

Abstract
Death-associated protein kinase (DAPK) has been found promoting cell death under stress conditions, including cell death during brain ischemia. However, little is known about the mechanisms how DAPK is involved in the neuronal death promoting process during ischemia. The present study was to examine the DAPK signal transduction pathways using an ischemia mimicking model, oxygen glucose deprivation (OGD). OGD was induced by incubating SH-SY5Y neuroblastoma cells in glucose free culture medium flushed with a mixture of N2 and CO2. DAPK expression was inhibited by transfection of SH-SY5Y cells with DAPK short hairpin RNA (shRNA). Cell death induced by OGD exposure was assessed by Annexin V-FITC and propidium iodide (PI) assay. Protein expressions were examined by Western blot and protein interactions were detected with immunoprecipitation followed by Western blot. OGD treatment resulted in neuronal death and led to DAPK activation as demonstrated by increase of DAPK (active form) and decrease of phospho-DAPK (inactive form). The activation of DAPK in turn led to BimEL up-regulation and ER stress activation. Further analyses showed that DAPK mediated BimEL expression through extracellular signal-regulated protein kinase1/2 (ERK1/2) inactivation and c-Jun-N-terminal kinase1/2 (JNK1/2) activation. These findings revealed novel signal transduction pathways leading to neuronal death in response to OGD.
Keywords: Death-associated protein kinase, ischemia, oxygen glucose deprivation, neuronal death
1
Death-associated protein kinase (DAPK), is a serine (Ser)/threonine (Thr) kinase. DAPK has a complex, multi-domain structure that includes a calcium/calmodulin-regulated kinase-domain, a series of ankyrin repeats and carboxyl-terminal death domain. Unlike many other kinases, DAPK is inactivated when it is phosphorylated at Ser308 since the phosphorylation blocks its calmodulin binding site (de Diego et al. 2010). DAPK has been shown to be involved in neuronal death. In PC12 cells, DAPK activity is highly increased upon exposing to apoptotic induction, and inhibition of DAPK activity rescues the cells (Yamamoto et al. 2002). In cultured hippocampal neurons, DAPK level is increased during apoptotic induction (Pelled et al. 2002). DAPK has also been found to be involved in neuronal death in animal ischemia studies. For example, DAPK mRNA expression is up-regulated in rat brain following global ischemia (Shamloo et al. 2005), and DAPK activity is found significantly increased in ischemic injured rat brain neurons (Schumacher et al. 2002). DAPK knockout mice show dramatic reduction in infarct volume and improved neurological function following cerebral ischemia (Tu et al. 2010).
Endoplasmic reticulum (ER) serves important functions in the cell. ER is responsible for post-translational modification of proteins, as well as sorting and exporting these proteins to appropriate destinations. ER is highly sensitive to stresses which result in unfolded protein accumulation in the ER. Upon the accumulation of unfolded proteins, a set of molecular signals for coping with ER stress, collectively termed unfolded protein response (UPR), are activated. The UPR is carried out by three trans-membrane initiator proteins: protein kinase RNA (PKR)-like ER kinase (PERK), inositol-requiring protein-1 (IRE1) and activating transcription factor-6 (ATF6). All these three effector proteins bind to the ER chaperone, Bip (GRP78), on their ER luminal domains, where Bip acts to repress their activities. When cells are under stress, Bip dissociates from the initiator proteins. The dissociation relieves the repression and allows activation of these initiator proteins. Whereas UPR is activated to protect cells, it becomes destructive when ER stress is sustained. With excess ER stress, all three initiator proteins mediate signaling pathways to induce the expression of C/EBP homologous protein (CHOP), also known as growth arrest- and DNA damage-inducible gene 153 (GADD153), which promotes apoptotic cell death (Tabas and Ron 2002; Szegezdi et al. 2006; Kim et al. 2008). Recently, it has shown that ER stress promotes catalytic activity of DAPK. Furthermore, DAPK plays a critical role in ER stress signaling, transmitting stress signals to two distinct directions, caspase activation and autophagy, and leading to cell death (Gozuacik et al. 2008).
Bim is a Bcl-2 homology-3 (BH-3) only pro-apoptotic Bcl-2 family member. Alternative splicing of Bim mRNA results in three isoforms: BimEL, BimL and BimS (O’Connor et al. 1998). Cells with low level of Bim expression are resistant to death induction, and Bim overexpression sensitizes cells to death signals (Leung et al. 2008). Bim has also been found mediating ER stress caused cell death (Puthalakath et al. 2007). Bim expression is regulated by several kinase signal transduction pathways. Extracellular signal-regulated protein kinase1/2 (ERK1/2) fosters BimEL degradation via phosphorylation on Ser65 (Ser69 in human) (Ewings et al. 2007). In contrast, c-Jun-N-terminal kinase (JNK) has been associated with Bim up-regulation. Treatment of human lung adenocarcinoma ASTC-a-1 cells with UV irradiation results in increase of Bim expression by activation of JNK1/2-Foxo3a signal transduction pathway (Wang et al. 2012). Interestingly, enhanced Bim expression by Foxo3a also results in apoptotic cell death in neurons (Gilley et al. 2003).
Although DAPK has been implicated in neuronal death in ischemic stroke and neurodegenerative diseases, such as Alzheimer’s disease (Shamloo et al. 2005; Duan et al. 2013), the mechanisms about how DAPK regulates cell death are still elusive. In the present study, we investigated the signal transduction pathways involved in DAPK mediated neuronal death in response to oxygen glucose deprivation (OGD).
2. Experimental procedures
2.1 Materials
All chemicals were purchased from Sigma-Aldrich (St. Louis, USA), except indicated.
2.2 Cell culture
Human SH-SY5Y neuroblastoma cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with penicillin (100 units/ml), streptomycin (100 μg/ml) (Fisher Scientific, Pittsburg, USA), and 10% (vol/vol) heat inactivated fetal bovine serum (FBS) (Atlanta Biologicals, Lawrenceville, USA). SH-SY5Y cells transfected with short hairpin RNA (shRNA) were maintained in DMEM supplemented with puromycin (2 ug/ml) and 10% (vol/vol) heat inactivated FBS. To inhibit JNK1/2 activity, cells were incubated with a specific JNK inhibitor SP600125 (Cell Signaling Technology, Danver, USA. cat #: 8177) at a concentration of 25 μM for 2 hours before OGD exposure.
2.3 OGD induction
OGD was induced as described previously (Kaushal and Schlichter 2008). Briefly, following replacement of culture medium with oxygen-glucose free DMEM (Invitrogen Corporation, Carlsbad, USA), the cells were placed in Modular Incubator Chamber (Billups-Rothenberg, Del Mar, USA). The oxygen-glucose free DMEM was prepared by bubbling glucose-free DMEM (Invitrogen Corporation, Carlsbad, USA) with a mixture of 95% N2 and 5% CO2 for 5 minutes. The chamber was sealed and flushed with a mixture of 95% N2 and 5% CO2 for 3 minutes with a flow rate of 40 liter per minute. Two ports of the chamber were then sealed and the chamber was placed at 37°C incubator for a desired time period. Control cells were maintained in a normoxic condition (5% CO2 and 95% air) with regular DMEM.
2.4 Western blot assay
Western blot analyses were carried out as described previously (Wang et al. 2006; Song et al. 2005). In brief, cells grown on 10 cm culture dish were harvested and lysed in cold lysis buffer [50 mM Tris (pH 7.4), 150 mM NaCl, 2 mM EDTA, 10% glycerol, 1% Triton X-100, 1 mM PMSF and 2% protease inhibitor mixture (Sigma-Aldrich, cat #: P8430)]. The cell lysates were centrifuged for 15 minutes at 18,000 x g at 4°C, supernatants were collected and protein concentrations in the supernatants were determined using Bio-Rad protein assay (Bio-Rad Laboratories, Hercules, USA). 25 μg proteins from cell lysates were subjected to electrophoresis on 12% SDS-polyacrylamide gels and transferred to polyvinylidene difluoride membrane. The membranes were blocked in TBS-T (Tris-buffered saline with 0.05% Tween-20) containing 5% milk and blotted overnight with primary antibodies. The membranes were washed and incubated for 1 hour with horseradish peroxidase (HRP) - conjugated secondary antibodies (Jackson ImmunoResearch, West Grove, USA). The membranes were washed and developed by chemiluminescence (Fisher Scientific). Primary antibodies include anti-CHOP (cat #: 2895), anti-eIf2α (cat #: 2103), anti-phospho-eIf2α (cat #: 3597), anti-phospho-Bim (Ser69) (cat #: 4585), anti-Bcl-xl (cat #: 2764), anti-JNK1/2 (cat #: 9258), anti-phospho-JNK1/2 (cat #: 4668), anti-ERK1/2 (cat #: 9102), anti-phospho-ERK1/2 (cat #: 9101) (Cell Signaling Technology); anti-BimEL (cat #: sc-27982), anti-β-actin (cat #: sc-130657), anti-Bcl-2 (cat #: sc-492), anti-Bax (cat #: sc-493) (Santa Cruz Biotechnology); anti-DAPK (cat #: D2178), anti-phospho-DAPK (cat #: D4941) (Sigma-Aldrich).
2.5 shRNA transfection
Cells grown in 6-well plate (600,000 cells/well) were transfected when cells reached around 50% confluence. Transfection was performed by adding polybrene as well as lentiviral particles encoding DAPK shRNA (Santa Cruz Biotechnology) to culture medium. After 16 hours transfection, the medium was replaced with DMEM containing 5% FBS. Two days later, puromycin dihydrochloride at a concentration of 2 μg/ml was added to the medium for selection. Cells that were not successfully transfected with the lentiviral particles were killed by puromycin dihydrochloride. The inhibition of DAPK expression was confirmed by Western blot analysis. Control cells received mock transfection with lentiviral particles encoding copGFP (Santa Cruz Biotechnology).
2.6 Immunoprecipitation
Cells were lysed in IP lysis buffer (Thermo Fisher Scientific, Rockford, USA) supplemented with protease inhibitor cocktail (Sigma-Aldrich, cat #: P8340), phosphatase inhibitor cocktail 2 (Sigma-Aldrich, cat #: P5726) & 3 (Sigma-Aldrich, cat #: P0044). Cell debris was removed by centrifugation and lysate was collected. Following pre-clearance with protein G agarose beads (Thermo Fisher Scientific), the lysate was incubated with antibody at 4°C rotating overnight. Then, protein G beads were added and rotated at 4°C for additional 2 hours. The bead complex was collected by centrifugation and washed with lysis buffer for 5 times. Antigen was eluted with elution buffer (Thermo Fisher Scientific), and was analyzed by Western blot.
2.7 Annexin V-FITC/propidium iodide (PI) assay
Cell death was detected with Annexin V-FITC and PI double staining kit (Invitrogen). After treatments, cells were washed with ice cold PBS twice. Cells were stained in 100 μl binding buffer containing 5 μl Annexin V-FITC and 1 μl PI, and were analyzed using BD FACScan flow cytometer with an aid of Cell Quest Pro software (BD Biosciences, San Jose, USA). Annexin V-FITC and PI signals were excited with 488 nm laser beam and emission was detected at 530 nm for Annexin V-FITC and 575 nm for PI. Apoptotic cells show Annexin V-FITC positive staining while late apoptotic and necrotic cells show both Annexin V-FITC and PI positive staining. Cell survival rate was calculated as Annexin V-FITC negative staining cell number/total cell number x100.
2.8 Immunocytochemistry
Cells were grown on microscope cover glass (Thermo Fisher Scientific). At the end of treatments, cells were fixed with 4% paraformaldehyde for 15 minutes, permeabilized in PBS with 0.3% Triton X-100 for 10 minutes and blocked with 1% bovine serum albumin (Song et al. 2005). Subsequently, the cells were incubated with anti-PDI (protein disulfide isomerase, an ER marker) antibody (Abcam, Cambridge, USA. cat #: ab-2792) and anti-BimEL antibody (Santa Cruz Biotechnology) overnight at 4°C and then followed by FITC anti-mouse and TRITC anti-goat secondary antibodies for 1 hour at room temperature. Fluorescence signal was examined under microscope.
2.9 Statistical analysis
Data of cell survival rates and Western blots were expressed as mean ± SEM, and were analyzed with one-way analysis of variance (ANOVA) followed by Tukey post hoc test when more than two groups were compared. For comparison between two groups, a t-test was used. A p value less than 0.05 was considered statistically significant.
3. Results
3.1 DAPK promoted neuronal death during OGD
To detect whether DAPK was involved in the cellular response to OGD treatment in SH-SY5Y cells, levels of DAPK and phospho-DAPK were measured following exposure of the cells to various periods of OGD (Figure 1A). Western blot analysis revealed a decrease of phospho-DAPK (inactive form of DAPK) when the cells were exposed to OGD for 0.5 hour and it further decreased when the cells were exposed to longer period of OGD. In contrast, the active form of DAPK increased following the OGD exposure. These results indicate that DAPK was activated following OGD treatment.
Figure 1.

DAPK promoted neuronal death during OGD. (A) DAPK expressions during OGD. SH-SY5Y cells were exposed to OGD for the indicated time period, lysed and analyzed with Western blot. β-actin was used as a loading control in this and subsequent figures. (B) DAPK shRNA transfection inhibited DAPK expression. SH-SY5Y cells were transfected with lentiviral particles encoding copGFP (mock) or DAPK shRNA. DAPK protein level was analyzed by Western blot. (C) Annexin-V FITC/PI assay of cell survival after OGD treatment. DAPK shRNA or mock transfected cells were treated with OGD for the indicated hours. Cell death was detected with Annexin-V FITC and PI staining and analysed by flow cytometry. Dots in the lower right quadrant represent population of apoptotic cells and dots in the upper right quadrant represent population of late stage of apoptotic cells and necrotic cells. Results are representative of one of three independent experiments. (D) Pooled data of Annexin-V FITC/PI assay (n=3). The bars represent mean±SEM. * (p<0.05) compared with mock transfected cells.
To determine the role of DAPK in OGD induced neuronal death, DAPK expression was inhibited by transfection of the cells with DAPK shRNA. The efficiency of transfection was evaluated by detecting DAPK expression with Western blot analysis. As shown in Figure 1B, DAPK expression decreased dramatically in the cells transfected with DAPK shRNA as compared to the cells received mock transfection. Subsequently, cell survival rates after OGD treatment were compared between DAPK shRNA and mock transfected cells (Figure 1C, 1D). Results from flow cytometry analysis showed that survival rate was 97.9% in shRNA transfected cells and 98% in mock transfected cells without OGD treatment. After exposing to OGD for 4 hours, the survival rate was 93.6% in shRNA transfected cells and 79.3% in mock transfected cells. Cell survival rate was 50.1% in shRNA transfected cells and 33.8% in mock transfected cells after 8 hour OGD exposure. The survival rate in DAPK shRNA transfected cells (31.5%) was almost doubled of that in mock transfected cells (18%) with exposure to OGD for 16 hours. DAPK shRNA transfection significantly increased cell survival when the cells were exposed to OGD for 8 hours (p<0.05) or 16 hours (p<0.05). These findings demonstrate that DAPK activation contributed to OGD-induced neuronal death.
3.2 DAPK induced ER stress during OGD
Activation of PERK/eIF2α ER stress signaling pathway has been detected during brain ischemia (Kumar et al. 2001). Upon dissociation from Bip, PERK phosphorylates eIF2α, which activates its downstream effectors and ultimately induces CHOP expression. To detect whether ER stress was involved in the OGD induced cell death, total eIF2α, phospho-eIF2α and CHOP levels were examined by Western blot analysis. As shown in Figure 2A, phospho-eIF2α was slightly elevated in the cells exposed to OGD for 2 hours and it was continuously increased with longer exposure periods. However, total eIF2α expression did not change following the OGD exposure. Increase of CHOP was found in cells exposed to OGD for 4 hours, and CHOP expression was higher with longer OGD exposure periods. These results indicate that ER stress was activated during OGD exposure.
Figure 2.

DAPK shRNA transfection reduced ER stress induced by OGD. (A) ER stress protein expressions after OGD treatment. SH-SY5Y cells were exposed to OGD for the indicated hours, lysed and analyzed with Western blot. (top) representative Western blot; (bottom) densitometric analysis of phospho-eIf2α and CHOP expressions (n=3). * (p <0.05) and † (p <0.01) compared with 0 hour group. (B) ER stress protein expressions in DAPK shRNA transfected cells. DAPK shRNA or mock transfected SH-SY5Y cells were exposed to OGD for indicated hours. Protein levels were analyzed by Western blot. (top) representative Western blot; (bottom) densitometric analysis of phospho-eIf2α and CHOP expressions in three independent experiments. Data from mock transfection was plotted as clear bars and from DAPK shRNA transfection as black bars. * (p <0.05) and † (p <0.01) compared with 8 hour mock transfection group.
Next, OGD-induced ER stress response was examined in DAPK shRNA transfected cells (Figure 2B). Although phospho-eIf2α and CHOP were detected in the DAPK shRNA transfected cells after 8 hour OGD exposure, the amount of both phospho-eIf2α and CHOP were significantly less than those in the mock transfected cells, which indicates that DAPK shRNA transfection alleviated OGD-induced ER stress.
3.3 DAPK up-regulated BimEL expression during OGD
The role of BimEL in DAPK mediated neuronal death during OGD was investigated. BimEL expression was measured in the cells received OGD treatment (Figure 3A). BimEL expression was remarkably increased in cells received 1 hour OGD exposure, and was maintained at high level following longer OGD exposures. However, expressions of several other Bcl-2 family members, Bax, Bcl-2 and Bcl-xl, did not change following the OGD exposures. Next, BimEL expression was examined in cells transfected with DAPK shRNA (Figure 3B). With 2 hour OGD treatment, BimEL was much less in DAPK shRNA transfected cells than in mock transfected cells. However, Bcl-2 expression was not affected by DAPK shRNA transfection. These results suggest that overexpression of BimEL during OGD was mediated by the DAPK signaling pathways.
Figure 3.

DAPK mediated BimEL overexpression during OGD treatment. (A) Bcl-2 family protein expressions after OGD treatment. SH-SY5Y cells were exposed to OGD for the indicated hours. Endogenous levels of BimEL, Bax, Bcl-2 and Bcl-xl were measured. (top) representative Western blot; (bottom) densitometric analysis of BimEL based on three independent experiments. * (p <0.05) and † (p <0.01) compared with 0 hour group. (B) BimEL expressions in DAPK shRNA transfected cells. DAPK shRNA or mock transfected SH-SY5Y cells were treated with OGD for the indicated hours. BimEL and Bcl-2 levels were detected with Western blot analysis. (top) representative Western blot; (bottom) densitometric analysis of BimEL in three independent experiments. Data from mock transfection was plotted as clear bars and from DAPK shRNA transfection as shaded bars. † (p <0.01) compared with mock transfected cells received 2 hour OGD treatment. (C) OGD treatment induced BimEL and ER colocalization. SH-SY5Y cells were treated without or with OGD for 2 hours. Cells were double stained with anti-PDI and anti-BimEL antibodies and visualized by microscopy equipped with proper filters. Representative images were shown. Scale bar: 10 um.
To examine whether the increased BimEL expression was associated with ER stress during OGD, subcellular localization of BimEL was examined (Figure 3C). Immunocytochemistry showed that 2 hour OGD exposure caused induction and translocation of BimEL to ER, suggesting BimEL was involved in OGD-induced ER stress.
3.4 phospho-ERK1/2 increased interaction with BimEL during OGD
Findings from the present study strongly implicated that BimEL expression was regulated by DAPK activation during OGD, however, immunoprecipitation study did not find physical association of DAPK and BimEL (data not shown). These findings thus suggest that DAPK regulated BimEL indirectly. This hypothesis is supported by the findings from previous studies that an interaction between DAPK and ERK is found in yeast two-hybrid screen (Chen et al. 2005) and phosphorylation of BimEL by ERK1/2 activation leads to BimEL ubiquitination and degradation (Ley et al. 2003; Weston et al. 2003). Therefore, experiments were carried out to examine if ERK1/2 was involved in the BimEL regulation by DAPK. Western blot showed that exposure of the cells to OGD resulted in decreased phospho-ERK1/2 (the active form) and it was nearly undetectable when the cells were exposed to OGD for 8 or 16 hours (Figure 4A). To further determine the relationship between DAPK and ERK1/2, total ERK1/2 and phospho-ERK1/2 expressions were examined in the cells transfected with DAPK shRNA. As shown in figure 4B, exposure of the mock transfected cells to OGD for 2 hours resulted in a significant decrease of phospho-ERK1/2. Transfection of DAPK shRNA completely prevented OGD induced decrease of phospho-ERK1/2. However, this transfection did not alter total ERK1/2 level. These results indicate DAPK inhibited ERK1/2 phosphorylation during OGD treatment.
Figure 4.
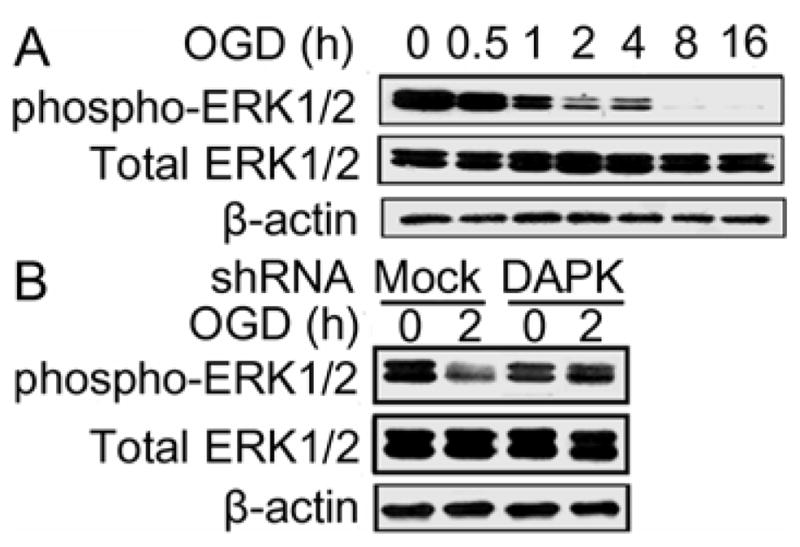
DAPK inhibited ERK1/2 activation during OGD exposure. (A) Total ERK1/2 and phospho-ERK1/2 expressions after OGD exposure. SH-SY5Y cells were exposed to OGD for the indicated hours. Total ERK1/2 and phospho-ERK1/2 levels were analyzed with Western blot. (B) Total ERK1/2 and phospho-ERK1/2 expressions in DAPK shRNA transfected cells. DAPK shRNA or mock transfected cells were treated with OGD for the indicated hours. Endogenous levels of total ERK1/2 and phospho-ERK1/2 were detected with Western blot analysis. The experiments were repeated two to three times with similar results.
Since phospho-ERK1/2 decreased after OGD treatment, we next studied whether interaction of BimEL with phospho-ERK1/2 was decreased accordingly. Immunoprecipitation result showed a direct interaction between phospho-ERK1/2 and BimEL (Figure 5A, B). Although phospho-ERK1/2 decreased in the cells received 2 hour OGD treatment, increased amount of BimEL associated with phospho-ERK1/2 was observed (Figure 5A). Furthermore, reverse immunoprecipitation confirmed this observation that increased phospho-ERK1/2 was found associated with BimEL following the treatment (Figure 5B). Consistent with these results, exposure of cells to OGD resulted in increased phospho-BimEL level with a maximal level observed in the cells treated with 2 hour OGD exposure (Figure 5C). These results suggest that phospho-ERK1/2 was involved in the increased phosphorylation of BimEL during OGD treatment.
Figure 5.
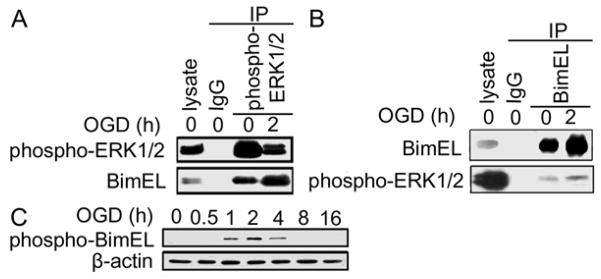
Interaction of BimEL with phospho-ERK1/2. (A) Co-immunoprecipitation of BimEL with phospho-ERK1/2. SH-SY5Y cells were exposed to OGD and then lysed and extracts were collected. The extracts were subjected to immunoprecipitation with IgG (control) or anti phospho-ERK1/2 and followed by Western blot analyses with anti-phospho-ERK1/2 or anti-BimEL antibody. (B) Reverse immunoprecipitation with BimEL and detected for phospho-ERK1/2. SH-SY5Y cells were exposed to OGD and then lysed and extracts were collected. The extracts were subjected to immunoprecipitation with IgG (control) or anti-BimEL and followed by Western blot analyses with anti-phospho-ERK1/2 or anti-BimEL antibody. (C) phospho-BimEL expression during OGD. Cells were treated with OGD for the indicated hours. Endogenous level of phospho-BimEL was detected with Western blot analysis. The experiments were repeated two to three times with similar results.
3.5 JNK activation up-regulated BimEL expression
The fact that BimEL level was increased and phospho-BimEL (destined for degradation) was also increased following OGD exposure suggested that BimEL synthesis was up-regulated. To test this hypothesis, the involvement of JNK1/2 signaling was studied in the DAPK induced BimEL up-regulation during OGD exposure. Total JNK1/2 level did not change following exposure of the cells to various periods of OGD, whereas phospho-JNK1/2 (active JNK1/2) level increased after OGD treatment (Figure 6A). Phospho-JNK1/2 expression increased in the cells exposed to OGD for 0.5 hour and it reached maximum in the cells treated with 2 hour OGD. Thereafter, phospho-JNK1/2 level started to decline. However, phospho-JNK1/2 level in cells treated with 16 hour OGD was still higher than that in the non-OGD control. Transfection of DAPK shRNA eliminated OGD induced JNK1/2 phosphorylation (Figure 6B), suggesting that DAPK promoted JNK1/2 activation during OGD treatment.
Figure 6.
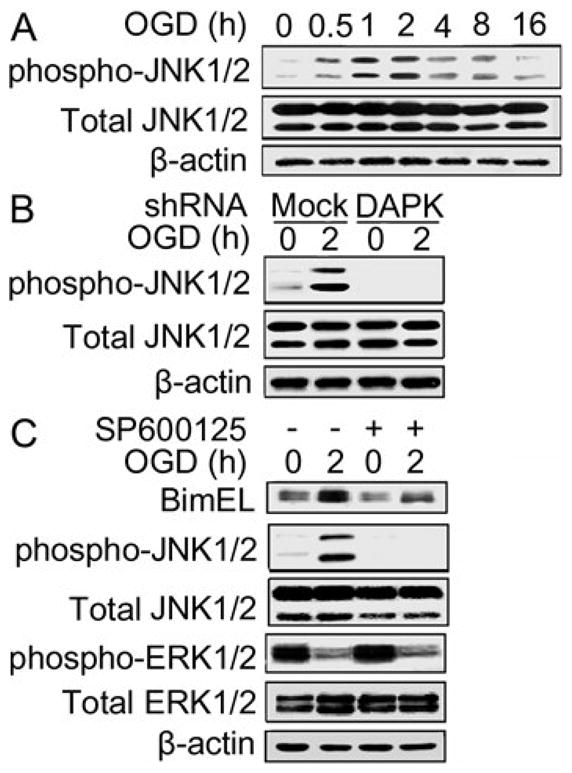
DAPK increased BimEL expression via JNK1/2 activation. (A) Total JNK1/2 and phospho-JNK1/2 expressions after OGD exposure. SH-SY5Y cells were exposed to OGD for the indicated hours. Total JNK1/2 and phospho-JNK1/2 expressions were detected with Western blot analysis. (B) Total JNK1/2 and phospho-JNK1/2 expressions in DAPK shRNA transfected cells. DAPK shRNA or mock transfected cells were treated with OGD for the indicated hours. Endogenous levels of total JNK1/2 and phospho-JNK1/2 were measured with Western blot analysis. (C) Effect of JNK1/2 inhibitor on BimEL expression during OGD. Cells were exposed to OGD in the presence or absence of JNK1/2 inhibitor for the indicated hours. Then the cells were collected, lysed and proteins were analyzed with Western blot. The experiments were repeated two to three times with similar results.
To detect whether JNK1/2 was playing a critical role in BimEL up-regulation, cells were treated with a JNK1/2 specific inhibitor, SP600125 (Bennett et al. 2001). As shown in Figure 6C, JNK1/2 inhibitor suppressed phospho-JNK1/2 expression, while it did not affect phospho-ERK1/2 expression. Furthermore, the treatment with SP600125 also reduced OGD induced increase of BimEL. Taken together, these findings support that DAPK activated JNK1/2 contributed to the BimEL up-regulation during OGD.
4. Discussion
It has been suggested that DAPK is involved in neuronal death during ischemic brain injury (Schumacher et al. 2002; Bialik and Kimchi 2004). However, molecular mechanisms about how DAPK mediates neuronal death are still unclear. Findings from the present study demonstrated that DAPK was activated during OGD and transfection of DAPK shRNA reduced neuronal death caused by OGD exposure. Findings also revealed that BimEL was a downstream effector of DAPK in its death promoting process. Further analysis indicated that DAPK regulated BimEL through inactivation of ERK1/2 and activation of JNK1/2. Thus, these findings revealed novel molecular pathways mediating neuronal death in response to OGD.
DAPK activation occurred rapidly after cells were exposed to OGD, with increased DAPK and decreased phospho-DAPK detected in cells received 0.5 hour OGD treatment. Although substantial cell death was observed in cells exposed to 4 hour or longer OGD exposure, no marked cell death was observed in cells treated with OGD for 2 hours or less (data not shown), suggesting DAPK activation occurred prior to the neuronal death. DAPK knockdown significantly increased cell survival in the cells treated with 8 or 16 hour OGD, strongly implying that DAPK signaling pathway was indispensible in the cell death during OGD.
A previous study has shown that DAPK induced cell death involves ER stress activation (Gozuacik et al. 2008). During ER stress, DAPK is activated through dephosphorylation at Ser308. Primary fibroblasts from DAPK knockout mice are protected from ER stress induced cell death. DAPK knockout mice are protected from kidney damage caused by ER stress (Gozuacik et al. 2008). In the experimental setting here, exposing the cells to OGD caused ER stress activation, as increased phospho-eIF2α and CHOP levels were detected. The increase of phospho-eIF2α and CHOP expressions were observed later than DAPK activation. Furthermore, DAPK shRNA transfection resulted in decreased phospho-eIF2α and CHOP expressions, suggesting ER stress activation was a downstream event of DAPK signaling upon OGD treatment.
Bim is a pro-apoptotic member of Bcl-2 family and has been found essential for ER stress induced cell death in different types of cells in in vitro or in vivo studies. For example, thymocytes and macrophages from Bim deficient mice are more resistant to ER stress caused cell death (Puthalakath et al. 2007). In the present study, BimEL level was highly increased following OGD treatment. BimS also increased after OGD treatment, but the level of BimS was very low. The basal level of BimL was very low in these cells and it did not change following the OGD treatment (data not shown). Therefore, the subsequent studies focused on the role of BimEL in DAPK mediated neuronal death. Bax is a pro-apoptotic Bcl-2 family member. In some studies, Bax is up-regulated in its protein level during apoptosis, while in others only Bax mitochondrial translocation is found (Lang-Rollin et al. 2005). In the present study, Bax expression was not changed following the OGD exposure. Whether Bax mitochondrial translocation contributed to the OGD induced cell death still needs to be investigated. Although Bcl-2 and Bcl-xl are the Bcl-2 members that protect cells from apoptosis, Bcl-2 and Bcl-xl levels did not change significantly following the OGD exposure. Similar results have also been observed during apoptosis in previous studies (Yang et al. 2000; Ribas et al. 2004), suggesting other mechanisms may be responsible for the cell death. A previous study also supports that Bim and Bcl-2 balance is critical in controlling memory T cell fate. Overexpression of Bim over Bcl-2 leads to more T cells being killed (Wojciechowski et al. 2007). In the present study, BimEL level was increased while Bcl-2 level was unchanged after OGD exposure, suggesting increased BimEL/Bcl-2 ratio may also played a role in the OGD induced neuronal death.
Findings from this study also indicate that DAPK regulated BimEL expression during OGD as DAPK shRNA transfection inhibited OGD induced increase of BimEL. A previous study shows that Bim is found in ER compartment in stressed cells but not in normal cells, indicating translocation of Bim during ER stress (Morishima et al. 2004). To detect whether the increased BimEL expression was associated with ER stress activation during OGD exposure, BimEL localization was examined. As expected, BimEL translocated to ER following OGD treatment, which suggests that BimEL was indeed involved in the ER stress during OGD. These data thus suggest that DAPK caused ER stress by up-regulating BimEL expression during OGD treatment.
ERK1/2 is a member of the mitogen activated protein kinase (MAPK) family and ERK1/2 can be activated by MEK through a cascade of upstream kinases (Wang et al. 2011). Once activated, ERK1/2 phosphorylates its substrates, leading to a number of cell processes including cell proliferation and cell survival. One of its substrates is BimEL. Phosphorylation of BimEL by ERK1/2 results in the ubiquitination and degradation of BimEL (Leung et al. 2008; Weston et al. 2003). Since a previous study reveals an interaction between DAPK and ERK1/2 in 293T cells (Chen et al. 2005), we hypothesized that DAPK regulated BimEL through ERK1/2 during OGD. Data from the present study showed that phospho-ERK1/2 decreased following the OGD treatment, and DAPK shRNA transfection prevented OGD induced decrease of phospho-ERK1/2, indicating that DAPK inhibited ERK1/2 activation during OGD. However, immunoprecipitation result revealed no direct interaction between DAPK and ERK1/2 (data not shown). Although phospho-ERK1/2 decreased following OGD treatment, there was increased BimEL associated with phospho-ERK1/2. Consistent with these results, treatment of cells with OGD also resulted in increased phospho-BimEL level. These findings suggest that phospho-ERK1/2 phosphorylated BimEL which was then designated for degradation, and DAPK inhibited phospho-ERK1/2 expression during OGD.
JNK1/2 is a member of MAPK family and studies have shown that JNK1/2 activation promotes neuronal apoptosis (Xia et al. 1995; Le-Niculescu et al. 1999). A previous study has revealed that DAPK physically interacts with protein kinase D (PKD) in response to oxidative stress, and PKD in turn activates JNK. Knockdown of either DAPK or PKD attenuates JNK activation (Eisenberg-Lerner and Kimchi 2007). To study if JNK was involved in the DAPK-BimEL signaling pathway during OGD treatment, we first examined how DAPK affected JNK1/2 expression. Knockdown of DAPK caused a marked reduction of phospho-JNK1/2, the active form of this enzyme, during OGD exposure, whereas no direct interaction was found between DAPK and JNK1/2 (data not shown). Whether PKD was involved in the DAPK-JNK1/2 signaling pathway during OGD treatment still needs to be investigated. Subsequently, effect of JNK1/2 on BimEL expression was studied. Inhibition of JNK1/2 with a specific inhibitor, SP600125, dramatically prevented BimEL increase during OGD exposure, suggesting that JNK1/2 played a critical role in BimEL up-regulation. These results are consistent with previous observations that JNK induces Bim expression following the treatment with UV irradiation (Ley et al. 2003). Interestingly, although JNK1/2 up-regulates Bim expression in cells treated with OGD or UV irradiation, the signaling pathways are differentially regulated in these two conditions. In the UV irradiation study, JNK up-regulates Bim through ERK activation. In the OGD condition, ERK activation was not affected with the presence of JNK inhibitor, suggesting ERK1/2 was not involved in the JNK1/2-BimEL signaling pathway (Wang et al. 2011). Collectively, these data indicate DAPK activated JNK1/2 which then increased BimEL expression during OGD. Although JNK1/2 inhibitor resulted in much lower BimEL expression, cells with the inhibitor still showed slightly increased BimEL expression following OGD treatment, suggesting some other signaling pathways might also be responsible for the BimEL increase during OGD.
We report here that DAPK signaling pathways play significant roles in OGD induced neuronal death. Exposure of the cells to OGD led to DAPK activation, which then promoted cell death through BimEL overexpression. Furthermore, DAPK induced BimEL overexpression was mediated by inactivating ERK1/2 and activating JNK1/2 signal transduction pathways (Figure 7). Thus, this study revealed new signal transduction pathways responsible for neuronal death during OGD treatment, which should assist our understanding of molecular mechanisms underlying the neuronal death during ischemic injury.
Figure 7.
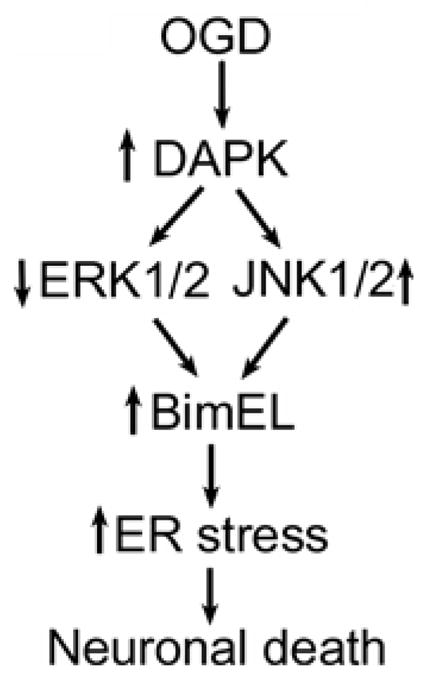
Schematic diagram of DAPK-BimEL signal transduction pathways in OGD induced neuronal death. Exposure of the cells to OGD activates DAPK which in turn increases BimEL expression through inactivation of ERK1/2 and activation of JNK1/2. BimEL over-expression then promotes ER stress and neuronal death.
Inhibition of DAPK reduced cell death induced by oxygen glucose deprivation.
DAPK inhibition decreased BimEL expression during oxygen glucose deprivation.
BimEL translocated to ER after treatment with oxygen glucose deprivation.
DAPK increased BimEL level through inactivation of ERK1/2 and activation of JNK1/2.
Acknowledgments
This work was supported by Kattner and Nita graduate scholarship and National Institutes of Health (R15NS072858).
ABBREVIATIONS
- DAPK
death-associated protein kinase
- OGD
oxygen glucose deprivation
- shRNA
short hairpin RNA
- PI
propidium iodide
- ERK1/2
extracellular signal-regulated protein kinase1/2
- JNK1/2
c-Jun-N-terminal kinase1/2
- ER
Endoplasmic reticulum
- UPR
unfolded protein response
- PERK
protein kinase RNA (PKR)-like ER kinase
- CHOP
C/EBP homologous protein
- DMEM
Dulbecco’s modified Eagle’s medium
- FBS
fetal bovine serum
- IP
Immunoprecipitation
- MAPK
mitogen activated protein kinase
- PKD
protein kinase D
Footnotes
The authors declare no conflict of interest
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bennett BL, Sasaki DT, Murray BW, O’Leary EC, Sakata ST, Xu W, Leisten JC, Motiwala A, Pierce S, Satoh Y, Bhagwat SS, Manning AM, Anderson DW. SP600125, an anthrapyrazolone inhibitor of Jun N-terminal kinase. Proc Natl Acad Sci U S A. 2001;98:13681–6. doi: 10.1073/pnas.251194298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bialik S, Kimchi A. DAP-kinase as a target for drug design in cancer and diseases associated with accelerated cell death. Semin Cancer Biol. 2004;14:283–94. doi: 10.1016/j.semcancer.2004.04.008. [DOI] [PubMed] [Google Scholar]
- Chen CH, Wang WJ, Kuo JC, Tsai HC, Lin JR, Chang ZF, Chen RH. Bidirectional signals transduced by DAPK-ERK interaction promote the apoptotic effect of DAPK. EMBO J. 2005;24:294–304. doi: 10.1038/sj.emboj.7600510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Diego I, Kuper J, Bakalova N, Kursula P, Wilmanns M. Molecular basis of the death-associated protein kinase-calcium/calmodulin regulator complex. Sci Signal. 2010;3:ra6. doi: 10.1126/scisignal.2000552. [DOI] [PubMed] [Google Scholar]
- Dennis SH, Jaafari N, Cimarosti H, Hanley JG, Henley JM, Mellor JR. Oxygen/glucose deprivation induces a reduction in synaptic AMPA receptors on hippocampal CA3 neurons mediated by mGluR1 and adenosine A3 receptors. J Neurosci. 2011;31:11941–52. doi: 10.1523/JNEUROSCI.1183-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan DX, Chai GS, Ni ZF, Hu Y, Luo Y, Cheng XS, Chen NN, Wang JZ, Liu GP. Phosphorylation of tau by death-associated protein kinase 1 antagonizes the kinase-induced cell apoptosis. J Alzheimers Dis. 2013;37:795–808. doi: 10.3233/JAD-130377. [DOI] [PubMed] [Google Scholar]
- Eisenberg-Lerner A, Kimchi A. DAP kinase regulates JNK signaling by binding and activating protein kinase D under oxidative stress. Cell Death Differ. 2007;14:1908–15. doi: 10.1038/sj.cdd.4402212. [DOI] [PubMed] [Google Scholar]
- Ewings KE, Hadfield-Moorhouse K, Wiggins CM, Wickenden JA, Balmanno K, Gilley R, Degenhardt K, White E, Cook SJ. ERK1/2-dependent phosphorylation of BimEL promotes its rapid dissociation from Mcl-1 and Bcl-xL. EMBO J. 2007;26:2856–67. doi: 10.1038/sj.emboj.7601723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilley J, Coffer PJ, Ham J. FOXO transcription factors directly activate bim gene expression and promote apoptosis in sympathetic neurons. J Cell Biol. 2003;162:613–22. doi: 10.1083/jcb.200303026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gozuacik D, Bialik S, Raveh T, Mitou G, Shohat G, Sabanay H, Mizushima N, Yoshimori T, Kimchi A. DAP-kinase is a mediator of endoplasmic reticulum stress-induced caspase activation and autophagic cell death. Cell Death Differ. 2008;15:1875–86. doi: 10.1038/cdd.2008.121. [DOI] [PubMed] [Google Scholar]
- Kaushal V, Schlichter LC. Mechanisms of microglia-mediated neurotoxicity in a new model of the stroke penumbra. J Neurosci. 2008;28(9):2221–30. doi: 10.1523/JNEUROSCI.5643-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim I, Xu W, Reed JC. Cell death and endoplasmic reticulum stress: disease relevance and therapeutic opportunities. Nat Rev Drug Discov. 2008;7:1013–30. doi: 10.1038/nrd2755. [DOI] [PubMed] [Google Scholar]
- Kritis A, Pourzitaki C, Klagas I, Chourdakis M, Albani M. Proteases inhibition assessment on PC12 and NGF treated cells after oxygen and glucose deprivation reveals a distinct role for aspartyl proteases. PloS One. 2011;6:e25950. doi: 10.1371/journal.pone.0025950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar R, Azam S, Sullivan JM, Owen C, Cavener DR, Zhang P, Ron D, Harding HP, Chen JJ, Han A, White BC, Krause GS, DeGracia DJ. Brain ischemia and reperfusion activates the eukaryotic initiation factor 2alpha kinase, PERK. J Neurochem. 2001;77:1418–21. doi: 10.1046/j.1471-4159.2001.00387.x. [DOI] [PubMed] [Google Scholar]
- Lang-Rollin I, Maniati M, Jabado O, Vekrellis K, Papantonis S, Rideout HJ, Stefanis L. Apoptosis and the conformational change of Bax induced by proteasomal inhibition of PC12 cells are inhibited by bcl-xL and bcl-2. Apoptosis. 2005;10:809–20. doi: 10.1007/s10495-005-0378-5. [DOI] [PubMed] [Google Scholar]
- Le-Niculescu H, Bonfoco E, Kasuya Y, Claret FX, Green DR, Karin M. Withdrawal of survival factors results in activation of the JNK pathway in neuronal cells leading to Fas ligand induction and cell death. Mol Cell Biol. 1999;19:751–63. doi: 10.1128/mcb.19.1.751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung KT, Li KK, Sun SS, Chan PK, Ooi VE, Chiu LC. Activation of the JNK pathway promotes phosphorylation and degradation of BimEL--a novel mechanism of chemoresistance in T-cell acute lymphoblastic leukemia. Carcinogenesis. 2008;29:544–51. doi: 10.1093/carcin/bgm294. [DOI] [PubMed] [Google Scholar]
- Ley R, Balmanno K, Hadfield K, Weston C, Cook SJ. Activation of the ERK1/2 signaling pathway promotes phosphorylation and proteasome-dependent degradation of the BH3-only protein, Bim. J Biol Chem. 2003;278:18811–6. doi: 10.1074/jbc.M301010200. [DOI] [PubMed] [Google Scholar]
- Morishima N, Nakanishi K, Tsuchiya K, Shibata T, Seiwa E. Translocation of Bim to the endoplasmic reticulum (ER) mediates ER stress signaling for activation of caspase-12 during ER stress-induced apoptosis. J Biol Chem. 2004;279:50375–81. doi: 10.1074/jbc.M408493200. [DOI] [PubMed] [Google Scholar]
- O’Connor L, Strasser A, O’Reilly LA, Hausmann G, Adams JM, Cory S, Huang DC. Bim: a novel member of the Bcl-2 family that promotes apoptosis. EMBO J. 1998;17:384–95. doi: 10.1093/emboj/17.2.384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelled D, Raveh T, Riebeling C, Fridkin M, Berissi H, Futerman AH, Kimchi A. Death-associated protein (DAP) kinase plays a central role in ceramide-induced apoptosis in cultured hippocampal neurons. J Biol Chem. 2002;277:1957–61. doi: 10.1074/jbc.M104677200. [DOI] [PubMed] [Google Scholar]
- Puthalakath H, O’Reilly LA, Gunn P, Lee L, Kelly PN, Huntington ND, Hughes PD, Michalak EM, McKimm-Breschkin J, Motoyama N, Gotoh T, Akira S, Bouillet P, Strasser A. ER stress triggers apoptosis by activating BH3-only protein Bim. Cell. 2007;129:1337–49. doi: 10.1016/j.cell.2007.04.027. [DOI] [PubMed] [Google Scholar]
- Ribas J, Boix J. Cell differentiation, caspase inhibition, and macromolecular synthesis blockage, but not BCL-2 or BCL-XL proteins, protect SH-SY5Y cells from apoptosis triggered by two CDK inhibitory drugs. Exp Cell Res. 2004;295:9–24. doi: 10.1016/j.yexcr.2003.12.019. [DOI] [PubMed] [Google Scholar]
- Schumacher AM, Velentza AV, Watterson DM, Wainwright MS. DAPK catalytic activity in the hippocampus increases during the recovery phase in an animal model of brain hypoxic-ischemic injury. Biochim Biophys Acta. 2002;1600:128–37. doi: 10.1016/s1570-9639(02)00453-3. [DOI] [PubMed] [Google Scholar]
- Shamloo M, Soriano L, Wieloch T, Nikolich K, Urfer R, Oksenberg D. Death-associated protein kinase is activated by dephosphorylation in response to cerebral ischemia. J Biol Chem. 2005;280:42290–9. doi: 10.1074/jbc.M505804200. [DOI] [PubMed] [Google Scholar]
- Song JH, Wang CX, Song DK, Wang P, Shuaib A, Hao C. Interferon gamma induces neurite outgrowth by up-regulation of p35 neuron-specific cyclin-dependent kinase 5 activator via activation of ERK1/2 pathway. J Biol Chem. 2005;280:12896–901. doi: 10.1074/jbc.M412139200. [DOI] [PubMed] [Google Scholar]
- Szegezdi E, Logue SE, Gorman AM, Samali A. Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep. 2006;7:880–5. doi: 10.1038/sj.embor.7400779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabas I, Ron D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat Cell Biol. 2002;13:184–90. doi: 10.1038/ncb0311-184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu W, Xu X, Peng L, Zhong X, Zhang W, Soundarapandian MM, Balel C, Wang M, Jia N, Zhang W, Lew F, Chan SL, Chen Y, Lu Y. DAPK1 interaction with NMDA receptor NR2B subunits mediates brain damage in stroke. Cell. 2010;140:222–34. doi: 10.1016/j.cell.2009.12.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang CX, Song JH, Song DK, Yong VW, Shuaib A, Hao C. Cyclin-dependent kinase-5 prevents neuronal apoptosis through ERK-mediated upregulation of Bcl-2. Cell Death Differ. 2006;13:1203–12. doi: 10.1038/sj.cdd.4401804. [DOI] [PubMed] [Google Scholar]
- Wang J, Zhou JY, Wu GS. Bim protein degradation contributes to cisplatin resistance. J Biol Chem. 2011;286:22384–92. doi: 10.1074/jbc.M111.239566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Chen WR, Xing D. A pathway from JNK through decreased ERK and Akt activities for FOXO3a nuclear translocation in response to UV irradiation. J Cell Physiol. 2012 Mar;227(3):1168–78. doi: 10.1002/jcp.22839. [DOI] [PubMed] [Google Scholar]
- Weston CR, Balmanno K, Chalmers C, Hadfield K, Molton SA, Ley R, Wagner EF, Cook SJ. Activation of ERK1/2 by deltaRaf-1:ER* represses Bim expression independently of the JNK or PI3K pathways. Oncogene. 2003;22:1281–93. doi: 10.1038/sj.onc.1206261. [DOI] [PubMed] [Google Scholar]
- Wojciechowski S, Tripathi P, Bourdeau T, Acero L, Grimes HL, Katz JD, Finkelman FD, Hildeman DA. Bim/Bcl-2 balance is critical for maintaining naive and memory T cell homeostasis. J Exp Med. 2007;204:1665–75. doi: 10.1084/jem.20070618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia Z, Dickens M, Raingeaud J, Davis RJ, Greenberg ME. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science. 1995;270:1326–31. doi: 10.1126/science.270.5240.1326. [DOI] [PubMed] [Google Scholar]
- Yamamoto M, Hioki T, Ishii T, Nakajima-Iijima S, Uchino S. DAP kinase activity is critical for C(2)-ceramide-induced apoptosis in PC12 cells. Eur J Biochem. 2002;269:139–47. doi: 10.1046/j.0014-2956.2002.00029.x. [DOI] [PubMed] [Google Scholar]
- Yang HL, Dong YB, Elliott MJ, Liu TJ, McMasters KM. Caspase activation and changes in Bcl-2 family member protein expression associated with E2F-1-mediated apoptosis in human esophageal cancer cells. Clin Cancer Res. 2000;6:1579–89. [PubMed] [Google Scholar]