

Abstract
We have assessed the efficacy of the recently developed CRISPR/Cas (clustered regularly interspaced short palindromic repeats/CRISPR-associated) system for genome modification in the amphibian Xenopus tropicalis. As a model experiment, targeted mutations of the tyrosinase gene were verified, showing the expected albinism phenotype in injected embryos. We further tested this technology by interrupting the six3 gene, which is required for proper eye and brain formation. Expected eye and brain phenotypes were observed when inducing mutations in the six3 coding regions, as well as when deleting the gene promoter by dual targeting. We describe here a standardized protocol for genome editing using this system. This simple and fast method to edit the genome provides a powerful new reverse genetics tool for Xenopus researchers.
Keywords: tyrosinase, albinism, six3, brain, eye, sgRNA
INTRODUCTION
Xenopus has long been a favored model organism for developmental embryology due to its unique combination of advantageous features, and the recent development of Xenopus tropicalis as a new model organism with a diploid genome, short generation time and sequenced genome information allows researchers to use genetic tools in frogs (Harland and Grainger 2011). Forward and reverse genetic approaches have identified developmental mutants and their responsible genes (Abu-Daya et al. 2012).
Recent technological advances have allowed researchers to perform rapid targeted gene editing in many organisms. Two methods, zinc-finger nucleases (ZFNs) and transcription activator-like effector nucleases (TALENs) (Joung and Sander 2013), have both been successfully applied in Xenopus (Ishibashi et al. 2012; Lei et al. 2012; Nakajima et al. 2012; Nakajima et al. 2013; Suzuki et al. 2013; Young et al. 2011). Another, simpler technology has also emerged: the CRISPR/Cas (clustered regularly interspaced short palindromic repeats/CRISPR-associated) system for genome modification, originally identified as part of the naturally occurring bacterial adaptive defense mechanism. CRISPR/Cas has now been successfully applied in major model organisms such as zebrafish (Hwang et al. 2013), mouse (Wang et al. 2013), fly (Bassett et al. 2013) and worm (Friedland et al. 2013) to effect targeted genome modification. Briefly, the original Cas9 in Streptococcus pyogenes is recruited to the target site by two RNAs, a CRISPR RNA (crRNA) that has complementary sequence to the target DNA, and the trans-activating CRISPR RNA (tracrRNA) that base pairs with the crRNA. For recognition, the target sequence must be followed by a protospacer adjacent motif (PAM) sequence (nGG, where n can be any nucleotide). For genome editing, the crRNA and tracrRNA can be fused into a single synthetic guide RNA (sgRNA) (Hwang et al. 2013), which functions efficiently with Cas9 to cause cleavage of the target site (~20 bp), which must be followed by the PAM sequence in the genome (Fig. 1a). A further constraint on the target sequence is due to the utilization of in vitro transcription promoters such as T7, T3 or SP6 to produce sgRNAs: since these promoters work optimally with a +1 guanine residue, the genomic target sequence must begin with a “G”. As with ZFNs and TALENs, these cleavages are then often imperfectly repaired via non-homologous end-joining (NHEJ), which can lead to frameshift-causing in-del mutations occurring mosaically throughout the injected embryo. Here we report the application of CRISPR/Cas system (hereafter called simply CRISPR) to edit the genome of Xenopus tropicalis, providing an additional tool for Xenopus researchers to achieve simple and efficient targeted mutagenesis.
FIG. 1.
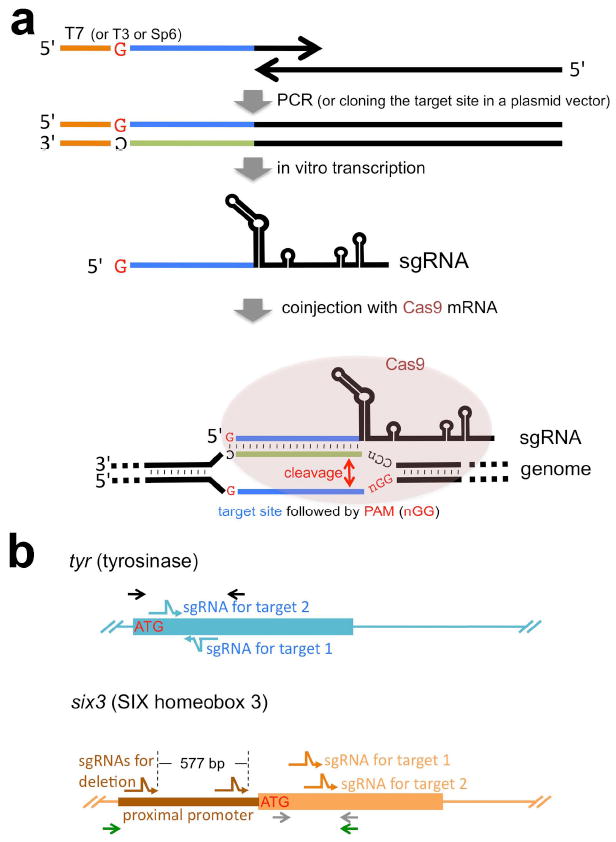
Strategy for CRISPR/Cas-mediated genome modification. (a) Schematic representation of experimental procedure. We found that conventional plasmid subcloning methods for making sgRNA templates was time-consuming and inconvenient; instead all sgRNAs used in this study are transcribed in vitro from double-stranded DNA templates that were made by PCR (except the sgRNA for tyr target site 1, which used a cloned template, pDR274-Xt-tyr). This PCR strategy uses a 5’ oligonucleotide (primer) that begins with the T7 promoter (shown as an orange line in this schematic; alternatively, T3 or SP6 promoters could be used) and contains the genomic target sequence (shown as a blue line in this schematic; note the genomic target sequence must begin with a G for proper transcriptional initiation using the T7 promoter) and a 3’ oligonucleotide (primer) that partly overlaps the 5’ primer and contains the sgRNA backbone sequence required for proper folding of sgRNAs. This rapid, easy way to make sgRNA templates by PCR takes less than two days to produce sgRNAs to inject once oligonucleotides are received. During the preparation of this manuscript, we have noticed that similar strategies have also been reported [e.g., (Bassett et al. 2013)] and thus this method has general versatility. The resulting sgRNAs were co-injected with Cas9 mRNA into one cell-stage Xenopus embryos. The bottom part of the scheme shows how sgRNA and Cas9 work to cleave the target site in the genome. Briefly, the sgRNA forms a complex with the Cas9 protein and identifies the target site via complementary basepairing. The Cas9 nuclease subsequently cleaves the genomic DNA at the target site, just upstream of the PAM sequence. (b) Targeting strategy for the two genes described in this study. Both tyr and six3 genes were targeted in exon 1 to cause frame-shifts after the translation initiation codon (ATG). Two independent sgRNAs were tested for both genes. The six3 gene was furthermore targeted in the proximal promoter region with two sgRNAs simultaneously to cause deletion of the promoter region. Bent arrows (blue for tyr, orange for six3 exons and brown for the six3 promoter) show sgRNA targets. Arrows (black for tyr, green and grey for six3) indicate genomic PCR primers for mutation analyses. Note that the 3’ primer for the six3 gene is common for both sets of PCR reactions but colored differently for clarity. Drawings are not to scale.
RESULTS and DISCUSSION
In order to establish a method for CRISPR-mediated genome editing of Xenopus tropicalis, as well as to determine the optimal conditions for inducing mutations, we first chose the tyrosinase (tyr) gene as a model for targeting because of the readily apparent mutant phenotype, albinism, as seen in previous work using ZFN or TALEN constructs (Ishibashi et al. 2012; Nakajima et al. 2012). The first tyr sgRNA was designed to target a sequence near the region that was used effectively with TALEN constructs (Fig. 1b, Fig.2) (Ishibashi et al. 2012).
FIG. 2.

Successful targeting of the tyrosinase gene caused albinism in Xenopus embryos. (a) Different dose combinations of Cas9 mRNA and sgRNA were tested. The severity of the phenotype was directly dependent on the amounts of RNAs injected. bacCas9, the original bacterial-codon Cas9 mRNA (which shows examples with a weak phenotype; a patchy loss of pigmentation in the RPE is indicated by white arrowheads). humCas9, the modified Cas9 using humanized-codons. sgRNA, targeting tyrosinase gene (first target, see Fig. 3b for location relative to ATG). The toxicity of sgRNA seems to vary depending on its sequence and also on the batch of embryos. For example, in one specific experiment, tyrosinase sgRNA (target1) was overall relatively non-toxic, with more than 75% of injected embryos developing normally one day after injection at all tested doses shown here (76.9-97% survivors [n=19-33] as opposed to 84.8% of uninjected embryos [n=46]), whereas in other experiments, survival was between 50-60% (see Supporting Information Table 1). (b) Representative results of conventional sequencing assays (top three panels). Each single embryo injected with indicated RNAs was lysed and the targeted genomic region was PCR-amplified; amplicons were then directly sequenced (DSP assay, see the text). Perturbation of peaks on the 3’ side of the PAM region (red arrows) suggests in-del events happen in-between the target sequence (shaded in purple) and the PAM region (shaded in red). PCR amplicons were re-cloned and sequenced to show the profile of individual mutations found in mosaic individuals (bottom sequence alignments). Dashes (-) indicate gaps. Lowercase green characters indicate insertions or substitutions. The numbers in parentheses indicate the frequency of each mutation pattern seen in total numbers of sequenced clones.
We initially examined various doses of injected sgRNA (25 pg - 200 pg) and Cas9 mRNA (550 pg - 2.2 ng). We also tested two Cas9 variants, one using the original bacterial codons (bacCas9 in Fig. 2a) (Hwang et al. 2013), and the other a “humanized” Cas9 (Chang et al. 2013) that instead uses codons optimized for mammalian genes, and which also includes nuclear localization signals at both ends of the protein (humCas9 in Fig. 2a), both of which have been shown to successfully edit the zebrafish genome. In Fig. 2a, since the phenotype is hard to score with a quantifiable scale, we show representative embryos as group to illustrate qualitative differences. Among the doses tested, a combination of the highest doses using humCas9 (i.e., 2.2 ng humCas9 mRNA and 200 pg sgRNA) showed almost complete albinism (Fig. 2a, left bottom panel), whereas at the same doses, bacCas9 showed much less activity, although it still could cause mutations (data not shown) and patchy loss of pigmentation in the retinal pigment epithelium (RPE) (white arrowheads in Fig. 2a). In general, at the doses using more than 100 pg sgRNA and 1.1 ng humCas9, we see the albino phenotype in essentially 100% of injected embryos although the phenotype may vary from the patchy loss of pigmentation in the RPE to almost complete albinism depending the doses used. As shown in Fig. 2b, we developed a quick genotyping method by simple direct sequencing of the PCR-amplified targeted genome region (hereafter, called DSP assay), namely, a single embryo was subjected to genomic PCR to amplify the target region and the resultant amplicon was directly sequenced without cloning. If mutations occur at relatively high frequency, the resultant PCR amplicon is a mixture of heterogeneous fragments with different mutations, and the reading of sequences becomes perturbed in the mutated region. Typical results of samples from a group injected with the lowest doses (humCas9 550 pg/sgRNA 25pg) or from a group injected with the highest doses (2.2 ng/200 pg) compared with wild-type sample are shown (Fig. 2b). Each sample was also subsequently re-cloned and individual clones per embryo were sequenced as shown in Fig. 2b (bottom sequencing alignments). Not surprisingly, since we saw almost completely albino embryos, indicative of highly efficient mutagenesis, the sample from the highest doses showed perturbation of peaks in the targeted region (Fig. 2b, the bottom chromatogram, arrow), suggesting the presence of in-del mutations in this region. Interestingly, a sample from the lowest doses also clearly showed perturbation of peaks in the targeted region (Fig. 2b, middle chromatogram) although more than 60% (7 out of 11 clones sequenced) of recovered sequences from an embryo were not mutated (Fig. 2b, top sequence alignment), suggesting that this method (DSP assay) is sensitive enough for quick screening of functional sgRNAs that have ability to cause mutations at the molecular level, even if relatively infrequent. Given that the phenotype at this dose was very mild, the DSP assay is also suitable for screening of putative carriers of potentially lethal mutations where a low mutation rate is necessary to be able to raise carriers. Although more sensitive screening methods could detect less efficiently mutated carriers, such carriers might not be practical for making mutant line(s) since one would need to screen so many offspring to identify heterozygotes in the next generation.
Since it has shorter recognition sequences compared to ZFNs or TALENs, CRISPR may potentially create more off-target mutations, and to overcome this problem several approaches are being used (Carroll 2013). In order to rule out the possibility that off-target effects are causing observed phenotypes, we have established a standard method for performing F0 embryo assays using Xenopus tropicalis: 1) target at least two independent regions of the gene of interest to see reproducibility of the phenotype (including its variations), and, if possible, 2) perform rescue to confirm the phenotype is specific to a particular gene. However, it remains valuable to consider the issue of off-target effects, and the bioinformatics approaches described in the accompanying paper (Blitz et. al., in press), and below, can be used to identify putative sites and then evaluate the degree of mutagenesis in such off-target sites as performed by Blitz, et. al. (in press).
To pursue the two-target strategy, as shown in Fig. 3, we designed a second sgRNA for a different region of the tyr gene (Fig. 1b, 3b). Hereafter, we only used humCas9 (therefore simply called Cas9 below). Using the combination of the highest doses of RNAs as used above, the second sgRNA also showed the expected phenotype in 100% of embryos injected in multiple experiments although the severity varied from patchy loss of pigmentation to almost complete albinism (Fig. 3a). Two independent sgRNAs targeting different regions of the gene showed the same phenotype, suggesting that the phenotype is likely due to inactivation of the targeted gene, and not due to off-target mutations. However, we failed to rescue the phenotype by co-injection of tyr mRNA (up to 200 pg) under conditions we tested (data not shown). This is likely due to the timing of expression of the tyr gene, which begins at late tailbud stages when injected mRNA is likely to be already degraded.
FIG. 3.

The mutation of a second tyr gene target site also caused albinism. (a) Phenotype of injected embryos (bottom five embryos; top five embryos are uninjected sibling controls) with Cas9 mRNA (2.2 ng) and sgRNA (200 pg) for the second target site. (b) Representative mutation profiles from two embryos sequenced after cloning. The second target sequence (shaded in purple) and the PAM sequence (in red) are shown together with first target and PAM sequences (in gray; see Fig. 2) relative to the ATG (Met) site. The alignment shown is labeled as described in Fig. 2b.
To demonstrate the general applicability of this approach we chose a second gene, six3, of direct interest to ongoing studies of eye determination in the Grainger lab. six3 is one of a group of eye-field transcription factors expressed in the anterior region of the vertebrate neural plate and is essential for eye formation (Zuber et al. 2003). We found that embryos injected with the first six3 targeted CRISPR (for target site1) showed consistent abnormalities that were similar to known loss-of-function phenotypes of Six3 protein. In X. laevis, loss-of-function of six3 using a dominant-negative construct or morpholino (MO) knock-down was reported to show eye and anterior head defects (Gestri et al. 2005). In humans, mutations in the homeodomain of SIX3 gene are known to cause holoprosencephaly (HPE) (Wallis et al. 1999), and haploinsufficiency of Six3 in the mouse causes HPE (Geng et al. 2008). Complete inactivation of Six3 results in the absence of the eyes and nose in mouse (Lagutin et al. 2003). What we have observed in X. tropicalis mutant embryos here is consistent with those reports (see Fig. 4).
FIG. 4.

Summary of phenotype caused by mutations of six3 gene. All targeted mutations showed a similar phenotype. (a-e) Phenotype variations seen in st.40 embryos targeted with the sgRNA for coding region site 1. By this stage, the obvious phenotype is significantly reduced eye size, but a brain defect (see below) is not yet obvious. The phenotype is six3-specific because it was partially rescued by co-injection of six3 mRNA (b, c). Severity of phenotype was scored as +++ (most severe, no eye or tiny piece of eye), ++ (severe, small and malformed eye), + (modest, small relatively normal looking eye), and - (no or little phenotype) (a-c). As observed with the tyr target injections, toxicity of injected RNAs varied greatly depending on batch of embryos. For example, in different experiments, the percentage of normal embryos recovered after the same RNA doses were injected (six3 target 1) varied from 43% to 87% (see Supporting Information Table 1). (d) Chromatograms of the DSP assay from an uninjected embryo (WT) and one of rescued embryos shown in (b). Perturbation of peaks is seen around the PAM region (two-headed red arrow), suggesting the presence of mutated sequences. At later stages, the brain phenotype becomes obvious (e-g). (e) Examples of the phenotype of embryos (emb1 to emb3) targeted to delete the proximal promoter region at st.42. Using the primer as schematically shown in Fig. 1b (green arrows), genomic PCR of sibling uninjected wild-type embryo showed an expected ~ 1.2 kbp band, whereas embryos injected with sgRNAs had an extra ~ 0.6 kbp band (emb1-3, white arrows). One band (marked by *) was recloned and sequenced; the results are shown in Supporting Information Fig. S1. The promoter deletion injections were on average more toxic, perhaps due to higher total amounts of RNAs being injected, but survival rates as high as 68% were observed in individual experiments. (f) Phenotype examples of st. 42 embryos injected with sgRNA targeting coding region site 1 (left panel) and site 2 (right panel), lateral views. Compared to wild-type embryos (bottom, WT, round shaped head), mutant embryo heads had a more angular and flattened shape. The dorsal view (second panel) and frontal sections (third panel) of the same embryos (emb1-1, 1-2, 1-3) in first left panel clearly show narrow heads (compare two-headed white arrows between WT and mutants) and smaller brains (black arrowheads in frontal sections), especially the forebrain (red arrowheads in second panel, see also Fig. 4g) in the mutant embryos. The sequencing summary of mutated loci of emb1-1 and 1-3 in the top left panel and emb2-1, 2-2 in the top right panel are shown in Supporting Information Fig. S1. (g) A mild phenotype seen in embryos (target site 2) at st. 46. Top panel shows a dorsal view of embryos. Second panel shows horizontal sections of equivalent embryos shown in the top panel. At early stages, these embryos could be scored as wild type. Later observation of mutant embryos reveals that the nasal pits (red arrows) are fused (shown here) or shifted medially (not shown), and the telencephalon (red arrowheads) is smaller and fused. The last panel shows chromatograms of the DSP assay from one of the sectioned embryos shown above to confirm genomic mutations as evidenced by perturbation of peaks around PAM region (two-headed red arrow) in the mutant embryo.
As described above, we tested two sgRNAs targeting different coding regions and one targeting the proximal promoter (Fig. 1b, Supporting Information Fig. S1). Although the efficiency of each sgRNA differed, all sgRNAs caused a similar phenotype, suggesting the phenotype is likely due to the mutations of six3 gene but not due to mutations in off-target sites. We used 3.2 ng Cas9 mRNA and 200 pg sgRNA for F0 mutant characterization for single sgRNAs. This dose combination induced reproducible phenotypes as described in detail below in essentially all embryos injected in multiple experiments although the severity varied from one embryo to another. In another set of experiments we attempted to delete an approximately 600bp cis-regulatory element, the six3 proximal promoter, by designing CRISPR target sites flanking the region (Fig. 1b). When CRISPR-mediated cleavage occurs at both sites surrounding this region, NHEJ may result in a deletion of the flanked region. For this experiment, 4.7 or 5.7 ng Cas9 mRNA and 200 pg of each sgRNA were injected, showing a consistent phenotype in up to 50% of the survivors (Fig. 4e). We could confirm that a deletion of approximately 600bp occurred in some cells of injected embryos as determined by PCR assay and subsequent cloning and sequencing of the targeted region (Fig. 4e and Supporting Information Fig. S1).
By st. 40, the majority of embryos injected with sgRNAs targeting six3 coding regions (essentially 100% in multiple experiments for target 1, and 60-100% for target 2) showed reduction of eye size, although the severity varied (Fig. 4a, c). Importantly, this phenotype was partially rescued by co-injection of X. laevis six3 mRNA (20 pg) as evidenced by shift of severity toward a milder phenotype (Fig. 4b, c). We confirmed that the rescued embryos were mutated by the DSP assay (Fig. 4d). By st. 42, the heads of mutants had a more angular and flattened shape from a lateral view (Fig. 4e,f, top panels) and narrower heads with smaller brain regions from the dorsal view (Fig. 4f, middle panel) or in a frontal section (Fig. 4f, bottom panel, black arrowhead) than wild-type embryos. When using the sgRNA for target 2, by st. 46 we also noticed a milder phenotype where the nasal pits were fused or shifted medially (not shown), and the telencephalon was smaller and fused in the mutant (Fig. 4g). Mutation profiles of mutant embryos described above are shown in Supporting Information Fig. S1. In summary, multiple sgRNAs showed common phenotypes and the phenotypes could be significantly rescued by co-injection of mRNA, suggesting that the observed phenotypes are due to partial inactivation of the six3 gene but not due to mutations of off-target sites. Also, importantly, we successfully showed that by simultaneously targeting two sites surrounding the promoter, the flanked region (up to 600 bp in this experiment) could be deleted. This strategy is particularly useful for studying promoter and enhancer regions of a gene.
In our experiments, as noted in Figure legends 2 and 4, there is some impact of injecting CRISPR RNAs on embryo survival, and we consider possibilities here to explain these results to help other investigators who may use this technology. Three obvious possibilities are: egg quality, toxicity of RNAs, and off-target effects of CRISPR RNAs. We are certain that egg quality issues come into play, since the same constructs are found to result in very different survival rates with eggs from different females (see Supporting Information Table 1). In injections involving all target sequences used in this work where there were low survival rates in one experiment (e.g. as low as 50%), there were other experiments where survival was significantly higher (e.g. 80%). To examine whether individual RNAs are themselves toxic, we injected either Cas9 or sgRNA’s alone at doses used in targeting experiments, and there we found significant mortality (up to 50%) in some egg batches where higher doses were used, suggesting that RNA toxicity is present to a significant degree. In addition to our multiple target and rescue strategies to minimize the chance of off target effects, both egg batch and single RNA injection results argue against off-target effects as an explanation for the lethality we see, since the latter is variable (and can be quite low) and can be elicited without biologically active CRISPR function (i.e. embryos injected with Cas9 or sgRNA alone).
As shown in the accompanying paper (Blitz et al., in press), one can bioinformatically identify and systematically evaluate off-target effects experimentally; for the tyr target studied, these are not significant. The same tyr target was used in our study (target 1), along with a second target that also induced albinism. In our experiments off-target effects were studied in two ways. First, possible target sites were evaluated bioinformatically during the design process for both tyr and six3 (see Methods) to eliminate any target that might have identical sequence or a single base mismatch in sequence elsewhere in the genome. Second, although our data strongly suggests we are not seeing off-target effects in our experiments for reasons noted above, we also searched for off-target sequences with up to four base mismatches in all of the targets used in our studies of six3, and have found none in coding sequences in genomic sequence (Supporting Information Table 2).
Here, we have demonstrated for two genes that CRISPR-mediated mutagenesis is useful for quickly analyzing loss-of-function phenotypes in genes of interest that function at later stages of development when conventional MO-type loss-of-function assays are not feasible. Because of mosaicism of mutant and wild-type cells in a whole embryo, we are probably seeing hypomorphic phenotype(s), which is actually an advantage of using F0 animals because we might not see subtle gene effects in the case of complete inactivation of a gene in an established mutant line. On the other hand, because of the mosaicism, phenotype variation could be significant from one animal to another. Therefore, we need to be careful in concluding that observed phenotypes are due to inactivation of the gene of interest. For detailed, precise analysis of loss-of-function phenotypes, mutant lines should be established, for which CRISPR methodology will be a very powerful and efficient tool. For the creation of mutant lines that may cause embryonic lethal phenotypes in F0 animals with high mutation rates, the use of lower doses of RNAs or use of the less efficient, bacterial Cas9, may be advantageous. Furthermore, the sensitivity of the DSP assay is sufficient to identify non-phenotypic putative founders, and can later be used to genotype heterozygous offspring. Maintenance of such lines through out-crossing also serves to breed out putative off-target mutations. In the future, more detailed analysis of six3 null mutant(s) will be performed using established mutant line(s) where the value of consistent, heritable phenotypes provides additional advantages over the more variable phenotypes (and issues like toxicity of injected embryos) that one sees in F0-injected embryos.
We should keep in mind that the availability of CRISPR target sequence is somewhat constrained and thus there will be situations where one needs TALENs for gene editing, since they have more flexibility in selection of target sequences. Additionally, the increased possibility of off-target effects when using CRISPR to study F0 phenotypes may be problematic; however, the guidelines presented here, that is, targeting different regions of the same gene as well as performing rescue experiments when they are feasible, should help address these concerns. Also, as has been shown by Blitz et al. (in press), the off-target sites may not be problematic in Xenopus for some target sequences, since they could not find evidence for mutations of possible off-target sites for the targets site 1 of tyr sgRNA. Furthermore, as suggested above, over the course of the creation of stable mutant lines, any off-target mutations will be bred out, and such lines will be powerful tools for studying gene function. The clear advantages of the CRISPR-mediated genome editing method, including the ability to easily and quickly design oligonucleotides to create sgRNA templates, as well as effect targeted deletions of larger regions of regulatory DNA such as promoters and enhancers, makes this method an important part of the growing Xenopus genetic-manipulation toolkit.
METHODS
Frogs, plasmids, oligonucleotides, and mRNA injections
Xenopus tropicalis eggs were fertilized, dejellied, and injected as described previously (Ogino, McConnell, and Grainger 2006). Embryos were staged according to Nieuwkoop and Faber (1967). The plasmids MLM3613 (bacterial Cas9 expression vector) and DR274 (sgRNA expression vector) were purchased from Addgene (Cambridge, MA, USA). The ORF of Cas9 from MLM3613 was PCR-cloned into the pCS2+ vector to yield pCS2-Cas9 (that encodes Cas9 based on bacterial codon usage). X. laevis six3 cDNA (a kind gift from Gaia Gestri) was recloned into pCS2+ to yield pCS2-Six3. The capped mRNAs were synthesized from pCS2-Cas9 and pCS2-Six3 using the mMESSAGE mMACHINE® Sp6 (Invitrogen) or pXT7-Cas9 encoding “humanized” Cas9 (Chang et al. 2013) using the mMESSAGE mMACHINE® T7 Kit (Invitrogen).
The sequence of each sgRNA was designed using the ZiFiT Targeter program website (http://zifit.partners.org/ZiFiT/ChoiceMenu.aspx) (Hwang et al. 2013) and resultant sequences used in this study are described in Fig. 2b, 3b, and Supporting Information Fig. S1. sgRNAs were evaluated for the probability of causing off-target effects by searching the Xenopus tropicalis genome, JGI 4.2/xenTro3 (Nov, 2009), using the site GGGenome (http://gggenome.dbcls.jp/), searching both genomic strands; possible sgRNAs that had up to a 1bp mismatch elsewhere in the genome were not considered for further experimental design. The sgRNAs were transcribed either from the template cloned into DR274 (for tyrosinase target1, the resultant clone pDR274-Xt-tyr) or PCR-amplified templates (see Fig. 1a and below) using the MEGAscript® T7 Kit (Invitrogen). We tested two combinations of different lengths of 5’ and 3’ oligonucleotides to make sgRNA templates and both combinations successfully worked. Here we only show our most used oligonucleotide design (shown schematically in Fig. 1a) in 5’ to 3’ orientation. The 5’ oligonucleotide sequence is: TAATACGACTCACTATAGnnnnnnnnnnnnnnnnnnnGTTTTAGAGCTAGAAATAGC AAG, where the T7 promoter, which can be replaced with a T3 or Sp6 promoter, is in bold and the sgRNA target sequence is in italic, the first G of which is required for transcription by T7 (T3 or Sp6) polymerase. Thus the target sequence must start from G followed by any nucleotide [n], that must be followed by PAM in the genome, namely, the target sequence in the genome must be 5’-G-n19-nGG-3′ where the last nGG is the PAM sequence. Note that, when using the cloning vector (DR274) to make the template, the availability of target sites is further limited by the addition of another G (i.e., 5’-GG-n18-nGG-3′) due to its cloning strategy (Hwang et al. 2013). The 3’ oligonucleotide sequence is: AAAAGCACCGACTCGGTGCCACTTTTTCAAGTTGATAACGGACTAGCCTTATT TTAACTTGCTATTTCTAGCTCTAAAAC.
More in-depth evaluation of possible off-target sites was performed using the GGGenome search site as described above, but allowing for up to 4 mismatches. Hits were aligned to each sgRNA target as the reference sequence, using Geneious 6.1.5 (Biomatters, http://www.geneious.com/), and the alignment was then manually inspected to remove sequences with indels. The genomic locations of all possible off target regions was further evaluated using the UCSC genome browser (http://genome.ucsc.edu/) to determine if any were found in known coding regions.
Genotyping of embryos
Embryos were lysed and lysates were used for genomic PCR as described previously (Fish et al. 2012). PCR amplicons were purified and sequenced directly for tentative genotyping (DSP assay) or after recloning for definitive genotyping. The primers used for genotyping are as follows (in 5’ to 3’ orientation). For tyrosinase (corresponding to black arrows in Fig. 1b): tyr-5’, TGATGTAAGCCTGCACATGTGA; tyr-3’, CAGTCTGCACAGTTATAGCCCA. For six3 coding (corresponding to grey arrows in Fig. 1b) and promoter (corresponding to green arrows in Fig. 1b) regions: six3 cod-5’, CTTCTTTCTCCCTGGCTCCT; six3 pro-5’, ATGGATAGCCAGGCAGACAG; six3 com-3’, CTTATTGATGGCCTCACACG (note that 3’ primer is common for both PCR reaction).
Histology
Embyros were cut in two so that heads could be processed for histology while the remainder of the embryo was used for genotyping. Heads were fixed overnight in Bouin’s fixative, washed in 70% ethanol then dehydrated through a graded ethanol series and cleared with xylene for paraffin embedding. Blocks were sectioned at 10 μm and sections were stained with hematoxylin.
Supplementary Material
Supporting Information Fig. S1 Mutation profiles of the six3 gene found in individual embryos injected with sgRNAs targeting different regions. Top sequence shows the positions of target 1 and 2 relative to the translation initiation codon (ATG, marked as Met) to cause frame-shifts. Note that target sites are partially overlapping. “Target 1” lists representative mutated sequences after recloning from two embryos (emb1-1 and emb1-3, see also Fig. 4f, six3 target 1) injected with sgRNA for target site 1. Eleven clones from one embryo (emb1-3) and ten clones from a second embryo (emb1-1) were analyzed. Note that no wild-type sequence (WT) was recovered. “Target 2” shows representative mutated sequences from two embryos (emb2-1 and emb2-2, see also Fig. 4f, six3 target 2) injected with sgRNA to target site 2. Nine clones from one embryo (emb2-1) and five clones from a second embryo (emb2-2) were analyzed. Note that WT sequence was recovered though at a relatively low frequency. “Proximal promoter targets” shows the -574 to +34 region with two sgRNA target sites and representative mutated sequences after recloning (14 clones) from an injected embryo (emb3, see also Fig. 4e). +1 is a tentative transcriptional start site according to the 5’ end of an EST clone (accession number: DC185735). Target sequences and corresponding PAM regions are shaded in purple and red, respectively. The alignment is shown as described in Fig. 2b.
Supporting Information Fig. S2 Confirmation of germline transmission of CRISPR-mediated mutagenesis targeting the tyr gene (target 1). An F0 mature male raised from albino tadpoles injected with the humCas9 (2.2 ng) and sgRNA (tyr target 1, 200 pg) was outcrossed to obtain F1 animals, which were subjected to the DSP assay. Representative chromatograms are shown (top, wild type; bottom, heterozygous mutant carrier). Out of 8 tadpoles tested, 3 showed germline transmission as evidenced by double peaks observed when reading sequence around the target site (red arrow), in contrast to the multiple peaks seen in F0 mosaic animals, see Fig. 2b.
Acknowledgments
We wish to thank Jianzhong Jeff Xi for sharing of plasmid pXT7-Cas9 (which should be requested directly from JJX because of Material Transfer Agreement restrictions) and Gaia Gestri for X. laevis six3 cDNA. The first preliminary result regarding knockdown of the tyrosinase gene (target 1) described in this paper was obtained during the 2013 Cold Spring Harbor Laboratory Xenopus Course, which we would like to acknowledge. We also thank Ira Blitz and Ken Cho for sharing information before publication. The authors would also like to gratefully acknowledge the support of the National Xenopus Resource and its staff. Due to the rapid accumulation of reports using the same methodologies and space limitations, we apologize for not being able to cite all relevant references. The plasmid pDR274-Xt-tyr described in the study can be obtained from GHT (gerald.h.thomsen@stonybrook.edu) and RMG (rmg9p@virginia.edu). pCS2-Cas9 is available from RMG.
This work was funded by NIH grants EY022954, EY018000, and OD010997 and a research award from the Sharon Stewart Aniridia Trust to RMG and NIH grant HD064908 to GHT. Support for JOH was provided by NIH predoctoral training grant T32GM007964.
Footnotes
Note added in proof
After acceptance of this paper, we have confirmed germline transmission of CRISPR-mediated mutagenesis targeting the tyr gene (target 1), see Supporting Information Fig. S2.
LITERATURE CITED
- Abu-Daya A, Khokha MK, Zimmerman LB. The Hitchhiker’s guide to Xenopus genetics. genesis. 2012;50:164–175. doi: 10.1002/dvg.22007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassett AR, Tibbit C, Ponting CP, Liu J-LL. Highly Efficient Targeted Mutagenesis of Drosophila with the CRISPR/Cas9 System. Cell Reports. 2013;4:220–228. doi: 10.1016/j.celrep.2013.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blitz IL, Biesinger J, Xie X, Cho KWY. Biallelic genome modification in F0 Xenopus tropicalis embryos using the CRISPR/Cas system. genesis. doi: 10.1002/dvg.22719. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll D. Staying on target with CRISPR-Cas. Nat Biotech. 2013;31:807–809. doi: 10.1038/nbt.2684. [DOI] [PubMed] [Google Scholar]
- Chang N, Sun C, Gao L, Zhu D, Xu X, Zhu X, Xiong J-W, Xi JJ. Genome editing with RNA-guided Cas9 nuclease in Zebrafish embryos. 2013;23:465–472. doi: 10.1038/cr.2013.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fish MB, Nakayama T, Grainger RM. Simple, fast, tissue-specific bacterial artificial chromosome transgenesis in Xenopus. genesis. 2012;50:307–315. doi: 10.1002/dvg.20819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedland AE, Tzur YB, Esvelt KM, Colaiácovo MP, Church GM, Calarco JA. Heritable genome editing in C. elegans via a CRISPR-Cas9 system. Nature Methods. 2013;10:741–743. doi: 10.1038/nmeth.2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geng X, Speirs C, Lagutin O, Inbal A, Liu W, Solnica-Krezel L, Jeong Y, Epstein DJ, Oliver G. Haploinsufficiency of Six3 fails to activate sonic hedgehog expression in the ventral forebrain and causes holoprosencephaly. Developmental Cell. 2008;15:236–247. doi: 10.1016/j.devcel.2008.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gestri G, Carl M, Appolloni I, Wilson SW, Barsacchi G, Andreazzoli M. Six3 functions in anterior neural plate specification by promoting cell proliferation and inhibiting Bmp4 expression. Development. 2005;132:2401–2413. doi: 10.1242/dev.01814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harland RM, Grainger RM. Xenopus research: metamorphosed by genetics and genomics. Trends in Genetics. 2011;27:507–515. doi: 10.1016/j.tig.2011.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang WY, Fu Y, Reyon D, Maeder ML, Tsai SQ, Sander JD, Peterson RT, Yeh J-RJ, Joung JK. Efficient genome editing in zebrafish using a CRISPR-Cas system. Nat Biotech. 2013;31:227–229. doi: 10.1038/nbt.2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishibashi S, Cliffe R, Amaya E. Highly efficient bi-allelic mutation rates using TALENs in Xenopus tropicalis. Biology Open. 2012;1:1273–1276. doi: 10.1242/bio.20123228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joung JK, Sander JD. TALENs: a widely applicable technology for targeted genome editing. Nat Rev Mol Cell Biol. 2013;14:49–55. doi: 10.1038/nrm3486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagutin OV, Zhu CC, Kobayashi D, Topczewski J, Shimamura K, Puelles L, Russell HRC, McKinnon PJ, Solnica-Krezel L, Oliver G. Six3 repression of Wnt signaling in the anterior neuroectoderm is essential for vertebrate forebrain development. Genes & Development. 2003;17:368–379. doi: 10.1101/gad.1059403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei Y, Guo X, Liu Y, Cao Y, Deng Y, Chen X, Cheng CHK, Dawid IB, Chen Y, Zhao H. Efficient targeted gene disruption in Xenopus embryos using engineered transcription activator-like effector nucleases (TALENs) Proceedings of the National Academy of Sciences. 2012;109:17484–17489. doi: 10.1073/pnas.1215421109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajima K, Nakai Y, Okada M, Yaoita Y. Targeted gene disruption in the Xenopus tropicalis genome using designed TALE Nucleases. Zoological Science. 2013;30:455–460. doi: 10.2108/zsj.30.455. [DOI] [PubMed] [Google Scholar]
- Nakajima K, Nakajima T, Takase M, Yaoita Y. Generation of albino Xenopus tropicalis using zinc-finger nucleases. Development, Growth & Differentiation. 2012;54:777–784. doi: 10.1111/dgd.12006. [DOI] [PubMed] [Google Scholar]
- Nieuwkoop PD, Faber J. Normal table of Xenopus laevis (Daudin) New York: Garland; 1967. reprinted in 1994. [Google Scholar]
- Ogino H, McConnell WB, Grainger RM. High-throughput transgenesis in Xenopus using I-SceI meganuclease. Nat Protocols. 2006;1:1703–1710. doi: 10.1038/nprot.2006.208. [DOI] [PubMed] [Google Scholar]
- Suzuki KT, Isoyama Y, Kashiwagi K, Sakuma T, Ochiai H, Sakamoto N, Furuno N, Kashiwagi A, Yamamoto T. High efficiency TALENs enable F0 functional analysis by targeted gene disruption in Xenopus laevis embryos. Biology Open. 2013;2:448–452. doi: 10.1242/bio.20133855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallis DE, Roessler E, Hehr U, Nanni L, Wiltshire T, Richieri-Costa A, Gillessen-Kaesbach G, Zackai EH, Rommens J, Muenke M. Mutations in the homeodomain of the human SIX3 gene cause holoprosencephaly. Nat Genet. 1999;22:196–198. doi: 10.1038/9718. [DOI] [PubMed] [Google Scholar]
- Wang H, Yang H, Shivalila CS, Dawlaty MM, Cheng AW, Zhang F, Jaenisch R. One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell. 2013;153:910–918. doi: 10.1016/j.cell.2013.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young JJ, Cherone JM, Doyon Y, Ankoudinova I, Faraji FM, Lee AH, Ngo C, Guschin DY, Paschon DE, Miller JC, Zhang L, Rebar EJ, Gregory PD, Urnov FD, Harland RM, Zeitler B. Efficient targeted gene disruption in the soma and germ line of the frog Xenopus tropicalis using engineered zinc-finger nucleases. 2011;108:7052–7057. doi: 10.1073/pnas.1102030108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuber ME, Gestri G, Viczian AS, Barsacchi G, Harris WA. Specification of the vertebrate eye by a network of eye field transcription factors. Development. 2003;130:5155–5167. doi: 10.1242/dev.00723. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information Fig. S1 Mutation profiles of the six3 gene found in individual embryos injected with sgRNAs targeting different regions. Top sequence shows the positions of target 1 and 2 relative to the translation initiation codon (ATG, marked as Met) to cause frame-shifts. Note that target sites are partially overlapping. “Target 1” lists representative mutated sequences after recloning from two embryos (emb1-1 and emb1-3, see also Fig. 4f, six3 target 1) injected with sgRNA for target site 1. Eleven clones from one embryo (emb1-3) and ten clones from a second embryo (emb1-1) were analyzed. Note that no wild-type sequence (WT) was recovered. “Target 2” shows representative mutated sequences from two embryos (emb2-1 and emb2-2, see also Fig. 4f, six3 target 2) injected with sgRNA to target site 2. Nine clones from one embryo (emb2-1) and five clones from a second embryo (emb2-2) were analyzed. Note that WT sequence was recovered though at a relatively low frequency. “Proximal promoter targets” shows the -574 to +34 region with two sgRNA target sites and representative mutated sequences after recloning (14 clones) from an injected embryo (emb3, see also Fig. 4e). +1 is a tentative transcriptional start site according to the 5’ end of an EST clone (accession number: DC185735). Target sequences and corresponding PAM regions are shaded in purple and red, respectively. The alignment is shown as described in Fig. 2b.
Supporting Information Fig. S2 Confirmation of germline transmission of CRISPR-mediated mutagenesis targeting the tyr gene (target 1). An F0 mature male raised from albino tadpoles injected with the humCas9 (2.2 ng) and sgRNA (tyr target 1, 200 pg) was outcrossed to obtain F1 animals, which were subjected to the DSP assay. Representative chromatograms are shown (top, wild type; bottom, heterozygous mutant carrier). Out of 8 tadpoles tested, 3 showed germline transmission as evidenced by double peaks observed when reading sequence around the target site (red arrow), in contrast to the multiple peaks seen in F0 mosaic animals, see Fig. 2b.