

Abstract
Methylation at histone H3 lysine 27 (H3K27me) is an evolutionarily conserved epigenetic mark associated with transcriptional repression and replication elongation. We have previously shown that in Tetrahymena thermophila, a unicellular eukaryote, the histone methyltransferases (HMTs) TXR1 and EZL2 are primarily responsible for H3K27 mono-methylation (H3K27me1) and di-/tri-methylation (H3K27me2/3), respectively. Using 15N metabolically labeled histones as the internal reference, we quantified global changes in histone post-translational modifications in ΔTXR1 and ΔEZL2 cells, to systematically identify potential crosstalk between H3K27 methylation and other PTMs across all four core histones as well as their variants. Most prominently, we observed hyper-acetylation of histones H2A, H2A.Z, and H4 in their N-terminal domains in response to decreased H3K27 methylation. We also provide additional evidence implicating hyper-acetylation in the DNA damage response pathway in replication-defective ΔTXR1 cells, in apparent contrast to the transcriptional role of hyper-acetylation in ΔEZL2 cells.
In eukaryotic cells, nuclear DNA wraps around an octamer of two copies each of the four core histones H2A, H2B, H3, and H4, to form the nucleosome, the basic unit of chromatin. Many post-translational modifications (PTMs)1, including methylation, acetylation, phosphorylation, biotinylation, citrullination, ADP-ribosylation, and ubiquitylation, occur at numerous sites on histones (1). Dynamic changes in the chromatin structure and architecture, including the switch between condensed and decondensed states as well as interactions with a wide range of protein complexes, are modulated by these PTMs, deposited by histone modifying enzymes in a combinatorial pattern that is still being actively deciphered (2, 3). Reflecting its crucial role in DNA-mediated transactions, the functionality of chromatin is relatively robust, tolerating substantial mutations in histone residues carrying PTMs as well as histone modifying enyzmes (4). The robustness is generally attributed to redundant or parallel roles played by many histone modifications, which often adapt compensatory changes to these perturbations. However, details of the crosstalk among histone PTMs and their physiological impacts are still incomplete (5).
Deposited by histone methyltransferases (HMTs) and removed by demethylases, histone lysine methylation (mono-, di-, and tri-methylated forms) is a dynamic epigenetic mark (6–8). Boosted by the identification and characterization of the enzymatic machinery, histone lysine methylation has been the target of considerable research efforts, reflecting its key role in modulating histone functions. Unlike acetylation, methylation does not directly affect histone/DNA interactions by altering the net charge of histones, and its physiological impacts depend on the exact form and site of modification, as well as the effector proteins recognizing them (9).
The evolutionarily conserved H3K27 methylation has been traditionally associated with heterochromatin formation and transcriptional repression (10). Enhancer of zeste (E(z)), initially identified by its role in Polycomb repression, is the HMT required for H3K27 methylation in Drosophila (11–13). E(z) homologs are found in protozoa, metazoa, and plants, though they are conspicuously missing in fungi. All three forms of H3K27 methylation (H3K27me1/2/3) are catalyzed by E(z) homologs in metazoa (14). However, in protozoa and plants, a different family of HMTs are specific for H3K27me1, examplified by Arabidopsis ATXR5/ATXR6 (15).
As a protozoan model organism, Tetrahymena thermophila features high levels of H3K27 methylation and several H3K27-specific HMTs, including EZL1, EZL2, EZL3 (all E(z) homologs), and TXR1 (ATXR5/ATXR6 homolog) (16–19). In vegetatively growing cells, only TXR1 and EZL2 are significantly expressed. Our previous studies show that they jointly regulate H3K27 methylation at this physiological stage: TXR1 preferentially deposits H3K27me1 and affects replication elongation, whereas EZL2 is mainly responsible for H3K27me2/3 and transcription repression (18). Although H3K27 methylation is extensively studied, its relationship with other histone PTMs has not been systematically explored. In this study, we used MS-based quantitative proteomics methods to compare the global histone PTM levels in wild-type (WT), ΔTXR1, and ΔEZL2 cells. We identified potential crosstalk between H3K27 methylation and other histone PTMs, including prominent hyper-acetylation of histones H2A, H2A.Z, and H4 in their N-terminal tails. We also propose that two different pathways underlie the hyper-acetylation events observed in ΔTXR1 and ΔEZL2 cells. The results provide further support for the functional differentiation between TXR1-dependent H3K27me1 and EZL2-dependent H3K27me2/3.
EXPERIMENTAL PROCEDURES
Construction of HMT Knockout Strains
HMT mutants, ΔTXR1 and ΔEZL2, were generated from Tetrahymena wild-type CU428 cells as described previously (18). Briefly, genomic sequences flanking TXR1 or EZL2 were PCR amplified and fused with the neo4 cassette, which confers paromomycin resistance (20). Transformants were generated by standard biolistic bombardment and selected by paromomycin (21, 22). After phenotypic assortment, complete replacement of endogenous copies in macronuclei was verified by quantitative PCR.
Cell Culture, Core Histone Preparation, and HPLC Purification
Media, cell culture conditions, and procedures used for nuclear preparation, acid extraction of histone, and histone purification were as described previously (17, 18). The overall experimental design is illustrated in supplemental Fig. S1. Briefly, Tetrahymena wild-type, ΔTXR1, and ΔEZL2 cells were grown in 1× SPP medium at 30 °C with gentle shaking. Logarithmic-phase (2 × 105/ml) cells were collected for subsequent experiments. To prepare the [15N] histones as internal standards, wild-type Tetrahymena cells were metabolically labeled by feeding on Escherichia coli BL21 cells grown in the [15N] M9 minimal medium supplemented with 15N-substituted Bioexpress (Cambridge Isotope Laboratories, Andover, MA). To isolate Tetrahymena macronuclei, 2 × 108 cells were collected by centrifugation, resuspended in 200 ml of medium A (0.1 m sucrose, 2 mm MgCl2, 4% gum arabic, 10 mm Tris, 5 mm EDTA, 10 mm butyric acid, and 1 mm PMSF, pH 6.75), and disrupted by vigorous blending in the presence of 1-octanol (∼1 ml); macronuclei were subsequently separated by differential centrifugation. The purified macronuclei (1 × 108) were extracted in 1 ml of sulfuric acid (0.4 N) overnight. The acid-extracted histone samples were precipitated by 20% trichloroacetic acid, washed with acetone, and dissolved in 500 μl of water. To further purify individual histones, the histone samples were resolved on a C8 reversed-phase high performance liquid chromatography (HPLC) column. HPLC fractions containing individual histones were combined after evaluation by SDS-PAGE and quantified by the Bradford method.
NanoLC-MS Analysis of Core Histone PTMs
Chemical derivatization, trypsinization, and nanoLC-MS/MS analysis of histone samples (in biological triplicates) were performed as previously described (17, 23). Briefly, 3 μg of each core histone from WT, ΔTXR1, or ΔEZL2 cells, were equally mixed with the internal standards, the [15N] core histones prepared from WT cells as described above. Before mass spectrometry analysis, samples were chemically derivatized with propionic anhydride before and after trypsin digestion (23). All histones were in-solution digested with sequencing-grade trypsin and the fully propionylated peptide mixtures were vacuum-dried and dissolved in 0.1% formic acid before the LC-MS analysis. Acid extracted bulk histones (for H2A.Y identification) and histone H2A.Z (for N-terminal acetylation quantification) collected from HPLC were directly submitted to trypsin digestion without propionylation. The peptide mixtures were separated with a C18 analytical column (3 μm, 300Å, 150 mm x 100 μm; CVC Technologies, Fontana, CA), eluted, and introduced into a Thermo Fisher Orbitrap XL mass spectrometer with a 15 μm PicoTip emitter (New Objective, Woburn, MA) at 200 nl/min flow rate. Mass spectrometer was connected with a manufacturer's nano-ESI source operated in both positive ion mode and data dependent mode at a resolution of 30,000 on MS1. The 8 strongest peaks were collision induced dissociation (CID) fragmented with 30% normalized collision energy. To achieve the best LC-MS/MS performance, a 2-min dynamic exclusion and internal mass lock were also executed.
Raw data processing, database searching, and peptide quantification were all done in Mascot Distiller (Matrix Sciences, Version 2.4 for Distiller and Version 2.2.07 for search engine). The NCBI Tetrahymena database (created from NCBI on 5/25/2010 with a total of 51,502 sequence entries) was used in performing the database searching. Precursor ion tolerance was 10 ppm and 0.8 Da for the fragment ions. N-terminal propionylation was set as fixed modification and the following PTMs were considered as variable modifications: Acetylation (Protein N terminus, K), Methylation (Protein N terminus, KR), Propionylation (K, unmodified or monomethylated), Phosphorylation (STY), Formylation (K), Citrullination (R), and Ubiquitination (K). Up to five missed cleavages were allowed for trypsin digestion as the propionylation and PTMs on lysyl residues blocks trypsin digestion. Several peptides from each of the core histones were selected for normalization by their average values: GKTASSKQVSR, GQASQDL, FLKHGR, AGLQFPVGR, HLLLAIR, ATIAGGGVIPHIHK from H2A and its variants; LLLPGELAR, IALESSKLVR, EVQTAVKLLLPGELAR from H2B; FRPGTVALR, VTIMTKDMQLAR, YQKSTDLLIR from H3; and SFLENVVR, QVLKSFLENVVR, TLYGFGG from H4. We assumed that the levels of those peptides were unchanged across different epigenetic features as their ratios (L/H) were close to 1 and quite consistent (coefficient of variation, CV <15%) in all experiments. Student t test was done in Microsoft Excel and heatmaps were generated from R software. All peptides with scores above mascot identity or homology threshold were considered as positive hits when a confidence threshold is set at p = 0.05. Spectra assigned with PTMs were manually validated based on the same criteria we published previously (17). Our unambiguous assignment of PTM sites in the H2B N-terminal tail was facilitated by a previous MS study of Tetrahymena H2B (24).
Analysis of Acetylation States in Histone H2A and H4 by Acetic Acid-Urea Polyacrylamide Gel Electrophoresis
To analyze the acetylated isoforms of H2A and H4, HPLC-purified intact proteins were separated by continuous acetic acid-urea (AU) gel system as described (25). H2A and H4 samples prepared from WT and mutants were treated with λ protein phosphatase (New England Biolabs, Ipswich, MA), re-pelleted, and dissolved in 10 μl of loading buffer containing 2 m urea, 2% β-mercaptoethanol, and 5% acetic acid, before they were loaded onto the gel. The gel was stained using the blue silver staining method for 30 min and visualized by destaining in deionized water for several times (26).
Chromatin Immunoprecipitation (ChIP)
Cells were fixed with 0.5% paraformaldehyde (PFA) and treated with micrococcal nuclease (MNase). Mono-nucleosomes were purified by sucrose gradient as described (27). ChIP was performed as described (16), using purified mono-nucleosome as input. The primary antibody (Penta, used at 1:500 dilution) has been validated to recognize acetylated-lysine in H2A.Z and H4 N-terminal tails (28). Salmon sperm DNA covered protein A beads (Millipore) were used to recover the immuno-complex.
RESULTS AND DISCUSSION
In Tetrahymena WT, ΔTXR1, and ΔEZL2 cells, we identified 72 PTMs in core histones and quantified 55 of them with low variance (CV<15%, after data normalization): 17/13 (identified/quantified) in H2A, 12/10 in H2B variants, 32/25 in H3 variants, and 11/7 in H4. Methylation and acetylation were the most common PTMs identified, whereas histone H3 exhibited the most complex modification profile (supplemental Fig. S2 and Table S1). The annotated spectra of all identified PTMs are provided (supplemental Fig. S3). Overall, at the Mascot identity threshold, the false discovery rate (FDR) is close to 1% on average as estimated from the decoy database. The identified tryptic peptides provided sequence coverages for H2A, H2B, H3, and H4 at 60%, 68%, 76%, and 97%, respectively (supplemental Fig. S2). Variants were identified for all histones except H4, including three H2A variants (H2A, H2A.X, and H2A.Z), two H2B variants (H2B.1, H2B.2), and three H3 variants (H3, H3.3, and H3.4).
Off-site Hyper-methylation Potentially Compensates H3K27 Methylation Deficiency in ΔTXR1 Cells
H3K27 methylation was significantly affected in ΔTXR1 and ΔEZL2 cells, as previously reported (supplemental Table S2-H3) (17, 18). In ΔTXR1 cells, the mono-methylation of H3K27 and H3K36 were reduced at least 80%, whereas the unmethylated form of the peptide increased significantly. The dimethylated form was also significantly reduced, whereas the trimethylated form was only slightly affected. In ΔEZL2 cells, H3K27me2/3 levels declined by at least 80%, whereas H3K27me1 levels increased, consistent with H3K27me1 being a feedstock for the HMT activity of EZL2. However, H3K27 methylation was not completely eliminated even in the ΔTXR1/ΔEZL2 double knockout cells (supplemental Table S3-EZL2/TXR1), suggesting the presence of other HMTs with similar specificity but reduced efficiency. In contrast to the decreased levels of H3K27me1, increased off-site mono-methylation was observed for H3 at K4, K23, and particularly K56 in ΔTXR1 (Table S2-H3). Changes in lysine mono-methylation on histones other than H3 were less significant (p ≥ 0.05, supplemental Table S2, H2A, H2B, H4). Also observed was increased arginine mono-methylation at H3R83 and possibly at H3R128 (p ≥ 0.05). It is possible that hyper-methylation at these sites at least partially compensates for H3K27me1 deficiency in ΔTXR1 cells.
Hyper-acetylation in Histone H2A, H2A.Z, and H4 in Response to H3K27 Methylation Deficiency
Controlled by histone acetyltransferases (HATs) and histone deacetylases (HDACs), histone acetylation contributes to a more open chromatin structure and recruits effector proteins leading to downstream events (29). Acetylation neutralizes the positively charged lysyl residues on histones and attenuates electrostatic interactions with the negatively charged DNA, facilitating chromatin decondensation. Acetyl-lysine is also recognized by bromodomains, which are ubiquitous in eukaryotes and widely distributed in pathways involving DNA transactions (30). Hyper-acetylation is traditionally linked to transcription activation (31, 32), and more recently implicated in DNA repair (33).
Elevated acetylation levels were observed on many sites in histones from ΔTXR1 and ΔEZL2 cells (supplemental Table S2). Hyper-acetylation of H2A (including the canonical H2A and the variant H2A.X, sharing the same N-terminal sequence), H2A.Z, and H4 in their N-terminal tails is the most prominent feature in the H3K27 methylation-deficient mutants (Figs. 1, 2, and 3). There are four acetylatable lysine residues in the N-terminal domains of H2A and H2A.X, which share the same amino acid sequence and are not distinguishable by MS. The presence of mono-, di-, tri-, and tetra-acetylated forms, with their various permutations, generated complicated tryptic peptides, which were resolved by MS (Fig. 1A, 1B). As determined from the quantitative proteomics results, the mutant/WT ratios of the acetylated forms were gradually increased from low to high degree of acetylation (Fig. 1A). The fully tetra-acetylated peptide showed a more than threefold increase in H2A from ΔTXR1 and ΔEZL2 cells (Fig. 1A). This increase in hyper-acetylated H2A was also clearly visualized by the acetic acid-urea gel electrophoresis (Fig. 4), which separates different protein isoforms by their net charges, directly reflecting acetylation levels in the absence of phosphorylation (on phosphatase treatment). Future analysis of separated H2A and H2A.X will address whether they have different patterns of N-terminal acetylation.
Fig. 1.

Hyper-acetylation of H2A N-terminal domain in ΔTXR1 and ΔEZL2 cells. Acetylation of H2A N-terminal domains exhibited progressive increases from low degree to high degree of acetylation. Levels of the fully, tetra-acetylated peptide were dramatically increased in the HMT mutants. A, combinatorial acetylation patterns in H2A N-terminal domains. B, H2A N-terminal tetra-acetylation pattern confirmed via tandem mass spectrum of 1172.628 Da precursor ion at 1 ppm (Z = 2+, Ion score = 54). The b- and y- fragment ions are consistent with the tetra-acetylated peptide. The ratio of tetra-acetylated peptide is determined by extracted ion chromatography in a matched time window (Retention time) and is greatly up-regulated in both ΔTXR1 and ΔEZL2 cells. Inset: relative intensities for heavy isotope labeled wild type versus mutant peptide by retention time. Note that PTM combinations in the heatmap are averaged over charge states and tryptic cleavage sites.
Fig. 2.

Hyper-acetylation of H2A.Z N-terminal domain in ΔTXR1 and ΔEZL2 cells. Acetylation in H2A.Z N-terminal domains exhibited progressive increases from low degree to high degree of acetylation. Levels of the fully, hexa-acetylated peptide were dramatically increased in the HMT mutants. A, combinatorial acetylation patterns in H2A.Z N-terminal domains. B, H2A.Z N-terminal hexa-acetylation. The first six lysine residues are all targets of acetylation as confirmed by tandem MS of 736.4026 Da precursor ion at 2 ppm (Z = 3+, Ion score = 91). The acetylated peptide is greatly increased in HTM knockout cells as quantified by 15N uniform labeling. Note that PTM combinations in the heatmap are averaged over charge states and tryptic cleavage sites.
Fig. 3.

Hyper-acetylation of H4 N-terminal domain in ΔTXR1 and ΔEZL2 cells. H4 N-terminal domains exhibited progressive increases from low degree to high degree of acetylation. Levels of the fully, tetra-acetylated peptide were dramatically increased in the HMT mutants. A, combinatorial acetylation patterns in H4 N-terminal domains. B, H4 N-terminal tetra-acetylation confirmed via tandem mass spectrum of 841.9559 Da precursor ion at 2 ppm (Z = 2+, Ion score = 51). Note that PTM combinations in the heatmap are averaged over charge states and tryptic cleavage sites.
Fig. 4.
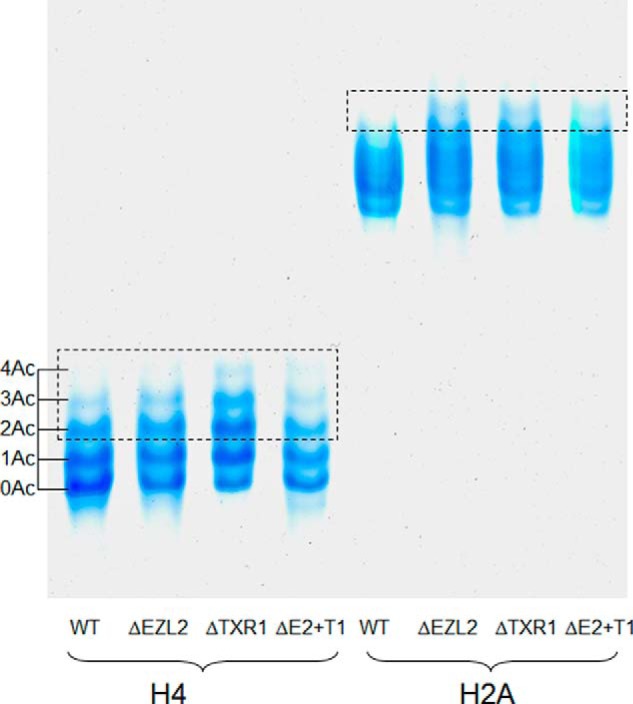
Validation of histone hyper-acetylation by acid-urea gel electrophoresis. Increased levels of acetylation from low-degree to high-degree in H2A and H4 N-terminal domains were also demonstrated by acid-urea gel electrophoresis in the HMT mutants. Histone samples were treated with λ protein phosphatase to remove phosphorylations before loading onto the gel. Five micrograms of total protein (either H4 or H2A) were loaded onto a 15% acid urea gel and run at 100V for 30 min followed by 400V for 10 h. Note: ΔE2+T1 is the double knockout strain of EZL2 and TXR1.
There are six acetylatable lysine residues in the N-terminal tail of H2A.Z. We observed a similar trend in the gradual buildup of the hyper-acetylated forms, culminating in dramatic accumulations of penta- and hexa-acetylated peptides in ΔTXR1 and ΔEZL2 cells (Fig. 2A, 2B). There are four acetylatable lysine residues in the conserved N-terminal tail of H4 (K4, K7, K11, and K15, corresponding to K5, K8, K12, and K16 in higher eukaryotes). Accumulations of hyper-acetylated forms in the two mutants were observed, coupled to corresponding diminishment of unacetylated forms (Fig. 3A, 3B). In addition to hyper-acetylation of H2A, H2A.Z, and H4 in their N-terminal domains, K9ac, K14ac, K18ac, K23ac, K27ac, and K56ac levels were also moderately elevated in H3 (Tab. S2-H3). It is worth noting that ΔTXR1 cells appeared to exhibit increased levels of hyper-acetylation over ΔEZL2 cells for H4, and to a lesser degree, H2A.Z, whereas the two mutants were virtually indistinguishable in H2A hyper-acetylation levels.
H3K27 methylation, especially the di- and tri- forms, is generally associated with heterochromatin and hypo-acetylation. It is therefore not surprising to see increased acetylation levels in the H3K27me-deficient mutants, especially ΔEZL2 cells. Nonetheless, we were intrigued by the specific hyper-acetylation of H2A, H2A.Z, and H4. The patterns of acetylation were very similar: an inverted gradient of accumulation tilting toward the highly acetylated forms rather than the less modified forms. This implies potential synergistic effects of lysine acetylation and a positive feedback mechanism. The reader-writer coupling, represented by the wide-spread association of bromodomains with HATs, may be a key contributing factor. In addition, genetic studies in yeast have demonstrated the hierarchical order of the lysine acetylation events on the conserved H4 N-terminal tail: relative to K5ac, K8ac, and K12ac, H4K16ac is especially important and has major effects on chromatin structure (34, 35). Generally, our observations on H4 N-terminal acetylation agree well with these studies but provide a more detailed picture of the combinatorial response. Importantly, we only detected H4K15ac (equivalent to yeast H4K16ac) in association with other acetylation events, further supporting that it plays a rate-limiting and possibly founding role in establishing hyper-acetylation. This hierarchical buildup of H4 hyper-acetylation may be physiologically significant, as the tetra-acetylated form of H4 is highly correlated with transcription and the most highly acetylated forms have the highest affinity for transcription factors (31, 36).
It is intriguing that we only observed hyper-acetylation on H2A, H2A.Z, and H4, but not on H2B and H3. This substrate specificity suggests the involvement of NuA4-family HATs. Initially characterized in yeast, the NuA4 complex is evolutionarily conserved (37) and NuA4-like HAT activities have been reported in Tetrahymena (38). Furthermore, the Tetrahymena genome encodes several homologs of the MYST/MOZ family HAT, as well as homologs to key structural components of NuA4 complexes (data not shown). NuA4 has been implicated in transcription activation and DNA repair (37, 39–41). The former is consistent with de-repression associated with inactivation of the Polycomb pathway, as in ΔEZL2 cells, whereas the latter is consistent with the observed chromatin decondensation associated with the DNA damage response to replication stress in ΔTXR1 cells (18).
Other Histone Modifications Potentially in Crosstalk with H3K27 Methylation
All core histones and the linker histone H1 have been found to be phosphorylated across species and implicated in gene activation, chromosome condensation, DNA repair, apoptosis, among other cellular functions (42–46). Phosphorylation of H1, H2A, and H2A.Z from Tetrahymena macronuclei has been previously reported (47–49). In this study, we identified phosphorylation in H2A and H2B from Tetrahymena macronuclei without prior enrichment of phospho-proteins/peptides. H2A and H2A.X are the most highly phosphorylated core histones in Tetrahymena macronuclei (47). Mutagenesis studies have pinpointed phosphorylation events at the C-terminal tail of H2A.X, including constitutive phosphorylation (at S122, S124, T127, and S129 of H2A.X; the equivalent modifications in H2A are not studied) as well as DNA damage signaling (at S134 of H2A.X; a conserved phosphorylation mark generally referred to as γH2A.X) (49). However, we were unable to recover the corresponding phosphopeptides in this study, possibly because of their poor retention on RP-HPLC, very low levels not easily observed in unenriched samples, or ion suppression effects. Intriguingly, we found that the N-terminal domain of H2A was also phosphorylated, although we could not identify the specific site because of incomplete ion fragmentation and we could not quantify effectively because of sample-to-sample variation. Our finding is consistent with the identification of H2A serine 1 phosphorylation in mammalian cells, with implications in transcription, mitosis and S phase-associated events (50, 51). We also identified H2B phosphorylation in the C-terminal tail, although again, we could not precisely localize the site (supplemental Fig. S4 and Table S2-H2B). This modification was significantly down-regulated (by as much as 80%) in the mutants in variant H2B.1. Mammalian H2B is phosphorylated at three sites (S113, T116, T120) near the C-terminal tail, but their biological functions remain unclear (52).
We also identified several formylation sites in H2A and H3, two of which (H2A K39, and H3 K23) were slightly elevated in the mutants (supplemental Table S2). N-formylation of lysine is a common but under-studied PTM in histones, high mobility group proteins, and other nonhistone proteins (53, 54). Similar to acetylation in structure and chemical properties, formylation can potentially share some common functions with acetylation and histone formylation has been implicated in the DNA damage response (53).
DNA Damage Response to Replication Stress May Reshape the Epigenetic Landscape in ΔTXR1 Cells
In metazoa, homologs of Drosophila E(z) control all three forms of H3K27 methylation, contributing to the Polycomb repression pathway essential for Hox gene silencing and X-inactivation (55). In plants, H3K27me1 is largely delegated to homologs of Arabidopsis ATXR5 and ATXR6, which constitute another pathway involved in heterochromatin formation and DNA replication (15, 56). The homologous HMTs and corresponding pathways responsible for H3K27 methylation have also been identified in the protozoan Tetrahymena (16–19). As described in our previous publications (17, 18), there is clear division of labor between TXR1-dependent H3K27me1 and EZL2-dependent H3K27me2/H3K27me3: the former is required for efficient replication elongation, whereas the latter is likely involved in transcriptional gene silencing. Here we focus on ΔTXR1 cells, and explore how the epigenetic landscape is reshaped by replication stress and DNA damage response.
We have previously reported that γH2A.X (H2A.X S134 phosphorylation) is highly induced in ΔTXR1 cells (18), as a sensitive indicator of replication stress (57). γH2A.X is also known as an upstream signal for the DNA damage response (DDR) (58). Recent studies in yeast and mammalian systems have demonstrated that the NuA4 HAT complex can be recruited to sites of DNA damage by γH2A.X (37, 39–41). Further, NuA4 targets nucleosomal H2A, H2A.Z, and H4, with further enhanced HAT activities for the latter two, and modulates local chromatin structure and accessibility to the DNA repair machinery (37). Indeed, we observed a small but consistent increase in hyper-acetylation levels in H2A.Z and H4, but not H2A, in ΔTXR1 over ΔEZL2 cells. We also analyzed the genomic distribution of H2A.Z and H4 hyper-acetylation by ChIP, using a well-established antibody specific for acetylated lysyl residues in the N-terminal domains of H2A.Z and H4 (28). We focused on the acetylation levels in the rDNA mini-chromosome. In Tetrahymena, the genomic region containing the 35S pre-rRNA (transcribed by RNA polymerase I) is rearranged to form a palindromic dimer and the ∼20 kb mini-chromosome is replicated from origins near the center (59, 60). rDNA is under severe replication stress in ΔTXR1 cells (18), but is not transcribed by the NuA4-coupled RNA polymerase II, allowing us to differentiate the DNA repair functionality of NuA4 from its transcriptional counterpart. Here we observed a significant enrichment of the acetylation signals in ΔTXR1 cells, but not in WT and ΔEZL2 cells (Fig. 5). This result is consistent with the general macronuclear chromatin decondensation and the specific nucleolar abnormality observed in ΔTXR1 cells (18). In conclusion, we posit that the particular hyper-acetylation pattern in ΔTXR1 cells can be attributed to γH2A.X-mediated recruitment of NuA4 to regions under replication stress.
Fig. 5.
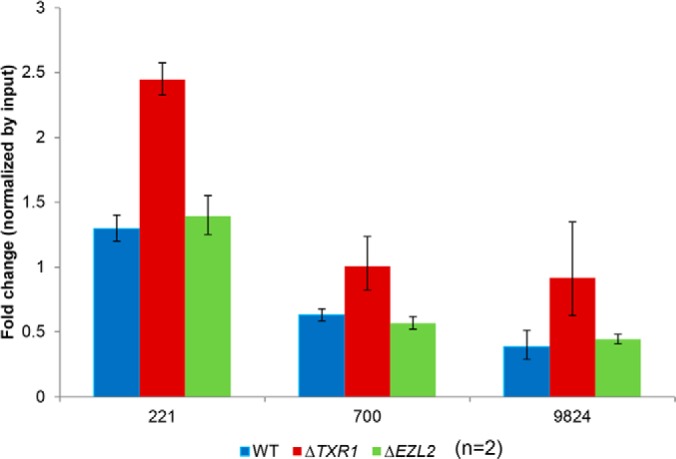
Preferential H2A.Z and H4 hyper-acetylation of rDNA in ΔTXR1 cells. Hyper-acetylation of H2A.Z and H4 was associated with the rDNA in ΔTXR1 cells, demonstrated by cross-linking chromatin immuno-precipitation (X-ChIP) using antisera recognizing acetylated H4 and H2A.Z. ChIP samples were quantified by Q-PCR and normalized with input. The numbers on the x-axis denote the median positions of the PCR products. rDNA mini-chromosome sequence (corresponding to half of the palindrome, numbered from the center to one end) was retrieved from GenBank (accession No. X54512).
We also quantified acid-extracted nuclear proteins (mostly histones) by MS. Intriguingly, we observed that H2A.Y, a recently identified H2A variant interacting with protein phosphatase 1 (PP1) (61), was highly induced only in ΔTXR1 cells (Fig. 6A). The MS result is consistent with our microarray data, which showed a ninefold increase of H2A.Y mRNA levels in ΔTXR1 cells (relative to WT or ΔEZL2 cells) (Fig. 6B). Indeed, H2A.Y was present at very low levels in WT cells and could not be detected in our MS analysis, despite repeated trials. It is important to note that levels of all other histones, including H2A.Z, were not significantly changed in the mutants (data not shown), consistent with our microarray results (Fig. 6B). H2A.Y has been reported to regulate H3 S10 phosphorylation in mitotic micronuclei (a mark completely missing in the amitotic macronuclei in this study) (61). However, the macronuclear target(s) of H2A.Y, which most likely accounts for its essential function (61), remains elusive. Based on our results, we propose that H2A.Y plays a critical role in regulating levels of γH2A.X. It may also play a role in other phosphorylation events associated with the DNA damage response (C.Z., P.C.A., and Y.L., unpublished results).
Fig. 6.

Preferential induction of H2A.Y in ΔTXR1 cells. Induction of the histone variant H2A.Y in ΔTXR1 cells, as demonstrated by MS and microarray analyses. A, Selected tandem MS of H2A.Y peptide precursor ion of 1031.0866 Da (Z = 2+, mass error = 1ppm, Ion score = 113). The peptide corresponds to H2A.Y residues 143–160: NLVFNLFPSLVILDTLDK. All fragment ions were accounted for. H2A.Y peptides were detected by MS only in ΔTXR1 cells. B, induction of H2A.Y mRNA levels in ΔTXR1 cells. HTAY gene (encoding H2A.Y protein) was significantly induced in ΔTXR1 cells (>9 folds, p < 0.01) whereas genes encoding all other histones remained mostly unchanged.
Conclusions and Implications
The highly conserved H3K27 methylation is intensively studied because of its long-established biological significance in heterochromatin formation (55), with implications in Polycomb repression (55), long noncoding RNA (lncRNA) biogenesis (62, 63), and DNA replication in heterochromatic regions (56). Its misregulation results in abnormal development and tumorigenesis (64, 65). This work represents a systematic investigation of the global level crosstalk between H3K27 methylation and other histone PTMs. Despite the dramatic deficiency in H3K27 methylation in the two HMT deletion Tetrahymena mutants, viability is not affected, implying the existence of effective compensatory mechanisms. In the context of ΔTXR1 cells, which are deficient in H3K27me1 and also defective in replication elongation, we want to re-emphasize several adaptations at the chromatin level (summarized in Fig. 7). First, there is increasing off-site lysine mono-methylation. Particularly interesting is H3K56me1, which has recently been shown to interact with PCNA and play a role in DNA replication (66). However, H3K56me1 in mammalian cells is reportedly catalyzed by G9a, which has no homolog in Tetrahymena. This may be another case for conserved histone PTMs with alternative modifying enzymes, to be sorted out in future studies. Second, the epigenetic landscape is reshaped by the DNA damage response. There is dramatic induction of γH2A.X (H2A.X S134 phosphorylation), which has been well-documented in our previous study (18), but was not detected by this MS study because of technical limitations. We also demonstrated H2A, H2A.Z, and H4 hyper-acetylation, which we infer is elicited by γH2A.X and mediated by NuA4-like HAT activities in Tetrahymena (38). In addition, H2A.Y is highly induced, potentially recruiting more protein phosphatase 1 (PP1) to chromatin (61). This may represent an effort to modulate levels of γH2A.X, as well as other phosphorylation events in the DNA damage response. In summary, our study has revealed possible mechanisms and potential pathways involved in crosstalk with H3K27 methylation, and raised more questions about the network relationships of histone PTMs.
Fig. 7.

Epigenetic changes in ΔTXR1 cells: response to replication stress. Multiple histone PTMs in crosstalk with H3K27 methylation, including hyper-acetylation (H2A, H2A.Z, and H4), hyper-methylation (particularly at H3K56), and γH2A.X (H2A.X S134 phosphorylation). They are potentially underlain by DNA damage response to replication stress in ΔTXR1 cells, modulated by ATR, NuA4, and H2A.Y. ac: acetylation; me1: mono-methylation; ph: phosphorylation. See text for details.
Supplementary Material
Footnotes
Author contributions: C.Z. and Y.L. designed research; C.Z., S.G., and A.J.M. performed research; C.Z., S.G., and P.C.A. analyzed data; C.Z., Y.L., and P.C.A. wrote the paper.
* This work was supported by National Institute of Health grants #1P41RR018627 (PCA) and R01GM087343 (YL).
 This article contains supplemental Figs. S1 to S4 and Tables S1 to S3.
This article contains supplemental Figs. S1 to S4 and Tables S1 to S3.
Data availability: The MS raw data can be downloaded from PeptideAtlas (http://www.peptideatlas.org/) with data set Identifier: PASS00209&Password: RH4425to, The microarray data can be downloaded from the National Center for Biotechnology Information (NCBI) GEO database with the accession number GSE44990.
1 The abbreviations used are:
- PTM
- Post-translational modification
- HMT
- Histone methyltransferase
- HAT
- Histone acetyltransferase
- SET
- Suppressor of variegation, Enhancer of Zeste, Trithorax
- ATXR5/6
- Arabidopsis Trithorax Related 5/6
- EZL2
- Enhancer of Zeste-like 2
- TXR1
- Tetrahymena Trithorax Related 1
- me(1/2/3)
- (Mono-, di-, and tri-) Methylation
- ac
- Acetylation
- form
- Formylation
- phos
- Phosphorylation
- pr
- Propionylation
- ub
- Ubiquitination.
REFERENCES
- 1. Sidoli S., Cheng L., Jensen O. N. (2012) Proteomics in chromatin biology and epigenetics: Elucidation of post-translational modifications of histone proteins by mass spectrometry. J. Proteomics 75, 3419–3433 [DOI] [PubMed] [Google Scholar]
- 2. Jenuwein T., Allis C. D. (2001) Translating the histone code. Science 293, 1074–1080 [DOI] [PubMed] [Google Scholar]
- 3. Strahl B. D., Allis C. D. (2000) The language of covalent histone modifications. Nature 403, 41–45 [DOI] [PubMed] [Google Scholar]
- 4. Roth S. Y., Denu J. M., Allis C. D. (2001) Histone acetyltransferases. Annu. Rev. Biochem. 70, 81–120 [DOI] [PubMed] [Google Scholar]
- 5. Yang X. J., Seto E. (2008) Lysine acetylation: codified crosstalk with other posttranslational modifications. Mol. Cell 31, 449–461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Greer E. L., Shi Y. (2012) Histone methylation: a dynamic mark in health, disease and inheritance. Nat. Rev. Genet. 13, 343–357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Klose R. J., Zhang Y. (2007) Regulation of histone methylation by demethylimination and demethylation. Nat. Rev. Mol. Cell Biol. 8, 307–318 [DOI] [PubMed] [Google Scholar]
- 8. Martin C., Zhang Y. (2005) The diverse functions of histone lysine methylation. Nat. Rev. Mol. Cell Biol. 6, 838–849 [DOI] [PubMed] [Google Scholar]
- 9. Taverna S. D., Li H., Ruthenburg A. J., Allis C. D., Patel D. J. (2007) How chromatin-binding modules interpret histone modifications: lessons from professional pocket pickers. Nat. Struct. Mol. Biol. 14, 1025–1040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cao R., Zhang Y. (2004) The functions of E(Z)/EZH2-mediated methylation of lysine 27 in histone H3. Curr. Opin. Genet. Dev. 14, 155–164 [DOI] [PubMed] [Google Scholar]
- 11. Cao R., Wang L., Wang H., Xia L., Erdjument-Bromage H., Tempst P., Jones R. S., Zhang Y. (2002) Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science 298, 1039–1043 [DOI] [PubMed] [Google Scholar]
- 12. Czermin B., Melfi R., McCabe D., Seitz V., Imhof A., Pirrotta V. (2002) Drosophila enhancer of Zeste/ESC complexes have a histone H3 methyltransferase activity that marks chromosomal Polycomb sites. Cell 111, 185–196 [DOI] [PubMed] [Google Scholar]
- 13. Muller J., Hart C. M., Francis N. J., Vargas M. L., Sengupta A., Wild B., Miller E. L., O'Connor M. B., Kingston R. E., Simon J. A. (2002) Histone methyltransferase activity of a Drosophila Polycomb group repressor complex. Cell 111, 197–208 [DOI] [PubMed] [Google Scholar]
- 14. Jacob Y., Michaels S. D. (2009) H3K27me1 is E(z) in animals, but not in plants. Epigenetics 4, 366–369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jacob Y., Feng S., LeBlanc C. A., Bernatavichute Y. V., Stroud H., Cokus S., Johnson L. M., Pellegrini M., Jacobsen S. E., Michaels S. D. (2009) ATXR5 and ATXR6 are H3K27 monomethyltransferases required for chromatin structure and gene silencing. Nat. Struct. Mol. Biol. 16, 763–768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Liu Y., Taverna S. D., Muratore T. L., Shabanowitz J., Hunt D. F., Allis C. D. (2007) RNAi-dependent H3K27 methylation is required for heterochromatin formation and DNA elimination in Tetrahymena. Genes Dev. 21, 1530–1545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhang C., Molascon A. J., Gao S., Liu Y., Andrews P. C. (2013) Quantitative proteomics reveals that the specific methyltransferases Txr1p and Ezl2p differentially affect the mono-, di- and trimethylation states of histone H3 lysine 27 (H3K27). Mol. Cell. Proteomics 12, 1678–1688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gao S., Xiong J., Zhang C., Berquist B. R., Yang R., Zhao M., Molascon A. J., Kwiatkowski S. Y., Yuan D., Qin Z., Wen J., Kapler G. M., Andrews P. C., Miao W., Liu Y. (2013) Impaired replication elongation in Tetrahymena mutants deficient in histone H3 Lys 27 monomethylation. Genes Dev. 27, 1662–1679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chung P. H., Yao M. C. (2012) Tetrahymena thermophila JMJD3 homolog regulates H3K27 methylation and nuclear differentiation. Eukaryot. Cell 11, 601–614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mochizuki K. (2008) High efficiency transformation of Tetrahymena using a codon-optimized neomycin resistance gene. Gene 425, 79–83 [DOI] [PubMed] [Google Scholar]
- 21. Hai B., Gorovsky M. A. (1997) Germ-line knockout heterokaryons of an essential alpha-tubulin gene enable high-frequency gene replacement and a test of gene transfer from somatic to germ-line nuclei in Tetrahymena thermophila. Proc. Natl. Acad. Sci. U.S.A. 94, 1310–1315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cassidy-Hanley D., Bowen J., Lee J. H., Cole E., VerPlank L. A., Gaertig J., Gorovsky M. A., Bruns P. J. (1997) Germline and somatic transformation of mating Tetrahymena thermophila by particle bombardment. Genetics 146, 135–147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Garcia B. A., Mollah S., Ueberheide B. M., Busby S. A., Muratore T. L., Shabanowitz J., Hunt D. F. (2007) Chemical derivatization of histones for facilitated analysis by mass spectrometry. Nat. Protoc. 2, 933–938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Medzihradszky K. F., Zhang X., Chalkley R. J., Guan S., McFarland M. A., Chalmers M. J., Marshall A. G., Diaz R. L., Allis C. D., Burlingame A. L. (2004) Characterization of Tetrahymena histone H2B variants and posttranslational populations by electron capture dissociation (ECD) Fourier transform ion cyclotron mass spectrometry (FT-ICR MS). Mol. Cell. Proteomics 3, 872–886 [DOI] [PubMed] [Google Scholar]
- 25. Shechter D., Dormann H. L., Allis C. D., Hake S. B. (2007) Extraction, purification and analysis of histones. Nat. Protoc. 2, 1445–1457 [DOI] [PubMed] [Google Scholar]
- 26. Candiano G., Bruschi M., Musante L., Santucci L., Ghiggeri G. M., Carnemolla B., Orecchia P., Zardi L., Righetti P. G. (2004) Blue silver: a very sensitive colloidal Coomassie G-250 staining for proteome analysis. Electrophoresis 25, 1327–1333 [DOI] [PubMed] [Google Scholar]
- 27. O'Neill L. P., Turner B. M. (2003) Immunoprecipitation of native chromatin: NChIP. Methods 31, 76–82 [DOI] [PubMed] [Google Scholar]
- 28. Lin R., Leone J. W., Cook R. G., Allis C. D. (1989) Antibodies specific to acetylated histones document the existence of deposition- and transcription-related histone acetylation in Tetrahymena. J. Cell Biol. 108, 1577–1588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Cheung W. L., Briggs S. D., Allis C. D. (2000) Acetylation and chromosomal functions. Curr. Opin. Cell Biol. 12, 326–333 [DOI] [PubMed] [Google Scholar]
- 30. Sanchez R., Zhou M. M. (2009) The role of human bromodomains in chromatin biology and gene transcription. Curr. Opin. Drug Discov. Devel. 12, 659–665 [PMC free article] [PubMed] [Google Scholar]
- 31. Shahbazian M. D., Grunstein M. (2007) Functions of site-specific histone acetylation and deacetylation. Annu. Rev. Biochem. 76, 75–100 [DOI] [PubMed] [Google Scholar]
- 32. Grunstein M. (1997) Histone acetylation in chromatin structure and transcription. Nature 389, 349–352 [DOI] [PubMed] [Google Scholar]
- 33. Price B. D., D'Andrea A. D. (2013) Chromatin remodeling at DNA double-strand breaks. Cell 152, 1344–1354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dion M. F., Altschuler S. J., Wu L. F., Rando O. J. (2005) Genomic characterization reveals a simple histone H4 acetylation code. Proc. Natl. Acad. Sci. U.S.A. 102, 5501–5506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Shogren-Knaak M., Ishii H., Sun J. M., Pazin M. J., Davie J. R., Peterson C. L. (2006) Histone H4-K16 acetylation controls chromatin structure and protein interactions. Science 311, 844–847 [DOI] [PubMed] [Google Scholar]
- 36. Vettese-Dadey M., Grant P. A., Hebbes T. R., Crane- Robinson C., Allis C. D., Workman J. L. (1996) Acetylation of histone H4 plays a primary role in enhancing transcription factor binding to nucleosomal DNA in vitro. EMBO J. 15, 2508–2518 [PMC free article] [PubMed] [Google Scholar]
- 37. Doyon Y., Cote J. (2004) The highly conserved and multifunctional NuA4 HAT complex. Curr. Opin. Genet. Dev. 14, 147–154 [DOI] [PubMed] [Google Scholar]
- 38. Ohba R., Steger D. J., Brownell J. E., Mizzen C. A., Cook R. G., Cote J., Workman J. L., Allis C. D. (1999) A novel H2A/H4 nucleosomal histone acetyltransferase in Tetrahymena thermophila. Mol. Cell. Biol. 19, 2061–2068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Downs J. A., Allard S., Jobin-Robitaille O., Javaheri A., Auger A., Bouchard N., Kron S. J., Jackson S. P., Cote J. (2004) Binding of chromatin-modifying activities to phosphorylated histone H2A at DNA damage sites. Mol. Cell 16, 979–990 [DOI] [PubMed] [Google Scholar]
- 40. Kusch T., Florens L., Macdonald W. H., Swanson S. K., Glaser R. L., Yates J. R., 3rd, Abmayr S. M., Washburn M. P., Workman J. L. (2004) Acetylation by Tip60 is required for selective histone variant exchange at DNA lesions. Science 306, 2084–2087 [DOI] [PubMed] [Google Scholar]
- 41. Murr R., Loizou J. I., Yang Y. G., Cuenin C., Li H., Wang Z. Q., Herceg Z. (2006) Histone acetylation by Trrap-Tip60 modulates loading of repair proteins and repair of DNA double-strand breaks. Nat. Cell Biol. 8, 91–99 [DOI] [PubMed] [Google Scholar]
- 42. Rossetto D., Avvakumov N., Cote J. (2012) Histone phosphorylation: a chromatin modification involved in diverse nuclear events. Epigenetics 7, 1098–1108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Fischle W., Tseng B. S., Dormann H. L., Ueberheide B. M., Garcia B. A., Shabanowitz J., Hunt D. F., Funabiki H., Allis C. D. (2005) Regulation of HP1-chromatin binding by histone H3 methylation and phosphorylation. Nature 438, 1116–1122 [DOI] [PubMed] [Google Scholar]
- 44. Cheung W. L., Ajiro K., Samejima K., Kloc M., Cheung P., Mizzen C. A., Beeser A., Etkin L. D., Chernoff J., Earnshaw W. C., Allis C. D. (2003) Apoptotic phosphorylation of histone H2B is mediated by mammalian sterile twenty kinase. Cell 113, 507–517 [DOI] [PubMed] [Google Scholar]
- 45. Dou Y., Mizzen C. A., Abrams M., Allis C. D., Gorovsky M. A. (1999) Phosphorylation of linker histone H1 regulates gene expression in vivo by mimicking H1 removal. Mol. Cell 4, 641–647 [DOI] [PubMed] [Google Scholar]
- 46. Wei Y., Mizzen C. A., Cook R. G., Gorovsky M. A., Allis C. D. (1998) Phosphorylation of histone H3 at serine 10 is correlated with chromosome condensation during mitosis and meiosis in Tetrahymena. Proc. Natl. Acad. Sci. U.S.A. 95, 7480–7484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Allis C. D., Gorovsky M. A. (1981) Histone phosphorylation in macro- and micronuclei of Tetrahymena thermophila. Biochemistry 20, 3828–3833 [DOI] [PubMed] [Google Scholar]
- 48. Mizzen C. A., Dou Y., Liu Y., Cook R. G., Gorovsky M. A., Allis C. D. (1999) Identification and mutation of phosphorylation sites in a linker histone. Phosphorylation of macronuclear H1 is not essential for viability in tetrahymena. J. Biol. Chem. 274, 14533–14536 [DOI] [PubMed] [Google Scholar]
- 49. Song X., Gjoneska E., Ren Q., Taverna S. D., Allis C. D., Gorovsky M. A. (2007) Phosphorylation of the SQ H2A.X motif is required for proper meiosis and mitosis in Tetrahymena thermophila. Mol. Cell. Biol. 27, 2648–2660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Barber C. M., Turner F. B., Wang Y., Hagstrom K., Taverna S. D., Mollah S., Ueberheide B., Meyer B. J., Hunt D. F., Cheung P., Allis C. D. (2004) The enhancement of histone H4 and H2A serine 1 phosphorylation during mitosis and S-phase is evolutionarily conserved. Chromosoma 112, 360–371 [DOI] [PubMed] [Google Scholar]
- 51. Zhang Y., Griffin K., Mondal N., Parvin J. D. (2004) Phosphorylation of histone H2A inhibits transcription on chromatin templates. J. Biol. Chem. 279, 21866–21872 [DOI] [PubMed] [Google Scholar]
- 52. Olsen J. V., Vermeulen M., Santamaria A., Kumar C., Miller M. L., Jensen L. J., Gnad F., Cox J., Jensen T. S., Nigg E. A., Brunak S., Mann M. (2010) Quantitative phosphoproteomics reveals widespread full phosphorylation site occupancy during mitosis. Sci. Signal. 3, ra3. [DOI] [PubMed] [Google Scholar]
- 53. Jiang T., Zhou X., Taghizadeh K., Dong M., Dedon P. C. (2007) N-formylation of lysine in histone proteins as a secondary modification arising from oxidative DNA damage. Proc. Natl. Acad. Sci. U.S.A. 104, 60–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Wisniewski J. R., Zougman A., Mann M. (2008) Nepsilon-formylation of lysine is a widespread post-translational modification of nuclear proteins occurring at residues involved in regulation of chromatin function. Nucleic Acids Res. 36, 570–577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Lanzuolo C., Orlando V. (2012) Memories from the polycomb group proteins. Annu. Rev. Genet. 46, 561–589 [DOI] [PubMed] [Google Scholar]
- 56. Jacob Y., Stroud H., Leblanc C., Feng S., Zhuo L., Caro E., Hassel C., Gutierrez C., Michaels S. D., Jacobsen S. E. (2010) Regulation of heterochromatic DNA replication by histone H3 lysine 27 methyltransferases. Nature 466, 987–991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Zou L., Elledge S. J. (2003) Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science 300, 1542–1548 [DOI] [PubMed] [Google Scholar]
- 58. Flynn R. L., Zou L. (2011) ATR: a master conductor of cellular responses to DNA replication stress. Trends Biochem. Sci. 36, 133–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Truett M. A., Gall J. G. (1977) The replication of ribosomal DNA in the macronucleus of Tetrahymena. Chromosoma 64, 295–303 [DOI] [PubMed] [Google Scholar]
- 60. Cech T. R., Brehm S. L. (1981) Replication of the extrachromosomal ribosomal RNA genes of Tetrahymena thermophilia. Nucleic Acids Res. 9, 3531–3543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Song X., Bowen J., Miao W., Liu Y., Gorovsky M. A. (2012) The nonhistone, N-terminal tail of an essential, chimeric H2A variant regulates mitotic H3-S10 dephosphorylation. Genes Dev. 26, 615–629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Wu S. C., Kallin E. M., Zhang Y. (2010) Role of H3K27 methylation in the regulation of lncRNA expression. Cell Res. 20, 1109–1116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Rinn J. L., Kertesz M., Wang J. K., Squazzo S. L., Xu X., Brugmann S. A., Goodnough L. H., Helms J. A., Farnham P. J., Segal E., Chang H. Y. (2007) Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell 129, 1311–1323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Chi P., Allis C. D., Wang G. G. (2010) Covalent histone modifications–miswritten, misinterpreted and mis-erased in human cancers. Nat. Rev. Cancer 10, 457–469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Martinez-Garcia E., Licht J. D. (2010) Deregulation of H3K27 methylation in cancer. Nat. Genet. 42, 100–101 [DOI] [PubMed] [Google Scholar]
- 66. Yu Y., Song C., Zhang Q., DiMaggio P. A., Garcia B. A., York A., Carey M. F., Grunstein M. (2012) Histone H3 lysine 56 methylation regulates DNA replication through its interaction with PCNA. Mol. Cell 46, 7–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.