

Abstract
Background
Cystic kidneys and vascular aneurysms are clinical manifestations seen in patients with polycystic kidney disease (PKD), a cilia-associated pathology (ciliopathy). Survivin overexpression is associated with cancer, but the clinical pathology associated with survivin down-regulation or knockout has never been studied before. The present studies aim to examine if and how cilia function (Pkd1 or Pkd2) and structure (Tg737) play a role in cystic kidney and aneurysm through survivin down-regulation.
Methods and Results
Cysts and aneurysms from PKD patients, Pkd mouse and zebrafish models are characterized by chromosome instability and low survivin expression. This triggers cytokinesis defects and formation of nuclear polyploidy or aneuploidy. In vivo conditional mouse and zebrafish models confirm that survivin gene deletion in the kidneys results in a cystic phenotype. As in hypertensive Pkd1, Pkd2 and Tg737 deletion, aneurysm formation can also be induced in vascular-specific normotensive survivin mice. Survivin knockout also contributes to abnormal oriented cell division in both kidney and vasculature. Furthermore, survivin expression and ciliary localization are regulated by flow-induced cilia activation through PKC, Akt and NF-κB. Circumventing ciliary function by re-expressing survivin can rescue PKD phenotypes.
Conclusions
For the first time, our studies offer a unifying mechanism that explains both renal and vascular phenotypes in PKD. Although primary cilia dysfunction accounts for aneurysm formation and hypertension, hypertension itself does not cause aneurysm. Furthermore, aneurysm and cyst formation share a common cellular and molecular pathway involving cilia function or structure, survivin expression, cytokinesis, cell ploidy, symmetrical cell division and tissue architecture orientation.
Keywords: aurora, cardiovascular, fluid-shear stress, renal epithelia, vascular endothelia
INTRODUCTION
Polycystic kidney disease (PKD) is the most common hereditary kidney disorder, and formation of bilateral cystic kidneys is the hallmark of the disease. Among other extra-renal phenotypes, aneurysm formation is one of the deadliest vascular abnormalities observed in PKD patients. Unfortunately, there is no study that explains the formation of these “bulb-like structures” in both vasculatures and renal tubules. Abnormalities in primary cilia1–4, polyploidy5, 6 and centrosomal number5, 7 have been independently studied in the vascular or renal systems. However, there is currently no unifying mechanism that explains these cellular phenotypes.
Survivin is a chromosomal passenger involved in coordinating proper chromosomal events during mitosis8. Due to its clinical manifestation in cancer, overexpression of survivin has always been the main focus of medical research. Whereas overexpression of survivin is associated with cancer formation and progression, Survivin knockout mouse model is not viable beyond 4.5 days post coitum9. Interestingly, survivin homozygote cells isolated at 4.5 days post coitum show a similar cellular polyploidy phenotype to Pkd cells.
Our previous in vitro studies showed that vascular endothelial Pkd cell lines are characterized by survivin down-regulation, resulting in abnormal spindle assembly checkpoint and polyploidy5. Here, we expanded our study through the use of in vivo mouse and zebrafish models to demonstrate that survivin knockout or knockdown is sufficient to induce the formation of bulb-like structures in the kidney tubule (cysts) and artery (aneurysms). Our studies further suggest that mechanosensory cilia regulate survivin expression and dictate the formation of cell ploidy. The asymmetric cell division resulting from abnormal ploidy further undermines the establishment of tissue polarity or planar cell polarity, which is believed to be the underlying mechanism for tubule or artery dilatation. We thus propose a common cellular mechanism through survivin to explain both vascular and renal phenotypes in PKD.
METHODS
Signed and informed consent to collect disposed PKD human kidneys was obtained from the patients, and kidney collection protocols were approved by the Department for Human Research Protections of the Biomedical Institutional Review Board of The University of Toledo. The use of animal tissues was approved by The University of Toledo animal care and use committee.
Mouse models
The following mouse models were used in our studies; Mx1Cre, PdgfβCre, Tie2Cre, Pkd1flox, Pkd2−, Tg737Orpk, and survivinflox. To accelerate experimental cystic model, unilateral ureteral obstruction (UUO) was generated by tying a 6-0 silk suture against a 28G needle in the mice. Standard histology analyses were used to examine the kidneys. To accelerate experimental aneurysm formation, 0.25 M calcium chloride was placed directly on the abdominal aorta of the mice for 10 minutes.
Cell culture
Human and mouse primary tubular cells from distal collecting tubules were used in the present studies. For cell lines, we used previously generated mouse endothelial cells3 and human renal epithelial cells4. In some experiments, human full-length survivin-GFP was used, in addition to siRNA knockdown on PKC, Akt, aurora-A and survivin. These cells were then subjected to cell cycle, live-imaging, karyotyping, immunostaining, and/or Western analysis.
Chromosomal analysis
Chromosomes from a single cell were spread and hybridized with a cocktail of mouse or human fluorescence-labeled probes specific for individual chromosomes10. Data were analyzed with automated SKY View software (Version 1.62). Because zebrafish chromosome-specific probes were not available, individual chromosomes were analyzed based on the ideogram derived from the replication banding of Danio rerio.
Live-imaging study
Primary renal epithelial cells or vascular endothelial cells were transfected with or without Survivin siRNA. Hoechst dye was use to indicate nucleus.
Zebrafish study
Wild-type (wt) zebrafish AB strains were used for knockdown experiments with either control morpholino (controlMO: 5′-CCT CTT ACC TCA GTT ACA ATT TAT A-3′) or Pkd2 morpholino (pkd2MO: 5′-AGG ACG AAC GCG ACT GGG CTC ATC-3′). For rescue experiments, 100 pg of purified full-length human survivin mRNA were either co-injected with pkd2 morpholinos or alone into the 1–2 cell-stage embryos. In another case, 2.5 ng vascular endothelial growth factor (VEGF) was co-injected with pkd2 morpholinos.
RNA isolation and RT-PCR
Effectiveness of the knockdown or overexpression in zebrafish was verified by RT-PCR. Total RNA was isolated from zebrafish embryos using TRIzol (Invitrogen, Inc.) followed by DNase treatment (Roche, Inc.). Superscript-II (Invitrogen, Inc.) was used for cDNA synthesis. RT-PCR is performed under the following cycling conditions: 95 °C for 15 min and then 40 cycles of 95 °C for 30s, 60 °C for 1 min and 72 °C for 1 min. The cDNAs were amplified using specific primers indicated in Table 111–14.
Table 1.
Primer sequences
| Description | Primer sequence | Reference |
|---|---|---|
| Zebrafish pkd2 | Forward: 5′-GGG ATA CGT GCT GTG GTT CTC-3′ Reverse: 5′-CAC GAT GAG CTC CAG TCG CGT-3′ |
11 |
| Human survivin | Forward: 5′-AAG AAC TGG CCC TTC TTG GA-3′ Reverse: 5′-CAA CCG GAC GAA TGC TTT TT-3′ |
12 |
| Zebrafish survivin | Forward: 5′-GGA GCG ACT TCG CAT CTA CAT-3′ Reverse: 5′-ACC TCA TCA CGA AAG TAG GCA ATC-3′ |
13 |
| Zebrafish α-tubulin | Forward: 5′-GGA GCT CAT TGA CCT TGT TTT AGA TA-3′ Reverse: 5′-GCT GTG GAA GAC CAG GAA ACC-3′ |
14 |
Pharmacological agents
The pharmacological agents used in our studies include PKC inhibitor (5 μM, Bisindolylmaleimide XI hydrochloride; Sigma Inc.), PKC activator (10 μM forskolin; Sigma Inc.), taxol (33.3 nM; Sigma, Inc.), nocodazole (0.1 μg/ml; Sigma, Inc.), VEGF (2.5 ng; Prospec, Inc.), and colcemid (50 μg/ml; Invitrogen Inc.).
Data analysis
Both surgical and non-surgical kidneys were studied and compared among the mouse groups. All quantifiable data were reported as mean ± SEM. Distribution analyses were performed on all data sets before any statistical comparisons to confirm normal data distribution (a bell shaped curve distribution). Homogeneity of variance (homoscedasticity) was also verified within each data set. When data set was not normally distributed or heterogeneous variance was detected, the distributions were normalized via log transformation. This approach produced normally distributed data sets. After distribution and variance analyses, data comparisons for more than two groups were performed using ANOVA test followed by Dunn’s Multiple Comparison posttest analysis. Comparison between two groups was carried out using student t-test.
Whenever possible, paired-experimental design was used in our studies to allow more powerful statistical analysis and fewer mice to be used in each study group. For all comparisons, power analyses were performed routinely to enable reliable conclusions, and comparisons with negative results had statistical powers of ≥0.8. Unless otherwise indicated, the difference between groups was statistically significant at p<0.05, and it is indicated in the graphs by the asterisk to denote comparison to the wild-type control, non-treated, non-induced or static group. The number of experimental replicates is indicated in the figures or figure legends. All statistical analysis was done with GraphPad Prism, version 5.0.
RESULTS
Human and mouse polycystic kidneys are characterized by abnormal ploidy and survivin down-regulation
Compared to non-cystic tissue (Fig. 1a), karyotyping data of a single renal epithelium from PKD patients showed an abnormal ploidy (Fig. 1b). We consistently observed an astonishingly high abnormality in the genetic composition in the samples acquired from PKD patients (Fig. 1c). We recently showed that survivin is down-regulated in Pkd-derived mouse vascular endothelia5. We therefore examined survivin expression levels in our patients’ samples. All freshly isolated kidney samples from PKD patients consistently show a down-regulation in survivin expression (Fig. 1d).
Figure 1.
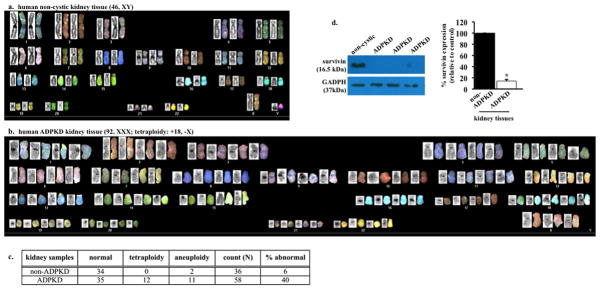
Autosomal dominant polycystic kidney disease (ADPKD) renal epithelia are characterized by abnormal ploidy level and survivin down-regulation. (a) Karyotyping was carried out in freshly isolated epithelial cells from non-ADPKD patients to visualize individual chromosomes (non-cytic kidney). (b) Characterization of individual chromosomes from a single renal epithelium isolated from ADPKD patient indicated tetraploid and abnormal chromosomal composition. (c) Overall karyotype analysis of individual cells confirmed the presence of abnormal genomic compositions (aneuploidy or polyploidy) in cells from ADPKD patients. (d) Kidney tissues from ADPKD patients were also used to confirm survivin expression, and GADPH was used as loading control. Bar graph shows relative survivin expression levels. N=3 each for freshly isolated non-ADPKD and ADPKD kidneys.
Survivin down-regulation is sufficient to promote cystic kidney ex vivo and in vivo
Because Survivin knockout mouse dies at 4.5 days post coitum9, we crossed survivin-flox mouse with kidney-specific Cre mouse (Mx1Cre). We also performed UUO surgery as a renal injury model to examine the relationship between renal injury and cyst formation. We inactivated survivin (Mx1Cre:Survivinflox/flox) in one-week old mice and analyzed the cystic kidney phenotypes in five-week (Fig. 2a-i) and three-month (Fig. 2a-ii) old mice. At five-weeks old, the effects of Survivin knockout were most apparent in injury model, in which the UUO kidneys were bulged and filled with fluid. Kidneys from three-month-old Mx1Cre:Survivinflox/flox mice showed severe gross anatomical kidney defects. Cross-section analysis further showed that inactivation of survivin at one-week old was sufficient to induce kidney cyst formation at five-weeks old, although it was not as severe as those with UUO surgery (Fig. 2b). Histology analysis using standard H&E and fluorescent lectin staining confirmed a gross structure abnormality in Survivin knockout kidney, especially in the injury model, compared to wild-type age-matched kidneys undergoing the same surgery. Survivin inactivation resulted in a progressively more severe cystic kidney phenotype in older mice.
Figure 2.

Survivin downregulation is sufficient to induce cystic kidney formation. (a-i) Unilateral ureteral obstruction (UUO) was performed on either wild-type or Survivin knockout (Mx1Cre:Survivinflox/flox) mice at one-month old, and mice were sacrificed a week later. (a-ii) Mx1Cre:Survivinflox/flox induced- and non-induced littermates were sacrificed, and their kidneys were compared three months later. Comparison of gross features revealed enlargement in Survivin knockout kidneys with an apparent bulged and fluid-filled kidney. (b) H&E-stained kidney sections from five-week old wild-type as well as Mx1Cre:survivinflox/flox mice with or without UUO surgery are shown. Florescence studies were performed with DBA staining (red) and counterstained with WGA (green) and DAPI (blue).
Survivin down-regulation exacerbates aneurysm formation
The occurrence of aneurysm represents a major risk factor for morbidity and mortality associated with PKD15. To examine whether Survivin knockout would result in aneurysm, we induced aneurysm formation in endothelial-specific Survivin knockout (PdgfβCre:Survivinflox/flox) mice. These mice were later sacrificed to measure the aorta diameter at the site of the aneurysm surgery. Unlike wild-type mice, in which aorta diameter was only slightly enlarged following aneurysm surgery, Survivin knockout mice displayed a gross aortic aneurysm similar to that of PdgfβCre:Pkd1flox/flox, Pkd2+/− or Tg737Orpk/Orpk mice following aneurysm surgery (Fig. 3a). Histological analysis of the cross sections further confirmed a marked arterial enlargement and aneurysm formation at the site of surgery from PdgfβCre:Survivinflox/flox, PdgfβCre:Pkd1flox/flox, Pkd2+/−, and Tg737Orpk/Orpk mice (Fig. 3b). Surprisingly, the Pkd2+/− mice also demonstrated a high propensity for aneurysm formation. Our data clearly indicated that similar to Pkd1, Pkd2 or Tg737 inactivation, Survivin knockout resulted in aneurysm formation (Fig. 3c). We next categorized the aneurysm types according to the classification by Daugherty16. Regardless of the genotypes, the mutant mice consistently showed a more severe grade than the wild-type mice (Fig. 3d). Taken together, we proposed that vascular and kidney phenotypes of PKD may share a similar cellular mechanism through survivin.
Figure 3.

Aneurysm formation is an extra-renal phenotype of PKD. (a) Aneurysm surgery was performed in mice at one-month-old and mice were sacrificed at three months of age. The aortas were isolated and their diameters were measured at the surgical site. Unlike wild-type, Pdgfβcre:Survivinflox/flox mice showed a severe aneurysm induction to a similar extent shown in Pdgfβcre:Pkd1flox/flox, Pkd2+/−, and Tg737orpk/orpk mice. (b) Representatives of cross-sections of aortas at the aneurysm surgery site are shown in control wild-type, Pdgfβcre:Pkd1flox/flox, Pkd2+/−, Tg737orpk/orpk, or Pdgfβcre:Survivinflox/flox mice with and without aneurysm surgery. Similar to Pdgfβcre:Pkd1flox/flox, Pkd2+/− and Tg737orpk/orpk mice, Pdgfβcre:Survflox/flox mice exhibited aneurysm formation and aortic dilation compared to wild-type mice. Arrows point to aneurysm formation, and double head arrows point to aorta diameter. N≥3 for each group and genotype. Bar=200 μm. (c) Bar graph shows averaged values for aorta diameter in wild-type, Pdgfβcre:Pkd1flox/flox, Pkd2+/−, Tg737orpk/orpk, or Pdgfβcre:Survivinflox/flox mice. (d) The grade of aneurysm is also tabulated.
Cellular mechanism of cystic and aneurysm formation involves polyploidy formation due to abnormal cell division
To examine the mechanism by which survivin down-regulation contributes to cystic kidney and vascular aneurysm, we performed live-cell imaging on renal epithelia (Fig. 4a). As expected, we observed a symmetric division in normal epithelial cell (Supplement Movie S1). Although survivin knockdown epithelium committed to enter cell division, severe cytokinesis defect was observed, resulting in failure to exit mitosis properly (Supplement Movie S2). This, in turn, led to polyploidy formation with cytomegaly and multi-nucleated phenotypes. Similar studies were performed on vascular endothelial cells (Fig. 4b). Likewise, similar observations were obtained in control (Supplement Movie S3) and survivin knockdown endothelia (Supplement Movie S4).
Figure 4.
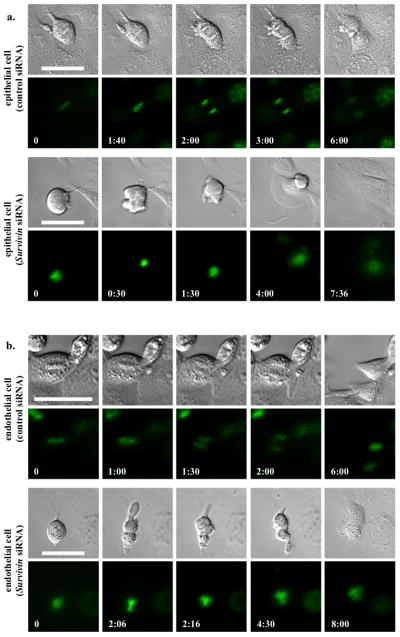
Survivin down-regulation is associated with abnormal cytokinesis in primary cells of renal epithelia and vascular endothelial cells. (a) To obtain the mechanistic insights of cytokinesis defect in renal epithelial cells, live-cell imaging analysis was performed. (b) A similar study was also done in vascular endothelial cells. Control and survivin knockdown cells were loaded with Hoechst dye to examine nuclear division (lower panels), while DIC images were used to study cytokinesis (top panels). Time stamps indicate hours and minutes as illustrated in Supplementary Materials, Movies S1–S4. Bar=50 μm. N≥3 for each group and treatment.
Polyploidy formation contributes to abnormal oriented cell division in cystic expansion and aneurysm formation
Oriented cell division dictates the maintenance of renal tubule diameter during tubular lengthening. Defects in this process will trigger renal tubular enlargement and cyst formation in Pkd rodent models17, 18. We thus examined this possibility in Survivin mouse. Unlike kidney sections from wild-type mice in which normal cell division orientation was parallel to the axis of kidney tubules, kidney sections from Survivin knockout mice (Mx1Cre:Survivinflox/flox) revealed abnormal cell division and orientation pattern (Fig. 5a). Both mitotic misorientation and abnormal cell division were very apparent in the Survivin knockout mice, particularly following UUO surgery. Abnormal cell divisions include enlarged nucleus, multi-nucleated cells, or asymmetric mitosis. Our data further strengthened the argument that survivin shared a similar cellular mechanism as previously reported in polycystic kidney models17–19.
Figure 5.

Abnormal cellular division orientation is associated with renal cystic and vascular aneurysm phenotypes. (a) Kidney tubular sections from both Mx1Cre:survivinflox/flox mice, with or without UUO surgery, showed abnormal cell division and division orientation with respect to the axis of the kidney tubule. ZO-1 staining was used to indicate renal tubule orientation, and cell-undergoing division within the region is further enlarged. (b) Longitudinal abdominal aortic sections in non-surgery (control) and aneurysm-induced (surgery) models were studied to analyze endothelial orientation and cell division. Nucleus from smooth muscle cells was shown in blue, and nucleus of a single intimal layer of endothelial tissue was pseudo-colored in yellow. Abnormal randomized cell orientation is clearly visible. In all figures, division orientation relative to tubule/artery axis is shown in green double head arrows, and abnormal cell division is indicated in green arrows. N≥3 for each group and genotype. N≥100 for distribution of spindle orientation angle for each genotype and each treatment. Bar=40 μm.
To further test our hypothesis that the pathogenesis of aneurysm and cystic kidney shared a common cellular mechanism, we examined for the first time how oriented cell division contributed to aneurysm formation in both Survivin and Pkd mouse models (Fig. 5b). We studied cell division, cell-cell orientation, and division orientation in aortas from wild-type, PdgfβCre:Pkd1flox/flox, Pkd2+/−, Tg737Orpk/Orpk and PdgfβCre:Survivinflox/flox mice. Aorta sections from control wild-type mice displayed normal cell division orientation patterns; however, cell division orientation was slightly perturbed following aneurysm surgery. Similarly, aorta sections from Pkd2+/− mice displayed normal cell or cell division orientation, but they showed abnormal cell division following aneurysm surgery. On the other hand, aorta sections from PdgfβCre:Pkd1flox/flox, Tg737Orpk/Orpk, and PdgfβCre:Survivinflox/flox mice displayed abnormal cell division, cell-cell orientation, and cell division orientation with or without aneurysm surgery.
Primary cilia regulate cell division through survivin expression
We previously showed that low survivin expression is associated with abnormal mitotic events in endothelial cells with cilia dysfunction5. To test our hypothesis that cilia function regulated survivin expression, we examined whether and how flow-induced cilia activation could regulate survivin expression. Wild-type, Pkd1−/− and Tg737Orpk/Orpk endothelial cells were subjected to fluid-shear stress, and survivin expression was analyzed (Fig. 6a). The differential expression of survivin between wild-type and cilia mutant cells was most obvious in the presence of fluid-shear stress. Survivin expression increased following fluid-flow in wild-type but not in cilia mutant cells. However, expression of aurora-A kinase was maintained at the same levels in all cells following fluid-flow, indicating the specificity of flow-induced survivin expression. More surprising is that fluid-flow induced survivin localization to primary cilia only in wild-type cells (Fig. 6b). Because increase in survivin expression and localization to cilia were not observed in cilia mutant cells in response to fluid-shear stress, this indicated that fluid-flow was acting directly on cilia to induce survivin expression.
Figure 6.

Cilia regulate cell division through survivin expression. (a) Western blot analysis was used to study survivin and aurora-A expressions in wild-type and cilia mutant cells (Pkd1−/− and Tg737Orpk/Orpk) in the presence or absence of fluid-shear stress. GAPDH and actin were used as loading controls. Bar graph represents averaged survivin and aurora-A expressions. (b) Acetylated-α-tubulin was used as a ciliary marker to indicate ciliary expression and localization of survivin in response to fluid-shear in wild-type but not mutant cells. (c) Cells treated with survivin or aurora-A inhibitors are characterized by multiple centrosomes, abnormal mitotic spindle, and mitotic arrest during cell division. Cells were stained with acetylated-α-tubulin (green) and pericentrin (red) and captured at resting (i) and dividing (ii) stages of cell cycle. Bar=10 μm. N=3 for each cell type and treatment. Statistics was performed by comparing individual group to their corresponding wild-type static control groups.
We next treated wild-type, Pkd1−/− and Tg737Orpk/Orpk cells with survivin or aurora-A inhibitors. At resting state (Fig. 6c-i), such an inhibition resulted in centrosome over-amplification with multiple stubby cilia formation. In dividing cells (Fig. 6c-ii), inhibiting survivin or aurora-A induced mitotic arrest with profound defects in the bi-polar spindle formation. Defects in resting and dividing cells were observed in wild-type cells, and they became more widespread in cilia mutant cells. Taken together, our data indicated that survivin expression was regulated by flow-induced cilia activation, and that both survivin and aurora-A played critical roles in centrosomal number and cell division regulation.
Survivin expression is regulated by PKC, Akt and NF-κB
We previously demonstrated that (PKC) and Akt are down-stream messengers of primary cilia5. Here, we asked whether cilia-induced survivin expression was also mediated by PKC and/or Akt. To assess whether Akt was downstream of PKC, we treated wild-type and cilia mutant cells with PKC inhibitor or PKC activator (Fig. 7a). Expression of phosphorylated Akt (p-Akt) was significantly down-regulated in all groups treated with PKC inhibitor compared to control non-treated groups. Moreover, p-Akt was significantly increased in cells treated with PKC activator. Consistent with a previous report20, our data support that Akt was downstream of PKC. We next analyzed whether Akt expression was dependent on cilia function (Fig. 7b). Cilia activation by fluid-flow caused a significant increase in p-Akt level in wild-type, but not in cilia mutant cells. Moreover, this increase in p-Akt expression by fluid-shear stress was repressed in wild-type cells treated with PKC inhibitor, indicating that Akt activation was dependent on cilia function and requires PKC activity. In mutant cells, p-Akt expression was consistently and significantly depressed by PKC inhibitor. On the other hand, changes in aurora-A expression was not consistently observed in all groups following fluid-shear stress or PKC inhibitor. We next examined whether aurora-A would regulate p-Akt, Akt or survivin expression levels (Fig. 7c). Our data demonstrated that while inhibiting aurora-A showed no apparent changes on p-Akt or Akt expression, survivin level was slightly but not significantly altered. This suggested that aurora-A was neither regulated by fluid-flow nor upstream of Akt.
Figure 7.
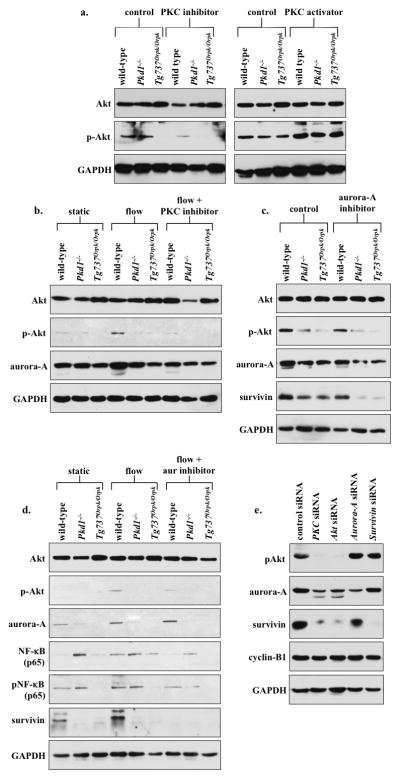
PKC/Akt/NF-κB signaling pathway regulates flow-induced survivin expression and cell division. (a) After wild-type and cilia mutant (Pkd1−/− and Tg737Orpk/Orpk) cells were treated with PKC inhibitor or activator, both Akt and p-Akt were analyzed. When treated with PKC inhibitor, all cell lines showed down-regulation of p-Akt, while PKC activator treatment showed an increase in p-Akt compared to non-treated control cells. (b) The effect of fluid-flow on Akt and aurora-A expression was analyzed in the presence or absence of PKC inhibitor. When subjected to fluid-shear, p-Akt expression was up-regulated only in wild-type cells. While p-Akt expression returned to basal levels following treatment with PKC inhibitor and fluid-shear stress in wild-type cells, it stayed repressed in mutant cells. (c) Treatment with aurora-A inhibitor resulted in a decrease in p-Akt, aurora-A and survivin expression; however, these decreases were not significant from the control, non-treated group. (d) While total Akt level was not changed, fluid-shear stress significantly induced expression of p-Akt in wild-type but not in mutant cells. Aurora-A expression was increased following fluid-shear stress in wild-type cells; however, this increase was not significant from control. Both NF-κB and pNF-κB expressions were increased following fluid-shear stress only in wild-type cells, while mutant cells maintained a high basal level of NF-κB compared to static wild-type cells. Survivin expression was increased following shear-stress in wild-type cells. (e) Western blot analyses were conducted to confirm the signaling mechanism involving survivin expression by siRNA-mediated knockdown of PKC, Akt, aurora A, or survivin. To further confirm the involvement of these signaling molecules in centrosome number and cell division abnormality, immunofluorescence and flow cytometry analyses were presented in the Supplementary Materials together with the statistics.
It has been reported that Akt can regulate NF-κB, which is known to regulate survivin expression21, 22. To investigate this possibility in our system, we studied both NF-κB and phosphorylated NF-κB (pNF-κB). We further inhibited aurora-A in the presence of fluid-flow to confirm our earlier results and to study the relationship between aurora-A and NF-κB (Fig. 7d). Our study corroborated our previous results that flow induces Akt phosphorylation5, and this induction was not affected by aurora-A inhibition. Consistent with our earlier studies, survivin expression was increased by flow, although this increase could be repressed by aurora-A inhibitor in the wild-type cells. More importantly, both NF-κB and pNF-κB expressions were significantly increased in the presence of fluid-flow in wild-type cells. No obvious changes of NF-κB and pNF-κB expressions in response to fluid-flow were observed in cilia mutant cells, although the basal level of NF-κB in the mutant cells was higher than in wild-type cells.
We thus far used various pharmacological agents to examine potential signaling pathways, amid some might have non-specific or off-target effects. Therefore, we next used siRNA knockdown approaches to verify our proposed pathway (Fig. 7e). Knockdown of PKC or Akt resulted in down-regulation of p-Akt, aurora-A and survivin expression. Aurora-A knockdown resulted in a decreased expression of aurora-A and survivin, whereas survivin knockdown showed a decrease in survivin expression only. The expression level of a common cell cycle marker, cyclin-B1, did not change following the siRNA studies, confirming that these knockdowns did not affect cell cycle and proliferation status in our cells. Taken together, we propose that the cilia-PKC-Akt-NFκB pathway was involved in survivin expression and cell division regulation.
Re-expression of survivin rescues PKD phenotypes
Because Survivin knockout results in PKD phenotypes, it is expected that re-expression of survivin to normal levels should alleviate those phenotypes. To test this hypothesis, we used a zebrafish model for our in vivo studies. It has been previously reported that morpholino (MO)-induced depletion of pkd2 causes profound developmental abnormalities including cystic kidneys, curly tails, and pericardiac edema in zebrafish embryos11. We determined whether we could rescue the Pkd2 morphants from these phenotypes by co-injecting mRNAs encoding the open reading frame of survivin. Because VEGF is known to induce survivin expression through Akt-NFκB pathway5, 21, we also tested if modulating this pathway by co-injecting VEGF would rescue PKD phenotypes. Our studies showed that co-injection of survivin mRNA or VEGF in morpholino knockdown of pkd2 rescued the curly tail and cystic kidney phenotypes (Fig. 8a). Overall data analysis showed that the rescue by survivin mRNA or VEGF was more apparent in younger (28 or 48 hpf) than older (72 hpf) fish. This was likely due to a decrease in the effectiveness or stability of injected survivin mRNA or VEGF as the fish developed to older stages. To examine if pkd2MO zebrafish was associated with survivin down-regulation as seen in human PKD and mouse models, we studied the levels of survivin transcript (Fig. 8b) and protein (Fig. 8c). The endogenous zebrafish Pkd2 transcript levels were decreased in pkd2MO, pkd2MO plus survivin mRNA- or pkd2MO plus VEGF-injected embryos, compared to controlMO fish. Injection of survivin mRNA alone did not alter zebrafish pkd2 transcript. Furthermore, the endogenous zebrafish survivin transcript levels were significantly decreased in the presence of pkd2MO, but this could be rescued by VEGF. Our data suggested that the PKD phenotypic rescued in pkd2MO plus VEGF-injected embryos was achieved via induction of endogenous zebrafish survivin, unlike in pkd2MO plus survivin mRNA-injected embryos, which rescue depended on exogenous human survivin. Survivin protein was then quantified using an antibody that recognizes both human and zebrafish forms. Our analysis confirmed the decrease of survivin expression in pkd2MO fish, and it further indicated that survivin expression could be rescued by human survivin mRNA or VEGF injection, leading to PKD phenotypic rescue.
Figure 8.

Survivin overexpression rescued PKD phenotypes in zebrafish. (a) Zebrafish embryos were scored for phenotypic observations at different developmental stages. Shown here are representative images of zebrafish at 24, 48 and 72 hours-post-fertilization (hpf) injected with either control morpholino (MO), pkd2 MO, pkd2 MO plus survivin mRNA, survivin mRNA alone, or pkd2 MO plus VEGF. Abnormal phenotypes associated with pkd2 MO injections such as curly tail and renal cyst were rescued by survivin mRNA or VEGF injection into zebrafish embryos. Representative images of 48-hpf zebrafish sections are shown. The sections were stained with H&E (bottom panels), and arrows point to pronephric structures. (b) RT-PCR was performed to examine zebrafish (zf) and human (hu) transcript levels for survivin and to confirm pkd2 knockdown. Human survivin was introduced through mRNA injection. α-tubulin was used as a loading control. (c) Expression levels of survivin were analyzed in all the groups, in which zebrafish and human survivin can be recognized by the same antibody. (d) Analyses of individual chromosomes were performed in all groups of fish embryos to study chromosomal number. Black bar=500 μm; red bar=50 μm. All quantification and statistical analyses on Western blot, RT-PCR and polyploidy level are presented in the Supplementary Materials.
In order to study how survivin recued the PKD phenotypes, we investigated if a similar molecular mechanism to human PKD and mouse models might involve abnormal polyploidy formation in zebrafish. Analysis of individual chromosomes confirmed a significant increase in polyploidy formation from cells derived from pkd2MO fish, compared to those derived from controlMO fish (Fig. 8d). This polyploidy increase could be partially rescued following co-injection with survivin mRNA. Overall, our data suggested that survivin played an important role in regulating ploidy, a common cellular contributor to PKD phenotypes.
DISCUSSION
Our studies show that abnormal function of mechanosensory cilia leads to survivin down-regulation, which is associated with abnormal ploidy formation and contributes to cystic kidney and vascular aneurysm phenotypes. We show for the very first time that Survivin conditional knockout in the kidney or vascular tissues is associated with cyst or aneurysm formation, respectively. At least in the zebrafish model, re-expression of survivin can partially rescue PKD phenotypes. Overall, our studies demonstrate that primary cilia control renal and vascular architectures through survivin expression and symmetrical cell division along the longitudinal axis of the tissues.
Data from our PKD patients are supported strongly by the mouse and zebrafish studies, indicating that survivin down-regulation triggers polyploidy formation, the predominant phenotype observed throughout our studies. In our controls, especially in human samples, some polyploidy was detected. This is most likely due to a physiological aging process characterized by cellular senescence23. Because a subpopulation of our pre-cystic cells exhibited 8N DNA content, it suggests that consecutive rounds of DNA replication without proper cell division is still possible in cells with a low survivin expression. This is consistent with the report that survivin deletion causes an overall decrease in cell number at the expense of DNA accumulation24. We further propose that polyploidy could potentially be used as an early marker in PKD. Of note is that the sensitivity of karyotyping technique is far more superior than flow cytometry in single-cell analyses10. Thus, changes in chromosome number can be easily identified with absolute certainty in PKD patients before end-stage renal failure.
To ensure the clinical relevance of our findings in age- and sex-matched patients, we utilized non-cystic and PKD human epithelial cell lines (SuppFig. 1). We also used Pkd2 and Tg737 mouse models to verify our clinical data. Our mouse data supported the idea that polyploidy formation precedes cystic expansion and contributes to vascular aneurysm. As in human cell lines, all kidneys from Pkd mouse model samples were characterized by polyploidy and also had a significant down-regulation in survivin expression.
Given the evidence that kidney injury will trigger tubular epithelial cell proliferation25, in which survivin is required, it is not surprising that Mx1Cre:Survivinflox/flox mice exhibited more severe cystic kidneys in UUO-induced injury compared to their non-surgical counterparts. In Mx1Cre:Survivinflox/flox mice with no surgery, the cystic kidney phenotype progressively became more severe with aging. Similar to Pkd-mouse models, inactivation of vascular Survivin exhibited no apparent or consistent phenotype in one-month-old adult mice. Especially during injury, however, survivin down-regulation genetically or pharmacologically was directly linked to cystic kidney formation (SuppFig. 2). To facilitate and accelerate vascular phenotype, we used a CaCl2 aneurysm-induction model. Not only in survivin but also in other Pkd-mouse models, abnormal cell division or cilia function is sufficient to exacerbate the aneurysm phenotype and atherosclerotic plaques following the surgery. Homozygous Survivin knockout mice are also characterized by multi-nucleation, polyploidy and apoptosis9, which are also seen in our renal epithelia and vascular endothelia of Survivin, Pkd1 and Tg737.
Down-regulation of survivin was associated with apoptosis (SuppFig. 3). More importantly, when blood pressure was monitored in our mutant mice, Survivin knockout mice surprisingly did not show an elevated blood pressure. This was consistent with general understanding that hypertension itself does not account for aneurysm development26. Supporting this view, patients with PKD have a significantly greater chance to develop aneurysm than a general population with hypertension27.
Oriented cell division is involved in a variety of processes that contribute to organ shape and morphogenesis and are involved in coordinated cell division, differentiation and spatial distribution. Elongation of the kidney tubule is also associated with oriented cell division, which when perturbed would result in cystic formation17–19. We show here that not only was oriented cell division perturbed in our survivin-inactivated renal tubules, but the asymmetric cell division phenotype was also evident in dividing tubular cells. The mechanism of disturbed cell division and mitotic orientation was also confirmed for the first time in the arteries of Survivin, Pkd1, Pkd2, and Tg737 mice. These mice show distorted cell division and mitotic spindle orientation, even before the aneurysm is formed, as also evidence from our studies on mitotic-stress test and ploidy level in survivin knockdown epithelial and endothelial cells (SuppFigs. 4 and 5). Overall, our comprehensive studies suggest that survivin down-regulation is involved in the control of cell division, polypoidy and asymmetric division orientation. Furthermore, the control of cell division orientation defined a common mechanism for both cystic expansion and aneurysm formation in both Pkd and survivin mouse models.
Survivin, together with aurora-A kinase, regulates several distinct mitotic events such as the formation of mitotic spindle and cytokinetic ring8. We examined aurora-A kinase in our study, because aurora-A has been shown to be localized to the basal body of primary cilia28 and expressed abnormally in the cyst-lining renal epithelia29. Generally, neither survivin nor aurora-A was mis-localized in the mutant cells at different stages of cell division (SuppFig. 6). Our current data also suggest that aurora-A expression is not regulated by cilia activation through fluid-flow. Nonetheless, our study reinforces the localization of aurora-A to the centriole in the resting stage and to the centrosome and mid-body during cell division. More importantly, we demonstrated for the first time that inhibiting aurora-A function or expression would result in defects in cell division, ploidy, centrosomal amplification, cytokinesis, and mitotic spindle formation; all of which were phenotypes associated with survivin down-regulation. This suggests that although aurora-A may not be part of the cilia-survivin pathway, aurora-A and survivin may function as molecular partners reflecting their common roles in the contraction of the cytokinetic ring in regulating cell division (SuppFig. 7).
Not only is survivin expression increased after cilia activation, our study also shows for the first time that its subcellular localization is differentially regulated from centriole to primary cilia following fluid-shear stress. These localizations may reflect a novel survivin function in non-dividing cells and may contribute to a larger pathological spectrum, other than cancer or PKD. Moreover, our immunofluorescence analysis reveals the localization pattern of survivin during mitotic division, specifically during cytokinesis, reflecting its role in cytokinesis. This is also supported by live imaging studies, in which survivin knockdown causes severe cytokinesis defects resulting in cytomegaly and polyploidy phenotypes in both renal epithelia and vascular endothelia (Supplemental Movies). Despite the finding that survivin expression was dysregulated in the cilia mutant cells, it is worth mentioning that survivin localization was not perturbed during cell division. To further decipher the signaling mechanisms between cilia and survivin expression, we examined the cilia-PKC-Akt-NFκB-survivin/aurora-A pathway.
In an attempt to elucidate the physiological relevance of survivin down-regulation in cystic kidney and vascular phenotypes in PKD, we used a zebrafish model to study the roles of survivin expression. Four novel insights are provided by these studies. First, pkd2 knockdown is associated with survivin down-regulation in zebrafish, which confirms our hypothesis that survivin expression is regulated by cilia function. Second, the rescue of PKD phenotypes associated with pkd2 knockdown by re-expression survivin provides further evidence for the importance of survivin roles in PKD. Third, VEGF is an attractive modulator to induce survivin expression in pkd2 knockdown fish. Fourth, pkd2 knockdown contributes to polyploidy, a common mechanism representing PKD phenotypes as also seen in PKD patients and mouse models (SuppFig. 8).
In summary, our studies provide a novel aspect towards understanding the mechanism of pathogeneses of cystic kidney and aneurysm formation in PKD. Our current study shows for the first time that these phenotypes are mainly contributed by abnormal cilia function, resulting in dysregulation of survivin expression. Abnormal survivin expression further causes abnormal cytokinesis, which results in cell polyploidy, multi-mitotic spindle formation and aberrant cell division orientation. The asymmetric cell division together with abnormal planar cell polarity contributes to the expansion of tissue architecture, resulting in the formation of cystic kidney and vascular aneurysm. All in all, data from this study suggest that improving survivin expression could be a promising therapeutic target for kidney and vascular complications associated with PKD. Overall, our current working model would be: primary cilia → PKC → Akt → NF-κB → survivin/aurora-A → cytokinesis → polyploidy → asymmetric cell division / planar cell polarity → cystic kidney and vascular aneurysm (architecture expansion).
Supplementary Material
Acknowledgments
We thank Charisse Montgomery and Maki Takahashi for their editing service and technical support. All supporting materials are available in the online data supplement at http://circ.ahajournals.org.
Funding Sources: This work was supported by awards from the NIH (DK080640).
Footnotes
Conflict of Interest Disclosures: None.
References
- 1.AbouAlaiwi WA, Takahashi M, Mell BR, Jones TJ, Ratnam S, Kolb RJ, Nauli SM. Ciliary polycystin-2 is a mechanosensitive calcium channel involved in nitric oxide signaling cascades. Circ Res. 2009;104:860–9. doi: 10.1161/CIRCRESAHA.108.192765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nauli SM, Alenghat FJ, Luo Y, Williams E, Vassilev P, Li X, Elia AE, Lu W, Brown EM, Quinn SJ, Ingber DE, Zhou J. Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells. Nat Genet. 2003;33:129–37. doi: 10.1038/ng1076. [DOI] [PubMed] [Google Scholar]
- 3.Nauli SM, Kawanabe Y, Kaminski JJ, Pearce WJ, Ingber DE, Zhou J. Endothelial cilia are fluid shear sensors that regulate calcium signaling and nitric oxide production through polycystin-1. Circulation. 2008;117:1161–71. doi: 10.1161/CIRCULATIONAHA.107.710111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Xu C, Shmukler BE, Nishimura K, Kaczmarek E, Rossetti S, Harris PC, Wandinger-Ness A, Bacallao RL, Alper SL. Attenuated, flow-induced ATP release contributes to absence of flow-sensitive, purinergic Cai2+ signaling in human ADPKD cyst epithelial cells. Am J Physiol Renal Physiol. 2009;296:F1464–76. doi: 10.1152/ajprenal.90542.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.AbouAlaiwi WA, Ratnam S, Booth RL, Shah JV, Nauli SM. Endothelial cells from humans and mice with polycystic kidney disease are characterized by polyploidy and chromosome segregation defects through survivin down-regulation. Hum Mol Genet. 2011;20:354–67. doi: 10.1093/hmg/ddq470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Battini L, Macip S, Fedorova E, Dikman S, Somlo S, Montagna C, Gusella GL. Loss of polycystin-1 causes centrosome amplification and genomic instability. Hum Mol Genet. 2008;17:2819–33. doi: 10.1093/hmg/ddn180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Burtey S, Riera M, Ribe E, Pennenkamp P, Rance R, Luciani J, Dworniczak B, Mattei MG, Fontes M. Centrosome overduplication and mitotic instability in PKD2 transgenic lines. Cell Biol Int. 2008;32:1193–8. doi: 10.1016/j.cellbi.2008.07.021. [DOI] [PubMed] [Google Scholar]
- 8.Terada Y. Role of chromosomal passenger complex in chromosome segregation and cytokinesis. Cell Struct Funct. 2001;26:653–7. doi: 10.1247/csf.26.653. [DOI] [PubMed] [Google Scholar]
- 9.Uren AG, Wong L, Pakusch M, Fowler KJ, Burrows FJ, Vaux DL, Choo KH. Survivin and the inner centromere protein INCENP show similar cell-cycle localization and gene knockout phenotype. Curr Biol. 2000;10:1319–28. doi: 10.1016/s0960-9822(00)00769-7. [DOI] [PubMed] [Google Scholar]
- 10.AbouAlaiwi WA, Rodriguez I, Nauli SM. Spectral karyotyping to study chromosome abnormalities in humans and mice with polycystic kidney disease. J Vis Exp. 2012;60:3887. doi: 10.3791/3887. pii. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sun Z, Amsterdam A, Pazour GJ, Cole DG, Miller MS, Hopkins N. A genetic screen in zebrafish identifies cilia genes as a principal cause of cystic kidney. Development. 2004;131:4085–93. doi: 10.1242/dev.01240. [DOI] [PubMed] [Google Scholar]
- 12.Falleni M, Pellegrini C, Marchetti A, Oprandi B, Buttitta F, Barassi F, Santambrogio L, Coggi G, Bosari S. Survivin gene expression in early-stage non-small cell lung cancer. J Pathol. 2003;200:620–6. doi: 10.1002/path.1388. [DOI] [PubMed] [Google Scholar]
- 13.Ko CY, Tsai MY, Tseng WF, Cheng CH, Huang CR, Wu JS, Chung HY, Hsieh CS, Sun CK, Hwang SP, Yuh CH, Huang CJ, Pai TW, Tzou WS, Hu CH. Integration of CNS survival and differentiation by HIF2alpha. Cell Death Differ. 2011;18:1757–70. doi: 10.1038/cdd.2011.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Muntean BS, Horvat CM, Behler JH, Aboualaiwi WA, Nauli AM, Williams FE, Nauli SM. A Comparative Study of Embedded and Anesthetized Zebrafish in vivo on Myocardiac Calcium Oscillation and Heart Muscle Contraction. Front Pharmacol. 2010;1:139. doi: 10.3389/fphar.2010.00139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ecder T, Schrier RW. Cardiovascular abnormalities in autosomal-dominant polycystic kidney disease. Nat Rev Nephrol. 2009;5:221–8. doi: 10.1038/nrneph.2009.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Daugherty A, Manning MW, Cassis LA. Antagonism of AT2 receptors augments angiotensin II-induced abdominal aortic aneurysms and atherosclerosis. Br J Pharmacol. 2001;134:865–70. doi: 10.1038/sj.bjp.0704331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fischer E, Legue E, Doyen A, Nato F, Nicolas JF, Torres V, Yaniv M, Pontoglio M. Defective planar cell polarity in polycystic kidney disease. Nat Genet. 2006;38:21–3. doi: 10.1038/ng1701. [DOI] [PubMed] [Google Scholar]
- 18.Patel V, Li L, Cobo-Stark P, Shao X, Somlo S, Lin F, Igarashi P. Acute kidney injury and aberrant planar cell polarity induce cyst formation in mice lacking renal cilia. Hum Mol Genet. 2008;17:1578–90. doi: 10.1093/hmg/ddn045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Delaval B, Bright A, Lawson ND, Doxsey S. The cilia protein IFT88 is required for spindle orientation in mitosis. Nat Cell Biol. 2011;13:461–8. doi: 10.1038/ncb2202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nazarewicz RR, Salazar G, Patrushev N, San Martin A, Hilenski L, Xiong S, Alexander RW. Early endosomal antigen 1 (EEA1) is an obligate scaffold for angiotensin II-induced, PKC-alpha-dependent Akt activation in endosomes. J Biol Chem. 2011;286:2886–95. doi: 10.1074/jbc.M110.141499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li W, Wang H, Kuang CY, Zhu JK, Yu Y, Qin ZX, Liu J, Huang L. An essential role for the Id1/PI3K/Akt/NFkB/survivin signalling pathway in promoting the proliferation of endothelial progenitor cells in vitro. Mol Cell Biochem. 2011;363:135–45. doi: 10.1007/s11010-011-1166-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lin J, Guan Z, Wang C, Feng L, Zheng Y, Caicedo E, Bearth E, Peng JR, Gaffney P, Ondrey FG. Inhibitor of differentiation 1 contributes to head and neck squamous cell carcinoma survival via the NF-kappaB/survivin and phosphoinositide 3-kinase/Akt signaling pathways. Clin Cancer Res. 2011;16:77–87. doi: 10.1158/1078-0432.CCR-08-2362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jones MR, Ravid K. Vascular smooth muscle polyploidization as a biomarker for aging and its impact on differential gene expression. J Biol Chem. 2004;279:5306–13. doi: 10.1074/jbc.M308406200. [DOI] [PubMed] [Google Scholar]
- 24.Levkau B, Schafers M, Wohlschlaeger J, von Wnuck Lipinski K, Keul P, Hermann S, Kawaguchi N, Kirchhof P, Fabritz L, Stypmann J, Stegger L, Flogel U, Schrader J, Fischer JW, Hsieh P, Ou YL, Mehrhof F, Tiemann K, Ghanem A, Matus M, Neumann J, Heusch G, Schmid KW, Conway EM, Baba HA. Survivin determines cardiac function by controlling total cardiomyocyte number. Circulation. 2008;117:1583–93. doi: 10.1161/CIRCULATIONAHA.107.734160. [DOI] [PubMed] [Google Scholar]
- 25.Yang L, Besschetnova TY, Brooks CR, Shah JV, Bonventre JV. Epithelial cell cycle arrest in G2/M mediates kidney fibrosis after injury. Nat Med. 2011;16:535–43. doi: 10.1038/nm.2144. 1p following 143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen J, Kuhlencordt PJ, Astern J, Gyurko R, Huang PL. Hypertension does not account for the accelerated atherosclerosis and development of aneurysms in male apolipoprotein e/endothelial nitric oxide synthase double knockout mice. Circulation. 2001;104:2391–4. doi: 10.1161/hc4501.099729. [DOI] [PubMed] [Google Scholar]
- 27.Chapman AB, Rubinstein D, Hughes R, Stears JC, Earnest MP, Johnson AM, Gabow PA, Kaehny WD. Intracranial aneurysms in autosomal dominant polycystic kidney disease. N Engl J Med. 1992;327:916–20. doi: 10.1056/NEJM199209243271303. [DOI] [PubMed] [Google Scholar]
- 28.Plotnikova OV, Nikonova AS, Loskutov YV, Kozyulina PY, Pugacheva EN, Golemis EA. Calmodulin activation of Aurora-A kinase (AURKA) is required during ciliary disassembly and in mitosis. Mol Biol Cell. 2012;23:2658–70. doi: 10.1091/mbc.E11-12-1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Plotnikova OV, Pugacheva EN, Golemis EA. Aurora A kinase activity influences calcium signaling in kidney cells. J Cell Biol. 2011;193:1021–32. doi: 10.1083/jcb.201012061. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.