamine expansion.
Why are the polyglutamine-containing proteins aggregating at all? Nearly two decades ago, it was described that polyQ repeats may form β-sheet-like structures enabling polyglutamine proteins to accumulate via polar zippers [113]. This process may be favoured by the release of a polyglutamine-containing fragment out of the context of the affected protein. The data from transgenic mice present a clear picture: the use of a protein fragment containing an expanded polyglutamine gave rise to a phenotype stronger than the full-length protein [114-116]. The amino acids surrounding the polyglutamine repeat seem to modify, retard or even prevent the aggregation of the expanded protein. Interestingly, the polyglutamine repeat is located in all polyglutamine SCA tendentially at the N- or C-terminus rather than in the middle of the protein [117], thereby facilitating the release of such fragments. In this line, for several polyglutamine diseases, cleavage of the affected protein was observed [118-120]. As cleaving enzymes, both caspases and calpains were described [121-126]. Importantly, for SCA3, in vitro and in vivo data did reveal that even protein domains of ATXN3 outside of the polyglutamine repeat are able to aggregate [127-129], indicating that not only the polyglutamine-containing fragment but also the remnant of the protein contribute to the aggregation process.
In addition to the affected, expanded protein, intranuclear aggregates also contain the non-affected normal allele of the respective protein; and other SCA-associated proteins are recruited to the aggregates as well [130-132]. Besides that, several other proteins including ubiquitin, chaperones, proteasomal subunits and transcription factors were detected within the aggregates [130, 133-135], indicating that interference of the proteasomal function and transcriptional regulation are involved in the pathogenesis. Indeed, it has been shown that the ubiquitin–proteasome system (UPS) is not able to efficiently degrade polyglutamine aggregates [136] and that the UPS might even be functionally impaired by polyglutamine aggregates [137-139]. In PD, for alpha-synuclein, it was shown that its aggregates are not degraded by the proteasome but through autophagosomes [140]. These results suggest that this might also be the case for protein aggregates associated with spinocerebellar ataxias.
Whether protein aggregates in polyglutamine diseases are something bad or something good is a long and still ongoing discussion [141]. On one hand, the large accumulation itself and the recruitment of additional proteins into the aggregates may mix up the nuclear homeostasis. On the other hand, the separation of the missfolded and toxic expanded polyglutamine repeat could relieve the intracellular refolding and disposal system and thereby contribute to the restoration of a normal homeostasis. Although (large) protein aggregates are without a doubt a hallmark of polyQ-caused SCAs, their formation turned out to be no prerequisite for the development of a phenotype: protein aggregates have also been observed in brain regions which are mildly or non-affected by degeneration as well as even in peripheral tissues [99-101, 142]. Furthermore, several transgenic models developed neurological symptoms long before the first protein aggregates could be detected microscopically [143-146]. Novel techniques for the visualization of aggregates on the sub-microscopic level (like sophisticated variations of classical gel electrophoresis) [147] confirmed, however, that the formation of smaller, still soluble, accumulations precedes the onset of symptoms. This means that not the large protein aggregates itself but oligomers or microaggregates seem to be the (initial) toxic species [147-149] leading to, e.g., transcriptional dysregulation [149], whilst the larger macroaggregates could be even protective. This concept is supported by the observation that an improved solubility of polyglutamines (via upregulated chaperone activity) enhanced their toxicity, whilst intensified aggregation was beneficial [150]. For this reason, an increased disposal of the toxic proteins via the proteasome or autophagy appears to be a more promising approach than the inhibition of aggregate formation in general. In fact, the induction of autophagy has turned out to be a promising therapeutic approach [151, 152], as is discussed in the following section.
Autophagy Upregulation as a Therapeutic Strategy for Certain Spinocerebellar Ataxias (Benjamin R. Underwood and David C. Rubinsztein)
Autophagy as a Cellular Process
The cellular process of macroautophagy, which we will call autophagy, was first described by the Nobel laureate Christian de Duve nearly 50 years ago, though it is only in the much more recent past that it has become the focus of intense research [153]. Autophagy is a process by which a variety of substrates, including large cargoes such as organelles, can be degraded. It can be stimulated by a wide range of physiological stimuli (like oxidative stress) or drugs (like rapamycin) [154, 155]. On initiation, autophagosomes form at random locations in the cytoplasm before moving towards the microtubule organising centre, where lysosomes are clustered. This brings them into proximity to lysosomes and facilitates autophagosome–lysosome fusion, after which the lysosomal hydrolases degrade autophagic contents. Work in the early 1990s identified the genes coding for essential autophagy proteins in yeast, and this was the first step towards elucidation of the cellular mechanisms underlying this process [156]. Whilst a detailed description of the molecular machinery of autophagy is beyond the scope of this manuscript, some elements are relevant and worth highlighting. Initiation of autophagy requires two large macromolecular complexes. The first of these contains ULK1/2, a phosphorylation substrate for the serine/threonine kinase mammalian target of rapamycin (mTOR). Phosphorylation of ULK1/2 by mTOR leads to inhibition of autophagy. Treatment with rapamycin results in inactivation of mTOR, decreased phosphorylation of ULK1/2 and subsequent induction of autophagy [157]. The second complex contains a number of proteins including Beclin-1 and the class 3 phosphoinositide 3 kinase (PI3K) Vps34. This is important since PI3K inhibitors such as 3-methyladenine are powerful inhibitors of autophagy and therefore useful tools for laboratory investigation of this process [158]. We discuss next how understanding of the machinery of autophagy has led to pharmacological interventions to both induce and inhibit the process.
Autophagy is not just regulated at initiation. Further important machinery is required for the expansion of the autophagosome. This includes two ubiquitin-like conjugation reactions, the second of which results in the incorporation of microtubule-associated protein 1 light chain 3 (LC3) to the autophagosomal membrane. This is important as LC3 is the only known specific marker of the autophagosome and, as a result, provides the basis for many autophagy assays [159]. Fusion is the last step of the process and again requires both specific molecular machinery and normal lysosomal physiology. Preventing normal acidification of the lysosome, for example with drugs such as the H+ ATPase antagonist bafilomycin A1, prevents fusion and thus blocks autophagic flux, again providing an essential laboratory tool [160]. A much more detailed review of the current understanding of the molecular basis of autophagy can be found in [161].
Autophagy as a Therapeutic Strategy in Neurodegenerative Disease
Interest in autophagy and neurodegeneration has developed from two key observations. Firstly, autophagy is required for neuronal health. Mice deficient for essential autophagic proteins exhibit progressive neurodegeneration and intra-neuronal protein aggregation, features seen in many neurodegenerative diseases [162]. Loss of autophagy in the central nervous system causes neurodegeneration in mice [163]. Subsequently, dysfunctional or abnormal autophagy has been found in a number of neurodegenerative disease models, including inborn errors of metabolism, Alzheimer’s disease, Huntington’s disease, Parkinson’s disease and frontotemporal dementia [164-168]. Changes in autophagy have been reported in models of spinocerebellar ataxia. For example, mutant ataxin-3, the causative protein abnormality in SCA3, has been shown to enhance the autophagic degradation of parkin, which may in turn explain some of the parkinsonian features seen in this condition [169].
The second observation, which has linked the fields of autophagy and neurodegeneration, is that mutant aggregate-prone cytoplasmic proteins which underpin many of these diseases are autophagic substrates. This was first shown for mutant huntingtin, but has been subsequently expanded to show that mutant forms of tau, ataxin-3 and α-synuclein are similarly degraded by autophagy [155, 170-172]. These substrates probably have a high dependency on autophagy because their oligomeric and higher-order species cannot be degraded by the proteasome as they cannot access its narrow opening. These findings suggest the possibility of ameliorating conditions caused by such mutant proteins by upregulating autophagy. Subsequently, autophagy upregulation with a variety of different drugs has shown improvement in the phenotype of cellular, fly, zebrafish and mouse models of Huntington’s disease [173, 174]. Similar results have been obtained in mouse models of Alzheimer’s disease and Parkinson’s disease, where autophagy has been upregulated by both drugs and via genetic upregulation of autophagy-inducing proteins [175-177]. Crucially, these findings in basic science and laboratory models of disease are beginning to be translated into potential therapies. Indeed, safety trials of autophagy-upregulating drugs have been initiated in Huntington’s disease.
Autophagy and Spinocerebellar Ataxia
Given the similarity between Huntington’s disease as a polyglutamine disorder and the wide involvement of autophagy in protein misfolding neurodegenerative disease, it is no surprise that autophagy has been investigated in the context of SCAs caused by polyglutamine expansions, where the mutant protein has a significant cytoplasmic residence time (nuclear proteins are inaccessible to autophagy). Genome-wide screens for modifiers of toxicity in SCA3 highlighted autophagy as an important pathway, where overexpression of key autophagy proteins suppressed toxicity in a Drosophila melanogaster model of SCA3 [178]. Similarly, ataxins 3 and 7 have been shown to be autophagy substrates in cell models, as have mutant proteins that lead to SCA but which do not exhibit polyglutamine expansions, such as mutant protein kinase c gamma which causes SCA14 [170, 179-181]. In these models, pharmacological induction of autophagy has been shown to decrease toxicity. The concept of ameliorating toxicity by inducing autophagy has been expanded to rodent models of SCA3, both using temserolimus, a rapamycin analogue, or by overexpression of beclin-1; in both cases, improvement in disease phenotype was seen [152, 182]. Given the body of evidence, both broadly in neurodegeneration and in several specific SCAs, and the identification of FDA-approved drugs which induce autophagy but have minimal side effects, there is now the real prospect of beginning trials of potential disease-modifying drugs in patient populations.
Ataxias as Channelopathies (Daniel R. Scoles and Stefan-M. Pulst)
Channelopathies are clinically definable syndromes that are caused by mutations affecting transmembrane channel functions. Transmembrane channels are important for the normal function of electrically excitable tissues including the heart, muscle, peripheral and central nervous system, and channelopathies can involve any of these tissues. Although the type of mutation and channel involved gives rise to significant phenotypic variability, channelopathies share some common phenotypic features: they are often associated with episodic or paroxysmal dysfunction such as seizures, paroxysmal paralyses or movement disorders, or episodic ataxias. Next, we discuss ataxia disorders that arise from transmembrane channel mutations and the resultant physiology and patient phenotypes, not all of which are episodic.
Transmembrane channels fall into two main categories, including “selectively permeable” or “gated” ion channels (both voltage-gated and ligand-gated) and active transporters. Ion channels and active transporters have complementary functions: active transporters maintain ion gradients of physiologically relevant ions (Na+, K+, Cl−, Ca2+, H+) in an energy-dependent fashion (usually ATP) and provide a driving force that moves ions through gated ion channels upon stimulation to open by voltage changes or ligand binding. Some electrogenic active transporters also utilize ion gradients to aid the movement of the transported ions, including glutamine, serotonin, norepinephrine and dopamine transporters [183, 184]. Mutations in these transmembrane channels disrupt electrical signals required for normal neuronal or muscle function, leading to clinically defined syndromes known as channelopathies (Table 2).
Table 2.
Channel genes of the ataxia channelopathies
| Gene | Ataxia | Mutation | Channel type |
|---|---|---|---|
| CACNA1A | SCA6 | CAG repeat | VG |
| EA−2 | Frameshift, splice site, missense |
||
|
KCNC3
(Kv3.3) |
SCA13 | Missense | VG |
|
KCNN3
(hSKCa3) |
Sporadic Ataxia | CAG repeat | VG |
| KCNA1 | EA-1 | Missense | VG |
| KCND3 | SCA19/22 | Missense, 3 nt in-frame deletion |
VG |
| ITPR1 | SCA15/16 | Heterozygous gene deletion |
LG |
| SLC1A3 | EA | Missense | LG/AT |
| ATP1A3 | EA | Missense | AT |
| ATP2B3 | XLCCA | Missense | AT |
SCA, spinocerebellar ataxia; EA, episodic ataxia; XLCCA, X-linked congenitalcerebellar ataxia. VG voltage gated; LG, ligand gated; AT, active transporter.
Voltage-Gated Ion Channels
Each of the voltage-gated ion channels (VGICs) has a pore-forming α-subunit that opens and closes at specific membrane potentials, resulting in a tightly timed passage of ions across the membrane. Voltage-gated sodium channels and voltage-gated calcium channels are typically closed at the resting potential and have active roles in neuronal depolarization, whilst voltage-gated potassium channels function in repolarization and repetitive firing. Defects in voltage-gated chloride channels are unrelated to ataxias, but cause renal failure and myotonia [185, 186]. There are five VGICs that when mutated give rise to ataxia disorders, including CACNA1A, KCNC3, KCNN3 (hSKCa3), KCNA1 and KCND3.
CACNA1A
CACNA1A encodes the α1a pore-forming subunit of the P/Q-type calcium channel Cav2.1. Distinctly different cerebellar diseases are caused by CACNA1A mutations, dependent upon the nature of the mutation.
SCA6 is an inherited progressive ataxia caused by CAG expansion repeat mutation in the CACNA1A gene [187]. The CAG repeat is located in a 3′-alternatively spliced exon and codes for a polyQ repeat. SCA6 patients have a relatively pure cerebellar phenotype and slow progression [187-189].
Episodic ataxia type 2 (EA-2) is caused most frequently by CACNA1A frameshift mutations or truncations as well as missense mutations that result in Cav2.1 loss of function [190-192]. Some patients will develop a progressive degenerative ataxia. Pace-making precision of Purkinje cells (PCs) is defective in mice harbouring a spontaneously occurring CACNA1A point mutation [193]. However, SCA6(84Q) knock-in mice had PCs with aggregated Cav2.1, but normal electrophysiological phenotype, suggesting that SCA6 is caused by an age-dependent partial toxic gain of function related to Cav2.1 aggregation [194].
Missense mutations in CACNA1A are also responsible for familial hemiplegic migraine type 1 (FHM1). These mutations have mixed effects on Cav2.1 channel function, commonly including channel activation at more negative potentials and either increases or decreases in channel current and speed of recovery or inactivation [195-199]. Of note is that some individuals with FHM1 may develop a progressive ataxia in late life.
KCNC3 (Kv3.3)
Spinocerebellar ataxia type 13 (SCA13) is caused by missense mutations in the voltage-gated potassium channel KCNC3 (Kv3.3). Depending on the nature of the mutation, SCA13 can present as a dominant late-onset progressive ataxia [200] or a relatively stationary early-onset ataxia at times associated with mental retardation [201]. Initially, two KCNC3 mutations were identified, including R420H in a Filipino and F448L in a French pedigree [202]. The R420H mutation changes a highly conserved amino acid that is localised inside the S4 transmembrane domain at the voltage-sensing region and leads to a dominant negative allele [202, 203]. The F448L mutation alters an amino acid just inside the S5 transmembrane domain and resulted in a gain-of-function allele [202]. More extensive studies of 260 familial ataxia patients in Europe and 327 sporadic and familial ataxia cases in the USA demonstrated that KCNC3 mutations were relatively rare, but showed that the R420H and R423H mutations were recurrent [204, 205].
KCNN3 (hSKCa3)
Upon expression of a truncated dominant negative allele of the slow calcium-activated potassium channel, mice developed an ataxia that could be ameliorated with riluzole [206]. This led Figueroa and colleagues [207] to examine two polyglutamine tracts in hSKCa3 for mutations in 122 patients with autosomal dominant or sporadic ataxias. The study did not reveal any clear CAG expansion mutations, but showed a possible association of long normal KCNN3 CAG alleles and the presence of ataxia. Further studies are needed to address the possible association of KCNN3 and ataxia in humans.
KCNA1
Episodic ataxia type 1 (EA-1) is a dominantly inherited disease caused by multiple missense mutations in the KCNA1 gene, which encodes the alpha subunit of the Kv1.1 channel [190, 208]. The disease is characterised by myokymia and episodic skeletal muscle contractions [209]. Mutations in KCNA1 cause loss of function and are associated with nerve hyperexcitability [210]. Some mutations cause impairment to both subunit tetramerisation and membrane targeting [211]. Rodent models utilizing Kv1 channel inhibitors demonstrated that normal Kv1 channels function to suppress PC hyperexcitability [212]. Consistent with this finding, a Kcna1 V208A/+ mouse model of EA-1 demonstrated increased GABAergic inhibitory postsynaptic currents of PCs in electrophysiological recordings compared to wild-type mice [213]. Additionally, a Kcna1 S309T/+ ENU mutagenized rat model of EA-1 with myokymia was characterised by motor incoordination and seizures [214].
KCND3
Recently, five groups in the Netherlands, France, US, Japan and Taiwan identified mutations in KCND3 encoding Kv4.3, a Shal-related voltage-gated potassium channel that gives rise to a transient outward A-type K+ current [215, 216]. Two of the five groups had independently mapped an ataxia locus to partially overlapping segments of human chromosome 1 in prior studies, but with designations of SCA19 and SCA22.
KCND3 mutations occurred worldwide and in individuals with different ethnic backgrounds. Two of the mutations were recurrent in different ethnic groups. Initial in vitro characterisation indicated that mutant alleles led to loss of function, although a dominant negative effect of these alleles has not yet been demonstrated.
Ligand-Gated Ion Channels
The ligand-gated ion channels share a common pore-forming architecture that consists of five protein subunits. These channels open after binding of a specific ligand, allowing passage of ions through the channel pore. Ligand/receptor pairs include nicotinic acetylcholine/nAChR, γ-aminobutyric acid (GABA)/GABAR, glycine/GlyR, serotonin/5-HT3, inositol-3-phosphate (IP3)/IP3R, and glutamate/(NMDAR, AMPAR and kainate receptor). Ataxia is an uncommon manifestation of mutations in ligand-gated ion channels.
ITPR1
Spinocerebellar ataxia type 15 (SCA15, and also SCA16) [217] is caused by heterozygous gene deletions of the inositol 1,4,5-triphosphate receptor gene type 1 (ITPR1) [218, 219]. SCA15/16 is a dominantly inherited slowly progressing ataxia characterised by head tremor and cerebellar atrophy. The ITPR1-encoded channel IP3R is an intracellular ligand-gated Ca2+ channel located to the membranes of the endoplasmic or sarcoplasmic reticulum.
Active Transporters
The active transporters share a common function as transmembrane ion pumps powered by a nucleotide triphosphate, typically ATP that pumps specific ions against a concentration gradient. The classic example is the Na+/K+ ATPase that generates a negative charge inside the plasma membrane that supports neuron or muscle resting potentials by pumping three Na+ ions out of the cell in exchange for two K+ ions that are pumped in from the outside. Mutations in exchange transporters can result in changes in the ratio of ions that are exchanged and loss of electrogenesis, or complete loss of function. Active transporter types exist that can pump multiple ions including Na+, K+, H+, Mg2+, Cu2+ and Ca2+, as well as amino acids, cholesterols, hormones and neurotransmitters. We discuss next syndromes associated with mutation in Ca2+ and glutamate transporters.
SLC1A3
SLC1A3 encodes a glutamate/aspartate transporter known as glutamate transporter excitatory amino acid transporter 1 (EAAT1), or glutamate aspartate transporter. EAAT1 has dual functions as a glutamate transporter as well as a ligand-gated anion channel [220]. Investigation on one patient with episodic ataxia, seizures, migraine and alternating hemiplegia identified the cause as a P290R missense mutation in the EAAT1 fifth transmembrane domain [221]. This mutation caused a dominant negative effect that lowered SLC1A3 expression and the transport of glutamate by EAAT1. Another SLC1A3 mutation, causing a V449C substitution in the hydrophobic C-terminal domain of EAAT1, resulted in loss of glutamate transport but no alteration on anion transport, demonstrating that the two functions are uncoupled [222].
ATP1A3
Mutation in the ATP1A3 gene is well established as the cause of early-onset alternating hemiplegia [223] and rapid-onset dystonia Parkinsonism (RPD) [224]. Genetic analysis of two unique initially misdiagnosed paediatric cases of RDP with motor delay and episodic ataxia identified causative mutations in ATP1A3 [225]. In one case, the mutation, a R756H substitution in the intracellular motif of the channel protein, was inherited, whilst the other mutation was a de novo D923N substitution that was previously shown to abrogate channel function by preventing binding of Na+ [226].
ATP2B3
X-linked congenital cerebellar ataxia is caused by mutation in the ATP2B3 gene encoding a calcium channel designated as plasma membrane Ca2+ ATPase 3 (PMCA3) [227]. PMCA3 functions to extrude Ca2+ from the cell and is inactivated by calmodulin binding. When intracellular Ca2+ rises, it binds calmodulin, releasing it from PMCA3 and activating the channel. The G1107D mutation in the calmodulin-binding domain of PMCA3 results in loss of function and abnormal elevation of intracellular Ca2+.
Secondary Channel Abnormalities
In addition to direct mutations of channel proteins, voltage-gated and ligand-gated channels have been implicated as secondary mediators of polyQ pathology. These studies were conducted in transgenic animal models of SCAs examining spontaneous PC firing in cerebellar slice preparation [228-230]. Whereas the presence of mutant ataxin-3 results in depolarization block of PCs, mutant ataxins 1 and 2 lead to a slowing of PC spontaneous firing. Hansen and colleagues [228] showed that decline in motor performance closely mirrored the progressive slowing of PC firing frequency in a transgenic ATXN2[Q127] animal model.
As shown by reciprocal deletion/duplication syndromes such as Charcot–Marie–Tooth disease (CMT1A) and hereditary neuropathy with liability to pressure palsy [231], loss of tight dosage control can be pathogenic for some genes. Haploinsufficiency at the ITPR1 locus causes SCA15/16, but functional activation of IP3R1 in the presence of mutant polyQ proteins is also involved in PC death. Using in vitro PC culture and animal models, the Bezprozvanny laboratory found that interaction of mutant ataxin 2 or 3 with the IP3R1 led to exaggerated Ca2+ release and subsequent PC death in culture and in vivo, which could be prevented by pretreatment with dantrolene [10, 11].
Therapeutics for Channelopathies
Channel proteins belong to a class of proteins in which the structure and function are extremely well understood. Thus, they offer great promise for the rational design of compounds that either up- or downregulate the function of the respective channel. Alternatively, downstream targets of signalling through the channel may be amenable to therapy. A challenge for these approaches is the abundant expression of channel proteins not only in different neuronal populations in the CNS but also in other tissues, thus increasing the potential for significant side effects. Direct modulation of gene expression in the CNS via viral gene delivery or use of antisense molecules may offer hope for targeted therapies in the future.
RNA Toxicity as an Underlying Mechanism of Spinocerebellar Ataxias (Karen N. McFarland and Tetsuo Ashizawa)
RNA toxicity in a number of spinocerebellar ataxias is proposed as a gain-of-function mechanism for neurodegeneration. In these cases, the mutation is the expansion of a microsatellite repeat sequence in non-coding regions of the mutated gene. Ataxic phenotypes result from CTG expansions in ATXN8OS in SCA8, ATTCT expansions in ATXN10 in SCA10 [232], TGGAA expansions in TK2/BEAN1 in SCA31 [233], GGCCTG expansions in the NOP56 gene in SCA36 [83] and CGG expansions in FMR1 in fragile X-associated tremor and ataxia syndrome (FXTAS) [234].
Numerous lines of evidence support a gain-of-function model at the RNA level, as we illustrate by example of spinocerebellar ataxia type 10 (SCA10). SCA10 results from the expansion of a normally polymorphic ATTCT pentanucleotide repeat in intron 9 of the ATXN10 gene on chromosome 22q13 [232]. With normal alleles ranging from 9 to 32 copies [235], the repeat expands up to 4,500 repeats in SCA10 patients [232]. The repeat expansion does not appear to cause a protein loss of function as mice heterozygous for an Atxn10 null allele are pathologically and behaviourally normal [236]. Additionally, individuals with an ATXN10 haploinsufficiency, caused by a balanced translocation of chromosome 22, which disrupts one copy of ATXN10, are phenotypically normal [237].
Ataxin-10 (ATXN10) transcript levels are produced to levels similar to controls in fibroblasts isolated from SCA10 patients [236]. Furthermore, ATXN10 pre-mRNA is processed normally with proper splicing of intron 9, which bears the repeat expansion [236]. Thus, the ATTCT expansion is transcribed into AUUCU RNA, which accumulates into RNA foci. The RNA foci are found in patient fibroblasts as well as in the brains of transgenic mice expressing untranslated AUUCU repeat expansions [238, 239]. The SCA10 RNA foci in these cells co-localise with the RNA-binding protein (RBP), heterogeneous nuclear ribonucleoprotein K (hnRNP K). We hypothesize that the normal functions of various RBPs are inhibited by binding and sequestration by the AUUCU RNA expansion, thereby resulting in a loss of function for the RBP.
hnRNP K is ubiquitously expressed and contains a nuclear-cytoplasmic shuttling “KNS” domain as well as three KH (K homology) domains, responsible for RNA and ssDNA binding [240, 241]. hnRNP K has multiple functions within the cell, including splicing regulation, translational control, chromatin remodelling, transcription regulation and mRNA stability, and is a component of stress granules [242]. hnRNP K also partners with diverse proteins including protein kinase C delta (PKCδ), which serves to phosphorylate hnRNP K [243, 244]. As our hnRNP K sequestration hypothesis suggests, preventing normal hnRNP K function by AUUCU binding will have a broad range of deleterious effects on cellular function (Fig. 2).
Fig. 2.
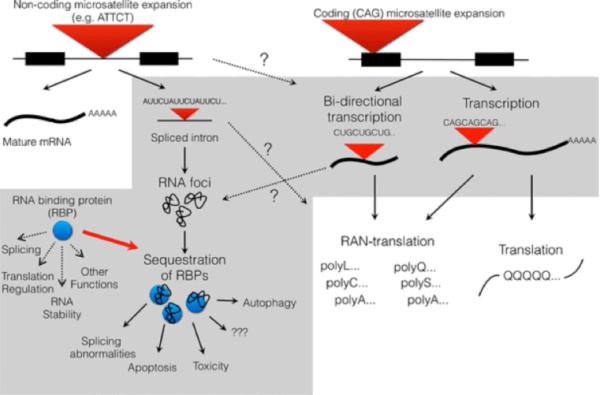
Toxic RNA species and their effects. Non-coding microsatellite expansions, such as the ATTCT expansion in SCA10, are transcribed, but not translated. The RNA form foci which bind and sequester RNA binding proteins (RBPs). Sequestration of the RBP is hypothesized to alter the normal function of RBP and have deleterious downstream effects on RNA splicing, translational regulation, apoptosis and autophagy, in addition to other uncharacterized and unknown effects. Coding microsatellite expansions have long been recognised as a protein gain of function producing polyglutamine-containing proteins. However, their gene loci can also serve as a substrate for bidirectional transcription. The natural sense and antisense transcripts may then be used in RAN translation.
In support of this hypothesis, we find that the normal functions of hnRNP K are impaired in SCA10 fibroblasts as well as in SCA10 transgenic mouse models expressing untranslated AUUCU repeat expansions. Normal hnRNP K–PKCδ interactions are decreased, resulting in apoptosis triggered by the increased localisation of PKCδ to the mitochondria [238]. The function of hnRNP K as a regulator of alternative splicing is perturbed, as seen in altered levels of splicing isoforms of hnRNP K target genes in SCA10 patient fibroblasts [243]. Additionally, these molecular phenotypes are mimicked by knockdown of hnRNP K using small inhibitory RNA (siRNA) in normal fibroblasts [243].
Similar mechanisms are proposed for the other SCAs that are caused by non-coding repeat expansions. RNA foci are formed in SCA31 Purkinje cells and the RNA repeat sequence binds the RBPs, splicing factor arginine/serine rich 1 and SRFS9, in vitro [233]. Similarly, RNA foci form from the GGCCUG expansion in SCA36, and these foci co-localise with the SRFS2 protein in lymphoblastoid cell lines from patients [83]. Sequestration of RBPs Sam68/KHDRBS1, hnRNP G and MBNL1 by CGG-containing RNA foci occurs in FXTAS [245].
Loss of Function of RNA-Binding Proteins Leads to Spliceopathy
Loss of RBP function via sequestration of the RBPs by the repeat-containing RNA foci alters the regulation of alternative splicing and downstream dysfunction of these proteins. Such spliceopathies are a common underlying theme amongst numerous non-coding repeat expansion disorders, including not only the SCAs as discussed above but also the neuromuscular disorders myotonic dystrophy types 1 and 2 (DM1 and DM2). DM1 represents the archetypical non-coding repeat disorder. CUG-containing RNA foci form within the nucleus of DM1 cells and co-localise with proteins from the muscleblind family (MBNL1) in cardiac and skeletal muscle as well as neurons of DM1 patient cells [246, 247]. Altered splicing of insulin receptor pre-mRNA in skeletal muscles from DM1 patients correlates with a decreased response to insulin in culture [248]. Alterations in the levels of splice isoforms of cardiac troponin T (TNNT2) pre-mRNA are found in DM1 heart and skeletal muscles [249]. Aberrant ClC-1 pre-mRNA in DM1 cells causes a loss of ClC-1 protein function [250, 251]. Correcting the splicing patterns of ClC-1 can reverse the problems of chloride conductance, hyperexcitability and myotonia [252]. Thus, preventing the interaction between RNA expansion and their interacting RBPs may represent a universal therapeutic target for many of these disorders.
Repeat Interruptions as a Phenotypic Modifier
The nucleotide composition of the repeat expansion may hypothetically influence this disease mechanism. Repeat interruptions in SCA10 expansion dramatically influence repeat stability and alter canonical rules of genetic anticipation [253]. Furthermore, as RNA-binding proteins are known to interact in a sequence-specific manner, repeat interruptions within the expansions may differentially interact with a variety of RNA-binding proteins. Such a hypothesis may explain phenotypic differences that are observed for a single SCA disorder, as well as other repeat expansion disorders, and is evidenced in SCA1, SCA2, SCA10, SCA31 and DM1 [254-265].
Bidirectional Transcription
Antisense transcripts from microsatellite repeat expansions are increasingly recognised as potential auxiliary toxic agents in the pathogenic process. Antisense transcripts result from bidirectional transcription at the expansion, a concept first recognised in SCA8 [266] and subsequently found in other trinucleotide repeat disorder including myotonic dystrophy type 1 [267], SCA7 [268], Huntington’s disease [269] and Huntington’s disease like-2 [270].
In SCA8, a non-coding CTG expansion was first identified in the ATXN8 gene in an ataxia patient [266]. Both CUG and CAG transcripts originate from the expansion and are found in patient tissue samples, indicating that transcription occurs from both strands of the microsatellite expansion [271]. CAG transcripts produce polyglutamine inclusions [272], whilst CUG transcripts result in RNA foci [110, 273]. As a result, understanding of the pathogenic mechanism in SCA8 is confounded by a gain of function at both protein and RNA levels.
Bidirectional transcription will likely become a more prominent mechanism in other ataxias that have long thought to be protein gain-of-function mutations. With the advent of next-generation sequencing technologies allowing for deep sequencing of RNA transcriptomes, an increasing number of coding trinucleotide repeat disorders (e.g. SCA1/ATXN1, SCA2/ATXN2, SCA3/ATXN3, SCA6/CACNA1A, SCA7/ATXN7, SCA17/TBP and DRPLA) are recognised as having natural antisense transcripts to varying degrees [274]. In addition, antisense transcripts to the huntingtin gene are produced in cells from the frontal cortex of HD patients and contain the CTG repeat [269].
Exact mechanisms downstream of the bidirectional transcription of repeat expansions have not been fully worked out. Antisense transcription can regulate gene expression through a variety of mechanisms, which may be perturbed by the repeat expansion [274]. Antisense transcripts to the CAG repeat expansion disorders could also follow the same downstream pathways described above (Fig. 2). Untranslated CUG and CAG repeat expansions can separately trigger the formation of RNA foci and have similar effects on spliceopathy phenotypes caused by the loss of RBP function [275].
Repeat-Associated Non-ATG-Mediated Translation (RAN Translation)
A new mechanism for neurodegeneration in SCA8 was found when polyglutamine proteins were produced after the only ATG site upstream of the CAG expansion was mutated [276]. Further experimentation found that proteins were translated in all three frames—polyglutamine, polyalanine and polyserine—from constructs that lacked the traditional ATG translation start site. Such a phenomenon was dubbed repeat-associated non-ATG (RAN) translation and is dependent on repeat length as well as the formation of an RNA hairpin secondary structure. In vivo evidence for polyalanine proteins was found in the Purkinje cells of SCA8 postmortem cerebellum and in SCA8 transgenic mice. More recently, evidence of RAN translation is seen from the GGGGCC expansion in the c9orf72 locus of ALS-FTD [277]. Furthermore, indications for RAN translation are also found in cardiomyocytes and myoblasts from mouse models of DM1. Whilst RAN translation has not yet been shown to have disease-causing effects, it may represent a new pathogenic mechanism as many of the expansion-containing RNA molecules, both from sense and antisense transcripts, have the potential to become substrates in this pathway [278].
Expansion-containing RNA molecules, whether through their native sense or antisense transcripts, are important players in the pathways that lead to disease phenotypes. The degree of interplay (Fig. 2) between these pathways, which are described above, has yet to be determined and will complicate further understanding of pathogenic processes. Yet as many of these disorders share similar pathways, the development of therapeutic targets may benefit from exploiting these common features.
Mitochondria and Neurodegeneration in SCAs (Franco Taroni and Stefania Magri)
In the last 20 years, it has become quite evident that mitochondrial dysfunction may severely affect the proper function of the cerebellum and its afferent and efferent connections. The occurrence of ataxia in mitochondrial disorders related to defined primary mutations of mitochondrial DNA (mtDNA) is well known. This is the case with myoclonic epilepsy with ragged red fibers caused by the 8344A>G mutation; mitochondrial encephalomyopathy, lactic acidosis and stroke-like episodes caused by the 3243A>G mutation; maternally inherited Leigh syndrome caused by point mutations in the mtDNA genes ATP6, ND1 and tRNA Lys ; NARP caused by ATP6 mutations at nt 8993; and Kearns–Sayre syndrome associated with large-scale rearrangements of mtDNA [279, 280]. In these cases, the impaired function of the mitochondria is that which traditionally characterises these organelles as the “powerhouses of the cell”: production of ATP via the concerted action of the tricarboxylic acid cycle and the respiratory chain/oxidative phosphorylation system. Another example of cerebellar ataxia caused by energy production defect is that associated with deficiency of coenzyme Q10 (CoQ10) [281]. CoQ10, also known as ubiquinone, is a small lipid-soluble carrier with important antioxidant properties which is required to transfer electrons in the respiratory chain from complexes I and II to complex III [282]. In addition to its electron transfer properties, as a well-known antioxidant, CoQ10 protects cells from reactive oxygen species (ROS) damage by scavenging free radicals [283]. In the autosomal recessive form SCAR9, mutations in the gene encoding the mitochondrial kinase ADCK3 [284, 285] block the biosynthesis of CoQ10, ultimately resulting in reduced activity of respiratory chain. Recently, cerebellar phenotypes recapitulating the human disease have been generated by blocking CoQ10 biosynthesis in two conditional knockout mice [286]. An impairment in energy production also underlies the pathomechanisms leading to cerebellar ataxia in some recessively inherited defects of nuclear DNA genes (POLG and C10orf2) encoding mitochondrial proteins involved in mtDNA replication and maintenance [287, 288]. Several ataxic phenotypes are associated with mutations of the POLG gene, encoding the catalytic subunit A of mitochondrial DNA polymerase g [289]. POLG-related ataxias have an overlapping clinical spectrum of disorders (ataxia neuropathy spectrum or ANS) organized around ataxia and neuropathy in the absence of significant muscle weakness or myopathy [280, 288, 290]. ANS embraces the phenotypes also referred to as mitochondrial recessive ataxia syndrome [291] and sensory ataxia neuropathy dysarthria and ophthalmoplegia [292] and may partly overlap with spinocerebellar ataxia with epilepsy/myoclonic epilepsy myopathy sensory ataxia phenotype [290]. As a consequence of mitochondrial DNA polymerase mutations, these disorders are characterised by qualitative (multiple deletions) or quantitative (depletion) alterations of mtDNA, which ultimately result in impairment of energy production. Depletion of mtDNA and energy defect is also the mechanism that underlies infantile-onset spinocerebellar ataxia, a recessively inherited disorder caused by mutation in the C10orf2 gene (previously PEO1/TWINKLE) which encodes twinkle, a specific mitochondrial helicase involved in DNA replication, and one of its smaller isoform, twinky, whose function is currently unknown [293]. More recently, a pathogenic mechanism involving defective mtDNA maintenance and mitochondrial dysfunction has been proposed also for ataxia with oculomotor apraxia type 1 and spinocerebellar ataxia with axonal neuropathy (SCAN1) by demonstrating mitochondrial localization and function of the proteins defective in these disorders, aprataxin [294] and tyrosyl-DNA phosphodiesterase [295], respectively. The pathogenesis of Friedreich ataxia, a paradigm for mitochondria-related neurodegeneration, is more complex and controversial [296-298]. The main pathophysiological consequences of frataxin deficiency are a severe impairment of iron–sulfur cluster (ISC) biosynthesis that affects ISC-containing enzymes localised in various cellular compartments including the mitochondria (complexes I, II and III of the respiratory chain and aconitase of the tricarboxylic acid cycle), cytosol and nucleus [299]; mitochondrial iron overload [300, 301]; and an increased sensitivity to oxidative stress [300, 302].
The above discussion emphasizes the sensitivity of the spinocerebellar system to bioenergetics perturbations. However, a large number of studies in late-onset neurodegenerative disorders including spinocerebellar ataxias have clearly shown that other mitochondrial functions are greatly important for the disease process, including organelle shaping and trafficking, mitochondrial quality control and apoptosis [303, 304].
Apoptosis via the intrinsic mitochondrial pathway is, in fact, the most common mechanism of cell death in neurodegenerative diseases [303, 305], and direct activation of apoptotic pathways has been demonstrated in several polyglutamine expansion diseases including spinocerebellar ataxias [306-309]. In the intrinsic pathway, several intracellular signals, including DNA damage, endoplasmic reticulum stress, or mitochondrial dysfunction itself, induce mitochondrial membrane permeabilization, which causes the release of pro-apoptotic proteins (cytochrome c, Smac/DIABLO and Omi/HtrA2) from the intermembrane space into the cytosol, a process regulated by pro-apoptotic (Bax, Bak) and anti-apoptotic (Bcl-xL) members of the Bcl-2 family. Cell death occurs by the final activation of effector caspases via maturation of caspase-9 (mediated by cytochrome c) or blockage of IAP (inhibitor of apoptosis) proteins (mediated by Smac/DIABLO and Omi/HtrA2) [305]. This mechanism is exemplified by cellular models of SCA3 and SCA7, in which both polyglutamine-expanded proteins, mutant ataxin-3Q79 and ataxin-7Q75, induce neuronal death by the activation of the intrinsic mitochondrial apoptotic pathway via upregulation of the mitochondrial pro-apoptotic factor Bax and downregulation of the anti-apoptotic factor Bcl-xL [308, 309]. More recently, however, expanded ataxin-3 (Q84 and Q104) has been shown to be associated with reduced activity of mitochondrial respiratory chain complex II with no evidence of activation of the apoptotic cascade [310], as previously reported for another polyglutamine disease [311].
Closely related to mitochondria-mediated apoptosis [312] and crucial for neuronal survival is the dynamic regulation of mitochondrial morphology and assembly of the mitochondria into a continuous network by the concerted action of opposing fission and fusion events [313, 314]. The importance of a proper mitochondrial fission/fusion balance in neurons is illustrated by two neurodegenerative disorders caused by mutations in the mitochondrial fusion GTPases MFN2 and OPA1 [315] and by a mouse model of cerebellar neurodegeneration generated by selectively removing Mfn2 from the cerebellum [316]. Unique among spinocerebellar ataxias, SCA12 has been proposed to be caused by a brain-specific regulatory subunit (Bß2) of the protein phosphatase PP2A that upon cell stress is rapidly targeted to the outer mitochondrial membrane, where it promotes apoptosis by inducing Drp1- and Fis1-dependent mitochondrial fission [317]. The SCA12 mutation is a CAG repeat expansion in the PPP2R2B 5′ untranslated region, which functions as a cis promoter element that upregulates PPP2R2B expression [318]. Therefore, the disease process would originate from an abnormally increased activity of a pro-apoptotic protein, resulting in severe fragmentation of the mitochondrial network. Interestingly, the opposite phenotype (mitochondrial hyperfusion) has been observed in autosomal recessive spastic ataxia of Charlevoix–Saguenay caused by loss-of-function mutations in a protein (sacsin) that co-localises with the mitochondria [319]. In a Drosophila model of SCA12, overexpression of ppp2r2b induced fission of the mitochondria accompanied by increases in cytosolic ROS, cytochrome c and caspase-3 activity [320]. Overexpression of Mn2+ SOD2 and antioxidant treatment reduced ROS and caspase-3 activity and extended the life span of SCA12 transgenic flies. Thus, SCA12 well illustrates the tight link that exists between mitochondrial apoptotic pathways, organelle dynamics and oxidative stress in the pathogenesis of mitochondria-mediated spinocerebellar degeneration.
Maintenance of an efficient mitochondrial network by balanced fusion and fission events is intimately connected to mitochondrial quality control. The cell has surveillance mechanisms to eliminate misfolded or unwanted proteins via autophagic and ubiquitin–proteasome systems located in the cytosol. In contrast, the mitochondria do not contain proteasomes and rely on a remarkable number of chaperones and proteases which promote folding of newly imported proteins, protect mitochondrial proteins against heat or oxidative stress and eliminate irreversibly damaged polypeptides [321]. Mutations in mitochondrial quality control genes could hamper the efficient degradation of potentially deleterious proteins, thus leading to neuronal dysfunction and, ultimately, to cell death. SCA28, caused by mutations in the AFG3L2 gene, provides an example of such perturbation of mitochondrial homeostasis [322]. Notably, it is the only autosomal dominant spinocerebellar ataxia caused by mutations affecting a mitochondrial-resident protein. AFG3L2 is a ubiquitous component of the m-AAA metalloprotease complex located in the internal mitochondrial membrane. Interestingly, the homologous protein paraplegin is also a component of the same mitochondrial complex and causes recessive spastic paraplegia type 7, a neurodegenerative disease closely related to spinocerebellar ataxia [323]. AFG3L2 can form both homo-oligomeric (AFG3L2/AFG3L2) and hetero-oligomeric (AFG3L2/paraplegin) complexes. The m-AAA complex is part of the mitochondrial inner membrane protein quality control system and participates in the maturation of precursor proteins, the degradation of incorrectly folded polypeptides and the assembly of components of the inner membrane, including complexes of the respiratory chain [324].
So far, only missense mutations have been described, the vast majority of which occur in the highly conserved proteolytic domain [322, 325]. In one consanguineous family, a homozygous mutation has been identified in two siblings with a distinct phenotype characterised clinically by lower extremity spasticity, peripheral neuropathy, apoptosis, oculomotor apraxia, dystonia, cerebellar atrophy and progressive myoclonic epilepsy [326]. In a cellular model, the mutations impair AFG3L2 proteolytic activity, respiratory chain complex IV activity and, eventually, cell respiration [322]. Although AFG3L2 is ubiquitously expressed, muscle histopathology in patients is normal with no ragged-red fibres, no mtDNA deletions and normal respiratory chain activity [327], which indicates a selective vulnerability of the cerebellum consistent with the high levels of AFG3L2 protein detected in human Purkinje cells [322].
In a conditional mouse model, restricted deletion of the Afg3l2 gene in Purkinje cells demonstrates that mitochondrial fragmentation is an early event in the disease process and is caused by defective protein synthesis associated with impaired mitochondrial assembly [328]. In a distinct mouse model of the disease, cells lacking Afg3l2 exhibit respiratory dysfunction and fragmentation of the mitochondrial network, which causes the decrease of mitochondrial Ca2+ uptake by impairing mitochondrial–ER communication [329]. Altogether, these data provide further evidence that mitochondria-mediated neurodegeneration is a complex multifaceted process in which bioenergetics impairment, albeit important, is only one of the key players [304].
Because the interrelated multilevel pathways that regulate mitochondrial homeostasis are still unclear, the quest for effective treatments that improve mitochondrial function is still an unresolved issue. This knowledge will likely provide clinicians with novel and highly effective therapeutics to, in the very least, delay ataxia neurodegeneration.
Therapeutic Targets in Inherited Ataxias (Massimo Pandolfo)
Hereditary ataxias are characterised by quite diverse genetic, clinical, pathophysiological, pathogenic and neuropathological features; hence, potential therapeutic targets for disease modification depend on the specific condition being considered. So far, even though clinical trials are quite advanced in some cases, like Friedreich’s ataxia [330], no molecules have been reported to show efficacy in slowing the progression or the amelioration of neurological symptoms. Furthermore, there is currently no effective symptomatic treatment for ataxia. Physical therapy will not be addressed here, but it should be noticed here that intensive coordinative training may improve functional performance in various cerebellar and afferent ataxias [331, 332] and remains, along with other rehabilitative approaches, the main therapeutic option that we can currently offer these patients.
Potential disease-modifying therapeutics for inherited ataxias can be divided into two main categories: (1) those that modulate gene expression and (2) those that target specific pathogenic mechanisms triggered by the genetic defect.
Modulating the Expression of the Mutated Gene
Therapies that modulate gene expression may target the pathogenic process in inherited ataxias at its roots. Several inherited ataxias are due to altered levels of expression of the causative gene without changes in the encoded protein sequence (FRDA and fragile × tremor–ataxia syndrome) or to expression of a mutated allele encoding a toxic protein (polyQ diseases). Increasing gene expression in FRDA, decreasing gene expression in FXTAS, or preventing the expression of the mutated allele in polyQ diseases represents potentially valuable therapeutic approaches for these disorders. Furthermore, induction of exon skipping may reestablish the reading frame after frameshift mutations in some recessive ataxias or prevent the incorporation of the exon containing the pathogenic mutation, as in SCA6. Ataxias due to loss-of-function mutations may in principle benefit from gene or protein replacement therapies.
In FRDA, the GAA repeat expansions in the FXN gene induce a closed chromatin conformation, characterised by increased DNA methylation and by specific posttranslational modifications in the core histones, in particular loss of acetylation at various lysines and increased trimethylation at histone H3 lysine 9 (H3K9) [333]. Pharmacological inhibitors of HDACs may counteract these chromatin changes and reactivate FXN expression. Experiments in peripheral blood mononuclear cells from FRDA patients and in knock-in animal models carrying expanded GAA repeats indicated that a specific family of benzamide HDAC inhibitors can increase frataxin levels, whilst most other HDAC inhibitors are ineffective [334-336]. Benzamide HDAC inhibitors specifically target HDAC1 and HDAC3 and have a slow-on/slow-off kinetic, and both characteristics are essential for frataxin upregulation [337]. Importantly, frataxin upregulation is obtained at doses that cause minimal overall gene expression changes, suggesting that toxicity due to gene expression dysregulation is unlikely [334]. A phase I study with one of these molecules is ongoing.
The current model of FXTAS pathogenesis is that of RNA toxicity [338] (discussed earlier in this manuscript). Pre-mutation fragile × (FRAXA) alleles containing 50–200 CGG triplets in the 5′ untranslated region (UTR) of the FMR1 gene are transcribed at higher levels than normal alleles. FMR1 RNA containing such FRAXA pre-mutation repeats accumulates in nuclear foci, probably as a consequence of the tendency of the repeats to form hairpin structures, and sequesters specific RNA binding proteins, whose normal function is then compromised. FXTAS patient-derived lymphoblasts and fibroblasts show increased histone acetylation at the FMR1 locus, which correlates with increased gene expression. Furthermore, experiments in Drosophila showed that the simultaneous overexpression of HDACs 3, 6, or 11 suppresses the neurodegeneration induced by a CGG-containing transgene [339]. Histone acetyltransferase (HAT) inhibitors have been shown to repress FMR1 mRNA expression in pre-mutation carrier cell lines and extend life span in CGG repeat-expressing Drosophila [339], providing a proof of principle that HAT inhibitors or HDAC activators might be used to selectively repress transcription at the FMR1 locus in individuals affected with FXTAS.
In polyQ diseases, altered normal functionality and the acquisition of novel toxic properties by the polyQ proteins are thought to contribute to pathogenesis [17]. Therefore, suppressing the expression of the mutated protein is an appealing therapeutic approach. In a transgenic mouse model of SCA1, recombinant AAV expressing short hairpin RNAs directed against different portions of the ataxin-1 mRNA improved motor coordination and restored cerebellar morphology, clearing the characteristic ataxin-1 containing nuclear inclusions in Purkinje cells [340]. Ataxins 3 and 7 have been similarly targeted [341]. However, this approach does not differentiate between the mutated and the normal allele of the target gene. The resulting downregulation of both mutated and normal protein may lead to functional consequences due to loss of normal function. This is more than a potential concern, as indicated by the appearance of pathological phenotypes in knockout animals for genes involved in polyQ diseases and particularly by the fact that partial loss of function is likely to contribute to the pathogenesis of several polyQ diseases. Thus, specific suppression of the mutated allele has been attempted by using RNAi molecules whose target sequences include single nucleotide polymorphisms (SNPs) that are in linkage disequilibrium with the mutated allele [342]. Whilst results in animal and cell culture models indicate that this approach may be effective, the need for a SNP that differentiates the alleles limits its application to a portion of patients with specific polyQ diseases. The possibility to target the CAG repeat is also being studied, but the abundance of CAG repeat-containing human transcripts (estimated at ~200) makes off-target effects likely. It has been suggested that modified siRNAs functioning like some naturally occurring miRNAs that inhibit translation rather than inducing mRNA cleavage may be used to achieve downregulation of a specific polyQ protein [341]. The key to success is therefore the identification of the best reagent to obtain specific allele suppression, in terms of type of RNAi and of choice of target sequence. A gene replacement strategy that combines non-allele-selective gene silencing with the expression of an exogenous normal allele is also being considered.
The use of antisense oligonucleotides (ASOs) has also been explored as a way of suppressing polyQ protein expression through translational blockade or RNaseH activation. A variety of chemical modifications that favour one or the other mechanism, stability and cell penetration, have been considered, particularly in models of Huntington’s disease [343]. A clinical trial in HD with intraventricularly administered ASOs is underway.
SCA6 is caused by an expanded CAG repeat in the gene encoding the Ca(V)2.1 voltage-gated calcium channel alpha subunit (CACNA1A). The corresponding polyQ tract is located in an intracytoplasmic portion of the protein near its carboxy-terminus, which does not participate in channel function and is encoded by an alternatively spliced exon, so only a fraction of Ca(V)2.1 molecules contains it. Interestingly, inclusion of this exon in the mature mRNA is enhanced by the presence of an expanded CAG repeat. Thus, suppressing the polyQ-encoding Ca(V)2.1 splice variant with a splice isoform-specific RNAi strategy may represent a potential therapy for SCA6. This was achieved in a variety of cell-based models including a human neuronal cell line using a human miR124-based RNAi construct [344].
Delivery to the CNS, which is protected by the blood–brain barrier, and across cell membranes remains a major problem that needs to be resolved to make RNAi and ASO approaches a viable option to treat polyQ diseases and other conditions that may benefit from specific suppression or modulation of gene expression. Chemical modifications and non-viral and viral vectors are all being considered for this purpose, but progress beyond animal models has so far been limited [345].
Targeting the Pathogenic Mechanisms Triggered by the Genetic Defects
Antioxidants have been proposed for the treatment of many neurodegenerative diseases, including inherited ataxias, but clinical results have been so far mostly disappointing. A reason for the limited success of antioxidant treatments is probably the lack of specificity of what has been attempted. Better understanding of the sources and mechanisms of oxidative damage in different conditions, including the role that oxidative stress has in each case and the specific pathways that are activated, is necessary to design more effective, targeted treatments [346].
Lipid-soluble antioxidants have been used to treat FRDA. In this disease, defective iron–sulphur (Fe–S) cluster biogenesis in the mitochondria causes iron accumulation in these organelles and impaired electron transport in the respiratory chain. Both processes lead to increased formation of toxic free radicals, hence the rationale for antioxidant use. Randomised, placebo-controlled trials (RCTs) have been conducted so far with the short-chain coenzyme Q analogue idebenone and with A0001 (alpha-tocopherylquinone), which is a lipid-soluble antioxidant and a respiratory chain stimulator molecule related to both coenzyme Q and vitamin E [330]. Despite some promising results in a phase 2 study, no efficacy of idebenone in improving ataxia or cardiomyopathy could be demonstrated in two phase 3 studies. A phase 2A trial with A0001 showed signs of efficacy on ataxia [347], but the short duration of the study (4 weeks) imposes caution, so further trials are necessary before a conclusion about efficacy can be reached. Deferiprone, an orally administered, membrane-permeable iron chelator, also functions as an antioxidant by removing excess redox-active iron from the mitochondria. A 6-month phase 2 RCT in FRDA, however, was inconclusive (unpublished data).
A role of oxidative stress is postulated in other ataxias as well. Oxidative stress is the primary cause of DNA damage in neurons and has a key role in the pathogenesis of ataxias due to defective DNA repair, providing a rationale for antioxidant treatment. No RCT has been performed to date, but encouraging results have been obtained in cellular and animal models. The observation of the amelioration of ataxia in AT patients treated with steroids, originally for lymphoma, prompted further consideration of glucocorticoids for this condition [348]. The neurological benefit is probably due to the fact that these molecules penetrate into the CNS, where they act as anti-inflammatory and antioxidant drugs. However, the side effects of glucocorticoids, particularly when life-long exposure starting in childhood is envisioned, including a possibly increased risk of infection in the already immunocompromised AT patients, impose caution before this treatment can be recommended.
In dominant SCAs, mitochondrial dysfunction and oxidative damage are also thought to play a role, supporting the use of antioxidants as potential therapeutics, but there is currently little clinical evidence of efficacy. Other proposed general therapeutic targets include the formation of toxic aggregates of polyQ proteins or protein fragments, transcriptional dysregulation by polyQ nuclear aggregates, Purkinje cell dysfunction due to altered Ca2+ signalling and homeostasis, and altered physiological properties of cerebellar neurons due to changes in ion channel expression at the cell membrane. Additional specific approaches depend on the function of the mutated gene.
Conformational therapeutics to counteract polyQ protein aggregation (discussed in earlier section) has mostly been attempted for HD. Utilised molecules include the polyphenol epigallocatechingallate and the sulfobenzoic acid derivative C2-8. Results in HD animal models were mildly encouraging [349], but further studies are necessary to refine this approach and determine its applicability to SCAs.
Transcriptional dysregulation is considered a likely pathogenic mechanism in polyQ diseases, possibly due to sequestration of transcription factors by toxic protein aggregates in the nucleus [17] (discussed in earlier sections). In particular, sequestration of the histone acetyl transferase CBP leads to diffuse histone hypoacetylation and transcriptional repression, suggesting that HDAC inhibitors may be beneficial [350]. Most supporting data come from HD animal models, but a beneficial effect of the HDAC inhibitor sodium butyrate was also shown in an SCA3 model [351].
Altered Ca2+ signalling in Purkinje cells is thought to be a general pathogenic mechanism in SCAs [352] (discussed in earlier sections). Inositol 1,4,5-trisphosphate receptor type 1, ITPR1, a ligand-gated ion channel that releases Ca2+ from the ER, is a key regulator of Purkinje cell activity, whose function may be disrupted by interaction with polyQ proteins or, in SCA15 and 29, directly by gene mutations. Proposed interventions include inhibition of Ca2+ release from the ER with dantrolene or similar drugs (in SCA2 and SCA3) [353], inhibition of the calcium-activated proteases calpains (in SCA2) [21] and modulation of calcium-activated potassium channels (in SCA2) [22].
Changes in potassium currents have been detected in Purkinje cells in an SCA1 mouse model. In this model, increased K+ conductance correlates with decreased Purkinje cell firing rate and motor impairment. Treatment with the K+ channel inhibitors 3-aminopyridine and 3,4-diaminopyridine corrected the electrophysiological abnormality, restored motor function and, if started early in the disease course, partially prevented cell loss [229]. No human trials have so far been conducted.
Modulators of Phenotype and Neurodegeneration in Spinocerebellar Ataxias (Giovanni Stevanin and Alexis Brice)
Spinocerebellar ataxias are clinically and neuropathologically extremely heterogeneous. For example, onset is generally observed during the third or fourth decade, but can also occur in childhood or old age. In addition, neuropathologically, prominent atrophy of the cerebellum and brain stem is usually observed, but other structures may also be affected, sometimes with only very slight lesions in the vermis. This leads to a considerable range of phenotypes. This clinical and pathological heterogeneity has been partially explained by various, mostly genetic, factors. Since SCAs are rare and those caused by polyglutamine/CAG expansions are by far the most frequent forms, potential modulators have been described so far mainly in these subtypes of SCAs.
The Nature of the Mutation
The major contributor to the phenotypical heterogeneity is the pathological mutation. Its nature affects disease progression. Indeed, disease progression is usually slower in patients with conventional mutations in SCA genes, in whom brain atrophy is often restricted to the vermis or hemispheres of the cerebellum, whilst the disease progresses faster and is multifocal in patients with CAG/polyQ expansions [24], therefore affecting neuropathology profiles. In addition, among SCAs caused by CAG repeat expansions, a prospective study of 526 patients showed that progression was faster in SCA1 patients compared to SCA2, SCA3 and SCA6 [2].
The Size of the Pathological Expansion
Among the frequent SCAs caused by CAG/polyglutamine expansions, age at onset varies as a negative function of the size of the expansion [354]. The number of CAG repeats on the expanded allele accounts for 59 % (SCA6) to 88 % (SCA7) of the variability of the age at onset, which is broad enough in any given SCA to preclude precise age at onset prediction on the basis of repeat size.
CAG repeat size also affects disease severity and frequency of several clinical signs, and therefore also partly accounts for phenotypic variability among patients [24, 355]. This is illustrated in DRPLA, in which longer repeats are associated with an earlier age at onset and a phenotype characterised by progressive myoclonus, epilepsy and dementia, whereas smaller repeats result in a high frequency of choreoathetosis and psychiatric manifestations in adults [356, 357]. Furthermore, SCA2 patients with small expansions may present with a parkinsonism or motor neuron disease, sometimes without ataxia [358, 359]. In SCA3/MJD patients, the frequency of pyramidal signs increases with the size of the expanded repeat, whereas the frequency of altered vibration sense decreases [360, 361]. In SCA7 patients, the frequency of decreased visual acuity, ophthalmoplegia and Babinski signs increases with the number of CAG repeats [362].
Disease duration until death is also negatively correlated with the number of CAG repeats on the expanded allele at the SCA7 locus and is limited to a few months or years in very early-onset patients [362].
Finally, somatic mosaicism of the mutation (i.e. its size varying among tissues) has been hypothesized to play a role in the specificity of the lesions, but this question remains unsolved.
Other Genetic Factors
The correlation factor between age at onset and CAG repeat size in polyglutamine SCAs ranges from 0.5 to 0.7 in most studies, suggesting that other genetic factors contribute to the variability. The involvement of other “familial”, and therefore likely genetic, factors that cluster within families has been suggested very early [363] and confirmed more recently [364]. Indeed, in SCA1, SCA3 and SCA6, the length of the non-expanded repeat in trans has a modest but significant influence on the age at onset [361, 364], and repeats in the RAI1 or CACNA1A/SCA6 genes affect the age at onset in SCA2 patients [188, 365, 366]. Age at onset in SCA3 patients is also dependent on SCA2 normal allele sizes [367]. However, most of these studies have not yet been replicated and the proportion of age at onset variance explained by these potential modifiers is usually very small. This calls for large collaborative worldwide efforts.
Besides the genes already described to impact on SCA pathology, microarray experiments and yeast two-hybrid screens have revealed a number of interesting genes that might modulate the ataxia phenotype, although this remains to be proven at the cellular level. The ataxia interactome that connects most of the known ataxia genes demonstrated that many ataxia-causing proteins share interacting partners, particularly among the transcription regulation, nuclear import, RNA splicing and ubiquitinylation processes [40]. These pathways have also been evidenced in genetic screens in SCA Drosophila models [178, 368-370]. The role of microRNA as potent phenotypic modulators has also been shown in fly models [92] and has been discussed in previous sections.
Non-genetic Factors
The clinical phenotype is influenced by disease duration in all forms of SCAs. The frequency of ophthalmoplegia, amyotrophy, sphincter disturbances and dysphagia increases with disease duration in the autosomal dominant cerebellar ataxias (ADCA) type I. In SCA6 patients, whilst the phenotype may remain pure during the first 10 years of disease duration, additional signs can occur later. Gender has also been shown to modulate SCA3 and SCA6 disease progression [2].
The cell context may also affect the toxicity of the proteins and then neurodegeneration. First, neurons are post-mitotic cells and are therefore thought to be more sensitive to this toxicity. Cell-specific properties may also affect the polyglutamine toxicity level. Somatic mosaicism, differences in DNA repair and protein degradation pathways have been proposed to explain the sensitivity of certain subcellular populations of neurons. Although spatiotemporal accumulation of the pathological protein occurs in polyglutamine SCAs, it seems that there is a disconnection between the presence of the inclusions and neurodegeneration. Indeed, the density of the neuronal intranuclear inclusions in a given structure does not seem to correlate with the degree of neurodegeneration. This is particularly striking in SCA17 in which the density in inclusions never reach 3 % of the neurons in affected or in unaffected brain regions. In SCA17, except for the cerebral cortex, there is an inverse correlation between the presence of NIIs and the severity of lesions in a given structure [371, 372], i.e. no NIIs are detected in the Purkinje [373] and granule cell layers or in the locus coeruleus, all of which degenerate, but they are detected in the putamen, dentate nucleus and pontine nuclei, which are unaffected or only mildly affected [371]. It is also becoming more and more evident that posttranslational modifications of the proteins (cleavage, phosphorylations, etc.) that may differ according to the structures are critical steps for protein toxicity. This is particularly evident for the ATXN1 protein, the phosphorylation of which is involved in its stabilization, interactions, and toxicity [49, 374] and opens the way to therapeutic assays [375]. Sumoylation of ATXN7 has also been connected to the pathological process in SCA7 [376]. Another point of interest is the abnormal conformation of the protein that benefits from the use of chaperones to improve the phenotype of fly, mouse and cell SCA models [51, 377-382].
Although these pathologies are inherited, the environment is also hypothesized to play a role in the modulation of polyglutamine toxicity. A study had shown that environmental enrichment of R6/1 mice, models of Huntington’s disease, delays the onset of motor symptoms [383]. Along the same line, mild exercise in the SCA1 mouse model improved survival and motor functions by compensation of the hyper-repression effect on transcription of the polyglutamine expansion [57]. We are just now learning the complex genetic interactions among the different factors involved including the environment taken place in the modulation of the disease phenotype.
Conclusions
Clearly, one of the seminal discoveries in the research of spinocerebellar ataxias is the identification of the causative gene defect as the initial step to further investigate the underlying molecular mechanisms in each specific ataxia subtype, with the ultimate goal of finding treatments. Identifying the mutation enables characterising the biological functions of the gene products, uncovering the molecular pathways implicated and understanding the effects of the mutation in the affected neuronal systems. A comprehensive understanding of the mechanisms that are fundamental for dysfunction and degeneration of the affected neural systems in spinocerebellar ataxias is essential for developing effective therapeutic approaches. The identification of earlier molecular dysfunction in SCAs helps to direct the study of mechanisms of neurotoxicity to earlier stages of the disease. Applying effective treatments to SCAs in the window of reversibility of early neuronal damage is now the major challenge, and the key barrier to effective therapy is that SCAs present clinically when neuronal loss is well advanced and thus irreversible. Current treatments are almost all directed at modifying symptoms; few address underlying pathogenic mechanisms and are inevitably delivered too late to rescue dying neurons. In this consensus manuscript, we have attempted to offer an overview and discuss the most relevant pathways and topics relevant in the neurodegeneration process in spinocerebellar ataxias, including transcription, aggregation, autophagy, ion channels, mitochondria, RNA toxicity, and their components and modulators of the disease phenotype, with the aim of discussing the current strategies being translated into treatments. Despite the many novel findings we have discussed in the preceding sections, a common view is shared that further basic research is needed, particularly that aimed at understanding and identifying the players and their interactions triggering the early neuronal dysfunction by each of the mutated genes, when neurodegenerative rescue is still possible. This would help direct the therapeutic strategies at earlier stages of the disease with the hope of not only halting the disease progression but also of the possibility of motor recovery in ataxia disorders.
Acknowledgments
The authors acknowledge the following agencies for funding: the Spanish Ministry of Science and Innovation (BFU2008-00527/BMC to AM-D and IS); the Carlos III Health Institute (CP08/00027 to AM-D); the Iberoamerican Programme for Science, Technology and Development (CYTED; RIBERMOV, 210RT0390 to AM-D and IS); the European Commission (EUROSCA project, LHSM-CT-2004-503304 to AB, AM-D, DCR, GS and OR; the NEUROMICS project 7th PCRD-305121 to AB, AM-D, DCR, GS, IS, OR); the Fundació de la Marató de TV3 (Televisió de Catalunya, 100730 to AM-D and IS); the French Association “Connaitre les Syndrômes Cérébelleux”, the Verum Foundation (to GS); and the program “Investissements d’Avenir” (to AB and GS). TA is supported by the US National Institutes of Health (grant R01NS083564). DCR is funded by a Wellcome Trust Principal Fellowship, a Wellcome Trust/MRC Strategic Grant on Alzheimer’s disease and the Biomedical Research Unit in Dementia at Addenbrooke’s Hospital. Funding was obtained from the National Institutes of Health, R01NS033123 to S.M.P. and RC4NS073009 to SMP and DRS. FT was funded by Telethon-Italia (GGP09301), the Italian Ministry of Health (RF-2009-1539841) and ERA-Net E-Rare-2 JTC2011 (Euro-SCAR). Antoni Matilla-Dueñas is a Miguel Servet Investigator in Neurosciences of the Spanish National Health System.
We apologize to those research groups whose contributions could not be referred in this review due to space constraints.
Abbreviations
- AFG3L2
ATPase family member 3-like 2
- ATN1
Human atrophin-1 protein
- Atxn
Mouse ataxin
- ATXN
Human ataxin
- BDNF
Brain-derived neurotrophic factor
- CAG
Codon that codes for glutamine
- Ced
C. elegans cell death gene
- DCD
Dark cell degeneration
- DRPLA
Dentatorubral–pallidoluysian atrophy
- EAAT1
Excitatory amino acid transporter 1
- FDA
US Food and Drug Administration
- FRDA
Friedreich’s ataxia
- FXTAS
Fragile X-associated tremor/ataxia syndrome
- GABA
γ-Aminobutyric acid
- HAT
Histone acetyltransferase
- HD
Huntington’s disease
- HDAC
Histone deacetylase
- hnRNP K
Heterogeneous nuclear ribonucleoprotein K
- IP3R1
Inositol 1,4,5-triphosphate receptor type 1
- miRNA
MicroRNA
- mTOR
Mammalian target of rapamycin
- NIIs
Neuronal intranuclear inclusions (bodies)
- PC
Purkinje cells
- PolyQ
Polyglutamine
- PPP2R2B
Human protein phosphatase 2, regulatory subunit B, beta
- RBP
RNA-binding protein
- RCT
Randomised, placebo-controlled trial
- ROS
Reactive oxygen species
- SCAs
Spinocerebellar ataxias
- TBP
TATA-binding protein
- UPS
Ubiquitin–proteasome system
- VGICs
Voltage-gated ion channels
Footnotes
The final publication is available at http://link.springer.com/article/10.1007/s12311-013-0539-y
Conflict of Interest
The authors declare no competing financial interests.
References
- 1.Orr H, Chung M-y, Banfi S, Kwiatkowski TJ, Jr, Servadio A, Beaudet AL, et al. Expansion of an unstable trinucleotide (CAG) repeat in spinocerebellar ataxia type 1. Nat Genet. 1993;4:221–6. doi: 10.1038/ng0793-221. [DOI] [PubMed] [Google Scholar]
- 2.Jacobi H, Bauer P, Giunti P, Labrum R, Sweeney MG, Charles P, et al. The natural history of spinocerebellar ataxia type 1, 2, 3, and 6: a 2-year follow-up study. Neurology. 2011;77:1035–41. doi: 10.1212/WNL.0b013e31822e7ca0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dohlinger S, Hauser TK, Borkert J, Luft AR, Schulz JB. Magnetic resonance imaging in spinocerebellar ataxias. Cerebellum. 2008;7:204–14. doi: 10.1007/s12311-008-0025-0. [DOI] [PubMed] [Google Scholar]
- 4.Reetz K, Costa AS, Mirzazade S, Lehmann A, Juzek A, Rakowicz M, et al. Genotype-specific patterns of atrophy progression are more sensitive than clinical decline in SCA1, SCA3 and SCA6. Brain. 2013;136:905–17. doi: 10.1093/brain/aws369. [DOI] [PubMed] [Google Scholar]
- 5.Manto MU. The wide spectrum of spinocerebellar ataxias (SCAs) Cerebellum. 2005;4:2–6. doi: 10.1080/14734220510007914. [DOI] [PubMed] [Google Scholar]
- 6.Schulz JB, Borkert J, Wolf S, Schmitz-Hubsch T, Rakowicz M, Mariotti C, et al. Visualization, quantification and correlation of brain atrophy with clinical symptoms in spinocerebellar ataxia types 1, 3 and 6. Neuroimage. 2010;49:158–68. doi: 10.1016/j.neuroimage.2009.07.027. [DOI] [PubMed] [Google Scholar]
- 7.Scherzed W, Brunt ER, Heinsen H, de Vos RA, Seidel K, Burk K, et al. Pathoanatomy of cerebellar degeneration in spinocerebellar ataxia type 2 (SCA2) and type 3 (SCA3) Cerebellum. 2012;11:749–60. doi: 10.1007/s12311-011-0340-8. [DOI] [PubMed] [Google Scholar]
- 8.Abraham MC, Lu Y, Shaham S. A morphologically conserved nonapoptotic program promotes linker cell death in Caenorhabditis elegans. Dev Cell. 2007;12:73–86. doi: 10.1016/j.devcel.2006.11.012. [DOI] [PubMed] [Google Scholar]
- 9.Yuan J, Kroemer G. Alternative cell death mechanisms in development and beyond. Genes Dev. 2010;24:2592–602. doi: 10.1101/gad.1984410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu J, Tang TS, Tu H, Nelson O, Herndon E, Huynh DP, et al. Deranged calcium signaling and neurodegeneration in spinocerebellar ataxia type 2. J Neurosci. 2009;29:9148–62. doi: 10.1523/JNEUROSCI.0660-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen X, Tang TS, Tu H, Nelson O, Pook M, Hammer R, et al. Deranged calcium signaling and neurodegeneration in spinocerebellar ataxia type 3. J Neurosci. 2008;28:12713–24. doi: 10.1523/JNEUROSCI.3909-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Custer SK, Garden GA, Gill N, Rueb U, Libby RT, Schultz C, et al. Bergmann glia expression of polyglutamine-expanded ataxin-7 produces neurodegeneration by impairing glutamate transport. Nat Neurosci. 2006;9:1302–11. doi: 10.1038/nn1750. [DOI] [PubMed] [Google Scholar]
- 13.Maltecca F, Magnoni R, Cerri F, Cox GA, Quattrini A, Casari G. Haploinsufficiency of AFG3L2, the gene responsible for spinocerebellar ataxia type 28, causes mitochondria-mediated Purkinje cell dark degeneration. J Neurosci. 2009;29:9244–54. doi: 10.1523/JNEUROSCI.1532-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Oppenheim RW, Flavell RA, Vinsant S, Prevette D, Kuan CY, Rakic P. Programmed cell death of developing mammalian neurons after genetic deletion of caspases. J Neurosci. 2001;21:4752–60. doi: 10.1523/JNEUROSCI.21-13-04752.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Blum ES, Abraham MC, Yoshimura S, Lu Y, Shaham S. Control of nonapoptotic developmental cell death in Caenorhabditis elegans by a polyglutamine-repeat protein. Science. 2012;335:970–3. doi: 10.1126/science.1215156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Blum ES, Schwendeman AR, Shaham S. PolyQ disease: misfiring of a developmental cell death program? Trends Cell Biol. 2013;23:168–74. doi: 10.1016/j.tcb.2012.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Orr HT. Cell biology of spinocerebellar ataxia. J Cell Biol. 2012;197:167–77. doi: 10.1083/jcb.201105092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chakrabarti L, Eng J, Ivanov N, Garden GA, La Spada AR. Autophagy activation and enhanced mitophagy characterize the Purkinje cells of pcd mice prior to neuronal death. Mol Brain. 2009;2:24. doi: 10.1186/1756-6606-2-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Strahlendorf J, Box C, Attridge J, Diertien J, Finckbone V, Henne WM, et al. AMPA-induced dark cell degeneration of cerebellar Purkinje neurons involves activation of caspases and apparent mitochondrial dysfunction. Brain Res. 2003;994:146–59. doi: 10.1016/j.brainres.2003.09.048. [DOI] [PubMed] [Google Scholar]
- 20.Barenberg P, Strahlendorf H, Strahlendorf J. Hypoxia induces an excitotoxic-type of dark cell degeneration in cerebellar Purkinje neurons. Neurosci Res. 2001;40:245–54. doi: 10.1016/s0168-0102(01)00234-6. [DOI] [PubMed] [Google Scholar]
- 21.Kasumu A, Bezprozvanny I. Deranged calcium signaling in Purkinje cells and pathogenesis in spinocerebellar ataxia 2 (SCA2) and other ataxias. Cerebellum. 2012;11:630–9. doi: 10.1007/s12311-010-0182-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kasumu AW, Hougaard C, Rode F, Jacobsen TA, Sabatier JM, Eriksen BL, et al. Selective positive modulator of calcium-activated potassium channels exerts beneficial effects in a mouse model of spinocerebellar ataxia type 2. Chem Biol. 2012;19:1340–53. doi: 10.1016/j.chembiol.2012.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Matilla-Dueñas A, Goold R, Giunti P. Molecular pathogenesis of spinocerebellar ataxias. Brain. 2006;129:1357–70. doi: 10.1093/brain/awl081. [DOI] [PubMed] [Google Scholar]
- 24.Durr A. Autosomal dominant cerebellar ataxias: polyglutamine expansions and beyond. Lancet Neurol. 2010;9:885–94. doi: 10.1016/S1474-4422(10)70183-6. [DOI] [PubMed] [Google Scholar]
- 25.Friedman MJ, Shah AG, Fang ZH, Ward EG, Warren ST, Li S, et al. Polyglutamine domain modulates the TBP-TFIIB interaction: implications for its normal function and neurodegeneration. Nat Neurosci. 2007;10:1519–28. doi: 10.1038/nn2011. [DOI] [PubMed] [Google Scholar]
- 26.Goold R, Hubank M, Hunt A, Holton J, Menon RP, Revesz T, et al. Down-regulation of the dopamine receptor D2 in mice lacking ataxin 1. Hum Mol Genet. 2007;16:2122–34. doi: 10.1093/hmg/ddm162. [DOI] [PubMed] [Google Scholar]
- 27.Crespo-Barreto J, Fryer JD, Shaw CA, Orr HT, Zoghbi HY. Partial loss of ataxin-1 function contributes to transcriptional dysregulation in spinocerebellar ataxia type 1 pathogenesis. PLoS Genet. 2010;6:e1001021. doi: 10.1371/journal.pgen.1001021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Matilla-Dueñas A, Sanchez I, Corral-Juan M, Davalos A, Alvarez R, Latorre P. Cellular and molecular pathways triggering neurodegeneration in the spinocerebellar ataxias. Cerebellum. 2010;9:148–66. doi: 10.1007/s12311-009-0144-2. [DOI] [PubMed] [Google Scholar]
- 29.Orr HT. Nuclear ataxias. Cold Spring Harb Perspect Biol. 2010;2:a000786. doi: 10.1101/cshperspect.a000786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Matilla-Dueñas A. The ever expanding spinocerebellar ataxias. Editorial. Cerebellum. 2012;11:821–7. doi: 10.1007/s12311-012-0376-4. [DOI] [PubMed] [Google Scholar]
- 31.Sanchez I, Piñol P, Corral-Juan M, Pandolfo M, Matilla-Dueñas A. A novel function of Ataxin-1 in the modulation of PP2A activity is dysregulated in the spinocerebellar ataxia type 1. Hum Mol Genet. 2013;22:3425–37. doi: 10.1093/hmg/ddt197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McCullough SD, Grant PA. Histone acetylation, acetyltransferases, and ataxia—alteration of histone acetylation and chromatin dynamics is implicated in the pathogenesis of polyglutamine-expansion disorders. Adv Protein Chem Struct Biol. 2010;79:165–203. doi: 10.1016/S1876-1623(10)79005-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Smith KT, Workman JL. Introducing the acetylome. Nat Biotechnol. 2009;27:917–9. doi: 10.1038/nbt1009-917. [DOI] [PubMed] [Google Scholar]
- 34.Matilla-Dueñas A, Goold R, Giunti P. Clinical, genetic, molecular, and pathophysiological insights into spinocerebellar ataxia type 1. Cerebellum. 2008;7:106–14. doi: 10.1007/s12311-008-0009-0. [DOI] [PubMed] [Google Scholar]
- 35.Mushegian AR, Bassett DE, Jr, Boguski MS, Bork P, Koonin EV. Positionally cloned human disease genes: patterns of evolutionary conservation and functional motifs. Proc Natl Acad Sci U S A. 1997;94:5831–6. doi: 10.1073/pnas.94.11.5831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.de Chiara C, Giannini C, Adinolfi S, de Boer J, Guida S, Ramos A, et al. The AXH module: an independently folded domain common to ataxin-1 and HBP1. FEBS Lett. 2003;551:107–12. doi: 10.1016/s0014-5793(03)00818-4. [DOI] [PubMed] [Google Scholar]
- 37.Chen YW, Allen MD, Veprintsev DB, Lowe J, Bycroft M. The structure of the AXH domain of spinocerebellar ataxin-1. J Biol Chem. 2004;279:3758–65. doi: 10.1074/jbc.M309817200. [DOI] [PubMed] [Google Scholar]
- 38.de Chiara C, Menon RP, Adinolfi S, de Boer J, Ktistaki E, Kelly G, et al. The AXH domain adopts alternative folds the solution structure of HBP1 AXH. Structure (Camb) 2005;13:743–53. doi: 10.1016/j.str.2005.02.016. [DOI] [PubMed] [Google Scholar]
- 39.Yue S, Serra HG, Zoghbi HY, Orr HT. The spinocerebellar ataxia type 1 protein, ataxin-1, has RNA-binding activity that is inversely affected by the length of its polyglutamine tract. Hum Mol Genet. 2001;10:25–30. doi: 10.1093/hmg/10.1.25. [DOI] [PubMed] [Google Scholar]
- 40.Lim J, Hao T, Shaw C, Patel AJ, Szabo G, Rual JF, et al. A protein–protein interaction network for human inherited ataxias and disorders of Purkinje cell degeneration. Cell. 2006;125:801–14. doi: 10.1016/j.cell.2006.03.032. [DOI] [PubMed] [Google Scholar]
- 41.Matilla A, Koshy BT, Cummings CJ, Isobe T, Orr HT, Zoghbi HY. The cerebellar leucine-rich acidic nuclear protein interacts with ataxin-1. Nature. 1997;389:974–8. doi: 10.1038/40159. [DOI] [PubMed] [Google Scholar]
- 42.Bowman AB, Lam YC, Jafar-Nejad P, Chen HK, Richman R, Samaco RC, et al. Duplication of Atxn1l suppresses SCA1 neuropathology by decreasing incorporation of polyglutamine-expanded ataxin-1 into native complexes. Nat Genet. 2007;39:373–9. doi: 10.1038/ng1977. [DOI] [PubMed] [Google Scholar]
- 43.Tong X, Gui H, Jin F, Heck BW, Lin P, Ma J, et al. Ataxin-1 and Brother of ataxin-1 are components of the Notch signalling pathway. EMBO Rep. 2011;12:428–35. doi: 10.1038/embor.2011.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lam YC, Bowman AB, Jafar-Nejad P, Lim J, Richman R, Fryer JD, et al. ATAXIN-1 interacts with the repressor Capicua in its native complex to cause SCA1 neuropathology. Cell. 2006;127:1335–47. doi: 10.1016/j.cell.2006.11.038. [DOI] [PubMed] [Google Scholar]
- 45.Tsuda H, Jafar-Nejad H, Patel AJ, Sun Y, Chen HK, Rose MF, et al. The AXH domain of ataxin-1 mediates neurodegeneration through its interaction with Gfi-1/Senseless proteins. Cell. 2005;122:633–44. doi: 10.1016/j.cell.2005.06.012. [DOI] [PubMed] [Google Scholar]
- 46.Tsai CC, Kao HY, Mitzutani A, Banayo E, Rajan H, McKeown M, et al. Ataxin 1, a SCA1 neurodegenerative disorder protein, is functionally linked to the silencing mediator of retinoid and thyroid hormone receptors. Proc Natl Acad Sci U S A. 2004;101:4047–52. doi: 10.1073/pnas.0400615101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Okazawa H, Rich T, Chang A, Lin X, Waragai M, Kajikawa M, et al. Interaction between mutant ataxin-1 and PQBP-1 affects transcription and cell death. Neuron. 2002;34:701–13. doi: 10.1016/s0896-6273(02)00697-9. [DOI] [PubMed] [Google Scholar]
- 48.Serra HG, Duvick L, Zu T, Carlson K, Stevens S, Jorgensen N, et al. RORalpha-mediated Purkinje cell development determines disease severity in adult SCA1 mice. Cell. 2006;127:697–708. doi: 10.1016/j.cell.2006.09.036. [DOI] [PubMed] [Google Scholar]
- 49.Lim J, Crespo-Barreto J, Jafar-Nejad P, Bowman AB, Richman R, Hill DE, et al. Opposing effects of polyglutamine expansion on native protein complexes contribute to SCA1. Nature. 2008;452:713–8. doi: 10.1038/nature06731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.de Chiara C, Menon RP, Strom M, Gibson TJ, Pastore A. Phosphorylation of S776 and 14-3-3 binding modulate ataxin-1 interaction with splicing factors. PLoS One. 2009;4:e8372. doi: 10.1371/journal.pone.0008372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fernandez-Funez P, Nino-Rosales ML, de Gouyon B, She WC, Luchak JM, Martinez P, et al. Identification of genes that modify ataxin-1-induced neurodegeneration. Nature. 2000;408:101–6. doi: 10.1038/35040584. [DOI] [PubMed] [Google Scholar]
- 52.Mizutani A, Wang L, Rajan H, Vig PJ, Alaynick WA, Thaler JP, et al. Boat, an AXH domain protein, suppresses the cytotoxicity of mutant ataxin-1. EMBO J. 2005;24:3339–51. doi: 10.1038/sj.emboj.7600785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gehrking KM, Andresen JM, Duvick L, Lough J, Zoghbi HY, Orr HT. Partial loss of Tip60 slows mid-stage neurodegeneration in a spinocerebellar ataxia type 1 (SCA1) mouse model. Hum Mol Genet. 2011;20:2204–12. doi: 10.1093/hmg/ddr108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Serra HG, Byam CE, Lande JD, Tousey SK, Zoghbi HY, Orr HT. Gene profiling links SCA1 pathophysiology to glutamate signaling in Purkinje cells of transgenic mice. Hum Mol Genet. 2004;13:2535–43. doi: 10.1093/hmg/ddh268. [DOI] [PubMed] [Google Scholar]
- 55.Ciani L, Salinas PC. WNTs in the vertebrate nervous system: from patterning to neuronal connectivity. Nat Rev Neurosci. 2005;6:351–62. doi: 10.1038/nrn1665. [DOI] [PubMed] [Google Scholar]
- 56.Glover JC, Renaud JS, Rijli FM. Retinoic acid and hindbrain patterning. J Neurobiol. 2006;66:705–25. doi: 10.1002/neu.20272. [DOI] [PubMed] [Google Scholar]
- 57.Fryer JD, Yu P, Kang H, Mandel-Brehm C, Carter AN, Crespo-Barreto J, et al. Exercise and genetic rescue of SCA1 via the transcriptional repressor Capicua. Science. 2011;334:690–3. doi: 10.1126/science.1212673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ralser M, Albrecht M, Nonhoff U, Lengauer T, Lehrach H, Krobitsch S. An integrative approach to gain insights into the cellular function of human ataxin-2. J Mol Biol. 2005;346:203–14. doi: 10.1016/j.jmb.2004.11.024. [DOI] [PubMed] [Google Scholar]
- 59.Nonis D, Schmidt MH, van de Loo S, Eich F, Dikic I, Nowock J, et al. Ataxin-2 associates with the endocytosis complex and affects EGF receptor trafficking. Cell Signal. 2008;20:1725–39. doi: 10.1016/j.cellsig.2008.05.018. [DOI] [PubMed] [Google Scholar]
- 60.Lastres-Becker I, Rub U, Auburger G. Spinocerebellar ataxia 2 (SCA2) Cerebellum. 2008;7:115–24. doi: 10.1007/s12311-008-0019-y. [DOI] [PubMed] [Google Scholar]
- 61.Satterfield TF, Pallanck LJ. Ataxin-2 and its Drosophila homolog, ATX2, physically assemble with polyribosomes. Hum Mol Genet. 2006;15:2523–32. doi: 10.1093/hmg/ddl173. [DOI] [PubMed] [Google Scholar]
- 62.Hallen L, Klein H, Stoschek C, Wehrmeyer S, Nonhoff U, Ralser M, et al. The KRAB-containing zinc-finger transcriptional regulator ZBRK1 activates SCA2 gene transcription through direct interaction with its gene product, ataxin-2. Hum Mol Genet. 2011;20:104–14. doi: 10.1093/hmg/ddq436. [DOI] [PubMed] [Google Scholar]
- 63.Urrutia R. KRAB-containing zinc-finger repressor proteins. Genome Biol. 2003;4:231. doi: 10.1186/gb-2003-4-10-231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Shimohata T, Nakajima T, Yamada M, Uchida C, Onodera O, Naruse S, et al. Expanded polyglutamine stretches interact with TAFII130, interfering with CREB-dependent transcription. Nat Genet. 2000;26:29–36. doi: 10.1038/79139. [DOI] [PubMed] [Google Scholar]
- 65.Takahashi J, Tanaka J, Arai K, Funata N, Hattori T, Fukuda T, et al. Recruitment of nonexpanded polyglutamine proteins to intranuclear aggregates in neuronal intranuclear hyaline inclusion disease. J Neuropathol Exp Neurol. 2001;60:369–76. doi: 10.1093/jnen/60.4.369. [DOI] [PubMed] [Google Scholar]
- 66.McCampbell A, Taylor JP, Taye AA, Robitschek J, Li M, Walcott J, et al. CREB-binding protein sequestration by expanded polyglutamine. Hum Mol Genet. 2000;9:2197–202. doi: 10.1093/hmg/9.14.2197. [DOI] [PubMed] [Google Scholar]
- 67.Chai Y, Shao J, Miller VM, Williams A, Paulson HL. Live-cell imaging reveals divergent intracellular dynamics of polyglutamine disease proteins and supports a sequestration model of pathogenesis. Proc Natl Acad Sci U S A. 2002;99:9310–5. doi: 10.1073/pnas.152101299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wang G, Sawai N, Kotliarova S, Kanazawa I, Nukina N. Ataxin-3, the MJD1 gene product, interacts with the two human homologs of yeast DNA repair protein RAD23, HHR23A and HHR23B. Hum Mol Genet. 2000;9:1795–803. doi: 10.1093/hmg/9.12.1795. [DOI] [PubMed] [Google Scholar]
- 69.Evert BO, Araujo J, Vieira-Saecker AM, de Vos RA, Harendza S, Klockgether T, et al. Ataxin-3 represses transcription via chromatin binding, interaction with histone deacetylase 3, and histone deacetylation. J Neurosci. 2006;26:11474–86. doi: 10.1523/JNEUROSCI.2053-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Costa Mdo C, Paulson HL. Toward understanding Machado–Joseph disease. Prog Neurobiol. 2012;97:239–57. doi: 10.1016/j.pneurobio.2011.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Evert BO, Vogt IR, Vieira-Saecker AM, Ozimek L, de Vos RA, Brunt ER, et al. Gene expression profiling in ataxin-3 expressing cell lines reveals distinct effects of normal and mutant ataxin-3. J Neuropathol Exp Neurol. 2003;62:1006–18. doi: 10.1093/jnen/62.10.1006. [DOI] [PubMed] [Google Scholar]
- 72.Araujo J, Breuer P, Dieringer S, Krauss S, Dorn S, Zimmermann K, et al. FOXO4-dependent upregulation of superoxide dismutase-2 in response to oxidative stress is impaired in spinocerebellar ataxia type 3. Hum Mol Genet. 2011;20:2928–41. doi: 10.1093/hmg/ddr197. [DOI] [PubMed] [Google Scholar]
- 73.Helmlinger D, Hardy S, Abou-Sleymane G, Eberlin A, Bowman AB, Gansmüller A, et al. Glutamine-expanded ataxin-7 alters TFTC/STAGA recruitment and chromatin structure leading to photoreceptor dysfunction. PLoS Biol. 2006;4:e67. doi: 10.1371/journal.pbio.0040067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Samara NL, Wolberger C. A new chapter in the transcription SAGA. Curr Opin Struct Biol. 2011;21:767–74. doi: 10.1016/j.sbi.2011.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chen YC, Gatchel JR, Lewis RW, Mao CA, Grant PA, Zoghbi HY, et al. Gcn5 loss-of-function accelerates cerebellar and retinal degeneration in a SCA7 mouse model. Hum Mol Genet. 2011;21:394–405. doi: 10.1093/hmg/ddr474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Koide R, Kobayashi S, Shimohata T, Ikeuchi T, Maruyama M, Saito M, et al. A neurological disease caused by an expanded CAG trinucleotide repeat in the TATA-binding protein gene: a new polyglutamine disease? Hum Mol Genet. 1999;8:2047–53. doi: 10.1093/hmg/8.11.2047. [DOI] [PubMed] [Google Scholar]
- 77.Nakamura K, Jeong SY, Uchihara T, Anno M, Nagashima K, Nagashima T, et al. SCA17, a novel autosomal dominant cerebellar ataxia caused by an expanded polyglutamine in TATA-binding protein. Hum Mol Genet. 2001;10:1441–8. doi: 10.1093/hmg/10.14.1441. [DOI] [PubMed] [Google Scholar]
- 78.Kalmar B, Greensmith L. Induction of heat shock proteins for protection against oxidative stress. Adv Drug Deliv Rev. 2009;61:310–8. doi: 10.1016/j.addr.2009.02.003. [DOI] [PubMed] [Google Scholar]
- 79.Williams AJ, Paulson HL. Polyglutamine neurodegeneration: protein misfolding revisited. Trends Neurosci. 2008;31:521–8. doi: 10.1016/j.tins.2008.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Arawaka S, Machiya Y, Kato T. Heat shock proteins as suppressors of accumulation of toxic prefibrillar intermediates and misfolded proteins in neurodegenerative diseases. Curr Pharm Biotechnol. 2010;11:158–66. doi: 10.2174/138920110790909713. [DOI] [PubMed] [Google Scholar]
- 81.Sajjad MU, Samson B, Wyttenbach A. Heat shock proteins: therapeutic drug targets for chronic neurodegeneration? Curr Pharm Biotechnol. 2010;11:198–215. doi: 10.2174/138920110790909641. [DOI] [PubMed] [Google Scholar]
- 82.Gautier T, Berges T, Tollervey D, Hurt E. Nucleolar KKE/D repeat proteins Nop56p and Nop58p interact with Nop1p and are required for ribosome biogenesis. Mol Cell Biol. 1997;17:7088–98. doi: 10.1128/mcb.17.12.7088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kobayashi H, Abe K, Matsuura T, Ikeda Y, Hitomi T, Akechi Y, et al. Expansion of intronic GGCCTG hexanucleotide repeat in NOP56 causes SCA36, a type of spinocerebellar ataxia accompanied by motor neuron involvement. Am J Hum Genet. 2011;89:121–30. doi: 10.1016/j.ajhg.2011.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zhang S, Xu L, Lee J, Xu T. Drosophila atrophin homolog functions as a transcriptional corepressor in multiple developmental processes. Cell. 2002;108:45–56. doi: 10.1016/s0092-8674(01)00630-4. [DOI] [PubMed] [Google Scholar]
- 85.Wang L, Rajan H, Pitman JL, McKeown M, Tsai CC. Histone deacetylase-associating atrophin proteins are nuclear receptor corepressors. Genes Dev. 2006;20:525–30. doi: 10.1101/gad.1393506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Shen Y, Lee G, Choe Y, Zoltewicz JS, Peterson AS. Functional architecture of atrophins. J Biol Chem. 2007;282:5037–44. doi: 10.1074/jbc.M610274200. [DOI] [PubMed] [Google Scholar]
- 87.Wang L, Tsai CC. Atrophin proteins: an overview of a new class of nuclear receptor corepressors. Nucl Recept Signal. 2008;6:e009. doi: 10.1621/nrs.06009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wood JD, Nucifora FC, Jr, Duan K, Zhang C, Wang J, Kim Y, et al. Atrophin-1, the dentato-rubral and pallido-luysian atrophy gene product, interacts with ETO/MTG8 in the nuclear matrix and represses transcription. J Cell Biol. 2000;150:939–48. doi: 10.1083/jcb.150.5.939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Napoletano F, Occhi S, Calamita P, Volpi V, Blanc E, Charroux B, et al. Polyglutamine atrophin provokes neurodegeneration in Drosophila by repressing fat. EMBO J. 2011;30:945–58. doi: 10.1038/emboj.2011.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lin X, Antalffy B, Kang D, Orr HT. Zoghbi HY. Polyglutamine expansion down-regulates specific neuronal genes before pathologic changes in SCA1. Nat Neurosci. 2000;3:157–63. doi: 10.1038/72101. [DOI] [PubMed] [Google Scholar]
- 91.Wiegert JS, Bading H. Activity-dependent calcium signaling and ERK-MAP kinases in neurons: a link to structural plasticity of the nucleus and gene transcription regulation. Cell Calcium. 2011;49:296–305. doi: 10.1016/j.ceca.2010.11.009. [DOI] [PubMed] [Google Scholar]
- 92.Bilen J, Liu N, Bonini NM. A new role for microRNA pathways: modulation of degeneration induced by pathogenic human disease proteins. Cell Cycle. 2006;5:2835–8. doi: 10.4161/cc.5.24.3579. [DOI] [PubMed] [Google Scholar]
- 93.Karres JS, Hilgers V, Carrera I, Treisman J, Cohen SM. The conserved microRNA miR-8 tunes atrophin levels to prevent neurodegeneration in Drosophila. Cell. 2007;131:136–45. doi: 10.1016/j.cell.2007.09.020. [DOI] [PubMed] [Google Scholar]
- 94.Lee Y, Samaco RC, Gatchel JR, Thaller C, Orr HT, Zoghbi HY. miR-19, miR-101 and miR-130 co-regulate ATXN1 levels to potentially modulate SCA1 pathogenesis. Nat Neurosci. 2008;11:1137–9. doi: 10.1038/nn.2183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Persengiev S, Kondova I, Otting N, Koeppen AH, Bontrop RE. Genome-wide analysis of miRNA expression reveals a potential role for miR-144 in brain aging and spinocerebellar ataxia pathogenesis. Neurobiol Aging. 2011;32(2316):e17–27. doi: 10.1016/j.neurobiolaging.2010.03.014. [DOI] [PubMed] [Google Scholar]
- 96.DiFiglia M, Sapp E, Chase KO, Davies SW, Bates GP, Vonsattel JP, et al. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science. 1997;277:1990–3. doi: 10.1126/science.277.5334.1990. [DOI] [PubMed] [Google Scholar]
- 97.Paulson HL, Perez MK, Trottier Y, Trojanowski JQ, Subramony SH, Das SS, et al. Intranuclear inclusions of expanded polyglutamine protein in spinocerebellar ataxia type 3. Neuron. 1997;19:333–44. doi: 10.1016/s0896-6273(00)80943-5. [DOI] [PubMed] [Google Scholar]
- 98.Skinner PJ, Koshy BT, Cummings CJ, Klement IA, Helin K, Servadio A, et al. Ataxin-1 with an expanded glutamine tract alters nuclear matrix-associated structures. Nature. 1997;389:971–4. doi: 10.1038/40153. [DOI] [PubMed] [Google Scholar]
- 99.Becher MW, Kotzuk JA, Sharp AH, Davies SW, Bates GP, Price DL, et al. Intranuclear neuronal inclusions in Huntington’s disease and dentatorubral and pallidoluysian atrophy: correlation between the density of inclusions and IT15 CAG triplet repeat length. Neurobiol Dis. 1998;4:387–97. doi: 10.1006/nbdi.1998.0168. [DOI] [PubMed] [Google Scholar]
- 100.Holmberg M, Duyckaerts C, Durr A, Cancel G, Gourfinkel-An I, Damier P, et al. Spinocerebellar ataxia type 7 (SCA7): a neurodegenerative disorder with neuronal intranuclear inclusions. Hum Mol Genet. 1998;7:913–8. doi: 10.1093/hmg/7.5.913. [DOI] [PubMed] [Google Scholar]
- 101.Li M, Miwa S, Kobayashi Y, Merry DE, Yamamoto M, Tanaka F, et al. Nuclear inclusions of the androgen receptor protein in spinal and bulbar muscular atrophy. Ann Neurol. 1998;44:249–54. doi: 10.1002/ana.410440216. [DOI] [PubMed] [Google Scholar]
- 102.Schmidt T, Landwehrmeyer GB, Schmitt I, Trottier Y, Auburger G, Laccone F, et al. An isoform of ataxin-3 accumulates in the nucleus of neuronal cells in affected brain regions of SCA3 patients. Brain Pathol. 1998;8:669–79. doi: 10.1111/j.1750-3639.1998.tb00193.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Huynh DP, Del Bigio MR, Ho DH, Pulst SM. Expression of ataxin-2 in brains from normal individuals and patients with Alzheimer’s disease and spinocerebellar ataxia 2. Ann Neurol. 1999;45:232–41. doi: 10.1002/1531-8249(199902)45:2<232::aid-ana14>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 104.Koyano S, Uchihara T, Fujigasaki H, Nakamura A, Yagishita S, Iwabuchi K. Neuronal intranuclear inclusions in spinocerebellar ataxia type 2: triple-labeling immunofluorescent study. Neurosci Lett. 1999;273:117–20. doi: 10.1016/s0304-3940(99)00656-4. [DOI] [PubMed] [Google Scholar]
- 105.Ishikawa K, Fujigasaki H, Saegusa H, Ohwada K, Fujita T, Iwamoto H, et al. Abundant expression and cytoplasmic aggregations of [alpha]1A voltage-dependent calcium channel protein associated with neurodegeneration in spinocerebellar ataxia type 6. Hum Mol Genet. 1999;8:1185–93. doi: 10.1093/hmg/8.7.1185. [DOI] [PubMed] [Google Scholar]
- 106.Munch C, Bertolotti A. Propagation of the prion phenomenon: beyond the seeding principle. J Mol Biol. 2012;421:491–8. doi: 10.1016/j.jmb.2011.12.061. [DOI] [PubMed] [Google Scholar]
- 107.Freundt EC, Maynard N, Clancy EK, Roy S, Bousset L, Sourigues Y, et al. Neuron-to-neuron transmission of alpha-synuclein fibrils through axonal transport. Ann Neurol. 2012;72:517–24. doi: 10.1002/ana.23747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Yang W, Dunlap JR, Andrews RB, Wetzel R. Aggregated polyglutamine peptides delivered to nuclei are toxic to mammalian cells. Hum Mol Genet. 2002;11:2905–17. doi: 10.1093/hmg/11.23.2905. [DOI] [PubMed] [Google Scholar]
- 109.Ren PH, Lauckner JE, Kachirskaia I, Heuser JE, Melki R, Kopito RR. Cytoplasmic penetration and persistent infection of mammalian cells by polyglutamine aggregates. Nat Cell Biol. 2009;11:219–25. doi: 10.1038/ncb1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Daughters RS, Tuttle DL, Gao W, Ikeda Y, Moseley ML, Ebner TJ, et al. RNA gain-of-function in spinocerebellar ataxia type 8. PLoS Genet. 2009;5:e1000600. doi: 10.1371/journal.pgen.1000600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Seidel K, Siswanto S, Brunt ER, den Dunnen W, Korf HW, Rub U. Brain pathology of spinocerebellar ataxias. Acta Neuropathol. 2012;124:1–21. doi: 10.1007/s00401-012-1000-x. [DOI] [PubMed] [Google Scholar]
- 112.Houlden H, Johnson J, Gardner-Thorpe C, Lashley T, Hernandez D, Worth P, et al. Mutations in TTBK2, encoding a kinase implicated in tau phosphorylation, segregate with spinocerebellar ataxia type 11. Nat Genet. 2007;39:1434–6. doi: 10.1038/ng.2007.43. [DOI] [PubMed] [Google Scholar]
- 113.Perutz MF, Johnson T, Suzuki M, Finch JT. Glutamine repeats as polar zippers: their possible role in inherited neurodegenerative diseases. Proc Natl Acad Sci U S A. 1994;91:5355–8. doi: 10.1073/pnas.91.12.5355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Ikeda H, Yamaguchi M, Sugai S, Aze Y, Narumiya S, Kakizuka A. Expanded polyglutamine in the Machado–Joseph disease protein induces cell death in vitro and in vivo. Nat Genet. 1996;13:196–202. doi: 10.1038/ng0696-196. [DOI] [PubMed] [Google Scholar]
- 115.Mangiarini L, Sathasivam K, Mahal A, Mott R, Seller M, Bates GP. Instability of highly expanded CAG repeats in mice transgenic for the Huntington’s disease mutation. Nat Genet. 1997;15:197–200. doi: 10.1038/ng0297-197. [DOI] [PubMed] [Google Scholar]
- 116.Cowan KJ, Diamond MI, Welch WJ. Polyglutamine protein aggregation and toxicity are linked to the cellular stress response. Hum Mol Genet. 2003;12:1377–91. doi: 10.1093/hmg/ddg151. [DOI] [PubMed] [Google Scholar]
- 117.Gusella JF, MacDonald ME. Molecular genetics: unmasking polyglutamine triggers in neurodegenerative disease. Nat Rev Neurosci. 2000;1:109–15. doi: 10.1038/35039051. [DOI] [PubMed] [Google Scholar]
- 118.Sieradzan KA, Mechan AO, Jones L, Wanker EE, Nukina N, Mann DM. Huntington’s disease intranuclear inclusions contain truncated, ubiquitinated huntingtin protein. Exp Neurol. 1999;156:92–9. doi: 10.1006/exnr.1998.7005. [DOI] [PubMed] [Google Scholar]
- 119.Garden GA, Libby RT, Fu YH, Kinoshita Y, Huang J, Possin DE, et al. Polyglutamine-expanded ataxin-7 promotes non-cell-autonomous Purkinje cell degeneration and displays proteolytic cleavage in ataxic transgenic mice. J Neurosci. 2002;22:4897–905. doi: 10.1523/JNEUROSCI.22-12-04897.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Goti D, Katzen SM, Mez J, Kurtis N, Kiluk J, Ben-Haiem L, et al. A mutant ataxin-3 putative-cleavage fragment in brains of Machado–Joseph disease patients and transgenic mice is cytotoxic above a critical concentration. J Neurosci. 2004;24:10266–79. doi: 10.1523/JNEUROSCI.2734-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Wellington CL, Ellerby LM, Hackam AS, Margolis RL, Trifiro MA, Singaraja R, et al. Caspase cleavage of gene products associated with triplet expansion disorders generates truncated fragments containing the polyglutamine tract. J Biol Chem. 1998;273:9158–67. doi: 10.1074/jbc.273.15.9158. [DOI] [PubMed] [Google Scholar]
- 122.Graham RK, Deng Y, Slow EJ, Haigh B, Bissada N, Lu G, et al. Cleavage at the caspase-6 site is required for neuronal dysfunction and degeneration due to mutant huntingtin. Cell. 2006;125:1179–91. doi: 10.1016/j.cell.2006.04.026. [DOI] [PubMed] [Google Scholar]
- 123.Haacke A, Hartl FU, Breuer P. Calpain inhibition is sufficient to suppress aggregation of polyglutamine-expanded ataxin-3. J Biol Chem. 2007;282:18851–6. doi: 10.1074/jbc.M611914200. [DOI] [PubMed] [Google Scholar]
- 124.Young JE, Gouw L, Propp S, Sopher BL, Taylor J, Lin A, et al. Proteolytic cleavage of ataxin-7 by caspase-7 modulates cellular toxicity and transcriptional dysregulation. J Biol Chem. 2007;282:30150–60. doi: 10.1074/jbc.M705265200. [DOI] [PubMed] [Google Scholar]
- 125.Hubener J, Weber JJ, Richter C, Honold L, Weiss A, Murad F, et al. Calpain-mediated ataxin-3 cleavage in the molecular pathogenesis of spinocerebellar ataxia type 3 (SCA3) Hum Mol Genet. 2013;22:508–18. doi: 10.1093/hmg/dds449. [DOI] [PubMed] [Google Scholar]
- 126.Simoes AT, Goncalves N, Koeppen A, Deglon N, Kugler S, Duarte CB, et al. Calpastatin-mediated inhibition of calpains in the mouse brain prevents mutant ataxin 3 proteolysis, nuclear localization and aggregation, relieving Machado–Joseph disease. Brain. 2012;135:2428–39. doi: 10.1093/brain/aws177. [DOI] [PubMed] [Google Scholar]
- 127.Masino L, Nicastro G, Menon RP, Dal Piaz F, Calder L, Pastore A. Characterization of the structure and the amyloidogenic properties of the Josephin domain of the polyglutamine-containing protein ataxin-3. J Mol Biol. 2004;344:1021–35. doi: 10.1016/j.jmb.2004.09.065. [DOI] [PubMed] [Google Scholar]
- 128.Ellisdon AM, Thomas B, Bottomley SP. The two-stage pathway of ataxin-3 fibrillogenesis involves a polyglutamine-independent step. J Biol Chem. 2006;281:16888–96. doi: 10.1074/jbc.M601470200. [DOI] [PubMed] [Google Scholar]
- 129.Hubener J, Vauti F, Funke C, Wolburg H, Ye Y, Schmidt T, et al. N-terminal ataxin-3 causes neurological symptoms with inclusions, endoplasmic reticulum stress and ribosomal dislocation. Brain. 2011;134:1925–42. doi: 10.1093/brain/awr118. [DOI] [PubMed] [Google Scholar]
- 130.Perez MK, Paulson HL, Pendse SJ, Saionz SJ, Bonini NM, Pittman RN. Recruitment and the role of nuclear localization in polyglutamine-mediated aggregation. J Cell Biol. 1998;143:1457–70. doi: 10.1083/jcb.143.6.1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Fujigasaki H, Uchihara T, Koyano S, Iwabuchi K, Yagishita S, Makifuchi T, et al. Ataxin-3 is translocated into the nucleus for the formation of intranuclear inclusions in normal and Machado–Joseph disease brains. Exp Neurol. 2000;165:248–56. doi: 10.1006/exnr.2000.7479. [DOI] [PubMed] [Google Scholar]
- 132.Uchihara T, Fujigasaki H, Koyano S, Nakamura A, Yagishita S, Iwabuchi K. Non-expanded polyglutamine proteins in intranuclear inclusions of hereditary ataxias—triple-labeling immunofluorescence study. Acta Neuropathol. 2001;102:149–52. doi: 10.1007/s004010100364. [DOI] [PubMed] [Google Scholar]
- 133.Chai Y, Wu L, Griffin JD, Paulson HL. The role of protein composition in specifying nuclear inclusion formation in polyglutamine disease. J Biol Chem. 2001;276:44889–97. doi: 10.1074/jbc.M106575200. [DOI] [PubMed] [Google Scholar]
- 134.Zander C, Takahashi J, El Hachimi KH, Fujigasaki H, Albanese V, Lebre AS, et al. Similarities between spinocerebellar ataxia type 7 (SCA7) cell models and human brain: proteins recruited in inclusions and activation of caspase-3. Hum Mol Genet. 2001;10:2569–79. doi: 10.1093/hmg/10.22.2569. [DOI] [PubMed] [Google Scholar]
- 135.Schmidt T, Lindenberg KS, Krebs A, Schols L, Laccone F, Herms J, et al. Protein surveillance machinery in brains with spinocerebellar ataxia type 3: redistribution and differential recruitment of 26S proteasome subunits and chaperones to neuronal intranuclear inclusions. Ann Neurol. 2002;51:302–10. doi: 10.1002/ana.10101. [DOI] [PubMed] [Google Scholar]
- 136.Verhoef LG, Lindsten K, Masucci MG, Dantuma NP. Aggregate formation inhibits proteasomal degradation of polyglutamine proteins. Hum Mol Genet. 2002;11:2689–700. doi: 10.1093/hmg/11.22.2689. [DOI] [PubMed] [Google Scholar]
- 137.Bence NF, Sampat RM, Kopito RR. Impairment of the ubiquitin–proteasome system by protein aggregation. Science. 2001;292:1552–5. doi: 10.1126/science.292.5521.1552. [DOI] [PubMed] [Google Scholar]
- 138.Waelter S, Boeddrich A, Lurz R, Scherzinger E, Lueder G, Lehrach H, et al. Accumulation of mutant huntingtin fragments in aggresome-like inclusion bodies as a result of insufficient protein degradation. Mol Biol Cell. 2001;12:1393–407. doi: 10.1091/mbc.12.5.1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Zhou H, Cao F, Wang Z, Yu ZX, Nguyen HP, Evans J, et al. Huntingtin forms toxic NH2-terminal fragment complexes that are promoted by the age-dependent decrease in proteasome activity. J Cell Biol. 2003;163:109–18. doi: 10.1083/jcb.200306038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Petroi D, Popova B, Taheri-Talesh N, Irniger S, Shahpasandzadeh H, Zweckstetter M, et al. Aggregate clearance of alpha-synuclein in Saccharomyces cerevisiae depends more on autophagosome and vacuole function than on the proteasome. J Biol Chem. 2012;287:27567–79. doi: 10.1074/jbc.M112.361865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Sisodia SS. Nuclear inclusions in glutamine repeat disorders: are they pernicious, coincidental, or beneficial? Cell. 1998;95:1–4. doi: 10.1016/s0092-8674(00)81743-2. [DOI] [PubMed] [Google Scholar]
- 142.Adachi H, Katsuno M, Minamiyama M, Waza M, Sang C, Nakagomi Y, et al. Widespread nuclear and cytoplasmic accumulation of mutant androgen receptor in SBMA patients. Brain. 2005;128:659–70. doi: 10.1093/brain/awh381. [DOI] [PubMed] [Google Scholar]
- 143.Klement IA, Skinner PJ, Kaytor MD, Yi H, Hersch SM, Clark HB, et al. Ataxin-1 nuclear localization and aggregation: role in polyglutamine-induced disease in SCA1 transgenic mice. Cell. 1998;95:41–53. doi: 10.1016/s0092-8674(00)81781-x. [DOI] [PubMed] [Google Scholar]
- 144.Saudou F, Finkbeiner S, Devys D, Greenberg ME. Huntingtin acts in the nucleus to induce apoptosis but death does not correlate with the formation of intranuclear inclusions. Cell. 1998;95:55–66. doi: 10.1016/s0092-8674(00)81782-1. [DOI] [PubMed] [Google Scholar]
- 145.Kretzschmar D, Tschape J, Bettencourt Da Cruz A, Asan E, Poeck B, Strauss R, et al. Glial and neuronal expression of polyglutamine proteins induce behavioral changes and aggregate formation in Drosophila. Glia. 2005;49:59–72. doi: 10.1002/glia.20098. [DOI] [PubMed] [Google Scholar]
- 146.Boy J, Schmidt T, Schumann U, Grasshoff U, Unser S, Holzmann C, et al. A transgenic mouse model of spinocerebellar ataxia type 3 resembling late disease onset and gender-specific instability of CAG repeats. Neurobiol Dis. 2010;37:284–93. doi: 10.1016/j.nbd.2009.08.002. [DOI] [PubMed] [Google Scholar]
- 147.Weiss A, Klein C, Woodman B, Sathasivam K, Bibel M, Regulier E, et al. Sensitive biochemical aggregate detection reveals aggregation onset before symptom development in cellular and murine models of Huntington’s disease. J Neurochem. 2008;104:846–58. doi: 10.1111/j.1471-4159.2007.05032.x. [DOI] [PubMed] [Google Scholar]
- 148.Bucciantini M, Giannoni E, Chiti F, Baroni F, Formigli L, Zurdo J, et al. Inherent toxicity of aggregates implies a common mechanism for protein misfolding diseases. Nature. 2002;416:507–11. doi: 10.1038/416507a. [DOI] [PubMed] [Google Scholar]
- 149.Takahashi T, Nozaki K, Tsuji S, Nishizawa M, Onodera O. Polyglutamine represses cAMP-responsive-element-mediated transcription without aggregate formation. Neuroreport. 2005;16:295–9. doi: 10.1097/00001756-200502280-00019. [DOI] [PubMed] [Google Scholar]
- 150.Chafekar SM, Wisen S, Thompson AD, Echeverria A, Walter GM, Evans CG, et al. Pharmacological tuning of heat shock protein 70 modulates polyglutamine toxicity and aggregation. ACS Chem Biol. 2012;7:1556–64. doi: 10.1021/cb300166p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Bauer PO, Goswami A, Wong HK, Okuno M, Kurosawa M, Yamada M, et al. Harnessing chaperone-mediated autophagy for the selective degradation of mutant huntingtin protein. Nat Biotechnol. 2010;28:256–63. doi: 10.1038/nbt.1608. [DOI] [PubMed] [Google Scholar]
- 152.Menzies FM, Huebener J, Renna M, Bonin M, Riess O, Rubinsztein DC. Autophagy induction reduces mutant ataxin-3 levels and toxicity in a mouse model of spinocerebellar ataxia type 3. Brain. 2010;133:93–104. doi: 10.1093/brain/awp292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Klionsky DJ. Autophagy: from phenomenology to molecular understanding in less than a decade. Nat Rev Mol Cell Biol. 2007;8:931–7. doi: 10.1038/nrm2245. [DOI] [PubMed] [Google Scholar]
- 154.Scherz-Shouval R, Shvets E, Fass E, Shorer H, Gil L, Elazar Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J. 2007;26:1749–60. doi: 10.1038/sj.emboj.7601623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Ravikumar B, Duden R, Rubinsztein DC. Aggregate-prone proteins with polyglutamine and polyalanine expansions are degraded by autophagy. Hum Mol Genet. 2002;11:1107–17. doi: 10.1093/hmg/11.9.1107. [DOI] [PubMed] [Google Scholar]
- 156.Yang Z, Klionsky DJ. An overview of the molecular mechanism of autophagy. Curr Top Microbiol Immunol. 2009;335:1–32. doi: 10.1007/978-3-642-00302-8_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Ganley IG, du Lam H, Wang J, Ding X, Chen S, Jiang X. ULK1.ATG13.FIP200 complex mediates mTOR signaling and is essential for autophagy. J Biol Chem. 2009;284:12297–305. doi: 10.1074/jbc.M900573200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Blommaart EF, Krause U, Schellens JP, Vreeling-Sindelarova H, Meijer AJ. The phosphatidylinositol 3-kinase inhibitors wortmannin and LY294002 inhibit autophagy in isolated rat hepatocytes. Eur J Biochem. 1997;243:240–6. doi: 10.1111/j.1432-1033.1997.0240a.x. [DOI] [PubMed] [Google Scholar]
- 159.Klionsky DJ, Abeliovich H, Agostinis P, Agrawal DK, Aliev G, Askew DS, et al. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy. 2008;4:151–75. doi: 10.4161/auto.5338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Yamamoto A, Tagawa Y, Yoshimori T, Moriyama Y, Masaki R, Tashiro Y. Bafilomycin A1 prevents maturation of autophagic vacuoles by inhibiting fusion between autophagosomes and lysosomes in rat hepatoma cell line, H-4-II-E cells. Cell Struct Funct. 1998;23:33–42. doi: 10.1247/csf.23.33. [DOI] [PubMed] [Google Scholar]
- 161.Ravikumar B, Sarkar S, Davies JE, Futter M, Garcia-Arencibia M, Green-Thompson ZW, et al. Regulation of mammalian autophagy in physiology and pathophysiology. Physiol Rev. 2010;90:1383–435. doi: 10.1152/physrev.00030.2009. [DOI] [PubMed] [Google Scholar]
- 162.Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006;441:885–9. doi: 10.1038/nature04724. [DOI] [PubMed] [Google Scholar]
- 163.Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006;441:880–4. doi: 10.1038/nature04723. [DOI] [PubMed] [Google Scholar]
- 164.Winslow AR, Chen CW, Corrochano S, Acevedo-Arozena A, Gordon DE, Peden AA, et al. Alpha-synuclein impairs macroautophagy: implications for Parkinson’s disease. J Cell Biol. 2010;190:1023–37. doi: 10.1083/jcb.201003122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Ju JS, Fuentealba RA, Miller SE, Jackson E, Piwnica-Worms D, Baloh RH, et al. Valosin-containing protein (VCP) is required for autophagy and is disrupted in VCP disease. J Cell Biol. 2009;187:875–88. doi: 10.1083/jcb.200908115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Fukuda T, Ewan L, Bauer M, Mattaliano RJ, Zaal K, Ralston E, et al. Dysfunction of endocytic and autophagic pathways in a lysosomal storage disease. Ann Neurol. 2006;59:700–8. doi: 10.1002/ana.20807. [DOI] [PubMed] [Google Scholar]
- 167.Sanchez-Varo R, Trujillo-Estrada L, Sanchez-Mejias E, Torres M, Baglietto-Vargas D, Moreno-Gonzalez I, et al. Abnormal accumulation of autophagic vesicles correlates with axonal and synaptic pathology in young Alzheimer’s mice hippocampus. Acta Neuropathol. 2012;123:53–70. doi: 10.1007/s00401-011-0896-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Martinez-Vicente M, Talloczy Z, Wong E, Tang G, Koga H, Kaushik S, et al. Cargo recognition failure is responsible for inefficient autophagy in Huntington’s disease. Nat Neurosci. 2010;13:567–76. doi: 10.1038/nn.2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Durcan TM, Fon EA. Mutant ataxin-3 promotes the autophagic degradation of parkin. Autophagy. 2011;7:233–4. doi: 10.4161/auto.7.2.14224. [DOI] [PubMed] [Google Scholar]
- 170.Berger Z, Ravikumar B, Menzies FM, Oroz LG, Underwood BR, Pangalos MN, et al. Rapamycin alleviates toxicity of different aggregate-prone proteins. Hum Mol Genet. 2006;15:433–42. doi: 10.1093/hmg/ddi458. [DOI] [PubMed] [Google Scholar]
- 171.Webb JL, Ravikumar B, Atkins J, Skepper JN, Rubinsztein DC. Alpha-synuclein is degraded by both autophagy and the proteasome. J Biol Chem. 2003;278:25009–13. doi: 10.1074/jbc.M300227200. [DOI] [PubMed] [Google Scholar]
- 172.Lunemann JD, Schmidt J, Schmid D, Barthel K, Wrede A, Dalakas MC, et al. Beta-amyloid is a substrate of autophagy in sporadic inclusion body myositis. Ann Neurol. 2007;61:476–83. doi: 10.1002/ana.21115. [DOI] [PubMed] [Google Scholar]
- 173.Williams A, Sarkar S, Cuddon P, Ttofi EK, Saiki S, Siddiqi FH, et al. Novel targets for Huntington’s disease in an mTOR-independent autophagy pathway. Nat Chem Biol. 2008;4:295–305. doi: 10.1038/nchembio.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Ravikumar B, Vacher C, Berger Z, Davies JE, Luo S, Oroz LG, et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet. 2004;36:585–95. doi: 10.1038/ng1362. [DOI] [PubMed] [Google Scholar]
- 175.Caccamo A, Majumder S, Richardson A, Strong R, Oddo S. Molecular interplay between mammalian target of rapamycin (mTOR), amyloid-beta, and tau: effects on cognitive impairments. J Biol Chem. 2010;285:13107–20. doi: 10.1074/jbc.M110.100420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Spilman P, Podlutskaya N, Hart MJ, Debnath J, Gorostiza O, Bredesen D, et al. Inhibition of mTOR by rapamycin abolishes cognitive deficits and reduces amyloid-beta levels in a mouse model of Alzheimer’s disease. PLoS One. 2010;5:e9979. doi: 10.1371/journal.pone.0009979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Spencer B, Potkar R, Trejo M, Rockenstein E, Patrick C, Gindi R, et al. Beclin 1 gene transfer activates autophagy and ameliorates the neurodegenerative pathology in alpha-synuclein models of Parkinson’s and Lewy body diseases. J Neurosci. 2009;29:13578–88. doi: 10.1523/JNEUROSCI.4390-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Bilen J, Bonini NM. Genome-wide screen for modifiers of ataxin-3 neurodegeneration in Drosophila. PLoS Genet. 2007;3:1950–64. doi: 10.1371/journal.pgen.0030177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Yamamoto K, Seki T, Adachi N, Takahashi T, Tanaka S, Hide I, et al. Mutant protein kinase C gamma that causes spinocerebellar ataxia type 14 (SCA14) is selectively degraded by autophagy. Genes Cells. 2010;15:425–38. doi: 10.1111/j.1365-2443.2010.01395.x. [DOI] [PubMed] [Google Scholar]
- 180.Yu X, Ajayi A, Boga NR, Strom AL. Differential degradation of full-length and cleaved ataxin-7 fragments in a novel stable inducible SCA7 model. J Mol Neurosci. 2012;47:219–33. doi: 10.1007/s12031-012-9722-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Mookerjee S, Papanikolaou T, Guyenet SJ, Sampath V, Lin A, Vitelli C, et al. Posttranslational modification of ataxin-7 at lysine 257 prevents autophagy-mediated turnover of an N-terminal caspase-7 cleavage fragment. J Neurosci. 2009;29:15134–44. doi: 10.1523/JNEUROSCI.4720-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Nascimento-Ferreira I, Santos-Ferreira T, Sousa-Ferreira L, Auregan G, Onofre I, Alves S, et al. Overexpression of the autophagic beclin-1 protein clears mutant ataxin-3 and alleviates Machado–Joseph disease. Brain. 2011;134:1400–15. doi: 10.1093/brain/awr047. [DOI] [PubMed] [Google Scholar]
- 183.Grewer C, Rauen T. Electrogenic glutamate transporters in the CNS: molecular mechanism, pre-steady-state kinetics, and their impact on synaptic signaling. J Membr Biol. 2005;203:1–20. doi: 10.1007/s00232-004-0731-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Ramamoorthy S, Leibach FH, Mahesh VB. Ganapathy V. Active transport of dopamine in human placental brush-border membrane vesicles. Am J Physiol. 1992;262:C1189–96. doi: 10.1152/ajpcell.1992.262.5.C1189. [DOI] [PubMed] [Google Scholar]
- 185.Fahlke C. Molecular mechanisms of ion conduction in ClC-type chloride channels: lessons from disease-causing mutations. Kidney Int. 2000;57:780–6. doi: 10.1046/j.1523-1755.2000.00915.x. [DOI] [PubMed] [Google Scholar]
- 186.Thakker RV. Chloride channels in renal disease. Adv Nephrol Necker Hosp. 1999;29:289–98. [PubMed] [Google Scholar]
- 187.Zhuchenko O, Bailey J, Bonnen P, Ashizawa T, Stockton DW, Amos C, et al. Autosomal dominant cerebellar ataxia (SCA6) associated with small polyglutamine expansions in the alpha 1A-voltage-dependent calcium channel. Nat Genet. 1997;15:62–9. doi: 10.1038/ng0197-62. [DOI] [PubMed] [Google Scholar]
- 188.Pulst SM, Santos N, Wang D, Yang H, Huynh D, Velazquez L, et al. Spinocerebellar ataxia type 2: polyQ repeat variation in the CACNA1A calcium channel modifies age of onset. Brain. 2005;128:2297–303. doi: 10.1093/brain/awh586. [DOI] [PubMed] [Google Scholar]
- 189.Geschwind DH, Perlman S, Figueroa KP, Karrim J, Baloh RW, Pulst SM. Spinocerebellar ataxia type 6. Frequency of the mutation and genotype-phenotype correlations. Neurology. 1997;49:1247–51. doi: 10.1212/wnl.49.5.1247. [DOI] [PubMed] [Google Scholar]
- 190.Baloh RW. Episodic ataxias 1 and 2. Handb Clin Neurol. 2012;103:595–602. doi: 10.1016/B978-0-444-51892-7.00042-5. [DOI] [PubMed] [Google Scholar]
- 191.Jodice C, Mantuano E, Veneziano L, Trettel F, Sabbadini G, Calandriello L, et al. Episodic ataxia type 2 (EA2) and spinocerebellar ataxia type 6 (SCA6) due to CAG repeat expansion in the CACNA1A gene on chromosome 19p. Hum Mol Genet. 1997;6:1973–8. doi: 10.1093/hmg/6.11.1973. [DOI] [PubMed] [Google Scholar]
- 192.Denier C, Ducros A, Vahedi K, Joutel A, Thierry P, Ritz A, et al. High prevalence of CACNA1A truncations and broader clinical spectrum in episodic ataxia type 2. Neurology. 1999;52:1816–21. doi: 10.1212/wnl.52.9.1816. [DOI] [PubMed] [Google Scholar]
- 193.Walter JT, Alvina K, Womack MD, Chevez C, Khodakhah K. Decreases in the precision of Purkinje cell pacemaking cause cerebellar dysfunction and ataxia. Nat Neurosci. 2006;9:389–97. doi: 10.1038/nn1648. [DOI] [PubMed] [Google Scholar]
- 194.Watase K, Barrett CF, Miyazaki T, Ishiguro T, Ishikawa K, Hu Y, et al. Spinocerebellar ataxia type 6 knockin mice develop a progressive neuronal dysfunction with age-dependent accumulation of mutant CaV2.1 channels. Proc Natl Acad Sci U S A. 2008;105:11987–92. doi: 10.1073/pnas.0804350105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Tonelli A, D‘Angelo MG, Salati R, Villa L, Germinasi C, Frattini T, et al. Early onset, non fluctuating spinocerebellar ataxia and a novel missense mutation in CACNA1A gene. J Neurol Sci. 2006;241:13–7. doi: 10.1016/j.jns.2005.10.007. [DOI] [PubMed] [Google Scholar]
- 196.Jen J, Wan J, Graves M, Yu H, Mock AF, Coulin CJ, et al. Loss-of-function EA2 mutations are associated with impaired neuromuscular transmission. Neurology. 2001;57:1843–8. doi: 10.1212/wnl.57.10.1843. [DOI] [PubMed] [Google Scholar]
- 197.Tottene A, Fellin T, Pagnutti S, Luvisetto S, Striessnig J, Fletcher C, et al. Familial hemiplegic migraine mutations increase Ca(2+) influx through single human CaV2.1 channels and decrease maximal CaV2.1 current density in neurons. Proc Natl Acad Sci U S A. 2002;99:13284–9. doi: 10.1073/pnas.192242399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Mullner C, Broos LA, van den Maagdenberg AM, Striessnig J. Familial hemiplegic migraine type 1 mutations K1336E, W1684R, and V1696I alter Cav2.1 Ca2+ channel gating: evidence for beta-subunit isoform-specific effects. J Biol Chem. 2004;279:51844–50. doi: 10.1074/jbc.M408756200. [DOI] [PubMed] [Google Scholar]
- 199.Ducros A, Denier C, Joutel A, Cecillon M, Lescoat C, Vahedi K, et al. The clinical spectrum of familial hemiplegic migraine associated with mutations in a neuronal calcium channel. N Engl J Med. 2001;345:17–24. doi: 10.1056/NEJM200107053450103. [DOI] [PubMed] [Google Scholar]
- 200.Waters MF, Fee D, Figueroa KP, Nolte D, Muller U, Advincula J, et al. An autosomal dominant ataxia maps to 19q13: allelic heterogeneity of SCA13 or novel locus? Neurology. 2005;65:1111–3. doi: 10.1212/01.wnl.0000177490.05162.41. [DOI] [PubMed] [Google Scholar]
- 201.Herman-Bert A, Stevanin G, Netter JC, Rascol O, Brassat D, Calvas P, et al. Mapping of spinocerebellar ataxia 13 to chromosome 19q13.3-q13.4 in a family with autosomal dominant cerebellar ataxia and mental retardation. Am J Hum Genet. 2000;67:229–35. doi: 10.1086/302958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Waters MF, Minassian NA, Stevanin G, Figueroa KP, Bannister JP, Nolte D, et al. Mutations in voltage-gated potassium channel KCNC3 cause degenerative and developmental central nervous system phenotypes. Nat Genet. 2006;38:447–51. doi: 10.1038/ng1758. [DOI] [PubMed] [Google Scholar]
- 203.Waters MF, Pulst SM. Sca13. Cerebellum. 2008;7:165–9. doi: 10.1007/s12311-008-0039-7. [DOI] [PubMed] [Google Scholar]
- 204.Figueroa KP, Minassian NA, Stevanin G, Waters M, Garibyan V, Forlani S, et al. KCNC3: phenotype, mutations, channel biophysics—a study of 260 familial ataxia patients. Hum Mutat. 2010;31:191–6. doi: 10.1002/humu.21165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Figueroa KP, Waters MF, Garibyan V, Bird TD, Gomez CM, Ranum LP, et al. Frequency of KCNC3 DNA variants as causes of spinocerebellar ataxia 13 (SCA13) PLoS One. 2011;6:e17811. doi: 10.1371/journal.pone.0017811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Shakkottai VG, Chou CH, Oddo S, Sailer CA, Knaus HG, Gutman GA, et al. Enhanced neuronal excitability in the absence of neurodegeneration induces cerebellar ataxia. J Clin Invest. 2004;113:582–90. doi: 10.1172/JCI20216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Figueroa KP, Chan P, Schols L, Tanner C, Riess O, Perlman SL, et al. Association of moderate polyglutamine tract expansions in the slow calcium-activated potassium channel type 3 with ataxia. Arch Neurol. 2001;58:1649–53. doi: 10.1001/archneur.58.10.1649. [DOI] [PubMed] [Google Scholar]
- 208.Browne DL, Gancher ST, Nutt JG, Brunt ER, Smith EA, Kramer P, et al. Episodic ataxia/myokymia syndrome is associated with point mutations in the human potassium channel gene, KCNA1. Nat Genet. 1994;8:136–40. doi: 10.1038/ng1094-136. [DOI] [PubMed] [Google Scholar]
- 209.D’Adamo MC, Imbrici P, Sponcichetti F, Pessia M. Mutations in the KCNA1 gene associated with episodic ataxia type-1 syndrome impair heteromeric voltage-gated K(+) channel function. FASEB J. 1999;13:1335–45. doi: 10.1096/fasebj.13.11.1335. [DOI] [PubMed] [Google Scholar]
- 210.Jan LY, Jan YN. Voltage-gated potassium channels and the diversity of electrical signalling. J Physiol. 2012;590:2591–9. doi: 10.1113/jphysiol.2011.224212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Rea R, Spauschus A, Eunson LH, Hanna MG, Kullmann DM. Variable K(+) channel subunit dysfunction in inherited mutations of KCNA1. J Physiol. 2002;538:5–23. doi: 10.1113/jphysiol.2001.013242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Khavandgar S, Walter JT, Sageser K, Khodakhah K. Kv1 channels selectively prevent dendritic hyperexcitability in rat Purkinje cells. J Physiol. 2005;569:545–57. doi: 10.1113/jphysiol.2005.098053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Herson PS, Virk M, Rustay NR, Bond CT, Crabbe JC, Adelman JP, et al. A mouse model of episodic ataxia type-1. Nat Neurosci. 2003;6:378–83. doi: 10.1038/nn1025. [DOI] [PubMed] [Google Scholar]
- 214.Ishida S, Sakamoto Y, Nishio T, Baulac S, Kuwamura M, Ohno Y, et al. Kcna1-mutant rats dominantly display myokymia, neuromyotonia and spontaneous epileptic seizures. Brain Res. 2012;1435:154–66. doi: 10.1016/j.brainres.2011.11.023. [DOI] [PubMed] [Google Scholar]
- 215.Lee YC, Durr A, Majczenko K, Huang YH, Liu YC, Lien CC, et al. Mutations in KCND3 cause spinocerebellar ataxia type 22. Ann Neurol. 2012;72:859–69. doi: 10.1002/ana.23701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Duarri A, Jezierska J, Fokkens M, Meijer M, Schelhaas HJ, den Dunnen WF, et al. Mutations in potassium channel kcnd3 cause spinocerebellar ataxia type 19. Ann Neurol. 2012;72:870–80. doi: 10.1002/ana.23700. [DOI] [PubMed] [Google Scholar]
- 217.Storey E, Gardner RJ. Spinocerebellar ataxia type 15. Handb Clin Neurol. 2012;103:561–5. doi: 10.1016/B978-0-444-51892-7.00037-1. [DOI] [PubMed] [Google Scholar]
- 218.Iwaki A, Kawano Y, Miura S, Shibata H, Matsuse D, Li W, et al. Heterozygous deletion of ITPR1, but not SUMF1, in spinocerebellar ataxia type 16. J Med Genet. 2008;45:32–5. doi: 10.1136/jmg.2007.053942. [DOI] [PubMed] [Google Scholar]
- 219.van de Leemput J, Chandran J, Knight MA, Holtzclaw LA, Scholz S, Cookson MR, et al. Deletion at ITPR1 underlies ataxia in mice and spinocerebellar ataxia 15 in humans. PLoS Genet. 2007;3:e108. doi: 10.1371/journal.pgen.0030108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Seal RP, Leighton BH, Amara SG. A model for the topology of excitatory amino acid transporters determined by the extracellular accessibility of substituted cysteines. Neuron. 2000;25:695–706. doi: 10.1016/s0896-6273(00)81071-5. [DOI] [PubMed] [Google Scholar]
- 221.Jen JC, Wan J, Palos TP, Howard BD, Baloh RW. Mutation in the glutamate transporter EAAT1 causes episodic ataxia, hemiplegia, and seizures. Neurology. 2005;65:529–34. doi: 10.1212/01.wnl.0000172638.58172.5a. [DOI] [PubMed] [Google Scholar]
- 222.Seal RP, Shigeri Y, Eliasof S, Leighton BH, Amara SG. Sulfhydryl modification of V449C in the glutamate transporter EAAT1 abolishes substrate transport but not the substrate-gated anion conductance. Proc Natl Acad Sci U S A. 2001;98:15324–9. doi: 10.1073/pnas.011400198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Heinzen EL, Swoboda KJ, Hitomi Y, Gurrieri F, Nicole S, de Vries B, et al. De novo mutations in ATP1A3 cause alternating hemiplegia of childhood. Nat Genet. 2012;44:1030–4. doi: 10.1038/ng.2358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.de Carvalho AP, Sweadner KJ, Penniston JT, Zaremba J, Liu L, Caton M, et al. Mutations in the Na+/K+-ATPase alpha3 gene ATP1A3 are associated with rapid-onset dystonia parkinsonism. Neuron. 2004;43:169–75. doi: 10.1016/j.neuron.2004.06.028. [DOI] [PubMed] [Google Scholar]
- 225.Brashear A, Mink JW, Hill DF, Boggs N, McCall WV, Stacy MA, et al. ATP1A3 mutations in infants: a new rapid-onset dystonia-parkinsonism phenotype characterized by motor delay and ataxia. Dev Med Child Neurol. 2012;54:1065–7. doi: 10.1111/j.1469-8749.2012.04421.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Einholm AP, Toustrup-Jensen MS, Holm R, Andersen JP, Vilsen B. The rapid-onset dystonia parkinsonism mutation D923N of the Na+, K+-ATPase alpha3 isoform disrupts Na+ interaction at the third Na+ site. J Biol Chem. 2010;285:26245–54. doi: 10.1074/jbc.M110.123976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Zanni G, Cali T, Kalscheuer VM, Ottolini D, Barresi S, Lebrun N, et al. Mutation of plasma membrane Ca2+ ATPase isoform 3 in a family with X-linked congenital cerebellar ataxia impairs Ca2+ homeostasis. Proc Natl Acad Sci U S A. 2012;109:14514–9. doi: 10.1073/pnas.1207488109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Hansen S, Pratap M, Otis T, Pulst SM. Changes in Purkinje cell firing and gene expression precede behavioral pathology in a mouse model of SCA2. Hum Mol Genet. 2013;22:271–83. doi: 10.1093/hmg/dds427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Hourez R, Servais L, Orduz D, Gall D, Millard I, de Kerchove d’Exaerde A, et al. Aminopyridines correct early dysfunction and delay neurodegeneration in a mouse model of spinocerebellar ataxia type 1. J Neurosci. 2011;31:11795–807. doi: 10.1523/JNEUROSCI.0905-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Shakkottai VG, do Carmo Costa M, Dell’Orco JM, Sankaranarayanan A, Wulff H, Paulson HL. Early changes in cerebellar physiology accompany motor dysfunction in the polyglutamine disease spinocerebellar ataxia type 3. J Neurosci. 2011;31:13002–14. doi: 10.1523/JNEUROSCI.2789-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Shchelochkov OA, Cheung SW, Lupski JR. Genomic and clinical characteristics of microduplications in chromosome 17. Am J Med Genet. 2010;152A:1101–10. doi: 10.1002/ajmg.a.33248. [DOI] [PubMed] [Google Scholar]
- 232.Matsuura T, Yamagata T, Burgess DL, Rasmussen A, Grewal RP, Watase K, et al. Large expansion of the ATTCT pentanucleotide repeat in spinocerebellar ataxia type 10. Nat Genet. 2000;26:191–4. doi: 10.1038/79911. [DOI] [PubMed] [Google Scholar]
- 233.Sato N, Amino T, Kobayashi K, Asakawa S, Ishiguro T, Tsunemi T, et al. Spinocerebellar ataxia type 31 is associated with “inserted” penta-nucleotide repeats containing (TGGAA)n. Am J Hum Genet. 2009;85:544–57. doi: 10.1016/j.ajhg.2009.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Hagerman RJ, Leehey M, Heinrichs W, Tassone F, Wilson R, Hills J, et al. Intention tremor, parkinsonism, and generalized brain atrophy in male carriers of fragile X. Neurology. 2001;57:127–30. doi: 10.1212/wnl.57.1.127. [DOI] [PubMed] [Google Scholar]
- 235.Wang JL, Wu YQ, Lei LF, Shen L, Jiang H, Zhou YF, et al. [Polynucleotide repeat expansion of nine spinocerebellar ataxia subtypes and dentatorubral–pallidoluysian atrophy in healthy Chinese Han population] Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 2010;27:501–5. doi: 10.3760/cma.j.issn.1003-9406.2010.05.006. [DOI] [PubMed] [Google Scholar]
- 236.Wakamiya M, Matsuura T, Liu Y, Schuster GC, Gao R, Xu W, et al. The role of ataxin 10 in the pathogenesis of spinocerebellar ataxia type 10. Neurology. 2006;67:607–13. doi: 10.1212/01.wnl.0000231140.26253.eb. [DOI] [PubMed] [Google Scholar]
- 237.Keren B, Jacquette A, Depienne C, Leite P, Durr A, Carpentier W, et al. Evidence against haploinsuffiency of human ataxin 10 as a cause of spinocerebellar ataxia type 10. Neurogenetics. 2010;11:273–4. doi: 10.1007/s10048-009-0227-8. [DOI] [PubMed] [Google Scholar]
- 238.White MC, Gao R, Xu W, Mandal SM, Lim JG, Hazra TK, et al. Inactivation of hnRNP K by expanded intronic AUUCU repeat induces apoptosis via translocation of PKCdelta to mitochondria in spinocerebellar ataxia 10. PLoS Genet. 2010;6:e1000984. doi: 10.1371/journal.pgen.1000984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.White M, Xia G, Gao R, Wakamiya M, Sarkar PS, McFarland K, et al. Transgenic mice with SCA10 pentanucleotide repeats show motor phenotype and susceptibility to seizure: a toxic RNA gain-of-function model. J Neurosci Res. 2012;90:706–14. doi: 10.1002/jnr.22786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Chan JY, Huang SM, Liu ST, Huang CH. The transactivation domain of heterogeneous nuclear ribonucleoprotein K overlaps its nuclear shuttling domain. Int J Biochem Cell Biol. 2008;40:2078–89. doi: 10.1016/j.biocel.2008.02.005. [DOI] [PubMed] [Google Scholar]
- 241.Dejgaard K, Leffers H. Characterisation of the nucleic-acid-binding activity of KH domains. Different properties of different domains. Eur J Biochem. 1996;241:425–31. doi: 10.1111/j.1432-1033.1996.00425.x. [DOI] [PubMed] [Google Scholar]
- 242.Bomsztyk K, Denisenko O, Ostrowski J. hnRNP K: one protein multiple processes. Bioessays. 2004;26:629–38. doi: 10.1002/bies.20048. [DOI] [PubMed] [Google Scholar]
- 243.Schullery DS, Ostrowski J, Denisenko ON, Stempka L, Shnyreva M, Suzuki H, et al. Regulated interaction of protein kinase Cdelta with the heterogeneous nuclear ribonucleoprotein K protein. J Biol Chem. 1999;274:15101–9. doi: 10.1074/jbc.274.21.15101. [DOI] [PubMed] [Google Scholar]
- 244.Brodie C, Blumberg PM. Regulation of cell apoptosis by protein kinase c delta. Apoptosis. 2003;8:19–27. doi: 10.1023/a:1021640817208. [DOI] [PubMed] [Google Scholar]
- 245.Sellier C, Rau F, Liu Y, Tassone F, Hukema RK, Gattoni R, et al. Sam68 sequestration and partial loss of function are associated with splicing alterations in FXTAS patients. EMBO J. 2010;29:1248–61. doi: 10.1038/emboj.2010.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Mankodi A, Urbinati CR, Yuan QP, Moxley RT, Sansone V, Krym M, et al. Muscleblind localizes to nuclear foci of aberrant RNA in myotonic dystrophy types 1 and 2. Hum Mol Genet. 2001;10:2165–70. doi: 10.1093/hmg/10.19.2165. [DOI] [PubMed] [Google Scholar]
- 247.Jiang H, Mankodi A, Swanson MS, Moxley RT, Thornton CA. Myotonic dystrophy type 1 is associated with nuclear foci of mutant RNA, sequestration of muscleblind proteins and deregulated alternative splicing in neurons. Hum Mol Genet. 2004;13:3079–88. doi: 10.1093/hmg/ddh327. [DOI] [PubMed] [Google Scholar]
- 248.Savkur RS, Philips AV, Cooper TA. Aberrant regulation of insulin receptor alternative splicing is associated with insulin resistance in myotonic dystrophy. Nat Genet. 2001;29:40–7. doi: 10.1038/ng704. [DOI] [PubMed] [Google Scholar]
- 249.Philips AV, Timchenko LT, Cooper TA. Disruption of splicing regulated by a CUG-binding protein in myotonic dystrophy. Science. 1998;280:737–41. doi: 10.1126/science.280.5364.737. [DOI] [PubMed] [Google Scholar]
- 250.Charlet BN, Savkur RS, Singh G, Philips AV, Grice EA, Cooper TA. Loss of the muscle-specific chloride channel in type 1 myotonic dystrophy due to misregulated alternative splicing. Mol Cell. 2002;10:45–53. doi: 10.1016/s1097-2765(02)00572-5. [DOI] [PubMed] [Google Scholar]
- 251.Mankodi A, Takahashi MP, Jiang H, Beck CL, Bowers WJ, Moxley RT, et al. Expanded CUG repeats trigger aberrant splicing of ClC-1 chloride channel pre-mRNA and hyperexcitability of skeletal muscle in myotonic dystrophy. Mol Cell. 2002;10:35–44. doi: 10.1016/s1097-2765(02)00563-4. [DOI] [PubMed] [Google Scholar]
- 252.Wheeler TM, Lueck JD, Swanson MS, Dirksen RT, Thornton CA. Correction of ClC-1 splicing eliminates chloride channelopathy and myotonia in mouse models of myotonic dystrophy. J Clin Invest. 2007;117:3952–7. doi: 10.1172/JCI33355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.McFarland KN, Liu J, Landrian I, Gao R, Sarkar PS, Raskin S, et al. Paradoxical effects of repeat interruptions on spinocerebellar ataxia type 10 expansions and repeat instability. Eur J Hum Genet. 2013;21:1272–6. doi: 10.1038/ejhg.2013.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Ranum LP, Chung MY, Banfi S, Bryer A, Schut LJ, Ramesar R, et al. Molecular and clinical correlations in spinocerebellar ataxia type I: evidence for familial effects on the age at onset. Am J Hum Genet. 1994;55:244–52. [PMC free article] [PubMed] [Google Scholar]
- 255.Quan F, Janas J, Popovich BW. A novel CAG repeat configuration in the SCA1 gene: implications for the molecular diagnostics of spinocerebellar ataxia type 1. Hum Mol Genet. 1995;4:2411–3. doi: 10.1093/hmg/4.12.2411. [DOI] [PubMed] [Google Scholar]
- 256.Matsuyama Z, Izumi Y, Kameyama M, Kawakami H, Nakamura S. The effect of CAT trinucleotide interruptions on the age at onset of spinocerebellar ataxia type 1 (SCA1) J Med Genet. 1999;36:546–8. [PMC free article] [PubMed] [Google Scholar]
- 257.Matsuura T, Fang P, Pearson CE, Jayakar P, Ashizawa T, Roa BB, et al. Interruptions in the expanded ATTCT repeat of spinocerebellar ataxia type 10: repeat purity as a disease modifier? Am J Hum Genet. 2006;78:125–9. doi: 10.1086/498654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Spaans F, Faber CG, Smeets HJ, Hofman PA, Braida C, Monckton DG, et al. Encephalopathic attacks in a family co-segregating myotonic dystrophy type 1, an intermediate Charcot–Marie–Tooth neuropathy and early hearing loss. J Neurol Neurosurg Psychiatry. 2009;80:1029–35. doi: 10.1136/jnnp.2008.170126. [DOI] [PubMed] [Google Scholar]
- 259.Braida C, Stefanatos RK, Adam B, Mahajan N, Smeets HJ, Niel F, et al. Variant CCG and GGC repeats within the CTG expansion dramatically modify mutational dynamics and likely contribute toward unusual symptoms in some myotonic dystrophy type 1 patients. Hum Mol Genet. 2010;19:1399–412. doi: 10.1093/hmg/ddq015. [DOI] [PubMed] [Google Scholar]
- 260.Elden AC, Kim HJ, Hart MP, Chen-Plotkin AS, Johnson BS, Fang X, et al. Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nature. 2010;466:1069–75. doi: 10.1038/nature09320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Sakai H, Yoshida K, Shimizu Y, Morita H, Ikeda S, Matsumoto N. Analysis of an insertion mutation in a cohort of 94 patients with spinocerebellar ataxia type 31 from Nagano, Japan. Neurogenetics. 2010;11:409–15. doi: 10.1007/s10048-010-0245-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Bonini NM, Gitler AD. Model organisms reveal insight into human neurodegenerative disease: ataxin-2 intermediate-length polyglutamine expansions are a risk factor for ALS. J Mol Neurosci. 2011;45:676–83. doi: 10.1007/s12031-011-9548-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Corrado L, Mazzini L, Oggioni GD, Luciano B, Godi M, Brusco A, et al. ATXN-2 CAG repeat expansions are interrupted in ALS patients. Hum Genet. 2011;130:575–80. doi: 10.1007/s00439-011-1000-2. [DOI] [PubMed] [Google Scholar]
- 264.Ishikawa K, Durr A, Klopstock T, Muller S, De Toffol B, Vidailhet M, et al. Pentanucleotide repeats at the spinocerebellar ataxia type 31 (SCA31) locus in Caucasians. Neurology. 2011;77:1853–5. doi: 10.1212/WNL.0b013e3182377e3a. [DOI] [PubMed] [Google Scholar]
- 265.Yu Z, Zhu Y, Chen-Plotkin AS, Clay-Falcone D, McCluskey L, Elman L, et al. PolyQ repeat expansions in ATXN2 associated with ALS are CAA interrupted repeats. PLoS One. 2011;6:e17951. doi: 10.1371/journal.pone.0017951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Koob MD, Moseley ML, Schut LJ, Benzow KA, Bird TD, Day JW, et al. An untranslated CTG expansion causes a novel form of spinocerebellar ataxia (SCA8) Nat Genet. 1999;21:379–84. doi: 10.1038/7710. [DOI] [PubMed] [Google Scholar]
- 267.Cho DH, Thienes CP, Mahoney SE, Analau E, Filippova GN, Tapscott SJ. Antisense transcription and heterochromatin at the DM1 CTG repeats are constrained by CTCF. Mol Cell. 2005;20:483–9. doi: 10.1016/j.molcel.2005.09.002. [DOI] [PubMed] [Google Scholar]
- 268.Libby RT, Hagerman KA, Pineda VV, Lau R, Cho DH, Baccam SL, et al. CTCF cis-regulates trinucleotide repeat instability in an epigenetic manner: a novel basis for mutational hot spot determination. PLoS Genet. 2008;4:e1000257. doi: 10.1371/journal.pgen.1000257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Chung DW, Rudnicki DD, Yu L, Margolis RL. A natural antisense transcript at the Huntington's disease repeat locus regulates HTT expression. Hum Mol Genet. 2011;20:3467–77. doi: 10.1093/hmg/ddr263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Rudnicki DD, Pletnikova O, Vonsattel JP, Ross CA, Margolis RL. A comparison of Huntington disease and Huntington disease-like 2 neuropathology. J Neuropathol Exp Neurol. 2008;67:366–74. doi: 10.1097/NEN.0b013e31816b4aee. [DOI] [PubMed] [Google Scholar]
- 271.Moseley ML, Zu T, Ikeda Y, Gao W, Mosemiller AK, Daughters RS, et al. Bidirectional expression of CUG and CAG expansion transcripts and intranuclear polyglutamine inclusions in spinocerebellar ataxia type 8. Nat Genet. 2006;38:758–69. doi: 10.1038/ng1827. [DOI] [PubMed] [Google Scholar]
- 272.Ikeda Y, Daughters RS, Ranum LP. Bidirectional expression of the SCA8 expansion mutation: one mutation, two genes. Cerebellum. 2008;7:150–8. doi: 10.1007/s12311-008-0010-7. [DOI] [PubMed] [Google Scholar]
- 273.Chen IC, Lin HY, Lee GC, Kao SH, Chen CM, Wu YR, et al. Spinocerebellar ataxia type 8 larger triplet expansion alters histone modification and induces RNA foci. BMC Mol Biol. 2009;10:9. doi: 10.1186/1471-2199-10-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Batra R, Charizanis K, Swanson MS. Partners in crime: bidirectional transcription in unstable microsatellite disease. Hum Mol Genet. 2010;19:R77–82. doi: 10.1093/hmg/ddq132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Mykowska A, Sobczak K, Wojciechowska M, Kozlowski P, Krzyzosiak WJ. CAG repeats mimic CUG repeats in the misregulation of alternative splicing. Nucleic Acids Res. 2011;39:8938–51. doi: 10.1093/nar/gkr608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Zu T, Gibbens B, Doty NS, Gomes-Pereira M, Huguet A, Stone MD, et al. Non-ATG-initiated translation directed by microsatellite expansions. Proc Natl Acad Sci U S A. 2011;108:260–5. doi: 10.1073/pnas.1013343108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Ash PE, Bieniek KF, Gendron TF, Caulfield T, Lin WL, Dejesus-Hernandez M, et al. Unconventional translation of C9ORF72 GGGGCC expansion generates insoluble polypeptides specific to c9FTD/ALS. Neuron. 2013;77:639–46. doi: 10.1016/j.neuron.2013.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Pearson CE. Repeat associated non-ATG translation initiation: one DNA, two transcripts, seven reading frames, potentially nine toxic entities! PLoS Genet. 2011;7:e1002018. doi: 10.1371/journal.pgen.1002018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Finsterer J. Mitochondrial ataxias. Can J Neurol Sci. 2009;36:543–53. doi: 10.1017/s0317167100008027. [DOI] [PubMed] [Google Scholar]
- 280.Di Donato S, Marmolino D, Taroni F. Mitochondrial disorders. In: Manto M, Schmahmann J, Rossi F, Gruol D, Koibuchi N, editors. Handbook of the cerebellum and cerebellar disorder. Springer; the Netherlands: 2013. pp. 2269–311. [Google Scholar]
- 281.Montero R, Pineda M, Aracil A, Vilaseca MA, Briones P, Sanchez-Alcazar JA, et al. Clinical, biochemical and molecular aspects of cerebellar ataxia and coenzyme Q10 deficiency. Cerebellum. 2007;6:118–22. doi: 10.1080/14734220601021700. [DOI] [PubMed] [Google Scholar]
- 282.Quinzii CM, Lopez LC, Naini A, DiMauro S, Hirano M. Human CoQ10 deficiencies. Biofactors. 2008;32:113–8. doi: 10.1002/biof.5520320113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Spindler M, Beal MF, Henchcliffe C. Coenzyme Q10 effects in neurodegenerative disease. Neuropsychiatr Dis Treat. 2009;5:597–610. doi: 10.2147/ndt.s5212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Lagier-Tourenne C, Tazir M, Lopez LC, Quinzii CM, Assoum M, Drouot N, et al. ADCK3, an ancestral kinase, is mutated in a form of recessive ataxia associated with coenzyme Q10 deficiency. Am J Hum Genet. 2008;82:661–72. doi: 10.1016/j.ajhg.2007.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Gerards M, van den Bosch B, Calis C, Schoonderwoerd K, van Engelen K, Tijssen M, et al. Nonsense mutations in CABC1/ADCK3 cause progressive cerebellar ataxia and atrophy. Mitochondrion. 2010;10:510–5. doi: 10.1016/j.mito.2010.05.008. [DOI] [PubMed] [Google Scholar]
- 286.Lu S, Lu LY, Liu MF, Yuan QJ, Sham MH, Guan XY, et al. Cerebellar defects in Pdss2 conditional knockout mice during embryonic development and in adulthood. Neurobiol Dis. 2012;45:219–33. doi: 10.1016/j.nbd.2011.08.006. [DOI] [PubMed] [Google Scholar]
- 287.Spinazzola A, Zeviani M. Disorders of nuclear-mitochondrial intergenomic signaling. Gene. 2005;354:162–8. doi: 10.1016/j.gene.2005.03.025. [DOI] [PubMed] [Google Scholar]
- 288.Copeland WC. Defects in mitochondrial DNA replication and human disease. Crit Rev Biochem Mol Biol. 2012;47:64–74. doi: 10.3109/10409238.2011.632763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Kaguni LS. DNA polymerase gamma, the mitochondrial replicase. Annu Rev Biochem. 2004;73:293–320. doi: 10.1146/annurev.biochem.72.121801.161455. [DOI] [PubMed] [Google Scholar]
- 290.Wong LJ, Naviaux RK, Brunetti-Pierri N, Zhang Q, Schmitt ES, Truong C, et al. Molecular and clinical genetics of mitochondrial diseases due to POLG mutations. Hum Mutat. 2008;29:E150–72. doi: 10.1002/humu.20824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Hakonen AH, Heiskanen S, Juvonen V, Lappalainen I, Luoma PT, Rantamaki M, et al. Mitochondrial DNA polymerase W748S mutation: a common cause of autosomal recessive ataxia with ancient European origin. Am J Hum Genet. 2005;77:430–41. doi: 10.1086/444548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Milone M, Brunetti-Pierri N, Tang LY, Kumar N, Mezei MM, Josephs K, et al. Sensory ataxic neuropathy with ophthalmoparesis caused by POLG mutations. Neuromuscul Disord. 2008;18:626–32. doi: 10.1016/j.nmd.2008.05.009. [DOI] [PubMed] [Google Scholar]
- 293.Nikali K, Suomalainen A, Saharinen J, Kuokkanen M, Spelbrink JN, Lonnqvist T, et al. Infantile onset spinocerebellar ataxia is caused by recessive mutations in mitochondrial proteins Twinkle and Twinky. Hum Mol Genet. 2005;14:2981–90. doi: 10.1093/hmg/ddi328. [DOI] [PubMed] [Google Scholar]
- 294.Sykora P, Croteau DL, Bohr VA, Wilson DM., 3rd Aprataxin localizes to mitochondria and preserves mitochondrial function. Proc Natl Acad Sci U S A. 2011;108:7437–42. doi: 10.1073/pnas.1100084108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Das BB, Dexheimer TS, Maddali K, Pommier Y. Role of tyrosyl-DNA phosphodiesterase (TDP1) in mitochondria. Proc Natl Acad Sci U S A. 2010;107:19790–5. doi: 10.1073/pnas.1009814107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Pandolfo M, Pastore A. The pathogenesis of Friedreich ataxia and the structure and function of frataxin. J Neurol. 2009;256(Suppl 1):9–17. doi: 10.1007/s00415-009-1003-2. [DOI] [PubMed] [Google Scholar]
- 297.Marmolino D. Friedreich’s ataxia: past, present and future. Brain Res Rev. 2011;67:311–30. doi: 10.1016/j.brainresrev.2011.04.001. [DOI] [PubMed] [Google Scholar]
- 298.Martelli A, Napierala M, Puccio H. Understanding the genetic and molecular pathogenesis of Friedreich’s ataxia through animal and cellular models. Dis Model Mech. 2012;5:165–76. doi: 10.1242/dmm.008706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Martelli A, Wattenhofer-Donze M, Schmucker S, Bouvet S, Reutenauer L, Puccio H. Frataxin is essential for extramitochondrial Fe–S cluster proteins in mammalian tissues. Hum Mol Genet. 2007;16:2651–8. doi: 10.1093/hmg/ddm163. [DOI] [PubMed] [Google Scholar]
- 300.Babcock M, de Silva D, Oaks R, Davis-Kaplan S, Jiralerspong S, Montermini L, et al. Regulation of mitochondrial iron accumulation by Yfh1p, a putative homolog of frataxin. Science. 1997;276:1709–12. doi: 10.1126/science.276.5319.1709. [DOI] [PubMed] [Google Scholar]
- 301.Puccio H, Simon D, Cossee M, Criqui-Filipe P, Tiziano F, Melki J, et al. Mouse models for Friedreich ataxia exhibit cardiomyopathy, sensory nerve defect and Fe–S enzyme deficiency followed by intramitochondrial iron deposits. Nat Genet. 2001;27:181–6. doi: 10.1038/84818. [DOI] [PubMed] [Google Scholar]
- 302.Wong A, Yang J, Cavadini P, Gellera C, Lonnerdal B, Taroni F, et al. The Friedreich’s ataxia mutation confers cellular sensitivity to oxidant stress which is rescued by chelators of iron and calcium and inhibitors of apoptosis. Hum Mol Genet. 1999;8:425–30. doi: 10.1093/hmg/8.3.425. [DOI] [PubMed] [Google Scholar]
- 303.DiMauro S, Schon EA. Mitochondrial disorders in the nervous system. Annu Rev Neurosci. 2008;31:91–123. doi: 10.1146/annurev.neuro.30.051606.094302. [DOI] [PubMed] [Google Scholar]
- 304.Schon EA, Przedborski S. Mitochondria: the next (neurode)generation. Neuron. 2011;70:1033–53. doi: 10.1016/j.neuron.2011.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305.Kroemer G, Galluzzi L, Brenner C. Mitochondrial membrane permeabilization in cell death. Physiol Rev. 2007;87:99–163. doi: 10.1152/physrev.00013.2006. [DOI] [PubMed] [Google Scholar]
- 306.Choo YS, Johnson GV, MacDonald M, Detloff PJ, Lesort M. Mutant huntingtin directly increases susceptibility of mitochondria to the calcium-induced permeability transition and cytochrome c release. Hum Mol Genet. 2004;13:1407–20. doi: 10.1093/hmg/ddh162. [DOI] [PubMed] [Google Scholar]
- 307.Lipinski MM, Yuan J. Mechanisms of cell death in polyglutamine expansion diseases. Curr Opin Pharmacol. 2004;4:85–90. doi: 10.1016/j.coph.2003.09.008. [DOI] [PubMed] [Google Scholar]
- 308.Chou AH, Yeh TH, Kuo YL, Kao YC, Jou MJ, Hsu CY, et al. Polyglutamine-expanded ataxin-3 activates mitochondrial apoptotic pathway by upregulating Bax and downregulating Bcl-xL. Neurobiol Dis. 2006;21:333–45. doi: 10.1016/j.nbd.2005.07.011. [DOI] [PubMed] [Google Scholar]
- 309.Wang HL, Yeh TH, Chou AH, Kuo YL, Luo LJ, He CY, et al. Polyglutamine-expanded ataxin-7 activates mitochondrial apoptotic pathway of cerebellar neurons by upregulating Bax and downregulating Bcl-x(L) Cell Signal. 2006;18:541–52. doi: 10.1016/j.cellsig.2005.05.024. [DOI] [PubMed] [Google Scholar]
- 310.Laco MN, Oliveira CR, Paulson HL, Rego AC. Compromised mitochondrial complex II in models of Machado–Joseph disease. Biochim Biophys Acta. 1822;2012:139–49. doi: 10.1016/j.bbadis.2011.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311.Ruan Q, Lesort M, MacDonald ME, Johnson GV. Striatal cells from mutant huntingtin knock-in mice are selectively vulnerable to mitochondrial complex II inhibitor-induced cell death through a non-apoptotic pathway. Hum Mol Genet. 2004;13:669–81. doi: 10.1093/hmg/ddh082. [DOI] [PubMed] [Google Scholar]
- 312.Youle RJ, Karbowski M. Mitochondrial fission in apoptosis. Nat Rev Mol Cell Biol. 2005;6:657–63. doi: 10.1038/nrm1697. [DOI] [PubMed] [Google Scholar]
- 313.Chan DC. Mitochondria: dynamic organelles in disease, aging, and development. Cell. 2006;125:1241–52. doi: 10.1016/j.cell.2006.06.010. [DOI] [PubMed] [Google Scholar]
- 314.Liesa M, Palacin M, Zorzano A. Mitochondrial dynamics in mammalian health and disease. Physiol Rev. 2009;89:799–845. doi: 10.1152/physrev.00030.2008. [DOI] [PubMed] [Google Scholar]
- 315.Chan DC. Mitochondrial dynamics in disease. N Engl J Med. 2007;356:1707–9. doi: 10.1056/NEJMp078040. [DOI] [PubMed] [Google Scholar]
- 316.Chen H, McCaffery JM, Chan DC. Mitochondrial fusion protects against neurodegeneration in the cerebellum. Cell. 2007;130:548–62. doi: 10.1016/j.cell.2007.06.026. [DOI] [PubMed] [Google Scholar]
- 317.Dagda RK, Merrill RA, Cribbs JT, Chen Y, Hell JW, Usachev YM, et al. The spinocerebellar ataxia 12 gene product and protein phosphatase 2A regulatory subunit Bbeta2 antagonizes neuronal survival by promoting mitochondrial fission. J Biol Chem. 2008;283:36241–8. doi: 10.1074/jbc.M800989200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318.Lin CH, Chen CM, Hou YT, Wu YR, Hsieh-Li HM, Su MT, et al. The CAG repeat in SCA12 functions as a cis element to up-regulate PPP2R2B expression. Hum Genet. 2010;128:205–12. doi: 10.1007/s00439-010-0843-2. [DOI] [PubMed] [Google Scholar]
- 319.Girard M, Lariviere R, Parfitt DA, Deane EC, Gaudet R, Nossova N, et al. Mitochondrial dysfunction and Purkinje cell loss in autosomal recessive spastic ataxia of Charlevoix–Saguenay (ARSACS) Proc Natl Acad Sci U S A. 2012;109:1661–6. doi: 10.1073/pnas.1113166109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320.Wang YC, Lee CM, Lee LC, Tung LC, Hsieh-Li HM, Lee-Chen GJ, et al. Mitochondrial dysfunction and oxidative stress contribute to the pathogenesis of spinocerebellar ataxia type 12 (SCA12) J Biol Chem. 2011;286:21742–54. doi: 10.1074/jbc.M110.160697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321.Rugarli EI, Langer T. Mitochondrial quality control: a matter of life and death for neurons. EMBO J. 2012;31:1336–49. doi: 10.1038/emboj.2012.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322.Di Bella D, Lazzaro F, Brusco A, Plumari M, Battaglia G, Pastore A, et al. Mutations in the mitochondrial protease gene AFG3L2 cause dominant hereditary ataxia SCA28. Nat Genet. 2010;42:313–21. doi: 10.1038/ng.544. [DOI] [PubMed] [Google Scholar]
- 323.Casari G, De Fusco M, Ciarmatori S, Zeviani M, Mora M, Fernandez P, et al. Spastic paraplegia and OXPHOS impairment caused by mutations in paraplegin, a nuclear-encoded mitochondrial metalloprotease. Cell. 1998;93:973–83. doi: 10.1016/s0092-8674(00)81203-9. [DOI] [PubMed] [Google Scholar]
- 324.Tatsuta T, Langer T. Quality control of mitochondria: protection against neurodegeneration and ageing. EMBO J. 2008;27:306–14. doi: 10.1038/sj.emboj.7601972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 325.Cagnoli C, Stevanin G, Brussino A, Barberis M, Mancini C, Margolis RL, et al. Missense mutations in the AFG3L2 proteolytic domain account for approximately 1.5% of European autosomal dominant cerebellar ataxias. Hum Mutat. 2010;31:1117–24. doi: 10.1002/humu.21342. [DOI] [PubMed] [Google Scholar]
- 326.Pierson TM, Adams D, Bonn F, Martinelli P, Cherukuri PF, Teer JK, et al. Whole-exome sequencing identifies homozygous AFG3L2 mutations in a spastic ataxia-neuropathy syndrome linked to mitochondrial m-AAA proteases. PLoS Genet. 2011;7:e1002325. doi: 10.1371/journal.pgen.1002325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327.Mariotti C, Brusco A, Di Bella D, Cagnoli C, Seri M, Gellera C, et al. Spinocerebellar ataxia type 28: a novel autosomal dominant cerebellar ataxia characterized by slow progression and ophthalmoparesis. Cerebellum. 2008;7:184–8. doi: 10.1007/s12311-008-0053-9. [DOI] [PubMed] [Google Scholar]
- 328.Almajan ER, Richter R, Paeger L, Martinelli P, Barth E, Decker T, et al. AFG3L2 supports mitochondrial protein synthesis and Purkinje cell survival. J Clin Invest. 2012;122:4048–58. doi: 10.1172/JCI64604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 329.Maltecca F, De Stefani D, Cassina L, Consolato F, Wasilewski M, Scorrano L, et al. Respiratory dysfunction by AFG3L2 deficiency causes decreased mitochondrial calcium uptake via organellar network fragmentation. Hum Mol Genet. 2012;21:3858–70. doi: 10.1093/hmg/dds214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 330.Perlman SL. A review of Friedreich ataxia clinical trial results. J Child Neurol. 2012;27:1217–22. doi: 10.1177/0883073812453872. [DOI] [PubMed] [Google Scholar]
- 331.Ilg W, Synofzik M, Brotz D, Burkard S, Giese MA, Schols L. Intensive coordinative training improves motor performance in degenerative cerebellar disease. Neurology. 2009;73:1823–30. doi: 10.1212/WNL.0b013e3181c33adf. [DOI] [PubMed] [Google Scholar]
- 332.Ilg W, Brotz D, Burkard S, Giese MA, Schols L, Synofzik M. Long-term effects of coordinative training in degenerative cerebellar disease. Mov Disord. 2010;25:2239–46. doi: 10.1002/mds.23222. [DOI] [PubMed] [Google Scholar]
- 333.Soragni E, Herman D, Dent SY, Gottesfeld JM, Wells RD, Napierala M. Long intronic GAA*TTC repeats induce epigenetic changes and reporter gene silencing in a molecular model of Friedreich ataxia. Nucleic Acids Res. 2008;36:6056–65. doi: 10.1093/nar/gkn604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 334.Rai M, Soragni E, Jenssen K, Burnett R, Herman D, Coppola G, et al. HDAC inhibitors correct frataxin deficiency in a Friedreich ataxia mouse model. PLoS One. 2008;3:e1958. doi: 10.1371/journal.pone.0001958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 335.Rai M, Soragni E, Chou CJ, Barnes G, Jones S, Rusche JR, et al. Two new pimelic diphenylamide HDAC inhibitors induce sustained frataxin upregulation in cells from Friedreich’s ataxia patients and in a mouse model. PLoS One. 2010;5:e8825. doi: 10.1371/journal.pone.0008825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 336.Herman D, Jenssen K, Burnett R, Soragni E, Perlman SL, Gottesfeld JM. Histone deacetylase inhibitors reverse gene silencing in Friedreich’s ataxia. Nat Chem Biol. 2006;2:551–8. doi: 10.1038/nchembio815. [DOI] [PubMed] [Google Scholar]
- 337.Xu C, Soragni E, Chou CJ, Herman D, Plasterer HL, Rusche JR, et al. Chemical probes identify a role for histone deacetylase 3 in Friedreich’s ataxia gene silencing. Chem Biol. 2009;16:980–9. doi: 10.1016/j.chembiol.2009.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 338.Shan G, Xu S, Jin P. FXTAS: a bad RNA and a hope for a cure. Expert Opin Biol Ther. 2008;8:249–53. doi: 10.1517/14712598.8.3.249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 339.Todd PK, Oh SY, Krans A, Pandey UB, Di Prospero NA, Min KT, et al. Histone deacetylases suppress CGG repeat-induced neurodegeneration via transcriptional silencing in models of fragile × tremor ataxia syndrome. PLoS Genet. 2010;6:e1001240. doi: 10.1371/journal.pgen.1001240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 340.Xia H, Mao Q, Eliason SL, Harper SQ, Martins IH, Orr HT, et al. RNAi suppresses polyglutamine-induced neurodegeneration in a model of spinocerebellar ataxia. Nat Med. 2004;10:816–20. doi: 10.1038/nm1076. [DOI] [PubMed] [Google Scholar]
- 341.Scholefield J, Greenberg LJ, Weinberg MS, Arbuthnot PB, Abdelgany A, Wood MJ. Design of RNAi hairpins for mutation-specific silencing of ataxin-7 and correction of a SCA7 phenotype. PLoS One. 2009;4:e7232. doi: 10.1371/journal.pone.0007232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 342.Hu J, Gagnon KT, Liu J, Watts JK, Syeda-Nawaz J, Bennett CF, et al. Allele-selective inhibition of ataxin-3 (ATX3) expression by antisense oligomers and duplex RNAs. Biol Chem. 2011;392:315–25. doi: 10.1515/BC.2011.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 343.Fiszer A, Olejniczak M, Switonski PM, Wroblewska JP, Wisniewska-Kruk J, Mykowska A, et al. An evaluation of oligonucleotide-based therapeutic strategies for polyQ diseases. BMC Mol Biol. 2012;13:6. doi: 10.1186/1471-2199-13-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 344.Tsou WL, Soong BW, Paulson HL, Rodriguez-Lebron E. Splice isoform-specific suppression of the Cav2.1 variant underlying spinocerebellar ataxia type 6. Neurobiol Dis. 2011;43:533–42. doi: 10.1016/j.nbd.2011.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 345.Bowers WJ, Breakefield XO, Sena-Esteves M. Genetic therapy for the nervous system. Hum Mol Genet. 2011;20:R28–41. doi: 10.1093/hmg/ddr110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 346.Pandolfo M. Drug insight: antioxidant therapy in inherited ataxias. Nat Clin Pract Neurol. 2008;4:86–96. doi: 10.1038/ncpneuro0704. [DOI] [PubMed] [Google Scholar]
- 347.Lynch DR, Willi SM, Wilson RB, Cotticelli MG, Brigatti KW, Deutsch EC, et al. A0001 in Friedreich ataxia: biochemical characterization and effects in a clinical trial. Mov Disord. 2012;27:1026–33. doi: 10.1002/mds.25058. [DOI] [PubMed] [Google Scholar]
- 348.Broccoletti T, Del Giudice E, Amorosi S, Russo I, Di Bonito M, Imperati F, et al. Steroid-induced improvement of neurological signs in ataxia–telangiectasia patients. Eur J Neurol. 2008;15:223–8. doi: 10.1111/j.1468-1331.2008.02060.x. [DOI] [PubMed] [Google Scholar]
- 349.Ross CA, Tabrizi SJ. Huntington’s disease: from molecular pathogenesis to clinical treatment. Lancet Neurol. 2011;10:83–98. doi: 10.1016/S1474-4422(10)70245-3. [DOI] [PubMed] [Google Scholar]
- 350.Gottesfeld JM, Pandolfo M. Development of histone deacetylase inhibitors as therapeutics for neurological disease. Future Neurol. 2009;4:775–84. doi: 10.2217/fnl.09.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 351.Chou AH, Chen SY, Yeh TH, Weng YH, Wang HL. HDAC inhibitor sodium butyrate reverses transcriptional downregulation and ameliorates ataxic symptoms in a transgenic mouse model of SCA3. Neurobiol Dis. 2011;41:481–8. doi: 10.1016/j.nbd.2010.10.019. [DOI] [PubMed] [Google Scholar]
- 352.Schorge S, van de Leemput J, Singleton A, Houlden H, Hardy J. Human ataxias: a genetic dissection of inositol triphosphate receptor (ITPR1)-dependent signaling. Trends Neurosci. 2010;33:211–9. doi: 10.1016/j.tins.2010.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 353.Kasumu AW, Liang X, Egorova P, Vorontsova D, Bezprozvanny I. Chronic suppression of inositol 1,4,5-triphosphate receptor-mediated calcium signaling in cerebellar Purkinje cells alleviates pathological phenotype in spinocerebellar ataxia 2 mice. J Neurosci. 2012;32:12786–96. doi: 10.1523/JNEUROSCI.1643-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 354.Stevanin G, Durr A, Brice A. Clinical and molecular advances in autosomal dominant cerebellar ataxias: from genotype to phenotype and physiopathology. Eur J Hum Genet. 2000;8:4–18. doi: 10.1038/sj.ejhg.5200403. [DOI] [PubMed] [Google Scholar]
- 355.Netravathi M, Pal PK, Purushottam M, Thennarasu K, Mukherjee M, Jain S. Spinocerebellar ataxias types 1, 2 and 3: age adjusted clinical severity of disease at presentation correlates with size of CAG repeat lengths. J Neurol Sci. 2009;277:83–6. doi: 10.1016/j.jns.2008.10.016. [DOI] [PubMed] [Google Scholar]
- 356.Ikeuchi T, Koide R, Tanaka H, Onodera O, Igarashi S, Takahashi H, et al. Dentatorubral-pallidoluysian atrophy: clinical features are closely related to unstable expansions of trinucleotide (CAG) repeat. Ann Neurol. 1995;37:769–75. doi: 10.1002/ana.410370610. [DOI] [PubMed] [Google Scholar]
- 357.Komure O, Sano A, Nishino N, Yamauchi N, Ueno S, Kondoh K, et al. DNA analysis in hereditary dentatorubral–pallidoluysian atrophy: correlation between CAG repeat length and phenotypic variation and the molecular basis of anticipation. Neurology. 1995;45:143–9. doi: 10.1212/wnl.45.1.143. [DOI] [PubMed] [Google Scholar]
- 358.Charles P, Camuzat A, Benammar N, Sellal F, Destee A, Bonnet AM, et al. Are interrupted SCA2 CAG repeat expansions responsible for parkinsonism? Neurology. 2007;69:1970–5. doi: 10.1212/01.wnl.0000269323.21969.db. [DOI] [PubMed] [Google Scholar]
- 359.Modoni A, Contarino MF, Bentivoglio AR, Tabolacci E, Santoro M, Calcagni ML, et al. Prevalence of spinocerebellar ataxia type 2 mutation among Italian parkinsonian patients. Mov Disord. 2007;22:324–7. doi: 10.1002/mds.21228. [DOI] [PubMed] [Google Scholar]
- 360.Maciel P, Gaspar C, DeStefano AL, Silveira I, Coutinho P, Radvany J, et al. Correlation between CAG repeat length and clinical features in Machado–Joseph disease. Am J Hum Genet. 1995;57:54–61. [PMC free article] [PubMed] [Google Scholar]
- 361.Durr A, Stevanin G, Cancel G, Duyckaerts C, Abbas N, Didierjean O, et al. Spinocerebellar ataxia 3 and Machado–Joseph disease: clinical, molecular, and neuropathological features. Ann Neurol. 1996;39:490–9. doi: 10.1002/ana.410390411. [DOI] [PubMed] [Google Scholar]
- 362.David G, Durr A, Stevanin G, Cancel G, Abbas N, Benomar A, et al. Molecular and clinical correlations in autosomal dominant cerebellar ataxia with progressive macular dystrophy (SCA7) Hum Mol Genet. 1998;7:165–70. doi: 10.1093/hmg/7.2.165. [DOI] [PubMed] [Google Scholar]
- 363.DeStefano AL, Cupples LA, Maciel P, Gaspar C, Radvany J, Dawson DM, et al. A familial factor independent of CAG repeat length influences age at onset of Machado–Joseph disease. Am J Hum Genet. 1996;59:119–27. [PMC free article] [PubMed] [Google Scholar]
- 364.van de Warrenburg BP, Hendriks H, Durr A, van Zuijlen MC, Stevanin G, Camuzat A, et al. Age at onset variance analysis in spinocerebellar ataxias: a study in a Dutch–French cohort. Ann Neurol. 2005;57:505–12. doi: 10.1002/ana.20424. [DOI] [PubMed] [Google Scholar]
- 365.Hayes S, Turecki G, Brisebois K, Lopes-Cendes I, Gaspar C, Riess O, et al. CAG repeat length in RAI1 is associated with age at onset variability in spinocerebellar ataxia type 2 (SCA2) Hum Mol Genet. 2000;9:1753–8. doi: 10.1093/hmg/9.12.1753. [DOI] [PubMed] [Google Scholar]
- 366.Chattopadhyay B, Ghosh S, Gangopadhyay PK, Das SK, Roy T, Sinha KK, et al. Modulation of age at onset in Huntington’s disease and spinocerebellar ataxia type 2 patients originated from eastern India. Neurosci Lett. 2003;345:93–6. doi: 10.1016/s0304-3940(03)00436-1. [DOI] [PubMed] [Google Scholar]
- 367.Jardim L, Silveira I, Pereira ML, do Ceu Moreira M, Mendonca P, Sequeiros J, et al. Searching for modulating effects of SCA2, SCA6 and DRPLA CAG tracts on the Machado–Joseph disease (SCA3) phenotype. Acta Neurol Scand. 2003;107:211–4. doi: 10.1034/j.1600-0404.2003.00046.x. [DOI] [PubMed] [Google Scholar]
- 368.Latouche M, Lasbleiz C, Martin E, Monnier V, Debeir T, Mouatt-Prigent A, et al. A conditional pan-neuronal Drosophila model of spinocerebellar ataxia 7 with a reversible adult phenotype suitable for identifying modifier genes. J Neurosci. 2007;27:2483–92. doi: 10.1523/JNEUROSCI.5453-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 369.Lessing D, Bonini NM. Polyglutamine genes interact to modulate the severity and progression of neurodegeneration in Drosophila. PLoS Biol. 2008;6:e29. doi: 10.1371/journal.pbio.0060029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 370.Vo SH, Butzlaff M, Pru SK, Ni Charthaigh RA, Karsten P, Lankes A, et al. Large-scale screen for modifiers of ataxin-3-derived polyglutamine-induced toxicity in Drosophila. PLoS One. 2012;7:e47452. doi: 10.1371/journal.pone.0047452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 371.Fujigasaki H, Martin JJ, De Deyn PP, Camuzat A, Deffond D, Stevanin G, et al. CAG repeat expansion in the TATA box-binding protein gene causes autosomal dominant cerebellar ataxia. Brain. 2001;124:1939–47. doi: 10.1093/brain/124.10.1939. [DOI] [PubMed] [Google Scholar]
- 372.Bruni AC, Takahashi-Fujigasaki J, Maltecca F, Foncin JF, Servadio A, Casari G, et al. Behavioral disorder, dementia, ataxia, and rigidity in a large family with TATA box-binding protein mutation. Arch Neurol. 2004;61:1314–20. doi: 10.1001/archneur.61.8.1314. [DOI] [PubMed] [Google Scholar]
- 373.Rolfs A, Koeppen AH, Bauer I, Bauer P, Buhlmann S, Topka H, et al. Clinical features and neuropathology of autosomal dominant spinocerebellar ataxia (SCA17) Ann Neurol. 2003;54:367–75. doi: 10.1002/ana.10676. [DOI] [PubMed] [Google Scholar]
- 374.Emamian ES, Kaytor MD, Duvick LA, Zu T, Tousey SK, Zoghbi HY, et al. Serine 776 of ataxin-1 is critical for polyglutamine-induced disease in SCA1 transgenic mice. Neuron. 2003;38:375–87. doi: 10.1016/s0896-6273(03)00258-7. [DOI] [PubMed] [Google Scholar]
- 375.Kaytor MD, Byam CE, Tousey SK, Stevens SD, Zoghbi HY, Orr HT. A cell-based screen for modulators of ataxin-1 phosphorylation. Hum Mol Genet. 2005;14:1095–105. doi: 10.1093/hmg/ddi122. [DOI] [PubMed] [Google Scholar]
- 376.Janer A, Werner A, Takahashi-Fujigasaki J, Daret A, Fujigasaki H, Takada K, et al. SUMOylation attenuates the aggregation propensity and cellular toxicity of the polyglutamine expanded ataxin-7. Hum Mol Genet. 2010;19:181–95. doi: 10.1093/hmg/ddp478. [DOI] [PubMed] [Google Scholar]
- 377.Warrick JM, Chan HY, Gray-Board GL, Chai Y, Paulson HL, Bonini NM. Suppression of polyglutamine-mediated neurodegeneration in Drosophila by the molecular chaperone HSP70. Nat Genet. 1999;23:425–8. doi: 10.1038/70532. [DOI] [PubMed] [Google Scholar]
- 378.Chan HY, Warrick JM, Gray-Board GL, Paulson HL, Bonini NM. Mechanisms of chaperone suppression of polyglutamine disease: selectivity, synergy and modulation of protein solubility in Drosophila. Hum Mol Genet. 2000;9:2811–20. doi: 10.1093/hmg/9.19.2811. [DOI] [PubMed] [Google Scholar]
- 379.Kazemi-Esfarjani P, Benzer S. Genetic suppression of polyglutamine toxicity in Drosophila. Science. 2000;287:1837–40. doi: 10.1126/science.287.5459.1837. [DOI] [PubMed] [Google Scholar]
- 380.Cummings CJ, Sun Y, Opal P, Antalffy B, Mestril R, Orr HT, et al. Over-expression of inducible HSP70 chaperone suppresses neuropathology and improves motor function in SCA1 mice. Hum Mol Genet. 2001;10:1511–8. doi: 10.1093/hmg/10.14.1511. [DOI] [PubMed] [Google Scholar]
- 381.Kimura Y, Koitabashi S, Kakizuka A, Fujita T. Initial process of polyglutamine aggregate formation in vivo. Genes Cells. 2001;6:887–97. doi: 10.1046/j.1365-2443.2001.00472.x. [DOI] [PubMed] [Google Scholar]
- 382.Higashiyama H, Hirose F, Yamaguchi M, Inoue YH, Fujikake N, Matsukage A, et al. Identification of ter94, Drosophila VCP, as a modulator of polyglutamine-induced neurodegeneration. Cell Death Differ. 2002;9:264–73. doi: 10.1038/sj.cdd.4400955. [DOI] [PubMed] [Google Scholar]
- 383.van Dellen A, Blakemore C, Deacon R, York D, Hannan AJ. Delaying the onset of Huntington’s in mice. Nature. 2000;404:721–2. doi: 10.1038/35008142. [DOI] [PubMed] [Google Scholar]
- 384.Imbert G, Saudou F, Yvert G, Devys D, Trottier Y, Garnier JM, et al. Cloning of the gene for spinocerebellar ataxia 2 reveals a locus with high sensitivity to expanded CAG/glutamine repeats. Nat Genet. 1996;14:285–91. doi: 10.1038/ng1196-285. [DOI] [PubMed] [Google Scholar]
- 385.Pulst SM, Nechiporuk A, Nechiporuk T, Gispert S, Chen XN, Lopes-Cendes I, et al. Moderate expansion of a normally biallelic trinucleotide repeat in spinocerebellar ataxia type 2. Nat Genet. 1996;14:269–76. doi: 10.1038/ng1196-269. [DOI] [PubMed] [Google Scholar]
- 386.Sanpei K, Takano H, Igarashi S, Sato T, Oyake M, Sasaki H, et al. Identification of the spinocerebellar ataxia type 2 gene using a direct identification of repeat expansion and cloning technique, DIRECT. Nat Genet. 1996;14:277–84. doi: 10.1038/ng1196-277. [DOI] [PubMed] [Google Scholar]
- 387.Kawaguchi Y, Okamoto T, Taniwaki M, Aizawa M, Inoue M, Katayama S, et al. CAG expansions in a novel gene for Machado–Joseph disease at chromosome 14q32 . Nat Genet. 1994;8:221–7. doi: 10.1038/ng1194-221. [DOI] [PubMed] [Google Scholar]
- 388.Hellenbroich Y, Pawlack H, Rub U, Schwinger E, Zuhlke C. Spinocerebellar ataxia type 4. Investigation of 34 candidate genes. J Neurol. 2005;252:1472–5. doi: 10.1007/s00415-005-0892-y. [DOI] [PubMed] [Google Scholar]
- 389.Ikeda Y, Dick KA, Weatherspoon MR, Gincel D, Armbrust KR, Dalton JC, et al. Spectrin mutations cause spinocerebellar ataxia type 5. Nat Genet. 2006;38:184–90. doi: 10.1038/ng1728. [DOI] [PubMed] [Google Scholar]
- 390.David G, Abbas N, Stevanin G, Durr A, Yvert G, Cancel G, et al. Cloning of the SCA7 gene reveals a highly unstable CAG repeat expansion. Nat Genet. 1997;17:65–70. doi: 10.1038/ng0997-65. [DOI] [PubMed] [Google Scholar]
- 391.Higgins JJ, Pho LT, Ide SE, Nee LE, Polymeropoulos MH. Evidence for a new spinocerebellar ataxia locus. Mov Disord. 1997;12:412–7. doi: 10.1002/mds.870120322. [DOI] [PubMed] [Google Scholar]
- 392.O'Hearn E, Holmes SE, Margolis RL. Spinocerebellar ataxia type 12. Handb Clin Neurol. 2012;103:535–47. doi: 10.1016/B978-0-444-51892-7.00034-6. [DOI] [PubMed] [Google Scholar]
- 393.Stevanin G, Durr A. Spinocerebellar ataxia 13 and 25. Handb Clin Neurol. 2012;103:549–53. doi: 10.1016/B978-0-444-51892-7.00035-8. [DOI] [PubMed] [Google Scholar]
- 394.Chen DH, Brkanac Z, Verlinde CL, Tan XJ, Bylenok L, Nochlin D, et al. Missense mutations in the regulatory domain of PKCgamma: a new mechanism for dominant nonepisodic cerebellar ataxia. Am J Hum Genet. 2003;72:839–49. doi: 10.1086/373883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 395.Brkanac Z, Fernandez M, Matsushita M, Lipe H, Wolff J, Bird TD, et al. Autosomal dominant sensory/motor neuropathy with ataxia (SMNA): linkage to chromosome 7q22-q32. Am J Med Genet. 2002;114:450–7. doi: 10.1002/ajmg.10361. [DOI] [PubMed] [Google Scholar]
- 396.Brkanac Z, Spencer D, Shendure J, Robertson PD, Matsushita M, Vu T, et al. IFRD1 is a candidate gene for SMNA on chromosome 7q22-q23. Am J Hum Genet. 2009;84:692–7. doi: 10.1016/j.ajhg.2009.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 397.Knight MA, Hernandez D, Diede SJ, Dauwerse HG, Rafferty I, van de Leemput J, et al. A duplication at chromosome 11q12.2-11q12.3 is associated with spinocerebellar ataxia type 20. Hum Mol Genet. 2008;17:3847–53. doi: 10.1093/hmg/ddn283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 398.Vuillaume I, Devos D, Schraen-Maschke S, Dina C, Lemainque A, Vasseur F, et al. A new locus for spinocerebellar ataxia (SCA21) maps to chromosome 7p21.3-p15.1. Ann Neurol. 2002;52:666–70. doi: 10.1002/ana.10344. [DOI] [PubMed] [Google Scholar]
- 399.Delplanque J, Devos D, Vuillaume I, De Becdelievre A, Vangelder E, Maurage CA, et al. Slowly progressive spinocerebellar ataxia with extrapyramidal signs and mild cognitive impairment (SCA21) Cerebellum. 2008;7:179–83. doi: 10.1007/s12311-008-0014-3. [DOI] [PubMed] [Google Scholar]
- 400.Verbeek DS, van de Warrenburg BP, Wesseling P, Pearson PL, Kremer HP, Sinke RJ. Mapping of the SCA23 locus involved in autosomal dominant cerebellar ataxia to chromosome region 20p13-12.3. Brain. 2004;127:2551–7. doi: 10.1093/brain/awh276. [DOI] [PubMed] [Google Scholar]
- 401.Hekman KE, Yu GY, Brown CD, Zhu H, Du X, Gervin K, et al. A conserved eEF2 coding variant in SCA26 leads to loss of translational fidelity and increased susceptibility to proteostatic insult. Hum Mol Genet. 2012;21:5472–83. doi: 10.1093/hmg/dds392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 402.Brusse E, de Koning I, Maat-Kievit A, Oostra BA, Heutink P, van Swieten JC. Spinocerebellar ataxia associated with a mutation in the fibroblast growth factor 14 gene (SCA27): a new phenotype. Mov Disord. 2006;21:396–401. doi: 10.1002/mds.20708. [DOI] [PubMed] [Google Scholar]
- 403.Mariotti C, Bella DD, Di Donato S, Taroni F. Spinocerebellar ataxia type 28. Handb Clin Neurol. 2012;103:575–9. doi: 10.1016/B978-0-444-51892-7.00039-5. [DOI] [PubMed] [Google Scholar]
- 404.Dudding TE, Friend K, Schofield PW, Lee S, Wilkinson IA, Richards RI. Autosomal dominant congenital non-progressive ataxia overlaps with the SCA15 locus. Neurology. 2004;63:2288–92. doi: 10.1212/01.wnl.0000147299.80872.d1. [DOI] [PubMed] [Google Scholar]
- 405.Storey E, Bahlo M, Fahey M, Sisson O, Lueck CJ, Gardner RJ. A new dominantly inherited pure cerebellar ataxia, SCA 30. J Neurol Neurosurg Psychiatry. 2009;80:408–11. doi: 10.1136/jnnp.2008.159459. [DOI] [PubMed] [Google Scholar]
- 406.Jiang H, Zhu H-P, Gomez CM. SCA32: an autosomal dominant cerebellar ataxia with azoospermia maps to chromosome 7q32-q33. Abstract. Mov Disord. 2010:S192. [Google Scholar]
- 407.Giroux JM, Barbeau A. Erythrokeratodermia with ataxia. Arch Dermatol. 1972;106:183–8. [PubMed] [Google Scholar]
- 408.Wang JL, Yang X, Xia K, Hu ZM, Weng L, Jin X, et al. TGM6 identified as a novel causative gene of spinocerebellar ataxias using exome sequencing. Brain. 2010;133:3510–8. doi: 10.1093/brain/awq323. [DOI] [PubMed] [Google Scholar]
- 409.Li M, Pang SY, Song Y, Kung MH, Ho SL, Sham PC. Whole exome sequencing identifies a novel mutation in the transglutaminase 6 gene for spinocerebellar ataxia in a Chinese family. Clin Genet. 2013;83:269–73. doi: 10.1111/j.1399-0004.2012.01895.x. [DOI] [PubMed] [Google Scholar]
- 410.Serrano-Munuera C, Corral-Juan M, Stevanin G, San Nicolas H, Roig C, Corral J, et al. New subtype of spinocerebellar ataxia with altered vertical eye movements mapping to chromosome 1p32. JAMA Neurol. 2013;70:764–71. doi: 10.1001/jamaneurol.2013.2311. [DOI] [PubMed] [Google Scholar]
- 411.Koide R, Ikeuchi T, Onodera O, Tanaka H, Igarashi S, Endo K, et al. Unstable expansion of CAG repeat in hereditary dentatorubral–pallidoluysian atrophy (DRPLA) Nat Genet. 1994;6:9–13. doi: 10.1038/ng0194-9. [DOI] [PubMed] [Google Scholar]
- 412.Nagafuchi S, Yanagisawa H, Sato K, Shirayama T, Ohsaki E, Bundo M, et al. Dentatorubral and pallidoluysian atrophy expansion of an unstable CAG trinucleotide on chromosome 12p. Nat Genet. 1994;6:14–8. doi: 10.1038/ng0194-14. [DOI] [PubMed] [Google Scholar]
- 413.Yabe I, Sasaki H, Kikuchi S, Nonaka M, Moriwaka F, Tashiro K. Late onset ataxia phenotype in dentatorubro-pallidoluysian atrophy (DRPLA) J Neurol. 2002;249:432–6. doi: 10.1007/s004150200034. [DOI] [PubMed] [Google Scholar]
- 414.Amino T, Ishikawa K, Toru S, Ishiguro T, Sato N, Tsunemi T, et al. Redefining the disease locus of 16q22.1-linked autosomal dominant cerebellar ataxia. J Hum Genet. 2007;52:643–9. doi: 10.1007/s10038-007-0154-1. [DOI] [PubMed] [Google Scholar]
- 415.Onodera Y, Aoki M, Mizuno H, Warita H, Shiga Y, Itoyama Y. Clinical features of chromosome 16q22.1 linked autosomal dominant cerebellar ataxia in Japanese. Neurology. 2006;67:1300–2. doi: 10.1212/01.wnl.0000238507.85436.20. [DOI] [PubMed] [Google Scholar]
- 416.Swartz BE, Burmeister M, Somers JT, Rottach KG, Bespalova IN, Leigh RJ. A form of inherited cerebellar ataxia with saccadic intrusions, increased saccadic speed, sensory neuropathy, and myoclonus. Ann N Y Acad Sci. 2002;956:441–4. doi: 10.1111/j.1749-6632.2002.tb02850.x. [DOI] [PubMed] [Google Scholar]