

Abstract
Recombinant neuronal nitric-oxide synthase (nNOS) expressed in baculovirus-infected Sf9 cells contains approximately 1 equiv of tightly bound tetrahydrobiopterin (BH4) per dimer and binds a second equivalent with a dissociation constant in the 10–7–10–6 M range. Less is known about the pterin-binding properties of nNOS originating from expression systems such as Escherichia coli that do not produce BH4. We determined the binding properties of E. coli-expressed nNOS for BH4 and several inhibitory pterins by monitoring their effects on enzyme activity. E. coli-expressed nNOS as isolated was activated by BH4 monophasically with EC50 ≈ 2 × 10–7 M, demonstrating a lack of tight pterin binding. However, overnight incubation with BH4 resulted in tight binding of one BH4 per dimer, yielding an enzyme that resembled Sf9-expressed nNOS. Tight pterin binding was also induced by preincubation with 4-amino-tetrahydrobiopterin, but not by 7,8-dihydrobiopterin or 4-amino-dihydrobiopterin, suggesting that tight-binding site formation requires preincubation with a fully reduced pteridine. Kinetic experiments showed that tight-binding site formation takes approximately 10 min with 1 μM BH4 (2 min with 1 μM 4-amino-BH4) at 4 °C. Anaerobic preincubation experiments demonstrated that O2 is not involved in the process. Gel electrophoretic studies suggest that tight-binding site formation is accompanied by an increase in the strength of the NOS dimer. We propose that incubation of pterin-free nNOS with BH4 creates one tight pterin-binding site per dimer, leaving the other site unaffected, in a reaction that involves redox chemistry.
Nitric oxide (NO) plays a plethora of physiological and pathophysiological roles in biology.1 The main source of NO in mammals is the enzyme nitric-oxide synthase (NOS, EC 1.14.13.39), which catalyzes the conversion of l-arginine (Arg) into l-citrulline and NO.2−4 The three isoforms, neuronal, endothelial, and inducible NOS (nNOS, eNOS, and iNOS), differ in tissue distribution and physiological function. All isoforms are dimeric and utilize O2 and the electron donor NADPH as cosubstrates. A key role in the mechanism of NO synthesis is played by the essential cofactor tetrahydrobiopterin ((6R)-5,6,7,8-tetrahydro-l-biopterin, BH4).5−8 In the absence of BH4, the enzyme still oxidizes NADPH and reduces O2, but the oxidation of Arg is blocked, a phenomenon known as uncoupling.3−6 Under these conditions, NOS produces O2– instead of NO, with major potential pathophysiological consequences: the bioavailability of NO decreases by diminished NO production and efficient scavenging of NO by O2–, and the reaction between O2– and NO results in the formation of the strongly oxidizing and presumably deleterious product peroxynitrite (ONOO–).
Recombinant nNOS expressed in baculovirus-infected Sf9 insect cells, which have the capacity to synthesize BH4 endogenously, contains a substoichiometric amount of BH4 after purification, usually amounting to approximately 50% per dimer or heme.9−13 Similar stoichiometries have been reported for the other isoforms as well.14−19 Addition of BH4 to such preparations causes full saturation of the enzyme with BH4, accompanied by increased NO formation and diminished O2–/H2O2 production.12,20−22 Addition of 7,8-dihydrobiopterin (BH2), which binds to NOS with comparable affinity as BH4 but does not support NO synthesis, to BH4-saturated nNOS lowers NO production to the level observed before BH4 addition but does not affect the basal activity.9,23 On the basis of these observations and detailed binding studies with radiolabeled BH4 and BH4-deficient nNOS, we proposed that BH4 binds to NOS anticooperatively.12,21 Accordingly, dimeric NOS has two identical binding sites with very high affinity for BH4 (≤1 nM), but binding of BH4 to one site lowers the affinity of the second site by several orders of magnitude.21,24−26 This concept implies reversible binding of BH4 to both sites, in line with the observation that all bound radioligand dissociated from the enzyme in the presence of unlabeled BH4.9,21,26
Although this model explained most of the data, some questions remained. Specifically, if high-affinity BH4 binding is reversible, it is unclear why inhibitory pteridines such as BH2 cannot block NOS activity completely.9,27,28 There also appears to be a discrepancy between the enzyme expressed in and purified from baculovirus-infected Sf9 cells and NOS from expression systems like Escherichia coli that do not synthesize BH4: with some exceptions,19,26,29 there is little evidence in the literature for tight BH4 binding to such enzyme species.30 In this respect, it may be relevant that iNOS was reported to form undisruptable dimers when expressed in Sf9 cells but not in E. coli, suggesting an effect of the expression system on the enzyme structure.31
To resolve these issues, we decided to revisit the topic of high- and low-affinity BH4 binding. Specifically, we searched for answers to the following questions: (i) Can we reproduce the previously published observations of high- and low-affinity binding of BH4 to baculovirus-infected Sf9-expressed nNOS that prompted us to propose anticooperative BH4 binding? (ii) Does E. coli-expressed BH4-free nNOS behave differently in this respect? (iii) If so, can these differences be offset by preincubation with BH4? The results of the present study confirm the heterogeneity of BH4 binding in nNOS derived from Sf9 cells. Such heterogeneity is absent in E. coli-derived nNOS initially but becomes evident after preincubation with BH4. The same effect is observed upon preincubation with 4-amino-tetrahydrobiopterin (4-amino-BH4) but not with the dihydropteridines BH2 and 4-amino-dihydrobiopterin (4-amino-BH2). We propose that incubation of pterin-free nNOS with BH4 results in one tight-binding site per dimer in a reaction that involves redox chemistry.
Materials and Methods
Materials
l-[2,3,4,5-3H]Arginine hydrochloride ([3H]-Arg, 57 Ci/mmol) was from American Radiolabeled Chemicals Inc., purchased through Humos Diagnostic GmbH (Maria Enzersdorf, Austria). [3′-3H](6R)-5,6,7,8-Tetrahydro-l-biopterin ([3H]-BH4, 14 Ci/mmol) was prepared as described.32 Pteridines were from Dr. B. Schircks Laboratories, Jona, Switzerland. Stock solutions of tetrahydropteridines were prepared in 10 mM HCl; dihydropteridines were dissolved in water. Oxyhemoglobin (oxyHb) was prepared from hemoglobin (Sigma-Aldrich, Cat. No. H2500) immediately before use.33 Other chemicals were from Sigma-Aldrich (Vienna, Austria).
Enzyme Expression and Purification
Rat recombinant (BH4-containing) nNOS, designated Sf9-nNOS hereafter, was purified from baculovirus-infected insect cells as described.34
To obtain E. coli-expressed nNOS, designated ECo-nNOS hereafter, we prepared the human enzyme as a glutathione-S-transferase fusion protein according to a standard protocol. Briefly, human nNOS cDNA sequences were obtained by PCR using a pcDNA3 plasmid with a human nNOS insert and primers that were designed to incorporate NdeI and NotI restriction sites. Subsequently this product was cloned into the NdeI–NotI sites of a pGEX-4T1 vector with the help of T4 DNA ligase (Invitrogen; #15224-017) to create a glutathione-S-transferase–nNOS fusion construct under transcriptional regulation by the inducible bacterial tac promoter. This construct was amplified in chemical competent BL21 cells (One Shot TOP-10 Chemical Competent E. coli; Life Technologies) and used to transfect BL21(DE3) E. coli. Expression of the fusion protein was induced for 20 h with 0.2 mM isopropyl-1-thio-β-d-galactopyranoside in a freshly diluted overnight bacterial culture. Bacteria were pelleted and resuspended in lysis buffer (50 mM tris(hydroxymethyl)-aminomethane (Tris, pH 7.5), 100 μg/mL lysozyme, 1 mM dithiothreitol (DTT), 150 mM NaCl, 0.1% Triton X-100, 1 mM phenylmethylsulfonyl fluoride), sonicated for 8 × 10 s on ice, and centrifuged (20 min, 17 000 rpm, 4 °C). The supernatant was incubated with glutathione-sepharose beads (Sigma-Aldrich, Cat. Nr. G4510) for 4 h at 4 °C. The beads were washed three times with 50 mM Tris (pH 7.5), 1 mM DTT, 500 mM NaCl, and 0.1% Triton X-100, and three times with 50 mM Tris (pH 7.5), 1 mM DTT, 150 mM NaCl, 0.1% Triton X-100, and 10% glycerol. The protein was eluted with the same buffer containing 10 mM glutathione. Enzyme concentrations were determined spectroscopically from the Soret absorbance peak and expressed as the concentration of the monomer assuming a molecular mass of 160 kDa.
The purified protein displayed a Soret band at 404 nm that shifted to 401 nm in the presence of 100 μM BH4 (Supplementary Figure 1, Supporting Information), indicative of a largely high-spin species. It contained 46 ± 1% FAD/heme and 31 ± 1% FMN/heme and no BH4. For comparison, Sf9-nNOS contained 49 ± 1% FAD/heme, 33 ± 1% FMN/heme, and 35 ± 3% BH4/heme. Flavin and pterin contents were determined by HPLC according to published procedures.35
Oxyhemoglobin Assay
NO formation was determined spectrophotometrically with a Hewlett-Packard 8452A diode array spectrophotometer as the conversion of oxyHb to methemoglobin by monitoring the absorbance difference between 420 and 401 nm. Purified nNOS (2 μg/mL, ∼11 nM) was incubated at 37 °C in 50 mM triethanolamine (TEA, pH 7.4) containing 0.2 mM Arg, 0.2 mM NADPH, 5 μM FAD, 5 μM FMN, 0.5 mM CaCl2, 5 μM oxyHb, 1000 U/mL superoxide dismutase (SOD), 50 mU/mL catalase (CAT), 0.2 mM 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate (CHAPS), 0.1 mM ethylenediaminetetraacetic acid (EDTA), and pteridines as indicated. The reaction was initiated by addition of 10 μg/mL calmodulin (CaM) and monitored for 3 min. NO concentrations were calculated with an absorbance difference coefficient of ΔΔε420–401 = 91.9 mM–1·cm–1.36 Values were corrected for oxyHb autoxidation by subtraction of blank rates obtained in the absence of enzyme.
Overnight preincubations were carried out at ∼8 °C with 5 μg/mL (∼27 nM) nNOS in 50 mM TEA (pH 7.4) containing 0.5 mM Arg, 0.5 mM CHAPS, 0.25 mM EDTA, and pteridines as indicated; these mixtures were diluted 2.5-fold at the start of the reaction to yield the assay conditions indicated above.
For the determination of the time dependence of the emergence of tight binding (Figure 7), all ingredients were present during preincubation at ∼4 °C, except for the second (“incubation”) pteridine (10 μM BH4 or 4-amino-BH4) and CaM (10 μg/mL), which were added at the start of the reaction.
Figure 7.

Kinetics of tight binding site formation for ECo-nNOS. Shown is the effect of the time interval between BH4 and 4-amino-BH4 administration on the rate of NO formation by ECo-nNOS, as measured with the oxyHb assay. Closed circles: the enzyme was preincubated with 1 μM 4-amino-BH4 for the indicated time, after which the activity was determined in the presence of 10 μM BH4. The curve through the data points is the best fit to a single exponential with parameters: kobs = 1.6 ± 0.3 min–1, Act0 = 151 ± 4 nmol·mg–1·min–1, Act∞ = 87 ± 2 nmol·mg–1·min–1. Open circles: the enzyme was preincubated with 1 μM BH4 for the indicated time, after which the activity was determined in the presence of 10 μM 4-amino-BH4. The curve through the data points is the best fit to a single exponential with parameters: kobs = 0.23 ± 0.10 min–1, Act0 = 16 ± 4 nmol·mg–1·min–1, Act∞ = 79 ± 11 nmol·mg–1·min–1. In both cases data points are shown ± SEM (n = 3). Preincubation conditions: 2 μg/mL (∼11 nM) ECo-nNOS, 0.2 mM Arg, 0.2 mM NADPH, 5 μM FAD, 5 μM FMN, 0.5 mM CaCl2, 5 μM oxyHb, 1000 U/mL SOD, 50 mU/mL CAT, 0.2 mM CHAPS, 0.1 mM EDTA, 50 mM TEA (pH 7.4), and 1 μM BH4 or 4-amino-BH4 at 4 °C. The assay conditions were the same except for the temperature (37 °C), the presence of 10 μM 4-amino-BH4 or BH4 (in the case of preincubation with BH4 and 4-amino-BH4, respectively), and the presence of 10 μg/mL CaM, which was added to start the reaction. In both cases, data points are shown ± SEM (n = 3).
Plots of the activity vs the pterin concentration were fitted to variations of the Hill equation:
or
where [I] is the concentrations of the inhibitory pterin, Act0 and Act∞ are the activities in the absence and presence of the pterin, respectively, and h is the Hill coefficient.
To obtain an indication for the affinity of the inhibitor in the absence of the activating pterin, we calculated apparent inhibition constants according to
Citrulline Assay
NOS activity was also determined as the formation of [3H]-citrulline from [3H]-Arg.37 Purified nNOS (2 μg/mL, ∼11 nM) was incubated for 10 min in 0.1 mL of 50 mM TEA (pH 7.4) containing 0.1 mM [3H]-Arg (∼60 000 cpm), 0.2 mM NADPH, 5 μM FAD, 5 μM FMN, 0.5 mM CaCl2, 0.2 mM CHAPS, 0.1 mM EDTA, 10 μg/mL CaM, and pteridines as indicated at 37 °C, followed by separation and detection of [3H]-citrulline. Blank values were determined in the absence of enzyme.
Radioligand Binding Studies
Radioligand binding experiments were performed as described previously.9,21 To determine the rate of association, purified nNOS (40 μg/mL, ∼216 nM) was incubated at 37, 25, and 8 °C with [3H]-BH4 (20 nM) in 50 mM TEA (pH 7.4) containing 0.1 mM Arg. For dissociation kinetics, the enzyme was preincubated in 50 mM TEA (pH 7.4) and 0.1 mM Arg for 10 min (37 °C), 20 min (25 °C), or 60 min (8 °C) with 20 nM [3H]-BH4, after which excess BH2 (1 mM) was added at t = 0. Separation of bound from free radioligand was performed by polyethylene glycol precipitation and rapid vacuum filtration over Whatman glass fiber filters (GF/B). Filters were treated at room temperature with 0.15 M H3PO4 for 3 h, followed by incubation with scintillation fluid (Ultima Gold XR, PerkinElmer Inc.) and scintillation counting.9 Rate constants were calculated by fitting the individual data to single exponentials.
Low-Temperature Polyacrylamide Gel Electrophoresis (LT-PAGE)
To test for tight dimer formation, the enzyme was analyzed by low-temperature sodium dodecyl sulfate (SDS)-PAGE.38 The protein (20 μg) was incubated for 5 min at 37 °C in 0.1 mL TEA (50 mM, pH 7.4) with Arg (1 mM) and BH4, 4-amino-BH4, BH2, or 4-amino-BH2 (0.2 mM each). After addition of 0.1 mL of chilled 0.125 M Tris-HCl (pH 6.8), containing 4% (w/v) SDS, 10% (v/v) 2-mercaptoethanol, 20% (w/v) glycerol, and 0.02% (w/v) bromophenol blue, samples containing 8 μg of nNOS were subjected to SDS-PAGE for 90 min at 120 V on discontinuous 4% SDS gels (1.5 mm). Gels and buffers were equilibrated at 4 °C, and the buffer tank was cooled during electrophoresis in an ice bath. Gels were stained with Coomassie Brilliant Blue and densitometrically analyzed with an E.A.S.Y. 440K camera (Herolab GmbH) and ImageJ 1.46r software (Wayne Rasband, National Institutes of Health, USA).
Separation and Determination of NOS-Bound and Free Pteridines after Overnight Incubation
ECo-nNOS (∼1 μM) was incubated overnight at ∼8 °C with 10 μM BH4 in 50 mM TEA (pH 7.4) containing 0.5 mM Arg, 0.5 mM CHAPS, and 0.25 mM EDTA in a total volume of 2.5 mL. This mixture was diluted 6-fold with the same buffer (50 mM TEA, pH 7.4, containing 0.5 mM Arg, 0.5 mM CHAPS, and 0.25 mM EDTA) and concentrated by ultrafiltration for 15 min at 4000g (Amicon Ultra-15, Merck Millipore, cutoff 50 kDa) to a final volume of ∼500 μL. The heme content was then estimated from the Soret peak (398 nm, ε = 91.9 mM–1·cm–1), and the pteridine content (BH4 and BH2/biopterin) was determined by HPLC as published.
Results
Effect of BH4 and BH2 on NO Production by Sf9-nNOS
We first determined the effect of BH4 on NO formation by Sf9-nNOS with the oxyHb assay. This enzyme already contains approximately 1 equivalent of BH4 per dimer, resulting in half-maximal activity in the absence of added BH4.10 Indeed, as illustrated in Figure 1A, BH4 induced an increase of the activity with an EC50 of (2.4 ± 0.6) × 10–7 M, and the activity in the absence of added BH4 was 44 ± 3% of the maximal activity, in line with published observations.9,12 As reported previously,9 BH2 inhibited the increase in activity induced by additional BH4 (10 μM), but not the basal activity (Figure 1B), with an IC50 of (1.3 ± 1.1) × 10–5 M, which, assuming simple competition, corresponds to a Kiapp of ∼3 × 10–7 M.
Figure 1.

Effect of BH4 (A) and BH2 (B) on NO production by Sf9-nNOS. Panel A shows the stimulation by BH4 of NO formation, measured with the oxyHb assay. The curve through the data is the best fit to the Hill equation with fitting parameters EC50 = 2.4 ± 0.6 × 10–7 M, Act0 = 147 ± 8 nmol·mg–1·min–1, Act∞ = 333 ± 9 nmol·mg–1·min–1, h = 1.1 ± 0.3. Panel B shows the inhibition by BH2 in the absence (closed symbols) and presence (open symbols) of 10 μM BH4. The curve through the data in the presence of BH4 is the best fit to the Hill equation with fitting parameters IC50 = (1.3 ± 1.1) × 10–5 M, Act0 = 283 ± 7 nmol·mg–1·min–1, Act∞ = 177 ± 17 nmol·mg–1·min–1, h = 0.5 ± 0.2. Experimental conditions: 2 μg/mL (∼13 nM) Sf9-nNOS, 0.2 mM Arg, 0.2 mM NADPH, 5 μM FAD, 5 μM FMN, 10 μg/mL CaM, 0.5 mM CaCl2, 5 μM oxyHb, 1000 U/mL SOD, 50 mU/mL CAT, 0.2 mM CHAPS, 0.1 mM EDTA, 50 mM TEA (pH 7.4), and pteridines as indicated at 37 °C. Data points are presented ± SEM (n = 3).
Effect of BH4 and BH2 on NO Production by ECo-nNOS
We next repeated these experiments with ECo-nNOS, which does not contain BH4 as isolated. This enzyme had no basal activity and was activated monophasically by BH4 with an EC50 of (2.0 ± 0.3) × 10–7 M (Figure 2A), in line with observations obtained with BH4-deficient Sf9-nNOS.21 Contrary to the result obtained with Sf9-nNOS, BH2 inhibited the activity in the presence of 10 μM BH4 completely with an IC50 of (2.42 ± 0.13) × 10–5 M, which corresponds to a Kiapp of (4.7 ± 0.4) × 10–7 M (Figure 2B). These results imply that ECo-nNOS does not exhibit biphasic BH4 binding under these conditions.
Figure 2.

Effect of BH4 (A) and BH2 (B) on NO production by ECo-nNOS. Panel A shows the stimulation by BH4 of NO formation, measured with the oxyHb assay. The curve through the data is the best fit to the Hill equation with fitting parameters EC50 = (2.0 ± 0.3) × 10–7 M, Act0 = 1 ± 5 nmol·mg–1·min–1, Act∞ = 178 ± 5 nmol·mg–1·min–1, h = 1.05 ± 0.15. Panel B shows the inhibition by BH2 in the presence of 10 μM BH4. The curve through the data is the best fit to the Hill equation with fitting parameters IC50 = (2.42 ± 0.13) × 10–5 M, Act0 = 197.6 ± 1.6 nmol·mg–1·min–1, Act∞ = −2 ± 3 nmol·mg–1·min–1, h = 0.96 ± 0.04. Experimental conditions: 2 μg/mL (∼11 nM) ECo-nNOS, 0.2 mM Arg, 0.2 mM NADPH, 5 μM FAD, 5 μM FMN, 10 μg/mL CaM, 0.5 mM CaCl2, 5 μM oxyHb, 1000 U/mL SOD, 50 mU/mL CAT, 0.2 mM CHAPS, 0.1 mM EDTA, 50 mM TEA (pH 7.4), and pteridines as indicated at 37 °C. Data points are presented ± SEM (n = 3).
Effect of BH4 and BH2 on NO Production by ECo-nNOS after Overnight Incubation with BH4
The lack of biphasicity might be a property of the expression system, or high-affinity BH4 binding might require preincubation with the pteridine. To discriminate between these possibilities, we incubated ECo-nNOS at ∼8 °C overnight with 25 μM BH4. This procedure yielded an enzyme with a basal activity (in the absence of BH4) about half as high as observed at saturating BH4 concentrations before preincubation (compare Figures 2 and 3A). Additional BH4 resulted in an activity increase to a maximal level of approximately 75% of that observed before overnight incubation, with an EC50 of (1.0 ± 0.9) × 10–6 M (Figure 3A, see Supplementary Figure S2A, Supporting Information for corresponding citrulline formation). The activity increase, but not the basal activity, was blocked by BH2 with an IC50 of (1.8 ± 1.0) × 10–7 M (Figure 3B and Figure S2B, Supporting Information). These results imply that overnight incubation with BH4 transforms ECo-nNOS into an enzyme with similar properties as the Sf9-expressed enzyme (apart from the exceptionally low IC50 for BH2, for which we currently have no explanation yet).
Figure 3.

Effect of BH4 (A) and BH2 (B) on NO production by ECo-nNOS after overnight incubation with BH4. Panel A shows the stimulation by BH4 of NO formation, measured with the oxyHb assay. The curve through the data is the best fit to the Hill equation with fitting parameters EC50 = (1.0 ± 0.9) × 10–6 M, Act0 = 90 ± 10 nmol·mg–1·min–1, Act∞ = 133 ± 8 nmol·mg–1·min–1, h = 1.3 ± 1.2. Panel B shows the inhibition by BH2 in the absence (closed symbols) and presence (open symbols) of 10 μM BH4. The curve through the data in the presence of BH4 is the best fit to the Hill equation with fitting parameters IC50 = (1.8 ± 1.0) × 10–7 M, Act0 = 146 ± 4 nmol·mg–1·min–1, Act∞ = 86 ± 3 nmol·mg–1·min–1, h = 0.96 ± 0.04. Overnight incubation conditions: 5 μg/mL (∼27 nM) ECo-nNOS, 0.5 mM Arg, 0.5 mM CHAPS, 0.25 mM EDTA, and 25 μM BH4 in 50 mM TEA (pH 7.4) at ∼8 °C. Assay conditions: 2 μg/mL (∼11 nM) ECo-nNOS, 0.2 mM Arg, 0.2 mM NADPH, 5 μM FAD, 5 μM FMN, 10 μg/mL CaM, 0.5 mM CaCl2, 5 μM oxyHb, 1000 U/mL SOD, 50 mU/mL CAT, 0.2 mM CHAPS, 0.1 mM EDTA, 50 mM TEA (pH 7.4), and pteridines as indicated at 37 °C. Note that the assay samples also contain 10 μM (autoxidized) BH4, carried over from the preincubation mixture. Data points are presented ± SEM (n = 3).
To investigate if BH2 might be able to replace tightly bound BH4 over a longer period, we incubated Sf9-nNOS for 24 and 48 h at 8 °C in the absence and presence of 100 μM BH2 (Supplementary Figure S3, Supporting Information). In the absence of BH4, the activity decreased from 215 ± 10 to 160 ± 19 and 128 ± 15 nmol·mg–1·min–1 after 24 and 48 h, respectively. Addition of 10 μM BH4 to these samples yielded approximately doubled these activities to 409, 357, and 235 nmol·mg–1·min–1 after 0, 24, and 48 h, demonstrating that the low-affinity binding site was still accessible. When these experiments were performed in the presence of 100 μM BH2, we observed residual activities of 208 ± 11, 156 ± 10, and 131 ± 3 nmol·mg–1·min–1 after 0, 24, and 48 h, respectively. These values are essentially indiscernible from the activities in the absence of BH2, indicating that no exchange between BH2 and tightly bound BH4 takes places even after 48 h.
Pteridine Content of ECo-nNOS after Overnight Incubation with BH4
In confirmation of these observations, HPLC determinations indicated that after overnight incubation with 10 μM BH4 ECo-nNOS contained 43 ± 5% BH4/heme, which compares well to the BH4 content of Sf9-nNOS (35 ± 3% BH4/heme). After overnight incubation, these enzyme samples contained a large excess of BH2 (4.6 ± 0.7 μM). However, after one cycle of dilution followed by ultrafiltration (see Materials & Methods) the BH2 content decreased to 7.3 ± 0.4% per heme, while the BH4 content remained fixed at 45 ± 3%, indicating that all BH4 was bound tightly to the enzyme, whereas BH2, originating from autoxidation of excess BH4, was not. These results also imply that there was no exchange between tightly bound BH4 and autoxidized BH2 in solution.
Effect of BH4 on NO Production by ECo-nNOS after Overnight Incubation with BH2
To investigate if BH2 can also induce high-affinity binding, we incubated the enzyme overnight in the presence of 25 μM BH2. In the case of high-affinity BH2 binding, this procedure should yield an enzyme that can only be activated to half the value observed before preincubation. However, as shown in Figure 4A (and Figure S2C, Supporting Information), the maximal activity was not affected by preincubation with BH2. Instead, preincubation with BH2 caused a rightward shift of the curve to an EC50 of (4.7 ± 0.3) × 10–6 M. Assuming that BH2 is stable during preincubation, the observed shift of the curve corresponds to a Kiapp for BH2 of (4.3 ± 1.2) × 10–7 M, in excellent agreement with the value calculated from Figure 2. These observations demonstrate that, unlike BH4, BH2 is not able to create a tight-binding site.
Figure 4.

Effect of BH4 on NO production by ECo-nNOS after overnight incubation with inhibitory pteridines. Panel A shows the stimulation by BH4 of NO formation, measured with the oxyHb assay, after overnight incubation with BH2. The curve through the data is the best fit to the Hill equation with fitting parameters EC50 = (4.7 ± 0.3) × 10–6 M, Act0 = 4 ± 2 nmol·mg–1·min–1, Act∞ = 185 ± 3 nmol·mg–1·min–1, h = 1.19 ± 0.08. For comparison, the curve obtained without overnight preincubation with BH2 (Figure 2A) is shown as well (gray small circles, dashed line). Panel B shows the stimulation by BH4 of NO formation, measured with the oxyHb assay, after overnight incubation with 4-amino-BH4 (closed circles) or 4-amino-BH2 (open circles). The curves through the data are best fits to the Hill equation with fitting parameters EC50 = (1.1 ± 0.3) × 10–5 M, Act0 = 3 ± 3 nmol·mg–1·min–1, Ac.∞ = 95 ± 6 nmol·mg–1·min–1, h = 1.0 ± 0.2 for 4-amino-BH4 preincubation and EC50 = (1.04 ± 0.19) × 10–5 M, Act0 = 9 ± 4 nmol·mg–1·min–1, Act∞ = 185 ± 8 nmol·mg–1·min–1, h = 0.82 ± 0.11 for 4-amino-BH2 preincubation. For comparison, the curve obtained without preincubation (Figure 2A) is included as well (small gray symbols, dashed line). Overnight incubation conditions: 5 μg/mL (∼27 nM) ECo-nNOS, 0.5 mM Arg, 0.5 mM CHAPS, 0.25 mM EDTA, and 25 μM pteridine (BH2, 4-amino-BH4, or 4-amino-BH2) in 50 mM TEA (pH 7.4) at ∼8 °C. Assay conditions: 2 μg/mL (∼11 nM) ECo-nNOS, 0.2 mM Arg, 0.2 mM NADPH, 5 μM FAD, 5 μM FMN, 10 μg/mL CaM, 0.5 mM CaCl2, 5 μM oxyHb, 1000 U/mL SOD, 50 mU/mL CAT, 0.2 mM CHAPS, 0.1 mM EDTA, 50 mM TEA (pH 7.4), and BH4 as indicated at 37 °C. Note that the assay samples also contain 10 μM BH2, (autoxidized) 4-amino-BH4, or 4-amino-BH2, carried over from the preincubation mixture. Data points are presented ± SEM (n = 4 or 3 for BH2 or 4-amino-BH4 preincubation, and for 4-amino-BH2 preincubation, respectively).
Effect of BH4 on NO Production by ECo-nNOS after Overnight Incubation with 4-Amino-BH4
To investigate why BH4 but not BH2 induced formation of tight pterin binding, we repeated the experiment with 4-amino-BH4. As illustrated by Figure 4B (and Figure S2D, Supporting Information), the maximal activity attained with BH4 was decreased by approximately 50% after overnight preincubation with 4-amino-BH4, concomitant with a rightward shift of the EC50 to 1.1 ± 0.3 × 10–5 M. These results suggest that 4-amino-BH4, like BH4, induces the formation of one tight pterin-binding site per dimer.
To ascertain that the inability of BH2 to replace tightly bound BH4 was not due to its inability to create a tight-binding site, we repeated the experiments of Figure 3B with 4-amino-BH4 instead of BH2. We found that 4-amino-BH4 did not significantly affect the residual activity after overnight incubation with BH4 (80 ± 4 and 67 ± 11 in the absence and presence of 1 mM 4-amino-BH4, respectively; results not shown), indicating that, like BH2, 4-amino-BH4 cannot displace BH4 from its tight-binding site.
Effect of BH4 on NO Production by ECo-nNOS after Overnight Incubation with 4-Amino-BH2
BH4 and 4-amino-BH4 differ from BH2 primarily by their redox state. To investigate if the redox state is critical for high-affinity binding, we repeated the experiment after overnight incubation with 4-amino-BH2, the 2-electron oxidized form of 4-amino-BH4. Like BH2 and in contrast to 4-amino-BH4, overnight incubation with 4-amino-BH2 did not affect the maximal activity with BH4; it only caused a rightward shift of the concentration–response curve (EC50 to (1.0 ± 0.2) × 10–5 M, Figure 4B, Figure S2D, Supporting Information). These results suggest that only fully reduced pteridines are able to induce tight pterin binding.35
Effect of BH4 and BH2 on NO Production by ECo-nNOS after Anaerobic Overnight Incubation with BH4
One way in which the pterin redox state might influence the results is by autoxidation, which will affect tetra- but not dihydropteridines. If autoxidation of BH4 were crucial for the formation of a high-affinity binding site, no high-affinity binding should be observed in the absence of oxygen during preincubation. As illustrated by Figure 5, overnight anaerobic preincubation of ECo-nNOS with BH4 yielded an enzyme that was maximally active. This can be explained by the fact that without autoxidation the concentration of BH4 in solution remains saturating overnight. However, BH2 could only inhibit about half of the activity (with IC50 (3.0 ± 1.5) × 10–5 M), indicating that despite the absence of oxygen a high-affinity site had formed. This demonstrates that the presence of O2 is no prerequisite for high-affinity binding site formation.
Figure 5.

Effect of BH2 on NO production by ECo-nNOS after anaerobic overnight incubation with BH4. The figure shows the stimulation by BH4 of NO formation, measured with the oxyHb assay. The curve through the data is the best fit to the Hill equation with fitting parameters IC50 = (3.0 ± 1.5) × 10–5 M, Act0 = 153 ± 4 nmol·mg–1·min–1, Act∞ = 94 ± 8 nmol·mg–1·min–1, h = 0.9 ± 0.4. For comparison, the curve obtained after aerobic preincubation with BH4 is included as well (small gray symbols, dashed line, from Figure 3B). Overnight incubation conditions: 5 μg/mL (∼27 nM) ECo-nNOS, 0.5 mM Arg, 0.5 mM CHAPS, 0.25 mM EDTA, and 25 μM BH4 in 50 mM TEA (pH 7.4) at ∼8 °C. Assay conditions: 2 μg/mL (∼11 nM) ECo-nNOS, 0.2 mM Arg, 0.2 mM NADPH, 5 μM FAD, 5 μM FMN, 10 μg/mL CaM, 0.5 mM CaCl2, 5 μM oxyHb, 1000 U/mL SOD, 50 mU/mL CAT, 0.2 mM CHAPS, 0.1 mM EDTA, 50 mM TEA (pH 7.4), and BH2 as indicated at 37 °C. Note that the assay samples also contain 10 μM BH4, carried over from the anaerobic preincubation mixture. Data points are presented ± SEM (n = 3).
Association and Dissociation Kinetics of ECo-nNOS and Radiolabeled BH4
Whereas the activity titration studies described above are characterized by virtually irreversible tight BH4 binding, prior radioligand binding studies suggested otherwise. Specifically, the complete displacement of radiolabeled BH4 from the enzyme in the presence of excess unlabeled BH4 implied that high-affinity BH4 binding is essentially reversible.9,21,26 To investigate this apparent discrepancy, we measured the association and dissociation kinetics of radiolabeled BH4 for ECo-nNOS. As shown in Figure 6A, binding of [3H]-BH4 appears to display two phases with about half of the label bound within seconds, and the other half binding with apparent rate constants of 0.91 ± 0.16, 0.55 ± 0.10, and 0.076 ± 0.006 min–1 at 37, 25, and 8 °C, respectively. At 8 and 25 °C, most of the label was still recovered after 24 h, whereas the enzyme tended to lose the label at 37 °C. In agreement with prior observations, BH2 displaced the label at 37 °C almost completely (96%) with an apparent rate constant of (1.70 ± 0.05) × 10–2 min–1 (Figure 6B). At lower temperatures, BH2 was able to displace about 60% of the label with apparent rate constants of (7.78 ± 0.13) × 10–3 and (2.9 ± 0.5) × 10–3 min–1 at 25 and 8 °C, respectively, with the remaining 40% still bound after 24 h. Virtually identical results were obtained when unlabeled BH4 was applied instead of BH2 to displace [3H]-BH4 (not shown). These results indicate that, at least at 37 °C, BH4 binding is reversible.
Figure 6.

Kinetics of BH4 binding to ECo-nNOS. (A) Association of radiolabeled BH4 to ECo-nNOS at 8 (open circles), 25 (closed circles), and 37 °C (squares, dotted line). Experimental conditions: 40 μg/mL (∼250 nM) ECo-nNOS, 20 nM [3H]-BH4, 0.1 mM Arg, 50 mM TEA (pH 7.4). The data points are presented ± SEM (n = 2). The curves through the data points are best single-exponential fits with parameters: at 37 °C: ka = 0.91 ± 0.16 min–1, Δ[3H]-BH4 = 264 ± 24 fmol, [3H]-BH4∞ = 444 ± 6 fmol; at 25 °C: ka = 0.55 ± 0.10 min–1, Δ[3H]-BH4 = 217 ± 17 fmol, [3H]-BH4∞ = 499 ± 6 fmol; at 8 °C: ka = 0.076 ± 0.006 min–1, Δ[3H]-BH4 = 314 ± 9 fmol, [3H]-BH4∞ = 554 ± 8 fmol. The data points are presented ± SEM (n = 2). (B) Dissociation of radiolabeled BH4 from ECo-nNOS at 8 (open circles), 25 (closed circles), and 37 °C (squares, dotted line). The procedure was the same as for association, except that excess BH2 (1 mM) was added after 10, 20, or 60 min incubation with [3H]-BH4 at 37, 25, and 8 °C, respectively. The time of BH2 addition was taken as t = 0. The curves through the data points are best single-exponential fits with parameters: at 37 °C: kd = (17.0 ± 0.5) × 10–3 min–1, Δ[3H]-BH4 = 485 ± 4 fmol, [3H]-BH4∞ = 19 ± 3 fmol; at 25 °C: kd = (7.8 ± 1.3) × 10–3 min–1, Δ[3H]-BH4 = 252 ± 14 fmol, [3H]-BH4∞ = 161 ± 13 fmol; at 8 °C: kd = (2.9 ± 0.5) × 10–3 min–1, Δ[3H]-BH4 = 266 ± 23 fmol, [3H]-BH4∞ = 184 ± 22 fmol. The data points are presented ± SEM (n = 2).
A possible explanation could be that the autoxidation product in the overnight incubation experiments is not necessarily BH2, since aerobic autoxidation has been reported to yield varying mixtures of BH2, 7,8-dihydropterin, and other products, depending on the reaction conditions.39 It is therefore conceivable that autoxidation under the present conditions yields a product mixture with low affinity for NOS. To check this possibility, we repeated the dissociation studies with [3H]-BH4 in the presence of autoxidized (overnight) BH4. The results were identical to those observed in the presence of BH2 or unlabeled BH4 (not shown). In line with previous observations with BH4-deficient Sf9-nNOS,21 these results suggest that BH4 binding is completely reversible at 37 °C in contradiction to the results obtained with the activity assays (see Discussion).
Kinetics of High-Affinity Binding Site Formation
In the present studies, we routinely incubated the enzyme overnight with an excess of pteridine to elicit the transformation from an enzyme that binds BH4 monophasically to a species that has one pterin bound (quasi-)irreversibly. In solution, free BH4 will be completely autoxidized within this period; in fact, we observed spectrophotometrically that BH4 autoxidized with t1/2 of ∼2 h, with complete autoxidation achieved after approximately 6–7 h under the conditions of preincubation (Supplementary Figure S4, Supporting Information). Consequently, binding site formation must also be complete within this period. To investigate the kinetics of the process, we performed a series of experiments, in which we preincubated the enzyme with 1.0 μM 4-amino-BH4 for varying periods (0–10 min), followed by the determination of the activity in the presence of 10 μM BH4. As shown in Figure 7, preincubation with 1 μM 4-amino-BH4 gradually reduced the activity that could be attained to a value slightly greater (58 ± 2%) than half the activity observed when both pterins were added simultaneously, with an apparent rate constant of 1.6 ± 0.3 min–1.
We also performed the reverse experiment, in which we added BH4 (1 μM) first and an excess of 4-amino-BH4 (10 μM) at the start of the assay. As shown in Figure 7, in this case the activity increased when the time interval between the addition of the pterins became longer, from a very low value when both pterins were added together, to a similar final value as in the reverse experiment (79 ± 11 compared to 87 ± 2 nmol·min–1·mg–1). However, the transition appears to be considerably slower under these conditions (0.23 ± 0.10 min–1).
Effect of Incubation with Pteridines on Dimer Stability of ECo-nNOS
It is well documented that BH4 enhances NOS dimer stability.15,25,28,38,40,41 To check if high-affinity BH4 binding-site formation correlates with the formation of a SDS-resistant NOS dimer, we performed LT-PAGE of ECo-nNOS with and without preincubation with different pteridines (Figure 8). It should be noted that in these experiments we are not monitoring the actual oligomeric state of the protein, which is expected to be almost completely dimeric,42 but rather the effects of tight pterin binding on the SDS-resistance of the dimer, which is anticipated to reflect structural changes at the dimer interface. Without pteridines, dimers were not detectable, but preincubation with BH4 or 4-amino-BH4 yielded 43 ± 3 and 44 ± 3% of SDS-resistant dimers, in line with published observations.29,43 Preincubation of the enzyme with BH2 or 4-amino-BH2 on the other hand did not produce any SDS-resistant dimers, suggesting a link with tight-binding site formation.
Figure 8.

Effect of pteridine preincubation on ECo-nNOS dimer stability. ECo-nNOS was preincubated with or without pteridines (BH4, BH2, 4-amino-BH4, or 4-amino-BH2) and Arg, followed by LT-PAGE. Shown is the relative content of SDS-resistant dimer, estimated from densitometric analysis. Preincubation conditions: 20 μg of ECo-nNOS in 0.1 mL of TEA (50 mM, pH 7.4) and 0.2 mM pteridines and 0.1 mM Arg as indicated, for 5 min at 37 °C. See experimental procedures for further details. 4AH4: 4-amino-BH4; 4AH2: 4-amino-BH2.
Discussion
Incubation of ECo-nNOS with BH4 Results in Tight Binding of 1 equiv of BH4 per NOS dimer
Our first goal was to establish if there is a difference in the response to BH4 between BH4-containing Sf9-nNOS and BH4-free ECo-nNOS. We found that both enzyme species exhibited a similar EC50 for added BH4 with no indication of biphasic behavior for ECo-nNOS, indicating that the enzyme does not display high-affinity pterin binding upon short-term exposure to BH4. One possible explanation for this would be that BH4 must be present during protein synthesis to be bound with high affinity. It is also conceivable that the expression system affects the structure of the enzyme in some way. Interestingly, it has been reported that iNOS forms undisruptable dimers when expressed in Sf9 cells but not in E. coli, suggesting that the expression system may indeed affect NOS structure.31 However, overnight incubation of ECo-nNOS with BH4 yielded an enzyme with very similar pterin-binding properties as the Sf9-expressed enzyme. This demonstrates that the expression system is not essential for high-affinity pterin binding and that BH4 need not be present during enzyme synthesis.
Whereas there is good agreement between the data obtained with Sf9- and ECo-nNOS, previous studies suggest that eNOS and iNOS behave differently. Similar to the present study, preincubation of BH4-free E. coli-expressed iNOS with BH4 yielded an enzyme species that still contained strongly bound BH4 (approximately 0.3–0.4 equiv), but, contrary to nNOS, BH4 was able to induce (almost) complete reactivation of iNOS after preincubation with 4-amino-BH4.29 Unlike nNOS, Sf9-expressed eNOS appears to contain very little BH4 after purification (≤10%)16 and to lose all bound BH4 in a few hours at 24 °C.44 These studies seem to indicate that the other isoforms show no (eNOS) or less (iNOS) tight binding. However, further studies are necessary to resolve this issue.
Tight Binding Is Induced by Tetra- but Not by Dihydropteridines
Intriguingly, tight pterin binding was induced in ECo-nNOS by BH4 and 4-amino-BH4 but not by the respective 2-electron oxidized species BH2 and 4-amino-BH2. Since all pterins appear to be bound comparably with similar allosteric and conformational consequences,5 this suggests that the redox state of the pteridines is directly involved in the process of tight binding. Consequently, the phenomenon is unlikely to represent a simple binding equilibrium (see below).
Interestingly, the different effects of the tetra- and dihydropteridines on tight pterin binding appear to be mirrored by the effects on the formation of SDS-resistant dimers, which also required the tetrahydro- derivatives. In apparent contrast to the present results, BH2 has been reported to stabilize NOS dimers.38,45 However, the study by Klatt et al. was performed with BH4-containing nNOS purified from porcine brain and did show a considerably weaker effect by BH2 compared to BH4.38 Presta et al. determined the effect on dimer formation per se for iNOS.45 Both studies may have reported on a more general effect of pterins on dimerization rather than the more specific redox-state dependent effect observed here.
Is High-Affinity Pterin Binding Reversible?
When we first noticed that BH2 is unable to expel endogenously bound BH4 from its binding site on nNOS, we explained this by heterogeneity, with part of the enzyme having BH4 irreversibly bound.9 On the basis of studies with BH4-deficient Sf9-nNOS, we later revised this interpretation to propose that nNOS has two identical but highly anticooperative binding sites per dimer.21 From the present observations for the association and dissociation rate constants, we can estimate values of 4–9 nM for the apparent equilibrium dissociation constants (Kd). As the enzyme is present in excess in the radiolabeling studies (250 nM nNOS, 20 nM [3H]-BH4), these values should reflect high-affinity binding only. The dissociation experiments at 37 °C imply that this process is essentially reversible. The observation that a significant fraction of [3H]-BH4 remained bound after 24 h incubation with excess unlabeled BH4 or BH2 at 8 and 25 °C is intriguing, considering that only half of the pterin binding sites is expected to exhibit tight binding. However, as mentioned above, because of the large excess of NOS over [3H]-BH4, all label should be bound with high affinity, and no heterogeneity in BH4 dissociation is expected.
Whereas the radioligand binding studies imply reversible high-affinity binding, the activity titrations seem to suggest otherwise. No exchange of (tightly bound) pterin occurred during the activity assays and in the experiments of Figure 7. In these cases, the apparent contradiction is explained by the different time scales of the experiments: with a dissociation rate constant at 37 °C of 0.017 min–1 one can estimate that exchange should occur with a half-life of ∼40 min, well beyond the time scale of the activity assays; similarly, for the experiment of Figure 7 the dissociation rate constant (<0.003 min–1) implies a half-life of 4 h or more. Less easy to explain is the fact that the enzyme with 1 equiv of BH4 per dimer seems not be inactivated by pterin autoxidation during overnight incubation. Assuming that NOS-bound BH4 is protected from autoxidation, the apparent autoxidation rate constant is expected to be the product of the actual autoxidation rate constant (∼1 × 10–4 s–1 under the present conditions; see Figure S4, Supporting Information) and the fraction of free BH4, which in turn depends on the tight-binding BH4 dissociation equilibrium constant (Kd) and the protein concentration (∼10–8 M). As detailed in the Supporting Information (Figure S5), the lack of enzyme inactivation implies a Kd in the picomolar range or lower. We arrived at similarly low upper estimates for the Kd by different methods earlier.21,24 Most problematic is the observation that the activity due to tight pterin binding is also retained when the enzyme is incubated overnight with an excess of BH4. In view of the rate of BH4 autoxidation, this procedure should result after ∼7 h in NOS with one tightly bound BH4 in the presence of a large excess of autoxidized BH4. According to the radiolabeling studies, and in clear contrast to the observations, the enzyme ought to lose most tightly bound BH4 in the remaining incubation time by exchange with autoxidized BH4 under these conditions.
In summary, the radiolabeling studies demonstrate that high-affinity pterin binding is reversible, but the discrepancies between those studies and the activity titrations suggest that the two series of experiments may not be monitoring the same process. Clearly, further studies will be required to resolve this issue.
Is High-Affinity Pterin Binding Anticooperative?
A second open question concerns the involvement of anticooperativity in the generation of heterogeneous BH4 binding. This concept was also based on analysis of the radioligand binding studies. Specifically, BH4-containing and -deficient nNOS exhibited virtually identical [3H]-BH4 dissociation rates in the presence of Arg and unlabeled BH4. As BH4-containing and -deficient nNOS were expected to have the radioligand bound at the low- and the high-affinity site respectively, this observation argued strongly in favor of two identical anticooperative binding sites.21,26 Essentially identical binding sites are also suggested by the fact that the two monomers are identical, and crystallographic studies of pterin-bound NOS oxygenase domains failed to show any major effect of BH4 or other pteridines on the enzyme structure or any obvious differences between the two subunits.46−48
However, if the manifestation of heterogeneous BH4 binding upon preincubation were the result of the emergence of anticooperativity, one would expect the affinity of the low-affinity site to decrease in the process. Instead, the EC50 values for the activation of ECo-nNOS before and after overnight incubation with BH4 were similar, which implies that preincubation does not modify the affinity of the “low-affinity” site. Consequently, anticooperativity may not be the origin of the heterogeneity of pterin binding.
Mechanistic Considerations
Tight binding was accomplished by tetra- but not by dihydropteridines. This is reminiscent of the differential effects of tetra- and dihydropteridines on NOS activity, with only the former supporting NO synthesis.5 Indeed, the inability of dihydropteridines to bind tightly suggests a redox-mediated process. We therefore propose that the first equivalent of BH4 that binds to nNOS is able to create one tight-binding site by a process that involves redox chemistry. The other site is not affected and binds a second equivalent of BH4 with unchanged affinity (Scheme 1). It is yet unclear why tight binding is only accomplished for the first pterin, but one may speculate that the Zn-binding site is involved, as it is the only unique structure in the dimer.46,49 Moreover, it is close to both pterin-binding sites and redox-sensitive. In view of the well-documented role of the Zn-site in dimerization, it may therefore be relevant that we observed formation of SDS-resistant dimers only with tetra- and not with dihydropteridines. Further studies will be necessary to clarify the mechanism of tight BH4 binding and why it allows only one BH4 to be bound tightly per dimer.
Scheme 1. Proposed Explanation for Tight BH4 Binding to nNOS.
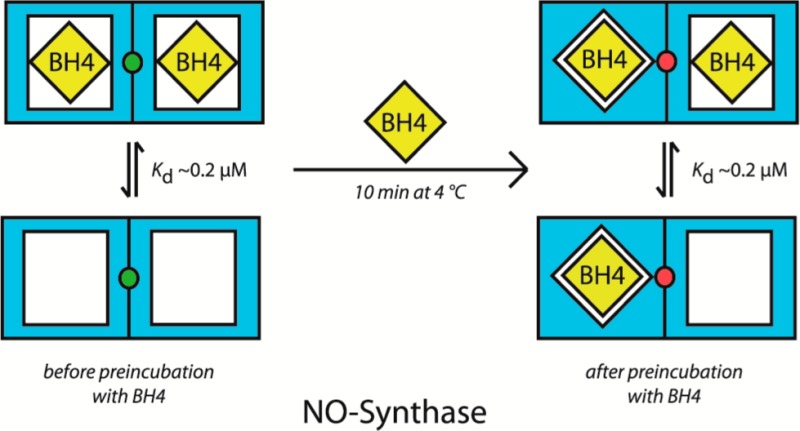
Dimeric nNOS contains two identical sites that bind BH4 with relatively low affinity, manifested by a EC50 of ∼0.2 μM in the activity assays. Binding of the first equivalent of BH4 causes a redox reaction that results in very tight binding of the pterin. The reaction may involve a unique site on the enzyme (the green dot in the scheme) that is not too far away from both binding sites, perhaps the Zn-binding site at the dimer interface. Upon tight binding of 1 equiv of BH4, this site is no longer able to assist in tight binding of the second equivalent of BH4 (illustrated in the scheme by the change in color from green to red). Consequently, only 1 equiv of BH4 is tightly bound, with the other site still exhibiting EC50 ≈ 0.2 μM. The radioligand binding studies suggest that BH4 binding is fully reversible, indicated by the short backward arrow in the middle of the scheme. However, it is presently unclear if the phenomenon of tight binding is covered by the radioligand binding studies under the applied conditions. It is also unclear if dissociation of tightly bound BH4, if it occurs, is accompanied by irreversible inactivation and/or changes in the quaternary structure of the enzyme.
Physiological Implications
In recent years, evidence has been mounting for NOS uncoupling as an important factor in a range of pathologies,50−54 and BH4 deficiency or its oxidation to BH2 is assumed to be a leading cause of uncoupling.50−55 An important implication of the results of the present study is that, at least in the case of the neuronal isoform, BH4 deficiency or oxidation will never lead to complete NOS uncoupling, since only enzyme that has never been exposed to BH4 at all will have no BH4 bound at its tight-binding site. Consequently, even under conditions of severe oxidative stress NOS will still produce O2– and NO simultaneously. However, it should be stressed that the present study was limited to nNOS. Notably, most studies on the pathophysiology of uncoupling focused on eNOS,50−54 with few studies involving nNOS.56−58 It will therefore be interesting to see if the results in this study can be extrapolated to the other isoforms. Another important open question concerns the relative rates with which BH4-containing and BH4-free subunits produce their respective products. Although it is conceivable that NO and O2– are produced with equal rates, other stoichiometries are possible.5
In the laboratory, the observations reported here imply that, without sufficient preincubation, results obtained with BH4-replenished recombinant NOS derived from sources that do not synthesize BH4 (bacteria, yeast, etc.) may not reflect the in vivo situation, since it takes some time to induce tight binding. The observations reported here may also be relevant for the studies by Tsai and co-workers, who took great pains to remove excess BH4 from their samples, distinguishing between BH4 as a “ligand” and a “cofactor”.59,60 The present results suggest that those studies may have been performed with preparations consisting mainly of dimers with one tightly bound BH4.
Summary and Conclusions
Tight binding of approximately 1 equiv of BH4 per nNOS dimer is confirmed in the present study. The phenomenon is not restricted to a specific expression system and does not require a cellular environment. It is not evident in radioligand binding studies, perhaps because of the far lower concentration of (labeled) BH4 in those studies. Tight binding occurs with tetra- but not with dihydropteridines, suggesting the involvement of redox chemistry. One implication of this study is that, as isolated, recombinant nNOS expressed in E. coli does not behave like the native enzyme, since the tight pterin-binding site must first be created, which takes place on a time scale of minutes. Another implication is that, at least in the case of the neuronal isoform, NOS will never be fully uncoupled, since only enzyme that is never exposed to BH4 at all will have no BH4 bound at its tight-binding site. Consequently, even under conditions of severe oxidative stress NOS will still produce O2– and NO simultaneously.
Glossary
Abbreviations
- NOS
nitric-oxide synthase
- nNOS
neuronal NOS
- eNOS
endothelial NOS
- iNOS
inducible NOS
- Sf9-nNOS
recombinant rat nNOS expressed in and purified from baculovirus-infected Sf9 insect cells
- ECo-nNOS
recombinant human nNOS expressed in and purified from Escherichia coli as a glutathione-S-transferase fusion protein
- BH4
tetrahydrobiopterin ((6R)-2-amino-6-[(1R,2S)-1,2-dihydroxypropyl]-5,6,7,8-tetrahydropteridin-4(1H)-one)
- BH2
7,8-dihydrobiopterin
- 4-amino-BH4
4-amino-tetrahydrobiopterin
- 4-amino-BH2
4-amino-dihydrobiopterin
- [3H]-BH4
[3′-3H](6R)-5,6,7,8-tetrahydro-l-biopterin
- [3H]-Arg
l-[2,3,4,5-3H]arginine hydrochloride
- oxyHb
oxyhemoglobin
- Tris
tris(hydroxymethyl)-aminomethane
- DTT
dithiothreitol
- TEA
triethanolamine
- SOD
Cu,Zn-superoxide dismutase
- CAT
catalase
- CHAPS
3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate
- EDTA
ethylenediaminetetraacetic acid
- CaM
calmodulin
- SDS
sodium dodecyl sulfate
- PAGE
polyacrylamide gel electrophoresis
- LT-PAGE
low temperature PAGE
- SEM
standard error of the mean
Supporting Information Available
Figure S1: UV/vis spectra of ECo-nNOS in the absence and presence of BH4; Figure S2A–D: This figure shows results from experiments similar to those of Figures 3 and 4, performed with the citrulline assay33 instead of the oxyhemoglobin assay; Figure S3: Effect of 48 h incubation with BH2 on NO production by BH4-preincubated Sf9-nNOS; Figure S4: Autoxidation of BH4 in solution; Figure S5: Estimation of the higher limit of the BH4 tight-binding dissociation constant. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
This work was supported by the Austrian Science Fund (FWF): P23135 (to A.C.F.G.), P24005, P24946 (to B.M.), and P22289 (to E.R.W.).
Supplementary Material
References
- Gao Y. (2010) The multiple actions of NO. Pfluegers Arch. 459, 829–839. [DOI] [PubMed] [Google Scholar]
- Alderton W. K.; Cooper C. E.; Knowles R. G. (2001) Nitric oxide synthases: Structure, function and inhibition. Biochem. J. 357, 593–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorren A. C. F.; Mayer B. (2007) Nitric-oxide synthase: A cytochrome P450 family foster child. Biochim. Biophys. Acta 1770, 432–445. [DOI] [PubMed] [Google Scholar]
- Förstermann U.; Sessa W. C. (2012) Nitric oxide synthases: Regulation and function. Eur. Heart J. 33, 829–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorren A. C. F.; Mayer B. (2002) Tetrahydrobiopterin in nitric oxide synthesis: A novel biological role for pteridines. Curr. Drug Metab. 3, 133–157. [DOI] [PubMed] [Google Scholar]
- Werner E. R.; Gorren A. C. F.; Heller R.; Werner-Felmayer G.; Mayer B. (2003) Tetrahydrobiopterin and nitric oxide: Mechanistic and pharmacological aspects. Exp. Biol. Med. 228, 1291–1302. [DOI] [PubMed] [Google Scholar]
- Wei C.-C.; Crane B. R.; Stuehr D. J. (2003) Tetrahydrobiopterin radical enzymology. Chem. Rev. 103, 2365–2383. [DOI] [PubMed] [Google Scholar]
- Werner E. R.; Blau N.; Thöny B. (2011) Tetrahydrobiopterin: Biochemistry and pathophysiology. Biochem. J. 438, 397–414. [DOI] [PubMed] [Google Scholar]
- Klatt P.; Schmid M.; Leopold E.; Schmidt K.; Werner E. R.; Mayer B. (1994) The pteridine binding site of brain nitric oxide synthase: Tetrahydrobiopterin binding kinetics, specificity, and allosteric interaction with the substrate domain. J. Biol. Chem. 269, 13861–13866. [PubMed] [Google Scholar]
- Roman L. J.; Sheta E. A.; Martasek P.; Gross S. S.; Liu Q.; Masters B. S. S. (1995) High-level expression of functional rat neuronal nitric oxide synthase in Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 92, 8428–8432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riveros-Moreno V.; Heffernan B.; Torres B.; Chubb A.; Charles I.; Moncada S. (1995) Purification to homogeneity and characterisation of rat brain recombinant nitric oxide synthase. Eur. J. Biochem. 230, 52–57. [DOI] [PubMed] [Google Scholar]
- List B. M.; Klatt P.; Werner E. R.; Schmidt K.; Mayer B. (1996) Overexpression of neuronal nitric oxide synthase in insect cells reveals requirement of haem for tetrahydrobiopterin binding. Biochem. J. 315, 57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abu-Soud H. M.; Presta A.; Mayer B.; Stuehr D. J. (1997) Analysis of neuronal NO synthase under single-turnover conditions: Conversion of Nω-hydroxyarginine to nitric oxide and citrulline. Biochemistry 36, 10811–10816. [DOI] [PubMed] [Google Scholar]
- Hevel J. M.; Marletta M. A. (1992) Macrophage nitric oxide synthase: Relationship between enzyme-bound tetrahydrobiopterin and synthase activity. Biochemistry 31, 7160–7165. [DOI] [PubMed] [Google Scholar]
- Baek K. J.; Thiel B. A.; Lucas S.; Stuehr D. J. (1993) Macrophage nitric oxide synthase subunits: Purification, characterization, and role of prosthetic groups and substrate in regulating their association into a dimeric enzyme. J. Biol. Chem. 268, 21120–21129. [PubMed] [Google Scholar]
- List B. M.; Klösch B.; Völker C.; Gorren A. C. F.; Sessa W. C.; Werner E. R.; Kukovetz W. R.; Schmidt K.; Mayer B. (1997) Characterization of bovine endothelial nitric oxide synthase as a homodimer with down-regulated uncoupled NADPH oxidase activity: Tetrahydrobiopterin binding kinetics and role of haem in dimerization. Biochem. J. 323, 159–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witteveen C. F. B.; Giovanelli J.; Yim M. B.; Gachhui R.; Stuehr D. J.; Kaufman S. (1998) Reactivity of the flavin semiquinone of nitric oxide synthase in the oxygenation of arginine to NG-hydroxyarginine, the first step of nitric oxide synthesis. Biochem. Biophys. Res. Commun. 250, 36–42. [DOI] [PubMed] [Google Scholar]
- Witteveen C. F. B.; Giovanelli J.; Kaufman S. (1999) Reactivity of tetrahydrobiopterin bound to nitric-oxide synthase. J. Biol. Chem. 274, 29755–29762. [DOI] [PubMed] [Google Scholar]
- Rafferty S. P.; Boyington J. C.; Kulansky R.; Sun P. D.; Malech H. J. (1999) Stoichiometric arginine binding in the oxygenase domain of inducible nitric oxide synthase requires a single molecule of tetrahydrobiopterin per dimer. Biochem. Biophys. Res. Commun. 257, 344–347. [DOI] [PubMed] [Google Scholar]
- Heinzel B.; John M.; Klatt P.; Böhme E.; Mayer B. (1992) Ca2+/calmodulin-dependent formation of hydrogen peroxide by brain nitric oxide synthase. Biochem. J. 281, 627–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorren A. C. F.; List B. M.; Schrammel A.; Pitters E.; Hemmens B.; Werner E. R.; Schmidt K.; Mayer B. (1996) Tetrahydrobiopterin-free neuronal nitric oxide synthase: Evidence for two identical highly anticooperative pteridine binding sites. Biochemistry 35, 16735–16745. [DOI] [PubMed] [Google Scholar]
- Stroes E.; Hijmering M.; van Zandvoort M.; Wever R.; Rabelink T. J.; van Faassen E. E. (1998) Origin of superoxide production by endothelial nitric oxide synthase. FEBS Lett. 438, 161–164. [DOI] [PubMed] [Google Scholar]
- Bömmel H. M.; Reif A.; Fröhlich L. G.; Frey A.; Hofmann H.; Marecak D. M.; Groehn V.; Kotsonis P.; La M.; Köster S.; Meinecke M.; Bernhardt M.; Weeger M.; Ghisla S.; Prestwich G. D.; Pfleiderer W.; Schmidt H. H. H. W. (1998) Anti-pterins as tools to characterize the function of tetrahydrobiopterin in NO synthase. J. Biol. Chem. 273, 33142–33149. [DOI] [PubMed] [Google Scholar]
- Gorren A. C. F.; Schrammel A.; Schmidt K.; Mayer B. (1997) Thiols and neuronal nitric oxide synthase: Complex formation, competitive inhibition, and enzyme stabilization. Biochemistry 36, 4360–4366. [DOI] [PubMed] [Google Scholar]
- Gorren A. C. F.; Schrammel A.; Schmidt K.; Mayer B. (1998) Effects of pH on the structure and function of neuronal nitric oxide synthase. Biochem. J. 331, 801–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alderton W. K.; Boyhan A.; Lowe P. N. (1998) Nitroarginine and tetrahydrobiopterin binding to the haem domain of neuronal nitric oxide synthase using a scintillation proximity assay. Biochem. J. 332, 195–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werner E. R.; Pitters E.; Schmidt K.; Wachter H.; Werner-Felmayer G.; Mayer B. (1996) Identification of the 4-amino-analogue of tetrahydrobiopterin as a dihydropteridine reductase inhibitor and a potent pteridine antagonist of rat neuronal nitric oxide synthase. Biochem. J. 320, 193–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reif A.; Fröhlich L. G.; Kotsonis P.; Frey A.; Bömmel H. M.; Wink D. A.; Pfleiderer W.; Schmidt H. H. H. W. (1999) Tetrahydrobiopterin inhibits monomerization and is consumed during catalysis in neuronal NO synthase. J. Biol. Chem. 274, 24921–24929. [DOI] [PubMed] [Google Scholar]
- Mayer B.; Wu C.; Gorren A .C. F.; Pfeiffer S.; Schmidt K.; Clark P.; Stuehr D. J.; Werner E. R. (1997) Tetrahydrobiopterin binding to macrophage inducible nitric oxide synthase: Heme spin shift and dimer stabilization by the potent pterin antagonist 4-amino-tetrahydrobiopterin. Biochemistry 36, 8422–8427. [DOI] [PubMed] [Google Scholar]
- Vásquez-Vivar J.; Hogg N.; Martásek P.; Karoui H.; Pritchard K. A. Jr.; Kalyanaraman B. (1999) Tetrahydrobiopterin-dependent inhibition of superoxide generation from neuronal nitric oxide synthase. J. Biol. Chem. 274, 26736–26742. [DOI] [PubMed] [Google Scholar]
- Kolodziejski P. J.; Rashid M. B.; Eissa N. T. (2003) Intracellular formation of “undisruptable” dimers of inducible nitric oxide synthase. Proc. Natl. Acad. Sci. U.S.A. 100, 14263–14268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werner E. R.; Schmid M.; Werner-Felmayer G.; Mayer B.; Wachter H. (1994) Synthesis and characterization of 3H-labelled tetrahydrobiopterin. Biochem. J. 304, 189–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer B.; Klatt P.; Böhme E.; Schmidt K. (1992) Regulation of neuronal nitric oxide and cyclic GMP formation by Ca2+. J. Neurochem. 59, 2024–2029. [DOI] [PubMed] [Google Scholar]
- Harteneck C.; Klatt P.; Schmidt K.; Mayer B. (1994) Expression of rat brain nitric oxide synthase in baculovirus-infected insect cells and characterization of the purified enzyme. Biochem. J. 304, 683–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer B.; John M.; Heinzel B.; Werner E. R.; Wachter H.; Schultz G.; Böhme E. (1991) Brain nitric oxide synthase is a biopterin- and flavin-containing multi-functional oxido-reductase. FEBS Lett. 288, 187–191. [DOI] [PubMed] [Google Scholar]
- Wenzl M. V.; Beretta M.; Griesberger M.; Russwurm M.; Koesling D.; Schmidt K.; Mayer B.; Gorren A. C. F. (2011) Site-directed mutagenesis of aldehyde dehydrogenase-2 suggests three distinct pathways of nitroglycerin biotransformation. Mol. Pharmacol. 80, 258–266. [DOI] [PubMed] [Google Scholar]
- Mayer B.; Klatt P.; Werner E. R.; Schmidt K. (1994) Molecular mechanisms of inhibition of porcine brain nitric oxide synthase by the antinociceptive drug 7-nitro-indazole. Neuropharmacology 33, 1253–1259. [DOI] [PubMed] [Google Scholar]
- Klatt P.; Schmidt K.; Lehner D.; Glatter O.; Bächinger H. P.; Mayer B. (1995) Structural analysis of porcine brain nitric oxide synthase reveals a role for tetrahydrobiopterin and L-arginine in the formation of an SDS-resistant dimer. EMBO J. 14, 3687–3695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis M. D.; Kaufman S.; Milstien S. (1988) The auto-oxidation of tetrahydrobiopterin. Eur. J. Biochem. 173, 345–351. [DOI] [PubMed] [Google Scholar]
- Hemmens B.; Goessler W.; Schmidt K.; Mayer B. (2000) Role of bound zinc in dimer stabilization but not enzyme activity of neuronal nitric-oxide synthase. J. Biol. Chem. 275, 35786–35791. [DOI] [PubMed] [Google Scholar]
- Kotsonis P.; Fröhlich L. G.; Shutenko Z. V.; Horejsi R.; Pfleiderer W.; Schmidt H. H. H. W. (2000) Allosteric regulation of neuronal nitric oxide synthase by tetrahydrobiopterin and suppression of auto-damaging superoxide. Biochem. J. 346, 767–776. [PMC free article] [PubMed] [Google Scholar]
- Panda K.; Rosenfeld R. J.; Ghosh S.; Meade A. L.; Getzoff E. D.; Stuehr D. J. (2002) Distinct dimer interaction and regulation in nitric-oxide synthase types I, II, and III. J. Biol. Chem. 277, 31020–31030. [DOI] [PubMed] [Google Scholar]
- Pfeiffer S.; Gorren A. C. F.; Pitters E.; Schmidt K.; Werner E. R.; Mayer B. (1997) Allosteric modulation of rat brain nitric oxide synthase by the pterin-site enzyme inhibitor 4-aminotetrahydrobiopterin. Biochem. J. 328, 349–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berka V.; Yeh H.-C.; Gao D.; Kiran F.; Tsai A.-L. (2004) Redox function of tetrahydrobiopterin and effect of l-arginine on oxygen binding in endothelial nitric oxide synthase. Biochemistry 43, 13137–13148. [DOI] [PubMed] [Google Scholar]
- Presta A.; Siddhanta U.; Wu C.; Sennequier N.; Huang L.; Abu-Soud H. M.; Erzurum S.; Stuehr D. J. (1998) Comparative functioning of dihydro- and tetrahydropterins in supporting electron transfer, catalysis, and subunit dimerization in inducible nitric oxide synthase. Biochemistry 37, 298–310. [DOI] [PubMed] [Google Scholar]
- Raman C. S.; Li H.; Martásek P.; Král V.; Masters B. S. S.; Poulos T. L. (1998) Crystal structure of constitutive endothelial nitric oxide synthase: A paradigm for pterin function involving a novel metal center. Cell 95, 939–950. [DOI] [PubMed] [Google Scholar]
- Crane B. R.; Arvai A. S.; Ghosh S.; Getzoff E. D.; Stuehr D. J.; Tainer J. A. (2000) Structures of the Nω- hydroxy-l-arginine complex of inducible nitric oxide synthase oxygenase dimer with active and inactive pterins. Biochemistry 39, 4608–4621. [DOI] [PubMed] [Google Scholar]
- Li H.; Igarashi J.; Jamal J.; Yang W.; Poulos T. L. (2006) Structural studies of constitutive nitric oxide synthases with diatomic ligands bound. J. Biol. Inorg. Chem. 11, 753–768. [DOI] [PubMed] [Google Scholar]
- Li H.; Raman C. S.; Glaser C. B.; Blasko E.; Young T. A.; Parkinson J. F.; Whitlow M.; Poulos T. L. (1999) Crystal structures of zinc-free and -bound heme domain of human inducible nitric-oxide synthase: Implications for dimer stability and comparison with endothelial nitric-oxide synthase. J. Biol. Chem. 274, 21276–21284. [DOI] [PubMed] [Google Scholar]
- Alp N. J.; Channon K. M. (2004) Regulation of endothelial nitric oxide synthase by tetrahydrobiopterin in vascular disease. Arterioscler. Thromb., Vasc. Biol. 24, 413–420. [DOI] [PubMed] [Google Scholar]
- Schulz E.; Jansen T.; Wenzel P.; Daiber A.; Münzel T. (2008) Nitric oxide, tetrahydrobiopterin, oxidative stress, and endothelial dysfunction in hypertension. Antioxid. Redox Signal. 10, 1115–1126. [DOI] [PubMed] [Google Scholar]
- Förstermann U.; Li H. (2011) Therapeutic effect of enhancing endothelial nitric oxide synthase (eNOS) expression and preventing eNOS uncoupling. Br. J. Pharmacol. 164, 213–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gielis J. F.; Lin J. Y.; Wingler K.; Van Schil P. E. Y.; Schmidt H. H.; Moens A. L. (2011) Pathogenetic role of eNOS uncoupling in cardiopulmonary disorders. Free Radic. Biol. Med. 50, 765–776. [DOI] [PubMed] [Google Scholar]
- Roe N. D.; Ren J. (2012) Nitric oxide synthase uncoupling: A therapeutic target in cardiovascular diseases. Vasc. Pharmacol. 57, 168–172. [DOI] [PubMed] [Google Scholar]
- Crabtree M. J.; Tatham A. L.; Al-Wakeel Y.; Warrick N.; Hale A. B.; Cai S.; Channon K. M.; Alp N. J. (2009) Quantitative regulation of intracellular endothelial nitric-oxide synthase (eNOS) coupling by both tetrahydrobiopterin-eNOS stoichiometry and biopterin redox status. Insights from cells with tet-regulated GTP cyclohydrolase I expression. J. Biol. Chem. 284, 1136–1144. [DOI] [PubMed] [Google Scholar]
- Shang T.; Kotamraju S.; Kalivendi S. V.; Kalyanaraman B. (2004) 1-Methyl-4-phenylpyridinium-induced apoptosis in cerebellar granule neurons is mediated by transferrin receptor iron-dependent depletion of tetrahydrobiopterin and neuronal nitric-oxide synthase-derived superoxide. J. Biol. Chem. 279, 19099–19112. [DOI] [PubMed] [Google Scholar]
- Shang T.; Kotamraju S.; Zhao H.; Kalivendi S. V.; Hillard C. J.; Kalyanaraman B. (2005) Sepiapterin attenuates 1-methyl-4-phenylpyridinium-induced apoptosis in neuroblastoma cells transfected with neuronal NOS: Role of tetrahydrobiopterin, nitric oxide, and proteasome activation. Free Radic. Biol. Med. 39, 1059–1074. [DOI] [PubMed] [Google Scholar]
- Sánchez A.; Contreras C.; Martínez M. P.; Climent B.; Benedito S.; Garciá-Sacristán A.; Hernández M.; Prieto D. (2012) Role of neural NO synthase (nNOS) uncoupling in the dysfunctional nitrergic vasorelaxation of penile arteries from insulin-resistant obese Zucker rats. PLoS One 7, e36027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berka V.; Tsai A.-L. (2000) Characterization of interactions among the heme center, tetrahydrobiopterin, and l-arginine binding sites of ferric eNOS using imidazole, cyanide, and nitric oxide as probes. Biochemistry 39, 9373–9383. [DOI] [PubMed] [Google Scholar]
- Berka V.; Wang L.-H.; Tsai A.-L. (2008) Oxygen-induced radical intermediates in the nNOS oxygenase domain regulated by l-arginine, tetrahydrobiopterin, and thiol. Biochemistry 47, 405–420. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.