

Abstract
The prevailing pathomechanistic paradigm for myotonic dystrophy (DM) is that aberrant expression of embryonic/fetal mRNA/protein isoforms accounts for most aspects of the pleiotropic phenotype. To identify aberrant isoforms in skeletal muscle of DM1 and DM2 patients, we performed exon-array profiling and RT-PCR validation on the largest DM sample set to date, including Duchenne, Becker and tibial muscular dystrophy (NMD) patients as disease controls, and non-disease controls. Strikingly, most expression and splicing changes in DM patients were shared with NMD controls. Comparison between DM and NMD identified almost no significant differences. We conclude that DM1 and DM2 are essentially identical for dysregulation of gene expression, and DM expression changes represent a subset of broader spectrum dystrophic changes. We found no evidence for qualitative splicing differences between DM1 and DM2. While some DM-specific splicing differences exist, most of the DM splicing differences were also seen in NMD controls. SSBP3 exon 6 missplicing was observed in all diseased muscle and led to reduced protein. We conclude there is no widespread DM-specific spliceopathy in skeletal muscle and suggest that missplicing in DM (and NMD) may not be the driving mechanism for the muscle pathology, since the same pathways show expression changes unrelated to splicing.
Keywords: Myotonic dystrophy, DM1, DM2, aberrant isoform expression, missplicing
1. Introduction
Myotonic dystrophy (DM) is the most common adult-onset muscular dystrophy and characterized by a pleiotropic phenotype with multisystem involvement [1, 2]. Although mutations have been identified, the pathomechanisms are incompletely understood, and presently there is no effective treatment. DM type 1 (DM1) and type 2 (DM2) are caused by different unstable, non-coding microsatellite expansions--(CTG)DM1 in DMPK [3–5] and (CCTG)DM2 in CNBP (ZNF9) [6, 7]. Despite similar mutations, DM1 and DM2 are clinically distinct diseases and the basis for these differences is as yet unknown [2]. Since transcription of the mutant repeats into (CUG)DM1/(CCUG)DM2 appears to be necessary and sufficient to cause disease [8, 9], the prevailing paradigm is that DM1 and DM2 are toxic RNA gain-of-function diseases [10–13]. Mutant RNAs form ribonuclear inclusions that sequester splice factors of the MBNL family, resulting in a number of embryonic/fetal isoforms being aberrantly expressed in adult tissues, with over 30 genes verified as misspliced to date [12, 14–17]. Thus, efforts to understand the pathomechanisms in DM have so far focused primarily on aberrant splicing. However, while missplicing of the chloride channel CLCN1 can account for the myotonia [18–22], the pathogenic roles of the other aberrantly spliced genes has not been experimentally demonstrated and remain circumstantial.
Evidence from several different animal models suggests that splicing, RNA foci, and muscle pathology are separable events [23–26] and strongly suggest the existence of additional pathomechanisms (recently reviewed in [2, 27]). Moreover, studying human patients and mouse models of muscular dystrophies not associated with toxic RNAs, we and others recently reported that splicing changes may be a much more general phenomenon of muscle disease and can be secondary to muscle regeneration [28, 29]. These findings raise questions concerning the extent to which missplicing plays a major role in the disease phenotype and whether expression or splicing changes in DM are specific to this disease or are common manifestations of dystrophic muscle in general. There is, as yet, no comprehensive study of expression and splicing for human DM skeletal muscle that addresses these issues, especially comparing DM1 to DM2, or DM (DM1 and/or DM2) to non-DM dystrophies. The purpose of our study was to elucidate the relative contributions of aberrant splicing and aberrant expression to the DM phenotype and to distinguish DM1- and DM2-specific events from those common to all dystrophic muscle. To this end, we performed global array-based expression and splicing profiling using the Affymetrix Exon 1.0 ST array. Along with skeletal muscle samples from DM1, DM2 and normal individuals, we also profiled samples from patients with non-DM inherited muscular dystrophies.
2. Materials and Methods
2.1 Overall experimental design
After confirming molecular diagnosis using patient DNA [7, 30], total RNA was extracted from skeletal muscle samples and used for both cDNA preparation and array hybridization [31]. Following analysis of the array data, pathway analysis was conducted on various gene lists resulting from these analyses and verification of aberrant splicing by RT-PCR was attempted for select genes identified as expressing alternative transcripts.
2.2 Patient Samples
For the exon array profiling we used a panel of 28 retrospective skeletal muscle biopsies from DM1 (n=8), DM2 (n=10), Becker muscular dystrophy (BMD, n=3), Duchenne muscular dystrophy (DMD, n=1), tibial muscular dystrophy (TMD, n=2) and normal skeletal muscle (n=4). Patient samples were obtained through the active collaboration and sharing of patient samples within the European Neuromuscular Centre (ENMC) consortium on DM2 and Other Myotonic Dystrophies [32]. Following informed consent samples were collected with the appropriate oversight at the institutions of the various collaborating investigators. Normal control RNAs were purchased commercially (Ambion, Biochain, and Stratagene). A subset of these samples was used for alternative splicing verification by RT-PCR. Several additional patient samples were included in the splice validation panel, but not the array experiment. Samples and assays run are described in more detail in Table 1.
Table 1.
Samples used for array analysis and RT-PCR validation
| No. | Sample ID* | Exon Array | RT-PCR |
|---|---|---|---|
| 1 | DM1_F_?_D | Y | N |
| 2 | DM1_?_?_? | N | Y |
| 3 | DM1_F_?_BB | Y | Y |
| 4 | DM1_F_54_? | Y | Y |
| 5 | DM1_F_29_BB | Y | N |
| 6 | DM1_M_25_? | Y | N |
| 7 | DM1**_M_21_? | Y | N |
| 8 | DM1_F_55_D | Y | N |
| 9 | DM1_?_?_? | Y | N |
| 10 | DM2_M_39_BB | Y | N |
| 11 | DM2_F_58_BB | Y | Y |
| 12 | DM2_F_50_BB | Y | Y |
| 13 | DM2_F_51_BB | Y | N |
| 14 | DM2_F_43_D | Y | Y |
| 15 | DM2_M_51_BB | Y | N |
| 16 | DM2_F_37_VL | Y | N |
| 17 | DM2_F_43_BB | Y | Y |
| 18 | DM2_F_65_BB | Y | Y |
| 19 | DM2_F_55_BB | Y | Y |
| 20 | BMD_M_50_D | Y | Y |
| 21 | BMD_M_45_BB | Y | Y |
| 22 | BMD_M_26_BB | Y | Y |
| 23 | DMD_M_20_G | Y | Y |
| 24 | DMD_M_5_BB | N | Y |
| 25 | DMD_M_8_BB | N | Y |
| 26 | TMD_M_58_EDL | Y | Y |
| 27 | TMD_F_88_EDL | Y | Y |
| 28 | N_F_61_? | Y | Y |
| 29 | N_M_43_? | Y | Y |
| 30 | N_M_85_? | Y | Y |
| 31 | N_M_43_? | Y | Y |
| 32 | N_M_26_? | Y | Y |
Sample identifier (ID) specifies diagnosis: DM1, DM2, BMD, DMD, TMD, or N. Sex: F, female; M, male. Age in years. Biopsy site: D, deltoid; BB, biceps brachii; VL, vastus lateralis; TA, tibialis anterior; G, gastrocnemius; EDL, extensor hallucis/digitorum longus. ?, information not available.
Homozygous DM1 patient.
2.3 Verification of diagnosis for DM1 and DM2
Presence or absence of DM1 or DM2 expansion mutations were verified by PCR-based molecular genetic diagnostic procedures as previously described [7, 30]. Two alleles observed at DMPK or CNBP/ZNF9 was considered sufficient reason for exclusion of a diagnosis of DM1 or DM2, respectively. Repeat-primed PCR (RP-PCR) was performed on all samples and was used to distinguish between homozygous samples and those with amplification-resistant expansions.
2.4 RNA extraction
RNA was extracted using the TriZol Reagent according to the manufacturer's suggestions (Invitrogen, Carlsbad, CA) and further purified using the RNeasy kit (Qiagen Inc., Valencia, CA). The quality and integrity of the RNA was then analyzed on an Agilent Bio- Analyzer (RNA 6000 Nano LabChip). Total cellular RNA samples with a RIN (RNA integrity number) >7 were used for further microarray studies [31].
2.5 Microarray exon profiling
Microarray experiments were performed with appropriate randomization in order to avoid confounding on one or more variables. For exon profiling on the Human Exon 1.0 ST array (Affymetrix Santa Clara, CA), RNA was processed for hybridization using a pre-commercial version of the Affymetrix GeneChip Whole Transcript (WT) Double-Stranded Target Labeling Assay for preparation of double-stranded (ds) target DNA (first generation protocol) or the Affymetrix GeneChip Whole Transcript (WT) Sense Target Labeling Assay for preparation of single-stranded (ss) target DNA (second generation protocol). Pre-commercial versions of the kits contained identical formulations to the commercial versions. Sample hybridization, washing and scanning was performed according to manufacturer's recommendations and as previously described [31].
2.6 Exon array data analysis
Exon arrays were analyzed by several methods. Initially, arrays were quantified using the PLIER algorithm introduced by Affymetrix as previously described [31]. The arrays were quantile-normalized, and GC-specific background was estimated and subtracted using the PM-GCBG option. All quantifications used log2(PLIER + 8) values, where the value “8” is an arbitrary shrinkage constant. PLIER summarizes groups of probe-level intensities, and we used two different groupings: (1) all probes within a single probe set region (PSR), and (2) all probes within a given gene. Genes were defined as RefSeq clusters using groupings of PSRs supplied by Affymetrix. Exon numbering within genes was checked by mapping the reported sequences against data from the UCSC genome browser (Build hg16).
Subsequently, arrays were reanalyzed using the Partek Genomics Suite (Partek Inc., St. Louise, MO), employing the workflow for exon arrays, and aligned to genome Build hg18. In this analysis only the core set of probes (as defined by Affymetrix) were included. To identify alternative PSR usage we calculated a Splice Index (SI), similar to that described by Du et al. [16], from the RMA normalized log2 probe intensities for each core PSR using the formula:
Individual PSR p-values were calculated using t-tests, and Benjamini-Hochberg correction for multiple testing was applied. In order to qualify as having alternative expression, a PSR had to meet three criteria: (1) SI≥1.5; (2) Benjamini-Hochberg adjusted p-value≤0.05; and (3) an overall gene expression fold-change FC<|3|. This latter exclusion was necessary because strong over- or under-expression distorts the SI.
Annotation of probe sets was extracted from the “UCSC Alt Events” tract from the UCSC Genome Browser (Build hg18). This track shows various types of alternative splicing and other events that result in more than a single transcript from the same gene, and is based on an analysis by the txgAnalyse program of splicing graphs produced by the txGraph program. Both of these programs were written by Jim Kent at UCSC.
2.7 cDNA synthesis
Total RNA from skeletal muscle was used to generate cDNA by standard methods. Briefly, for each sample 5 μg of total RNA were DNaseI treated (Ambion, Austin, TX) according to the manufacturer's suggestions. Absence of contaminating DNA in the RNA samples was confirmed by performing PCR on total RNA and appropriate genomic DNA controls using primers designed to amplify only an untranscribed genomic sequence. DnaseI-treated RNA was split in half, and first-strand cDNA synthesis was performed on each half in separate reactions using either random hexamer or oligo-(dT) priming according to the manufacturer's protocol (SuperScript™ III First-strand cDNA Synthesis Kit, Invitrogen, Carlsbad, CA). All cDNAs were then Rnase H treated. Equal amounts of random hexamer and oligo-(dT) primed cDNA were pooled and diluted to 200 μl with molecular grade RNAse-free H2O for use in RT-PCR reactions.
2.8 RT-PCR for isoforms quantification
Assays were designed to validate and quantify preferential isoform usage for selected genes. A total of 17 previously published events were tested along with 27 novel events, predicted by the array analysis. A subset of the samples used for the exon array comprised this panel (DM1, n=3; DM2, n=6; NMD, n=5; and N, n=5). Specific samples included are indicated in Table 1. We also verified aberrant splicing of INSR and MAPT by gel electrophoresis for these samples. Primers for each assay are described in Table S1. Several of the assays were designed to detect multiple predicted events. To estimate the relative amounts of each isoform present, we performed RT-PCR in a three-primer reaction incorporating a universal primer with a fluorescent label for quantification [28, 30]. Each RT-PCR was performed in log-linear range (28–32 cycles, determined empirically) with 2–3 μl of cDNA as template (12.5–18.75 ng starting total RNA-equivalents). Capillary electrophoresis on an ABI3100 Genetic Analyzer allowed peak heights for each isoform to be measured. For each sample, peak heights were added together to determine the total signal for the transcript. The Inclusion Index was calculated for each peak as a percent of the total signal for that sample. Differences between groups were tested for significance using two-sample t-tests.
2.9 Pathway analysis and functional annotation
Pathway analysis of differentially regulated genes identified by expression profiling was performed using the Ingenuity Pathway Analysis software (IPA, Ingenuity Systems, Release Number: 5.5 – 2233, http://www.ingenuity.com). IPA analysis uses the right-tailed Fisher Exact Test to define the most relevant functional categories and canonical pathways (Ingenuity Pathway Analysis-Canonical Pathways, IPA-CP).
2.10 Western blot analysis of SSBP3
Western blotting for SSBP3 protein was performed on skeletal muscle whole cell lysates using standard procedures [33]. Anti-SSBP3 polyclonal antibodies were raised in rabbits against unique peptide epitopes. The specificity of these antisera to detect SSBP3, and not the related members SSBP2 and SSBP4, was verified. None of the samples used for Western blotting were the same as those used for either the exon microarray or RT-PCR validation experiments and have not been included in Table 1.
3. Results
3.1 Array QC and overview
For initial quality control all samples were analyzed together as one group. Intensity distributions across all 228,872 core probes for the 28 skeletal muscle biopsies are shown in Fig. S1A. Tight distribution of signal was indicative of uniform quality across all samples. Principle component analysis (PCA) showed no clustering by scan date, indicating that there were no batch effects (Fig. S1B). Samples clustered primarily by their status as diseased or normal; all diseased muscle samples clustered together by PCA (Fig. S1C). Sources of variation in the experiment, as determined by ANOVA, are depicted in Fig. S1D. Disease vs. normal status accounted for the majority of the variation, while contributions of specific diagnosis, age, gender, and biopsy site were negligible and comparable to that of error. This finding suggested that all the dystrophic samples were very similar to each other and that mRNA expression in DM muscle was not notably different from other dystrophic muscles. We noticed that there seemed to be more sample-to-sample heterogeneity in the DM1 group, compared to the DM2 and NMD groups (Fig. S1E). All microarray data is publicly available (GEO, http://www.ncbi.nlm.nih.gov/geo/, accession number GSE48828).
3.2 Expression analysis
In order to address the question of how similar or different these dystrophies are with respect to mRNA expression, and whether there are significant disease-specific differences between DM1 and DM2, we used the Partek Genomics Suite to perform ANOVA-based expression analyses comparing each disease group to normal (DM1 vs. N, DM2 vs. N, and NMD vs. N), as well as each disease group to the others (DM1 vs. DM2, DM1 vs. NMD, and DM2 vs. NMD).
Table 2 summarizes the results of the various expression analyses and presents counts of transcripts with p-value<0.05 and |FC|>2 at FDR=0.05. Fig. 1A shows a Venn diagram depicting the intersections of the disease vs. normal comparisons for transcripts passing these filters. A complete list of transcripts detected in all comparisons, along with their fold change, p-value, and whether they are flagged as passing all filters can be found in Table S2.
Table 2.
Summary of dysregulated genes (|FC|>2 and p-value<0.05 with FDR=0.05)
|
|
||||||
|---|---|---|---|---|---|---|
| DM1 vs. N | DM2 vs. N | NMD vs. N | DM1 vs. DM2 | DM1 vs. NMD | DM2 vs. NMD | |
| p-value <0.05, FDR=0.05, FC>2 | 269 | 414 | 348 | 0 | 1 | 18 |
| p-value<0.05, FDR=0.05, FC<−2 | 340 | 484 | 332 | 2 | 0 | 11 |
| All dysregulated >2-fold | 609 | 898 | 680 | 2 | 0 | 29 |
| p-value (up vs. down) | 0.0040 | 0.0195 | 0.5395 | NA | NA | NA |
Fig. 1.
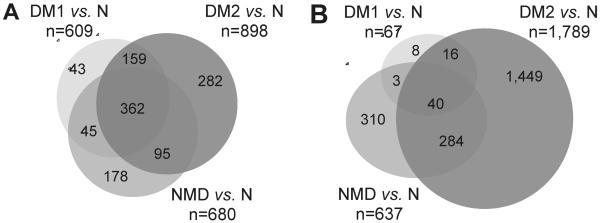
Venn diagrams depicting shared dysregulated (A) or misspliced (B) transcripts in pairwise comparisons between disease and normal samples. (A) Transcripts with |FC|>2 and p-value<0.05 with FDR=0.05 (Table 2). (B) Probe sets dysregulated with SI>1.5 and adjusted p-value<0.05 for genes with |FC|<3 (Table 3). Diagrams not to scale.
While the ratio of up- and down-regulated transcripts was about 50:50 for NMD vs. N, in DM vs. N over-expression was more frequent than under-expression. This deviation from the expected 50:50 ratio in both DM1 and DM2 was highly significant by X2 test (DM1, p-values=0.0040; and DM2, p-values=0.0195; Table 2). Importantly, very few genes (n=2; <0.1%) were significantly dysregulated in the direct comparisons between the different disease categories (Table 2), underlining the extensive molecular similarities between these various dystrophies.
3.3 Expression differences between DM1 and DM2
While intersection of the lists of dysregulated genes in the comparisons DM1 vs. N and DM2 vs. N identified a number of genes unique to each list, a direct comparison between DM1 and DM2 by ANOVA identified only three genes that passed FDR=0.05 and only two had a fold-change FC>|2| (EGR1, FC −7.35; FOS, FC −8.14). For these two genes the variation between DM1 samples was high, while variation between DM2 samples was not. It is not clear whether this heterogeneity was the primary driver of the observed difference. Dot plots showing log-intensities for all samples for EGR1 and FOS are shown in Fig. S2A–B.
3.4 Expression differences between DM and NMD
Intersection of gene lists from all three neuromuscular disease vs. normal comparisons identified 362 shared transcripts, encompassing 344 unique annotated genes with shared dysregulation between all three groups, highlighting their similarity. Similar to the DM1 vs. DM2 comparison, there were many non-overlapping transcripts. However, direct comparisons of disease vs. disease showed a different picture. For DM1 vs. NMD, only four genes pass the FDR=0.05 filter. These were C21ORF82 (FC 2.46), DYNC1I1 (FC −1.57), SOX6 (FC 1.47), and C1QTNF3 (FC 1.49). Dot plot for C21ORF82 is shown in Fig. S2C. In the case of DM2 vs. NMD, only 11 genes were under-expressed in DM2 and 18 over-expressed by ≥2-fold. The most over-expressed gene was HCN1 (FC 3.65) and the most under-expressed was IGF2 (FC −2.72). Dot plots for several of these genes are shown in Fig. S2D–H.
3.5 Dysregulation of genes containing (CAG)n/(CUG)n repeats
It has been shown that the expanded (CUG)DM1 repeat in the DMPK transcript is a target of Dicer [34] and, as such, complementary sequences in other mRNAs may render them subject to degradation via RNA-induced silencing complex (RISC) pathways. Because bidirectional transcription is sometimes a feature in repeat expansion diseases [35], we considered the possibility of both CAG and CUG fragments resulting from the actions of Dicer. To determine whether steady-state levels of transcripts containing polymorphic (CAG)n or (CUG)n repeats are disproportionally down-regulated in DM1, we identified 160 unique human genes containing at least 6 tandem CAG or CUG repeats that were detected in our skeletal muscle samples. Altogether, this represented about 1% of all detected transcripts. Representative repeat lengths varied from 6 to 42 repeats (RefSeq). The majority of transcripts (82% overall) had the repeat in the open reading frame (ORF). This was interesting and unexpected, given the polymorphic nature of many of these repeats and the inevitable effect on the resultant protein sequence. (CAG)n repeats were more numerous than (CUG)n repeats and had a wider range of sizes.
The transcripts and their repeats are described in Supplementary Table S3, along with the fold-changes observed in DM1 vs. N, DM2 vs. N and NMD vs. N for each. We determined whether fold-change relative to normal tissue (trichotomized to FC<1.5 down-regulated, unchanged, and FC>1.5 up-regulated) was related to the presence of a (CAG)n or a (CUG)n repeat in the transcript. Checking for association with both χ2 tests and Fisher's exact tests showed no significant association for (CUG)n-containing transcripts, but a marginal association between fold-change and (CAG)n-containing transcripts for DM2 and NMD, which did not remain significant after correction for multiple testing. Plotting the fold-change against the representative repeat length for CAG and CUG separately, we observed a slight bias towards under-expression for transcripts with (CUG)n repeats and over-expression for those containing (CAG)n, which was not correlated with repeat length (Fig. S5). Similar investigation was conducted by Osborne and colleagues [36] using the HSALR mouse model of DM1. They identified 11 transcripts containing (CAG)n tracts longer than 19 nucleotides that were dysregulated in HSALR mice but not Mbnl1−/− or Clcn1-deficient mice. For seven of these 11, the (CAG)n tract was conserved in humans. Of these seven transcripts, we detected six in our samples, none of which were dysregulated at FC>|1.5|. MED12 showed a tendency towards up-regulation with FC>1.4, but this was observed in all three sample groups, thus could not have been related to the presence of a repeat expansion mutation.
3.6 Pathway analysis of expression results
We used the Ingenuity Pathway Analysis program (IPA) to explore functional relationships between genes dysregulated (≥2-fold) in common between DM1 and DM2. The 521 transcripts in the intersection of (DM1 vs. N) and (DM2 vs. N) (Fig. 1A) represent 475 unique annotated genes, 468 of which were mapped to pathways and used by IPA in the analysis. To further understand the relationship between DM-specific and generic dystrophic changes, we also conducted pathway analysis using only the genes common to all 3 groups and those unique to NMD. Genes central to networks that were prominently dysregulated in DM1 and DM2 included RNA polymerases, growth arrest and DNA damage inducible genes (GADD45A and GADD45B), CDKN1A (Fig. 2), the collagen network, and genes regulated by MYC (Fig. S4B). One NMD-specific network featured muscle structural genes, including multiple myosin genes and ABLIM1, which were not dysregulated in the DM1/DM2 intersection group (Fig. S4A). Numerous networks dysregulated in the NMD group were also shared with the DM (DM1 and DM2) groups. Figure S4C shows one of these networks, centered on MYC, and Fig. S4D shows another shared network that includes NFKB and SSBP3. These data support the hypothesis that, while entry points may differ for individual diseases, the downstream pathomechanisms are shared among different genetically distinct muscular dystrophies.
Fig. 2.

SSBP3 missplicing in neuromuscular diseases. (A) Splice Index (SI) for each probe used in the original array analysis. Exon 6 is included more frequently in DM and NMD than in normal muscle. (B) Assay design for quantitative RT-PCR validation: primers are indicated by arrows; alternative exon 6 is shown in grey. (C) Inclusion indices (Inclusion/Total) from quantitative fluorescent RT-PCR. (D) Kruskal-Wallis plot of isoform ratios, p-value=0.0023. t-test p-values are shown below groups. (E–F) Western blots showing overall reduced protein expression of SSBP3 in DM1 (E) and, to a lesser extent, DM2 (F) compared to normal skeletal muscle. The same normal control is used on both blots. DM samples are different from those used for RT-PCR validation. CBB, Coomassie brilliant blue staining.
3.7 Splice variant analysis of array data
We initially took several approaches to the identification of genes containing differentially spliced exons. Results varied considerably depending on which probe sets (all, extended, or core) were used, and how outliers were identified. We first used the approach we had previously implemented and used successfully to identify missplicing events in glioblastoma brain tumors [31], another approach was the ANOVA-based method employed in the Partek Genomic Suite. Both methods identified a very high proportion of false-positive events, as evidenced by the failure to validate microarray predicted events by RT-PCR assays. Additionally, for the Partek analysis, it was not clear at what p-value significance should be defined, since over 50% of the transcripts had p-values≤0.05. For all methods, large expression differences or the presence of a large number of probes in a transcript tended to distort results, and many false-positives were encountered when validation was attempted. Finally, we settled on a system using only the Affymetrix core probe set and the splice index calculation described in the Materials and Methods section. This algorithm eliminated one source of false-positive signal, namely the effect of expression differences, while correcting for multiple testing. Overall, this method appeared to be conservative rather than inclusive and identified far fewer significant events than did the previous methods. Thus, while we expected to encounter many false-negatives, we predicted that false-positives would be lower than with the previous methods. Results obtained with this method are summarized in Table 3. The Venn diagram describing the overlap between the sample groups is shown in Fig. 1B; Table S4 contains the corresponding splice indices (SI) and adjusted p-values for all 228,872 core PSRs detected in this sample set for the three disease vs. normal comparisons. It is worth noting that the number of PSRs passing the three filters was small for DM1 vs. N--only 67 probe sets in 60 genes. For DM2 vs. N, there were 1,789 flagged PSRs, the majority of which do not overlap with the other two comparisons. Out of 33 missplicing events that have been reported to be associated with DM, none were flagged as significant in DM1 and just three in DM2 (Table 5). There was also considerable differential isoform usage for the NMD sample set, when compared to normal, with many flagged probe sets shared between DM2 and NMD.
Table 3.
Summary of alternatively spliced probe sets
| Comparison | No. Flagged Genes | No. Flagged PSRs* | No. Exonic | % Exonic | No. Intronic | % Intronic | No. Cassette PSRs | % Cassette | No. 5' PSRs | % 5' | No. 3' PSRs | % 3' |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| DM1 vs. N | 60 | 67 | 27 | 40.3 | 37 | 55.2 | 12 | 17.9 | 14 | 20.9 | 11 | 16.4 |
| DM2 vs. N | 1,289 | 1,789 | 1,301 | 72.7 | 435 | 24.3 | 161 | 9.0 | 171 | 9.6 | 108 | 6.0 |
| NMD vs. N | 541 | 637 | 481 | 75.5 | 134 | 21.0 | 45 | 7.1 | 67 | 10.5 | 46 | 7.2 |
PSRs (n=228,871) with adjusted p-value≤0.05 and SI≥1.5 in genes with |FC|<3.
Table 5.
Performance of Human Exon 1.0 ST array (Affymetrix) for 33 published missplicing events
| DM1 vs. N$ |
DM2 vs. N$ |
NMD vs. N$ |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Gene | In/Out | Exon | Ref. | On Array* | In Core Set* | Predicted PSR* | Flag* | SI | Adjusted p-value | SI | Adjusted p-value | SI | Adjusted p-value |
| ANK2 | Out | 21 | [37] | Y | N | 2740265 (ext) | na | na | na | na | na | na | |
| APP | Out | 6 | [38] | Y | Y | 3927282 | 1.084 | 1 | −1.021 | 0.982 | 1.255 | 0.481 | |
| ATP2A1 | Out | 22 | [39] | Y | Y | 3655096 | −1.526 | 0.866 | −1.427 | 0.642 | 1.191 | 0.898 | |
| CAPN3 | Out | 19 | [14] | Y | N | na | na | na | na | na | na | na | |
| CAPZB | Out | 8 | [37] | Y | Y | 2399783 | −1.107 | 1 | −1.087 | 0.92 | 1.129 | 0.695 | |
| CLCN1 | Out | 6, 6b, 7 | [18] | Y | Y | 3029080 (E6) | 1.275 | 0.969 | 1.273 | 0.651 | 1.571 | 0.476 | |
| DMD | Out | 71 | [40] | Y | Y | 4004072 | −1.553 | 0.933 | −1.645 | 0.673 | −1.306 | 0.932 | |
| Out | 78 | [40] | Y | Y | 4004053 | −1.957 | 0.919 | −1.385 | 0.657 | −1.1 | 0.979 | ||
| DTNA | In | 11a–12 | [41] | N | N | na | na | na | na | na | na | na | |
| FHOD1 | Out | 11a, 12 | [14] | Y | Y | 3695563 | −1.323 | 0.981 | 1.105 | 0.897 | 1.282 | 0.666 | |
| GFPT2 | Out | 10 | [14] | Y | Y | 2558088 | nd | nd | nd | nd | nd | nd | |
| GNAS | In | 3 | [16] | Y | Y | 3891224 | nd | nd | nd | nd | nd | nd | |
| GRIN1 | In | 5 | [38] | N | N | na | na | na | na | na | na | na | |
| INSR | Out | 11 | [39, 42] | Y | N | 3848288 | nd | nd | nd | nd | nd | nd | |
| LDB3 | In | 4 | [33] | Y | Y | 3255989 | 1.158 | 1 | 1.355 | 0.73 | −1.188 | 0.869 | |
| In | 7 | [14, 33] | Y | Y | 3256033 | 2.158 | 0.328 | 2.868 | 0.004 | −1.357 | 0.265 | ||
| MAPT | Out | 3,4 | [43] | Y | Y | 3723710 | −1.11 | 0.929 | −1.038 | 0.93 | −1.36 | 0.237 | |
| Out | 3,4 | [43] | Y | Y | 3723712 | −1.179 | 1 | −1.094 | 0.726 | −1.278 | 0.397 | ||
| MBNL1 | In | 7 | [14] | Y | Y | 2648197 | 1.062 | 1 | 1.055 | 0.946 | 1.073 | 0.899 | |
| In | 7 | [14] | Y | Y | 2648200 | −1.223 | 0.953 | −1.369 | 0.562 | 1.296 | 0.548 | ||
| MBNL2 | In | 7 | [14] | Y | Y | 3497640 | DM2 | 3.923 | 0.075 | 3.287 | 0.005 | 2.889 | 0.063 |
| MEF2A | In | 5a | [28] | Y | Y | 3611221 | DM2 | 1.664 | 0.214 | 1.56 | 0.033 | 1.559 | 0.202 |
| MEF2C | In | 5a | [28] | Y | Y | 2866255 | 1.994 | 0.391 | 1.857 | 0.073 | 1.528 | 0.316 | |
| In | 5a | [28] | Y | Y | 2866256 | 1.333 | 0.982 | 1.418 | 0.599 | 1.371 | 0.61 | ||
| MTDH | In | 7 | [16] | Y | Y | 3108463 | −1.023 | 1 | 1.127 | 0.632 | 1.186 | 0.458 | |
| MTMR1 | Out | 2.1, 2.2 | [44] | N | N | na | na | na | na | na | na | na | |
| NFIX | In | 7 | [16] | Y | Y | 3822162 | 1.165 | 0.928 | 1.158 | 0.647 | 1.181 | 0.497 | |
| NRAP | Out | 12 | [14] | Y | Y | 3307631 | −1.003 | 1 | −1.139 | 0.797 | −1.015 | 0.982 | |
| PDLIM3 | In | 5a | [14] | Y | Y | 2796971 | DM2 | 3.458 | 0.327 | 4.141 | 0.003 | 2.99 | 0.059 |
| PPP2R5C | Out | 12 | [16] | Y | Y | 3552821 | −1.564 | 0.742 | −1.518 | 0.369 | −2.189 | 0.227 | |
| RYR1 | Out | 70 | [39] | N | N | na | na | na | na | na | na | na | |
| SMYD1 | In | 5 | [16] | Y | Y | 2492767 | 1.896 | 0.788 | 2.014 | 0.158 | 2.085 | 0.263 | |
| SPAG9 | Out | 26a | [16] | Y | N | 3762534 (ext) | na | na | na | na | na | na | |
| TNNT2 | In | 5 | [45] | N | N | na | na | na | na | na | na | na | |
| TNNT3 | In | 7, 8, F | [23] | N | N | na | na | na | na | na | na | na | |
| TTN | In | 11 | [14] | Y | N | 2589834 (full) | na | na | na | na | na | na | |
Y, yes; N, no. na, not applicable; nd, not detected.
Flag criteria: (1) SI≥|1.5|, (2) adjusted p-value≤0.05, and (3) FC<|3|.
3.8 Independent verification of alternative splicing in dystrophic muscle samples
Because most published missplicing events in DM were not identified by the array analysis, it was necessary to confirm that the samples we used did indeed show differential isoform usage for the reported genes. Therefore, we performed isoform specific RT-PCR using the quantitative fluorescent method previously described [28, 33] for seventeen published missplicing events using a subset (n=19) of the array samples (DM1, n=3; DM2, n=6; NMD, n=5; and N, n=5; Table 1). A summary of the results, including the p-values obtained from t-tests comparing the three groups (DM1, DM2 and NMD) to normal samples, is shown in Table 4. All of the predicted aberrant splicing events were confirmed for DM2. Eleven events (65%) were confirmed for the DM1 samples; however, the DM1 validation sample set was smaller (n=3) and not all samples could be successfully amplified for all genes. Thus, the failure of statistical confirmation was likely due to technical issues. Of particular interest was the observation of significant missplicing in the NMD set for seven of the 17 (41%) genes. We previously reported missplicing of MEF2A and MEF2C in NMD samples [28]. However, we did not expect missplicing of MBNL1, NRAP, TNNT2, or TTN, which had previously been reported for DM1 and are generally considered to be DM-specific. Although splicing of ATP2A1 (SERCA1) in NMD samples was also significantly different from normal, the data show that while some exclusion of exon 22 is present in all NMD samples, the percent exclusion is lower than in DM (Fig. S3A). Thus exclusion of exon 22 of ATP2A1 is most pronounced in DM, but with some tendency towards more generic occurrence. Genes where aberrant isoform usage was completely DM-specific were FHOD1, LDB3 (exon 7), MBNL2, PDLIM3, RYR1 and TNNT3. No NMD RT-PCR data was available for ANK2, CAPZB, or the CLCN1 fetal exon. However, retention of CLCN1 intron 2 was not DM-specific and was seen by gel electrophoresis for all dystrophic samples, including NMD (Fig. S3B).
Table 4.
Validation of published missplicing events in dystrophic muscle (t-test)
| Gene | Event | p-value (DM1 vs. N) | p-value (DM2 vs. N) | p-value (NMD vs. N)* |
|---|---|---|---|---|
| ANK2 | Exclusion exon 43a | 0.297000 | 6.91e-03 | nd |
| ATP2A1 | Exclusion exon 22 | 0.021100 | 2.68e-11 | 0.001050 |
| CAPZB | Exclusion exon 8a | 0.158000 | 6.05e-03 | nd |
| CLCN1 | Inclusion fetal exon | 0.011300 | 4.25e-03 | nd |
| FHOD1 | Exclusion exon 11a–12 | 0.007780 | 3.41e-10 | 0.510000 |
| LDB3 | Inclusion exon 4 | 0.064300 | 5.18e-06 | 0.526000 |
| LDB3 | Inclusion exon 7 | 0.000291 | 1.57e-06 | 0.316000 |
| MBNL1 | Inclusion exon 6a/7 | 0.004010 | 9.60e-06 | 0.044800 |
| MBNL2 | Inclusion exon 7 | 0.007990 | 3.38e-04 | 0.288000 |
| MEF2A | Inclusion exon 5a | 0.007620 | 2.73e-05 | 0.009280 |
| MEF2C | Inclusion exon 5a | 0.005230 | 1.59e-07 | 0.000883 |
| NRAP | Exclusion exon 12 | 0.953000 | 3.37e-03 | 0.017000 |
| PDLIM3 | Inclusion exon 5a | 0.047500 | 3.92e-04 | 0.859000 |
| RYR1 | Exclusion exon 70 | 0.251000 | 2.39e-02 | 0.528000 |
| TNNT2 | Inclusion exon 5 | 0.012200 | 5.99e-02 | 0.033700 |
| TNNT3 | Inclusion fetal exon | 0.077700 | 9.88e-05 | 0.439000 |
| TTN | Inclusion exon 11 | 0.009280 | 6.75e-04 | 0.000719 |
nd, not done.
3.9 Array results for confirmed aberrant events
Using the published information for 33 reported missplicing events in DM (Table 5), we identified the corresponding PSRs on the exon array, when present. Table 5 summarizes this information along with the splice indices (SI) and p-values for each of the 36 PSRs (three events are interrogated by two PSRs), if detected in our samples. Of the 33 reported events, six were not represented at all on the array, five additional PSRs were not in the core set, and three of these core probes did not detect the interrogated sequence in our samples. Of the remaining 22 detected PSRs, only three were flagged as significant by our criteria: MBNL2 (exon 7), MEF2A (exon 5a), and PDLIM3 (exon 5a), and those only in DM2. LDB3 exon 7 passed the SI and p-value thresholds in DM2, but was overexpressed >3-fold and therefore was not flagged. Several other events had significant SI and unadjusted p-values, but failed to pass the correction for multiple testing. These included events in MBNL2, MEF2A, MEF2C and PDLIM3 for DM1, MBNL1, SMYD1 and MEF2C for DM2, and MBNL2, MEF2A, MEF2C and PDLIM3 for NMD. If we relaxed our criteria accordingly, the analysis could identify up to six of 22 potentially detectable PSRs (27%). None of the other methods identified more than two of the published missplicing events. Therefore, we concluded that, while the SI method of splice variant analysis was relatively insensitive, in that it only detected a small fraction (3 of 22) of the known events, its positive predictive value (PPV), the fraction of events it predicted which we were actually able to validate, was fairly good (100% of the DM2 events, 65% of the DM1 events, and 41% of the NMD events). We further concluded that novel flagged missplicing events actually represented a subset of true events and therefore reasonable generalizations about pathways could be made.
3.10 Validation of novel DM missplicing events
We attempted validation on 27 predicted missplicing events identified using our first analysis method. PSR sequence from the array annotation was used to identify the exons to be tested. Primers are described in Table S1. While amplification was successful for all assays, only one was positive for the predicted differential splicing, namely exon 6 of SSBP3 (Fig. 2). The single validated event, inclusion of exon 6 of SSBP3, was identified by our initial splice variant analysis algorithm [31], but is not represented in the core probe set used for the SI calculation. We therefore performed calculation of SI using additional probes (Fig. 2A). Upon validation by quantitative RT-PCR (Fig 2A–B), this exon was seen to be differentially spliced in DM1 (p-value=0.00126) and DM2 (p-value=0.00348; Fig. 2C). Although no validation data is available for NMD, the SI calculation confirmed that this alternative splicing event also occurs in NMD.
In addition to missplicing, SSBP3 also showed a trend towards downregulation in all three-disease groups, with fold-changes ranging from FC-1.25 in NMD to FC-1.45 in DM2. To determine whether the missplicing correlated with reduced protein expression, we performed western blotting on both DM1 and DM2 skeletal muscle extracts. While the size difference between the two isoforms precluded resolution, we observed reduction of overall protein levels compared to normal skeletal muscle (Fig. 2D–E). Fig. S4D shows one network of genes, including SSBP3, dysregulated in all three groups. Several known interacting protein partners of SSBP3 in this network, including LDB1, TAL1, TRIM33, ANKRD5 and ZNF226, are upregulated at the mRNA level.
For the 27 candidate missplicing events, only 15 showed the predicted isoforms. In the remaining 12 cases, RT-PCR products were monomorphic and the predicted alternative isoforms were not observed. While there was significance for two events, 13 of 15 showed no significant difference between the groups by quantitative RT-PCR. For TPD52L2 exon 4, significant differential splicing was observed, but in the opposite direction to that predicted by the array. Results are presented in Table 6. Because of the poor prediction of known events by the exon array (Table 5) and the high failure rate for validation of novel candidate splicing events predicted by our first analysis method (Table 6), we elected not to commit our limited RNA/cDNA from patient biopsies for further validations. Instead, the high RT-PCR validation failure rate prompted us to search for a more reliable predictive algorithm for the splice variant analysis of exon array data. The resulting SI algorithm, which gave us improved prediction of known events, is described in the Materials and Methods section.
Table 6.
Results of validation attempts for predicted novel missplicing events*
| Gene | Exon | In/Out | DM1 vs. N | DM2 vs. N | NMD vs. N | Result |
|---|---|---|---|---|---|---|
| ATP9B | 8 | Out | 0.5668 | 0.3534 | 0.1657 | no difference |
| 9b | Out | 0.3849 | 0.4133 | 0.8619 | no difference | |
| 11 | Out | 0.7261 | 0.6737 | 0.7798 | no difference | |
| BTRC | 2a | Out | 0.9578 | 0.6980 | 0.6051 | no difference |
| 2a, 2b | Out | 0.6641 | 0.5674 | 0.5452 | no difference | |
| 2b | Out | 0.3555 | 0.6376 | 0.5807 | no difference | |
| CD46 | 7 | In | 0.9039 | 0.8601 | 0.1370 | no difference |
| IL28RA | 2 | Out | 0.8594 | 0.6413 | 0.3470 | no difference |
| MASP1 | 2a | In | 0.7558 | 0.6472 | 0.1647 | no difference |
| SGCD | 2 | In | 0.6140 | 0.3070 | 0.9961 | no difference |
| SMN1 | 8 | In | 0.0626 | 0.9995 | 0.0738 | no difference |
| SSBP3 | 6 | In | 0.0013 | 0.0035 | na | DM vs. N significant |
| STAU | 1b | In | 0.2834 | 0.6541 | na | no difference |
| 2b | In | 0.7469 | 0.2206 | na | no difference | |
| TPD52L2 | 4 | In | 0.1647 | 0.0037 | 0.0051 | DM2 vs. N significant |
| NMD vs. N significant |
Predicted events with monomorphic/invariant PCR products: ANKD1 (E2), BIN1 (E11), CCNB1IP1 (E5), CTSB (E2b), FGD4 (E6, 14, 18), MKNK1 (E8), SPRY4 (E2), TMC4 (E11), TPM1 (E9b), and YY1AP1 (E4).
3.11 IPA analysis of differential PSRs in DM
We performed pathway analysis on the genes containing the flagged PSRs shared in the comparisons (DM1 + DM2) vs. N. IPA analysis was performed separately on the probes annotated as 5', 3', or cassette. In general, irrespective of whether we analyzed genes with alternative splicing common to DM1 and DM2, or to all three disease groups, we obtained the same set of pathways for 5', 3', or cassette exon probe sets that were seen as dysregulated in the expression analysis. The majority of genes where diseased muscle exhibited alternative isoforms were functionally related to development, gene expression and signaling.
3.12 Comparison of dysregulated genes to genes mutated in inherited neuromuscular disorders
The World Muscle Society (WMS; http://www.worldmusclesociety.org/) lists 333 unique genes that have been identified as harboring mutations that cause 685 inherited neuromuscular disorders (http://www.musclegenetable.fr). In order to further identify unifying themes in dystrophic muscle, we compared this list to the lists of genes dysregulated in our DM1, DM2 and NMD samples. The complete list of overlapping genes and their mutated phenotypes are presented in Table S5. For DM1, 19 genes were shared between both lists; for DM2 there were 29 shared genes, and for NMD 24 shared genes. Ten genes appeared on all lists: AKAP9, ANK2, CLCN1, LAMA4, LDB3, MYLK2, SCN4A, SGCD, SLC22A5, and SYNE1. In all, 40 genes from the WMS list were dysregulated in one or more of the three disease categories. The diseases caused by mutations in many of these shared genes share common features with the DM phenotype, including not just skeletal muscle myopathies, but also cardiomyopathies, arrhythmias and myotonia.
4. Discussion
The prevailing pathomechanistic paradigm for DM is that the aberrant presence of embryonic isoforms is responsible for many, if not most, aspects of the pleiotropic DM phenotype. Our objective in conducting this study was to identify such aberrantly expressed isoforms in skeletal muscle. Our sample set was the largest collection of DM samples to be analyzed to date using the Affymetrix Human Exon 1.0 ST array, and included a number of non-DM neuromuscular disease samples as disease controls. Inclusion of non-DM neuromuscular disease allowed for distinction of DM-specific changes from more generic muscle disease manifestations.
Overall, we found the Affymetrix exon array to be an excellent platform for expression analysis, in that it uses probes spanning the whole gene, thus avoiding the bias often encountered with previous generation 3-prime arrays and potentially detects more dysregulated genes. Our single most striking finding was that most of the expression changes associated with DM were also seen in the non-DM dystrophies. Direct comparison between DM and the other dystrophies in this study identified almost no significant differences. Our findings support the premise that there are few changes in expression that are specific to DM muscle, but rather that expression changes in DM muscle represent a subset of a broader spectrum of dystrophic changes.
Interestingly, we identified many more dysregulated genes in DM2 than in DM1. This was unexpected, and suggested the possibility that DM2 may be either phenotypically more heterogeneous at the cellular level, or mechanistically more complex than DM1. It is likely that additional pathomechanisms are indeed operative in DM2 muscle, possibly due to reduced expression of CNBP [46] or to a reported translational defect [47], but whether these are sufficient to explain the observed differences between DM1 and DM2 remains to be determined. We noted that the DM1 group had, in general, more sample-to-sample heterogeneity, possibly related to variation in repeat length. This heterogeneity could have confounded the identification of dysregulated genes, thus contributing to the larger list of genes dysregulated in DM2, where repeat length has not been reported to be an issue in phenotypic variation [2]. A direct comparison of DM1 to DM2 identified only three genes significantly different between them. Therefore, we concluded that DM1 and DM2 are qualitatively identical with respect to dysregulation of gene expression. While the ratio of up- and down-regulated transcripts in NMD vs. N was about 50:50, overexpression was significantly more frequent in DM than underexpression. This overall bias towards increased steady-state mRNA levels in DM, but not NMD, could be a reflection of the proposed role of MBNL depletion in reduced translation of a subset of mRNAs [17].
Osborne and colleagues [36] performed expression profiling with the Affymetrix Mouse Genome 430 microarray on HSALR DM1 transgenic mice expressing (CUG)250, and compared their results to profiles of Clcn1 or Mbnl1 null mice. They found that the majority of expression changes in the HSALR mice could be attributed to Mbnl1 sequestration. Similar to our findings, genes related to calcium signaling and homeostasis were among the most dysregulated. However, among their top 20-dysregulated genes only five were found to be dysregulated (FC>|1.3|) in our DM1 vs. N comparison. These were PLEKHO1 (FC -1.96), UCHL1 (FC -1.71), GDAP1 (FC 1.49), EDA2R (FC 1.56), and CILP (FC 1.94). All five were similarly dysregulated in DM2, while PLEKHO1 and CILP were also dysregulated in NMD (FC -1.89 and FC 2.69, respectively). It is worth noting that PLEKHO1 and UCHL1 were dysregulated in the opposite direction in our human samples from the HSALR mice. Overall, it does not appear as if the HSALR mice are a close model for gene expression in human DM1 patients. Nevertheless, the shared dysregulated genes deserve closer attention.
Using differential display RT-PCR, Zhang and colleagues [48] examined differential expression between normal and pathological skeletal muscles from the same autosomal recessive LGMD patient. Overall, they observed down-regulation of structural proteins of skeletal muscle fibers and up-regulation of proteins involved in signal transduction and regulation of gene expression. Of the 27-dysregulated genes identified by them and also detected in our skeletal muscle samples, 10 were concordantly dysregulated in both DM1 and DM2 and nine of these were similarly dysregulated in NMD. This high proportion of shared dysregulated genes between LGMD, DM and the other dystrophies studied here provide further evidence for the generic nature of many expression changes in dystrophic muscle.
Not surprisingly, a number of the dysregulated genes were those which, when mutated, are responsible for other neuromuscular diseases. We compared our lists of dysregulated genes with the World Muscle Society list of neuromuscular inherited diseases (http://www.worldmusclesociety.org/). This analysis showed that there are basically two groups of genes that are both dysregulated in these dystrophies and mutated in neuromuscular diseases. The first group, dysregulated primarily in DM, codes for muscle structural proteins, represented by myosins, tropomyosins and other cytoskeletal proteins (e.g., dystrophin and nexin). The second group, dysregulated to some extent in all disease groups, was primarily regulatory and related to the NFKB complex.
We also examined the possibility that Dicer-mediated degradation of the DM1 repeat transcript or its antisense counterpart might influence the steady-state levels of other mRNAs containing normally polymorphic CAG or CUG repeats [34]. As reported previously [36], we found no evidence for altered steady-state levels for (CAG)n/(CUG)n containing transcripts as a class. However, from our study it was not possible to determine whether repeat fragments resulting from the increased Dicer activity had any effect on translation.
Because of the location of probe sets in most exons, the Affymetrix Exon Array should provide an excellent platform to detect alternative isoform usage [49, 50]. We and others have successfully used it for this purpose in a number of studies [31, 49, 51–56]. We were, however, disappointed to discover that aberrantly spliced exons in DM were not readily identified using this exon array platform. We failed to identify a number of known and validated events, even when probes on the array represented the relevant exon. There are several possible explanations for these false-negative results, including technical issues relating to probe sequence and hybridization kinetics as well as biological issues such as sample heterogeneity and subtle isoform differences. However, in our opinion, the problem of false-positive results is the most serious limitation of this platform. Large transcripts (with >20 PSRs) have so much probe-to-probe variability that small differences between sample groups become significant, even in the absence of true alternative splicing. Skeletal muscle contains many such large structural genes, and these nearly always gave false-positive results for differential splicing. Since simple ANOVA cannot filter out this “noise,” we and other groups developed splice index calculations using several distinct yet similar approaches [57]. However, it appears that such analytical approaches cannot sufficiently reduce this complexity to unequivocally identify differently spliced genes in disease muscle.
One novel missplicing event identified in this study occurred in SSBP3. Human SSBP3 was first identified as a member of an evolutionarily conserved and ubiquitously expressed gene family with possible involvement in cancer [58]. The best characterized biochemical activity of SSBP3 is its ability to directly bind and protect the Lim Domain Binding Protein 1 (LDB1) from degradation [59–62]. SSBP3 was recently also shown to be required for neuronal morphology [63]. SSBP3 expression is high in skeletal muscle, heart, brain, kidney and hematopoietic tissues, but little is known about either its function in muscle or its isoform-specific functions. Here, we have demonstrated missplicing of exon 6 of SSBP3 at variable levels in DM1, DM2 and NMD, and reduced overall protein levels in DM. Several known targets or interacting partners of SSBP3, including COL1A1, COL1A2, COL3A1, LDB1, TAL1, TRIM33, ANKRD5 and ZNF226, were upregulated at the mRNA level, consistent with a possible repressive role for SSBP3 in transcription of these genes. Pathway analysis also suggests that SSBP3 may have links to the NFKB network, persistent inhibition of which has been linked directly to apoptosis, inappropriate immune cell development, and delayed cell growth [64]. Many of the downstream targets of NFKB are down-regulated in our patient samples, suggesting a possible role for SSBP3 in wasting or inflammation in dystrophic muscle.
Overall, our results conclusively show that most of the differential isoform usage seen in DM is also seen in NMD patient muscle. DM-specific missplicing certainly occurs for a few genes, and may be important for specific aspects of the phenotype. One prominent example is the inclusion of a fetal exon of CLCN1, which results in myotonia [18–22]. Aberrant presence of fetal isoforms of other proteins such as TNNT3, TNNT2, and ATP2A1 [33, 39, 65, 66] are also probably important in skeletal or cardiac muscle pathology in DM, and these were not seen, at least to the same extent, in the NMD samples. Some of the aberrant splicing reported in other tissues, such as brain, may also play a role in the DM-specific phenotype. However, we found no evidence for a widespread DM-specific spliceopathy in skeletal muscle and therefore suggest that alternative splicing in DM (and NMD) may not be the driving mechanism for the muscle pathology, since the same pathways also show expression changes unrelated to splicing. We also found no evidence to support the notion of splicing differences between DM1 and DM2. Differently spliced genes in the comparison DM1 vs. N were essentially a subset of the DM2 vs. N comparison. Most of the probes in the non-intersecting lists satisfy two, but not all three, of our criteria and reducing the stringency for DM1 to |SI|>1.3 produced essentially complete overlap with DM2.
Other studies have been conducted in an attempt to identify splicing events driving the DM phenotype. Orengo and colleagues [29] examined eight, previously associated with DM, missplicing events in four mouse models of muscular dystrophy (DM1, limb girdle muscular dystrophy, congenital merosin-deficient muscular dystrophy, and Duchenne muscular dystrophy) and two myotoxin (cardiotoxin and notexin) muscle injury models. They concluded that expression of neonatal alternative splicing isoforms in adult skeletal muscle occurs in several diverse models of both muscle degeneration and muscle damage as a result of a robust regenerative process. Our results are consistent with this observation. In a recent review, Kalsotra and Cooper [67] noted that during skeletal muscle differentiation, genes involved in actin binding, integrin signaling, nucleic acid metabolism and splicing are generally regulated through alternative splicing, while genes involved in muscle contraction, development, cell cycle and ion transport are generally regulated through transcription. We observed similar relationships within our data set.
Using the same Affymetrix Human Exon 1.0 ST array, Koebis and colleagues [68] investigated muscle samples from six DM1, mostly very young or congenital, and seven age-/sex-matched control cases. Using a slightly different splice index calculation from ours, they reported 54 differentially expressed probes, eight of which were concordantly dysregulated with our DM1 samples. Two were for previously reported genes (PDLIM3 and LDB3), while six were in novel genes (MFF, CD47, ABLIM1, NCOR2, ARHGEF7 and MYOM1). With the exception of LDB3 and MFF, all were similarly dysregulated in our NMD samples. Interestingly, three probe sets in BNIP1, RPA3 and ABCD4 were dysregulated in the opposite direction (inclusion rather than exclusion, or vice versa).
Studying the HSALR and Mbnl1−/− DM1 mouse models using a custom splice microarray, Du and colleagues [16] determined that loss of Mbnl1 accounts for >80% of the splicing pathology due to expression of the mutant (CUG)DM1 RNA and identified over 200 splicing events that were altered by the loss of Mbnl1. They validated that DM1 patients suffer many of the same missplicing events, including six novel missplicing events in NFIX, SMYD1, SPAG9, PPP2Y5D, MTDH and GNAS. On the commercial exon array we used, the equivalent probe in GNAS was not detected and the one in NFIX did not appear to be alternatively spliced in our DM1 samples. The corresponding probe sets in the other four genes had splice indices and unadjusted p-values indicative of alternative splicing in DM1 but were not significant after correction for multiple testing. Interestingly, similar to the reported mRNA dysregulation of extracellular matrix (ECM) genes in the DM1 mouse models [16], we saw transcriptional dysregulation of numerous ECM genes, including the same procollagen genes COL1A2 and COL15A1, as well as numerous other collagen genes in the human DM patients.
In summary, we characterized the largest collection of DM patient samples analyzed to date for altered splicing using the Affymetrix Human Exon 1.0 ST array and included non-DM neuromuscular disease patient samples as disease controls. We found that DM1 and DM2 skeletal muscles were essentially identical to each other for both expression and splicing. We found no evidence for widespread missplicing as a DM-specific pathomechanism in skeletal muscle. Rather, most expression and splicing changes were shared between multiple muscular dystrophies, as previously suggested [28, 29]. We also found no evidence to support the hypothesis that steady-state levels of transcripts containing (CAG)n/(CUG)n repeats are affected adversely by the presence of mutant (C/CUG)DM transcripts. Surprisingly, we found little similarity between genes dysregulated or misspliced in our DM1 patient muscle biopsies and results reported for various mouse models of DM1. Finally, we conclude that the Affymetrix exon array is not the most efficient platform for identification of alternative missplicing events in skeletal muscle. Although exon array and RNA-seq profiling have shown strong concordance at both the gene and exon mRNA levels [69], it remains to be seen if similar studies as ours on more uniformly, prospectively collected patient tissues using RNA-seq with a wider dynamic range will provide the same or different insights with respect to missplicing in DM.
Supplementary Material
Acknowledgements
We wish to acknowledge Tamer Ahmed, Shohrae Hajibashi, and Shodimu-Emmanuel Olufemi for technical support, Spiridon Tsavachidis for additional statistical and bioinformatics support, and Gil Cote for helpful discussions. We also thank Wlodzimierz J. Kryzosiak for sharing the annotated gene table with polymorphic (CAG)n/(CTG)n repeats. RK was supported by grants from NIH (AR48171), Muscular Dystrophy Association, USA, and the Kleberg Foundation. LTT was supported by a grant from NIH (AR44387) and JG by FIS-FEDER grant 13/1272. BU was supported by funding from the Folkhälsan Research Foundation, and grants from the Liv & Hälsa Foundation, the Vasa Central Hospital District Medical Research funds and Kung Gustav V Adolfs och Drottning Victorias minnesfond Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Data deposition: GEO Accession Number GSE48828
References
- [1].Harper PS. Myotonic Dystrophy. 2nd ed W.B. Saunders Co; London, UK: 1989. [Google Scholar]
- [2].Udd B, Krahe R. The myotonic dystrophies: molecular, clinical, and therapeutic challenges. Lancet Neurol. 2012;11:891–905. doi: 10.1016/S1474-4422(12)70204-1. [DOI] [PubMed] [Google Scholar]
- [3].Brook JD, McCurrach ME, Harley HG, et al. Molecular basis of myotonic dystrophy: expansion of a trinucleotide (CTG) repeat at the 3' end of a transcript encoding a protein kinase family member. Cell. 1992;68:799–808. doi: 10.1016/0092-8674(92)90154-5. [DOI] [PubMed] [Google Scholar]
- [4].Fu YH, Pizzuti A, Fenwick RG, Jr., et al. An unstable triplet repeat in a gene related to myotonic muscular dystrophy. Science. 1992;255:1256–1258. doi: 10.1126/science.1546326. [DOI] [PubMed] [Google Scholar]
- [5].Mahadevan M, Tsilfidis C, Sabourin L, et al. Myotonic dystrophy mutation: an unstable CTG repeat in the 3' untranslated region of the gene. Science. 1992;255:1253–1255. doi: 10.1126/science.1546325. [DOI] [PubMed] [Google Scholar]
- [6].Liquori CL, Ricker K, Moseley ML, et al. Myotonic dystrophy type 2 caused by a CCTG expansion in intron 1 of ZNF9. Science. 2001;293:864–867. doi: 10.1126/science.1062125. [DOI] [PubMed] [Google Scholar]
- [7].Bachinski LL, Udd B, Meola G, et al. Confirmation of the type 2 myotonic dystrophy (CCTG)n expansion mutation in patients with proximal myotonic myopathy/proximal myotonic dystrophy of different European origins: a single shared haplotype indicates an ancestral founder effect. Am J Hum Genet. 2003;73:835–848. doi: 10.1086/378566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Amack JD, Mahadevan MS. The myotonic dystrophy expanded CUG repeat tract is necessary but not sufficient to disrupt C2C12 myoblast differentiation. Hum Mol Genet. 2001;10:1879–1887. doi: 10.1093/hmg/10.18.1879. [DOI] [PubMed] [Google Scholar]
- [9].Mankodi A, Logigian E, Callahan L, et al. Myotonic dystrophy in transgenic mice expressing an expanded CUG repeat. Science. 2000;289:1769–1773. doi: 10.1126/science.289.5485.1769. [DOI] [PubMed] [Google Scholar]
- [10].Cooper TA. Molecular biology. Neutralizing toxic RNA. Science. 2009;325:272–273. doi: 10.1126/science.1177452. [DOI] [PubMed] [Google Scholar]
- [11].La Spada AR, Taylor JP. Repeat expansion disease: progress and puzzles in disease pathogenesis. Nat Rev Genet. 2010;11:247–258. doi: 10.1038/nrg2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Osborne RJ, Thornton CA. RNA-dominant diseases. Hum Mol Genet. 2006;15(Spec No 2):R162–169. doi: 10.1093/hmg/ddl181. [DOI] [PubMed] [Google Scholar]
- [13].Wheeler TM, Thornton CA. Myotonic dystrophy: RNA-mediated muscle disease. Curr Opin Neurol. 2007;20:572–576. doi: 10.1097/WCO.0b013e3282ef6064. [DOI] [PubMed] [Google Scholar]
- [14].Lin X, Miller JW, Mankodi A, et al. Failure of MBNL1-dependent postnatal splicing transitions in myotonic dystrophy. Hum Mol Genet. 2006;15:2087–2097. doi: 10.1093/hmg/ddl132. [DOI] [PubMed] [Google Scholar]
- [15].Kalsotra A, Xiao X, Ward AJ, et al. A postnatal switch of CELF and MBNL proteins reprograms alternative splicing in the developing heart. Proc Natl Acad Sci USA. 2008;105:20333–20338. doi: 10.1073/pnas.0809045105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Du H, Cline MS, Osborne RJ, et al. Aberrant alternative splicing and extracellular matrix gene expression in mouse models of myotonic dystrophy. Nat Struct Mol Biol. 2010;17:187–193. doi: 10.1038/nsmb.1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Wang ET, Cody NA, Jog S, et al. Transcriptome-wide regulation of pre-mRNA splicing and mRNA localization by muscleblind proteins. Cell. 2012;150:710–724. doi: 10.1016/j.cell.2012.06.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Charlet-B N, Savkur RS, Singh G, Philips AV, Grice EA, Cooper TA. Loss of the muscle-specific chloride channel in type 1 myotonic dystrophy due to misregulated alternative splicing. Mol Cell. 2002;10:45–53. doi: 10.1016/s1097-2765(02)00572-5. [DOI] [PubMed] [Google Scholar]
- [19].Lueck JD, Lungu C, Mankodi A, et al. Chloride channelopathy in myotonic dystrophy resulting from loss of posttranscriptional regulation for CLCN1. Am J Physiol Cell Physiol. 2007;292:C1291–1297. doi: 10.1152/ajpcell.00336.2006. [DOI] [PubMed] [Google Scholar]
- [20].Lueck JD, Mankodi A, Swanson MS, Thornton CA, Dirksen RT. Muscle chloride channel dysfunction in two mouse models of myotonic dystrophy. J Gen Physiol. 2007;129:79–94. doi: 10.1085/jgp.200609635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Mankodi A, Takahashi MP, Jiang H, et al. Expanded CUG repeats trigger aberrant splicing of ClC-1 chloride channel pre-mRNA and hyperexcitability of skeletal muscle in myotonic dystrophy. Mol Cell. 2002;10:35–44. doi: 10.1016/s1097-2765(02)00563-4. [DOI] [PubMed] [Google Scholar]
- [22].Wheeler TM, Lueck JD, Swanson MS, Dirksen RT, Thornton CA. Correction of ClC-1 splicing eliminates chloride channelopathy and myotonia in mouse models of myotonic dystrophy. J Clin Invest. 2007;117:3952–3957. doi: 10.1172/JCI33355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Kanadia RN, Johnstone KA, Mankodi A, et al. A muscleblind knockout model for myotonic dystrophy. Science. 2003;302:1978–1980. doi: 10.1126/science.1088583. [DOI] [PubMed] [Google Scholar]
- [24].Mahadevan MS, Yadava RS, Yu Q, et al. Reversible model of RNA toxicity and cardiac conduction defects in myotonic dystrophy. Nat Genet. 2006;38:1066–1070. doi: 10.1038/ng1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Timchenko NA, Patel R, Iakova P, Cai ZJ, Quan L, Timchenko LT. Overexpression of CUG triplet repeat-binding protein, CUGBP1, in mice inhibits myogenesis. J Biol Chem. 2004;279:13129–13139. doi: 10.1074/jbc.M312923200. [DOI] [PubMed] [Google Scholar]
- [26].Gomes-Pereira M, Cooper TA, Gourdon G. Myotonic dystrophy mouse models: towards rational therapy development. Trends Mol Med. 2011;17:506–517. doi: 10.1016/j.molmed.2011.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Sicot G, Gourdon G, Gomes-Pereira M. Myotonic dystrophy, when simple repeats reveal complex pathogenic entities: new findings and future challenges. Hum Mol Genet. 2011;20:R116–123. doi: 10.1093/hmg/ddr343. [DOI] [PubMed] [Google Scholar]
- [28].Bachinski LL, Sirito M, Bohme M, Baggerly KA, Udd B, Krahe R. Altered MEF2 isoforms in myotonic dystrophy and other neuromuscular disorders. Muscle Nerve. 2010;42:856–863. doi: 10.1002/mus.21789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Orengo JP, Ward AJ, Cooper TA. Alternative splicing dysregulation secondary to skeletal muscle regeneration. Ann Neurol. 2011;69:681–690. doi: 10.1002/ana.22278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Sallinen R, Vihola A, Bachinski LL, et al. New methods for molecular diagnosis and demonstration of the (CCTG)n mutation in myotonic dystrophy type 2 (DM2) Neuromuscul Disord. 2004;14:274–283. doi: 10.1016/j.nmd.2004.01.002. [DOI] [PubMed] [Google Scholar]
- [31].Cheung HC, Baggerly KA, Tsavachidis S, et al. Global analysis of aberrant premRNA splicing in glioblastoma using exon expression arrays. BMC Genomics. 2008;9:216. doi: 10.1186/1471-2164-9-216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Udd B, Meola G, Krahe R, et al. Myotonic Dystrophy DM2/PROMM and other myotonic dystrophies: 140th ENMC International Workshop with guidelines on management. Neuromuscul Disord. 2006 doi: 10.1016/j.nmd.2006.03.010. [DOI] [PubMed] [Google Scholar]
- [33].Vihola A, Bachinski LL, Sirito M, et al. Differences in aberrant expression and splicing of sarcomeric proteins in the myotonic dystrophies DM1 and DM2. Acta Neuropathol. 2010;119:465–479. doi: 10.1007/s00401-010-0637-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Krol J, Fiszer A, Mykowska A, Sobczak K, de Mezer M, Krzyzosiak WJ. Ribonuclease dicer cleaves triplet repeat hairpins into shorter repeats that silence specific targets. Mol Cell. 2007;25:575–586. doi: 10.1016/j.molcel.2007.01.031. [DOI] [PubMed] [Google Scholar]
- [35].Batra R, Charizanis K, Swanson MS. Partners in crime: bidirectional transcription in unstable microsatellite disease. Hum Mol Genet. 2010;19:R77–82. doi: 10.1093/hmg/ddq132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Osborne RJ, Lin X, Welle S, et al. Transcriptional and post-transcriptional impact of toxic RNA in myotonic dystrophy. Hum Mol Genet. 2009;18:1471–1481. doi: 10.1093/hmg/ddp058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Orengo JP, Chambon P, Metzger D, Mosier DR, Snipes GJ, Cooper TA. Expanded CTG repeats within the DMPK 3' UTR causes severe skeletal muscle wasting in an inducible mouse model for myotonic dystrophy. Proc Natl Acad Sci USA. 2008;105:2646–2651. doi: 10.1073/pnas.0708519105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Jiang H, Mankodi A, Swanson MS, Moxley RT, Thornton CA. Myotonic dystrophy type 1 is associated with nuclear foci of mutant RNA, sequestration of muscleblind proteins and deregulated alternative splicing in neurons. Hum Mol Genet. 2004;13:3079–3088. doi: 10.1093/hmg/ddh327. [DOI] [PubMed] [Google Scholar]
- [39].Kimura T, Nakamori M, Lueck JD, et al. Altered mRNA splicing of the skeletal muscle ryanodine receptor and sarcoplasmic/endoplasmic reticulum Ca2+-ATPase in myotonic dystrophy type 1. Hum Mol Genet. 2005;14:2189–2200. doi: 10.1093/hmg/ddi223. [DOI] [PubMed] [Google Scholar]
- [40].Nakamori M, Kimura T, Fujimura H, Takahashi MP, Sakoda S. Altered mRNA splicing of dystrophin in type 1 myotonic dystrophy. Muscle Nerve. 2007;36:251–257. doi: 10.1002/mus.20809. [DOI] [PubMed] [Google Scholar]
- [41].Nakamori M, Kimura T, Kubota T, et al. Aberrantly spliced alpha-dystrobrevin alters alpha-syntrophin binding in myotonic dystrophy type 1. Neurology. 2008;70:677–685. doi: 10.1212/01.wnl.0000302174.08951.cf. [DOI] [PubMed] [Google Scholar]
- [42].Savkur RS, Philips AV, Cooper TA. Aberrant regulation of insulin receptor alternative splicing is associated with insulin resistance in myotonic dystrophy. Nat Genet. 2001;29:40–47. doi: 10.1038/ng704. [DOI] [PubMed] [Google Scholar]
- [43].Sergeant N, Sablonniere B, Schraen-Maschke S, et al. Dysregulation of human brain microtubule-associated tau mRNA maturation in myotonic dystrophy type 1. Hum Mol Genet. 2001;10:2143–2155. doi: 10.1093/hmg/10.19.2143. [DOI] [PubMed] [Google Scholar]
- [44].Buj-Bello A, Furling D, Tronchere H, et al. Muscle-specific alternative splicing of myotubularin-related 1 gene is impaired in DM1 muscle cells. Hum Mol Genet. 2002;11:2297–2307. doi: 10.1093/hmg/11.19.2297. [DOI] [PubMed] [Google Scholar]
- [45].Philips AV, Timchenko LT, Cooper TA. Disruption of splicing regulated by a CUG-binding protein in myotonic dystrophy. Science. 1998;280:737–741. doi: 10.1126/science.280.5364.737. [DOI] [PubMed] [Google Scholar]
- [46].Raheem O, Olufemi SE, Bachinski LL, et al. Mutant (CCTG)n expansion causes abnormal expression of zinc finger protein 9 (ZNF9) in myotonic dystrophy type 2. Am J Pathol. 2010;177:3025–3036. doi: 10.2353/ajpath.2010.100179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Schneider-Gold C, Timchenko LT. CCUG repeats reduce the rate of global protein synthesis in myotonic dystrophy type 2. Rev Neurosci. 2010;21:19–28. doi: 10.1515/revneuro.2010.21.1.19. [DOI] [PubMed] [Google Scholar]
- [48].Zhang Y, Ye J, Chen D, et al. Differential expression profiling between the relative normal and dystrophic muscle tissues from the same LGMD patient. J Transl Med. 2006;4:53. doi: 10.1186/1479-5876-4-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Clark TA, Schweitzer AC, Chen TX, et al. Discovery of tissue-specific exons using comprehensive human exon microarrays. Genome Biol. 2007;8:R64. doi: 10.1186/gb-2007-8-4-r64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Ha K, Coulombe-Huntington J, Majewski J. Comparison of Affymetrix Gene Array with the Exon Array shows potential application for detection of transcript isoform variation. BMC Genomics. 2009;10:519. doi: 10.1186/1471-2164-10-519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Izaguirre DI, Zhu W, Hai T, Cheung HC, Krahe R, Cote GJ. PTBP1-dependent regulation of USP5 alternative RNA splicing plays a role in glioblastoma tumorigenesis. Mol Carcinogenesis. 2012;51:895–906. doi: 10.1002/mc.20859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Cheung HC, Hai T, Zhu W, et al. Splicing factors PTBP1 and PTBP2 promote proliferation and migration of glioma cell lines. Brain. 2009;132:2277–2288. doi: 10.1093/brain/awp153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Hu J, He X, Cote GJ, Krahe R. Singular Value Decomposition-based Alternative Splicing Detection. J Am Stat Assoc. 2009;104:944–953. doi: 10.1198/jasa.2009.ap08283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].de la Grange P, Gratadou L, Delord M, Dutertre M, Auboeuf D. Splicing factor and exon profiling across human tissues. Nucl Acids Res. 2010;38:2825–2838. doi: 10.1093/nar/gkq008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Menghi F, Jacques TS, Barenco M, et al. Genome-wide analysis of alternative splicing in medulloblastoma identifies splicing patterns characteristic of normal cerebellar development. Cancer Res. 2011;71:2045–2055. doi: 10.1158/0008-5472.CAN-10-2519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Kong SW, Hu YW, Ho JW, et al. Heart failure-associated changes in RNA splicing of sarcomere genes. Circ Cardiovasc Genet. 2010;3:138–146. doi: 10.1161/CIRCGENETICS.109.904698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Yamashita Y, Matsuura T, Shinmi J, et al. Four parameters increase the sensitivity and specificity of the exon array analysis and disclose 25 novel aberrantly spliced exons in myotonic dystrophy. J Hum Genet. 2012 doi: 10.1038/jhg.2012.37. [DOI] [PubMed] [Google Scholar]
- [58].Castro P, Liang H, Liang JC, Nagarajan L. A novel, evolutionarily conserved gene family with putative sequence-specific single-stranded DNA-binding activity. Genomics. 2002;80:78–85. doi: 10.1006/geno.2002.6805. [DOI] [PubMed] [Google Scholar]
- [59].Xu Z, Meng X, Cai Y, Liang H, Nagarajan L, Brandt SJ. Single-stranded DNA-binding proteins regulate the abundance of LIM domain and LIM domain-binding proteins. Genes Dev. 2007;21:942–955. doi: 10.1101/gad.1528507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Gungor C, Taniguchi-Ishigaki N, Ma H, et al. Proteasomal selection of multiprotein complexes recruited by LIM homeodomain transcription factors. Proc Natl Acad Sci USA. 2007;104:15000–15005. doi: 10.1073/pnas.0703738104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Cai Y, Xu Z, Nagarajan L, Brandt SJ. Single-stranded DNA-binding proteins regulate the abundance and function of the LIM-homeodomain transcription factor LHX2 in pituitary cells. Biochem Biophys Res Commun. 2008;373:303–308. doi: 10.1016/j.bbrc.2008.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Wang Y, Klumpp S, Amin HM, et al. SSBP2 is an in vivo tumor suppressor and regulator of LDB1 stability. Oncogene. 2010;29:3044–3053. doi: 10.1038/onc.2010.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Hashimoto Y, Muramatsu K, Kunii M, et al. Uncovering genes required for neuronal morphology by morphology-based gene trap screening with a revertible retrovirus vector. FASEB J. 2012;26:4662–4674. doi: 10.1096/fj.12-207530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Romashkova JA, Makarov SS. NF-kappaB is a target of AKT in anti-apoptotic PDGF signalling. Nature. 1999;401:86–90. doi: 10.1038/43474. [DOI] [PubMed] [Google Scholar]
- [65].Ho TH, Bundman D, Armstrong DL, Cooper TA. Transgenic mice expressing CUG-BP1 reproduce splicing mis-regulation observed in myotonic dystrophy. Hum Mol Genet. 2005;14:1539–1547. doi: 10.1093/hmg/ddi162. [DOI] [PubMed] [Google Scholar]
- [66].Vicente-Crespo M, Pascual M, Fernandez-Costa JM, et al. Drosophila muscleblind is involved in troponin T alternative splicing and apoptosis. PLoS One. 2008;3:e1613. doi: 10.1371/journal.pone.0001613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Kalsotra A, Cooper TA. Functional consequences of developmentally regulated alternative splicing. Nat Rev Genet. 2011;12:715–729. doi: 10.1038/nrg3052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Koebis M, Ohsawa N, Kino Y, Sasagawa N, Nishino I, Ishiura S. Alternative splicing of myomesin 1 gene is aberrantly regulated in myotonic dystrophy type 1. Genes Cells. 2011;16:961–972. doi: 10.1111/j.1365-2443.2011.01542.x. [DOI] [PubMed] [Google Scholar]
- [69].Raghavachari N, Barb J, Yang Y, et al. A systematic comparison and evaluation of high density exon arrays and RNA-seq technology used to unravel the peripheral blood transcriptome of sickle cell disease. BMC Med Genomics. 2012;5:28. doi: 10.1186/1755-8794-5-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.