

Abstract
Several members of the APOBEC3 family of cellular restriction factors provide intrinsic immunity to the host against viral infection. Specifically, APOBEC3DE, APOBEC3F, APOBEC3G, and APOBEC3H haplotypes II, V, and VII provide protection against HIV-1Δvif through hypermutation of the viral genome, inhibition of reverse transcription, and inhibition of viral DNA integration into the host genome. HIV-1 counteracts APOBEC3 proteins by encoding the viral protein Vif, which contains distinct domains that specifically interact with these APOBEC3 proteins to ensure their proteasomal degradation, allowing virus replication to proceed. Here, we review our current understanding of APOBEC3 structure, editing and non-editing mechanisms of APOBEC3-mediated restriction, Vif-APOBEC3 interactions that trigger APOBEC3 degradation, and the contribution of APOBEC3 proteins to restriction and control of HIV-1 replication in infected patients.
Keywords: APOBEC3G, APOBEC3F, APOBEC3H, cytidine deamination, Vif, restriction factor
Introduction
Over millions of years, eukaryotes have been exposed to bacterial and viral infections. In response to these pressures, mammalian hosts have not only developed powerful immune systems to detect and neutralize foreign invaders, but also acquired host restriction factors, i.e. intrinsic immunity, to defend against these infections. Restriction factors provide a first line of defense for the host to suppress replication of the pathogens. One important family of cellular cytidine deaminases is the apolipoprotein B messenger RNA (mRNA)-editing enzyme catalytic polypeptide (APOBEC) family of proteins, which act on single-stranded DNA (ssDNA) or RNA substrates. 1 In the human genome, this family of proteins consists of 11 members: activation-induced cytidine deaminase (AID), APOBEC1 (A1), APOBEC2 (A2), APOBEC3 (A3:A3A, A3B, A3C, A3DE, A3F, A3G, A3H) and APOBEC4 (A4), all of which contain a conserved zinc-coordinating cytidine deamination domain (Figure 1).
Figure 1. Functional roles of the APOBEC family of proteins.

Each APOBEC protein is shown, containing one or two conserved Zn2+ coordinating catalytic domains (green); active catalytic domains (red star). Numbers correspond to length of amino acid sequence. Both the catalytically active and non-active domains contain a conserved sequence His-X-Glu-X23-28-Pro-Cys-X2-4Cys, in which X represents any amino acid. Ch. 1, 6, 12, and 22 refer to human chromosome numbers.
The main function of the APOBEC family of proteins is to alter the nucleotide sequence through cytidine deamination, converting cytidines to uridines (C-to-U) or deoxycytidines to deoxyuridines (dC-to-dU). It is known that A1 is involved in editing the apolipoprotein B pre-mRNA to introduce a stop codon to create two distinct mRNAs important in lipid metabolism. 2; 3 AID functions in somatic hypermutation and antibody diversification by class-switch recombination of immunoglobulin genes. 4 A2, a muscle-specific APOBEC family member, has recently been shown to have a role in normal muscle differentiation and maintenance of muscle fiber-type ratios. 5; 6; 7; 8 In addition, A2 is involved in regulating TGF-β signaling to specify the left-right axis in vertebrates during embryogenesis, although gene loss is not lethal. 5; 9 The function of A4, primarily expressed in testis, is still not known. 10 In 2002, A3G was discovered to inhibit HIV-1 infection in the absence of the virally encoded protein, virion infectivity factor (Vif), marking the A3 proteins as important restriction factors against viral infection. 11 A3G was further shown to induce C-to-U deamination in the minus-strand DNA of the replicating virus, resulting in G-to-A hypermutation in the HIV-1 double-stranded DNA (dsDNA) genome, which introduces substitution mutations and stop codons that ultimately disrupt expression of functional viral proteins (Figure 2). 12; 13 HIV-1 has evolved to counteract the effects of A3G by coding for the accessory protein Vif. Vif binds A3G in the cytoplasm, directing it for polyubiquitination and proteasomal degradation, allowing HIV-1 to exclude A3G from the nascent virions, and thereby overcoming A3G-mediated inhibition of viral replication in infected cells. 14; 15; 16; 17; 18; 19; 20
Figure 2. Mechanism of action of A3G during infection with HIV-1Δvif virions.
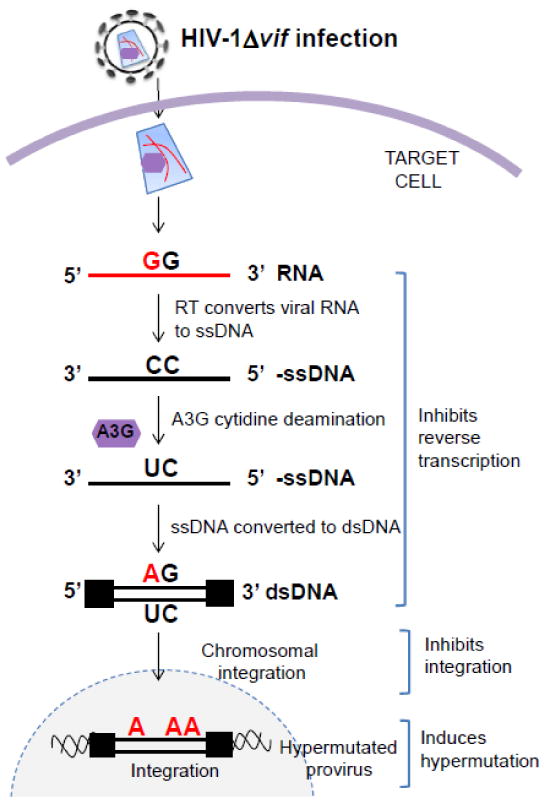
In virus producer cells, in the absence of a functional Vif protein, A3G (purple hexagon) is packaged into the viral particles. In the target cell, A3G exerts its antiviral activity by inhibiting reverse transcription, blocking integration, and inducing G-to-A hypermutation. In the hypermutation process, A3G mainly deaminates deoxycytidines in minus-strand DNA to deoxyuridines, which ultimately results in G-to-A hypermutation in the plus-strand DNA. Although hypermutated viral DNA may integrate into the host chromosomal DNA to form proviruses, they are largely defective.
The A3 proteins are true restriction factors because they block HIV-1 infectivity, they are induced by the innate immune response, and they exhibit positive selection due to their direct interactions with the viral “counter-restriction factor” Vif, reflecting an ongoing host-pathogen arms race resulting in co-evolution. 21 Since this discovery, the A3 proteins have been intensely studied. The human A3 locus, found on chromosome 22, encodes 7 A3 proteins with A3A, A3C, and A3H containing a single cytidine deaminase domain and A3B, A3DE, A3F and A3G containing two deaminase domains (CD1 and CD2), which likely formed as a result of gene duplication (Figure 1).1; 22 The A3 proteins are widely expressed in different tissues and cell types, especially targets of HIV-1 such as CD4+ T cells, macrophages and dendritic cells. 23; 24 The fact that A3 proteins act on ssDNA intermediates implies that their antiviral activity is not limited to HIV but extends to other retroviruses including gammaretroviruses (murine leukemia virus, mouse mammary tumor virus), 25; 26 simian immunodeficiency virus, 27 equine infectious anemia virus, 28 human T-cell leukemia virus type-1, 29; 30; 31 Rous sarcoma virus, 32 and foamy viruses. 33; 34; 35 In addition, A3 proteins also inhibit both LTR- and non-LTR retrotransposons, 36 and other viruses such as hepatitis B virus 37 and human papillomavirus. 38
In this review, we summarize our current knowledge of A3 proteins and their mechanisms of HIV-1 inhibition, with special emphasis on A3G, which seems to be the most important, and most-studied, inhibitor of HIV-1. We survey which A3 proteins are the main players in blocking HIV-1 infection, either through hypermutation or non-editing mechanisms (e.g. inhibition of reverse transcription and integration). We analyze the current structural and biochemical data examining how A3 proteins function and interact with Vif, and how they are regulated not only in cells, but also in HIV-1 infected patients.
Domain organization and tertiary structures of A3 proteins
The presence of either one (A3A, A3C, A3H) or two (A3B, A3DE, A3F, A3G) catalytic domains (CD1 and CD2) sandwiched between an N-terminal short α-helical domain and a C-terminal short linker peptide is a hallmark of the structural arrangement of A3 proteins (Figure 1). 22 A3B, A3DE, A3F, and A3G have a duplication of the entire unit consisting of 6 α-helices and 5 β-strands. 22 Each CD is organized as a HXEX23-28-PCX2-4C motif containing four invariable residues where X represents any amino acid and the histidine and the two cysteines are the essential residues that coordinate Zn2+, while the glutamic acid is important for the deaminase activity of the proteins by shuttling protons in the catalytically active domains. 22 Although structures of full-length A3G or A3F are not yet available, high resolution crystal and nuclear magnetic resonance (NMR) structures of human A3G-CTD, A3F-CTD, A3A, A3C, and A2 (the closest paralog of A3) have been determined. 39; 40; 41; 42; 43; 44; 45; 46; 47 Moreover, the NMR structure of murine A2 has been determined through the RIKEN Structural Genomics/Proteomics Initiative (see PDB entry 2RPZ; http://www.rcsb.org/pdb/explore/explore.do?structureId=2RPZ).
A3G structure
Several groups have successfully resolved high resolution crystal 39; 40; 44 or NMR 46; 48 structures of the C-terminal fragment of A3G (A3G-CTD), using either a wild-type or a soluble mutant of A3G-CTD (A3-2K3A) containing L234K, F310K, C234A, C321A, and C356A substitutions. The structures of the A3G-CTD exhibit a canonical polynucleotide cytidine deaminase fold that is composed of a five β-strand core surrounded by six α-helices with the catalytic site composed of two α-helices (α1 and α2) and a β3-strand that is accessible for large RNA or ssDNA binding (Figure 3A, B), common to all APOBEC proteins whose structures are known; i.e., A2, A3A, A3C, and A3F-CTD proteins (Figure 3C) 39; 40; 41; 42; 43; 44; 45 Despite this progress, many important aspects of the structures of A3 proteins have not been elucidated. Currently, it is unknown how the two domains of the A3 proteins are positioned relative to each other. In addition, the assignments of residues that are involved in ssDNA binding in different models have little correlation with each other and are speculative largely because none of the structures incorporated a substrate.
Figure 3. Structural comparison of APOBEC family members' crystal structures.

(A) The APOBEC proteins have a canonical deaminase core composed of five β strands and six α helices. As a prototype, the wild-type A3G-CTD monomer structure (PDB entry 3IQS) with the indicated α-helices, β-strands and loops is shown in a ribbon representation. Loop 3, 5, and 7 (L3, L5, and L7, respectively) are shown in blue, orange, and magenta colors, respectively. (B) The active site of A3G-CTD. A zinc atom is coordinated by the three residues H257, C288 and C291, and indirectly via a water molecule (view occluded by zinc atom) with E259. (C) Comparison of ribbon representations of A2 (PDB entry 2NYT), A3A (PDB entry 2M65), A3C (PDB entry 3VOW), A3F-CTD (PDB entry 4IOU), A3G-CTD (PDB entry 3ISQ) and A3G-CTD-2K3A (PDB entry 3IR2) crystal structures. The β1/β2 region is highlighted (blue oval) to emphasize similarities and the conformational plasticity in the β1-β2 loop region across all A3 proteins. The α1 helix (red box) exhibits a similar orientation across all A3 proteins but differs in A2 crystal structure. The corresponding loops (L3, L5, and L7) in Fig. 3A are also labeled in A3A and A3G structures.
A3C structure
Recently, a high resolution crystal structure of full-length A3C has also been determined (Figure 3C). 49 Interestingly, the crystal structure of A3C provided an important glimpse on the essential Vif-interacting residues on A3C protein, which forms a shallow cavity where Vif binds. 49 Using structure-guided mutagenesis, Kitamura et al. identified 10 hydrophobic or negatively charged residues between the α2 and α3 helices, which are conserved in the homologous domain of A3F and A3DE proteins but not in A3G (Figure 4).49 Notably, the shallow cavity in the interface is composed of the hydrophobic (L72, I79 and L80), aromatic (F75, Y86, F107 and H111) and hydrophilic (C76, S81 and E106) residues of A3C. Furthermore, mutational analysis of the equivalent ten residues in A3F (L255, F258, C259, I262, L263, S264, Y269, E289, F290 and H294) and A3DE (L268, F271, C272, I275, L276, S277, Y282, E302, F303 and H307) revealed clustering of these residues that form a Vif-binding cavity homologous to that observed in the A3C structure. 49
Figure 4. Ribbon diagrams and surface representations of the A3C, A3F-CTD and A3G-NTD model structures.

(A) Ribbon diagrams of the structure of A3C (PDB entry 3VOW), A3F-CTD (PDB entry 4IOU) and A3G-NTD, a possible model generated through homology modeling based on the A3C structure (PDB entry 3VOW) by using SwissModel Worksplace (http://swissmodel.expasy.org/), 276; 277 showing the known Vif-binding residues labeled in red (critical) and in pink (less critical). The equivalent critical residues in A3G are shown in yellow in A3C and A3F-CTD. The designations of critical and less critical residues are based on mutational analyses. 49; 51; 52; 65; 66 (B) Surface representation of the Vif-binding groove delineated by the dashed line in A3C (PDB entry 3VOW), A3F-CTD (PDB entry 4IOU), and A3G-NTD depicted with the same colors used as in (A).
A3F structure
Similar to A3G, A3F is notoriously insoluble and readily precipitates in solution, making it unsuitable for structural studies. Therefore a minimal A3F-CTD construct was engineered in which 11 amino acids were substituted (A3F185-373-11X) to increase the solubility of the protein, while maintaining its catalytic activity and susceptibility to HIV-1 Vif-mediated degradation (Figure 3C).45 The A3F-CTD crystallizes as an elongated rod-shaped asymmetric unit composed of four molecules assembled through two different monomer-monomer interfaces that can be viewed as two homodimers joined head-to-head. 45 Such structural configurations, distinct from the canonical square-shaped tetramer structure of free nucleotide cytidine deaminase where the loops from two neighboring monomers cover the active site so that only small free nucleotides can bind to the buried sites, 50 could provide an explanation for the ability of APOBEC family members to bind and deaminate long polynucleotide substrates. The head-to-head and the two head-to-tail interfaces of the crystal structure of A3F-CTD are held together by extensive salt bridges, hydrogen bonding and hydrophobic packing. The largest interface, a symmetric/isologous head-to-head interface, is principally formed as a result of hydrophobic β-sheet interactions of identical regions of homodimers. 45 The second interface, an asymmetric/heterologous interface, is a head-to-tail interaction between monomers within the homodimer unit cells, which involves the catalytic sites. 45
Unlike the A3G-CTD region, the CTD of A3F is involved both in the cytidine deaminase activity and Vif interaction (Figure 1). Therefore, the A3F-CTD crystal structure further elucidated an HIV-1 Vif binding surface. Interestingly, the majority of the A3F-CTD structure surface is negatively charged including the previously identified motif composed of 289EFLARH294 51 and E324 52 that determine the interaction between A3F and HIV-1 Vif (Figure 4). Consistent with this, a shallow groove was identified in the crystal structure of A3C as a binding pocket of HIV-1 Vif, 49 albeit significant differences were observed between the two structures (Figure 4). Another important observation was that the intermolecular contact surfaces involving 305RLYYFWD311 and 223EVVKHHSPVS232 regions of the asymmetric tetramer crystal structure of A3F were suggested as a potential oligomerization surfaces. 45 Interestingly, these motifs showed certain sequence similarities with 108HLYYFD113 and 23SLVKHHMYVS32 regions of HIV-1 Vif sequence. This suggests that perhaps HIV-1 Vif evolved to mimic this oligomerization interface so that it could bind to A3F and induce its degradation. It is necessary to point out however, that the interfaces between A3F-CTDs in the crystal structure may not represent the protein-protein interactions that occur with the intact A3F protein and could form as a result of removal of A3F-NTD. In this regard, the A2 crystal structure, 42 obtained from a truncated A2, indicated that it is a dimer, but the NMR structure of the intact A2 protein indicates that it is a monomer. 47 Further mutational analyses of these regions of Vif and A3F should verify these intriguing predictions and provide novel insights into Vif-A3F interactions.
A3A structure
The recently reported NMR structure of A3A 43 closely resembles the crystal structure of A3G-CTD quintuple mutant (A3G-2K3A) (Figure 3C). 44 Detailed analysis of the A3A NMR structure with particular emphasis around the active site, where the sequences significantly differ from the A3G-CTD, 39 reveals the presence of additional hydrophobic residues that distort the conformation of loop 7 (Figure 3C, magenta color) situated next to the active site. 43 A3A and A3G purportedly possess a different substrate specificity, which is believed to be due to certain structural differences at the substrate binding surface. 39; 43; 48; 53 Although A3A can bind to both 5′-TC- and 5′-CC-containing oligonucleotides using the same interaction surface, 5′-TC is the preferred dinucleotide substrate. Byeon et al. used NMR to map ssDNA substrate binding to A3A to a large surface covering loop 3, loop 5, loop 7 (Figure 3C, blue, orange, and magenta colors, respectively), and helix α4 clustered around the active site. 43 Interestingly, the clustering of the A3A substrate-interacting residues and the conformational changes of the ssDNA permit the reactive cytosine and only the immediate neighboring (-1, -2 and +1) nucleotides access the active site. Earlier studies also demonstrate DNA contraction during A3G scanning of ssDNA substrate 54 and RNA bending in a co-crystal structure of tRNA adenosine deaminase TadA/RNA complex, 55 suggesting that ssDNA/RNA substrate bending or extensive conformational changes are required because the active site of polynucleotide cytidine deaminases can only accommodate few nucleotides.
Unlike A3A, A3G preferentially binds to the 5′-CC dinucleotide in the ssDNA and the interaction surface is localized in loops spanning residues 207 to 217 and 313 to 320. 39; 40; 56; 57 Although the proposed ssDNA binding surfaces of A3G are not clustered, a recent observation demonstrates that protonation of H216, which is spatially close to the active site and is located in loop 1 composed of residues 207 to 217, was found to be important for determining substrate binding and HIV restriction. 58 Moreover, using mutagenesis and domain swapping, several groups have identified several amino acids, which include loop 7 residues that govern the intrinsic dinucleotide specificity (5′-CC vs 5′-TC context) of several cytidine deaminases. 55; 57; 59; 60; 61 Notably, a recent study showed that replacing the aspartic acid for tyrosine at position 317 (the corresponding residue in A3A is Y132) in loop 7 in A3G was sufficient to alter A3G's substrate specificity from 5′-CC to 5′-TC. 59
In summary, the structural similarities of A3 proteins parallel the similarities in their primary sequences. 22 The core structures, composed of six helices (α1-α6) intermixed with five β-strands (β1-β5), containing the zinc ion coordinated in the active site are highly conserved structural hallmarks of the APOBEC family of proteins. 62 The catalytic loop of α1 helix of all A3 proteins exhibits the same conformation, whereas it is oriented perpendicularly in the A2 crystal structure (Figure 3C). In both the A2 and A3F-CTD asymmetric tetrameric crystal structures, the β1-β2 loop exists in two distinct conformations depending on the interface involved in the asymmetric tetramer unit and the long continuous β2 strand was found to mediate homodimerization. 42; 45 However, this structural organization is not a common feature of all A3 proteins structures (Figure 3C). For instance, the NMR structures of A3A, 43 and wild-type A3G-CTD, 48 and the crystal and NMR structures of A3G-CTD (A3G-2K3A) 39; 44; 46 exhibit a discontinuous β2 strand, whereas the crystal and NMR structures of A2, 42; 47 as well as crystal structures of A3C, 49 A3G-CTD, 40 and A3F-CTD 45 revealed a long continuous and extended β2 strand (Figure 3C). While these structural differences may reflect the structures of the proteins in solution, they may also be the result of differences in the methods used to determine the structure or to the engineered protein constructs used for the structural studies. Nevertheless, a combination of these structural and sequence differences are believed to dictate the differential substrate specificity of the APOBEC family of cytidine deaminases. The remarkable advances that were made on the structural biology of the APOBEC family of proteins in the past few years have led to (i) an understanding of the architecture of their catalytic core, (ii) the identification of a determinant of their substrate specificity, and (iii) elucidation of the structures of A3 protein determinants involved in interaction with HIV-1 Vif. These advances will hopefully facilitate structure-based drug design and perhaps identification of small molecule inhibitors that specifically target A3-Vif interactions and allow the A3 proteins to potently block HIV replication.
Structural determinants of Vif-A3 interactions, virion incorporation, and substrate preference
Vif-A3 interaction determinants
As described earlier, due to the poor solubility of these proteins, detailed views of the A3G/A3F-Vif and/or A3G/A3F-ssDNA complexes are lacking. 39; 40; 44; 45; 46; 48 To achieve a more comprehensive picture of the enzymatic activities of A3 proteins, their interaction with HIV-1 Vif, and their regulation in cells, co-crystal structures that encompass full-length protein and their ssDNA substrate as well as Vif are essential. Nevertheless, the available structural data afford important glimpses into the antiviral mechanisms of the A3 proteins, their interaction with HIV-1 Vif, and their substrate specificity. 39; 43; 44; 45; 46; 49 Furthermore, using site-directed mutagenesis along with functional and biochemical analyses, the critical residues involved in Vif-A3 protein interactions have been revealed. 14; 49; 51; 63; 64; 65; 66; 67
A3 residues critical for A3-Vif interactions
Mapping critical residues in A3 proteins has demonstrated the clustering of critical residues in well-defined patches on the surface of structural models 45; 46; 48; 49; 51; 66; 67; 68 (Figure 4). Initially, we and others identified D128 as a critical residue in human A3G that was involved in interaction with HIV-1 Vif. 69; 70; 71; 72 Subsequently, we and others demonstrated that the 128DPD130 motif is critical for the interaction between A3G and Vif, and Vif-mediated degradation of A3G (Figure 4).65; 66 More precisely, HIV-1 Vif fails to neutralize A3G from African green monkey (agm) or rhesus macaque that contains a lysine (K) instead of aspartic acid (D) at position 128; however, the agm and rhesus macaque simian immunodeficiency virus (SIVagm and SIVmac, respectively) can induce the degradation of human A3G. Moreover, substitution of D for K in human A3G confers resistance to HIV-1 Vif. 69; 70; 71; 72 Interestingly, recent studies demonstrated that substitution of the aspartic acid at position 121 to lysine in A3H haplotype II, the equivalent residue of D128 A3G, render A3H resistant to HIV-1 Vif-mediated degradation. 73; 74; 75
Even though D128 in A3G and the equivalent residue in A3H are critical for interaction with HIV-1 Vif, they are not critical in all human A3 proteins. Our analysis using functional A3G and A3F chimeras identified amino acids 283-300 in the C-terminus of A3F as critically important residues for A3F-Vif interaction. 66 Further analysis of this region of A3F led to the identification of the 289EFLARH294 region in A3F, with E289 being the most critical residue for interaction with HIV-1 Vif, and 292ARH294 having a significant effect on Vif binding when all three residues are simultaneously mutated to alanines (Figure 4).51 The E324 residue of A3F was also shown to be critical for Vif interaction (Figure 4).52 Interestingly, these residues are conserved in A3C, A3DE and A3F but not in A3G. 49; 51
HIV-1 Vif residues important for A3-Vif interactions
Since Vif is also an extremely difficult protein to purify and maintain in solution for structural analysis, structural features of Vif that enable interactions with its partners have not been elucidated. Moreover, recombinant Vif is highly dynamic and likely to populate an ensemble of heterogeneous conformational states in solution, which is not amenable for structural studies. 76 Nevertheless, using site-directed mutagenesis several domains in Vif have been identified as being important for interactions with proteasomal components to induce degradation of A3 proteins. For example, several investigators have identified an 144SLQYLA149 domain that is important for interaction with elongin C (EloC) and an 108HX5-CX17-18-CX3-5-H139 domain critical for docking with cullin 5 (CUL5) of the E3 ubiquitin ligase complex. 77; 78; 79; 80; 81 In addition, through mutational analysis of the N-terminal regions of Vif, we and others have identified two distinct regions in HIV-1 Vif that specifically contribute to interactions with A3G or A3F proteins. 67; 82 The first region, 14DRMR17, and to a lesser extent W11 and Q12, are important for degradation of A3F. Interestingly, its substitution with 14SERQ17or 14SEMQ17(equivalent residues in SIVagm Vif) endows the mutant HIV-1 Vif to degrade agmA3G as well as D128K-A3G. 67; 69; 70; 71; 72; 83 This species specificity was attributed to the two polar amino acids (E15 and Q17) in Vif and their interaction with the negatively charged D128 in human A3G. However, Russell et al. demonstrated that a second region, the 40YRHHY44 domain, is actually the region that is essential for the binding of Vif to A3G in co-immunoprecipitation experiments. 67 Others also independently identified this same region to be essential for its interaction with A3G. 82 These two distinct domains of Vif show a differential effect on interaction of Vif with A3G or A3F, which are the two most potent antiviral A3 proteins. Interestingly, the amino acid variation at position 48 was found to dictate the differential sensitivity of A3H haplotype II to the two commonly used laboratory strains, NL4-3 and LAI, which contain N48 and H48 residues, respectively. While Vif containing N48 cannot neutralize the antiviral activity of A3H haplotype II, substitution to H48 as in the case of LAI, induces proteasomal degradation of A3H. 84
Alanine scanning mutagenesis in other regions of Vif identified Y69 and L72 amino acids in the 69YXXL72 motif to be critical for neutralizing the antiviral activity of both A3G and A3F. 82; 85; 86 In these studies, Vif proteins with mutations in this region were shown to retain binding to CUL5, confirming that the mutations did not result in expression of misfolded proteins that had lost all functions associated with Vif. Because of the absence of crystal or NMR structures of Vif and the complexity of its interaction in the context of E3 ubiquitin ligase, the identification of different regions of Vif involved in polyubiquitination and degradation of A3 proteins provided important insights in understanding the details of the Vif-A3 complex. 67; 82; 85; 86; 87; 88; 89; 90; 91; 92 However, much still remains to be determined including elucidation of the order of interaction of Vif, A3G, CBF-β - a cellular cofactor of Vif that facilitates A3G degradation, and E3 ubiquitin ligase complex. It is also important to note that in the absence of a structure, it cannot be determined whether the amino acids critical for interaction in A3 proteins or Vif are directly involved in the interactions, or they result in conformational changes that indirectly facilitate the interactions. Nonetheless, identification of these determinants that are critical for Vif-A3 protein interactions will hopefully assist rational design of inhibitors that inhibit Vif's capacity to induce degradation of the A3 proteins, thereby preserving their anti-HIV-1 function.
Structural determinants of A3G virion incorporation
Mutational analysis of A3 proteins has demonstrated the clustering of critical residues in well-defined patches on the surface of structural models. 45; 46; 48; 49; 51; 66; 67; 68 A3G from the producer cells, not the target cells, blocks HIV-1 replication in the subsequent round of infection; thus, it is the A3G carried by the virion that exerts the antiviral effects. 18; 28 To understand this essential aspect of the antiviral actions of A3G, we determined the stoichiometry of A3G in Vif-deficient virions produced from activated CD4+ T cells. 93 We found that about seven molecules of A3G were incorporated per virion; yet the infectivity of the virions is severely impaired in this physiologically relevant system, suggesting that the catalytic activity of A3G may play an important role in its antiviral activity. In the absence of Vif, A3G was shown to be incorporated into nascent virions through interactions with viral and non-viral RNAs and none of the viral proteins appeared to be required for virion incorporation. 94 Some studies have suggested that A3G interacts with the nucleocapsid (NC) domain of Gag in an RNA-dependent manner, which contributes to A3G virion incorporation. 95; 96; 97; 98; 99; 100 A3G encapsidation was also analyzed with HIV-1 Gag constructs in which the NC domain was replaced with a heterologous leucine zipper dimerization motif. 97 These Gag constructs produce viral particles that contain little or no RNA, and it was observed that these particles contain very little if any A3G, suggesting that the primary mechanism by which A3G is incorporated into virions is through its interaction with viral or non-viral RNA. Hydrophobic residues 124YYFW127 in the linker region between CD1 and CD2, and adjacent to the D128 residue that is critical for interaction with Vif, have been shown to be important for A3G virion encapsidation and were identified to interact with RNA and nucleocapsid in an RNA-dependent manner. 65; 101 Moreover, the W94/W127 residues in A3G were recently reported to be critical for its interaction with RNA, HMM complex assembly, and virion incorporation. 102
It is known that A3G is an RNA-binding protein 103; 104 and is associated with an array of cellular ribonucleoprotein (RNP) complexes in the cytosol. 56; 105; 106 Although the identity of the RNA(s) that promote the virion encapsidation of A3G remains unclear, HIV-1 genomic RNA and 7SL RNA have been implicated to play essential roles in the process.96; 98; 107; 108 Interactions of A3G with cellular 7SL RNA was suggested to be necessary for its virion incorporation.108 However, other studies have suggested that binding to 7SL RNA is dispensable for A3G virion incorporation. 109 We recently examined the role of 7SL RNA in A3G virion incorporation by sequestering the available 7SL RNA in cells by overexpression of SRP19, a protein that binds to 7SL RNA, and found that 7SL RNA availability did not have any effect on the efficiency of A3G virion incorporation. 110 Although binding to viral and/or non-viral RNAs by A3 proteins is essential for their virion incorporation, it is thought that given the vast amount of RNA present in cells, simple RNA binding cannot explain virion incorporation of A3G, and that some unidentified mechanisms may contribute to the specificity of the interactions of A3G with viral RNA, leading to their efficient virion incorporation.
Host proteins that regulate A3G functions and its Vif-mediated degradation
As recounted above, the anti-HIV action of A3G was discovered through efforts to understand the function of Vif that preserves viral infectivity. Subsequent studies revealed that Vif induces polyubiquitination and proteasomal degradation of A3G by hijacking the E3 ubiquitin ligase complex composed of CUL5, EloB, EloC and a RING-box subunit 2 (RBX2) and CBF-β, the allosteric regulator of Vif (Figure 5).14; 16; 18; 19; 20; 111; 112 A few studies have also reported that Vif binds to A3G mRNA and downregulates its expression and stability, and directly inhibits the deaminase activity of A3G. 19; 113; 114; 115. However, it is generally accepted that the primary mechanism by which Vif overcomes the antiviral activity of A3 proteins is by inducing their proteasomal degradation. In addition to these proteasomal components, several cellular factors including NEDD8, IFNs, HSP70, protein kinase A and others have been reported to modulate the expression of A3G and/or its antiviral activities (Table 1).
Figure 5. The role of CBF-β in Vif-mediated polyubiquitination and degradation of A3G.

Schematic depiction of different components of the A3G polyubiquitination and degradation pathway. CUL5 (scaffold) directly interacts with EloC (a component of the EloB/C adaptors) and RING finger protein 2 (RBX2; regulator of the stepwise cascade of substrate polyubiquitination), which also associates with E2 (ubiquitin conjugating enzyme). Vif (a substrate receptor for A3G) associates with the CUL5-EloB/C-RBX2-E2 complex by binding directly to CUL5 via a conserved Zinc ion coordinating HCCH motif and to EloC via its SOCS box motif. A3G is recruited by Vif to this complex. Subsequently, A3G is polyubiquitinated and is targeted for 26S proteasomal degradation. CBF-β, a recently discovered cofactor of Vif, has been proposed to be required to facilitate the Vif-CUL5 interaction and to suppress the antiviral activity of A3G in HIV-1 infection. 111; 112
Table 1. Host factors that influence the A3G antiviral activity.
| Host Proteins | Effects on A3G Expression, Function, or Degradation | Possible Mechanism | Ref |
|---|---|---|---|
| CBF-β | Promotes the polyubiquitination and degradation of A3G | Unclear | 111; 112 |
| IFN-α, β, γ | Upregulate the expression of A3G | Activation of JAK-STAK signaling pathway; activation of NFATc1/NFATc2 and IRF-4 | 129; 130; 131; 132; 133; 263 |
| HSP70 | Inhibits Vif-mediated A3G degradation | Unclear | 138; 139 |
| NEDD8 | Promotes the polyubiquitination and degradation of A3G | Activates the cullin-RING ubiquitin ligases by Neddylation | 125 |
| FLIP | Upregulates A3G expression | Unclear | 145 |
| IL-2, -15, -7 | Enhance the expression of A3G | Unclear | 146; 264 |
| PKA | Reduces the binding of A3G with Vif | Binds and phosphorylates A3G | 143 |
| RPA | Protects ssDNA from A3G binding and blocks the processivity of A3G | Non-specific steric hindrance | 265 |
| TLR3 | Induction of A3G expression | Unclear | 266 |
| PKCα/βI/MEK/ERK | Control A3G's mRNA and protein levels | Activates transcription | 267 |
| SP1, SP3 | Control A3G's mRNA levels | Binds to GC BOX of A3G's promoter | 268 |
| IL-1α, TNF-α | Upregulate the expression of A3G | Unclear | 269; 270; 271 |
| PIN1 | Reduces the expression of A3G | Interacts with A3G | 272 |
| C/EBP-β | Inhibits the activity of A3G | Interacts with A3G | 273 |
| TLR2 | Downregulates the expression of APOBEC3G | Unclear | 274 |
CBF-β: core binding factor β; HSP70: heat shock protein 70; FLIP: Fas-associated death domain (FADD) interleukin-1β-converting enzyme (FLICE)-like inhibitory protein; PKA: protein kinase A; RPA: replication protein A; TLR: Toll-like receptor; MEK: mitogen-activated extracellular signal-related kinase; ERK: extracellular signal-regulated kinases; SP1, SP3: specificity Protein 1/3; PIN1: peptidyl-prolyl cis-trans isomerase NIMA-interacting 1; NF-IL6 (C/EBP-β): CCAAT/enhancer-binding protein β; IRF-4: interferon regulatory factor 4; NFATc1/2: nuclear factor of activated T-cells, cytoplasmic 1 or 2.
CBF-β
Core binding factor-β (CBF-β) normally forms functional heterodimers with one of the three DNA-binding RUNX transcription factors and is involved in the regulation of hematopoiesis as well as development and differentiation of T lymphocytes. 116 Recently, two groups simultaneously reported that CBF-β can interact with Vif and this interaction is required for Vif-induced A3G polyubiquitination and degradation. 111; 112 Several lines of evidence suggest that Vif hijacks CBF-β, presumably before it recruits A3G to the Vif-CUL5-EloB/C-E3 ubiquitin ligase complex, to promote polyubiquitination and proteasomal degradation of A3G (Figure 5).111; 112 Moreover, stable knockdown of CBF-β in 293T cells reduces the stability of Vif and abolishes the assembly of Vif-CUL5-EloB/C-E3 ubiquitin ligase complex resulting in reduced HIV-1 infectivity primarily by preventing A3G degradation and preserving its antiviral activity. 111; 112
Vif binds to both isoforms of CBF-β 117; 118 and the N-terminus of CBF-β containing amino acids 15-126 was identified as the Vif binding region, 117 which appears to be distinct from its domain that interacts with the RUNX transcription factors. 112; 117; 118 However, when the N-terminal domain of CBF-β was extended to include residues 15-130, both Vif and RUNX were found to interact with CBF-β, suggesting that both proteins interact with CBF-β in a mutually exclusive manner. 117; 119 In fact, Hultquist et al. identified a single amino acid substitution, F68D, in CBF-β, which was sufficient to abrogate the interaction between CBF-β and Vif, while preserving its ability to form CBF-β-RUNX heterodimers. 118 Moreover, it has been shown that CBF-β prevents Vif oligomerization and promotes an ordered binding of Vif to the CUL5-E3 ubiquitin ligase complex. 119; 120 Although the molecular basis of Vif-CBF-β interaction is still unclear, a dual hijacking mechanism was recently proposed. 119 First, Vif recruits CBF-β to efficiently evade the innate immunity by promoting A3G degradation. Second, Vif sequesters CBF-β into the CUL5-Vif complex and perturbs the transcription of genes governed by RUNX-associated transcription factors. Disrupting the interaction between Vif and CBF-β may be an attractive pharmacological intervention for the development of anti-HIV therapies. 112 Furthermore, co-expression of Vif and CBF-β profoundly improves full-length Vif purification and solubility which will facilitate future structural and biochemical studies. 121
Nedd8
HIV-1 Vif recruits host factors to form an E3 ubiquitin ligase complex including CUL5, RBX, EloB, EloC, and CBF-β to degrade A3G (Figure 5).20; 111; 112 The cullin-RING ubiquitin ligase must be activated by the covalent modification of a conserved lysine in the C-terminal domain of all cullins with the ubiquitin-like protein NEDD8, 122 which requires the action of a three-enzyme E1-E2-E3 cascade much like that of ubiquitin. 123 Given that CUL5 NEDD8ylation is required for the activity of Vif, pharmacological inhibition of the NEDD8 E1 by a small molecule MLN4924 124 or knockdown of NEDD8-conjugating enzyme E2 UBE2F abrogates the action of Vif-induced degradation of A3G and restores the restriction of HIV. 125 Thus NEDD8 is also required to regulate the turnover of A3G.
Interferons
Understanding the regulation of the innate immune response, which includes cellular restriction factors such as A3 proteins, upon exposure to HIV-1 is essential. In fact, exposure to any foreign invaders including viral infections induces the innate immune response. One of the essential arms of the immune response is the type I interferon (IFN-α and -β) pathway that senses foreign dsDNA via its intracellular receptor, activated Toll-like receptor-3 (TLR-3), and generates an overwhelming antiviral state and curbs propagation of infection in immune cells.126In vitro studies showed that IFN-α is a potent inhibitor of HIV-1, particularly in the early stages of infection. 127; 128; 129 Interestingly, the IFN-α response seems to modulate the expression levels of restriction factors in response to exposure to HIV. For example, in macrophages, IFN-α and -β upregulate the expression of A3G, TRIM5α, tetherin, and anti-HIV miRNAs, and block viral replication.130 Furthermore, it has been shown recently that even low levels of IFN-α expression can induce expression of A3 proteins that is enough to restrict HIV-1 replication in monocyte-derived dendritic cells. 131 The induction of type I IFN-inducible antiviral factors, including A3G and tetherin, is believed to be through the JAK-STAT signaling pathway that fine-tunes the transcription of several essential cellular proteins. 132 In addition, it has been reported that A3G expression is critically dependent on the partnering of NFATc1/NFATc2 and IRF-4 that regulate the A3G promoter activity in T cells. When either NFATc1 or NFATc2 and IRF-4 were co-expressed in CEM-SS cells that normally do not express A3G and challenged with HIV-1Δvif, these cells avert infection primarily by inducing the expression of A3G. 133 Conversely, other host factors such as nerve growth factor (NGF) that can be released from HIV-1 infected macrophages were found to blunt IFN-γ-induced expression of A3G, favoring the propagation of HIV-1 infection in macrophages.134
Hsp70
Prostaglandin A1 (PGA1) inhibits the replication of a wide variety of DNA and RNA viruses, including HIV-1. 135 The proposed underlying mechanism is modulation of the expression of chaperone heat-shock proteins (HSP), especially HSP70 by regulating the action of nuclear factor-κB(NF-κB) and proliferator-activated receptor γ (PPARγ) in infected cells. 136; 137 Interestingly, HSP70 over-expression abrogates Vif-mediated degradation of A3G, 138 which suggests a potential role for PGA1 in innate immune response by upregulating the expression of HSP70 that prevents Vif-mediated A3G degradation. 139
Serine/threonine kinases and A3G phosphorylation
The archetypal mammalian cytidine deaminases, AID and A3G, are believed to be regulated by post-translational modifications, more specifically through phosphorylation by protein kinase A with functional consequences. For example, phosphorylation of certain serine/threonine residues of AID and A3G was claimed to be important in fine-tuning their role in adaptive and innate immune responses, respectively, and proposed to prevent potential deleterious mutagenic effects on the host genome.140; 141; 142 Additionally, Shirakawa et al. reported that phosphorylation of A3G at position T27 renders it less susceptible to Vif-mediated polyubiquitination and degradation.143 However, others showed that the serine/threonine phosphorylation of Vif and A3G is dispensable for Vif-mediated degradation of A3G. 144 Thus, the role of phosphorylation in regulating the antiviral activity of A3 proteins remains an open question that requires further study.
Other host factors that modulate A3G expression and function
The regulation of A3G gene expression and Vif-mediated degradation are affected by many other host factors. For example, recently, FLIP, also known as Fas-associated death domain (FADD) interleukin-1β-converting enzyme (FLICE)-like inhibitory protein, an inhibitor of caspase, was reported to inhibit HIV-1 replication in T cells including primary CD4+ T-cells and peripheral blood mononuclear cells (PBMCs). 145 Cellular FLIP (cFLIP) was found to regulate the expression of multiple HIV restriction factors including A3G, TRIM5, and Bst2/tetherin. 145 Interestingly, Oguariri et al. observed an anti-HIV-1 activity of interleukin-2 (IL-2) in HTLV-1 transformed laboratory T-cell lines such as MT-2, MT-4, SLB-1 and ATL-2 but not in other cell lines. 146 Although the loss of infectivity of the virus seems to be due to upregulation of A3G and possible incorporation into virions produced in these cell lines treated with IL-2, the relevance of this observation to the physiologically relevant target cells remains unknown.
Other cellular factors that are also believed to influence the function and/or expression of A3G include the human replication protein A (RPA), TLR2/3, PKC, SP3, PIN1, C/EBP-β, IFN type III, TNF-α, and others (Table 1). Although a considerable amount of knowledge is emerging in understanding the global landscape of host-pathogen interactions, much still remains to be determined, particularly in unraveling the delicate balance between the mechanism of evasion of host restriction factors and the hijacking of essential cellular cofactors by the pathogens to promote their own replication.
Mechanisms by which A3 proteins inhibit viral replication: hypermutation, inhibition of reverse transcription, and integration
The current weight of evidence indicates that A3G, and to a lesser extent A3DE, A3F and haplotypes II, V, and VII of A3H inhibit HIV-1 in the absence of Vif. 147; 148; 149; 150; 151; 152 A3A, A3B, and A3C have also been reported to inhibit retroviral replication in a limited number of studies. 60; 148; 153; 154; 155; 156
The phenomenon of extensive G-to-A mutations called hypermutation, initially described in a simple retrovirus, 157 is the result of C-to-U deamination of the viral minus-strand DNA by A3 proteins during reverse transcription. 158; 159 This results in G-to-A substitution in the complementary plus-strand often at a frequency that can exceed 10% of all G residues in the viral genome. The global hypermutation in the viral transcripts results in lethal mutational loads that preclude the subsequent generation of infectious particles and propagation of infection. 12; 28; 158; 160; 161; 162 Although hypermutation clearly results in lethal mutagenesis and inactivation of the viral genomes, 18; 28; 160 there is accumulating evidence that A3G and A3F also inhibit viral replication by reducing viral DNA synthesis 102; 163; 164; 165; 166; 167; 168; 169; 170; 171; 172; 173; 174 and integration of the viral DNA into the host genome. 172; 173; 175
Biochemical mechanisms of cytidine deamination and viral inhibition
Several lines of evidence, including recent data from primary target cells infected with HIV-1, 147 indicate that editing and non-editing mechanisms are separate and distinct modes of action of A3 proteins in blocking HIV-1 infection. 28; 167; 168; 172; 176; 177
Editing mechanism: hypermutation
As minus-strand DNA synthesis progresses during reverse transcription, the RNase H activity of reverse transcriptase (RT) degrades the RNA template, allowing A3G access to the newly synthesized ssDNA for cytidine deamination. A3G prefers to deaminate C residues in 5′-CCC or 5′-CC motifs on ssDNA, deaminating the 3′ C (5′-CCU or 5′-CU); A3G lacks cytidine deaminase activity on dsDNA or RNA templates. 56; 148; 162; 178 Site preferences for hypermutation are not fully understood, but the sequence context of the cDNA as well as the secondary structure (stem loops) of the substrate are known to influence cytidine deamination patterns. 179 For example, numerous studies have shown that A3G favors the consensus sequence 5′-(T/C)CC(G/A/C)- 3′. 148; 179; 180 Although A3G prefers to deaminate the second C in 5′-CC, resulting in 5′-GG-to-AG mutations on the viral cDNA, 148; 161 the remaining A3 proteins prefer to deaminate the C in the 5′-TC dinucleotide context, resulting in 5′-GA-to-AA mutations on the viral cDNA. 60; 148; 152; 154; 155; 156; 181; 182; 183 As discussed in later sections, patient-derived HIV-1 proviral sequences contain G-to-A hypermutations in either the GG-to-AG 184; 185; 186 or GA-to-AA dinucleotide context, 181; 187; 188 suggesting that A3G and at least one of the other A3 proteins contribute to HIV-1 restriction in vivo. The overrepresentation of GA-to-AA dinucleotide hypermutation reported from one set of patients was based in part on the sequence data using a short fragment of HIV-1 proviral region coding for protease and on the observation that A3F is partially resistant to HIV-1 Vif. 181; 188 A3F is the most likely A3 protein that is responsible for the GA-to-AA hypermutation, 181 although possible contributions from A3H haplotypes II, V, and VII and A3DE cannot be excluded. A3H haplotype II likely restricts HIV-1 in a deaminase-independent manner, 73 although A3H haplotype II (RDD mutant) and even the macaque A3H have shown GA-to-AA hypermutation profiles. 155; 189 It is unclear at this time whether hypermutations are more frequent in the GA or GG dinucleotide context in patients, and a systematic analysis of large proviral sequence databases derived from patient cohorts which include infections with different subtypes is needed to answer this question.
Insights into the biochemical mechanisms by which A3 proteins induce cytidine deamination can be gained by analyzing the patterns of G-to-A hypermutations in proviral genomes. First, hypermutation is often observed throughout the viral genome, suggesting that the A3 proteins must be able to access the viral minus-strand DNA to access multiple target sites. Second, some regions of the genome are hypermutated more extensively than others. Hypermutated proviral DNAs exhibit an overall “twin gradient”, one gradient with increasing frequency of mutation from the primer binding site to the central polypurine tract (cPPT), and a second gradient with increasing frequency of mutation from the cPPT to the polypurine tract (PPT) 190. These twin gradients are thought to be largely the direct result of increased access of the minus-strand ssDNA by the A3 proteins during reverse transcription. As minus-strand DNA synthesis is extended in the 5′ to 3′ direction by RT, RNase H removes the genomic RNA from the RNA-DNA hybrids, increasing access to the minus-strand DNA by the A3 proteins.
A deamination bias within very short stretches of substrate DNA has been observed in vitro, whereby the deamination occurs more frequently at the 5′ end of the ssDNA, while the 3′ end of the ssDNA is never deaminated (“dead zone”). 56; 191 A3G bound in the active orientation with the CD1 domain oriented at the 3′ end of the template would physically block deamination in this region. Recent in vitro data using FRET analysis shows that the observed 3′ to 5′ polarity is due to the binding of A3G to the ssDNA in an asymmetric catalytically active orientation with its CD2 active domain facing the 5′ end of the ssDNA. 54 A3G bound in the active orientation deaminates the 3′ C in the 5′-CCC-3′ motif more efficiently than the A3G bound in the inactive orientation. 162; 191 It has been suggested that this intrinsic directionality to the processive movement of A3G could partly explain the twin gradient observed over the entire viral genome.
Processivity refers to the ability of A3 proteins to catalyze multiple cytidine deamination reactions before dissociating from the template. It has been proposed that A3G induces hypermutation by binding its target through facilitated diffusion scanning mechanisms involving sliding and jumping motions, 56; 191; 192; 193 resulting in directional deamination. Recent results suggest that A3G can slide 69 nucleotides per second on ssDNA. 194 During reverse transcription, RNase H leaves fragments of RNA still attached to the newly synthesized DNA, which are likely to impede the sliding motions of A3 proteins. Perhaps to overcome this barrier, the A3 proteins have evolved to jump from one region of the template to another. When A3 physically leaves the template and reassociates elsewhere on the template (i.e. jumping); these jumps can be micro- or macroscopic depending upon the size of the jump. When A3G binds two ssDNA templates simultaneously, and switches to the alternate template, this is termed intersegmental transfer.195 Whether A3G can undergo sliding motions, jumping, and/or intersegmental transfers is currently being investigated. 196; 197
The catalytically inactive CD1 of A3G, which can bind RNA and DNA, 103; 178 mediates the processivity of A3G, 56; 196 while the catalytically active CD2 mediates cytidine deamination and hypermutation. 103; 178 As described earlier, the A3G mutant W127A is deficient in binding to RNA and remains monomeric. The monomeric A3G double mutant F126A/W127A, which is unable to jump efficiently, and mutant H186R, which is unable to slide efficiently, both deaminate ssDNA less efficiently that wild-type A3G, supporting the notion that a dimer or oligomer is the functional form of A3G, and the sliding and jumping movements are essential for efficient cytidine deamination. 193
Non-editing mechanisms: blocking reverse transcription and integration
In addition to inducing lethal mutagenesis, the A3 proteins employ non-editing mechanisms to inhibit retroviral replication. It was observed early that catalytic site mutants of A3G could inhibit viral replication, 171 and A3F catalytic site mutants could inhibit accumulation of reverse transcripts. 170 Other studies showed that A3G can inhibit reverse transcription by physically blocking RT template binding, reducing tRNA binding, inhibiting efficient tRNA processing, impairing elongation of cDNA synthesis, and blocking minus- and plus-strand transfer events. 165; 166; 167; 168; 169; 172; 198. Furthermore, the presence of dU in the minus-strand DNA was shown to decrease the efficiency of plus-strand DNA synthesis during reverse transcription. 199 In these in vitro studies and transient transfection assays, it was not clear whether the high levels of A3 proteins resulted in the non-editing modes of viral inhibition that were absent under physiological levels of the A3 proteins.
Two distinct mechanisms were thought to contribute to the reduction in accumulation of viral DNA products; first, A3G could bind to viral RNA or cDNA and inhibit the initiation or elongation of viral DNA synthesis; second, removal of the uracil base by uracil DNA glycosylase (UDG), followed by cleavage of the abasic site by apurinic-apyrimidinic endonucleases could result in degradation of reverse transcripts. To gain insights into the mechanisms by which A3G inhibits HIV-1 replication, we performed transient transfection assays in which the transfected cells expressed A3G at levels similar to those observed in primary activated CD4+ T cells, and quantified the synthesis and integration of viral DNA using quantitative real-time PCR.172 In agreement with other studies, 200; 201 our results showed that the inhibition of host UDG had no impact on the amount of viral DNA synthesized, suggesting that UDG was not inducing degradation of viral DNA subjected to cytidine deamination by A3G. We also observed that a 35-fold decrease in viral infectivity was accompanied by a 5 to 7-fold reduction in viral DNA synthesis, and in agreement with another study, 173 a 5-fold reduction in integration of the viral DNA. Moreover, the inhibition of reverse transcription and integration required the catalytic activity of A3G, and was the result of aberrant primer tRNA processing that subsequently reduced the efficiency of plus-strand DNA transfer and integration. Subsequently, we found that A3F also inhibits viral DNA integration, but it does so by inhibiting the 3′ processing reaction catalyzed by integrase.175 Overall, the results indicated most of the 35-fold decrease in viral infectivity could be explained by the 5 to 7-fold reduction in viral DNA synthesis and a 5-fold reduction in integration.
Recently, a detailed analysis of the editing and non-editing mechanisms of viral inhibition in primary CD4+ T cells was performed.147 The results indicated that both hypermutation and reduction of viral DNA synthesis occurred when physiological levels of A3 proteins were present, and that A3G was predominantly responsible for the observed cytidine deamination.
A3G Oligomerization
Several lines of evidence indicate that A3G oligomerization and binding to RNA or ssDNA are inextricably linked. 105; 191; 202; 203; 204; 205; 206; 207 Chelico et al. concluded that A3G oligomerization is primarily dependent on its interaction with ssDNA. Co-immunoprecipitation and cross-linking studies indicated that the Y124 and W127 residues were important for A3G's ability to bind to RNA and to form oligomers in an RNA-dependent manner. Consistent with the models of dimer interaction, 42; 45; 204 it has been shown that A3G, A3F and A3DE form homodimers and heterodimers in cells.152; 182 We used a bimolecular fluorescence complementation system to investigate A3G oligomerization and RNA binding in situ, and found that wild-type A3G molecules oligomerized and reconstituted fluorescence, while RNA-binding defective mutants of A3G were monomeric and failed to reconstitute fluorescence. 207 In these and other studies, it is unclear whether the RNA-dependent oligomers of A3G form protein-protein interactions after binding to RNA, or whether they represent single A3G molecules that are bound to the same RNA. Interestingly, we found that a wild-type A3G can weakly reconstitute fluorescence with an RNA-binding defective mutant of A3G, suggesting a model in which conformational changes in the wild-type A3G that occur after RNA binding can expose protein determinants that can interact with another A3G molecule.
McDougall et al. have analyzed the composition of oligomers formed in vitro in the presence of ssDNA and correlated the oligomeric forms with their deaminase activity; their results suggest that the homodimeric forms had low deaminase activity whereas homotetramers and higher order multimers were more efficient at carrying out cytidine deamination 208. Recently, high-speed atomic force microscopy (AFM) has been used to study the association of A3G with ssDNA; 194; 203 it was observed that the stoichiometry of A3G binding to ssDNA was independent of its 5′ to 3′ polarity and bound to gapped ssDNA substrates as well as tail-DNA, indicating that a DNA end is not required for binding. The authors observed that the assembled multimeric complexes could dissociate from the ssDNA as a whole complex or as monomers in a stepwise fashion. These results confirm the in situ results 207 and provide direct biochemical evidence to support the view that A3G is primarily monomeric in cells in the absence of RNA binding, and forms higher order multimers upon binding to single-stranded nucleic acid. Because of the abundance of RNA in cells, A3G largely exists as a high molecular mass complex (HMM) in association with RNA. 105
Anti-HIV activities of other APOBEC3 proteins: A3DE, A3F and A3H
A3G has been identified as a potent restriction factor of HIV-1Δvif in the natural target cells of infection - PBMCs, monocyte-derived macrophages, and dendritic cells; however, the role of A3DE, A3F, and A3H in HIV-1 restriction is more controversial. A3DE, A3F, and A3G are expressed in T cells and B cells, whereas A3H is expressed at higher levels in T cells than in B cells. 23 The expression levels of these proteins vary in different tissues (e.g., spleen, lymph node, lung, etc), and are largely dependent on the size of the lymphocyte population in each tissue. 23 Depletion of A3G from the nonpermissive T cell line CEM2n by gene targeting resulted in a significant decrease in the 5′-GG-to-AG hypermutation and a 10-fold increase in virus replication. However, restoration of the replication capacity of the virus remained 2-fold less compared to that in the permissive T cell line CEM-SS, indicating the presence of additional restriction factors that induce 5′-GA-to-AA hypermutations.
Although A3F was initially found to restrict HIV when overexpressed in virus producer cells, 182 in some studies it was observed to have little or no effect on HIV-1 replication under physiological conditions. Mulder et al.209 reported no difference in HIV replication in PBMCs using wild-type virus (which can degrade both A3F and A3G) and the W11R Vif mutant (which can degrade only A3G), whereas replication of the ΔSLQ Vif mutant (which does not degrade A3G) was significantly restricted. Moreover, A3F had no significant effect on virus infectivity when it was stably expressed in HeLa cells, even though a significant amount of A3F was packaged into progeny virions. 210 Interestingly, a selection experiment using HIV-1Δvif virus in stably transfected T cell lines that express A3F resulted in the selection of a mutant that restored Vif function, suggesting that A3F can impose selection pressure to maintain Vif function in vivo.211 Moreover, HIV-1 containing a Vif mutant (DRMR>4A) that could degrade A3G, but not A3F, was restricted in PBMCs, albeit to a lesser extent than virus that contains Vif (YRHHY>5A) that is capable of degrading A3F, but not A3G. 150 Overall, these studies indicate that both A3G and A3F can restrict HIV-1 replication in their natural target cells of infection, but A3G is a more potent inhibitor of HIV-1 than A3F.
Subsequent studies have revealed that A3DE and A3H haplotypes II, V, and VII, have restriction activity when stably expressed, further expanding the antiviral A3 repertoire in the producer cells. 151; 212 Viruses produced from a CEM cell line in which A3F was specifically knocked down showed increased infectivity compared to viruses produced from the parental cell line with normal A3F expression. 212 Interestingly, the 5′-GA-to-AA mutational pattern was unchanged, consistent with the idea that other A3 proteins are responsible for the 5′-GA-to-AA mutations.
Virus produced from A3F-null CEM cells in which A3DE was also depleted by shRNA had increased infectivity and decreased 5′-GA-to-AA mutations relative to virus produced from A3F-null CEM cells, indicating that A3DE is likely a true restriction factor in this cell line, potentially expanding the antiviral A3 proteins repertoire in T cells. Virus produced from A3F-null CEM cells in which A3H was also depleted by shRNA had increased infectivity, but no difference in mutation rate or pattern relative to virus produced from A3F-null CEM cells. Interestingly, the A3H mRNA level was 20-fold lower in the A3F-null CEM cell line compared to primary CD4+ T cells, indicating that A3H can restrict virus even at subphysiological levels. 212 A3H haplotype II-, but not haplotype I-, overexpression in producer cells has been shown to restrict the replication of some wild-type laboratory adapted HIV-1 strains. 153; 155 Moreover, only A3H haplotypes II, V, and VII that express stable proteins could incorporate into virions and restrict virus replication in 293T cells. 153 Interestingly, A3H Haplotype II is not completely neutralized by Vif primarily due to a single amino acid polymorphism at position 121 which is prevalent in people of African descent. 74; 75; 153; 155 Overall, while A3G is the most potent restriction factor, these members of A3 proteins can restrict HIV-1 replication. Their contribution to the inhibition of HIV-1 replication appears to be quantitatively less than that of A3G, but it is possible that they play important roles in certain physiological conditions or in specific HIV-1 infected populations.
Inhibition of HIV-1 replication by A3 proteins in humanized mice
Animal models for HIV-1 infection are essential tools for studying the in vivo mechanism of host-virus interactions such as transmission, replication, and pathogenesis. Nonhuman primates such as rhesus macaques infected with SIVmac or SIV/HIV chimeric viruses (SHIVs) represent well-characterized animal models of immunodeficiency virus infection. However, there are key limitations to these primate studies, including the inherent genetic variability of the host animals, and the fact that SIVmac, while closely related to HIV-2, is genetically distinct from HIV-1. Although chimpanzee immunodeficiency virus (SIVcpz) is believed to be a closely related ancestor of HIV-1, chimpanzees are not a suitable animal model for HIV-1 infection because of ethical concerns, availability of chimpanzees, and high cost. Therefore, humanized mouse models have been developed that support HIV-1 infection and recapitulate many aspects of the human infection. Three different strains of mice have been transplanted with human hematopoietic stem cells and have been confirmed to be permissive to HIV-1 infection: NOD/SCID, Rag2nullγCnull (double knockout; DKO), and NOD/SCID/γCnull (NOG of NSG). 213; 214; 215; 216; 217; 218; 219
Results from HIV-1 infected humanized mice have been shown to positively correlate with infection in humans. Ince et al. reported that the divergence rate of the HIV-1 env gene in the virus population in DKO-humanized mice was similar to that observed in patients during a similar period of infection. 220 The observed mutations in the env region were associated with increased CD4 binding efficiency, with one mouse even exhibiting a CCR5 to CXCR4 coreceptor switch. Moreover, A3G-associated hypermutation in proviral DNA was reported in wild-type HIV-1 infected NOG humanized mice, 221 indicating that A3G mediated G-to-A hypermutation abrogated HIV-1 replication in vivo.
Interestingly, Krisko et al. recently demonstrated that A3G and A3F proteins efficiently restrict R5-tropic HIV-1Δvif in bone marrow liver thymus (BLT) mice. 222 In stark contrast, the X4-tropic HIV-1Δvif virus escaped the actions of A3G and A3F in this murine model. BLT mice were developed by transplanting human hematopoietic stem cells along with human fetal thymus and liver tissue into NOD/SCID mice so that the T lymphocyte progenitors migrate and undergo the maturation process in the thymus. To date, BLT mice represent the only murine system designed to encompass the physiological T cell maturation process in the implanted human thymus. 223; 224 Interestingly, the expression levels of A3F and A3G in the thymus are much lower compared to PBMCs or other organs. Moreover, whereas 30-40% of thymocytes express CXCR4 coreceptor, <5% of the population was found to be CCR5 positive, 222 indicating that R5-tropic viruses have far fewer target cells in the thymus compared to the X4-tropic virus. Together, the reduced expression of A3G and A3F proteins may explain the differential resistance of the X4-tropic virus in thymocytes, with or without Vif expression. This was further corroborated by the sensitivity of the X4-tropic Vif-deficient virus to A3 restriction as extensive G-to-A hypermutation was observed in PBMCs and other organs. These data strongly indicate that the BLT mouse model may provide an excellent tool to study the impact of the antiviral actions of A3 proteins on HIV-1 infection in vivo and further aid in understanding the role of A3 proteins in the pathogenesis of HIV infection. Importantly, Vif-independent HIV-1 replication in the thymus highlights the complexity of the mechanisms by which HIV-1 can escape inhibition by A3 proteins.
A3 restriction in HIV-1 infected patients
The diversification of the HIV-1 population in an infected individual and its continuous adaptation to its host is the result of a combination of several factors: a large viral population, high replication and mutation rates, recombination, and various host selective pressures. 225 The high mutation rate is mainly attributed to error prone reverse transcription. 225 Although sublethal G-to-A hypermutations induced by the A3 proteins have also been proposed to contribute to viral variation, 226; 227; 228 its contribution to viral diversification and potential accumulation of resistance mutations in patients on therapy is controversial; 159; 229; 230; 231; 232; 233 and other studies have suggested that even a single incorporated A3G is likely to cause extensive and lethal hypermutation and may be an “all or nothing” phenomenon. 233 It is important to determine whether A3G has the capacity to contribute to viral variation, because potential therapeutic measures targeting the interaction between Vif and A3G may contribute to viral variation and selection of drug resistant variants.
A3 protein expression levels and/or natural variation of A3 proteins can influence their antiviral activity in patients. In agreement with this, several investigators reported that A3G expression levels correlate inversely with HIV-1 viral load and disease progression and directly with the mutational loads in the viral genome (Table 2). 234; 235; 236; 237; 238; 239 Interestingly, A3G mRNA levels are higher in long-term nonprogressors than in uninfected controls, while the lowest A3G mRNA levels are found in HIV progressors.234 In a South African cohort, significantly higher A3G expression in PBMCs was shown for HIV-1-exposed seronegative individuals compared to HIV-1-seropositive patients or healthy controls, 240 but another recent study could not demonstrate any difference in the expression levels of A3G in HIV-1-exposed seronegative patients.241 Contrary to the aforementioned cohorts, Gandhi et al. showed that the frequency of hypermutated viral genomes in elite controllers was not significantly different from that observed in patients on highly active antiretroviral therapy, and no correlation was found between A3G expression and the rate of hypermutation.242 Moreover, recent studies found no correlation between the expression levels of A3G and A3F and disease progression in HIV-1 infected children.243; 244; 245
Table 2. The effect of A3G natural variation on HIV-1 pathogenesis.
| A3G Expression or Polymorphisms | Findings | Ref |
|---|---|---|
| Increased expression is associated with | Increased proviral hypermutation; higher CD4+ T-cell count | 237; 238; 246 |
| Lower viral set point | 235 | |
| Resistance to HIV-1 infection after exposure | 240 | |
| Lower viral load in long term nonprogressors | 234 | |
| Higher CD4+ cell count in highly exposed seronegative individuals compared to healthy controls and HIV+ patients | 236 | |
| Unaffected expression | No correlation with control of viremia in elite controllers | 242 |
| In HIV-1 exposed seronegative subjects | 241 | |
| Decreased expression | Observed upon HIV-1 infection | 251 |
| No correlation with CD4+ T cell count or viral load | ||
| H186R polymorphism | Association with significantly increased viral load and decreased CD4+ T cell count with rapid progression to AIDS | 244; 250; 251 |
| C40693T polymorphism | An intronic sequence polymorphism associated with increased risk of infection | 275 |
| F119F | Associated with protection against disease progression in pediatric patients | 249 |
Sporadic G-to-A hypermutation is readily detected in significant proportions of the integrated proviral DNA from patient samples. 89; 246; 247 Interestingly, such mutational loads could not be found in viral RNA from plasma samples of the same HIV-1 infected patients, indicating that the hypermutated proviruses do not generate progeny virions. These in vivo results are in agreement with our analysis in cell culture using HIV-1Δvif virus, which showed a gradient hypermutation from proviral DNA>cellular RNA>viral RNA. 248 This hypermutation gradient results from purifying selection acting at the level of transcription and expression of functional viral proteins, leading to the preferential packaging of mostly unmutated viral genomes into virions.
Genetic variation in A3G and vif
Single nucleotide polymorphisms (SNPs) in A3G can alter its antiviral activity and hence the rate of HIV-1 disease progression. An SNP in A3G (H186R) in exon 4, which is abundant in African Americans (37%) but rare in Caucasians, is strongly associated with a more rapid CD4+ T cell loss and accelerated disease progression (Table 2).249; 250 This mutation was also found at a similar frequency in ART-naïve African HIV-infected adults and was significantly associated with increased viral load and a rapid decline in CD4+ T-cell count. 251 Singh et al. also identified another mutation in A3G (F119F), a synonymous genetic variant, which is associated with reduced rate of disease progression in HIV-1 infected children. 249 Although it is possible that this SNP may be in linkage disequilibrium with other as yet unidentified SNPs that alter the expression and/or function of A3G, the underlying mechanism is not completely understood. Other studies indicate that A3G- and A3F-induced hypermutation is associated with increased CD4+ T-cell count 246 and a reduction in viremia. 252 However, some studies have not found a correlation between A3G and A3F mRNA levels or polymorphisms and viral load and/or CD4+ T-cell count (Table 2). 242; 251; 253; 254; 255
Not surprisingly, the vif gene has high genetic variability 89; 256; 257; 258; 259 and subtype dependent amino acid substitutions 188; 260; 261 in patient samples with varying degrees of activity against A3 proteins. Interestingly, defective Vif alleles such as K22H that cannot efficiently neutralize A3G and A3F are readily detected in HIV-1 infected patients. 89; 259 Recently, a Vif mutant (I107T) was identified in one long-term nonprogressor African patient that failed to interact with A3G and induce A3G degradation.257 Given that I107 is in the 96T(Q/D/E)X5ADX2(I/L)107 motif of HIV-1 Vif that has been shown to be critical for Vif activity to induce A3G and A3F degradation as well as its interaction with CUL5, 262 the I107T mutant likely attenuates viral infection and profoundly impacts disease progression. Overall, both host and viral genetic diversity play important roles in viral fitness and host innate immunity.
Summary
The main function of A3 proteins is to provide intrinsic immunity to the host by preventing the spread of foreign nucleic acids either through editing (G-to-A hypermutation in the retroviral DNA) and non-editing mechanisms (inhibition of reverse transcription and integration). The host innate immune response is a first line of defense against foreign invaders. A3G, A3F, A3H haplotypes II, V, and VII, (and possibly A3DE) are the main human A3 proteins that block HIV-1 infection. Understanding the interactions between A3 proteins and Vif could provide a unique opportunity to develop novel therapeutic strategies for HIV-1 infected patients.
Highlights.
APOBEC3DE, APOBEC3F, APOBEC3G, and APOBEC3H haplotypes II, V, and VII inhibit HIV-1 replication.
HIV-1 Vif protein antagonizes the antiviral activity of APOBEC3 proteins by targeting them for proteasomal degradation.
Vif-APOBEC3 protein interactions provide a novel target for antiviral drug development.
Acknowledgments
We especially thank Hiroshi Matsuo and Wei-Shau Hu for their valuable comments on the manuscript. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Smith HC, Bennett RP, Kizilyer A, McDougall WM, Prohaska KM. Functions and regulation of the APOBEC family of proteins. Semin Cell Dev Biol. 2012;23:258–68. doi: 10.1016/j.semcdb.2011.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Teng B, Burant CF, Davidson NO. Molecular cloning of an apolipoprotein B messenger RNA editing protein. Science. 1993;260:1816–9. doi: 10.1126/science.8511591. [DOI] [PubMed] [Google Scholar]
- 3.Navaratnam N, Morrison JR, Bhattacharya S, Patel D, Funahashi T, Giannoni F, Teng BB, Davidson NO, Scott J. The p27 catalytic subunit of the apolipoprotein B mRNA editing enzyme is a cytidine deaminase. J Biol Chem. 1993;268:20709–12. [PubMed] [Google Scholar]
- 4.Muramatsu M, Kinoshita K, Fagarasan S, Yamada S, Shinkai Y, Honjo T. Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell. 2000;102:553–63. doi: 10.1016/s0092-8674(00)00078-7. [DOI] [PubMed] [Google Scholar]
- 5.Vonica A, Rosa A, Arduini BL, Brivanlou AH. APOBEC2, a selective inhibitor of TGFbeta signaling, regulates left-right axis specification during early embryogenesis. Dev Biol. 2011;350:13–23. doi: 10.1016/j.ydbio.2010.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Etard C, Roostalu U, Strahle U. Lack of Apobec2-related proteins causes a dystrophic muscle phenotype in zebrafish embryos. J Cell Biol. 2010;189:527–39. doi: 10.1083/jcb.200912125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sato Y, Probst HC, Tatsumi R, Ikeuchi Y, Neuberger MS, Rada C. Deficiency in APOBEC2 leads to a shift in muscle fiber type, diminished body mass, and myopathy. J Biol Chem. 2010;285:7111–8. doi: 10.1074/jbc.M109.052977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liao W, Hong SH, Chan BH, Rudolph FB, Clark SC, Chan L. APOBEC-2, a cardiac- and skeletal muscle-specific member of the cytidine deaminase supergene family. Biochem Biophys Res Commun. 1999;260:398–404. doi: 10.1006/bbrc.1999.0925. [DOI] [PubMed] [Google Scholar]
- 9.Mikl MC, Watt IN, Lu M, Reik W, Davies SL, Neuberger MS, Rada C. Mice deficient in APOBEC2 and APOBEC3. Mol Cell Biol. 2005;25:7270–7. doi: 10.1128/MCB.25.16.7270-7277.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rogozin IB, Basu MK, Jordan IK, Pavlov YI, Koonin EV. APOBEC4, a new member of the AID/APOBEC family of polynucleotide (deoxy)cytidine deaminases predicted by computational analysis. Cell Cycle. 2005;4:1281–5. doi: 10.4161/cc.4.9.1994. [DOI] [PubMed] [Google Scholar]
- 11.Sheehy AM, Gaddis NC, Choi JD, Malim MH. Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature. 2002;418:646–50. doi: 10.1038/nature00939. [DOI] [PubMed] [Google Scholar]
- 12.Lecossier D, Bouchonnet F, Clavel F, Hance AJ. Hypermutation of HIV-1 DNA in the absence of the Vif protein. Science. 2003;300:1112. doi: 10.1126/science.1083338. [DOI] [PubMed] [Google Scholar]
- 13.Harris RS, Sheehy AM, Craig HM, Malim MH, Neuberger MS. DNA deamination: not just a trigger for antibody diversification but also a mechanism for defense against retroviruses. Nat Immunol. 2003;4:641–3. doi: 10.1038/ni0703-641. [DOI] [PubMed] [Google Scholar]
- 14.Conticello SG, Harris RS, Neuberger MS. The Vif protein of HIV triggers degradation of the human antiretroviral DNA deaminase APOBEC3G. Curr Biol. 2003;13:2009–13. doi: 10.1016/j.cub.2003.10.034. [DOI] [PubMed] [Google Scholar]
- 15.Kobayashi M, Takaori-Kondo A, Miyauchi Y, Iwai K, Uchiyama T. Ubiquitination of APOBEC3G by an HIV-1 Vif-Cullin5-Elongin B-Elongin C complex is essential for Vif function. J Biol Chem. 2005;280:18573–8. doi: 10.1074/jbc.C500082200. [DOI] [PubMed] [Google Scholar]
- 16.Marin M, Rose KM, Kozak SL, Kabat D. HIV-1 Vif protein binds the editing enzyme APOBEC3G and induces its degradation. Nat Med. 2003;9:1398–403. doi: 10.1038/nm946. [DOI] [PubMed] [Google Scholar]
- 17.Mehle A, Strack B, Ancuta P, Zhang C, McPike M, Gabuzda D. Vif overcomes the innate antiviral activity of APOBEC3G by promoting its degradation in the ubiquitin-proteasome pathway. J Biol Chem. 2004;279:7792–8. doi: 10.1074/jbc.M313093200. [DOI] [PubMed] [Google Scholar]
- 18.Sheehy AM, Gaddis NC, Malim MH. The antiretroviral enzyme APOBEC3G is degraded by the proteasome in response to HIV-1 Vif. Nat Med. 2003;9:1404–7. doi: 10.1038/nm945. [DOI] [PubMed] [Google Scholar]
- 19.Stopak K, de Noronha C, Yonemoto W, Greene WC. HIV-1 Vif blocks the antiviral activity of APOBEC3G by impairing both its translation and intracellular stability. Mol Cell. 2003;12:591–601. doi: 10.1016/s1097-2765(03)00353-8. [DOI] [PubMed] [Google Scholar]
- 20.Yu X, Yu Y, Liu B, Luo K, Kong W, Mao P, Yu XF. Induction of APOBEC3G ubiquitination and degradation by an HIV-1 Vif-Cul5-SCF complex. Science. 2003;302:1056–60. doi: 10.1126/science.1089591. [DOI] [PubMed] [Google Scholar]
- 21.Harris RS, Hultquist JF, Evans DT. The restriction factors of human immunodeficiency virus. J Biol Chem. 2012;287:40875–83. doi: 10.1074/jbc.R112.416925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jarmuz A, Chester A, Bayliss J, Gisbourne J, Dunham I, Scott J, Navaratnam N. An anthropoid-specific locus of orphan C to U RNA-editing enzymes on chromosome 22. Genomics. 2002;79:285–96. doi: 10.1006/geno.2002.6718. [DOI] [PubMed] [Google Scholar]
- 23.Koning FA, Newman EN, Kim EY, Kunstman KJ, Wolinsky SM, Malim MH. Defining APOBEC3 expression patterns in human tissues and hematopoietic cell subsets. J Virol. 2009;83:9474–85. doi: 10.1128/JVI.01089-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Refsland EW, Stenglein MD, Shindo K, Albin JS, Brown WL, Harris RS. Quantitative profiling of the full APOBEC3 mRNA repertoire in lymphocytes and tissues: implications for HIV-1 restriction. Nucleic Acids Res. 2010;38:4274–84. doi: 10.1093/nar/gkq174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nitta T, Lee S, Ha D, Arias M, Kozak CA, Fan H. Moloney murine leukemia virus glyco-gag facilitates xenotropic murine leukemia virus-related virus replication through human APOBEC3-independent mechanisms. Retrovirology. 2012;9:58. doi: 10.1186/1742-4690-9-58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Okeoma CM, Lovsin N, Peterlin BM, Ross SR. APOBEC3 inhibits mouse mammary tumour virus replication in vivo. Nature. 2007;445:927–30. doi: 10.1038/nature05540. [DOI] [PubMed] [Google Scholar]
- 27.Mussil B, Sauermann U, Motzkus D, Stahl-Hennig C, Sopper S. Increased APOBEC3G and APOBEC3F expression is associated with low viral load and prolonged survival in simian immunodeficiency virus infected rhesus monkeys. Retrovirology. 2011;8:77. doi: 10.1186/1742-4690-8-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mangeat B, Turelli P, Caron G, Friedli M, Perrin L, Trono D. Broad antiretroviral defence by human APOBEC3G through lethal editing of nascent reverse transcripts. Nature. 2003;424:99–103. doi: 10.1038/nature01709. [DOI] [PubMed] [Google Scholar]
- 29.Sasada A, Takaori-Kondo A, Shirakawa K, Kobayashi M, Abudu A, Hishizawa M, Imada K, Tanaka Y, Uchiyama T. APOBEC3G targets human T-cell leukemia virus type 1. Retrovirology. 2005;2:32. doi: 10.1186/1742-4690-2-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mahieux R, Suspene R, Delebecque F, Henry M, Schwartz O, Wain-Hobson S, Vartanian JP. Extensive editing of a small fraction of human T-cell leukemia virus type 1 genomes by four APOBEC3 cytidine deaminases. J Gen Virol. 2005;86:2489–94. doi: 10.1099/vir.0.80973-0. [DOI] [PubMed] [Google Scholar]
- 31.Fan J, Ma G, Nosaka K, Tanabe J, Satou Y, Koito A, Wain-Hobson S, Vartanian JP, Matsuoka M. APOBEC3G generates nonsense mutations in human T-cell leukemia virus type 1 proviral genomes in vivo. J Virol. 2010;84:7278–87. doi: 10.1128/JVI.02239-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wiegand HL, Cullen BR. Inhibition of alpharetrovirus replication by a range of human APOBEC3 proteins. J Virol. 2007;81:13694–9. doi: 10.1128/JVI.01646-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lochelt M, Romen F, Bastone P, Muckenfuss H, Kirchner N, Kim YB, Truyen U, Rosler U, Battenberg M, Saib A, Flory E, Cichutek K, Munk C. The antiretroviral activity of APOBEC3 is inhibited by the foamy virus accessory Bet protein. Proc Natl Acad Sci U S A. 2005;102:7982–7. doi: 10.1073/pnas.0501445102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Delebecque F, Suspene R, Calattini S, Casartelli N, Saib A, Froment A, Wain-Hobson S, Gessain A, Vartanian JP, Schwartz O. Restriction of foamy viruses by APOBEC cytidine deaminases. J Virol. 2006;80:605–14. doi: 10.1128/JVI.80.2.605-614.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Russell RA, Wiegand HL, Moore MD, Schafer A, McClure MO, Cullen BR. Foamy virus Bet proteins function as novel inhibitors of the APOBEC3 family of innate antiretroviral defense factors. J Virol. 2005;79:8724–31. doi: 10.1128/JVI.79.14.8724-8731.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Arias JF, Koyama T, Kinomoto M, Tokunaga K. Retroelements versus APOBEC3 family members: No great escape from the magnificent seven. Front Microbiol. 2012;3:275. doi: 10.3389/fmicb.2012.00275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Turelli P, Mangeat B, Jost S, Vianin S, Trono D. Inhibition of hepatitis B virus replication by APOBEC3G. Science. 2004;303:1829. doi: 10.1126/science.1092066. [DOI] [PubMed] [Google Scholar]
- 38.Vartanian JP, Guetard D, Henry M, Wain-Hobson S. Evidence for editing of human papillomavirus DNA by APOBEC3 in benign and precancerous lesions. Science. 2008;320:230–3. doi: 10.1126/science.1153201. [DOI] [PubMed] [Google Scholar]
- 39.Chen KM, Harjes E, Gross PJ, Fahmy A, Lu Y, Shindo K, Harris RS, Matsuo H. Structure of the DNA deaminase domain of the HIV-1 restriction factor APOBEC3G. Nature. 2008;452:116–9. doi: 10.1038/nature06638. [DOI] [PubMed] [Google Scholar]
- 40.Holden LG, Prochnow C, Chang YP, Bransteitter R, Chelico L, Sen U, Stevens RC, Goodman MF, Chen XS. Crystal structure of the anti-viral APOBEC3G catalytic domain and functional implications. Nature. 2008;456:121–4. doi: 10.1038/nature07357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kitamura S, Ode H, Iwatani Y. Structural Features of Antiviral APOBEC3 Proteins are Linked to Their Functional Activities. Front Microbiol. 2011;2:258. doi: 10.3389/fmicb.2011.00258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Prochnow C, Bransteitter R, Klein MG, Goodman MF, Chen XS. The APOBEC-2 crystal structure and functional implications for the deaminase AID. Nature. 2007;445:447–51. doi: 10.1038/nature05492. [DOI] [PubMed] [Google Scholar]
- 43.Byeon IJ, Ahn J, Mitra M, Byeon CH, Hercik K, Hritz J, Charlton LM, Levin JG, Gronenborn AM. NMR structure of human restriction factor APOBEC3A reveals substrate binding and enzyme specificity. Nat Commun. 2013;4:1890. doi: 10.1038/ncomms2883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shandilya SM, Nalam MN, Nalivaika EA, Gross PJ, Valesano JC, Shindo K, Li M, Munson M, Royer WE, Harjes E, Kono T, Matsuo H, Harris RS, Somasundaran M, Schiffer CA. Crystal structure of the APOBEC3G catalytic domain reveals potential oligomerization interfaces. Structure. 2010;18:28–38. doi: 10.1016/j.str.2009.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bohn MF, Shandilya SM, Albin JS, Kouno T, Anderson BD, McDougle RM, Carpenter MA, Rathore A, Evans L, Davis AN, Zhang J, Lu Y, Somasundaran M, Matsuo H, Harris RS, Schiffer CA. Crystal Structure of the DNA Cytosine Deaminase APOBEC3F: The Catalytically Active and HIV-1 Vif-Binding Domain. Structure. 2013;21:1042–50. doi: 10.1016/j.str.2013.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Harjes E, Gross PJ, Chen KM, Lu Y, Shindo K, Nowarski R, Gross JD, Kotler M, Harris RS, Matsuo H. An extended structure of the APOBEC3G catalytic domain suggests a unique holoenzyme model. J Mol Biol. 2009;389:819–32. doi: 10.1016/j.jmb.2009.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Krzysiak TC, Jung J, Thompson J, Baker D, Gronenborn AM. APOBEC2 is a monomer in solution: implications for APOBEC3G models. Biochemistry. 2012;51:2008–17. doi: 10.1021/bi300021s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Furukawa A, Nagata T, Matsugami A, Habu Y, Sugiyama R, Hayashi F, Kobayashi N, Yokoyama S, Takaku H, Katahira M. Structure, interaction and real-time monitoring of the enzymatic reaction of wild-type APOBEC3G. EMBO J. 2009;28:440–51. doi: 10.1038/emboj.2008.290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kitamura S, Ode H, Nakashima M, Imahashi M, Naganawa Y, Kurosawa T, Yokomaku Y, Yamane T, Watanabe N, Suzuki A, Sugiura W, Iwatani Y. The APOBEC3C crystal structure and the interface for HIV-1 Vif binding. Nat Struct Mol Biol. 2012;19:1005–10. doi: 10.1038/nsmb.2378. [DOI] [PubMed] [Google Scholar]
- 50.Xie K, Sowden MP, Dance GS, Torelli AT, Smith HC, Wedekind JE. The structure of a yeast RNA-editing deaminase provides insight into the fold and function of activation-induced deaminase and APOBEC-1. Proc Natl Acad Sci U S A. 2004;101:8114–9. doi: 10.1073/pnas.0400493101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Smith JL, Pathak VK. Identification of specific determinants of human APOBEC3F, APOBEC3C, and APOBEC3DE and African green monkey APOBEC3F that interact with HIV-1 Vif. J Virol. 2010;84:12599–608. doi: 10.1128/JVI.01437-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Albin JS, LaRue RS, Weaver JA, Brown WL, Shindo K, Harjes E, Matsuo H, Harris RS. A single amino acid in human APOBEC3F alters susceptibility to HIV-1 Vif. J Biol Chem. 2010;285:40785–92. doi: 10.1074/jbc.M110.173161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bransteitter R, Prochnow C, Chen XS. The current structural and functional understanding of APOBEC deaminases. Cell Mol Life Sci. 2009;66:3137–47. doi: 10.1007/s00018-009-0070-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Senavirathne G, Jaszczur M, Auerbach PA, Upton TG, Chelico L, Goodman MF, Rueda D. Single-stranded DNA scanning and deamination by APOBEC3G cytidine deaminase at single molecule resolution. J Biol Chem. 2012;287:15826–35. doi: 10.1074/jbc.M112.342790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Losey HC, Ruthenburg AJ, Verdine GL. Crystal structure of Staphylococcus aureus tRNA adenosine deaminase TadA in complex with RNA. Nat Struct Mol Biol. 2006;13:153–9. doi: 10.1038/nsmb1047. [DOI] [PubMed] [Google Scholar]
- 56.Chelico L, Pham P, Calabrese P, Goodman MF. APOBEC3G DNA deaminase acts processively 3′ --> 5′ on single-stranded DNA. Nat Struct Mol Biol. 2006;13:392–9. doi: 10.1038/nsmb1086. [DOI] [PubMed] [Google Scholar]
- 57.Kohli RM, Abrams SR, Gajula KS, Maul RW, Gearhart PJ, Stivers JT. A portable hot spot recognition loop transfers sequence preferences from APOBEC family members to activation-induced cytidine deaminase. J Biol Chem. 2009;284:22898–904. doi: 10.1074/jbc.M109.025536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Harjes S, Solomon WC, Li M, Chen KM, Harjes E, Harris RS, Matsuo H. Impact of H216 on the DNA binding and catalytic activities of the HIV restriction factor APOBEC3G. J Virol. 2013;87:7008–14. doi: 10.1128/JVI.03173-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rathore A, Carpenter MA, Demir O, Ikeda T, Li M, Shaban NM, Law EK, Anokhin D, Brown WL, Amaro RE, Harris RS. The Local Dinucleotide Preference of APOBEC3G Can Be Altered from 5′-CC to 5′-TC by a Single Amino Acid Substitution. J Mol Biol. 2013 Aug 11; doi: 10.1016/j.jmb.2013.07.040. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Langlois MA, Beale RC, Conticello SG, Neuberger MS. Mutational comparison of the single-domained APOBEC3C and double-domained APOBEC3F/G anti-retroviral cytidine deaminases provides insight into their DNA target site specificities. Nucleic Acids Res. 2005;33:1913–23. doi: 10.1093/nar/gki343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wang M, Rada C, Neuberger MS. Altering the spectrum of immunoglobulin V gene somatic hypermutation by modifying the active site of AID. J Exp Med. 2010;207:141–53. doi: 10.1084/jem.20092238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Betts L, Xiang S, Short SA, Wolfenden R, Carter CW., Jr Cytidine deaminase. The 2.3 A crystal structure of an enzyme: transition-state analog complex. J Mol Biol. 1994;235:635–56. doi: 10.1006/jmbi.1994.1018. [DOI] [PubMed] [Google Scholar]
- 63.Zhang L, Saadatmand J, Li X, Guo F, Niu M, Jiang J, Kleiman L, Cen S. Function analysis of sequences in human APOBEC3G involved in Vif-mediated degradation. Virology. 2008;370:113–21. doi: 10.1016/j.virol.2007.08.027. [DOI] [PubMed] [Google Scholar]
- 64.Zhang W, Chen G, Niewiadomska AM, Xu R, Yu XF. Distinct determinants in HIV-1 Vif and human APOBEC3 proteins are required for the suppression of diverse host anti-viral proteins. PLoS One. 2008;3:e3963. doi: 10.1371/journal.pone.0003963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Huthoff H, Malim MH. Identification of amino acid residues in APOBEC3G required for regulation by human immunodeficiency virus type 1 Vif and Virion encapsidation. J Virol. 2007;81:3807–15. doi: 10.1128/JVI.02795-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Russell RA, Smith J, Barr R, Bhattacharyya D, Pathak VK. Distinct domains within APOBEC3G and APOBEC3F interact with separate regions of human immunodeficiency virus type 1 Vif. J Virol. 2009;83:1992–2003. doi: 10.1128/JVI.01621-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Russell RA, Pathak VK. Identification of two distinct human immunodeficiency virus type 1 Vif determinants critical for interactions with human APOBEC3G and APOBEC3F. J Virol. 2007;81:8201–10. doi: 10.1128/JVI.00395-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhang KL, Mangeat B, Ortiz M, Zoete V, Trono D, Telenti A, Michielin O. Model structure of human APOBEC3G. PLoS One. 2007;2:e378. doi: 10.1371/journal.pone.0000378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Xu H, Svarovskaia ES, Barr R, Zhang Y, Khan MA, Strebel K, Pathak VK. A single amino acid substitution in human APOBEC3G antiretroviral enzyme confers resistance to HIV-1 virion infectivity factor-induced depletion. Proc Natl Acad Sci U S A. 2004;101:5652–7. doi: 10.1073/pnas.0400830101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Schrofelbauer B, Chen D, Landau NR. A single amino acid of APOBEC3G controls its species-specific interaction with virion infectivity factor (Vif) Proc Natl Acad Sci U S A. 2004;101:3927–32. doi: 10.1073/pnas.0307132101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mangeat B, Turelli P, Liao S, Trono D. A single amino acid determinant governs the species-specific sensitivity of APOBEC3G to Vif action. J Biol Chem. 2004;279:14481–3. doi: 10.1074/jbc.C400060200. [DOI] [PubMed] [Google Scholar]
- 72.Bogerd HP, Doehle BP, Wiegand HL, Cullen BR. A single amino acid difference in the host APOBEC3G protein controls the primate species specificity of HIV type 1 virion infectivity factor. Proc Natl Acad Sci U S A. 2004;101:3770–4. doi: 10.1073/pnas.0307713101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Dang Y, Siew LM, Wang X, Han Y, Lampen R, Zheng YH. Human cytidine deaminase APOBEC3H restricts HIV-1 replication. J Biol Chem. 2008;283:11606–14. doi: 10.1074/jbc.M707586200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Li MM, Wu LI, Emerman M. The range of human APOBEC3H sensitivity to lentiviral Vif proteins. J Virol. 2010;84:88–95. doi: 10.1128/JVI.01344-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhen A, Wang T, Zhao K, Xiong Y, Yu XF. A single amino acid difference in human APOBEC3H variants determines HIV-1 Vif sensitivity. J Virol. 2010;84:1902–11. doi: 10.1128/JVI.01509-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Marcsisin SR, Narute PS, Emert-Sedlak LA, Kloczewiak M, Smithgall TE, Engen JR. On the solution conformation and dynamics of the HIV-1 viral infectivity factor. J Mol Biol. 2011;410:1008–22. doi: 10.1016/j.jmb.2011.04.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bergeron JR, Huthoff H, Veselkov DA, Beavil RL, Simpson PJ, Matthews SJ, Malim MH, Sanderson MR. The SOCS-box of HIV-1 Vif interacts with ElonginBC by induced-folding to recruit its Cul5-containing ubiquitin ligase complex. PLoS Pathog. 2010;6:e1000925. doi: 10.1371/journal.ppat.1000925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Luo K, Xiao Z, Ehrlich E, Yu Y, Liu B, Zheng S, Yu XF. Primate lentiviral virion infectivity factors are substrate receptors that assemble with cullin 5-E3 ligase through a HCCH motif to suppress APOBEC3G. Proc Natl Acad Sci U S A. 2005;102:11444–9. doi: 10.1073/pnas.0502440102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Mehle A, Goncalves J, Santa-Marta M, McPike M, Gabuzda D. Phosphorylation of a novel SOCS-box regulates assembly of the HIV-1 Vif-Cul5 complex that promotes APOBEC3G degradation. Genes Dev. 2004;18:2861–6. doi: 10.1101/gad.1249904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Mehle A, Thomas ER, Rajendran KS, Gabuzda D. A zinc-binding region in Vif binds Cul5 and determines cullin selection. J Biol Chem. 2006;281:17259–65. doi: 10.1074/jbc.M602413200. [DOI] [PubMed] [Google Scholar]
- 81.Yu Y, Xiao Z, Ehrlich ES, Yu X, Yu XF. Selective assembly of HIV-1 Vif-Cul5-ElonginB-ElonginC E3 ubiquitin ligase complex through a novel SOCS box and upstream cysteines. Genes Dev. 2004;18:2867–72. doi: 10.1101/gad.1250204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Yamashita T, Kamada K, Hatcho K, Adachi A, Nomaguchi M. Identification of amino acid residues in HIV-1 Vif critical for binding and exclusion of APOBEC3G/F. Microbes Infect. 2008;10:1142–9. doi: 10.1016/j.micinf.2008.06.003. [DOI] [PubMed] [Google Scholar]
- 83.Schrofelbauer B, Senger T, Manning G, Landau NR. Mutational alteration of human immunodeficiency virus type 1 Vif allows for functional interaction with nonhuman primate APOBEC3G. J Virol. 2006;80:5984–91. doi: 10.1128/JVI.00388-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ooms M, Letko M, Binka M, Simon V. The resistance of human APOBEC3H to HIV-1 NL4-3 molecular clone is determined by a single amino acid in Vif. PLoS One. 2013;8:e57744. doi: 10.1371/journal.pone.0057744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.He Z, Zhang W, Chen G, Xu R, Yu XF. Characterization of conserved motifs in HIV-1 Vif required for APOBEC3G and APOBEC3F interaction. J Mol Biol. 2008;381:1000–11. doi: 10.1016/j.jmb.2008.06.061. [DOI] [PubMed] [Google Scholar]
- 86.Pery E, Rajendran KS, Brazier AJ, Gabuzda D. Regulation of APOBEC3 proteins by a novel YXXL motif in human immunodeficiency virus type 1 Vif and simian immunodeficiency virus SIVagm Vif. J Virol. 2009;83:2374–81. doi: 10.1128/JVI.01898-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Santa-Marta M, da Silva FA, Fonseca AM, Goncalves J. HIV-1 Vif can directly inhibit apolipoprotein B mRNA-editing enzyme catalytic polypeptide-like 3G-mediated cytidine deamination by using a single amino acid interaction and without protein degradation. J Biol Chem. 2005;280:8765–75. doi: 10.1074/jbc.M409309200. [DOI] [PubMed] [Google Scholar]
- 88.Tian C, Yu X, Zhang W, Wang T, Xu R, Yu XF. Differential requirement for conserved tryptophans in human immunodeficiency virus type 1 Vif for the selective suppression of APOBEC3G and APOBEC3F. J Virol. 2006;80:3112–5. doi: 10.1128/JVI.80.6.3112-3115.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Simon V, Zennou V, Murray D, Huang Y, Ho DD, Bieniasz PD. Natural variation in Vif: differential impact on APOBEC3G/3F and a potential role in HIV-1 diversification. PLoS Pathog. 2005;1:e6. doi: 10.1371/journal.ppat.0010006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Chen G, He Z, Wang T, Xu R, Yu XF. A patch of positively charged amino acids surrounding the human immunodeficiency virus type 1 Vif SLVx4Yx9Y motif influences its interaction with APOBEC3G. J Virol. 2009;83:8674–82. doi: 10.1128/JVI.00653-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Dang Y, Wang X, Zhou T, York IA, Zheng YH. Identification of a novel WxSLVK motif in the N terminus of human immunodeficiency virus and simian immunodeficiency virus Vif that is critical for APOBEC3G and APOBEC3F neutralization. J Virol. 2009;83:8544–52. doi: 10.1128/JVI.00651-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Dang Y, Davis RW, York IA, Zheng YH. Identification of 81LGxGxxIxW89 and 171EDRW174 domains from human immunodeficiency virus type 1 Vif that regulate APOBEC3G and APOBEC3F neutralizing activity. J Virol. 2010;84:5741–50. doi: 10.1128/JVI.00079-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Xu H, Chertova E, Chen J, Ott DE, Roser JD, Hu WS, Pathak VK. Stoichiometry of the antiviral protein APOBEC3G in HIV-1 virions. Virology. 2007;360:247–56. doi: 10.1016/j.virol.2006.10.036. [DOI] [PubMed] [Google Scholar]
- 94.Svarovskaia ES, Xu H, Mbisa JL, Barr R, Gorelick RJ, Ono A, Freed EO, Hu WS, Pathak VK. Human apolipoprotein B mRNA-editing enzymecatalytic polypeptide-like 3G (APOBEC3G) is incorporated into HIV-1 virions through interactions with viral and nonviral RNAs. J Biol Chem. 2004;279:35822–8. doi: 10.1074/jbc.M405761200. [DOI] [PubMed] [Google Scholar]
- 95.Doehle BP, Schafer A, Wiegand HL, Bogerd HP, Cullen BR. Differential sensitivity of murine leukemia virus to APOBEC3-mediated inhibition is governed by virion exclusion. J Virol. 2005;79:8201–7. doi: 10.1128/JVI.79.13.8201-8207.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Luo K, Liu B, Xiao Z, Yu Y, Yu X, Gorelick R, Yu XF. Aminoterminal region of the human immunodeficiency virus type 1 nucleocapsid is required for human APOBEC3G packaging. J Virol. 2004;78:11841–52. doi: 10.1128/JVI.78.21.11841-11852.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Zennou V, Perez-Caballero D, Gottlinger H, Bieniasz PD. APOBEC3G incorporation into human immunodeficiency virus type 1 particles. J Virol. 2004;78:12058–61. doi: 10.1128/JVI.78.21.12058-12061.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Khan MA, Kao S, Miyagi E, Takeuchi H, Goila-Gaur R, Opi S, Gipson CL, Parslow TG, Ly H, Strebel K. Viral RNA is required for the association of APOBEC3G with human immunodeficiency virus type 1 nucleoprotein complexes. J Virol. 2005;79:5870–4. doi: 10.1128/JVI.79.9.5870-5874.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Alce TM, Popik W. APOBEC3G is incorporated into virus-like particles by a direct interaction with HIV-1 Gag nucleocapsid protein. J Biol Chem. 2004;279:34083–6. doi: 10.1074/jbc.C400235200. [DOI] [PubMed] [Google Scholar]
- 100.Schafer A, Bogerd HP, Cullen BR. Specific packaging of APOBEC3G into HIV-1 virions is mediated by the nucleocapsid domain of the gag polyprotein precursor. Virology. 2004;328:163–8. doi: 10.1016/j.virol.2004.08.006. [DOI] [PubMed] [Google Scholar]
- 101.Bulliard Y, Turelli P, Rohrig UF, Zoete V, Mangeat B, Michielin O, Trono D. Functional analysis and structural modeling of human APOBEC3G reveal the role of evolutionarily conserved elements in the inhibition of human immunodeficiency virus type 1 infection and Alu transposition. J Virol. 2009;83:12611–21. doi: 10.1128/JVI.01491-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Belanger K, Savoie M, Rosales Gerpe MC, Couture JF, Langlois MA. Binding of RNA by APOBEC3G controls deamination-independent restriction of retroviruses. Nucleic Acids Res. 2013;41:7438–52. doi: 10.1093/nar/gkt527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Navarro F, Bollman B, Chen H, Konig R, Yu Q, Chiles K, Landau NR. Complementary function of the two catalytic domains of APOBEC3G. Virology. 2005;333:374–86. doi: 10.1016/j.virol.2005.01.011. [DOI] [PubMed] [Google Scholar]
- 104.Iwatani Y, Takeuchi H, Strebel K, Levin JG. Biochemical activities of highly purified, catalytically active human APOBEC3G: correlation with antiviral effect. J Virol. 2006;80:5992–6002. doi: 10.1128/JVI.02680-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Chiu YL, Soros VB, Kreisberg JF, Stopak K, Yonemoto W, Greene WC. Cellular APOBEC3G restricts HIV-1 infection in resting CD4+ T cells. Nature. 2005;435:108–14. doi: 10.1038/nature03493. [DOI] [PubMed] [Google Scholar]
- 106.Chiu YL, Witkowska HE, Hall SC, Santiago M, Soros VB, Esnault C, Heidmann T, Greene WC. High-molecular-mass APOBEC3G complexes restrict Alu retrotransposition. Proc Natl Acad Sci U S A. 2006;103:15588–93. doi: 10.1073/pnas.0604524103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Wang T, Tian C, Zhang W, Sarkis PT, Yu XF. Interaction with 7SL RNA but not with HIV-1 genomic RNA or P bodies is required for APOBEC3F virion packaging. J Mol Biol. 2008;375:1098–112. doi: 10.1016/j.jmb.2007.11.017. [DOI] [PubMed] [Google Scholar]
- 108.Wang T, Tian C, Zhang W, Luo K, Sarkis PT, Yu L, Liu B, Yu Y, Yu XF. 7SL RNA mediates virion packaging of the antiviral cytidine deaminase APOBEC3G. J Virol. 2007;81:13112–24. doi: 10.1128/JVI.00892-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Bach D, Peddi S, Mangeat B, Lakkaraju A, Strub K, Trono D. Characterization of APOBEC3G binding to 7SL RNA. Retrovirology. 2008;5:54. doi: 10.1186/1742-4690-5-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Izumi T, Burdick R, Shigemi M, Plisov S, Hu WS, Pathak VK. Mov10 and APOBEC3G Localization to Processing Bodies is not Required for Virion Incorporation and Antiviral Activity. J Virol. 2013 Aug 7; doi: 10.1128/JVI.02070-13. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Jager S, Kim DY, Hultquist JF, Shindo K, LaRue RS, Kwon E, Li M, Anderson BD, Yen L, Stanley D, Mahon C, Kane J, Franks-Skiba K, Cimermancic P, Burlingame A, Sali A, Craik CS, Harris RS, Gross JD, Krogan NJ. Vif hijacks CBF-beta to degrade APOBEC3G and promote HIV-1 infection. Nature. 2012;481:371–5. doi: 10.1038/nature10693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Zhang W, Du J, Evans SL, Yu Y, Yu XF. T-cell differentiation factor CBF-beta regulates HIV-1 Vif-mediated evasion of host restriction. Nature. 2012;481:376–9. doi: 10.1038/nature10718. [DOI] [PubMed] [Google Scholar]
- 113.Mercenne G, Bernacchi S, Richer D, Bec G, Henriet S, Paillart JC, Marquet R. HIV-1 Vif binds to APOBEC3G mRNA and inhibits its translation. Nucleic Acids Res. 2010;38:633–46. doi: 10.1093/nar/gkp1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Kao S, Khan MA, Miyagi E, Plishka R, Buckler-White A, Strebel K. The human immunodeficiency virus type 1 Vif protein reduces intracellular expression and inhibits packaging of APOBEC3G (CEM15), a cellular inhibitor of virus infectivity. J Virol. 2003;77:11398–407. doi: 10.1128/JVI.77.21.11398-11407.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Feng Y, Love RP, Chelico L. HIV-1 viral infectivity factor (Vif) alters processive single-stranded DNA scanning of the retroviral restriction factor APOBEC3G. J Biol Chem. 2013;288:6083–94. doi: 10.1074/jbc.M112.421875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Wong WF, Kohu K, Chiba T, Sato T, Satake M. Interplay of transcription factors in T-cell differentiation and function: the role of Runx. Immunology. 2011;132:157–64. doi: 10.1111/j.1365-2567.2010.03381.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Du J, Zhao K, Rui Y, Li P, Zhou X, Zhang W, Yu XF. Differential requirements for HIV-1 Vif-mediated APOBEC3G degradation and RUNX1-mediated transcription by core binding factor beta. J Virol. 2013;87:1906–11. doi: 10.1128/JVI.02199-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Hultquist JF, McDougle RM, Anderson BD, Harris RS. HIV type 1 viral infectivity factor and the RUNX transcription factors interact with core binding factor beta on genetically distinct surfaces. AIDS Res Hum Retroviruses. 2012;28:1543–51. doi: 10.1089/aid.2012.0142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Kim DY, Kwon E, Hartley PD, Crosby DC, Mann S, Krogan NJ, Gross JD. CBFbeta stabilizes HIV Vif to counteract APOBEC3 at the expense of RUNX1 target gene expression. Mol Cell. 2013;49:632–44. doi: 10.1016/j.molcel.2012.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Salter JD, Lippa GM, Belashov IA, Wedekind JE. Core-binding factor beta increases the affinity between human Cullin 5 and HIV-1 Vif within an E3 ligase complex. Biochemistry. 2012;51:8702–4. doi: 10.1021/bi301244z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Evans SL, Han X, Liu Y, Yu XF. Characterization of the Interaction of Full-Length HIV-1 Vif Protein with its Key Regulator CBFβ and CRL5 E3 Ubiquitin Ligase Components. PLoS ONE. 2012;7:e33495. doi: 10.1371/journal.pone.0033495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Hori T, Osaka F, Chiba T, Miyamoto C, Okabayashi K, Shimbara N, Kato S, Tanaka K. Covalent modification of all members of human cullin family proteins by NEDD8. Oncogene. 1999;18:6829–34. doi: 10.1038/sj.onc.1203093. [DOI] [PubMed] [Google Scholar]
- 123.Petroski MD, Deshaies RJ. Function and regulation of cullin-RING ubiquitin ligases. Nat Rev Mol Cell Biol. 2005;6:9–20. doi: 10.1038/nrm1547. [DOI] [PubMed] [Google Scholar]
- 124.Soucy TA, Smith PG, Milhollen MA, Berger AJ, Gavin JM, Adhikari S, Brownell JE, Burke KE, Cardin DP, Critchley S, Cullis CA, Doucette A, Garnsey JJ, Gaulin JL, Gershman RE, Lublinsky AR, McDonald A, Mizutani H, Narayanan U, Olhava EJ, Peluso S, Rezaei M, Sintchak MD, Talreja T, Thomas MP, Traore T, Vyskocil S, Weatherhead GS, Yu J, Zhang J, Dick LR, Claiborne CF, Rolfe M, Bolen JB, Langston SP. An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature. 2009;458:732–6. doi: 10.1038/nature07884. [DOI] [PubMed] [Google Scholar]
- 125.Stanley DJ, Bartholomeeusen K, Crosby DC, Kim DY, Kwon E, Yen L, Cartozo NC, Li M, Jager S, Mason-Herr J, Hayashi F, Yokoyama S, Krogan NJ, Harris RS, Peterlin BM, Gross JD. Inhibition of a NEDD8 Cascade Restores Restriction of HIV by APOBEC3G. PLoS Pathog. 2012;8:e1003085. doi: 10.1371/journal.ppat.1003085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Pitini V, Arrigo C, Altavilla G. How Cells Respond to Interferons. J Clin Oncol. 2010;28:e439. doi: 10.1200/JCO.2010.28.9603. [DOI] [PubMed] [Google Scholar]
- 127.Meylan PRA, Guatelli JC, Munis JR, Richman DD, Kornbluth RS. Mechanisms for the Inhibition of HIV Replication by Interferons-α -β and -γ in Primary Human Macrophages. Virology. 1993;193:138–148. doi: 10.1006/viro.1993.1110. [DOI] [PubMed] [Google Scholar]
- 128.Goujon C, Malim MH. Characterization of the Alpha Interferon-Induced Postentry Block to HIV-1 Infection in Primary Human Macrophages and T Cells. J Virol. 2010;84:9254–9266. doi: 10.1128/JVI.00854-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Cheney KM, McKnight Á. Interferon-Alpha Mediates Restriction of Human Immunodeficiency Virus Type-1 Replication in Primary Human Macrophages at an Early Stage of Replication. PLoS ONE. 2010;5:e13521. doi: 10.1371/journal.pone.0013521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Cobos Jimenez V, Booiman T, de Taeye SW, van Dort KA, Rits MA, Hamann J, Kootstra NA. Differential expression of HIV-1 interfering factors in monocyte-derived macrophages stimulated with polarizing cytokines or interferons. Sci Rep. 2012;2:763. doi: 10.1038/srep00763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Mohanram V, Skold AE, Bachle SM, Pathak SK, Spetz AL. IFN-alpha induces APOBEC3G, F, and A in immature dendritic cells and limits HIV-1 spread to CD4+ T cells. J Immunol. 2013;190:3346–53. doi: 10.4049/jimmunol.1201184. [DOI] [PubMed] [Google Scholar]
- 132.Chen H, Wang LW, Huang YQ, Gong ZJ. Interferon-alpha Induces High Expression of APOBEC3G and STAT-1 in Vitro and in Vivo. Int J Mol Sci. 2010;11:3501–12. doi: 10.3390/ijms11093501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Farrow MA, Kim EY, Wolinsky SM, Sheehy AM. NFAT and IRF Proteins Regulate Transcription of the Anti-HIV Gene, APOBEC3G. J Biol Chem. 2011;286:2567–2577. doi: 10.1074/jbc.M110.154377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Souza TML, Rodrigues DQ, Passaes CPB, Barreto-de-Souza V, Aguiar RS, Temerozo JR, Morgado MG, Fontes CFL, Araujo EG, Bou-Habib DC. The nerve growth factor reduces APOBEC3G synthesis and enhances HIV-1 transcription and replication in human primary macrophages. Blood. 2011;117:2944–2952. doi: 10.1182/blood-2010-05-287193. [DOI] [PubMed] [Google Scholar]
- 135.Ankel H, Mittnacht S, Jacobsen H. Antiviral activity of prostaglandin A on encephalomyocarditis virus-infected cells: a unique effect unrelated to interferon. J Gen Virol. 1985;66(Pt 11):2355–64. doi: 10.1099/0022-1317-66-11-2355. [DOI] [PubMed] [Google Scholar]
- 136.Rossi A, Elia G, Santoro MG. Inhibition of nuclear factor κB by prostaglandin A1: An effect associated with heat shock transcription factor activation. Proc Natl Acad Sci U S A. 1997;94:746–750. doi: 10.1073/pnas.94.2.746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Hayes MM, Lane BR, King SR, Markovitz DM, Coffey MJ. Peroxisome proliferator-activated receptor gamma agonists inhibit HIV-1 replication in macrophages by transcriptional and post-transcriptional effects. J Biol Chem. 2002;277:16913–9. doi: 10.1074/jbc.M200875200. [DOI] [PubMed] [Google Scholar]
- 138.Sugiyama R, Nishitsuji H, Furukawa A, Katahira M, Habu Y, Takeuchi H, Ryo A, Takaku H. Heat shock protein 70 inhibits HIV-1 Vif-mediated ubiquitination and degradation of APOBEC3G. J Biol Chem. 2011;286:10051–7. doi: 10.1074/jbc.M110.166108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Sugiyama R, Abe M, Nishitsuji H, Murakami Y, Takeuchi H, Takaku H. Induction of heat-shock protein 70 by prostaglandin A inhibits HIV-1 Vif-mediated degradation of APOBEC3G. Antiviral Res. 2013;99:307–311. doi: 10.1016/j.antiviral.2013.06.017. [DOI] [PubMed] [Google Scholar]
- 140.Basu U, Chaudhuri J, Alpert C, Dutt S, Ranganath S, Li G, Schrum JP, Manis JP, Alt FW. The AID antibody diversification enzyme is regulated by protein kinase A phosphorylation. Nature. 2005;438:508–11. doi: 10.1038/nature04255. [DOI] [PubMed] [Google Scholar]
- 141.Pasqualucci L, Kitaura Y, Gu H, Dalla-Favera R. PKA-mediated phosphorylation regulates the function of activation-induced deaminase (AID) in B cells. Proc Natl Acad Sci U S A. 2006;103:395–400. doi: 10.1073/pnas.0509969103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Demorest ZL, Li M, Harris RS. Phosphorylation directly regulates the intrinsic DNA cytidine deaminase activity of activation-induced deaminase and APOBEC3G protein. J Biol Chem. 2011;286:26568–75. doi: 10.1074/jbc.M111.235721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Shirakawa K, Takaori-Kondo A, Yokoyama M, Izumi T, Matsui M, Io K, Sato T, Sato H, Uchiyama T. Phosphorylation of APOBEC3G by protein kinase A regulates its interaction with HIV-1 Vif. Nat Struct Mol Biol. 2008;15:1184–91. doi: 10.1038/nsmb.1497. [DOI] [PubMed] [Google Scholar]
- 144.Kopietz F, Jaguva Vasudevan AA, Kramer M, Muckenfuss H, Sanzenbacher R, Cichutek K, Flory E, Munk C. Interaction of human immunodeficiency virus type 1 Vif with APOBEC3G is not dependent on serine/threonine phosphorylation status. J Gen Virol. 2012;93:2425–30. doi: 10.1099/vir.0.043273-0. [DOI] [PubMed] [Google Scholar]
- 145.Tan J, Wang X, Devadas K, Zhao J, Zhang P, Hewlett I. Some mechanisms of FLIP expression in inhibition of HIV-1 replication in Jurkat cells, CD4+ T cells and PBMCs. J Cell Physiol. 2013 May 21; doi: 10.1002/jcp.24397. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 146.Oguariri RM, Dai L, Adelsberger JW, Rupert A, Stevens R, Yang J, Huang D, Lempicki RA, Zhou M, Baseler MW, Lane HC, Imamichi T. Interleukin-2 inhibits HIV-1 replication in some human T cell lymphotrophic virus-1-infected cell lines via the induction and incorporation of APOBEC3G into the virion. J Biol Chem. 2013;288:17812–22. doi: 10.1074/jbc.M113.468975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Gillick K, Pollpeter D, Phalora P, Kim EY, Wolinsky SM, Malim MH. Suppression of HIV-1 infection by APOBEC3 proteins in primary human CD4(+) T cells is associated with inhibition of processive reverse transcription as well as excessive cytidine deamination. J Virol. 2013;87:1508–17. doi: 10.1128/JVI.02587-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Bishop KN, Holmes RK, Sheehy AM, Davidson NO, Cho SJ, Malim MH. Cytidine deamination of retroviral DNA by diverse APOBEC proteins. Curr Biol. 2004;14:1392–6. doi: 10.1016/j.cub.2004.06.057. [DOI] [PubMed] [Google Scholar]
- 149.Zennou V, Bieniasz PD. Comparative analysis of the antiretroviral activity of APOBEC3G and APOBEC3F from primates. Virology. 2006;349:31–40. doi: 10.1016/j.virol.2005.12.035. [DOI] [PubMed] [Google Scholar]
- 150.Chaipan C, Smith JL, Hu WS, Pathak VK. APOBEC3G restricts HIV-1 to a greater extent than APOBEC3F and APOBEC3DE in human primary CD4+ T cells and macrophages. J Virol. 2013;87:444–53. doi: 10.1128/JVI.00676-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Hultquist JF, Lengyel JA, Refsland EW, LaRue RS, Lackey L, Brown WL, Harris RS. Human and rhesus APOBEC3D, APOBEC3F, APOBEC3G, and APOBEC3H demonstrate a conserved capacity to restrict Vif-deficient HIV-1. J Virol. 2011;85:11220–34. doi: 10.1128/JVI.05238-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Dang Y, Wang X, Esselman WJ, Zheng YH. Identification of APOBEC3DE as another antiretroviral factor from the human APOBEC family. J Virol. 2006;80:10522–33. doi: 10.1128/JVI.01123-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Wang X, Abudu A, Son S, Dang Y, Venta PJ, Zheng YH. Analysis of human APOBEC3H haplotypes and anti-human immunodeficiency virus type 1 activity. J Virol. 2011;85:3142–52. doi: 10.1128/JVI.02049-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Doehle BP, Schafer A, Cullen BR. Human APOBEC3B is a potent inhibitor of HIV-1 infectivity and is resistant to HIV-1 Vif. Virology. 2005;339:281–8. doi: 10.1016/j.virol.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 155.Harari A, Ooms M, Mulder LC, Simon V. Polymorphisms and splice variants influence the antiretroviral activity of human APOBEC3H. J Virol. 2009;83:295–303. doi: 10.1128/JVI.01665-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Aguiar RS, Lovsin N, Tanuri A, Peterlin BM. Vpr.A3A chimera inhibits HIV replication. J Biol Chem. 2008;283:2518–25. doi: 10.1074/jbc.M706436200. [DOI] [PubMed] [Google Scholar]
- 157.Pathak VK, Temin HM. Broad spectrum of in vivo forward mutations, hypermutations, and mutational hotspots in a retroviral shuttle vector after a single replication cycle: substitutions, frameshifts, and hypermutations. Proc Natl Acad Sci U S A. 1990;87:6019–23. doi: 10.1073/pnas.87.16.6019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Suspene R, Sommer P, Henry M, Ferris S, Guetard D, Pochet S, Chester A, Navaratnam N, Wain-Hobson S, Vartanian JP. APOBEC3G is a single-stranded DNA cytidine deaminase and functions independently of HIV reverse transcriptase. Nucleic Acids Res. 2004;32:2421–9. doi: 10.1093/nar/gkh554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Berkhout B, de Ronde A. APOBEC3G versus reverse transcriptase in the generation of HIV-1 drug-resistance mutations. AIDS. 2004;18:1861–3. doi: 10.1097/00002030-200409030-00022. [DOI] [PubMed] [Google Scholar]
- 160.Zhang H, Yang B, Pomerantz RJ, Zhang C, Arunachalam SC, Gao L. The cytidine deaminase CEM15 induces hypermutation in newly synthesized HIV-1 DNA. Nature. 2003;424:94–8. doi: 10.1038/nature01707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Harris RS, Bishop KN, Sheehy AM, Craig HM, Petersen-Mahrt SK, Watt IN, Neuberger MS, Malim MH. DNA deamination mediates innate immunity to retroviral infection. Cell. 2003;113:803–9. doi: 10.1016/s0092-8674(03)00423-9. [DOI] [PubMed] [Google Scholar]
- 162.Yu Q, Konig R, Pillai S, Chiles K, Kearney M, Palmer S, Richman D, Coffin JM, Landau NR. Single-strand specificity of APOBEC3G accounts for minus-strand deamination of the HIV genome. Nat Struct Mol Biol. 2004;11:435–42. doi: 10.1038/nsmb758. [DOI] [PubMed] [Google Scholar]
- 163.Shindo K, Takaori-Kondo A, Kobayashi M, Abudu A, Fukunaga K, Uchiyama T. The enzymatic activity of CEM15/Apobec-3G is essential for the regulation of the infectivity of HIV-1 virion but not a sole determinant of its antiviral activity. J Biol Chem. 2003;278:44412–6. doi: 10.1074/jbc.C300376200. [DOI] [PubMed] [Google Scholar]
- 164.Yang Y, Guo F, Cen S, Kleiman L. Inhibition of initiation of reverse transcription in HIV-1 by human APOBEC3F. Virology. 2007;365:92–100. doi: 10.1016/j.virol.2007.03.022. [DOI] [PubMed] [Google Scholar]
- 165.Guo F, Cen S, Niu M, Yang Y, Gorelick RJ, Kleiman L. The interaction of APOBEC3G with human immunodeficiency virus type 1 nucleocapsid inhibits tRNA3Lys annealing to viral RNA. J Virol. 2007;81:11322–31. doi: 10.1128/JVI.00162-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Iwatani Y, Chan DS, Wang F, Maynard KS, Sugiura W, Gronenborn AM, Rouzina I, Williams MC, Musier-Forsyth K, Levin JG. Deaminase-independent inhibition of HIV-1 reverse transcription by APOBEC3G. Nucleic Acids Res. 2007;35:7096–108. doi: 10.1093/nar/gkm750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Bishop KN, Verma M, Kim EY, Wolinsky SM, Malim MH. APOBEC3G inhibits elongation of HIV-1 reverse transcripts. PLoS Pathog. 2008;4:e1000231. doi: 10.1371/journal.ppat.1000231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Anderson JL, Hope TJ. APOBEC3G restricts early HIV-1 replication in the cytoplasm of target cells. Virology. 2008;375:1–12. doi: 10.1016/j.virol.2008.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Li XY, Guo F, Zhang L, Kleiman L, Cen S. APOBEC3G inhibits DNA strand transfer during HIV-1 reverse transcription. J Biol Chem. 2007;282:32065–74. doi: 10.1074/jbc.M703423200. [DOI] [PubMed] [Google Scholar]
- 170.Holmes RK, Koning FA, Bishop KN, Malim MH. APOBEC3F can inhibit the accumulation of HIV-1 reverse transcription products in the absence of hypermutation. Comparisons with APOBEC3G. J Biol Chem. 2007;282:2587–95. doi: 10.1074/jbc.M607298200. [DOI] [PubMed] [Google Scholar]
- 171.Newman EN, Holmes RK, Craig HM, Klein KC, Lingappa JR, Malim MH, Sheehy AM. Antiviral function of APOBEC3G can be dissociated from cytidine deaminase activity. Curr Biol. 2005;15:166–70. doi: 10.1016/j.cub.2004.12.068. [DOI] [PubMed] [Google Scholar]
- 172.Mbisa JL, Barr R, Thomas JA, Vandegraaff N, Dorweiler IJ, Svarovskaia ES, Brown WL, Mansky LM, Gorelick RJ, Harris RS, Engelman A, Pathak VK. Human immunodeficiency virus type 1 cDNAs produced in the presence of APOBEC3G exhibit defects in plus-strand DNA transfer and integration. J Virol. 2007;81:7099–110. doi: 10.1128/JVI.00272-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Luo K, Wang T, Liu B, Tian C, Xiao Z, Kappes J, Yu XF. Cytidine deaminases APOBEC3G and APOBEC3F interact with human immunodeficiency virus type 1 integrase and inhibit proviral DNA formation. J Virol. 2007;81:7238–48. doi: 10.1128/JVI.02584-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Guo F, Cen S, Niu M, Saadatmand J, Kleiman L. Inhibition of formula-primed reverse transcription by human APOBEC3G during human immunodeficiency virus type 1 replication. J Virol. 2006;80:11710–22. doi: 10.1128/JVI.01038-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Mbisa JL, Bu W, Pathak VK. APOBEC3F and APOBEC3G inhibit HIV-1 DNA integration by different mechanisms. J Virol. 2010;84:5250–9. doi: 10.1128/JVI.02358-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Bishop KN, Holmes RK, Sheehy AM, Malim MH. APOBEC-mediated editing of viral RNA. Science. 2004;305:645. doi: 10.1126/science.1100658. [DOI] [PubMed] [Google Scholar]
- 177.Bishop KN, Holmes RK, Malim MH. Antiviral potency of APOBEC proteins does not correlate with cytidine deamination. J Virol. 2006;80:8450–8. doi: 10.1128/JVI.00839-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Hache G, Liddament MT, Harris RS. The retroviral hypermutation specificity of APOBEC3F and APOBEC3G is governed by the C-terminal DNA cytosine deaminase domain. J Biol Chem. 2005;280:10920–4. doi: 10.1074/jbc.M500382200. [DOI] [PubMed] [Google Scholar]
- 179.Holtz CM, Sadler HA, Mansky LM. APOBEC3G cytosine deamination hotspots are defined by both sequence context and single-stranded DNA secondary structure. Nucleic Acids Res. 2013;41:6139–48. doi: 10.1093/nar/gkt246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Beale RC, Petersen-Mahrt SK, Watt IN, Harris RS, Rada C, Neuberger MS. Comparison of the differential context-dependence of DNA deamination by APOBEC enzymes: correlation with mutation spectra in vivo. J Mol Biol. 2004;337:585–96. doi: 10.1016/j.jmb.2004.01.046. [DOI] [PubMed] [Google Scholar]
- 181.Liddament MT, Brown WL, Schumacher AJ, Harris RS. APOBEC3F properties and hypermutation preferences indicate activity against HIV-1 in vivo. Curr Biol. 2004;14:1385–91. doi: 10.1016/j.cub.2004.06.050. [DOI] [PubMed] [Google Scholar]
- 182.Wiegand HL, Doehle BP, Bogerd HP, Cullen BR. A second human antiretroviral factor, APOBEC3F, is suppressed by the HIV-1 and HIV-2 Vif proteins. EMBO J. 2004;23:2451–8. doi: 10.1038/sj.emboj.7600246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Chen H, Lilley CE, Yu Q, Lee DV, Chou J, Narvaiza I, Landau NR, Weitzman MD. APOBEC3A is a potent inhibitor of adeno-associated virus and retrotransposons. Curr Biol. 2006;16:480–5. doi: 10.1016/j.cub.2006.01.031. [DOI] [PubMed] [Google Scholar]
- 184.Pillai SK, Abdel-Mohsen M, Guatelli J, Skasko M, Monto A, Fujimoto K, Yukl S, Greene WC, Kovari H, Rauch A, Fellay J, Battegay M, Hirschel B, Witteck A, Bernasconi E, Ledergerber B, Gunthard HF, Wong JK. Role of retroviral restriction factors in the interferon-alpha-mediated suppression of HIV-1 in vivo. Proc Natl Acad Sci U S A. 2012;109:3035–40. doi: 10.1073/pnas.1111573109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Vartanian JP, Henry M, Wain-Hobson S. Sustained G-->A hypermutation during reverse transcription of an entire human immunodeficiency virus type 1 strain Vau group O genome. J Gen Virol. 2002;83:801–5. doi: 10.1099/0022-1317-83-4-801. [DOI] [PubMed] [Google Scholar]
- 186.Armitage AE, Katzourakis A, de Oliveira T, Welch JJ, Belshaw R, Bishop KN, Kramer B, McMichael AJ, Rambaut A, Iversen AK. Conserved footprints of APOBEC3G on Hypermutated human immunodeficiency virus type 1 and human endogenous retrovirus HERV-K(HML2) sequences. J Virol. 2008;82:8743–61. doi: 10.1128/JVI.00584-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Fitzgibbon JE, Mazar S, Dubin DT. A new type of G-->A hypermutation affecting human immunodeficiency virus. AIDS Res Hum Retroviruses. 1993;9:833–8. doi: 10.1089/aid.1993.9.833. [DOI] [PubMed] [Google Scholar]
- 188.Janini M, Rogers M, Birx DR, McCutchan FE. Human immunodeficiency virus type 1 DNA sequences genetically damaged by hypermutation are often abundant in patient peripheral blood mononuclear cells and may be generated during near-simultaneous infection and activation of CD4(+) T cells. J Virol. 2001;75:7973–86. doi: 10.1128/JVI.75.17.7973-7986.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.OhAinle M, Kerns JA, Malik HS, Emerman M. Adaptive evolution and antiviral activity of the conserved mammalian cytidine deaminase APOBEC3H. J Virol. 2006;80:3853–62. doi: 10.1128/JVI.80.8.3853-3862.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Suspene R, Rusniok C, Vartanian JP, Wain-Hobson S. Twin gradients in APOBEC3 edited HIV-1 DNA reflect the dynamics of lentiviral replication. Nucleic Acids Res. 2006;34:4677–84. doi: 10.1093/nar/gkl555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Chelico L, Prochnow C, Erie DA, Chen XS, Goodman MF. Structural model for deoxycytidine deamination mechanisms of the HIV-1 inactivation enzyme APOBEC3G. J Biol Chem. 2010;285:16195–205. doi: 10.1074/jbc.M110.107987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Berg OG, Winter RB, von Hippel PH. Diffusion-driven mechanisms of protein translocation on nucleic acids. 1. Models and theory. Biochemistry. 1981;20:6929–48. doi: 10.1021/bi00527a028. [DOI] [PubMed] [Google Scholar]
- 193.Feng Y, Chelico L. Intensity of deoxycytidine deamination of HIV-1 proviral DNA by the retroviral restriction factor APOBEC3G is mediated by the noncatalytic domain. J Biol Chem. 2011;286:11415–26. doi: 10.1074/jbc.M110.199604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Shlyakhtenko LS, Lushnikov AY, Miyagi A, Li M, Harris RS, Lyubchenko YL. Nanoscale structure and dynamics of ABOBEC3G complexes with single-stranded DNA. Biochemistry. 2012;51:6432–40. doi: 10.1021/bi300733d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Nowarski R, Britan-Rosich E, Shiloach T, Kotler M. Hypermutation by intersegmental transfer of APOBEC3G cytidine deaminase. Nat Struct Mol Biol. 2008;15:1059–66. doi: 10.1038/nsmb.1495. [DOI] [PubMed] [Google Scholar]
- 196.Chelico L, Pham P, Goodman MF. Mechanisms of APOBEC3G-catalyzed processive deamination of deoxycytidine on single-stranded DNA. Nat Struct Mol Biol. 2009;16:454–5. doi: 10.1038/nsmb0509-454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Nowarski R, Britan-Rosich E, Kotler M. Reply to Mechanisms of APOBEC3G-catalyzed processive deamination of deoxycytidine on single-stranded DNA. Nat Struct Mol Biol. 2009:455–456. doi: 10.1038/nsmb0509-454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Adolph MB, Webb J, Chelico L. Retroviral restriction factor APOBEC3G delays the initiation of DNA synthesis by HIV-1 reverse transcriptase. PLoS One. 2013;8:e64196. doi: 10.1371/journal.pone.0064196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Klarmann GJ, Chen X, North TW, Preston BD. Incorporation of uracil into minus strand DNA affects the specificity of plus strand synthesis initiation during lentiviral reverse transcription. J Biol Chem. 2003;278:7902–9. doi: 10.1074/jbc.M207223200. [DOI] [PubMed] [Google Scholar]
- 200.Kaiser SM, Emerman M. Uracil DNA glycosylase is dispensable for human immunodeficiency virus type 1 replication and does not contribute to the antiviral effects of the cytidine deaminase Apobec3G. J Virol. 2006;80:875–82. doi: 10.1128/JVI.80.2.875-882.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Langlois MA, Neuberger MS. Human APOBEC3G can restrict retroviral infection in avian cells and acts independently of both UNG and SMUG1. J Virol. 2008;82:4660–4. doi: 10.1128/JVI.02469-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Salter JD, Krucinska J, Raina J, Smith HC, Wedekind JE. A hydrodynamic analysis of APOBEC3G reveals a monomer-dimer-tetramer selfassociation that has implications for anti-HIV function. Biochemistry. 2009;48:10685–7. doi: 10.1021/bi901642c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Shlyakhtenko LS, Lushnikov AY, Li M, Lackey L, Harris RS, Lyubchenko YL. Atomic force microscopy studies provide direct evidence for dimerization of the HIV restriction factor APOBEC3G. J Biol Chem. 2011;286:3387–95. doi: 10.1074/jbc.M110.195685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Wedekind JE, Gillilan R, Janda A, Krucinska J, Salter JD, Bennett RP, Raina J, Smith HC. Nanostructures of APOBEC3G support a hierarchical assembly model of high molecular mass ribonucleoprotein particles from dimeric subunits. J Biol Chem. 2006;281:38122–6. doi: 10.1074/jbc.C600253200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Huthoff H, Autore F, Gallois-Montbrun S, Fraternali F, Malim MH. RNA-dependent oligomerization of APOBEC3G is required for restriction of HIV-1. PLoS Pathog. 2009;5:e1000330. doi: 10.1371/journal.ppat.1000330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Chelico L, Sacho EJ, Erie DA, Goodman MF. A model for oligomeric regulation of APOBEC3G cytosine deaminase-dependent restriction of HIV. J Biol Chem. 2008;283:13780–91. doi: 10.1074/jbc.M801004200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Friew YN, Boyko V, Hu WS, Pathak VK. Intracellular interactions between APOBEC3G, RNA, and HIV-1 Gag: APOBEC3G multimerization is dependent on its association with RNA. Retrovirology. 2009;6:56. doi: 10.1186/1742-4690-6-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.McDougall WM, Okany C, Smith HC. Deaminase activity on single-stranded DNA (ssDNA) occurs in vitro when APOBEC3G cytidine deaminase forms homotetramers and higher-order complexes. J Biol Chem. 2011;286:30655–61. doi: 10.1074/jbc.M111.269506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Mulder LC, Ooms M, Majdak S, Smedresman J, Linscheid C, Harari A, Kunz A, Simon V. Moderate influence of human APOBEC3F on HIV-1 replication in primary lymphocytes. J Virol. 2010;84:9613–7. doi: 10.1128/JVI.02630-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Miyagi E, Brown CR, Opi S, Khan M, Goila-Gaur R, Kao S, Walker RC, Jr, Hirsch V, Strebel K. Stably expressed APOBEC3F has negligible antiviral activity. J Virol. 2010;84:11067–75. doi: 10.1128/JVI.01249-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Albin JS, Hache G, Hultquist JF, Brown WL, Harris RS. Long-term restriction by APOBEC3F selects human immunodeficiency virus type 1 variants with restored Vif function. J Virol. 2010;84:10209–19. doi: 10.1128/JVI.00632-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Refsland EW, Hultquist JF, Harris RS. Endogenous origins of HIV-1 G-to-A hypermutation and restriction in the nonpermissive T cell line CEM2n. PLoS Pathog. 2012;8:e1002800. doi: 10.1371/journal.ppat.1002800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Berges BK, Wheat WH, Palmer BE, Connick E, Akkina R. HIV-1 infection and CD4 T cell depletion in the humanized Rag2-/-gamma c-/- (RAG-hu) mouse model. Retrovirology. 2006;3:76. doi: 10.1186/1742-4690-3-76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Gorantla S, Sneller H, Walters L, Sharp JG, Pirruccello SJ, West JT, Wood C, Dewhurst S, Gendelman HE, Poluektova L. Human immunodeficiency virus type 1 pathobiology studied in humanized BALB/c-Rag2-/-gammac-/- mice. J Virol. 2007;81:2700–12. doi: 10.1128/JVI.02010-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Sun Z, Denton PW, Estes JD, Othieno FA, Wei BL, Wege AK, Melkus MW, Padgett-Thomas A, Zupancic M, Haase AT, Garcia JV. Intrarectal transmission, systemic infection, and CD4+ T cell depletion in humanized mice infected with HIV-1. J Exp Med. 2007;204:705–14. doi: 10.1084/jem.20062411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Watanabe S, Terashima K, Ohta S, Horibata S, Yajima M, Shiozawa Y, Dewan MZ, Yu Z, Ito M, Morio T, Shimizu N, Honda M, Yamamoto N. Hematopoietic stem cell-engrafted NOD/SCID/IL2Rgamma null mice develop human lymphoid systems and induce long-lasting HIV-1 infection with specific humoral immune responses. Blood. 2007;109:212–8. doi: 10.1182/blood-2006-04-017681. [DOI] [PubMed] [Google Scholar]
- 217.Zhang L, Kovalev GI, Su L. HIV-1 infection and pathogenesis in a novel humanized mouse model. Blood. 2007;109:2978–81. doi: 10.1182/blood-2006-07-033159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.An DS, Poon B, Ho Tsong Fang R, Weijer K, Blom B, Spits H, Chen IS, Uittenbogaart CH. Use of a novel chimeric mouse model with a functionally active human immune system to study human immunodeficiency virus type 1 infection. Clin Vaccine Immunol. 2007;14:391–6. doi: 10.1128/CVI.00403-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Baenziger S, Tussiwand R, Schlaepfer E, Mazzucchelli L, Heikenwalder M, Kurrer MO, Behnke S, Frey J, Oxenius A, Joller H, Aguzzi A, Manz MG, Speck RF. Disseminated and sustained HIV infection in CD34+ cord blood cell-transplanted Rag2-/-gamma c-/- mice. Proc Natl Acad Sci U S A. 2006;103:15951–6. doi: 10.1073/pnas.0604493103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Ince WL, Zhang L, Jiang Q, Arrildt K, Su L, Swanstrom R. Evolution of the HIV-1 env gene in the Rag2-/- gammaC-/- humanized mouse model. J Virol. 2010;84:2740–52. doi: 10.1128/JVI.02180-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Sato K, Izumi T, Misawa N, Kobayashi T, Yamashita Y, Ohmichi M, Ito M, Takaori-Kondo A, Koyanagi Y. Remarkable lethal G-to-A mutations in vif-proficient HIV-1 provirus by individual APOBEC3 proteins in humanized mice. J Virol. 2010;84:9546–56. doi: 10.1128/JVI.00823-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Krisko JF, Martinez-Torres F, Foster JL, Garcia JV. HIV restriction by APOBEC3 in humanized mice. PLoS Pathog. 2013;9:e1003242. doi: 10.1371/journal.ppat.1003242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Lan P, Tonomura N, Shimizu A, Wang S, Yang YG. Reconstitution of a functional human immune system in immunodeficient mice through combined human fetal thymus/liver and CD34+ cell transplantation. Blood. 2006;108:487–92. doi: 10.1182/blood-2005-11-4388. [DOI] [PubMed] [Google Scholar]
- 224.Melkus MW, Estes JD, Padgett-Thomas A, Gatlin J, Denton PW, Othieno FA, Wege AK, Haase AT, Garcia JV. Humanized mice mount specific adaptive and innate immune responses to EBV and TSST-1. Nat Med. 2006;12:1316–22. doi: 10.1038/nm1431. [DOI] [PubMed] [Google Scholar]
- 225.Rambaut A, Posada D, Crandall KA, Holmes EC. The causes and consequences of HIV evolution. Nat Rev Genet. 2004;5:52–61. doi: 10.1038/nrg1246. [DOI] [PubMed] [Google Scholar]
- 226.Sadler HA, Stenglein MD, Harris RS, Mansky LM. APOBEC3G contributes to HIV-1 variation through sublethal mutagenesis. J Virol. 2010;84:7396–404. doi: 10.1128/JVI.00056-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Neogi U, Shet A, Sahoo PN, Bontell I, Ekstrand ML, Banerjea AC, Sonnerborg A. Human APOBEC3G-mediated hypermutation is associated with antiretroviral therapy failure in HIV-1 subtype C-infected individuals. J Int AIDS Soc. 2013;16:18472. doi: 10.7448/IAS.16.1.18472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Hache G, Mansky LM, Harris RS. Human APOBEC3 proteins, retrovirus restriction, and HIV drug resistance. AIDS Rev. 2006;8:148–57. [PubMed] [Google Scholar]
- 229.Jern P, Russell RA, Pathak VK, Coffin JM. Likely role of APOBEC3G-mediated G-to-A mutations in HIV-1 evolution and drug resistance. PLoS Pathog. 2009;5:e1000367. doi: 10.1371/journal.ppat.1000367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Deforche K, Camacho R, Laethem KV, Shapiro B, Moreau Y, Rambaut A, Vandamme AM, Lemey P. Estimating the relative contribution of dNTP pool imbalance and APOBEC3G/3F editing to HIV evolution in vivo. J Comput Biol. 2007;14:1105–14. doi: 10.1089/cmb.2007.0073. [DOI] [PubMed] [Google Scholar]
- 231.Ebrahimi D, Anwar F, Davenport MP. APOBEC3 has not left an evolutionary footprint on the HIV-1 genome. J Virol. 2011;85:9139–46. doi: 10.1128/JVI.00658-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Wood N, Bhattacharya T, Keele BF, Giorgi E, Liu M, Gaschen B, Daniels M, Ferrari G, Haynes BF, McMichael A, Shaw GM, Hahn BH, Korber B, Seoighe C. HIV evolution in early infection: selection pressures, patterns of insertion and deletion, and the impact of APOBEC. PLoS Pathog. 2009;5:e1000414. doi: 10.1371/journal.ppat.1000414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Armitage AE, Deforche K, Chang CH, Wee E, Kramer B, Welch JJ, Gerstoft J, Fugger L, McMichael A, Rambaut A, Iversen AK. APOBEC3G-induced hypermutation of human immunodeficiency virus type-1 is typically a discrete “all or nothing” phenomenon. PLoS Genet. 2012;8:e1002550. doi: 10.1371/journal.pgen.1002550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Jin X, Brooks A, Chen H, Bennett R, Reichman R, Smith H. APOBEC3G/CEM15 (hA3G) mRNA levels associate inversely with human immunodeficiency virus viremia. J Virol. 2005;79:11513–6. doi: 10.1128/JVI.79.17.11513-11516.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Ulenga NK, Sarr AD, Thakore-Meloni S, Sankale JL, Eisen G, Kanki PJ. Relationship between human immunodeficiency type 1 infection and expression of human APOBEC3G and APOBEC3F. J Infect Dis. 2008;198:486–92. doi: 10.1086/590212. [DOI] [PubMed] [Google Scholar]
- 236.Vazquez-Perez JA, Ormsby CE, Hernandez-Juan R, Torres KJ, Reyes-Teran G. APOBEC3G mRNA expression in exposed seronegative and early stage HIV infected individuals decreases with removal of exposure and with disease progression. Retrovirology. 2009;6:23. doi: 10.1186/1742-4690-6-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Eyzaguirre LM, Charurat M, Redfield RR, Blattner WA, Carr JK, Sajadi MM. Elevated hypermutation levels in HIV-1 natural viral suppressors. Virology. 2013 doi: 10.1016/j.virol.2013.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Kourteva Y, De Pasquale M, Allos T, McMunn C, D'Aquila RT. APOBEC3G expression and hypermutation are inversely associated with human immunodeficiency virus type 1 (HIV-1) burden in vivo. Virology. 2012;430:1–9. doi: 10.1016/j.virol.2012.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Zhao M, Geng W, Jiang Y, Han X, Cui H, Dai D, Bao M, Pan Y, Wang Y, Zhang X, Zhang M, Qi G, Shang H. The associations of hA3G and hA3B mRNA levels with HIV disease progression among HIV-infected individuals of China. J Acquir Immune Defic Syndr. 2010;53(Suppl 1):S4–9. doi: 10.1097/QAI.0b013e3181c7d349. [DOI] [PubMed] [Google Scholar]
- 240.Biasin M, Piacentini L, Lo Caputo S, Kanari Y, Magri G, Trabattoni D, Naddeo V, Lopalco L, Clivio A, Cesana E, Fasano F, Bergamaschi C, Mazzotta F, Miyazawa M, Clerici M. Apolipoprotein B mRNA-editing enzyme, catalytic polypeptide-like 3G: a possible role in the resistance to HIV of HIV-exposed seronegative individuals. J Infect Dis. 2007;195:960–4. doi: 10.1086/511988. [DOI] [PubMed] [Google Scholar]
- 241.Mous K, Jennes W, Camara M, Seydi M, Daneau G, Mboup S, Kestens L, Van Ostade X. Expression analysis of LEDGF/p75, APOBEC3G, TRIM5alpha, and tetherin in a Senegalese cohort of HIV-1-exposed seronegative individuals. PLoS One. 2012;7:e33934. doi: 10.1371/journal.pone.0033934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Gandhi SK, Siliciano JD, Bailey JR, Siliciano RF, Blankson JN. Role of APOBEC3G/F-mediated hypermutation in the control of human immunodeficiency virus type 1 in elite suppressors. J Virol. 2008;82:3125–30. doi: 10.1128/JVI.01533-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Amoedo ND, Afonso AO, Cunha SM, Oliveira RH, Machado ES, Soares MA. Expression of APOBEC3G/3F and G-to-A hypermutation levels in HIV-1-infected children with different profiles of disease progression. PLoS One. 2011;6:e24118. doi: 10.1371/journal.pone.0024118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Bunupuradah T, Imahashi M, Iampornsin T, Matsuoka K, Iwatani Y, Puthanakit T, Ananworanich J, Sophonphan J, Mahanontharit A, Naoe T, Vonthanak S, Phanuphak P, Sugiura W. Association of APOBEC3G genotypes and CD4 decline in Thai and Cambodian HIV-infected children with moderate immune deficiency. AIDS Res Ther. 2012;9:34. doi: 10.1186/1742-6405-9-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.De Maio FA, Rocco CA, Aulicino PC, Bologna R, Mangano A, Sen L. APOBEC3-mediated editing in HIV type 1 from pediatric patients and its association with APOBEC3G/CUL5 polymorphisms and Vif variability. AIDS Res Hum Retroviruses. 2012;28:619–27. doi: 10.1089/AID.2011.0291. [DOI] [PubMed] [Google Scholar]
- 246.Land AM, Ball TB, Luo M, Pilon R, Sandstrom P, Embree JE, Wachihi C, Kimani J, Plummer FA. Human immunodeficiency virus (HIV) type 1 proviral hypermutation correlates with CD4 count in HIV-infected women from Kenya. J Virol. 2008;82:8172–82. doi: 10.1128/JVI.01115-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Kieffer TL, Kwon P, Nettles RE, Han Y, Ray SC, Siliciano RF. G-->A hypermutation in protease and reverse transcriptase regions of human immunodeficiency virus type 1 residing in resting CD4+ T cells in vivo. J Virol. 2005;79:1975–80. doi: 10.1128/JVI.79.3.1975-1980.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Russell RA, Moore MD, Hu WS, Pathak VK. APOBEC3G induces a hypermutation gradient: purifying selection at multiple steps during HIV-1 replication results in levels of G-to-A mutations that are high in DNA, intermediate in cellular viral RNA, and low in virion RNA. Retrovirology. 2009;6:16. doi: 10.1186/1742-4690-6-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Singh KK, Wang Y, Gray KP, Farhad M, Brummel S, Fenton T, Trout R, Spector SA. Genetic variants in the host restriction factor APOBEC3G are associated with HIV-1-related disease progression and central nervous system impairment in children. J Acquir Immune Defic Syndr. 2013;62:197–203. doi: 10.1097/QAI.0b013e31827ab612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.An P, Bleiber G, Duggal P, Nelson G, May M, Mangeat B, Alobwede I, Trono D, Vlahov D, Donfield S, Goedert JJ, Phair J, Buchbinder S, O'Brien SJ, Telenti A, Winkler CA. APOBEC3G genetic variants and their influence on the progression to AIDS. J Virol. 2004;78:11070–6. doi: 10.1128/JVI.78.20.11070-11076.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Reddy K, Winkler CA, Werner L, Mlisana K, Abdool Karim SS, Ndung'u T. APOBEC3G expression is dysregulated in primary HIV-1 infection and polymorphic variants influence CD4+ T-cell counts and plasma viral load. AIDS. 2010;24:195–204. doi: 10.1097/QAD.0b013e3283353bba. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Pace C, Keller J, Nolan D, James I, Gaudieri S, Moore C, Mallal S. Population level analysis of human immunodeficiency virus type 1 hypermutation and its relationship with APOBEC3G and vif genetic variation. J Virol. 2006;80:9259–69. doi: 10.1128/JVI.00888-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Cho SJ, Drechsler H, Burke RC, Arens MQ, Powderly W, Davidson NO. APOBEC3F and APOBEC3G mRNA levels do not correlate with human immunodeficiency virus type 1 plasma viremia or CD4+ T-cell count. J Virol. 2006;80:2069–72. doi: 10.1128/JVI.80.4.2069-2072.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Do H, Vasilescu A, Diop G, Hirtzig T, Heath SC, Coulonges C, Rappaport J, Therwath A, Lathrop M, Matsuda F, Zagury JF. Exhaustive genotyping of the CEM15 (APOBEC3G) gene and absence of association with AIDS progression in a French cohort. J Infect Dis. 2005;191:159–63. doi: 10.1086/426826. [DOI] [PubMed] [Google Scholar]
- 255.Piantadosi A, Humes D, Chohan B, McClelland RS, Overbaugh J. Analysis of the percentage of human immunodeficiency virus type 1 sequences that are hypermutated and markers of disease progression in a longitudinal cohort, including one individual with a partially defective Vif. J Virol. 2009;83:7805–14. doi: 10.1128/JVI.00280-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Jacobs GB, Nistal M, Laten A, van Rensburg EJ, Rethwilm A, Preiser W, Bodem J, Engelbrecht S. Molecular analysis of HIV type 1 vif sequences from Cape Town, South Africa. AIDS Res Hum Retroviruses. 2008;24:991–4. doi: 10.1089/aid.2008.0077. [DOI] [PubMed] [Google Scholar]
- 257.Peng J, Ao Z, Matthews C, Wang X, Ramdahin S, Chen X, Li J, Chen L, He J, Ball B, Fowke K, Plummer F, Embree J, Yao X. A Naturally Occurring Vif Mutant (I107T) Attenuates Anti-APOBEC3G Activity and HIV-1 Replication. J Mol Biol. 2013;425:2840–52. doi: 10.1016/j.jmb.2013.05.015. [DOI] [PubMed] [Google Scholar]
- 258.De Maio FA, Rocco CA, Aulicino PC, Bologna R, Mangano A, Sen L. Effect of HIV-1 Vif variability on progression to pediatric AIDS and its association with APOBEC3G and CUL5 polymorphisms. Infect Genet Evol. 2011;11:1256–62. doi: 10.1016/j.meegid.2011.04.020. [DOI] [PubMed] [Google Scholar]
- 259.Fourati S, Malet I, Binka M, Boukobza S, Wirden M, Sayon S, Simon A, Katlama C, Simon V, Calvez V, Marcelin AG. Partially active HIV-1 Vif alleles facilitate viral escape from specific antiretrovirals. AIDS. 2010;24:2313–21. doi: 10.1097/QAD.0b013e32833e515a. [DOI] [PubMed] [Google Scholar]
- 260.Iwabu Y, Kinomoto M, Tatsumi M, Fujita H, Shimura M, Tanaka Y, Ishizaka Y, Nolan D, Mallal S, Sata T, Tokunaga K. Differential anti-APOBEC3G activity of HIV-1 Vif proteins derived from different subtypes. J Biol Chem. 2010;285:35350–8. doi: 10.1074/jbc.M110.173286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Lisovsky I, Schader SM, Sloan RD, Oliveira M, Coutsinos D, Bernard NF, Wainberg MA. HIV-1 subtype variability in Vif derived from molecular clones affects APOBEC3G-mediated host restriction. Intervirology. 2013;56:258–64. doi: 10.1159/000348513. [DOI] [PubMed] [Google Scholar]
- 262.Dang Y, Wang X, York IA, Zheng YH. Identification of a critical T(Q/D/E)x5ADx2(I/L) motif from primate lentivirus Vif proteins that regulate APOBEC3G and APOBEC3F neutralizing activity. J Virol. 2010;84:8561–70. doi: 10.1128/JVI.00960-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Hou W, Wang X, Ye L, Zhou L, Yang ZQ, Riedel E, Ho WZ. Lambda interferon inhibits human immunodeficiency virus type 1 infection of macrophages. J Virol. 2009;83:3834–42. doi: 10.1128/JVI.01773-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Stopak KS, Chiu YL, Kropp J, Grant RM, Greene WC. Distinct patterns of cytokine regulation of APOBEC3G expression and activity in primary lymphocytes, macrophages, and dendritic cells. J Biol Chem. 2007;282:3539–46. doi: 10.1074/jbc.M610138200. [DOI] [PubMed] [Google Scholar]
- 265.Lada AG, Waisertreiger IS, Grabow CE, Prakash A, Borgstahl GE, Rogozin IB, Pavlov YI. Replication protein A (RPA) hampers the processive action of APOBEC3G cytosine deaminase on single-stranded DNA. PLoS One. 2011;6:e24848. doi: 10.1371/journal.pone.0024848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Zhou Y, Wang X, Liu M, Hu Q, Song L, Ye L, Zhou D, Ho W. A critical function of toll-like receptor-3 in the induction of anti-human immunodeficiency virus activities in macrophages. Immunology. 2010;131:40–9. doi: 10.1111/j.1365-2567.2010.03270.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Rose KM, Marin M, Kozak SL, Kabat D. Transcriptional regulation of APOBEC3G, a cytidine deaminase that hypermutates human immunodeficiency virus. J Biol Chem. 2004;279:41744–9. doi: 10.1074/jbc.M406760200. [DOI] [PubMed] [Google Scholar]
- 268.Muckenfuss H, Kaiser JK, Krebil E, Battenberg M, Schwer C, Cichutek K, Munk C, Flory E. Sp1 and Sp3 regulate basal transcription of the human APOBEC3G gene. Nucleic Acids Res. 2007;35:3784–96. doi: 10.1093/nar/gkm340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.McFarland KL, Klingenberg JM, Boyce ST, Supp DM. Expression of genes encoding antimicrobial proteins and members of the toll-like receptor/nuclear factor-kappaB pathways in engineered human skin. Wound Repair Regen. 2008;16:534–41. doi: 10.1111/j.1524-475X.2008.00401.x. [DOI] [PubMed] [Google Scholar]
- 270.Komohara Y, Suekane S, Noguchi M, Matsuoka K, Yamada A, Itoh K. Expression of APOBEC3G in kidney cells. Tissue antigens. 2007;69:95–8. doi: 10.1111/j.1399-0039.2006.00725.x. [DOI] [PubMed] [Google Scholar]
- 271.Wang YJ, Wang X, Zhang H, Zhou L, Liu S, Kolson DL, Song L, Ye L, Ho WZ. Expression and regulation of antiviral protein APOBEC3G in human neuronal cells. J Neuroimmunol. 2009;206:14–21. doi: 10.1016/j.jneuroim.2008.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Watashi K, Khan M, Yedavalli VR, Yeung ML, Strebel K, Jeang KT. Human immunodeficiency virus type 1 replication and regulation of APOBEC3G by peptidyl prolyl isomerase Pin1. J Virol. 2008;82:9928–36. doi: 10.1128/JVI.01017-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Kinoshita SM, Taguchi S. NF-IL6 (C/EBPbeta) induces HIV-1 replication by inhibiting cytidine deaminase APOBEC3G. Proc Natl Acad Sci U S A. 2008;105:15022–7. doi: 10.1073/pnas.0807269105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Ogawa Y, Kawamura T, Kimura T, Ito M, Blauvelt A, Shimada S. Gram-positive bacteria enhance HIV-1 susceptibility in Langerhans cells, but not in dendritic cells, via Toll-like receptor activation. Blood. 2009;113:5157–66. doi: 10.1182/blood-2008-10-185728. [DOI] [PubMed] [Google Scholar]
- 275.Valcke HS, Bernard NF, Bruneau J, Alary M, Tsoukas CM, Roger M. APOBEC3G genetic variants and their association with risk of HIV infection in highly exposed Caucasians. AIDS. 2006;20:1984–6. doi: 10.1097/01.aids.0000247124.35129.e1. [DOI] [PubMed] [Google Scholar]
- 276.Kiefer F, Arnold K, Kunzli M, Bordoli L, Schwede T. The SWISS-MODEL Repository and associated resources. Nucleic Acids Res. 2009;37:D387–92. doi: 10.1093/nar/gkn750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Arnold K, Bordoli L, Kopp J, Schwede T. The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics. 2006;22:195–201. doi: 10.1093/bioinformatics/bti770. [DOI] [PubMed] [Google Scholar]