

Abstract
14-3-3ζ is overexpressed in over 40% of breast cancers but its pathophysiological relevance to tumorigenesis has not been established. Here we show that 14-3-3ζ overexpression is sufficient to induce tumorigenesis in a transgenic mouse model of breast cancer. MMTV-LTR promoter driven HA-14-3-3ζ transgenic mice (MMTV-HA-14-3-3ζ) developed mammary tumors whereas control mice did not. Whey acidic protein promoter driven HA-14-3-3ζ transgenic mice (WAP-HA-14-3-3ζ) developed hyperplastic lesions and showed increased susceptibility to carcinogen-induced tumorigenesis. When crossed with MMTV-neu transgenic mice, 14-3-3ζ.neu transgenic mice exhibited accelerated mammary tumorigenesis and metastasis compared to MMTV-neu mice. Mechanistically, 14-3-3ζ overexpression enhanced MAPK/c-Jun signaling leading to increased miR-221 transcription, which inhibited p27 CDKI translation, and consequently, promoted cell proliferation. Importantly, this 14-3-3ζ/miR-221/p27/proliferation axis is also functioning in patients' breast tumors and associates with high grade cancers. Taken together, our findings show that 14-3-3ζ overexpression has a causal role in mammary tumorigenesis and progression, acting through miR-221 in cooperation with known oncogenic events to drive neoplastic cell proliferation.
Keywords: Breast cancer, 14-3-3ζ, microRNA, transgenic mice
Introduction
The 14-3-3 proteins comprise a family of highly conserved dimeric proteins in eukaryotic organisms and have seven isoforms in humans (1). They play a pivotal role in signal transduction as they interact with target proteins via phospho-serine/threonine-containing motifs and regulate proteins involved in diverse cellular processes (1-3). Importantly, overexpression of the 14-3-3ζ isoform is an early event in several types of cancers, including more than 40% of breast cancers (4, 5). 14-3-3ζ protein expression is dramatically increased at early stages of breast disease, the atypical ductal hyperplasia (ADH) and ductal carcinoma in situ (DCIS) stages (4). It is also associated with poor prognosis, metastatic disease recurrence and poor patient survival in breast and other cancers (4, 6). Furthermore, 14-3-3ζ cooperates with ErbB2 overexpression to promote the lethal transition of non-invasive DCIS to invasive ductal carcinoma (IDC) (7). These findings suggest that 14-3-3ζ plays a critical role in breast cancer initiation and progression.
In vitro studies revealed that 14-3-3ζ is a key regulator of multiple oncogenic signaling networks. For example, 14-3-3ζ overexpression promoted epithelial to mesenchymal transition (EMT) in non-transformed mammary epithelial cells (MECs) and severely disrupted acinar architecture of MECs grown in 3D culture, partly due to apoptotic resistance via p53 downregulation (8). In cancer cells, 14-3-3ζ enhanced Akt phosphorylation through phosphoinositide 3-kinase (PI3K) activation, conferring anchorage independent growth and apoptosis resistance, thus promoting cell survival (9, 10).
p27, a cyclin dependent kinase inhibitor, impedes cell cycle progression and maintains cells in the resting state by regulating the G1/S phase checkpoint. p27 knockout mice manifested multi-organ hyperplasia, pituitary tumors, and increased body size, suggesting that p27 plays a vital role in regulating cell growth (11). Low or absent p27 protein expression is reported in various tumors, including breast, and p27 expression is regulated through multiple mechanisms including microRNAs (miRNAs) (12-16). By binding to the 3’-untranslated region (3’UTR) of protein coding mRNAs, miRNAs control gene expression post-transcriptionally resulting in translation inhibition and, in some cases, transcript destabilization and degradation (17, 18). MiRNAs regulate many cellular processes and their aberrant expression has been observed in every disease studied to date (19).
While studies in patient tumor samples and cell lines imply a correlation between 14-3-3ζ and tumorigenicity, no direct in vivo evidence of 14-3-3ζ in promoting mammary tumorigenesis and metastasis has been presented to date. Here, we report the generation of two mammary gland-directed 14-3-3ζ transgenic mouse models and demonstrate for the first time, to our knowledge, that 14-3-3ζ plays a causal role in tumorigenesis. Mammary gland-directed 14-3-3ζ overexpressing transgenic mice had late onset of mammary tumors and 14-3-3ζ overexpression accelerated tumorigenesis induced by chemical carcinogens and other oncogenes. In addition to apoptosis inhibition, 14-3-3ζ overexpression enhanced tumor cell proliferation partly via the c-Jun/miR-221/p27 signaling axis. Human breast cancer tissue microarray analysis corroborated our in vivo findings. In addition, combining the expression information of 14-3-3ζ, miR-221, p27 and Ki-67 enabled better prediction of the tumor grade and prognosis compared to Ki-67 alone. Our findings establish 14-3-3ζ overexpression as an initiating and promoting event in breast cancer through deregulation of an integrative signaling network.
Material and Methods
Transgenic Mice
Generation of 14-3-3ζ transgenic mice explained in detail in Supplementary Materials and Methods. MMTV-neu (MMTV-neu-NDL) transgenic mice, previously described (20).
Pituitary Isografting and Carcinogen Treatment
Mice for DMBA (Sigma) treatment (WAP-HA-14-3-3ζ or nontransgenic B6D2 controls) were given a pituitary isograft for continuous hormonal stimulation of WAP-HA-14-3-3ζ expression (21). One 6-26-week-old donor pituitary gland was implanted under the kidney capsule of each mouse (5-week-old). Two DMBA doses were given weekly by gavage (0.5mg/dose dissolved in 0.1ml cottonseed oil) for 2 weeks beginning at 8 weeks of age.
Histological analysis
Immunohistochemistry (IHC) was performed as previously described (4, 7). For IHC analysis – expression levels were semi-quantified using immunoreactive scores (IRS range 0-12) (9). Score definitions: 0 (score 0-3), 1+ (score 4-6), 2+ (score 7-9), 3+ (score 10-12). 10 fields were randomly chosen and the average percentage of positive cells determined. Angiogenesis was evaluated by CD34 IHC and counting blood vessels in three areas of the section at 200x magnification, expressed as mean vessel number. In situ hybridization (ISH) was performed using miRNA-221 probe (Exiqon, miRCURY LNA 5′-DIG-labelled) and detected as previously described (22).
Tissue microarray (TMA)
Breast cancer TMA, BR208 (US Biomax Inc.) contained normal, premalignant and malignant breast tissues (total 208 cores). 18 cores were omitted from final statistical analysis due to lost cores during staining procedure.
Cell culture
Cell lines were purchased from American Type Culture Collection. Cell lines were authenticated using short tandem repeat profiling by MD Anderson Cancer Center (MDACC; Houston, TX) Cell Line Characterization Core Facility.
Statistical Analyses
Programs used: GraphPad Prism5, IBM SPSS Statistics19 and GNU-R. Tumor latency (time to mammary tumor onset, tumor-free survival) was measured to the date that first mammary tumor was palpable (at approximately 1mm diameter) and was analyzed using the Kaplan-Meier method. Survival differences were determined using log-rank test. Ordered logistic regression models were built and compared using marker combinations of the 14-3-3ζ/miR-221/p27/Ki-67 axis, or the linear combination of all markers to test association with deviance in tumor grades (23). Support Vector Machine (SVM) model was established as previously described (24). 14-3-3ζ, miR-221, p27 and Ki-67 expression levels were used as features, and tumor grade was used as the prediction target variable (normal tissue: Grade 0; tumor tissues: Grade 1, Grade 2 or Grade 3). All data: Bars – SD, *, P < 0.05, **, P < 0.01, ***, P<0.001.
Results
Generation and characterization of WAP-HA-14-3-3ζ and MMTV-HA-14-3-3ζ transgenic mice
To study the role of 14-3-3ζ overexpression in mammary tumorigenesis, we generated transgenic mice with mammary gland-directed HA-tagged 14-3-3ζ gene expression. In the B6D2 background, transgene expression was driven by either the rat WAP (rWAP) or the mouse WAP (mWAP) promoter, noted as WAP-HA-14-3-3ζ (Figure 1A). Founder mice were identified by PCR (Figure S1A). Varying levels of 14-3-3ζ transgene expression were detected in the different WAP-driven lines by immunohistochemical (IHC) staining and immunoblotting (IB) during lactation (Figures 1B, S1B). Transgene expression was specific to mammary gland epithelial cells and was not different between WAP promoter species. Higher HA-14-3-3ζ expressing transgenic strains (m26 and r10) were expanded for further investigation.
Figure 1. Generation of 14-3-3ζ Transgenic Mice and Analysis of Mammary Tumorigenesis.
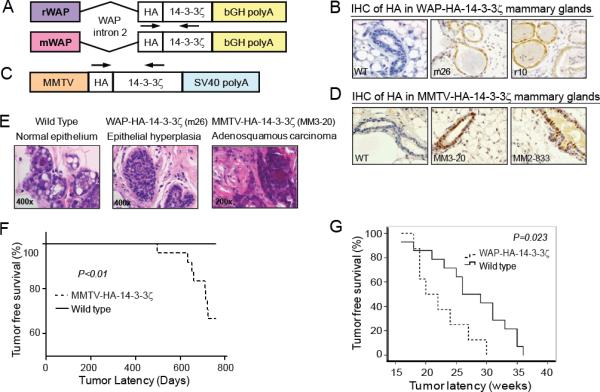
A, C, Schematic of the pWAP-HA-14-3-3ζ and pMMTV-HA-14-3-3ζ constructs. Arrows – genotyping primers locations. B, D, HA-14-3-3ζ protein IHC in lactating WAP-HA-14-3-3ζ and MMTV-HA-14-3-3ζ transgenic mice mammary glands (X400). E, H&E of lesions in multiparous wildtype and WAP-HA-14-3-3ζ mice (X400, both at 24 months of age) and MMTV-HA-14-3-3ζ tumor histology (X200, at 711 days old). F, MMTV-HA-14-3-3ζ mice (n=24) mammary tumor onset latency is shorter than wild-type mice (n=20). G, WAP-HA-14-3-3ζ mice (n=8) mammary tumor onset latency is shorter than wild-type (n=14) littermates following DMBA treatment.
WAP-driven transgenes are expressed in the mammary epithelium during mid-pregnancy through lactation (25). This limits studying the effects of 14-3-3ζ in other developmental stages of the mammary gland. MMTV-LTR promoter drives expression of heterologous genes primarily in the mammary epithelium starting during puberty and continuing throughout mammary gland development (26). Therefore, we generated MMTV–LTR-driven HA-tagged 14-3-3ζ transgenic mice in FVB background (Figure 1C). Founder mice were identified by PCR (Figure S1C) and 14-3-3ζ transgene expression in the mammary gland was detected by IHC and IB (Figures 1D, S1D). Higher HA-14-3-3ζ expressing transgenic strains (MM3-20 and MM2-833) were expanded for further investigation.
14-3-3ζ overexpression results in late-onset mammary tumors
To study the potential effect of 14-3-3ζ overexpression on mammary tumorigenesis, multiparous (3-4 pregnancies) wild-type, WAP-HA-14-3-3ζ and MMTV-HA-14-3-3ζ female transgenic mice were monitored for tumor development and subjected to histopathological analysis. As expected, wild-type mice had normal breast epithelium histology (Figure 1E). While no mammary tumors were detected in the multiparous WAP-HA-14-3-3ζ mice, epithelial hyperplastic lesions with the potential to develop into tumors were identified (Figure 1E). Of the 24 multiparous female MMTV-HA-14-3-3ζ mice monitored for tumor incidence (12 mice each from the MM3-20 and MM2-833 lines), 8 mice (33.3%) developed mammary tumors with a mean tumor latency of 662 days and demonstrated typical adenosquamous carcinoma characteristics (Supplementary Table 1, Figure 1E). MMTV-HA-14-3-3ζ mice exhibited significantly decreased tumor free survival compared to wild-type mice (Figure 1F). Transgene expression in mammary tumors of MMTV-HA-14-3-3ζ mice was confirmed by IB (Figure S1E). The tumor incidence was 41.7% for the MM3-20 line with higher 14-3-3ζ expression and 25% in the MM2-833 transgenic mice with lower 14-3-3ζ expression (Supplementary Table 2), suggesting that tumor development was related to HA-14-3-3ζ transgene expression level. Mammary tumors were of an invasive nature and lung metastasis was found in one tumor-bearing MMTV-HA-14-3-3ζ mouse at 711 days of age (Supplementary Table 2). Neither mammary tumors nor mammary intraepithelial neoplasia (MIN) were observed in a cohort of 20 FVB/N wild-type mice. Together, these data demonstrated that MMTV-driven 14-3-3ζ overexpression in FVB mice can induce late-onset mammary tumors.
14-3-3ζ increases susceptibility to carcinogen-induced mammary tumorigenesis
Since multiparous WAP-HA-14-3-3ζ transgenic mice did not develop tumors, we tested if the ectopic 14-3-3ζ expression promoted carcinogen-induced mammary tumorigenesis. After receiving pituitary isografts from donor mice to induce WAP promoter-driven transgene expression, WAP-HA-14-3-3ζ and B6D2 non-transgenic female littermates were treated with the chemical carcinogen 7,12 dimethylbenzanthracene (DMBA) and monitored for tumor formation. Resulting tumors were analyzed for transgene expression by IB and IHC (Figures S1F, S1G). WAP-HA-14-3-3ζ females developed mammary tumors with a significantly (P=0.023) shorter median tumor latency (20 weeks) than non-transgenic controls (26 weeks) (Figure 1G). Hence, mammary gland specific 14-3-3ζ overexpression enhanced susceptibility to carcinogen-induced tumorigenesis.
14-3-3ζ accelerates ErbB2/Neu-mediated mammary tumor development and progression
ErbB2 (Neu/HER2) is overexpressed in 25-30% of human breast carcinomas resulting in poor patient survival (27). To investigate whether 14-3-3ζ cooperates with oncogenes, such as neu, to accelerate mammary tumorigenesis in vivo, we backcrossed WAP-HA-14-3-3ζ mice (m26 line) into the FVB/N background and then crossed with MMTV-neu transgenic mice, which ectopically overexpress neu in the mammary glands and develops tumors with well-characterized incidence and latency (20). Having both strains in the FVB background ensures optimal MMTV-neu transgene expression and allows comparisons between non-siblings. Since WAP-driven transgene expression depends on hormonal stimulation of pregnancy and lactation, WAP-ζ.neu female mice and MMTV-neu controls were continuously mated to induce and maintain WAP-HA-14-3-3ζ expression. Multiparous WAP-ζ.neu mice developed mammary tumors with a significantly (P=0.018) shorter median tumor latency compared to multiparous MMTV-neu mice (137 versus 160 days) (Figure 2A).
Figure 2. 14-3-3ζ accelerated Neu-mediated mammary tumorigenesis and metastasis.
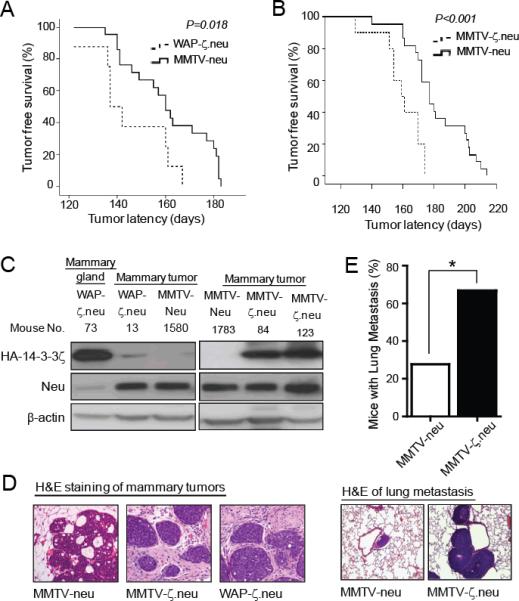
A, Multiparous WAP-ζ.neu mice (n=8) had a shorter mammary tumor onset latency compared to multiparous MMTV-neu mice (n=21). B, Virgin MMTV-ζ.neu mice (n=9) had a shorter mammary tumor onset latency compared to virgin MMTV-neu mice (n=22). C, Neu and HA-14-3-3ζ protein IB in mammary glands and tumors from WAP-ζ.neu, MMTV-ζ.neu and MMTV-neu mice. D, H&E of mammary tumors from MMTV-neu (DCIS), MMTV-ζ.neu and WAP-ζ.neu (invasive) mice (X100). E, Incidence of lung metastases in MMTV-ζ.neu (6/9) mice is higher than MMTV-neu (5/22) mice. Lung metastases H&E (X200).
We also crossed MMTV-HA-14-3-3ζ (MM3-20 line) with MMTV-neu mice to generate MMTV-ζ.neu mice and compared their tumorigenicity with MMTV-neu mice. Both strains of nulliparous mice had 100% mammary tumor incidence. MMTV-ζ.neu bitransgenic female mice exhibited significantly (P < 0.001) shorter median tumor latency compared to MMTV-neu transgenic mice (161 versus 182 days) (Figure 2B). In mammary tumors of WAP-ζ.neu mice, weak HA-14-3-3ζ transgene expression was detected by IB (Figure 2C, left). After tumor onset, it was difficult to keep the tumor-bearing WAP-ζ.neu bitransgenic female mice lactating, which explains the weak transgene expression. To determine whether the HA-14-3-3ζ transgene was expressed during tumor initiation, we collected lactating mammary glands one month before expected tumor development and found that HA-14-3-3ζ was expressed, contributing to the accelerated mammary tumorigenesis (Figure 2C, left, mouse no. 73). Transgene expression was readily detectable in mammary tumors of MMTV-ζ.neu bitransgenic mice (Figure 2C, right). Histological assessment revealed that mammary tumors from WAP-ζ.neu and MMTV-ζ.neu mice were more invasive compared to tumors from MMTV-neu mice (Figure 2D), characterized by tumor foci that invaded into the interlobular stroma with loss of the myoepithelial cell layer.
Given the invasive morphology of mammary tumors from bitransgenic mice, we assessed whether 14-3-3ζ overexpression enhanced neu-induced metastasis. Virgin MMTV-ζ.neu mice had a significantly (P< 0.05) increased incidence of lung metastases (66.7%) with high HA-14-3-3ζ expression compared to MMTV-neu mice (27.7%), 3-4 weeks after palpable mammary tumor detection (Figures 2E, S2A). Breast carcinomas require angiogenesis to sustain their growth and develop metastasis (28). Surely, increased angiogenesis in MMTV-ζ.neu mammary tumors, indicated by increased CD34 expression marker and blood vessel index, correlated with the increased metastases in MMTV-ζ.neu mice compared to MMTV-neu (Figure S2B). Increased angiogenesis results from production of pro-angiogenic factors, such as vascular endothelial growth factor (VEGF) (29). We had identified that 14-3-3ζ overexpression increases Akt activity (Figure S3C), resulting in p53 loss (Figure S3E) and increased VEGF transcription (8). Indeed, MMTV-ζ.neu mice tumors had a significant (P<0.001) increase in VEGF expression than MMTV-neu mice tumors (Figure S2C). Furthermore, the EMT phenotype acquisition is critical for 14-3-3ζ overexpressing MECs to gain invasive ability for subsequent metastases (7). Consistently, IHC and IB analysis showed a significant loss of E-cadherin expression (epithelial marker) and an increase in N-cadherin expression (mesenchymal marker) and TGFβR1 in primary tumors of MMTV-ζ.neu mice (Figure S2D, S2E). In tumors of MMTV-ζ.neu mice, 14-3-3ζ-mediated increase in angiogenesis, inhibition of apoptosis and EMT facilitates accelerated mammary tumor development and progression.
14-3-3ζ accelerates tumor proliferation via microRNA-221-mediated p27 down-regulation
Apoptosis inhibition and increased proliferation of tumor cells are two hallmarks of cancer (30). Apoptotic cell analysis revealed that MIN lesions and mammary tumors of bitransgenic mice had significantly (P<0.001) reduced TUNEL positive staining compared to those of MMTV-neu mice (Figures S3A, S3B). Mechanistically, 14-3-3ζ overexpression in the bitransgenic lesions led to increased Akt activity (Figure S3C), that enhanced FKHRL1 (Foxo3A) phosphorylation. Bound by 14-3-3ζ, phospho-FKHRL1 is retained in the cytosol and inhibited from transcribing pro-apoptotic genes (Figure S3D). Furthermore, decreased p53 expression in 14-3-3ζ overexpressing bitransgenic lesions contributes to inhibition of apoptosis (Figure S3E). These findings demonstrate the anti-apoptotic role of 14-3-3ζ as previously reported (8-10).
Interestingly, proliferating cell analysis showed that MIN lesions and mammary tumors from both bitransgenic mice strains had a significant (P<0.001) increase in Ki-67 staining compared to those from MMTV-neu mice (Figures 3A, 3B). Moreover, mammary tumor cells of MMTV-HA-14-3-3ζ mice showed increased proliferation compared to mammary epithelial cells from age-matched wild type mice (Figure S4A). The data indicates that 14-3-3ζ overexpression promotes cell proliferation in vivo.
Figure 3. 14-3-3ζ enhances tumor cell proliferation.
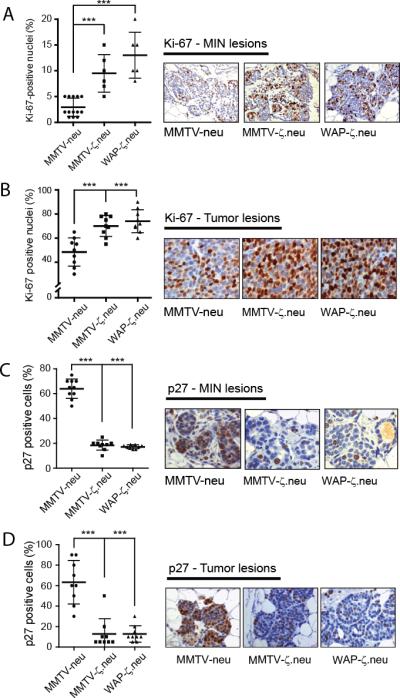
Age matched MMTV-neu, MMTV-ζ.neu and WAP-ζ.neu mice. A,B, Quantitative analysis of Ki-67-positive nuclei (left) and representative Ki-67 IHC (right) showed significant increase in Ki-67 positive staining in bitransgenic MIN lesions (X200, n=13,6,6) and tumors (X400, all n=9) compared to MMTV-neu. C,D, Quantitative analysis of p27-positive nuclei (left) and representative p27 IHC (right, X200) showed significant decrease in p27 positive staining in bitransgenic MIN lesions and tumors compared to MMTV-neu (all n=9).
To decipher the mechanism of 14-3-3ζ-mediated proliferation, we examined expression of cell cycle regulators. While only a moderate decrease in p21 expression was detected (Figures S4B, S4C), p27 expression was significantly (P< 0.001) decreased in MIN lesions and mammary tumors of bitransgenic mice relative to MMTV-neu mice (Figures 3C, 3D). Since low p27 expression has been associated with aggressive tumor behavior (15), it is conceivable that p27 down-regulation contributes to 14-3-3ζ-mediated tumorigenesis.
Expression of p27 is subjected to transcriptional and post-transcriptional regulation (31-33). We compared p27 protein and mRNA levels in MMTV-ζ.neu and MMTV-neu tumors. A notable decrease in p27 protein expression was detected in MMTV-ζ.neu tumors compared to MMTV-neu tumors with no significant (P>0.05) change in p27 mRNA expression (Figures 4A, S5A). To decipher mechanisms of 14-3-3ζ-induced p27 downregulation in breast cancer cells, we modulated 14-3-3ζ expression in two human breast cancer cell lines, MCF7 and DCIS.com, and a murine mammary tumor cell line, McNeuA. Stable HA-14-3-3ζ overexpression in these cell lines (MCF7-ζ, DCIS.com-ζ and McNeuA-ζ) led to decreased p27 protein expression while p27 mRNA expression remained unchanged (Figures 4B, S5B). Conversely, 14-3-3ζ knockdown by two independent shRNAs in MCF7 cells increased p27 protein expression with no impact on p27 mRNA expression (Figures 4C, S5C). Together, the results indicated an intriguing 14-3-3ζ-mediated post-transcriptional repression of p27.
Figure 4. p27 Protein Expression is Post-transcriptionally Regulated by miR-221.
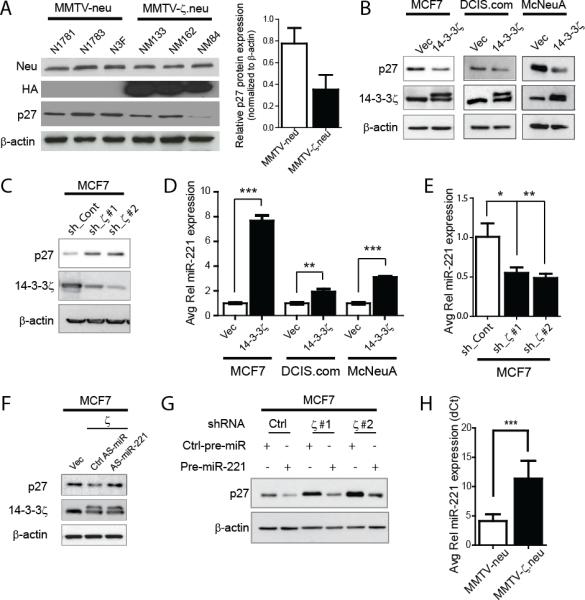
A, Representative p27 IB (left) shows significant p27 protein expression decrease in MMTV-ζ.neu tumors compared to MMTV-neu. Protein quantification is relative to β-actin (right). B, 14-3-3-ζ overexpressing cells showed significant decrease in p27 protein by IB. C, 14-3-3ζ knockdown in MCF7 cells rescued p27 protein expression by IB. D, miR-221 expression was increased in 14-3-3-ζ overexpressing cells versus control cells by qRT-PCR. E, 14-3-3ζ knockdown in MCF7 cells led to decreased miR-221 expression analyzed by qRT-PCR. F, AntagomiR-221 rescued p27 protein expression in MCF7-ζ cells by IB. G, Pre-miR-221 overexpression decreased p27 protein in MCF7 cells with 14-3-3ζ knockdown. H, qRT-PCR shows significant increase in miR-221 in MMTV-ζ.neu mammary tumors compared to MMTV-neu (all n=5).
Recently, it was reported that p27 could be regulated by miRNAs, miR-221, miR-222 and miR-181a (16, 34). We analyzed the expression of these miRNAs by qRT-PCR in MCF7-ζ, DCIS.com-ζ and McNeuA-ζ cells and found a consistent increase in miR-221 expression with 14-3-3ζ overexpression (Figure 4D). MiR-222 was not consistently increased and miR-181a remained unchanged (Figure S5D). Conversely, 14-3-3ζ knockdown in MCF7 cells resulted in a significant miR-221 expression decrease (Figure 4E) affirming that miR-221 expression is regulated by 14-3-3ζ.
To investigate whether miR-221 is critical in mediating 14-3-3ζ-induced p27 protein down-regulation, we examined p27 protein expression upon miR-221 modulating. MiR-221 knockdown in MCF7-ζ cells by antagomiR (AS-miR-221) rescued p27 protein expression (Figures 4F, S5E). Ectopic miR-221 expression by transfecting precursor miRNA-221 (pre-miR-221) into MCF7 or MCF7.shζ cells, resulted in a dramatic downregulation of p27 protein (Figure 4G). In vivo, miR-221 expression was significantly upregulated in mammary tumors of DMBA-treated WAP-HA-14-3-3ζ mice compared to those of wildtype mice (Figure S5F). Consistently, miR-221 expression, but not miR-222 and miR-181a, was increased in MMTV-ζ.neu tumors compared to MMTV-neu tumors by qRT-PCR (Figures 4H, S5G, S5H). Indeed, in 14-3-3ζ overexpressing mammary tumors, increased miR-221 expression by in situ hybridization (ISH) correlated with reduced p27 expression and increased Ki67 expression (Figure 5G). These data indicate that 14-3-3ζ-overexpression promotes tumor proliferation partly through miR-221-mediated p27 down-regulation.
Figure 5. MiRNA-221 expression is transcriptionally upregulated by 14-3-3ζ-mediated c-Jun activation.

A, Pri-miR-221, pre-miR-221 and mature miR-221 qRT-PCR analysis in MCF7-ζ versus control cells. B, Pri- and mature miR-221 qRT-PCR in MCF7 cells with 14-3-3ζ knockdown. C, IB analysis revealed increased JNK signaling and c-Jun expression in 14-3-3-ζ overexpressing cells versus control cells. D, Nucleo-cytoplasmic fractionation IB showed increased nuclear c-Jun localization in 14-3-3-ζ overexpressing cells. E, qRT-PCR analysis showed a decrease in pri-miR-221 expression with c-Jun knockdown (IB inset) in MCF7-ζ cells. F, ChIP analysis showed increased c-Jun binding to miR-221 promoter in MCF7-ζ cells versus control cells. G, Representative IHC of HA-14-3-3ζ, phospho-JNK, phospho-c-Jun, total-c-Jun, p27, Ki-67 and miR-221 ISH in MMTV-neu and MMTV-ζ.neu tumors (X400).
14-3-3ζ upregulates microRNA-221 transcription via c-Jun
Several studies reported that miR-221 is transcriptionally regulated (35-37). To elucidate whether and how miR-221 transcription is regulated by 14-3-3ζ, we analyzed the expression of the primary transcripts (pri-miR-221), the intermediate product (pre-miR-221) and the mature miR-221 in MCF7-ζ cells compared to control cells. MCF7-ζ cells had significantly increased pri-miR-221 expression, consistent with increased mature-miR-221 expression (Figure 5A), suggesting that 14-3-3ζ transcriptionally upregulates miR-221. Conversely, pri-miR-221 expression was significantly decreased in MCF7.shζ cells compared to control (Figure 5B).
MiR-221 is known to be independently transcribed and is not a by-product of host-gene transcription (37). To identify the transcription factors responsible for miR-221 transcription in 14-3-3ζ overexpressing cells, we examined expression of predicted miR-221 promoter-binding transcription factors (NF-κB and c-Jun) in MCF7-ζ, DCIS.com-ζ and McNeuA-ζ cells. No significant change in NF-κB activation was detected, making it an unlikely mediator of miR-221 transcription (Figure S6A). Interestingly, 14-3-3ζ overexpression lead to an increase in phospho-JNK levels leading to enhanced c-Jun phosphorylation (which stabilizes c-Jun) and nuclear localization compared to control cells (Figures 5C, 5D). c-Jun knockdown in MCF7-ζ cells resulted in pri-miR-221 expression decrease (Figure 5E), indicating that the JNK/c-Jun pathway is esssential in 14-3-3ζ-induced miR-221 transcriptional upregulation. We performed a chromatin immunoprecipitation (ChIP) assay using c-Jun antibody and found a robust enhancement of c-Jun binding to miR-221 promoter in the MCF7-ζ cells compared to MCF7-Vec cells (Figure 5F). Together, our data revealed that 14-3-3ζ-overexpression leads to miR-221 transcriptional upregulation via c-Jun. 14-3-3ζ overexpression in MMTV-ζ.neu mammary tumors correlated with increased phospho-JNK, phospho-c-Jun, total c-Jun, miR-221 expression, Ki-67 positivity and reduced p27 expression compared to MMTV-neu tumors (Figure 5G). These data indicated that c-Jun contributes to 14-3-3ζ-mediated miR-221 transcriptional upregulation and p27 protein downregulation in vivo.
C-Fos is known to partner with c-Jun in transcriptional regulation (38). Notably, 14-3-3ζ overexpressing cells had different degrees of increased c-Fos phosphorylation and Erk1/2 activation (Figure S6A). Inhibition of Erk1/2 in MCF7-ζ cells by chemical inhibitor (AZD6244) or knockdown of Erk1/2 or c-Fos by siRNAs led to a decrease in pri-miR-221 expression (Figures S6B, S6C, S6D). Hence, the Erk1/2/c-Fos pathway also contributes to miR-221 transcriptional upregulation in 14-3-3ζ-overexpressing breast cancer cells.
14-3-3ζ/miR-221/p27/proliferation axis is functioning in patients’ breast tumors and associates with high grade cancers
We examined whether the 14-3-3ζ/miR-221/p27 axis identified in our mouse models and human breast cancer cell lines is functioning in patients’ breast tumor tissues. Using tissue microarrays (TMA) containing 180 breast cancer tissue cores and 27 normal breast tissues cores, we detected 14-3-3ζ, p27, ErbB2, miR-221 and Ki-67 expression (Figure 6A, Supplementary Tables 3A, 3B, 3C). Notably, 14-3-3ζ overexpression correlated with increased miR-221 expression, validating that 14-3-3ζ overexpression is associated with increased miR-221 expression in human breast cancer tissues (P<0.01, Supplementary Table 3D).
Figure 6. 14-3-3ζ, miR-221, p27 CDKI and Ki-67 expressions in human breast cancer tissues.

A, Representative 14-3-3ζ, p27 and Ki-67 IHC, and miR-221 ISH in human breast cancer tissues versus normal mammary gland (X400). B, Proposed model.
To test the clinical applicability of our findings, we examined whether the expression of the components of the 14-3-3ζ/miR-221/p27axis, individually or together, could predict tumor progression, considering tumor grade as the read-out. Ki-67 is a well characterized molecular marker of tumor cell proliferation, known to be associated with tumor grade and poor prognosis (39). Our TMA tumor cohort revealed that increased miR-221 expression levels significantly correlated with increased percentage of Ki-67 positivity (low = <20% positive, high = >20% positive, P<0.01, Supplementary Table 3D). We asked if combining markers of the 14-3-3ζ/miR-221/p27 axis with Ki-67 would better predict tumor grade compared to Ki-67's predictive power as a sole parameter. Specifically, we tested whether the deviance in tumor grades were associated with 14-3-3ζ, miR-221, p27 expression and percentage of Ki-67 positivity in the TMA cases. We built and compared ordered logistic regression models (23) using single- and double-variable combinations of markers of the 14-3-3ζ/miR-221/p27/Ki-67 axis, or the linear combination of all four markers. Clearly, using all four markers predicted tumor grades with more than 30% of deviance compared to over 10% by Ki-67 alone (P<0.001, Supplementary Figure S7).
To test the possibility of predicting tumor grades in a patient cohort with Ki-67 as well as the expression of the 14-3-3ζ/miR-221/p27 axis, we built a model using Ki-67, 14-3-3ζ, miR-221 and p27 expression levels as features, and tumor grade as the prediction target variable (normal breast tissue – Grade 0; tumor tissues – Grade 1, Grade 2, Grade 3). We first established the SVM model on 80% of the cases (training set) from the TMA and then used the remaining 20% of the cases (test set) to assess the performance of the prediction model. The four-feature model (Ki-67+/14-3-3ζ+/miR-221+/p27 low expression) achieved an accuracy of 75.6% (28/37) to predict the tumor grade, compared to 37.8% (14/37) using only Ki-67 as the input variable or 56.7% (21/37) using only 14-3-3ζ as the input variable (Supplementary Table 4). All single-, double- and triple-variable marker combination analyses showed less accuracy than the four-feature model (Supplementary Table 4). This indicates that the combined marker expression of 14-3-3ζ, miR-221 and p27 with Ki-67 improves the prediction power of tumor grade and, potentially, of tumor prognosis.
Discussion
Transgenic mouse models are invaluable for studying the roles of various potential oncogenes (“oncomice”) in vivo (40). Here, we generated two HA-14-3-3ζ transgenic mouse models and demonstrated that 14-3-3ζ plays a causal role in mammary tumorigenesis. Thus far, the oncogenic nature of 14-3-3ζ protein had only been characterized in cell lines and its role in tumorigenesis had never been tested in a transgenic mouse model. Mammary tumors developed in MMTV-HA-14-3-3ζ transgenic mice but not in wild-type mice, implicating 14-3-3ζ as an onco-protein. Some oncogenes, such as neu, are vigorous and self-sufficient to induce tumor in transgenic mice (20). However, most transgenic mouse models that overexpress oncogenes in their mammary glands develop mammary tumors in a stochastic fashion with long tumor latencies, such as MMTV-c-myc transgenic mice (41, 42). In our studies, 14-3-3ζ presents itself as an oncogene with a long tumor latency. The 14-3-3ζ transgene overexpression and tumor phenotype seen in our mouse models is similar to that seen in human breast cancer patients, signifying the clinical relevance of these mouse models.
Latent oncogenes can cooperate with chemical carcinogens to induce tumorigenesis. In cooperation with a tumor inducing carcinogen (DMBA), 14-3-3ζ accelerated mammary tumorigenesis. Similarly, oncogenes cooperate to achieve a transformed phenotype (43). We generated MMTV-ζ.neu and WAP-ζ.neu bitransgenic mice to investigate the effect of 14-3-3ζ overexpression on neu-induced mammary tumorigenesis. 14-3-3ζ not only accelerated the initiation of neu-induced mammary tumors, but also conferred an invasive histology, indicative of metastases-prone tumors. A higher percentage of MMTV-ζ.neu tumor-bearing mice developed lung metastasis compared to MMTV-neu, indicating that 14-3-3ζ overexpression promoted neu-mediated mammary tumor metastatic progression. Our results corroborate clinical observations that breast cancers overexpressing both ErbB2 and 14-3-3ζ are more aggressive with a higher metastatic potential (7).
The role of 14-3-3ζ in anti-apoptosis is well characterized, conferring survival advantage upon cancerous cells and contributing to tumor progression (30) as seen in 14-3-3ζ overexpressing MIN lesions and mammary tumors. However, 14-3-3ζ overexpression increased tumor cell proliferation in MIN lesions and tumors, indicated by increased Ki-67 staining and p27 protein loss. This is the first report that shows 14-3-3ζ promotes cell proliferation.
The 14-3-3 family of proteins was reported to inhibit p27 nuclear import to prevent G1/S phase cell cycle checkpoint (12). This was not the case in our models because p27 protein expression was exclusively nuclear in 14-3-3ζ overexpressing MIN lesions and tumors (Figure 3C, 3D, 6A). Instead, we identified that 14-3-3ζ promoted proliferation via miRNA-221-mediated p27 post-transcriptional downregulation. MiRNA-221 was transcriptionally regulated by c-Jun, conferring a proliferative advantage in 14-3-3ζ overexpressing tumors. Consistent with previous reports that the 14-3-3ζ-Ras-Raf signaling axis activates the MAPK pathway (44), 14-3-3ζ overexpressing cell lines had different degrees of Erk1/ 2 and c-Fos activation. Thus, c-Fos may cooperate with c-Jun in transcribing miR-221. To our knowledge, this is the first report of miRNA regulation by 14-3-3ζ. While miR-221 upregulation has been associated with tamoxifen and fulvestrant resistance in breast cancers, other reports have implicated 14-3-3ζ in mediating endocrine resistance (36, 45, 46, 47). Our findings suggest that 14-3-3ζ could mediate some effects through miR-221, making our transgenic mouse models ideal for studying therapeutic resistance in vivo.
Patient breast cancer tissue analysis demonstrated that the 14-3-3ζ/miR-221/p27/Ki67 axis we identified in our experimental models is clinically relevant. That 14-3-3ζ and miR-221 correlated positively with Ki-67 and high grade breast cancers supports our findings that 14-3-3ζ overexpression mediates tumor cell proliferation and promotes malignant phenotypes. Furthermore, the combined power of 14-3-3ζ, miR-221, p27 and Ki-67 expression in predicting tumor grade is greater than using single marker or varying combinations of these markers. Combined, these markers could serve as an effective prognostic ‘signature’ to predict clinical outcomes in patients with 14-3-3ζ-overexpressing breast tumors.
We have demonstrated for the first time, that 14-3-3ζ overexpression plays a causal role in mammary tumors with long tumor latency, and cooperates with chemical carcinogens and other oncogenes to accelerate mammary tumorigenesis. 14-3-3ζ overexpression increased tumor cell proliferation via miR-221-mediated p27 downregulation (Figure 6B). Together, increased Ki-67 and miR-221 expression and decreased p27 expression can serve as powerful prognostic markers for 14-3-3ζ overexpressing breast cancer. Overall, our finding of the 14-3-3ζ/miR-221/p27/proliferation axis is a clinically relevant mechanism of 14-3-3ζ-overexpression-mediated breast cancer onset and progression.
Supplementary Material
Acknowledgements
We thank Dr. Dexing Fang for input and help in generating the transgenic mice, Dr. Jan Parker (MDACC GEM Facility) for pro-nuclei injections, the MDACC Core Histology Lab for tissue preparations, Dr. Qingling Wang for her help with microRNA-221 analysis, Ms. Kelli Cook for technical assistance and Mr. David Edwards for teaching the pituitary isografting technique.
Financial support: This work was supported by NIH grants P30-CA 16672 (MDACC), PO1-CA099031 project 4 (DY), RO1-CA112567-06 (DY), DOD Center of Excellence grant subproject W81XWH-06-2-0033 (DY), Susan G. Komen Breast Cancer Foundation Promise Grant KG091020 (DY), Cancer Prevention Research Institute of Texas Grant RP100726 (DY) and DOD Pre-doctoral Fellowship W81XWH-10-1-0260 (SR). S-H.L. was partially sponsored by Kaohsiung Chang Gung Memorial Hospital and Chang Gung University College of Medicine, Kaohsiung, Taiwan.
Footnotes
Conflicts of interest: No
References
- 1.Aitken A. 14-3-3 proteins: a historic overview. Semin Cancer Biol. 2006;16:162–72. doi: 10.1016/j.semcancer.2006.03.005. [DOI] [PubMed] [Google Scholar]
- 2.Pozuelo Rubio M, Geraghty KM, Wong BH, Wood NT, Campbell DG, Morrice N, et al. 14-3-3-affinity purification of over 200 human phosphoproteins reveals new links to regulation of cellular metabolism, proliferation and trafficking. Biochem J. 2004;379:395–408. doi: 10.1042/BJ20031797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hermeking H, Benzinger A. 14-3-3 proteins in cell cycle regulation. Semin Cancer Biol. 2006;16:183–92. doi: 10.1016/j.semcancer.2006.03.002. [DOI] [PubMed] [Google Scholar]
- 4.Neal CL, Yao J, Yang W, Zhou X, Nguyen NT, Lu J, et al. 14-3-3zeta overexpression defines high risk for breast cancer recurrence and promotes cancer cell survival. Cancer Res. 2009;69:3425–32. doi: 10.1158/0008-5472.CAN-08-2765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Matta A, Bahadur S, Duggal R, Gupta SD, Ralhan R. Over-expression of 14-3-3zeta is an early event in oral cancer. BMC Cancer. 2007;7:169. doi: 10.1186/1471-2407-7-169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yang X, Cao W, Zhou J, Zhang W, Zhang X, Lin W, et al. 14-3-3zeta positive expression is associated with a poor prognosis in patients with glioblastoma. Neurosurgery. 2011;68:932–8. doi: 10.1227/NEU.0b013e3182098c30. [DOI] [PubMed] [Google Scholar]
- 7.Lu J, Guo H, Treekitkarnmongkol W, Li P, Zhang J, Shi B, et al. 14-3-3zeta Cooperates with ErbB2 to promote ductal carcinoma in situ progression to invasive breast cancer by inducing epithelial mesenchymal transition. Cancer Cell. 2009;16:195–207. doi: 10.1016/j.ccr.2009.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Danes CG, Wyszomierski SL, Lu J, Neal CL, Yang W, Yu D. 14-3-3 zeta down-regulates p53 in mammary epithelial cells and confers luminal filling. Cancer Res. 2008;68:1760–7. doi: 10.1158/0008-5472.CAN-07-3177. [DOI] [PubMed] [Google Scholar]
- 9.Neal CL, Xu J, Li P, Mori S, Yang J, Neal NN, et al. Overexpression of 14-3-3zeta in cancer cells activates PI3K via binding the p85 regulatory subunit. Oncogene. 2012;31:897–906. doi: 10.1038/onc.2011.284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, et al. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell. 1999;96:857–68. doi: 10.1016/s0092-8674(00)80595-4. [DOI] [PubMed] [Google Scholar]
- 11.Fero ML, Rivkin M, Tasch M, Porter P, Carow CE, Firpo E, et al. A syndrome of multiorgan hyperplasia with features of gigantism, tumorigenesis, and female sterility in p27(Kip1)-deficient mice. Cell. 1996;85:733–44. doi: 10.1016/s0092-8674(00)81239-8. [DOI] [PubMed] [Google Scholar]
- 12.Sekimoto T, Fukumoto M, Yoneda Y. 14-3-3 suppresses the nuclear localization of threonine 157-phosphorylated p27(Kip1). EMBO J. 2004;23:1934–42. doi: 10.1038/sj.emboj.7600198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Halvorsen OJ, Haukaas SA, Akslen LA. Combined loss of PTEN and p27 expression is associated with tumor cell proliferation by Ki-67 and increased risk of recurrent disease in localized prostate cancer. Clin Cancer Res. 2003;9:1474–9. [PubMed] [Google Scholar]
- 14.Fredersdorf S, Burns J, Milne AM, Packham G, Fallis L, Gillett CE, et al. High level expression of p27(kip1) and cyclin D1 in some human breast cancer cells: inverse correlation between the expression of p27(kip1) and degree of malignancy in human breast and colorectal cancers. Proc Natl Acad Sci U S A. 1997;94:6380–5. doi: 10.1073/pnas.94.12.6380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hershko DD, Shapira M. Prognostic role of p27Kip1 deregulation in colorectal cancer. Cancer. 2006;107:668–75. doi: 10.1002/cncr.22073. [DOI] [PubMed] [Google Scholar]
- 16.le Sage C, Nagel R, Egan DA, Schrier M, Mesman E, Mangiola A, et al. Regulation of the p27(Kip1) tumor suppressor by miR-221 and miR-222 promotes cancer cell proliferation. EMBO J. 2007;26:3699–708. doi: 10.1038/sj.emboj.7601790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–97. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 18.Wu L, Fan J, Belasco JG. MicroRNAs direct rapid deadenylation of mRNA. Proc Natl Acad Sci U S A. 2006;103:4034–9. doi: 10.1073/pnas.0510928103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lu J, Getz G, Miska EA, Alvarez-Saavedra E, Lamb J, Peck D, et al. MicroRNA expression profiles classify human cancers. Nature. 2005;435:834–8. doi: 10.1038/nature03702. [DOI] [PubMed] [Google Scholar]
- 20.Ursini-Siegel J, Schade B, Cardiff RD, Muller WJ. Insights from transgenic mouse models of ERBB2-induced breast cancer. Nat Rev Cancer. 2007;7:389–97. doi: 10.1038/nrc2127. [DOI] [PubMed] [Google Scholar]
- 21.Medina D. Mammary tumorigenesis in chemical carcinogen-treated mice. II. Dependence on hormone stimulation for tumorigenesis. J Natl Cancer Inst. 1974;53:223–6. doi: 10.1093/jnci/53.1.223. [DOI] [PubMed] [Google Scholar]
- 22.Chang CJ, Chao CH, Xia WY, Yang JY, Xiong Y, Li CW, et al. p53 regulates epithelial-mesenchymal transition and stem cell properties through modulating miRNAs (vol 13, pg 317, 2011). Nature Cell Biology. 2011;13:1466. doi: 10.1038/ncb2173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Agresti A, editor. Categorical Data Analysis. 2nd ed. John Wiley & Sons Inc., Wiley-Interscience; Hoboken: 2002. [Google Scholar]
- 24.Chang CaL C. LIBSVM : a library for support vector machines. ACM Transactions on Intelligent Systems and Technology. 2011 [Google Scholar]
- 25.Andres AC, Schonenberger CA, Groner B, Hennighausen L, LeMeur M, Gerlinger P. Ha-ras oncogene expression directed by a milk protein gene promoter: tissue specificity, hormonal regulation, and tumor induction in transgenic mice. Proc Natl Acad Sci U S A. 1987;84:1299–303. doi: 10.1073/pnas.84.5.1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pattengale PK, Stewart TA, Leder A, Sinn E, Muller W, Tepler I, et al. Animal models of human disease. Pathology and molecular biology of spontaneous neoplasms occurring in transgenic mice carrying and expressing activated cellular oncogenes. Am J Pathol. 1989;135:39–61. [PMC free article] [PubMed] [Google Scholar]
- 27.Slamon DJ, Clark GM, Wong SG, Levin WJ, Ullrich A, McGuire WL. Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science. 1987;235:177–82. doi: 10.1126/science.3798106. [DOI] [PubMed] [Google Scholar]
- 28.Weidner N, Semple JP, Welch WR, Folkman J. Tumor angiogenesis and metastasis--correlation in invasive breast carcinoma. N Engl J Med. 1991;324:1–8. doi: 10.1056/NEJM199101033240101. [DOI] [PubMed] [Google Scholar]
- 29.Ravi R, Mookerjee B, Bhujwalla ZM, Sutter CH, Artemov D, Zeng Q, et al. Regulation of tumor angiogenesis by p53-induced degradation of hypoxia-inducible factor 1alpha. Genes Dev. 2000;14:34–44. [PMC free article] [PubMed] [Google Scholar]
- 30.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 31.Medema RH, Kops GJ, Bos JL, Burgering BM. AFX-like Forkhead transcription factors mediate cell-cycle regulation by Ras and PKB through p27kip1. Nature. 2000;404:782–7. doi: 10.1038/35008115. [DOI] [PubMed] [Google Scholar]
- 32.Pagano M, Tam SW, Theodoras AM, Beer-Romero P, Del Sal G, Chau V, et al. Role of the ubiquitin-proteasome pathway in regulating abundance of the cyclin-dependent kinase inhibitor p27. Science. 1995;269:682–5. doi: 10.1126/science.7624798. [DOI] [PubMed] [Google Scholar]
- 33.Malek NP, Sundberg H, McGrew S, Nakayama K, Kyriakides TR, Roberts JM. A mouse knock-in model exposes sequential proteolytic pathways that regulate p27Kip1 in G1 and S phase. Nature. 2001;413:323–7. doi: 10.1038/35095083. [DOI] [PubMed] [Google Scholar]
- 34.Cuesta R, Martinez-Sanchez A, Gebauer F. miR-181a regulates cap-dependent translation of p27(kip1) mRNA in myeloid cells. Mol Cell Biol. 2009;29:2841–51. doi: 10.1128/MCB.01971-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Davis BN, Hilyard AC, Nguyen PH, Lagna G, Hata A. Induction of microRNA-221 by platelet- derived growth factor signaling is critical for modulation of vascular smooth muscle phenotype. J Biol Chem. 2009;284:3728–38. doi: 10.1074/jbc.M808788200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Di Leva G, Gasparini P, Piovan C, Ngankeu A, Garofalo M, Taccioli C, et al. MicroRNA cluster 221-222 and estrogen receptor alpha interactions in breast cancer. J Natl Cancer Inst. 2010;102:706–21. doi: 10.1093/jnci/djq102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Galardi S, Mercatelli N, Farace MG, Ciafre SA. NF-kB and c-Jun induce the expression of the oncogenic miR-221 and miR-222 in prostate carcinoma and glioblastoma cells. Nucleic Acids Res. 2011;39:3892–902. doi: 10.1093/nar/gkr006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chiu R, Boyle WJ, Meek J, Smeal T, Hunter T, Karin M. The c-Fos protein interacts with c-Jun/AP-1 to stimulate transcription of AP-1 responsive genes. Cell. 1988;54:541–52. doi: 10.1016/0092-8674(88)90076-1. [DOI] [PubMed] [Google Scholar]
- 39.de Azambuja E, Cardoso F, de Castro G, Colozza M, Mano MS, Durbecq V, et al. Ki-67 as prognostic marker in early breast cancer: a meta-analysis of published studies involving 12 155 patients. Brit J Cancer. 2007;96:1504–13. doi: 10.1038/sj.bjc.6603756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hanahan D, Wagner EF, Palmiter RD. The origins of oncomice: a history of the first transgenic mice genetically engineered to develop cancer. Genes Dev. 2007;21:2258–70. doi: 10.1101/gad.1583307. [DOI] [PubMed] [Google Scholar]
- 41.Cardiff RD, Anver MR, Gusterson BA, Hennighausen L, Jensen RA, Merino MJ, et al. The mammary pathology of genetically engineered mice: the consensus report and recommendations from the Annapolis meeting. Oncogene. 2000;19:968–88. doi: 10.1038/sj.onc.1203277. [DOI] [PubMed] [Google Scholar]
- 42.Leder A, Pattengale PK, Kuo A, Stewart TA, Leder P. Consequences of widespread deregulation of the c-myc gene in transgenic mice: multiple neoplasms and normal development. Cell. 1986;45:485–95. doi: 10.1016/0092-8674(86)90280-1. [DOI] [PubMed] [Google Scholar]
- 43.Weinberg RA. Oncogenes, antioncogenes, and the molecular bases of multistep carcinogenesis. Cancer Res. 1989;49:3713–21. [PubMed] [Google Scholar]
- 44.Freed E, Symons M, Macdonald SG, McCormick F, Ruggieri R. Binding of 14-3-3 proteins to the protein kinase Raf and effects on its activation. Science. 1994;265:1713–6. doi: 10.1126/science.8085158. [DOI] [PubMed] [Google Scholar]
- 45.Miller TE, Ghoshal K, Ramaswamy B, Roy S, Datta J, Shapiro CL, et al. MicroRNA-221/222 confers tamoxifen resistance in breast cancer by targeting p27Kip1. J Biol Chem. 2008;283:29897–903. doi: 10.1074/jbc.M804612200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rao X, Di Leva G, Li M, Fang F, Devlin C, Hartman-Frey C, et al. MicroRNA-221/222 confers breast cancer fulvestrant resistance by regulating multiple signaling pathways. Oncogene. 2011;30:1082–97. doi: 10.1038/onc.2010.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bergamaschi A, Katzenellenbogen BS. Tamoxifen downregulation of miR-451 increases 14-3-3zeta and promotes breast cancer cell survival and endocrine resistance. Oncogene. 2012;31:39–47. doi: 10.1038/onc.2011.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.