

ABSTRACT
As with most life on Earth, the transition metal copper (Cu) is essential for the viability of the human pathogen Mycobacterium tuberculosis. However, infected hosts can also use Cu to control microbial growth. Several Cu-responsive pathways are present in M. tuberculosis, including the regulated in copper repressor (RicR) regulon, which is unique to pathogenic mycobacteria. In this work, we describe the contribution of each RicR-regulated gene to Cu resistance in vitro and to virulence in animals. We found that the deletion or disruption of individual RicR-regulated genes had no impact on virulence in mice, although several mutants had Cu hypersensitivity. In contrast, a mutant unable to activate the RicR regulon was not only highly susceptible to Cu but also attenuated in mice. Thus, these data suggest that several genes of the RicR regulon are required simultaneously to combat Cu toxicity in vivo or that this regulon is also important for resistance against Cu-independent mechanisms of host defense.
IMPORTANCE Mycobacterium tuberculosis is the causative agent of tuberculosis, killing millions of people every year. Therefore, understanding the biology of M. tuberculosis is crucial for the development of new therapies to treat this devastating disease. Our studies reveal that although host-supplied Cu can suppress bacterial growth, M. tuberculosis has a unique pathway, the RicR regulon, to defend against Cu toxicity. These findings suggest that Cu homeostasis pathways in both the host and the pathogen could be exploited for the treatment of tuberculosis.
IMPORTANCE
Mycobacterium tuberculosis is the causative agent of tuberculosis, killing millions of people every year. Therefore, understanding the biology of M. tuberculosis is crucial for the development of new therapies to treat this devastating disease. Our studies reveal that although host-supplied Cu can suppress bacterial growth, M. tuberculosis has a unique pathway, the RicR regulon, to defend against Cu toxicity. These findings suggest that Cu homeostasis pathways in both the host and the pathogen could be exploited for the treatment of tuberculosis.
INTRODUCTION
Mycobacterium tuberculosis is one of the most devastating microbial agents, as it infects nearly one-third of the world’s population and kills nearly two million people annually (http://www.who.int/en/). Currently available chemotherapies are lengthy and potentially toxic (1). In addition, the numbers of multiresistant, extensively resistant, and totally drug-resistant strains are rising (2–4). Thus, an improved understanding of M. tuberculosis pathogenesis is urgently needed in order to develop improved treatments for tuberculosis.
It has recently been determined that host-derived Cu is important for controlling M. tuberculosis infections in two animal models of infection (5). Cu is a well-known antimicrobial agent, but only in the last few years has its role been appreciated with regard to microbial infections in mammals (6). Previous studies found that Cu levels transiently increase in gamma interferon-activated macrophages infected with mycobacteria (7). In another study, it was shown that Cu accumulates within phagolysosomal compartments via the Cu-transporting ATPase ATP7A (8). Additionally, in a guinea pig model of infection, Cu accumulates in the granulomatous lesions of infected lungs (5). Perhaps because of this host response, it appears that M. tuberculosis has acquired several independent mechanisms to defend itself against Cu toxicity (6). These include mycobacterial Cu transport protein B (MctB) (5), the Cu-sensitive operon repressor (CsoR) operon (9, 10), and the regulated in Cu repressor (RicR) regulon (11).
The RicR regulon was discovered in an attempt to understand the link between M. tuberculosis proteasome function and pathogenesis, as M. tuberculosis strains defective for proteasomal degradation are highly attenuated in mice (11–14). This regulon includes ricR (encodes a transcriptional repressor), lpqS (encodes a putative lipoprotein), mymT (encodes a mycobacterial metallothionein), socAB (small open reading frame induced by copper A and B), and Rv2963 (a putative permease gene) (11). All five loci are transcriptionally repressed in strains defective for proteolysis by the M. tuberculosis proteasome (11). Interestingly, with the exception of ricR itself, all of these genes are found only in pathogenic mycobacteria, suggesting that they are important during infections of a vertebrate host. All of these genes have a palindromic motif in their promoters that is recognized by the transcriptional repressor RicR. Like its closely related paralog CsoR, RicR is presumed to bind Cu+ and is released from DNA (9, 11). The only previously characterized RicR-regulated gene other than ricR itself is mymT. Although a mymT mutant is hypersensitive to Cu, this mutant has no virulence defect in mice (15).
In this study, we sought to determine the contribution of every RicR-regulated gene to Cu resistance and virulence. We found that most of the genes conferred no to variable Cu resistance in vitro. Furthermore, none of the single mutants had an attenuated phenotype in mice. In contrast, repression of the entire RicR regulon resulted in a strong Cu-sensitive phenotype in vitro and severely attenuated growth in vivo. Thus, it appears that multiple members of the RicR regulon are required for Cu resistance during infections.
RESULTS
Most RicR-regulated genes are individually dispensable for Cu resistance in vitro and growth in mice.
The RicR regulon is presumed to be important for Cu resistance because a ricR null mutant, which constitutively expresses all of the genes in the RicR regulon (Fig. 1A), is resistant to high levels of Cu (11). However, the contributions of individual RicR-regulated genes to Cu resistance and virulence had not been determined. Therefore, we sought to quantify the Cu resistance of mutants lacking each RicR-regulated gene. Mutants with three RicR-regulated genes disrupted were identified in our lab collection of more than 10,000 ΦMycoMarT7 mutants in the M. tuberculosis H37Rv strain background (16). We also received a previously reported H37Rv mymT deletion-disruption strain (15). The genotypes of all of the strains used in this study are described in Table 1.
FIG 1 .

Contribution of RicR-regulated genes to Cu resistance. (A) Model of the RicR regulon in M. tuberculosis. Cytoplasmic MymT can bind with up to six Cu+ ions (black circles). LpqS and Rv2963 are predicted to be membrane-associated proteins. RicR is autoregulated and also represses socAB under low-Cu conditions. MmcO is an MCO. (B) Cu sensitivity assays assessing the ability of RicR regulon mutants to survive at the indicated concentrations of CuSO4 after 10 days. CFU were enumerated after 14 to 21 days of incubation on solid medium with trace amounts of Cu. Data are representative of at least two experiments, each done in triplicate. Abbreviations: comp., complemented; <L.O.D., below the limit of detection (which was 100 CFU). (C) Agar plate assay evaluating the Cu susceptibility of RicR regulon mutants. Serial dilutions of M. tuberculosis cultures were spotted onto 7H11-OADC agar plates with the CuSO4 concentrations indicated. The contrast was adjusted to make the images clearer here and in Fig. 5 and 6. Data are representative of two independent experiments.
TABLE 1 .
Strains, plasmids, and primers used in this study
| Strain, plasmid, or primer | Phenotype, genotype, or sequence | Source or reference |
|---|---|---|
| M. tuberculosis strains | ||
| H37Rv | WT | ATCC 25618 |
| MHD18 | Hygr WT/pMV306 | 12 |
| MHD22 | Kanr Hygr mpa::ΦMycoMarT7/pMV306 | 12 |
| MHD23 | Kanr Hygr mpa::ΦMycoMarT7/pMV-mpa | 12 |
| MHD62 | Kanr Hygr pafA::ΦMycoMarT7/pMV306 | 16 |
| MHD63 | Kanr Hygr pafA::ΦMycoMarT7/pMV-pafA | 16 |
| MHD131 | Kanr lpqS::ΦMycoMarT7 (transposon inserted in codon 44) | This work |
| MHD188 | Kanr Rv2963::ΦMycoMarT7 (transposon inserted in codon 351) | This work |
| MHD696 | Kanr socA::ΦMycoMarT7 (transposon inserted in codon 22) | This work |
| MHD701 | Hygr ΔmymT::hyg | 15 |
| MHD702 | Kanr Hygr ΔmymT::hyg/pMV306.kan-mymT | 15 |
| MHD752 | Hygr ΔmmcO::hyg | This work |
| MHD764 | Kanr Hygr lpqS::ΦMycoMarT7 ΔmmcO::hyg | This work |
| MHD794 | Kanr WT/pMV306.kan | This work |
| MHD795 | Kanr Hygr ΔmmcO::hyg/pMV306.kan | This work |
| MHD796 | Kanr Hygr ΔmmcO::hyg/pMV306.kan-mmcO | This work |
| MHD840 | Kanr Hygr ΔmymT::hyg ΔmmcO::kan | This work |
| MHD867 | Kanr Hygr ΔmymT::hyg ΔmmcO::kan/pMVstrep-mmcO mymT | This work |
| CDC1551 | WT | W. Bishai lab collection |
| MHD557 | Kanr ricR::ΦMycoMarT7 | 14 |
| MHD583 | Hygr WT/pMV306 | 11 |
| MHD589 | Kanr Hygr ricR::ΦMycoMarT7/pMV306 | 11 |
| MHD590 | Kanr Hygr ricR::ΦMycoMarT7/pMV-ricR | 11 |
| MHD694 | Kanr Hygr ricR::ΦMycoMarT7/pMV-ricRC38A | This work |
| MHD707 | Kanr Hygr ricR::ΦMycoMarT7/pMV-ricRpc-ricR | This work |
| MHD708 | Kanr Hygr ricR::ΦMycoMarT7/pMV-ricRpc-ricRC38A | This work |
| MHD755 | Hygr ΔmmcO::hyg | This work |
| E. coli strains | ||
| DH5α | F− ϕ80dlacZΔM15 Δ(lacZYA-argF)U169 deoR recA1 endA1 hsdR17 (rK− mK+) phoA supE44 λ-thi-1 gyrA96 relA1 | Gibco BRL |
| ER2566 | F− λ− fhuA2 [lon] ompT lacZ::T7 gene1 gal sulA11 Δ(mcrC-mrr)114::IS10 R(mcr-73::miniTn10)2 R(zgb-210::Tn10) (Tets) endA1 [dcm] | 35 |
| Plasmids | ||
| pET24b(+) | Kanr, for production of C-terminally His6 epitope-tagged protein | Novagen |
| pET24b(+)-mmcO | Kanr, for production of MmcO-His6 in E. coli | This work |
| pET24b(+)-ricR | Kanr, for production of untagged RicR in E. coli | 11 |
| pET24b(+)-ricRC38A | Kanr, for production of untagged RicRC38A in E. coli | This work |
| pMV306.kan | Kanr, integrates at attB site in mycobacterial chromosome | 36 |
| pMV306 | Hygr, integrates at attB site in mycobacterial chromosome | 36 |
| pMV306.strep | Strepr, integrates at attB site in mycobacterial chromosome | Gift from J. McKinney lab |
| pMV306.kan-mmcO | Kanr, for complementation of mmcO mutant | This work |
| pMV-ricR | Hygr, for complementation of ricR mutant | 11 |
| pMV-pc-ricR | Hygr, WT ricR expressed from mutated ricRp (see text) | This work |
| pMV-pc-ricRC38A | Hygr, ricRC38A allele expressed from mutated ricRp | This work |
| pMVstrep-mmcO mymT | Strepr, for complementation of mmcO mymT double mutant | This work |
| pYUB854 | Hygr, allelic-exchange vector | 32 |
| pYUB854.kan | Kanr, allelic-exchange vector | This work |
| pYUB854-mmcO | Hygr, M. tuberculosis ΔmmcO::hyg (deletion-disruption plasmid) | This work |
| pYUB854.kan-mmcO | Kanr, M. tuberculosis ΔmmcO::kan (deletion-disruption plasmid) | This work |
| Primers | ||
| NdeI Rv0846c F1 | GGCATATGCCCGAGCTGGCCACGAGCGGTAAC | |
| XhoI Rv0846c R1 | GGCTCGAGCAGAATGTAGTCCAGGCGGGTCGC | |
| Rv0846cF2seq | GGCACCGAGCCCGCGACTGCGAACATC | |
| Rv0846cR2seq | CCCGGCCAGCGCGATGCGGAACGCGGT | |
| KpnI 846cMutantF1 | GAGGGTACCTATCTGCGGGTTGGAGGTGATGCTTGTTG | |
| XbaI 846cMutantR1 | GTTACCGCTCGTGGCTCTAGACAGCTCGGGCATCGATC | |
| HindIII 846cMutantF2 | ACCCGCCTGGACAAGCTTTACATTCTGTGACAGGCGG | |
| SpeI 846cMutantR2 | TCAGGAGCTCATCGAGTTACTAGTGGATGCCGTAACC | |
| deltammcOF | AGGAGTGACTTGATATCCCTCCGGG | |
| deltammcOR | ATTGCGGAAGCCATTCACGATGGAC | |
| HindIIImmcOF2 | CAAAGCTTACGTGCCCGCTTTCCACGTGGCCC | |
| mmcOcompHpaIR | CTGTTAACTCACAGAATGTAGTCCAGGCGGGTC | |
| C38A SOE F | GTACGCCATTGACGTTCTGACC | |
| C38A SOE R | GGTCAGAACGTCAATGGCGTAC | |
| F1ricRpromsoe | CCGATACCCCGCTGTTGTACAAGATATGAT | |
| R1ricRpromsoe | ATCATATCTTGTACAACAGCGGGGTATCGG | |
| Rv0190 comp F-HindIII | GACAAGCTTCATTGTTCAAGTATGCGGCCCAAG | |
| Rv0190 comp R-KpnI | GACGGTACCTCAGGAACGAACCAGGCGCGCG | |
| Rv0190 F NdeI | GACCATATGACAGCAGCACACGGCTACAC | |
| Rv0190 Rev. Stop EcoRI | GACGAATTCTCAGGAACGAACCAGGCGCGCGATTG | |
| lpqS affinity F-BIO | BioTEG-ATCGCTCCTCGTCTGGATTT | |
| lpqS affinity R | AGCGCGACCGCGACAATC | |
| AgeI pYUBhygtokanfor | CAACCGGTCCCTCCCAAGGACACTGAGTCCTAAAG | |
| NcoI pYUBhygtokanrev | GTCCATGGTTAGAAAAACTCATCGAGCATCAAATG | |
| HpaI-mymT-for | GCGGTTAACGGGCGGTTGGGTTGCTGG | |
| MfeI-mymT-rev | GGCAATTGATAGGTCTACTTGACCGGGGCC |
We used a quantitative liquid-based assay to measure the Cu sensitivity of lpqS, Rv2963, socA, and mymT mutants compared to that of wild-type (WT) M. tuberculosis (11). As previously reported, the mymT mutant was more sensitive to Cu than WT M. tuberculosis or the complemented strain was (Fig. 1B) (15). The socA transposon mutant showed WT Cu resistance, while the Rv2963 transposon mutant was slightly (and not always reproducibly) more resistant to Cu (Fig. 1B). Perhaps most interestingly, the lpqS transposon mutant was consistently extremely hyperresistant to Cu. We also used a semiquantitative agar plate assay (15, 17) that showed similar Cu susceptibility results for the mymT mutant but not the lpqS mutant (Fig. 1C).
We next assessed the phenotypes of several mutants in mice. No single mutation attenuated bacterial growth in mice (Fig. 2A). Interestingly, the Rv2963 and lpqS mutant bacteria showed increased growth in vivo (Fig. 2A). In experiments where we inadvertently used a moderately large inoculum of bacteria (~2,000 CFU/mouse), we unexpectedly observed that mice infected with the lpqS mutant were moribund within 4 weeks (Fig. 2B). However, neither gene could restore WT virulence to the respective mutant (data not shown), assuming that the introduction of the WT allele of either gene expressed from its native promoter resulted in appropriate protein synthesis. Thus, it is unclear how (or if) disruption of either gene resulted in the observed hypervirulence phenotypes.
FIG 2 .

Contribution of RicR-regulated genes to virulence in mice. (A) CFU from lungs and spleens harvested on days 1 (n = 3) and 21 (n = 4) and at ~8 weeks (n = 4) from WT C57BL/6 mice infected with M. tuberculosis H37Rv RicR regulon single mutants. The initial dose of infection was ~500 to 1,000 CFU/mouse. Each datum point represents the average number of CFU from organs of three or four mice and the standard deviation. (B) Infection of WT C57BL/6 mice with a moderately large dose (~2,000 CFU/mouse) of WT or lpqS::ΦMycoMarT7 (MHD131) M. tuberculosis. Because of the slow movement and labored breathing of the mice infected with the lpqS mutant, they were sacrificed at day 26. N.D., not determined.
mmcO overexpression results in Cu hyperresistance but not hypervirulence.
Upon closer inspection of the lpqS locus, we hypothesized that the transposon insertion in the lpqS mutant somehow increased the expression of the divergently expressed gene mmcO (mycobacterial multicopper oxidase [MCO]) (17) (Fig. 1A and 3A). MCOs exist in all kingdoms of life and play a critical role in Cu and iron homeostasis (18–22). MCOs can oxidize substrates, including reduced metals such as Cu+ or Fe2+, as well as phenolic compounds (23–26). Recently it was shown that MmcO can oxidize Fe2+ to Fe3+ and perhaps can also convert Cu+ to Cu2+ (17). Additionally, Rowland and Niederweis observed the induction of MmcO production in M. tuberculosis upon Cu treatment (17). A microarray analysis determined that mmcO is more highly expressed in a ricR mutant than in WT M. tuberculosis, suggesting that it is Cu and RicR regulated (11). Consistent with these previously published data, we confirmed that MmcO levels are elevated upon Cu treatment and now show that it is also highly abundant in a ricR mutant (Fig. 3B) (11).
FIG 3 .
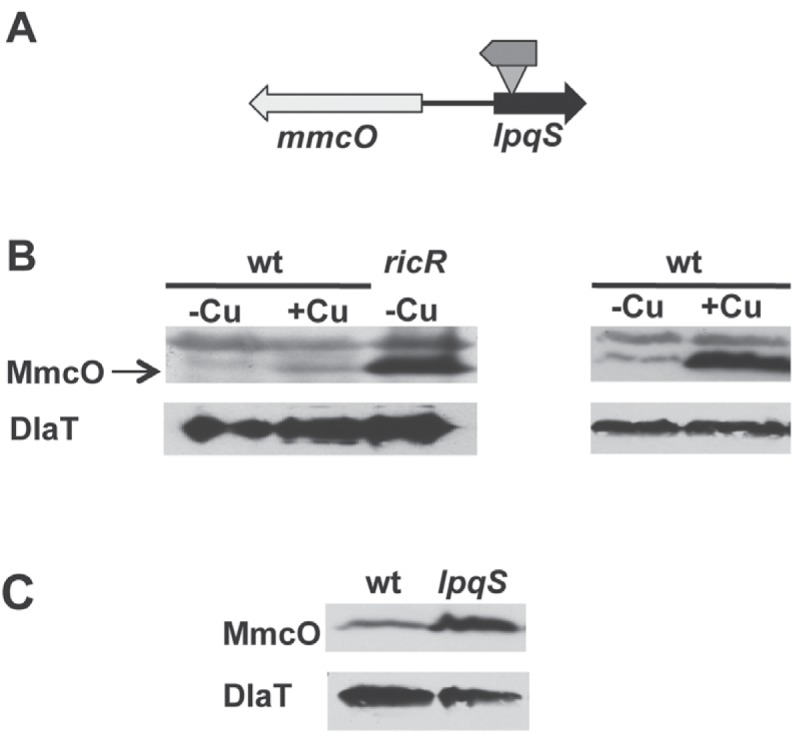
The transposon insertion in MHD131 increased the expression of mmcO. (A) Map of the lpqS locus in strain MHD131. The ΦMycoMarT7 transposon was inserted at nucleotide (nt) 130 of lpqS. lpqS is 393 nt long. (B) Immunoblot analysis showed that MmcO increased upon 4 h of Cu treatment or in a ricR mutant (left). Data are representative of three biological replicates. In another experiment, robust induction of MmcO was observed after 24 h of treatment with 50 µM CuSO4 (right). (C) MmcO levels were higher in the lpqS mutant than in WT M. tuberculosis. Immunoblotting for dihydrolipoamide acetyltransferase (DlaT) was used as a loading control for all experiments.
On the basis of the orientation of the neo gene in the ΦMycoMarT7 transposon in lpqS, we hypothesized that the neo promoter could increase mmcO expression in the absence of Cu (Fig. 3A), potentially resulting in the Cu hyperresistance and hypervirulence of this particular strain. We examined MmcO levels in the WT and lpqS mutant strains and found that MmcO was more abundant in the lpqS mutant than in the WT strain (Fig. 3C).
To determine if MmcO overproduction was responsible for the Cu hyperresistance and hypervirulence phenotypes of the lpqS mutant, we deleted and disrupted mmcO in this strain. We showed by immunoblotting that MmcO was absent from the double mutant strain (Fig. 4A). A Cu susceptibility assay revealed that deletion of mmcO from the lpqS mutant restored Cu sensitivity to WT levels, suggesting that MmcO contributed to the Cu hyperresistance of this strain (Fig. 4A). Importantly, the double mutant was no more sensitive to Cu than WT M. tuberculosis was, suggesting that lpqS itself has a little or no role in Cu resistance. Although Cu hyperresistance was eliminated upon the deletion of mmcO, the double mutant was as hypervirulent as the parental lpqS strain (Fig. 4B). Thus, MmcO overproduction was not responsible for the hypervirulence of this strain.
FIG 4 .

Overexpression of mmcO resulted in high Cu resistance. (A) Deletion of mmcO from the lpqS mutant resulted in WT Cu resistance. A Cu sensitivity assay (top) was performed with the WT strain and the lpqS and lpqS mmcO mutant strains. Data are representative of two experiments each done in triplicate. <L.O.D., below the limit of detection (which was 100 CFU). Immunoblot analysis (bottom) shows that disruption of mmcO in the lpqS mmcO strain eliminates MmcO protein from this strain. Data are representative of three biological replicates. (B) Deletion of mmcO from the lpqS mutant did not alleviate the hypervirulence phenotype. The CFU counts in the lungs and spleens of C57BL/6 mice infected with the WT, lpqS, and lpqS mmcO strains are plotted. The initial dose of infection was ~500 CFU/mouse. Measurements were made on days 1 (n = 3), 21 (n = 4), and 56 (n = 4). Two-way analysis of variance and a Bonferroni posttest showed that at day 21, both the lpqS and lpqS mmcO mutant strains were statistically significantly different from the WT strain (***, P < 0.001) and that at day 56, the lpqS mutant strain was statistically significantly different from the WT strain (**, P < 0.01). The data represent the mean ± standard deviation of a typical experiment that was done twice.
We do not understand the nature of the lpqS hypervirulence phenotype; complementation with lpqS or lpqS plus two downstream genes (cysK2, Rv0849) could not reduce bacterial growth to WT levels in mice (data not shown). Additionally, whole-genome sequencing of this strain did not reveal any differences from the parental WT strain except for the transposon insertion in lpqS.
Deletion of mmcO has no effect on virulence in mice.
Following the above observation that MmcO can confer Cu hyperresistance when overproduced, we wanted to determine the effect of deleting mmcO from WT M. tuberculosis. We deleted and disrupted mmcO in both the H37Rv and CDC1551 strains. Deletion was confirmed by PCR amplification of the region surrounding the deleted locus (data not shown) and immunoblot detection of MmcO protein (Fig. 5A). Interestingly, when we tested these strains for Cu sensitivity, we found that WT H37Rv was more sensitive to Cu than WT CDC1551 was and we thus had to use higher concentrations of CuSO4 for the CDC1551 strains in our assays. Nonetheless, in both strain backgrounds, the mmcO mutants showed WT Cu resistance in our quantitative Cu susceptibility assay (Fig. 5A). However, during the preparation of this report, Rowland and Niederweis showed than an mmcO mutant is hypersensitive to Cu by using the semiquantitative agar-based assay (17). We tested our mutants in the agar plate assay and indeed observed that both of our mmcO mutants were more sensitive to Cu than the WT or complemented strain was (Fig. 5B).
FIG 5 .

Deletion of mmcO had no effect on virulence. (A) Cu sensitivity assay (top) of WT and mmcO mutant strains in the H37Rv and CDC1551 backgrounds. We also complemented the mmcO mutation in the H37Rv background. This is representative of two independent experiments, each done in triplicate. At 150 µM, no CFU of the H37Rv strains were detected. Immunoblot analysis (bottom) of the same strains with polyclonal antibodies to MmcO. Antibodies to dihydrolipoamide acetyltransferase (DlaT) were used to show even loading of cell lysates. (B) Agar plate assay assessing the Cu sensitivity of the M. tuberculosis strains in panel A. Serial dilutions of M. tuberculosis cultures were spotted onto agar with the indicated CuSO4 concentrations. Data are representative of two independent experiments. (C) CFU counts in the lungs and spleens of C57BL/6 mice infected with WT, mmcO, and mmcO-complemented (comp.) M. tuberculosis strain H37Rv. The results shown are for days 1 (n = 3), 21 (n = 4), and 56 (n = 4). ns, not significant. The data represent the mean ± SD of a typical experiment that was done twice.
To determine the importance of mmcO in vivo, we infected mice with the H37Rv WT, mmcO mutant, and mmcO-complemented strains. All of these strains displayed WT growth in the lungs and spleens of WT C57BL/6 mice (Fig. 5C). Thus, mmcO alone does not significantly contribute to the virulence of M. tuberculosis in a mouse model of infection.
Deletion of both mmcO and mymT is not sufficient to attenuate M. tuberculosis in vivo.
Deletion of mmcO did not result in robust Cu hypersensitivity, suggesting that there might be other Cu-binding proteins that contribute to strong Cu resistance. Because mymT is the only other known RicR-regulated gene that significantly contributes to Cu resistance, we deleted and disrupted mmcO in the mymT deletion-disruption mutant and quantified the Cu resistance and virulence of this double mutant strain. We confirmed that MmcO protein was no longer produced by the double mutant strain (Fig. 6A). In the agar plate assay, the mmcO mymT double mutant showed greater Cu susceptibility than the mymT or mmcO single mutant, a phenotype that could be partially complemented with an integrative plasmid encoding mymT and mmcO expressed from their native promoters (Fig. 6B, top). However, in the liquid-based Cu assay, the double mutant showed Cu sensitivity similar to that of the mymT single mutant (Fig. 6B, bottom).
FIG 6 .

Deletion of both mmcO and mymT was not sufficient to attenuate M. tuberculosis in vivo. (A) Immunoblot analysis shows that MmcO protein was absent from an mmcO mymT double mutant. The WT, mmcO, mymT, mmcO mymT, and complemented (comp.) double mutant strains were used. Whole-cell lysates were analyzed with antibodies to MmcO. Dihydrolipoamide acetyltransferase (DlaT) was used as a loading control. (B) Agar plate assay (top) determining the Cu susceptibility of the WT, mmcO, mymT, mmcO mymT, and complemented double mutant strains. Data are representative of two independent experiments. The results of a liquid-based Cu sensitivity assay of the aforementioned M. tuberculosis strains are also shown (bottom). Data are representative of two experiments done in triplicate. (C) CFU counts in the lungs and spleens of C57BL/6 mice infected with the WT and mmcO; mymT, and mmcO mymT mutant M. tuberculosis strains. The results shown are for days 1 (n = 3), 21 (n = 4), and 56 (n = 4). The data represent means ± standard deviations. ns, not significant.
We next infected mice with the WT and single and double mutant strains. None of the mutants demonstrated an attenuated phenotype based on the bacterial burdens found in mouse lungs and spleens up to 8 weeks after aerosol infection (Fig. 6C). Thus, these data suggest that mmcO and mymT alone are not required for normal replication of M. tuberculosis in mice.
Constitutive repression of the RicR regulon results in robust Cu sensitivity in vitro and attenuation of M. tuberculosis in mice.
None of the genes of the RicR regulon seemed individually important for virulence. Therefore, we hypothesized that either several of the RicR-regulated genes are needed or the entire regulon is needed to play a significant role in virulence. Ideally, we would construct an M. tuberculosis strain that has all five RicR-regulated loci mutated. Because of the arduous process of deleting multiple genes from M. tuberculosis, we developed an alternative method to repress all of the genes in the RicR regulon by producing a “Cu-blind” allele of RicR in a ricR null mutant strain. RicR is a homologue of CsoR and has conserved residues that are predicted to bind Cu+ (9). Cysteine 38 of RicR is predicted to be required for Cu+ binding (D. Giedroc, personal communication); thus, conversion of cysteine 38 to alanine (RicRC38A) would prevent RicR from sensing Cu+ and detaching from DNA. We previously showed by using a DNA affinity chromatography assay that recombinant RicR binds to a specific sequence in vitro under low-Cu conditions and elutes from DNA with increasing amounts of Cu (11) (Fig. 7A, top). In contrast, RicRC38A was unresponsive to Cu and could only be eluted from DNA with a high-salt buffer (Fig. 7A, bottom). Because ricR is autoregulated, RicRC38A could also constitutively repress its own production, leading to reduced repressor levels. Therefore, two point mutations that disrupt RicR binding to DNA (11) were introduced into the ricR promoter (ricRp) of the ricRC38A construct to allow the constitutive expression of the ricRC38A allele (pc-ricRC38A) (see Table 1 and Materials and Methods). This allele was introduced into a CDC1551 ricR mutant on a plasmid that integrates into the attB site of the M. tuberculosis chromosome. As a control, we also introduced a plasmid expressing WT ricR from ricRpc (pc-ricR+) into the ricR mutant.
FIG 7 .

Repression of the entire RicR regulon sensitized M. tuberculosis to Cu and attenuated M. tuberculosis in mice. (A) DNA affinity chromatography shows that RicR dissociates from the lpqS promoter in the presence of Cu as previously described (top) while RicRC38A constitutively bound to DNA regardless of the presence of Cu (bottom). Protein was eluted from the DNA with sequential increasing amounts of CuSO4 or a high salt concentration (last). (B) A Cu sensitivity assay revealed that the pc-ricRC38A strain was hypersensitive to Cu. The WT CDC1551/pMV306 (empty vector), ricR/pMV306, ricR/pMV-ricR+, ricR/pMV-pc-ricR+, and ricR/pMV-pc-ricRC38A strains were tested for Cu sensitivity. Data represent the mean ± standard deviation of one experiment that was done three times. (C) Constitutive repression of the ricR regulon attenuated M. tuberculosis growth in mice. CFU counts in the lungs of C57BL/6 mice infected with the WT/pMV306, ricR/pMV-pc-ricR+, ricR/pMV-pc-ricRC38A, ricR/pMV306, or ricR/pMV-ricR+ (ricR comp.) strain are shown. The data were separated into two graphs for clarity but represent infections done within the same week. The WT infection in both panels represents the same experiment. The initial dose of infection was ~500 CFU/mouse. The data represent the means ± standard deviations at days 1 (n = 3), 21 (n = 4), and 63 (n = 4) postinfection. The WT and pc-ricRC38A data are representative of two independent infections. ****, P < 0.0001; ***, P < 0.001 (two-way analysis of variance with a Bonferroni posttest). (D) Proteasomal-degradation-defective strains were hypersensitive to Cu. The WT/pMV306 and mpa/pMV306, mpa/pMV-mpa+, pafA/pMV306, and pafA/pMV-pafA+ mutant H37Rv strains were exposed to Cu for 10 days. The data represent the means ± standard deviations of one typical experiment that was done three times. <L.O.D., below the limit of detection.
Expression of pc-ricRC38A resulted in extreme sensitivity to Cu (Fig. 7B, far right). Interestingly and in contrast, the pc-ricR+-expressing strain was hyperresistant to Cu (Fig. 7B). Because RicR is a Cu-binding protein, it is possible that it sequesters Cu and protects against Cu toxicity when constitutively overproduced. Importantly, we found that the pc-ricRC38A-expressing strain, but not the pc-ricR+-expressing strain, was highly attenuated in mice (Fig. 7C). Taken together, our data support a model where several gene products of the RicR regulon, perhaps including RicR itself, are likely critical for WT Cu resistance and full virulence in a mouse model of infection.
It is worth noting that although a ricR null mutant is hyperresistant to Cu in vitro, it is not hypervirulent in mice. This mutant had a subtle growth defect in the lungs of infected mice compared to the WT and ricR-complemented strains (Fig. 7C, right panel).
Finally, we tested if M. tuberculosis strains defective in proteasome function were more sensitive to Cu. Proteasomal degradation of a protein requires PafA (proteasome accessory factor A), which ligates the posttranslational modifier Pup (prokaryotic ubiquitin-like protein) to protein substrates, and Mpa (mycobacterial proteasome ATPase), which delivers pupylated protein substrates into the proteasome core for degradation (reviewed in reference 27). Mutations that reduce proteasomal degradation have repressed the expression of all RicR regulon genes; however, RicR itself does not appear to be a proteasome substrate (11). Nonetheless, because the RicR regulon is repressed in proteasome-defective M. tuberculosis strains, we predicted that these strains would be more sensitive to Cu than WT bacteria are. Indeed, we found that M. tuberculosis strains lacking mpa or pafA were more sensitive to Cu than the WT and complemented strains were (Fig. 7D). Taken together, this suggests that the in vivo attenuated phenotype of proteasomal degradation mutants could be partly due to reduced Cu resistance.
DISCUSSION
The discovery of several Cu-responsive regulons in a human-exclusive pathogen suggests that M. tuberculosis faces host-supplied Cu during infections. In this work, we sought to understand the contribution of the RicR regulon to Cu resistance and virulence in mice. We determined that, with the exception of mymT, the disruption of any single RicR-regulated gene was insufficient to sensitize M. tuberculosis to Cu. We also established in this study that mmcO is a member of the RicR regulon. Overexpression of mmcO resulted in Cu hyperresistance but did not impact virulence. We also determined that the contributions of individual RicR-regulated genes to pathogenesis appear to be minimal. Single mutations in mmcO, lpqS, Rv2963, and socA did not attenuate M. tuberculosis growth in mice, and curiously, lpqS and Rv2963 mutants were hypervirulent for reasons that remain to be determined. We also tested the idea that deletion of the two RicR-regulated genes directly implicated in Cu resistance (mymT, mmcO) might have a more robust effect on bacterial survival in vivo. However, an mmcO mymT mutant was as virulent as WT M. tuberculosis in mice.
We did not detect robust Cu-associated phenotypes with the lpqS and Rv2963 mutants. LpqS and Rv2963 are putative membrane proteins each predicted to have several histidines that localize just outside the cytoplasmic membrane. These residues may be potential candidates to coordinate metal ions. Rv2963 is predicted to be a permease the disruption of which may perhaps alter either the import or the export of Cu or other metal ions under certain conditions.
It has been reported that a ΔlpqS::hyg mutant is hypersensitive to Cu in vitro (28). As in our study, the authors of that previous study could not complement their mutation, suggesting that their Cu-sensitive phenotype might be unlinked to LpqS. A possibility is that the disruption of lpqS in the study of Sakthi and Narayanan was polar on the expression of genes that are important for Cu resistance. Two uncharacterized genes, cysK2 and Rv0849, are cotranscribed with lpqS and may perhaps have a role in Cu resistance. Another possibility is that disruption of lpqS results in the dysregulation of the divergently expressed gene mmcO, which is important for Cu resistance. The mechanism of RicR repression of mmcO expression is not fully understood but may involve the bending of DNA between mmcO and lpqS to simultaneously repress both genes with a single RicR-binding site. On the basis of the published lpqS data and our data, we strongly hypothesize that LpqS itself is not critical for Cu resistance.
socAB is perhaps the most mysterious RicR-regulated locus; these genes are found only in the M. tuberculosis complex and do not resemble sequences in any other organism sequenced to date. Because of the lack of robust phenotypes associated with disruptions in this locus, it is unclear what role it plays, if any, in Cu homeostasis.
Although the lpqS and Rv2963 transposon mutants lacked clear Cu resistance phenotypes, both had very intriguing hypervirulence phenotypes in mice; however, we could not complement these mutations to restore WT virulence. We hypothesized that overexpression of mmcO was responsible for the hypervirulence of the lpqS strain, but this was not the case (Fig. 4). Currently, we can only speculate as to why the lpqS and Rv2963 mutants are hypervirulent. A possibility is that the absence of these putative membrane proteins permits the bacteria to grow more rapidly in vivo, which appears to be the reason for the increased virulence of these strains. Alternatively, it is possible that mutant or truncated proteins that alter the course of infection are produced by these strains. Yet another possibility is that the transposon insertions in these mutants change the expression of other genes that increase the growth of the bacteria in vivo. Needless to say, we are very interested in understanding why these mutants rapidly kill their hosts and are testing several of these hypotheses.
None of the genes of the RicR regulon individually showed a role in promoting virulence. It is possible that in the absence of one or more of the RicR-regulated genes, other genes encoding Cu-binding proteins or efflux systems could be induced to compensate for their absence. Nonetheless, a ricR mutant that constitutively represses all RicR-dependent promoters was highly attenuated in mice. These data strongly suggest that the RicR regulon, either in its entirety or in part, is required for the full virulence of M. tuberculosis. Our in vitro and in vivo data also suggest that RicR itself may sequester Cu like a metallothionein because the constitutive overexpression of WT ricR (as opposed to ricRC38A, which is expected not to bind Cu) resulted in increased Cu resistance (Fig. 7B). Another possible reason that single mutations had little to no impact in vivo is that mice may not be the best model for testing the role of these genes; some genes may show importance in models of infection that more closely resemble human disease.
It is notable that we observed considerable differences in Cu susceptibility, depending on the assay we used. We observed robust differences in Cu resistance when using the liquid-based quantitative assay for the mymT (hypersensitive), ricR, and lpqS (hyperresistant) mutant strains, while in contrast, we could detect a phenotype for mmcO mutants only by using an agar plate-based assay. Interestingly, both assays revealed that M. tuberculosis strain CDC1551 is inherently more resistant to Cu than H37Rv is. The results from the different assays suggest that different Cu-binding proteins are important under different conditions. Furthermore, it has yet to be determined which Cu regulon, CsoR or RicR, responds first to Cu stress. It is also possible that the repressors respond to different concentrations of Cu. In the agar-based assay, bacteria were exposed to Cu throughout the experiment (14 to 21 days), whereas in the liquid-based assay we exposed the bacteria for a defined time period (10 days) before inoculation onto agar. Additionally, the oxygen tension, which has a critical impact on the redox status of Cu, could impact the effective Cu+ concentration during the experiment. Finally, the media used for agar versus broth cultures are also slightly different and may impact Cu susceptibility in unknown ways.
At the forefront of our remaining questions is what the link is between RicR regulon expression and M. tuberculosis proteasomal degradation. A simple explanation would be that RicR is a proteasome substrate and that the accumulation of this repressor in proteasome degradation-defective mutants results in repression of the regulon. However, we have no evidence that RicR accumulates in proteasome-defective mutants. Another possibility is that one or several Cu-binding proteins are proteasome substrates the accumulation of which in a proteasome degradation-defective strain sequesters Cu away from RicR, leading to gene repression. These and other hypotheses are currently being tested.
Our work supports previous observations that Cu homeostasis is critical for the pathogenesis of M. tuberculosis (5, 10). As in other organisms, too little accessible Cu is detrimental while too much Cu can be toxic. Taken together, our findings affirm that the careful control of Cu homeostasis is essential for M. tuberculosis virulence and that the RicR regulon plays an important and nonredundant role in this process.
MATERIALS AND METHODS
Bacterial strains, growth conditions, plasmids, and primers.
The bacterial strains, plasmids, and primers used in this work are listed in Table 1. M. tuberculosis strains were grown in 7H9 broth (Difco) supplemented with 0.2% glycerol, 0.05% Tween 80, 0.5% bovine serum albumin, 0.2% dextrose, and 0.085% sodium chloride (ADN) or Sauton’s minimal medium (3.7 mM potassium phosphate, 2.5 mM magnesium sulfate, 30 mM l-asparagine, 3.5 mM zinc sulfate, 9.5 mM citric acid, 6.0% glycerol, 0.005% ferric ammonium citrate, 0.05% Tween 80). Cultures were grown at 37°C without agitation in vented flasks. For M. tuberculosis growth on solid medium, Middlebrook 7H11 agar (Difco) was prepared with Middlebrook OADC (oleic acid, albumin, dextrose, and catalase; BBL) supplementation. M. tuberculosis strains were grown in 50 µg ml−1 kanamycin, 50 µg ml−1 hygromycin, and/or 25 µg ml−1 streptomycin when necessary. Escherichia coli cultures were grown in Luria-Bertani (LB) broth (Difco) or on LB agar at 37°C. Antibiotics were added at final concentrations of 100 µg ml−1 kanamycin, 150 µg ml−1 hygromycin, and 50 µg ml−1 streptomycin.
To clone the pc-ricRC38A allele into pMV306, site-directed mutagenesis was performed by splicing by overlap extension by PCR (29) (the primers used are described in Table 1). Cysteine 38 of RicR was first changed to alanine. A second mutation was then introduced into the ricRC38A plasmid; 2 nucleotides (nt) were changed in the ricR promoter (ricRp) (TACCCCGCTGGGTA → TACCCCGCTGTTTA = ricRpc) to reduce repressor binding.
With the exception of the biotin tetraethylene glycol (BioTEG; Operon)-labeled primer, all primers were purchased from Invitrogen (Table 1). Phusion polymerase and restriction enzymes from New England Biolabs were used for cloning. All plasmid inserts generated by PCR were sequenced by GENEWIZ (Plainfield, NJ).
Production of MmcO antibodies and MmcO immunoblot analysis.
For antibody production, MmcO-His6 was overproduced in E. coli and purified under denaturing conditions by following the manufacturer’s instructions (Qiagen). Polyclonal rabbit antibodies were generated by Covance (Denver, PA). DlaT (used for loading controls) and RicR antibodies were described previously (11, 30, 31).
For immunoblot analysis, equivalent cell numbers based on optical density at 580 nm (OD580) were collected for each relevant strain at the same growth phase. For Cu-treated protein samples, bacteria were treated with CuSO4 for 4 h at a final concentration of 500 µM. Bacteria were washed once with phosphate-buffered saline (PBS) with 0.05% Tween 80 and resuspended in 300 µl of lysis buffer (100 mM Tris-Cl, 1 mM EDTA, pH 8). Bacteria were lysed by bead beating with zirconia beads three times for 30 s each. Whole-cell lysates were mixed with 4× sodium dodecyl sulfate (SDS) sample buffer and boiled for 10 min at 100°C. Proteins were separated by 10% SDS-polyacrylamide gel electrophoresis, transferred to nitrocellulose, incubated with rabbit polyclonal antibodies to MmcO at a dilution of 1:1,000 in 3% BSA in Tris-buffered saline with Tween 20, and visualized with horseradish peroxidase coupled with anti-rabbit secondary antibodies (Thermo Scientific). Detection of horseradish peroxidase was performed with SuperSignal West Pico or West Femto Chemiluminescent Substrate (Thermo Scientific).
Construction of M. tuberculosis mutants.
We made M. tuberculosis mutants by using a method previously described by our lab (11). Briefly, pYUB854, which was originally developed for specialized transduction mutagenesis (32), was used to clone ~700 bp of sequence both upstream (5′) and downstream (3′) of a gene of interest. The 5′ and 3′ sequences, including the start and stop codons, respectively, were cloned to flank the hygromycin resistance cassette in pYUB854. We also constructed a kanamycin resistance-marked version of this plasmid to make the ΔmmcO::kan mutant. The hygromycin resistance cassette in pYUB854 was replaced with the kanamycin resistance cassette from pMV306.kan to generate pYUB854.kan (the primers and plasmids used are described in Table 1). Because the ricR mutant is in the CDC1551 background, we chose to mutate mmcO in both the H37Rv and CDC1551 strains.
Plasmids were digested with PacI, and ~1 µg of linearized, gel-purified DNA was used for electroporation into M. tuberculosis. M. tuberculosis strains were grown to an OD580 of ~0.4 to 1, washed, and resuspended in 10% glycerol to make electrocompetent cells as described in detail elsewhere (33). Bacteria were inoculated onto 7H11 solid medium with 50 µg ml−1 hygromycin or 50 µg ml−1 kanamycin as needed. A no-DNA control electroporation was always done to check for spontaneous antibiotic-resistant mutants. After 2 weeks, colonies were picked and inoculated into 200 µl of 7H9 medium with antibiotics. The 200-µl 7H9 starter cultures were inoculated into 5-ml cultures for further analysis. Potential mutants were tested by immunoblot analysis, PCR (Taq polymerase; Qiagen), and sequencing.
DNA affinity chromatography.
Experiments were performed as described previously (11). DNA probes were made by amplifying the lpqS promoter with a forward primer containing 5′ BioTEG (Operon) modifications. Clarified lysates of E. coli producing either WT RicR or RicRC38A were incubated with a DNA-Dynabead mixture. Protein interacting with the coupled DNA was sequentially eluted with increasing concentrations of CuSO4 as indicated and finally with high-salt buffer (1 M NaCl in 50 mM Tris, pH 7.5). RicR immunoblotting was performed as previously described (11).
Genome sequencing.
MHD131 (lpqS::ΦMycoMarT7) and the parental H37Rv strain were sequenced with an Illumina GenomeAnalyzer IIx. Approximately 5 µg of DNA was processed by the standard Illumina sample preparation protocol for genomic DNA (Illumina, Inc.), and the samples were sequenced in paired-end mode with a read length of 51 nt. The genomes were assembled by mapping reads to the public H37Rv reference sequence (GenBank accession no. NC_000962) and single-nucleotide polymorphisms and insertions/deletions as described in reference 34. The mean depths of coverage (number of reads covering each site) were 59.6 and 63.1 times, respectively, for the two strains.
Cu sensitivity assays.
CuSO4 powder was dissolved in water and sterilized through 0.45-µm-pore-size filters to make a 1 M stock solution. M. tuberculosis strains were grown in 7H9-ADN medium to an OD580 of ~0.5 to 1. Bacteria were harvested, washed once with Sauton’s medium (no added Cu), and then subjected to a low-speed spin (800 g for 8 min) to remove clumped cells. Bacteria were diluted to an OD580 of 0.08 in Sauton’s medium. Each culture was placed into the wells of a 96-well plate at 194 µl per well and treated with 6 µl of CuSO4 at different concentrations, and the plate was incubated for 10 days. Bacteria were then diluted and inoculated onto 7H11 agar and CFU were enumerated 2 to 3 weeks later. Because we noticed some variability in the minimum bactericidal Cu concentration between experiments, we always used a range of Cu concentrations for every experiment and drew conclusions based on reproducible trends. All experiments were done at least in duplicate.
The agar plate assay was performed as described previously (15, 17). M. tuberculosis strains were grown in 7H9-ADN medium to an OD580 of ~0.5 to 1. Cells were harvested by centrifugation, washed once with PBS-Tween (0.05%), and then spun slowly at 800 × g. Declumped cell suspensions were diluted to an OD580 of 0.1 with PBS-Tween. Three-microliter samples of serial dilutions were spotted onto M. tuberculosis 7H11-OADC agar plates containing either no additional CuSO4 or different concentrations thereof. Plates were incubated at 37°C for ~2 weeks.
Mouse infections.
Mouse infections were performed essentially as described previously (12). M. tuberculosis strains were grown in 7H9-ADN medium to an OD580 of ~0.4. Bacterial clumps were removed from the cultures as described above. Female C57BL6/J mice (Jackson Laboratories) were infected with a Glas-Col Inhalation Exposure System to inoculate ~200 to 400 CFU/mouse. Three (day 1) or four (days 21 and ~56) mice were humanely sacrificed, and their lungs and spleens were removed, homogenized, and inoculated onto 7H11 agar. CFU were enumerated after 2 to 3 weeks. All procedures were performed with the approval of the NYU Institutional Animal Care and Use Committee.
ACKNOWLEDGMENTS
We thank Andrew Darwin for critical review of a draft version of the manuscript. We are grateful to Carl Nathan, Ben Gold, and Bill Bishai for strains and antibodies. We thank Tamara Reyes-Robles for MmcO purification and Michael Niederweis, Jennifer Rowland, and David Giedroc for helpful suggestions.
This work was supported by NIH grant R01 HL92774 awarded to K.H.D. J.C.S. was supported by Robert A. Welch Foundation grant A-0015. We are grateful to Charlie Rice and the Rockefeller University for providing space and support for the year after Superstorm Sandy.
Footnotes
Citation Shi X, Festa RA, Ioerger TR, Butler-Wu S, Sacchettini JC, Darwin KH, Samanovic MI. 2014. The copper-responsive RicR regulon contributes to Mycobacterium tuberculosis virulence. mBio 5(1):e00876-13. doi:10.1128/mBio.00876-13.
REFERENCES
- 1. Blumberg HM, Burman WJ, Chaisson RE, Daley CL, Etkind SC, Friedman LN, Fujiwara P, Grzemska M, Hopewell PC, Iseman MD, Jasmer RM, Koppaka V, Menzies RI, O’Brien RJ, Reves RR, Reichman LB, Simone PM, Starke JR, Vernon AA, American Thoracic Society, Centers for Disease Control and Prevention, Infectious Diseases Society 2003. American Thoracic Society/centers for disease control and Prevention/infectious diseases society of America: treatment of tuberculosis. Am. J. Respir. Crit. Care Med. 167:603–662. 10.1164/rccm.167.4.603 [DOI] [PubMed] [Google Scholar]
- 2. Gandhi NR, Nunn P, Dheda K, Schaaf HS, Zignol M, van Soolingen D, Jensen P, Bayona J. 2010. Multidrug-resistant and extensively drug-resistant tuberculosis: a threat to global control of tuberculosis. Lancet 375:1830–1843. 10.1016/S0140-6736(10)60410-2 [DOI] [PubMed] [Google Scholar]
- 3. Zignol M, Hosseini MS, Wright A, Weezenbeek CL, Nunn P, Watt CJ, Williams BG, Dye C. 2006. Global incidence of multidrug-resistant tuberculosis. J. Infect. Dis. 194:479–485. 10.1086/505877 [DOI] [PubMed] [Google Scholar]
- 4. Velayati AA, Masjedi MR, Farnia P, Tabarsi P, Ghanavi J, ZiaZarifi AH, Hoffner SE. 2009. Emergence of new forms of totally drug-resistant tuberculosis bacilli: super extensively drug-resistant tuberculosis or totally drug-resistant strains in Iran. Chest 136:420–425. 10.1378/chest.08-2427 [DOI] [PubMed] [Google Scholar]
- 5. Wolschendorf F, Ackart D, Shrestha TB, Hascall-Dove L, Nolan S, Lamichhane G, Wang Y, Bossmann SH, Basaraba RJ, Niederweis M. 2011. Copper resistance is essential for virulence of Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. U. S. A. 108:1621–1626. 10.1073/pnas.1009261108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Samanovic MI, Ding C, Thiele DJ, Darwin KH. 2012. Copper in microbial pathogenesis: meddling with the metal. Cell Host Microbe 11:106–115. 10.1016/j.chom.2012.01.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wagner D, Maser J, Lai B, Cai Z, Barry CE, Höner zu Bentrup K, Russell DG, Bermudez LE. 2005. Elemental analysis of Mycobacterium avium-, Mycobacterium tuberculosis-, and Mycobacterium smegmatis-containing phagosomes indicates pathogen-induced microenvironments within the host cell’s endosomal system. J. Immunol. 174:1491–1500 http://www.jimmunol.org/content/174/3/1491.long [DOI] [PubMed] [Google Scholar]
- 8. White C, Lee J, Kambe T, Fritsche K, Petris MJ. 2009. A role for the ATP7A copper-transporting ATPase in macrophage bactericidal activity. J. Biol. Chem. 284:33949–33956. 10.1074/jbc.M109.070201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Liu T, Ramesh A, Ma Z, Ward SK, Zhang L, George GN, Talaat AM, Sacchettini JC, Giedroc DP. 2007. CsoR is a novel Mycobacterium tuberculosis copper-sensing transcriptional regulator. Nat. Chem. Biol. 3:60–68. 10.1038/nchembio844 [DOI] [PubMed] [Google Scholar]
- 10. Ward SK, Abomoelak B, Hoye EA, Steinberg H, Talaat AM. 2010. CtpV: a putative copper exporter required for full virulence of Mycobacterium tuberculosis. Mol. Microbiol. 77:1096–1110. 10.1111/j.1365-2958.2010.07273.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Festa RA, Jones MB, Butler-Wu S, Sinsimer D, Gerads R, Bishai WR, Peterson SN, Darwin KH. 2011. A novel copper-responsive regulon in Mycobacterium tuberculosis. Mol. Microbiol. 79:133–148. 10.1111/j.1365-2958.2010.07431.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Darwin KH, Ehrt S, Gutierrez-Ramos JC, Weich N, Nathan CF. 2003. The proteasome of Mycobacterium tuberculosis is required for resistance to nitric oxide. Science 302:1963–1966. 10.1126/science.1091176 [DOI] [PubMed] [Google Scholar]
- 13. Cerda-Maira FA, Pearce MJ, Fuortes M, Bishai WR, Hubbard SR, Darwin KH. 2010. Molecular analysis of the prokaryotic ubiquitin-like protein (pup) conjugation pathway in Mycobacterium tuberculosis. Mol. Microbiol. 77:1123–1135. 10.1111/j.1365-2958.2010.07276.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lamichhane G, Zignol M, Blades NJ, Geiman DE, Dougherty A, Grosset J, Broman KW, Bishai WR. 2003. A postgenomic method for predicting essential genes at subsaturation levels of mutagenesis: application to Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. U. S. A. 100:7213–7218. 10.1073/pnas.1231432100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gold B, Deng H, Bryk R, Vargas D, Eliezer D, Roberts J, Jiang X, Nathan C. 2008. Identification of a copper-binding metallothionein in pathogenic mycobacteria. Nat. Chem. Biol. 4:609–616. 10.1038/nchembio.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Festa RA, Pearce MJ, Darwin KH. 2007. Characterization of the proteasome accessory factor (paf) operon in Mycobacterium tuberculosis. J. Bacteriol. 189:3044–3050. 10.1128/JB.01597-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Rowland JL, Niederweis M. 2013. A multicopper oxidase is required for copper resistance in Mycobacterium tuberculosis. J. Bacteriol. 195:3724–3733. 10.1128/JB.00546-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hellman NE, Gitlin JD. 2002. Ceruloplasmin metabolism and function. Annu. Rev. Nutr. 22:439–458. 10.1146/annurev.nutr.22.012502.114457 [DOI] [PubMed] [Google Scholar]
- 19. Nakamura K, Go N. 2005. Function and molecular evolution of multicopper blue proteins. Cell. Mol. Life Sci. 62:2050–2066. 10.1007/s00018-004-5076-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kosman DJ. 2003. Molecular mechanisms of iron uptake in fungi. Mol. Microbiol. 47:1185–1197. 10.1046/j.1365-2958.2003.03368.x [DOI] [PubMed] [Google Scholar]
- 21. Huston WM, Jennings MP, McEwan AG. 2002. The multicopper oxidase of Pseudomonas aeruginosa is a ferroxidase with a central role in iron acquisition. Mol. Microbiol. 45:1741–1750. 10.1046/j.1365-2958.2002.03132.x [DOI] [PubMed] [Google Scholar]
- 22. Huston WM, Naylor J, Cianciotto NP, Jennings MP, McEwan AG. 2008. Functional analysis of the multi-copper oxidase from Legionella pneumophila. Microbes Infect. 10:497–503. 10.1016/j.micinf.2008.01.011 [DOI] [PubMed] [Google Scholar]
- 23. Kim C, Lorenz WW, Hoopes JT, Dean JF. 2001. Oxidation of phenolate siderophores by the multicopper oxidase encoded by the Escherichia coli yacK gene. J. Bacteriol. 183:4866–4875. 10.1128/JB.183.16.4866-4875.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Roberts SA, Weichsel A, Grass G, Thakali K, Hazzard JT, Tollin G, Rensing C, Montfort WR. 2002. Crystal structure and electron transfer kinetics of CueO, a multicopper oxidase required for copper homeostasis in Escherichia coli. Proc. Natl. Acad. Sci. U. S. A. 99:2766–2771. 10.1073/pnas.052710499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Djoko KY, Chong LX, Wedd AG, Xiao Z. 2010. Reaction mechanisms of the multicopper oxidase CueO from Escherichia coli support its functional role as a cuprous oxidase. J. Am. Chem. Soc. 132:2005–2015. 10.1021/ja9091903 [DOI] [PubMed] [Google Scholar]
- 26. Kosman DJ. 2010. Multicopper oxidases: a workshop on copper coordination chemistry, electron transfer, and metallophysiology. J. Biol. Inorg. Chem. 15:15–28. 10.1007/s00775-009-0590-9 [DOI] [PubMed] [Google Scholar]
- 27. Samanovic MI, Li H, Darwin KH. 2013. The pup-proteasome system of Mycobacterium tuberculosis. Subcell. Biochem. 66:267–295. 10.1007/978-94-007-5940-4_10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sakthi S, Narayanan S. 2013. The lpqS knockout mutant of Mycobacterium tuberculosis is attenuated in macrophages. Microbiol. Res. 168:407–414. 10.1016/j.micres.2013.02.007 [DOI] [PubMed] [Google Scholar]
- 29. Ho SN, Hunt HD, Horton RM, Pullen JK, Pease LR. 1989. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene 77:51–59 [DOI] [PubMed] [Google Scholar]
- 30. Tian J, Bryk R, Itoh M, Suematsu M, Nathan C. 2005. Variant tricarboxylic acid cycle in Mycobacterium tuberculosis: identification of α-ketoglutarate decarboxylase. Proc. Natl. Acad. Sci. U. S. A. 102:10670–10675. 10.1073/pnas.0501605102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Darwin KH, Lin G, Chen Z, Li H, Nathan CF. 2005. Characterization of a Mycobacterium tuberculosis proteasomal ATPase homologue. Mol. Microbiol. 55:561–571. 10.1111/j.1365-2958.2004.04403.x [DOI] [PubMed] [Google Scholar]
- 32. Bardarov S, Bardarov S, Jr, Pavelka MS, Jr, Sambandamurthy V, Larsen M, Tufariello J, Chan J, Hatfull G, Jacobs WR., Jr. 2002. Specialized transduction: an efficient method for generating marked and unmarked targeted gene disruptions in Mycobacterium tuberculosis, M. bovis BCG and M. smegmatis. Microbiology 148:3007–3017 http://mic.sgmjournals.org/content/148/10/3007.long [DOI] [PubMed] [Google Scholar]
- 33. Hatfull GF, Jacobs WR. 2000. Molecular genetics of mycobacteria. ASM Press, Washington, DC [Google Scholar]
- 34. Ioerger TR, Feng Y, Ganesula K, Chen X, Dobos KM, Fortune S, Jacobs WR, Jr, Mizrahi V, Parish T, Rubin E, Sassetti C, Sacchettini JC. 2010. Variation among genome sequences of H37Rv strains of Mycobacterium tuberculosis from multiple laboratories. J. Bacteriol. 192:3645–3653. 10.1128/JB.00166-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chong YH, Jung JM, Choi W, Park CW, Choi KS, Suh YH. 1994. Bacterial expression, purification of full length and carboxyl terminal fragment of Alzheimer amyloid precursor protein and their proteolytic processing by thrombin. Life Sci. 54:1259–1268 [DOI] [PubMed] [Google Scholar]
- 36. Stover CK, de la Cruz VF, Fuerst TR, Burlein JE, Benson LA, Bennett LT, Bansal GP, Young JF, Lee MH, Hatfull GF, Snapper SB, Barletta RG, Jacobs WR, Bloom BR. 1991. New use of BCG for recombinant vaccines. Nature 351:456–460 [DOI] [PubMed] [Google Scholar]