

Abstract
People with cystic fibrosis (CF) sinus disease have developmental sinus abnormalities with airway bacterial infection, inflammation, impaired mucociliary clearance and thick obstructive mucus. The pathophysiology of airway disease in CF is not completely understood, and current treatments in CF sinus disease ameliorate symptoms but do not provide a cure.
This manuscript reviews the history of CF, its manifestations in sinus disease, and the potential impact and relationship of CF on the upper and lower airway. We discuss recent discoveries in the pathophysiology of CF using the CF porcine animal model and exciting treatments that address the primary gene defect that may translate into improved outcomes in CF and non-CF sinusitis in humans.
Keywords: airway biology, electrophysiology, ion transport, gene therapy, CFTR
Introduction
Cystic fibrosis (CF) is a common, lethal genetic disorder in the United States, with 1 in 3,500 babies born with CF and 30,000 people in the US having the disease. Although advances in the clinical care of the CF patient have increased the average lifespan, the most common cause of mortality continues to be airway disease.1 CF was first named in 1938 by Dorothy Andersen to describe the pathologic changes of the pancreas in children with CF.2 The CF phenotype has expanded to multiple organ systems including the intestine, lung, sweat gland, liver, gallbladder, male genital tract, and the paranasal sinus.3–6 Although the association between early death and salty sweat was first made in the 18th century,7 a severe hot spell in New York in 1948 prompted Kessler’s observation that 5 of 10 children admitted for dehydration had CF.8 Di Sant’Agnese tested this association through an elegant set of experiments. He measured the electrolyte composition of sweat by applying occlusive gauze to the skin of subjects in a heated room and discovered that people with CF had increased sweat sodium (Na+) and chloride (Cl−) levels.9 In 1983, Paul Quinton isolated and microperfused the sweat glands from the skin of humans and found that low Cl− transport was the primary defect in CF.10 Although inheritance patterns in CF suggested an autosomal recessive disorder, it was not until 1989 that a collaborative effort from the labs of Drs. Collins, Riordan, and Tsui identified the CF transmembrane conductance regulator (CFTR) gene.11 The CFTR gene encodes an anion channel expressed on the cell surface that allows the transport of Cl− and bicarbonate (HCO3−). If the channel is analogous to a door, cyclic adenosine monophosphate (cAMP)-agonists open the lock and adenosine triphosphate (ATP) widens the doorway. Mutations in CFTR are responsible for CF and can be classified into six different classes: defects in protein production (Class I), processing (Class II), regulation (Class III), conduction (Class IV), reduced number of CFTR transcripts (Class V) and accelerated protein turnover (Class VI).5,12 The most common CF mutation is the ΔF508 mutation representing a deletion of three nucleotides resulting in absence of phenylalanine at position 508, leading to a misfolded protein that cannot be transported to the cell surface.
The unified airway in CF
As we breathe, air is passed through the conducting upper airway via our sinonasal passages, through the larynx, and enters the lower airway consisting of the trachea, bronchi, and bronchioles. The upper and lower conducting respiratory airway epithelia consist of pseudostratified ciliated epithelia with glandular epithelial cells and submucosal glands that produce mucus that coats the airway and provides a medium for mucociliary clearance. In general, the size of the airway progressively decreases from the sinonasal passage to the small bronchioles, which then progress to the microscopic alveoli that participate in gas exchange (Table 1). The unified airway model suggests that disease processes of the upper airway can influence that of the lower airway, and vice versa.13 In CF, loss of CFTR in the sinonasal and lower airway epithelia reduces Cl− and HCO3− transport 14 and results in the common end result of airway bacterial infection, inflammation, impaired mucociliary clearance and thick obstructive mucus.
Table 1.
Characteristics of the upper and lower airway
| Airway | Function | Cell types | Diameter (mm) |
|---|---|---|---|
| Nasal/Turbinates | Humidification, temperature regulation, filtration, mucociliary clearance | Pseudostratified columnar ciliary epithelia | 10 |
| Sinus | Pneumatized airway lumen in the skull, immunologic defense, patterened mucociliary clearance to ostial openings | Pseudostratified columnar ciliary epithelia | Ostial diameter = 2 |
| Olfactory | Specialized neuro-epithelia to detect odor | Olfactory cells, supporting cells, basal cells, brush cells | |
| Trachea | Cartilaginous tube that connects the upper airway to the pulmonary system | Pseudostratified columnar ciliary epithelia | 18 |
| Bronchi | Division of the trachea that conducts air in to the lungs | Pseudostratified columnar ciliary epithelia | 5–12 |
| Bronchioles | Division of the bronchi that conducts air to the alveoli | Pseudostratified columnar ciliary epithelia, Clara cells | 0.5–3.5 |
| Alveoli | Site of gas exchange, terminal division of respiratory tree | Type 1 and 2 alveolar cells | 0.4 |
Similar pathogens in upper and lower airway infection: Could the sinus be infecting the lungs in CF?
There are several studies that have looked at the similarities between bacterial pathogens in the sinus and lung in CF. In early disease, both the sinus and the lungs are infected with common bacteria including Staphylococcus aureus, Haemophilus influenzae, and Streptococcus pneumoniae. In chronic disease, Pseudomonas aeruginosa is the major pathogen in both the sinus and the lung.15,16 Early aggressive treatment to eradicate P. aeruginosa in the lung has been found to be the biggest factor in improving lifespan in CF. Unfortunately, the lungs eventually become recolonized with P. aeruginosa, and the bacteria can adapt to become drug-resistant and mucoid, forming biofilms that can be difficult to eradicate. Those who become recolonized with P. aeruginosa after eradication therapy often have similar strains as bacteria cultured from the sinus.16,17 Researchers have compared the genotypes of P. aeruginosa in the sinus and in the lungs of CF patients after lung transplant and found similarities in both genotype and gene expression phenotypes.18–20,21 There are various reports of the effect of sinus surgery in reducing CF lung disease, which may be attributed to the lack of a standard criteria for success in CF sinus surgery.22–24 These observational findings suggest that the sinus and upper airway can act as a bacterial reservoir and transmit disease to the lower airway. Therefore aggressive eradication of CF infection in the upper airway may improve treatment of lung disease.
Use of a CF porcine model to understand the pathophysiology of CF disease
A major obstacle in the study of CF has been the lack of a suitable animal model that replicates human CF disease. Previous animal models, including the CF knockout mouse, exhibit gastrointestinal abnormalities but do not spontaneously develop airway disease.25 The expression of a calcium-activated chloride channel (CaCC) may explain the lack of phenotype in CF mice.26 Prospective human studies of infants with CF to investigate the pathogenesis and pathophysiology of disease are difficult to perform due to ethical concerns. We have recently reported a novel, CF porcine knockout model that exhibits altered anion transport in airway epithelia, defective bacterial killing, and spontaneous development of airway disease similar to CF including airway inflammation, remodeling, and infection.27 Here we review recent discoveries from the CF pig model that have shed light on the pathophysiology of CF disease.
CF electrophysiology
The primary defect in CF is due to lack of CFTR anion transport. The main tools for examining ion transport in a cell are electrophysiology studies in humans (in vivo), in cultured airway epithelia (in vitro), and in freshly dissected tissue (ex vivo). If we assume only transcellular transport, we can oversimplify the ion transport of an airway epithelial cell into a sodium–potassium adenosine triphosphatase (Na+/K+-ATPase) pump and channels/transporters on the basolateral side, and the epithelial Na+ (ENaC) and Cl− (CFTR) channels on the apical surface. Voltage (Vt) represents the electric potential difference between the apical and basolateral surface of the cell, current (i) the flow of electrical charge through the cell, and conductance (Gt) the ease at which the current flows through the cell via ion channels (Figure 1). We can measure Vt changes in humans in vivo by measuring the potential difference between electrodes placed on the nasal epithelium and ground electrode. In 1983, Knowles noted characteristic voltage differences in people with CF: elevated basal Vt levels and an increased Vt change after blocking the ENaC channel with amiloride.28
Figure 1.
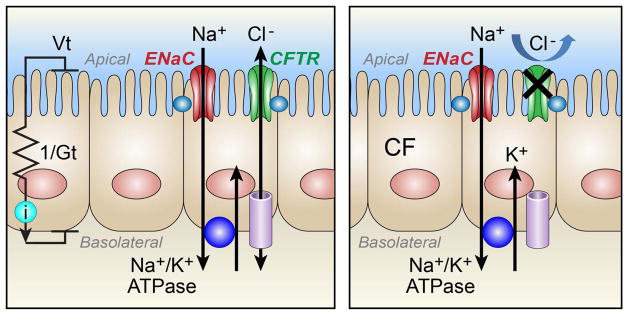
Transcellular ion transport in airway epithelia. Measurements of voltage (Vt), current (i), and conductance (Gt) are drawn in relation to an electric circuit. Vt measures the electric potential difference between the apical and basolateral surface of the cell, i the flow of electrical charge through the cell, and Gt the ease at which the current flows through the cell via ion channels. In this simplified diagram airway epithelia have a sodium–potassium adenosine triphosphatase (Na+/K+-ATPase) pump and channels/transporters on the basolateral side, and the epithelial Na+ (ENaC) and Cl− (CFTR) channels on the apical surface (A). In CF epithelia loss of the CFTR channel reduces Cl transport with increased basal Vt levels.
According to Ohm’s law, Vt = i/Gt. Therefore two possibilities can account for the increased Vt in CF. Increased Na+ transport could increase current (i) or lack of CFTR channels could decrease conductance (Gt). In order to distinguish between these two differences, Gt or (i) need to be measured. Measurements in conductance cannot be performed in vivo, but are commonly measured in freshly isolated explants or in vitro epithelial cultures in Ussing chambers. Electrophysiology studies of CF human ex vivo tissue “are confusing and difficult to interpret” according to a review by Geddes.29 Some studies have reported decreased basal Gt and a decrease in Gt after amiloride, suggesting that the Vt changes in CF were due to lack of Cl− transport.30 Other studies have found the opposite result of increased Na+ transport and Gt in CF epithelia.31 The investigators believed there were several reasons for these discrepancies: impaired function of ex vivo tissue due to hypoxia, physical damage of ex vivo tissue once mounted in the Ussing chamber, variability in the age and quality of the tissue, and the time to study thereby highlighting the technical difficulties of using ex vivo human airway tissue.31
The CF pig has enabled us to investigate the changes in Vt, (i) and Gt of airway epithelia in vivo, ex vivo, and in vitro cultures and thereby address controversies in CF electrophysiology. Chen et al. found that similar to in vivo studies in humans, CF pigs showed increased amiloride-sensitive Vt and I. However, in contrast to a widely held view, lack of CFTR did not increase transepithelial Na+ transport or liquid absorption, nor did it reduce periciliary liquid depth. Lack of apical Cl− conductance caused the change in voltage, not increased Na+ transport.32 This study identified loss of CFTR anion permeability as the primary transport defect at birth, which could contribute to the development of airway disease in CF.
Decreased HCO3− transport and pH in CF impairs innate immunity against bacteria
Although loss of CFTR results in chronic inflammation and infection of human airways, the mechanism of impaired immunity is unknown. One important question in CF has been which appears first: inflammation or infection of the airway? Stoltz et al. found that the airway of newborn CF pigs did not have inflammatory changes, yet bacterial challenges to the airway resulted in impaired killing in CF versus non-CF pigs.27 This finding suggests that CF pigs have a host defense defect against bacteria that results in airway inflammation. In a set of experiments to investigate the pathogenesis of the impaired host defense defect in CF porcine airways, Pezzulo et al. used immobilized bacteria on grids applied to the airway surface in vivo and quantified bacterial killing using a live/dead stain. By utilizing this method, they found that CF airway epithelia exhibited a bacteria killing defect independent of mucociliary clearance, phagocyte killing, or inflammation. They hypothesized that the bacteria killing defect was due to impaired HCO3− transport resulting in decreased airway surface liquid (ASL) pH in CF airway epithelia. Subsequent studies manipulating the ASL pH confirmed that decreased ASL pH impaired bacteria killing by decreasing the function of antimicrobial proteins in the ASL.33 These results suggest that increasing ASL pH and antimicrobial activity by HCO3− delivery may be an effective strategy in treating airway infection in CF.
The CF pig as a model to test hypotheses in CF sinusitis
There have been several unanswered questions in CF sinusitis. First, does sinus disease precede lung disease in CF? Second, what treatments are effective in CF sinusitis? And third, will early aggressive treatment of CF sinus disease slow progression of lung disease? The pig is a good model to study sinus development and disease, as pigs are born with ethmoid and maxillary sinuses and develop frontal and sphenoid sinuses later in life, similar to humans. Their relatively large size also allows endoscopic visualization and sampling of the sinonasal cavities (Figure 2). We have investigated the phenotype of the CF pigs and found that CF pigs have defective anion transport, newborn CF pigs have sinus hypoplasia, and CF pigs spontaneously develop CF sinus disease similar to that of humans with CF (Figure 3).34
Figure 2.

Piglet anatomy. Sagittal split of piglet head. A. Right hemisagittal section : unc = Uncinate process, mt = Middle turbinate, eth = ethmoid sinus. C, Left hemisagittal section with the uncinate process removed and wire into the middle meatus and in the maxillary sinus (mx). The endoscopic view of the anterior (B) nasal passageway showing the septum (s), inferior turbinate (it), and middle turbinate (mt). Further posterior (D) is the ethmoid sinus (eth).
Figure 3.
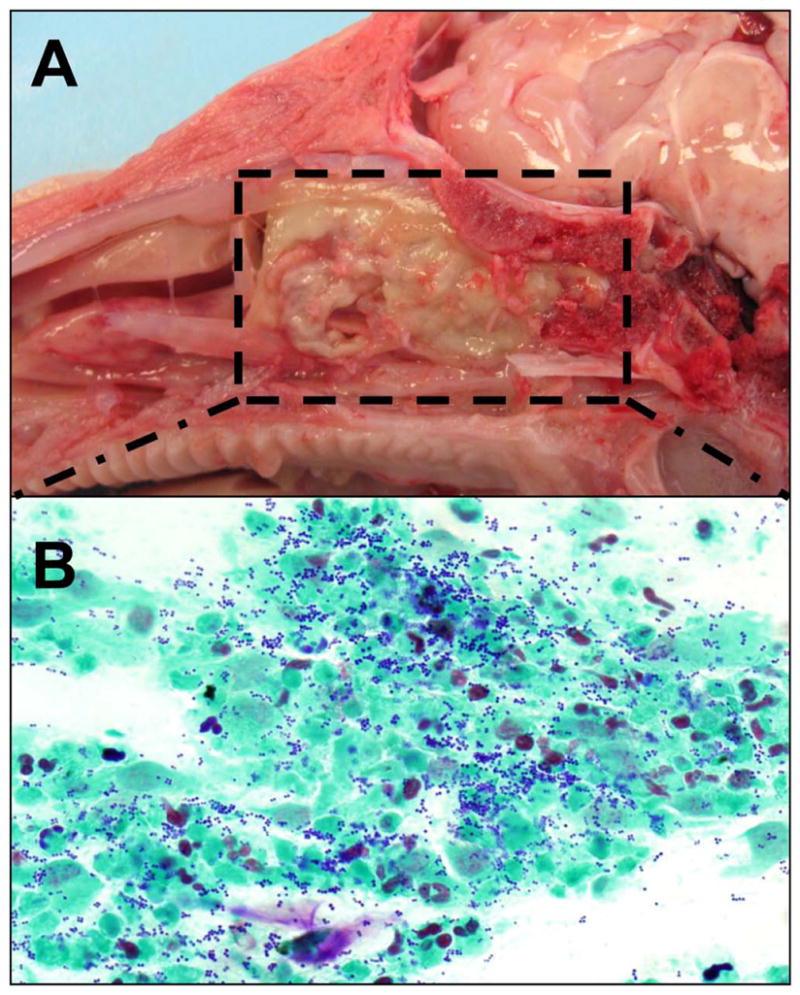
CF piglet with thick mucus occluding the ethmoid and maxillary sinus (A). Histology of the mucus consistent with cellular debris and gram positive cocci consistent with Staph aureus (B).
Does sinus disease precede lung disease in CF? We have followed several young children with CF whose first presenting symptom was due to sinonasal disease. These children’s sinus CT scans showed severe sinonasal disease, yet their screening lung CT exams in the same period did not identify chronic lung infection (Figure 4). Supporting this hypothesis are studies of children with positive upper airway but negative lower airway cultures in CF.35 This condition may be secondary to anatomical factors where disease occurs in the lumen of the sinus and must be cleared through the narrow ostia, whereas in the lung disease occurs in the small airways and is expelled by the cough mechanism and mucociliary transport through progressively larger airways. There may also be differences in antimicrobial factors, immune defense systems, and ion transport that account for disease in the upper and lower airway. Human studies to investigate the onset of sinus to lung disease in CF are difficult due to the ethical issues of repeated radiologic imaging in young children.36,37,38 The CF pig allows us to perform longitudinal studies of endoscopy, bronchoscopy, microbiologic cultures, and radiologic imaging from birth and throughout disease.
Figure 4.
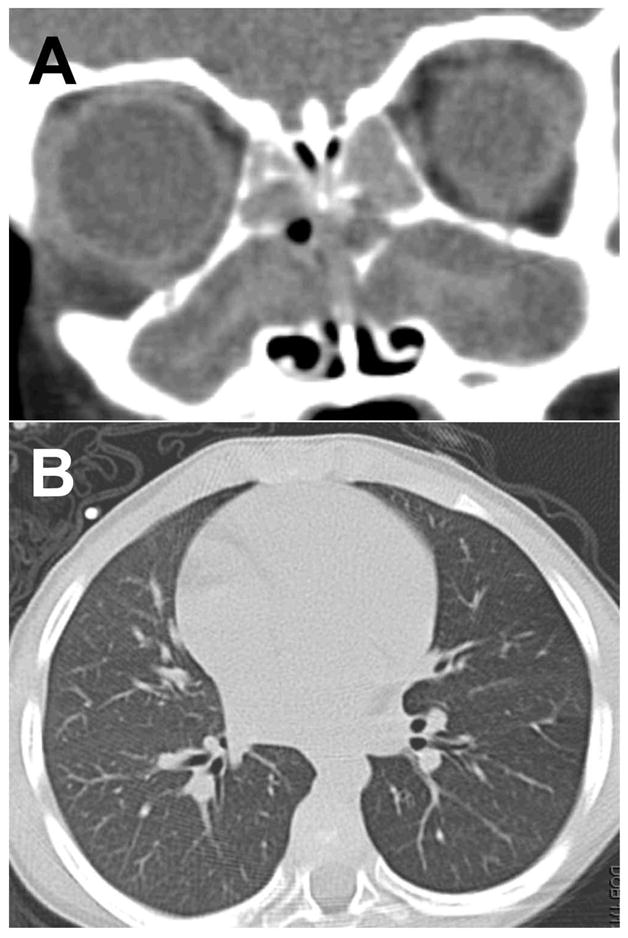
Sinus disease without lung disease in a child with CF. Sinus CT scan taken at 4 years of age consistent with chronic sinusitis with thick hyperdense mucus completely occluding the ethmoid and maxillary sinus (A). Lung CT scan taken at the same time without radiologically apparent lung disease (B).
Second, what treatments are effective in treating CF sinus disease? In humans, there is no clear evidence that sinus surgery improves outcomes in CF. Some studies have shown improvement in sinus disease as well as lung disease,22,39 while others have shown no effect.24 Although sinus surgery may improve symptoms of CF sinusitis, the disease invariably recurs evident as chronic bacterial infection and sinus opacification on CT.40 Therefore, we are unlikely to implement sinus surgery as a widespread treatment in CF due to the limited evidence of surgery in effectively treating disease. Gene therapy trials in humans directed to the sinus have shown transient gene expression and ion transport changes,41 but have not changed primary and secondary outcomes of sinonasal disease.42 These studies may be confounded by the advanced stage of sinus disease with associated inflammation and infection. We have investigated topical gene therapy to the porcine sinus with the goal of correcting CFTR expression and treating the primary gene defect. The relative symmetry of the sinus allows us to use one animal as its own control, and we have shown that we can selectively transduce sinus epithelial cells in the pig.43 In an in vitro model, we can also correct the CFTR defect in CF porcine sinus epithelia, as evidenced by increased anion transport.44
One of the biggest advances in CF medical therapy for a select group of patients has been the drug VX-770, also known as Kalydeco or Ivacaftor. VX-770 is the first medical drug that facilitates the opening of defective CFTR channels, and is thereby a “potentiator” of CFTR activity. The use of VX-770 is limited to people with the G551D mutation (Class III mutation), where CFTR protein is present at the cell surface but inactive due to defective regulation of CFTR. The first randomized trial of VX-770 in people with the G551D mutation showed improvements in anion transport measured by nasal electrophysiology and sweat Cl−, as well as clinical improvements in lung function.45 These promising findings led to U.S. Food and Drug Administration approval of the medication for people with CF and the G551D mutation. Unfortunately, this group represents only 4–5% of people with CF, and the medication is costly and likely needs to be taken long-term.46 Regardless, this has been a major medical advance in the treatment of CF, and novel therapies to address more common mutations of CFTR including ΔF508 are being investigated as “correctors” of CFTR. As medical therapies advance to provide options for cure in CF disease, there is a need to quantify the CF sinus phenotype so we can monitor sub-clinical levels of disease with the goal of prevention. Future studies in the CF pig will allow us to investigate if standard interventions including lavage, antibiotics, and surgery, or novel therapies including gene therapy and CFTR potentiators and correctors, can treat CF sinus disease.
Third, will the early aggressive treatment of CF sinus disease slow the progression of lung disease? Early eradication therapy of CF lung disease has been the greatest factor in reducing morbidity in CF, yet eventual recolonization of the lung occurs with bacteria that have adapted to become drug-resistant and form biofilms. If the sinus did act as a bacterial reservoir for lung recolonization, early aggressive therapy to eradicate CF sinus disease could improve outcomes in CF. Because of the paucity of symptoms and early onset of disease, treatment of the CF sinus occurs after chronic inflammatory changes in the sinus epithelia have already developed. The CF pig allows us to test therapies to the airway before the onset of disease in a window of opportunity where treatment may be most effective. We can also investigate if early treatment of CF sinus disease can delay the progression of CF lung disease.
Conclusion
CF is a common, fatal disease involving the airway. There is increasing evidence that CF sinus disease may affect CF lung disease, the most common cause of mortality in CF. We have been limited in treating the symptoms of advanced CF sinus disease with our current therapies. The recent advances of an animal model that replicates CF sinus disease, and novel therapies including gene therapy and potentiators and correctors of CFTR, offer promise to treat the primary gene defect and improve treatment of human CF disease.
Acknowledgments
We thank Joseph Zabner, David Stoltz, Peter Taft for critical review of the manuscript and the Cystic Fibrosis center at the University of Iowa. We thank Shawn Roach for the illustrations in Figs 1 and 2.
Footnotes
There is no conflict of interest to disclose
Financial disclosure: Supported by the National Institutes of Health/National Institute of Dental and Craniofacial Research (1K08DE021413-01A1) and the Cystic Fibrosis Foundation
References
- 1.Cystic Fibrosis Foundation Patient Registry. Bethesda, Maryland: 2011. 2011 Annual Data Report. [Google Scholar]
- 2.Andersen DH. Cystic fibrosis of the pancreas and its relation to celiac disease: a clinical and pathological study. Am J Dis Child. 1938;56:344–399. [Google Scholar]
- 3.Gysin C, Alothman GA, Papsin BC. Sinonasal disease in cystic fibrosis: clinical characteristics, diagnosis, and management. Pediatr Pulmonol. 2000;30:481–489. doi: 10.1002/1099-0496(200012)30:6<481::aid-ppul8>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 4.Quinton PM. Physiological basis of cystic fibrosis: a historical perspective. Physiol Rev. 1999;79:S3–S22. doi: 10.1152/physrev.1999.79.1.S3. [DOI] [PubMed] [Google Scholar]
- 5.Rowe SM, Miller S, Sorscher EJ. Cystic fibrosis. N Engl J Med. 2005;352:1992–2001. doi: 10.1056/NEJMra043184. [DOI] [PubMed] [Google Scholar]
- 6.Welsh MJ, Ramsey BW, Accurso F, Cutting GR. Cystic Fibrosis. In: Scriver CR, Beaudet AL, Sly WS, Valle D, Childs B, Vogelstein B, editors. The Metabolic and Molecular Basis of Inherited Disease. New York: McGraw-Hill; 2001. [Google Scholar]
- 7.Busch R. On the history of cystic fibrosis. Acta Univ Carol Med (Praha) 1990;36:13–15. [PubMed] [Google Scholar]
- 8.Kessler WR, Andersen DH. Heat prostration in fibrocystic disease of the pancreas and other conditions. Pediatrics. 1951;8:648–656. [PubMed] [Google Scholar]
- 9.Di Sant’Agnese PA, Darling RC, Perera GA, Shea E. Abnormal electrolyte composition of sweat in cystic fibrosis of the pancreas; clinical significance and relationship to the disease. Pediatrics. 1953;12:549–563. [PubMed] [Google Scholar]
- 10.Quinton PM. Chloride impermeability in cystic fibrosis. Nature. 1983;301:421–422. doi: 10.1038/301421a0. [DOI] [PubMed] [Google Scholar]
- 11.Riordan JR, Rommens JM, Kerem B, et al. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science. 1989;245:1066–1073. doi: 10.1126/science.2475911. [DOI] [PubMed] [Google Scholar]
- 12.Welsh MJ, Smith AE. Molecular mechanisms of CFTR chloride channel dysfunction in cystic fibrosis. Cell. 1993;73:1251–1254. doi: 10.1016/0092-8674(93)90353-r. [DOI] [PubMed] [Google Scholar]
- 13.Tos M. Distribution of mucus producing elements in the respiratory tract. Differences between upper and lower airway. Eur J Respir Dis Suppl. 1983;128 (Pt 1):269–279. [PubMed] [Google Scholar]
- 14.Sheppard DN, Welsh MJ. Structure and function of the CFTR chloride channel. Physiol Rev. 1999;79:S23–45. doi: 10.1152/physrev.1999.79.1.S23. [DOI] [PubMed] [Google Scholar]
- 15.Burns JL, Gibson RL, McNamara S, et al. Longitudinal assessment of Pseudomonas aeruginosa in young children with cystic fibrosis. J Infect Dis. 2001;183:444–452. doi: 10.1086/318075. [DOI] [PubMed] [Google Scholar]
- 16.Koch C. Early infection and progression of cystic fibrosis lung disease. Pediatr Pulmonol. 2002;34:232–236. doi: 10.1002/ppul.10135. [DOI] [PubMed] [Google Scholar]
- 17.Munck A, Bonacorsi S, Mariani-Kurkdjian P, et al. Genotypic characterization of Pseudomonas aeruginosa strains recovered from patients with cystic fibrosis after initial and subsequent colonization. Pediatr Pulmonol. 2001;32:288–292. doi: 10.1002/ppul.1121. [DOI] [PubMed] [Google Scholar]
- 18.Dosanjh A, Lakhani S, Elashoff D, Chin C, Hsu V, Hilman B. A comparison of microbiologic flora of the sinuses and airway among cystic fibrosis patients with maxillary antrostomies. Pediatr Transplant. 2000;4:182–185. doi: 10.1034/j.1399-3046.2000.00114.x. [DOI] [PubMed] [Google Scholar]
- 19.Mainz JG, Naehrlich L, Schien M, et al. Concordant genotype of upper and lower airways P aeruginosa and S aureus isolates in cystic fibrosis. Thorax. 2009;64:535–540. doi: 10.1136/thx.2008.104711. [DOI] [PubMed] [Google Scholar]
- 20.Taylor CJ, McGaw J, Howden R, Duerden BI, Baxter PS. Bacterial reservoirs in cystic fibrosis. Archives of disease in childhood. 1990;65:175–177. doi: 10.1136/adc.65.2.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ciofu O, Johansen HK, Aanaes K, et al. P. aeruginosa in the paranasal sinuses and transplanted lungs have similar adaptive mutations as isolates from chronically infected CF lungs. J Cyst Fibros. 2013 doi: 10.1016/j.jcf.2013.02.004. [DOI] [PubMed] [Google Scholar]
- 22.Holzmann D, Speich R, Kaufmann T, et al. Effects of sinus surgery in patients with cystic fibrosis after lung transplantation: a 10-year experience. Transplantation. 2004;77:134–136. doi: 10.1097/01.TP.0000100467.74330.49. [DOI] [PubMed] [Google Scholar]
- 23.Jones JW, Parsons DS, Cuyler JP. The results of functional endoscopic sinus (FES) surgery on the symptoms of patients with cystic fibrosis. Int J Pediatr Otorhinolaryngol. 1993;28:25–32. doi: 10.1016/0165-5876(93)90143-q. [DOI] [PubMed] [Google Scholar]
- 24.Rosbe KW, Jones DT, Rahbar R, Lahiri T, Auerbach AD. Endoscopic sinus surgery in cystic fibrosis: do patients benefit from surgery? Int J Pediatr Otorhinolaryngol. 2001;61:113–119. doi: 10.1016/s0165-5876(01)00556-0. [DOI] [PubMed] [Google Scholar]
- 25.Grubb BR, Boucher RC. Pathophysiology of gene-targeted mouse models for cystic fibrosis. Physiol Rev. 1999;79:S193–214. doi: 10.1152/physrev.1999.79.1.S193. [DOI] [PubMed] [Google Scholar]
- 26.Clarke LL, Grubb BR, Yankaskas JR, Cotton CU, McKenzie A, Boucher RC. Relationship of a non-cystic fibrosis transmembrane conductance regulator-mediated chloride conductance to organ-level disease in Cftr(−/−) mice. Proc Natl Acad Sci U S A. 1994;91:479–483. doi: 10.1073/pnas.91.2.479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stoltz DA, Meyerholz DK, Pezzulo AA, et al. Cystic fibrosis pigs develop lung disease and exhibit defective bacterial eradication at birth. Sci Transl Med. 2010;2:29ra31. doi: 10.1126/scitranslmed.3000928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Knowles M, Gatzy J, Boucher R. Relative ion permeability of normal and cystic fibrosis nasal epithelium. J Clin Invest. 1983;71:1410–1417. doi: 10.1172/JCI110894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Geddes D. Progress in research on cystic fibrosis. Thorax. 1984;39:721–725. doi: 10.1136/thx.39.10.721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Knowles MR, Stutts MJ, Spock A, Fischer N, Gatzy JT, Boucher RC. Abnormal ion permeation through cystic fibrosis respiratory epithelium. Science. 1983;221:1067–1070. doi: 10.1126/science.6308769. [DOI] [PubMed] [Google Scholar]
- 31.Boucher RC, Stutts MJ, Knowles MR, Cantley L, Gatzy JT. Na+ transport in cystic fibrosis respiratory epithelia. Abnormal basal rate and response to adenylate cyclase activation. J Clin Invest. 1986;78:1245–1252. doi: 10.1172/JCI112708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen JH, Stoltz DA, Karp PH, et al. Loss of anion transport without increased sodium absorption characterizes newborn porcine cystic fibrosis airway epithelia. Cell. 2010;143:911–923. doi: 10.1016/j.cell.2010.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pezzulo AA, Tang XX, Hoegger MJ, et al. Reduced airway surface pH impairs bacterial killing in the porcine cystic fibrosis lung. Nature. 2012;487:109–113. doi: 10.1038/nature11130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chang EH, Pezzulo AA, Meyerholz DK, et al. Sinus hypoplasia precedes sinus infection in a porcine model of cystic fibrosis. Laryngoscope. 2012;122:1898–1905. doi: 10.1002/lary.23392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Roby BB, McNamara J, Finkelstein M, Sidman J. Sinus surgery in cystic fibrosis patients: comparison of sinus and lower airway cultures. Int J Pediatr Otorhinolaryngol. 2008;72:1365–1369. doi: 10.1016/j.ijporl.2008.05.011. [DOI] [PubMed] [Google Scholar]
- 36.Neely JG, Harrison GM, Jerger JF, Greenberg SD, Presberg H. The otolaryngologic aspects of cystic fibrosis. Trans Am Acad Ophthalmol Otolaryngol. 1972;76:313–324. [PubMed] [Google Scholar]
- 37.Rosenfeld M, Emerson J, Accurso F, et al. Diagnostic accuracy of oropharyngeal cultures in infants and young children with cystic fibrosis. Pediatr Pulmonol. 1999;28:321–328. doi: 10.1002/(sici)1099-0496(199911)28:5<321::aid-ppul3>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 38.Burns JL, Gibson RL, McNamara S, et al. Longitudinal assessment of Pseudomonas aeruginosa in young children with cystic fibrosis. J Infect Dis. 2001;183:444–452. doi: 10.1086/318075. [DOI] [PubMed] [Google Scholar]
- 39.Moss RB, King VV. Management of sinusitis in cystic fibrosis by endoscopic surgery and serial antimicrobial lavage. Reduction in recurrence requiring surgery. Arch Otolaryngol Head Neck Surg. 1995;121:566–572. doi: 10.1001/archotol.1995.01890050058011. [DOI] [PubMed] [Google Scholar]
- 40.McMurphy AB, Morriss C, Roberts DB, Friedman EM. The usefulness of computed tomography scans in cystic fibrosis patients with chronic sinusitis. Am J Rhinol. 2007;21:706–710. doi: 10.2500/ajr.2007.21.3104. [DOI] [PubMed] [Google Scholar]
- 41.Wagner JA, Messner AH, Moran ML, et al. Safety and biological efficacy of an adeno-associated virus vector-cystic fibrosis transmembrane regulator (AAV-CFTR) in the cystic fibrosis maxillary sinus. Laryngoscope. 1999;109:266–274. doi: 10.1097/00005537-199902000-00017. [DOI] [PubMed] [Google Scholar]
- 42.Wagner JA, Nepomuceno IB, Messner AH, et al. A phase II, double-blind, randomized, placebo-controlled clinical trial of tgAAVCF using maxillary sinus delivery in patients with cystic fibrosis with antrostomies. Hum Gene Ther. 2002;13:1349–1359. doi: 10.1089/104303402760128577. [DOI] [PubMed] [Google Scholar]
- 43.Sinn PL, Cooney AL, Oakland M, et al. Lentiviral vector gene transfer to porcine airways. Mol Ther Nucleic Acids. 2012;1:e56. doi: 10.1038/mtna.2012.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Potash AE, Wallen TJ, Karp PH, et al. Adenoviral Gene Transfer Corrects the Ion Transport Defect in the Sinus Epithelia of a Porcine CF Model. Molecular therapy : the journal of the American Society of Gene Therapy. 2013 doi: 10.1038/mt.2013.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Accurso FJ, Rowe SM, Clancy JP, et al. Effect of VX-770 in persons with cystic fibrosis and the G551D-CFTR mutation. N Engl J Med. 2010;363:1991–2003. doi: 10.1056/NEJMoa0909825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kaiser J. Personalized medicine. New cystic fibrosis drug offers hope, at a price. Science. 2012;335:645. doi: 10.1126/science.335.6069.645. [DOI] [PubMed] [Google Scholar]