

Abstract
The gammaherpesviruses are unique for their capacity to establish a variety of gene expression programs during latent and lytic infection. This capacity enables the virus to control host-cell proliferation, prevent programmed cell death, elude immune cell detection, and ultimately adapt to a wide range of environmental and developmental changes in the host cell. This remarkable plasticity of gene expression results from the combined functionalities of viral and host factors that biochemically remodel and epigenetically modify the viral chromosome. These epigenetic modifications range from primary DNA methylations, to chromatin protein post-translational modifications, to higher-order chromosome conformations. In addition, gammaherpesviruses have acquired specialized tools to modulate the epigenetic processes that promote viral genome propagation and host-cell survival.
Variations of Gene Expression During Latency
The human gammaherpesviruses, Epstein-Barr Virus (EBV, also HHV4) and Kaposi’s Sarcoma-Associated Herpesvirus (KSHV, also HHV8) can establish latent infections in multiple cell types. During latency, the virus can express a restricted, but variable pattern of viral genes. These different gene expression patterns are essential for viral adaptation to the host and contribute significantly to viral-associated pathogenesis, including carcinogenesis. Epigenetic factors contribute substantially to the formation and heritability of these viral gene expression patterns.
EBV can assume at least four different gene expression programs during latent infection [1]. These “latency types” correlate with the host cell type (e.g. epithelial or lymphoid) or tumor origin (e.g. NPC or BL). In tissue culture, the latency types are metastable, in so far as they can maintain a stable pattern for multiple generations, but can also drift over time or switch in response to environmental signals or pharmacological manipulation. Cells that express all of the latency-associated transcripts (EBNA-LP, 2, 3a, 3b, 3c, 1, LMP-1, 2a, 2b, EBERS, miRNAs, BARTs) are referred to as type III, while those with a more restricted gene expression program are referred to as either Type 0, I, IIa, or IIb. Type 0 refers to the persistence of viral genomes in the absence of viral gene expression, a condition associated with latency in resting memory B-cells. The different latency types have distinct gene expression patterns and corresponding epigenetic modifications, which have been referred to as latency epigenotypes [2].
KSHV also shows variations in gene expression that depend on the cell and tumor type (reviewed in [3]). Most KSHV positive pleural effusion lymphoma (PEL) derived cell lines, express LANA, vCyclin, vFLIP, vmiRNAs, K12, and some variable expression of vIRF-3/LANA-2 [4-8]. In PEL tumor biopsies, rather than established cell lines, KSHV can express additional pathogenic genes, like vGPCR [9]. More sensitive methods using micro-arrays and proteomics have identified additional viral sense and anti-sense transcripts, as well as small peptides that may further refine our understanding of viral gene expression during latency [10]. Similar to PEL cells, KS lesions express the major latency transcripts, but a subset of cells also express some lytic genes, including the multicistronic transcript encoding vGPCR and K14 [11]. Interestingly, in PEL cell lines co-infected with EBV and KSHV, EBV adopts a type II restricted gene expression program [12,13].
Epigenetic Controls of Viral Gene Expression
DNA Methylation
Among the viral genes differentially regulated during EBV latency are those encoded by the long (~100 kb) alternatively spliced transcript initiating at the BamHI C promoter (Cp). These genes include EBNA-LP, EBNA-2, EBNA-3A, EBNA-3B, EBNA-3C, and EBNA1. It is well-established that differential DNA methylation upstream of Cp can lead to stable epigenetic repression [14-19](reviewed in [20]. When Cp DNA is methylated, transcription initiates at Qp to generate a smaller transcript that produces EBNA1, but none of the other EBNA genes. This switch from Cp to Qp is thought to occur during the natural infection as B-cells differentiate from proliferating centroblasts to resting memory B-cells [21]. Concurrently, methylation occurs at the promoter regions for the latency membrane proteins (LMP1 and LPM2), which results in their stable repression. Although BL tumors typically express type I latency pattern, BL derived cell lines can drift into a less restricted type III latency during laboratory cell culture. This shift to type III is thought to be driven by selective advantages of expressing type III genes that promote proliferation and survival, but compensatory changes in c-Myc expression may also be involved [22]. Changes in CpG methylation correlates strongly with the changes in viral promoter selection and latency type [14-16,23,24]. Importantly, treatment with DNA methylation inhibitors, like 5’azacytidine, can reverse repression of Cp transcripts in type I latency, indicating that CpG methylation can be a determinant of latency type [24]. In addition, some regions of the EBV genome are spared DNA methylation. This is particularly true of the regions surrounding Qp and OriP [18,25,26]. Methylation sparing of these regions may be essential for genome persistence during latency since Qp is the default promoter for EBNA1 in type I latency, and OriP is the episome maintenance element [27]. Since EBNA1 binds to both of these regions, it has been proposed that EBNA1 prevents local CpG methylation [28].
Methylation patterns have been examined genome-wide for KSHV [29,30]. Various peaks and valleys of CpG methylation were observed, with remarkable similarity among different cell types, ranging from B-cell derived PELs to in vitro infected SLK cells [29]. In all cases, regions of the viral genome were spared CpG methylation. These include the region immediately upstream of the latency promoter for LANA (LANAp) and several other locations including K9, ORF45/50, K7, and ORF8. The methylation pattern required significant time to establish, since KSHV genomes at 6 days post infection in SLK cells failed to establish any significant patterns of CpG methylation.
Viral-encoded factors can alter DNA methylation machinery by targeting DNA methyltransferases, DNA methyl binding proteins (MBPs), MBP-associated corepressor complexes, and direct binding to methylated DNA [31]. KSHV LANA binds to the de novo methyltransferase DNMT3a [32] and the methyl cytosine binding protein MeCP2 [33]. LANA can stabilize the repression of KSHV lytic cycle gene expression and its interactions with DNA methylation machinery may partly account for this repressing activity. Consistent with this finding is the observation that DNMT3a and 3b mediate CpG methylation and transcription repression of MHV68 ORF50 promoter in latently infected B-lymphocytes in vivo [34]. A remarkable series of findings have revealed a complex and paradoxical role of DNA methylation in the regulation of EBV lytic reactivation (reviewed in more detail in this journal series) [35-40]. The EBV lytic activator Zta has the unusual capacity to bind selectively to methylated DNA, enabling the establishment and reactivation of methylated genomes [35,41-43]. Viral DNA methylation patterns can also be regulated by non-coding RNAs [30]. In one study, deletion of the KSHV miRNA cluster led to a loss of DNA methylation at the ORF50 locus. At least one viral miRNA, miRK12-4-5p, affected viral DNA methylation through the indirect targeting of DNMT3b through the global repressor Rbl2.
Histone Modifications
Viral genomes appear to utilize cellular transcription factors and chromatin regulators indistinguishably from host chromosomes. Most viral promoters share conventional features of cellular epigenetic regulation, with histone acetylation and H3K4me3 methylation occurring at transcribed promoters. Increase histone H3 and H4 acetylation has been demonstrated for the activated Cp in response to EBNA2 activation in type III latency [44], and for activation of the EBV BZLF1/Zta [45,46], and KSHV ORF50/Rta immediate early gene promoters [47]. However, it remains possible that viral episomes are distinguished from cellular chromosomes by some as yet unknown, viral-specific epigenetic features.
Genome-wide studies of histone modifications have revealed several unexpected observations. Initial studies using low resolution PCR arrays, suggested that EBV genomes may be partitioned into relatively large active and inactive chromatin domains, with Cp and Qp representing distinct boundaries for different latency types [48]. More recent studies, using higher resolution tiling arrays or ChIP-Seq methods reveal a more discrete and mottled pattern of epigenetic regulatory marks [29,49,50]. For KSHV genomes, several loci including the immediate early promoter region for ORF50/Rta were found to have a bivalent epigenetic state, similar to that described for embryonic stem cell regulatory genes [51]. The bivalent marks consist of an overlap for H3K4me3, correlating with transcription activity, and H3K27me3, correlating with transcription repression[51]. The EZH2 methylase, a member of the polycomb complex (PcG), was shown to be critical for histone H3K27me3 and repression of KSHV lytic gene reactivation. Interestingly, a pharmacological inhibitor of H3K27 methylation, DZNep, stimulated reactivation of KSHV, but not EBV in dually infected JSC-1 PEL cells, suggesting that KSHV and EBV have different epigenetic controls over lytic reactivation [50].
Mapping of the human ENCODE data base for epigenetic modifications and transcription factor binding sites to the EBV genomes in human LCLs revealed important differences from previous low resolution studies, as well as comparative analysis with KSHV epigenetic marks [49]. Unlike KSHV, levels of H3K27me3 are surprisingly under-represented on the EBV genome, relative to other histone modifications or to the KSHV genome. Examination of the EBV epigenome revealed sharp peaks of histone H3K4me3 and acetylated histones (H3K27ac and H3K9ac) at sites of transcription initiation for EBERs, Cp, LMP1/LMP2, and RPMS1p (the promoter for the BART transcripts and miRNAs). Another feature of the EBV epigenome was the clustering of multiple transcription factors at the sites of active promoter regions, and very few interactions at transcriptionally silent genomic locations.
Chromatin Boundaries
The latent genomes of EBV and KSHV have meta-stable epigenetic modification patterns. The localization of histone H3K4me3 and H3K27ac at sites of transcription initiation are mechanistically explained by the recruitment of RNA polymerase II or III transcription and initiation factors to their cognate DNA recognition sites. High levels of the TATA binding protein (TBP) are found at the EBER promoter, while TBP-associated factors (TAF-1) and histone acetyltransferase co-activator p300 are elevated at the major RNA Pol II initiation sites at Cp, LMP1/LMP2, and RPMS1p [49]. Histone H3K4me3 is elevated at all of these sites, but it is not yet known what histone methylase is responsible for each of these promoter modifications. More cryptic is the mechanism for restricting repressive marks, like H3K9me3 and DNA methylation, to only a few subdomains. As noted above, DNA methylation and H3K9m3 are never found at some protected sites, like the EBV Qp [18] and the KSHV LANA promoter region [29,50]. Chromatin boundary elements may prevent these repressive marks from spreading into protected regions. Among the best-characterized metazoan chromatin boundary factor is the 11-zinc finger DNA binding protein CTCF[52,53]. CTCF binds to a region upstream of the EBV Qp [54] and surrounding the LANA promoter region [55]. Deletion of CTCF from the Qp region leads to an increase in histone H3K9m3, followed by encroaching CpG methylation. Deletion of CTCF from the LANA promoter region results in a loss of episome stability, but there is no evidence that this loss corresponds to an increase in H3K9m3 or DNA methylation. Thus, CTCF appears to function as a chromatin insulator that prevent histone H3K9m3 and DNA methylation at Qp, while it may serve a more complex function at the LANA promoter region. Chromatin boundaries may also be generated by other factors, including the viral encoded maintenance proteins, EBNA1 and LANA, that control nucleosome positioning [56,57] and DNA replication fork stalling [58,59], which may regulate the processive spreading of histone modifications.
Genome Conformation
Higher order DNA structures, like those involving DNA looping that mediate enhancer-promoter interactions, have been identified on both EBV and KSHV chromosomes [60,61]. For EBV, these DNA loop structures correlate with promoter selection, and therefore are likely to be coordinated with epigenetic patterning associated with each latency type. Therefore, higher-order chromatin structure is likely to be a heritable epigenetic regulatory feature. The DNA loops for EBV were found between OriP and either Cp in type III latency or Qp in type I latency [60]. CTCF binding sites located upstream of Qp and Cp were found to be important for mediating the long-distance looping between OriP-Qp in type I latency. These loops may also be mediated by EBNA1, which binds to both OriP and Qp. In KSHV, a DNA loop was identified for the CTCF binding sites in the LANA promoter region. In this case, a loop was formed with the 3’ end of the KSHV latency transcripts ending at K12, suggesting that the entire latency transcription area is contained within a DNA loop mediated in part by CTCF binding. Additionally, a longer DNA loop was also found between the LANA promoter region and the ORF50/Rta promoter control region. DNA loops are stabilized by the cellular factors involved in sister-chromatid cohesion, namely cohesin subunits SMC1, SMC3, RAD21, and SCC2. The cohesins form a ring like structure that can either embrace or hand-cuff two separate DNA molecules to stabilize a non-contiguous DNA-DNA interaction [62-65]. Deletion of the Rad21 subunit leads to a loss of the DNA loop between the latent and lytic control regions, and the reactivation of lytic cycle transcription [66]. These findings suggest that DNA loops are important for maintaining a stable gene expression program during latent infection, but that EBV and KSHV have very different conformations and control mechanisms.
Viral-Encoded Epigenomic Regulators
Viral-encoded factors can modulate the viral and host epigenomes through various mechanisms, including direct interactions with host epigenetic regulatory factors [67]. Here, we consider the contribution of the viral encoded episome maintenance proteins that bind directly to the viral genome and directly influence viral chromosome organization.
EBNA1 binds with high affinity to the FR, DS, and Qp regions of EBV. It can also bind with lower affinity to Rep*[68], and to several sites in the host chromosome [69,70]. At Qp, EBNA1 can function as a repressor of transcription [71], while at OriP it functions as an essential enhancer of Cp and LMP1/LMP2 promoters [72]. At cellular chromosomal sites, EBNA1 binding has no clear effect on transcription of neighboring cellular genes [69,70]. EBNA1 can activate some cellular genes, including NOX2 [73], but its mechanism of activation appears less direct than many well-characterized transcriptional activators and repressors. EBNA1 does not interact with any well-characterized co-activators or co-repressors of transcription. However, EBNA1 does interact with nucleosome assembly proteins and bind to chromatinized DNA [57,74], suggesting that it may play a role in organizing nucleosome position or higher-order chromatin structure. EBNA1 mediates a loop between the FR and DS in vitro, and also mediates transcription activation of Cp during primary infection [72]. Thus, EBNA1 may function as a chromosome architectural protein that organizes nucleosome position and long-distance DNA interactions. EBNA1 is subject to many post-translational modifications, including phosphorylation and poly-ADP ribosylation (PARylation) [75,76]. PARylation of CTCF has been implicated in DNA-looping, and it has been suggested that PARylation of EBNA1 may also contribute to the chromosome architectural functions of EBNA1.
LANA is a functional orthologue of EBNA1, and shares several important features, including the ability to bind to its respective viral genome with high affinity, and attach to metaphase chromosomes [77]. LANA binds the KSHV TR with high affinity, but does not show similar levels of site-specific enrichment at host chromosome sites as does EBNA1[78]. LANA can bind to several sequence specific DNA binding proteins, the best-characterized interaction being with RBP-jK[79]. RBP-jK (also referred to as CBF1 or CSL) is the major target of intracellular Notch, and a common interactor of gammaherpesvirus regulators, including KSHV Rta and LANA, and EBV EBNA2 and EBNA3C. LANA can prevent Rta activation of lytic replication through its competitive interactions with RBP-jK [79-82].
LANA interacts directly with numerous chromatin-associated and epigenetic regulatory factors, including histone H2A/H2B [83], BRD2/4[84], DEK [85], MeCP2 [85], nucleophosmin [86], CENP-F [87], DNMT3a [32], TRF1 [88], p53 [89], TopoII beta [90], and TIP60[88]. Interaction with TIP60, a histone acetyltransferase, may also mediate DNA damage response through acetylation of ATM and other DNA damage response proteins [88]. Interaction with MeCP2 occurs independently of its binding to nucleosomes and functions to localize LANA to chromocenters where it facilitates the transcriptional repression and heterochromatinization of the TRs [33]. However, MeCP2 can also cooperate with LANA to activate some promoters, suggesting that LANA has complex functionality at different chromosomal locations.
LANA is subject to several post-translation modifications that may confer epigenetic regulation of the viral chromosome. These include lysine acetylation[91], arginine methylation by PRMT1 [92], and phosphorylation by PIM1[93,94], CK1, and RSK3 [95]. EBNA1 is also subject to phosphorylation by CK2[96], and CDKs[97], which control important replication and maintenance functions. LANA and EBNA1 can also interact with HAUSP7, a ubiquitin protease that can stabilize p53 and modulate transcription through regulation of histone ubiquitylation cycles [98-100].
Metaphase Attachment Functions
Both LANA and EBNA1 tether the viral episome to the metaphase chromosome, a feature that has correlated strongly with episome maintenance. A potent metaphase attachment motif can be mapped to a stretch of basic amino acids in the amino terminal domain of LANA. This domain interacts with histone H2A/H2B and the interaction has been solved by X-ray crystallography[83]. EBNA1 interacts with metaphase chromosomes through two distinct motifs, an AT-hook motif that can interact with AT-rich DNA [101], and a separate domain that interacts with EBP2, an RNA binding protein that associates with metaphase chromosomes [102]. Both EBNA1 and LANA can interact with the BET family members, BRD4 for EBNA1[103], and BRD2 and 4 for LANA [84,104]. BET family proteins have bromodomains implicated in recognizing acetylated lysine on histone tails [105]. BRD family members have been implicated in the metaphase attachment of HPV E2 proteins [106,107], although they may also play a significant role in transcription regulation [108,109]. BET family proteins can interact with and regulate RNA polymerase II elongation factor pTEFb [110]. BET proteins have also been implicated in mitotic attachment and cell cycle bookmarking of transcriptionally active cellular genes [111,112]. How these viral factors integrate mitotic attachment with episome stability and transcriptional elongation will be an area of important future research.
Conclusions
The epigenetic control of gammaherpesvirus latency is an integral feature of chromosome biology. Epigenetic factors are essential for the complex orchestration of transcription, replication, chromosome mobilization and segregation at the appropriate time and place. Epigenetic features coordinate regulatory events between different genetic locations, through common protein complexes and higher order chromatin structures, including long-intervening DNA loops. These epigenetic regulators play important and significant roles in determining gammaherpesvirus gene regulation during latent infections. More importantly, these epigenetic controls provide both stability and plasticity to the viral genome that enables a programmed response to cellular and environmental changes. Much remains to be discovered for understanding gammaherpesvirus epigenetic controls and how these distinguish latency types. More urgently, will be to determine how epigenetic controls contribute to the formation of aberrant latent epigenotypes that increase risk of virus-driven carcinogenesis [113].
Figure 1. Progressive Epigenetic Modifications Regulate Gammaherpesvirus Latency.
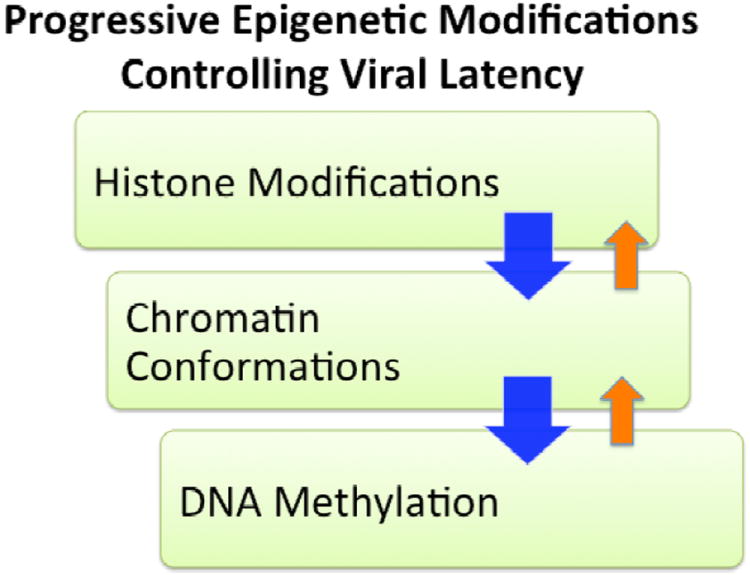
Early stage histone modifications lead to metastable gene expression programs, which are reinforced by secondary higher-order chromosome conformations, including DNA looping. DNA methylation occurs more gradually and may take the place of repressive histone modifications to increase epigenetic stability.
Figure 2. Viral Encoded Epigenetic Modulators.
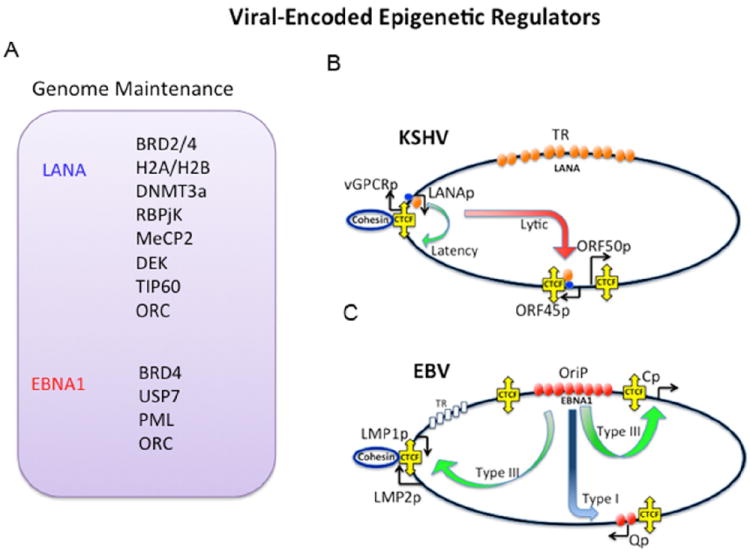
(A) Cellular epigenetic regulators that interact with gammaherpesvirus chromosome organizing/episome maintenance proteins LANA and EBNA1. (B) Schematic of promoter regulatory interactions mediated by viral chromosome organizing factors for KSHV, LANA, and CTCF. (C) Promoter regulatory interactions for EBV in type I or type III latency conformations. OriP, EBNA1, and CTCF are highlighted.
Highlights.
Gammaherpesviruses can adopt a variety of gene expression patterns during latent infection.
Gene expression patterns correlate with changes in epigenetic modifications, including DNA methylation, histone modifications, and chromosome conformations.
Viral episome maintenance proteins play a central role in epigenetic regulation and chromosome organization.
Acknowledgments
Funding was provided by NIH grants (RO1 DE 017336, R01 CA 117830, and RO1 CA 093606).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Rowe M, Lear AL, Croom-Carter D, Davies AH, Rickinson AB. Three pathways of Epstein-Barr virus gene activation from EBNA1-positive latency in B lymphocytes. J Virol. 1992;66:122–131. doi: 10.1128/jvi.66.1.122-131.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Takacs M, Segesdi J, Banati F, Koroknai A, Wolf H, Niller HH, Minarovits J. The importance of epigenetic alterations in the development of epstein-barr virus-related lymphomas. Mediterr J Hematol Infect Dis. 2009;1:e2009012. doi: 10.4084/MJHID.2009.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pantry SN, Medveczky PG. Epigenetic regulation of Kaposi’s sarcoma-associated herpesvirus replication. Semin Cancer Biol. 2009;19:153–157. doi: 10.1016/j.semcancer.2009.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fakhari FD, Dittmer DP. Charting latency transcripts in Kaposi’s sarcoma-associated herpesvirus by whole-genome real-time quantitative PCR. J Virol. 2002;76:6213–6223. doi: 10.1128/JVI.76.12.6213-6223.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jenner RG, Alba MM, Boshoff C, Kellam P. Kaposi’s sarcoma-associated herpesvirus latent and lytic gene expression as revealed by DNA arrays. J Virol. 2001;75:891–902. doi: 10.1128/JVI.75.2.891-902.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Paulose-Murphy M, Ha NK, Xiang C, Chen Y, Gillim L, Yarchoan R, Meltzer P, Bittner M, Trent J, Zeichner S. Transcription program of human herpesvirus 8 (kaposi’s sarcoma-associated herpesvirus) J Virol. 2001;75:4843–4853. doi: 10.1128/JVI.75.10.4843-4853.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li H, Komatsu T, Dezube BJ, Kaye KM. The Kaposi’s sarcoma-associated herpesvirus K12 transcript from a primary effusion lymphoma contains complex repeat elements, is spliced, and initiates from a novel promoter. J Virol. 2002;76:11880–11888. doi: 10.1128/JVI.76.23.11880-11888.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pearce M, Matsumura S, Wilson AC. Transcripts encoding K12, v-FLIP, v-cyclin, and the microRNA cluster of Kaposi’s sarcoma-associated herpesvirus originate from a common promoter. J Virol. 2005;79:14457–14464. doi: 10.1128/JVI.79.22.14457-14464.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nador RG, Milligan LL, Flore O, Wang X, Arvanitakis L, Knowles DM, Cesarman E. Expression of Kaposi’s sarcoma-associated herpesvirus G protein-coupled receptor monocistronic and bicistronic transcripts in primary effusion lymphomas. Virology. 2001;287:62–70. doi: 10.1006/viro.2001.1016. [DOI] [PubMed] [Google Scholar]
- 10*.Dresang LR, Teuton JR, Feng H, Jacobs JM, Camp DG, 2nd, Purvine SO, Gritsenko MA, Li Z, Smith RD, Sugden B, et al. Coupled transcriptome and proteome analysis of human lymphotropic tumor viruses: insights on the detection and discovery of viral genes. BMC Genomics. 2011;12:625. doi: 10.1186/1471-2164-12-625. Identification of new viral transcripts and polypeptides during KSHV and EBV latency. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dittmer DP. Transcription profile of Kaposi’s sarcoma-associated herpesvirus in primary Kaposi’s sarcoma lesions as determined by real-time PCR arrays. Cancer Res. 2003;63:2010–2015. [PubMed] [Google Scholar]
- 12.Trivedi P, Takazawa K, Zompetta C, Cuomo L, Anastasiadou E, Carbone A, Uccini S, Belardelli F, Takada K, Frati L, et al. Infection of HHV-8+ primary effusion lymphoma cells with a recombinant Epstein-Barr virus leads to restricted EBV latency, altered phenotype, and increased tumorigenicity without affecting TCL1 expression. Blood. 2004;103:313–316. doi: 10.1182/blood-2003-05-1710. [DOI] [PubMed] [Google Scholar]
- 13.Fassone L, Bhatia K, Gutierrez M, Capello D, Gloghini A, Dolcetti R, Vivenza D, Ascoli V, Lo Coco F, Pagani L, et al. Molecular profile of Epstein-Barr virus infection in HHV-8-positive primary effusion lymphoma. Leukemia. 2000;14:271–277. doi: 10.1038/sj.leu.2401651. [DOI] [PubMed] [Google Scholar]
- 14.Tao Q, Swinnen LJ, Yang J, Srivastava G, Robertson KD, Ambinder RF. Methylation status of the Epstein-Barr virus major latent promoter C in iatrogenic B cell lymphoproliferative disease. Application of PCR-based analysis. Am J Pathol. 1999;155:619–625. doi: 10.1016/S0002-9440(10)65157-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Robertson KD, Manns A, Swinnen LJ, Zong JC, Gulley ML, Ambinder RF. CpG methylation of the major Epstein-Barr virus latency promoter in Burkitt’s lymphoma and Hodgkin’s disease. Blood. 1996;88:3129–3136. [PubMed] [Google Scholar]
- 16.Ambinder RF, Robertson KD, Tao Q. DNA methylation and the Epstein-Barr virus. Semin Cancer Biol. 1999;9:369–375. doi: 10.1006/scbi.1999.0137. [DOI] [PubMed] [Google Scholar]
- 17.Minarovits J. Epigenotypes of latent herpesvirus genomes. Curr Top Microbiol Immunol. 2006;310:61–80. doi: 10.1007/3-540-31181-5_5. [DOI] [PubMed] [Google Scholar]
- 18.Fejer G, Koroknai A, Banati F, Gyory I, Salamon D, Wolf H, Niller HH. Minarovits J: Latency type-specific distribution of epigenetic marks at the alternative promoters Cp and Qp of Epstein-Barr virus. J Gen Virol. 2008;89:1364–1370. doi: 10.1099/vir.0.83594-0. [DOI] [PubMed] [Google Scholar]
- 19.Bakos A, Banati F, Koroknai A, Takacs M, Salamon D, Minarovits-Kormuta S, Schwarzmann F, Wolf H, Niller HH, Minarovits J. High-resolution analysis of CpG methylation and in vivo protein-DNA interactions at the alternative Epstein-Barr virus latency promoters Qp and Cp in the nasopharyngeal carcinoma cell line C666-1. Virus Genes. 2007;35:195–202. doi: 10.1007/s11262-007-0095-y. [DOI] [PubMed] [Google Scholar]
- 20.Smuk G, Illes A, Keresztes K, Kereskai L, Marton B, Nagy Z, Lacza A, Pajor L. Pheno- and genotypic features of Epstein-Barr virus associated B-cell lymphoproliferations in peripheral T-cell lymphomas. Pathol Oncol Res. 2009;16:377–383. doi: 10.1007/s12253-009-9233-2. [DOI] [PubMed] [Google Scholar]
- 21.Thorley-Lawson DA, Gross A. Persistence of the Epstein-Barr virus and the origins of associated lymphomas. N Engl J Med. 2004;350:1328–1337. doi: 10.1056/NEJMra032015. [DOI] [PubMed] [Google Scholar]
- 22.Kelly G, Bell A, Rickinson A. Epstein-Barr virus-associated Burkitt lymphomagenesis selects for downregulation of the nuclear antigen EBNA2. Nat Med. 2002;8:1098–1104. doi: 10.1038/nm758. [DOI] [PubMed] [Google Scholar]
- 23.Robertson KD, Ambinder RF. Methylation of the Epstein-Barr virus genome in normal lymphocytes. Blood. 1997;90:4480–4484. [PubMed] [Google Scholar]
- 24.Robertson KD, Hayward SD, Ling PD, Samid D, Ambinder RF. Transcriptional activation of the Epstein-Barr virus latency C promoter after 5-azacytidine treatment: evidence that demethylation at a single CpG site is crucial. Mol Cell Biol. 1995;15:6150–6159. doi: 10.1128/mcb.15.11.6150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Salamon D, Takacs M, Ujvari D, Uhlig J, Wolf H, Minarovits J, Niller HH. Protein-DNA binding and CpG methylation at nucleotide resolution of latency-associated promoters Qp, Cp, and LMP1p of Epstein-Barr virus. J Virol. 2001;75:2584–2596. doi: 10.1128/JVI.75.6.2584-2596.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schaefer BC, Strominger JL, Speck SH. Host-cell-determined methylation of specific Epstein-Barr virus promoters regulates the choice between distinct viral latency programs. Mol Cell Biol. 1997;17:364–377. doi: 10.1128/mcb.17.1.364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Paulson EJ, Speck SH. Differential methylation of Epstein-Barr virus latency promoters facilitates viral persistence in healthy seropositive individuals. J Virol. 1999;73:9959–9968. doi: 10.1128/jvi.73.12.9959-9968.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hsieh CL. Evidence that protein binding specifies sites of DNA demethylation. Mol Cell Biol. 1999;19:46–56. doi: 10.1128/mcb.19.1.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29**.Gunther T, Grundhoff A. The epigenetic landscape of latent Kaposi sarcoma-associated herpesvirus genomes. PLoS Pathog. 2010;6:e1000935. doi: 10.1371/journal.ppat.1000935. Genome-wide study of the KSHV methylation reveals conserved patterns of DNA methylation in different cell type models of latency. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lu F, Stedman W, Yousef M, Renne R, Lieberman PM. Epigenetic regulation of Kaposi’s sarcoma-associated herpesvirus latency by virus-encoded microRNAs that target Rta and the cellular Rbl2-DNMT pathway. J Virol. 2010;84:2697–2706. doi: 10.1128/JVI.01997-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dawson MA, Kouzarides T. Cancer epigenetics: from mechanism to therapy. Cell. 2012;150:12–27. doi: 10.1016/j.cell.2012.06.013. [DOI] [PubMed] [Google Scholar]
- 32.Shamay M, Krithivas A, Zhang J, Hayward SD. Recruitment of the de novo DNA methyltransferase Dnmt3a by Kaposi’s sarcoma-associated herpesvirus LANA. Proc Natl Acad Sci U S A. 2006;103:14554–14559. doi: 10.1073/pnas.0604469103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Matsumura S, Persson LM, Wong L, Wilson AC. The latency-associated nuclear antigen interacts with MeCP2 and nucleosomes through separate domains. J Virol. 2009;84:2318–2330. doi: 10.1128/JVI.01097-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gray KS, Forrest JC, Speck SH. The de novo methyltransferases DNMT3a and DNMT3b target the murine gammaherpesvirus immediate-early gene 50 promoter during establishment of latency. J Virol. 2010;84:4946–4959. doi: 10.1128/JVI.00060-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35*.Bergbauer M, Kalla M, Schmeinck A, Gobel C, Rothbauer U, Eck S, Benet-Pages A, Strom TM, Hammerschmidt W. CpG-methylation regulates a class of Epstein-Barr virus promoters. PLoS Pathog. 2010;6:e1001114. doi: 10.1371/journal.ppat.1001114. Paradoxical regulatory role of DNA methylation in the control of EBV latency and reactivation through selective recognition of cytosine methylation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kalla M, Schmeinck A, Bergbauer M, Pich D, Hammerschmidt W. AP-1 homolog BZLF1 of Epstein-Barr virus has two essential functions dependent on the epigenetic state of the viral genome. Proc Natl Acad Sci U S A. 2010;107:850–855. doi: 10.1073/pnas.0911948107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kalla M, Gobel C, Hammerschmidt W. The lytic phase of Epstein-Barr virus requires a viral genome with 5-methylcytosine residues in CpG sites. J Virol. 2011 doi: 10.1128/JVI.06314-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sinclair AJ. Unexpected structure of Epstein-Barr virus lytic cycle activator Zta. Trends Microbiol. 2006;14:289–291. doi: 10.1016/j.tim.2006.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Karlsson QH, Schelcher C, Verrall E, Petosa C, Sinclair AJ. Methylated DNA recognition during the reversal of epigenetic silencing is regulated by cysteine and serine residues in the Epstein-Barr virus lytic switch protein. PLoS Pathog. 2008;4:e1000005. doi: 10.1371/journal.ppat.1000005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Karlsson QH, Schelcher C, Verrall E, Petosa C, Sinclair AJ. The reversal of epigenetic silencing of the EBV genome is regulated by viral bZIP protein. Biochem Soc Trans. 2008;36:637–639. doi: 10.1042/BST0360637. [DOI] [PubMed] [Google Scholar]
- 41.Bhende PM, Seaman WT, Delecluse HJ, Kenney SC. The EBV lytic switch protein, Z, preferentially binds to and activates the methylated viral genome. Nat Genet. 2004;36:1099–1104. doi: 10.1038/ng1424. [DOI] [PubMed] [Google Scholar]
- 42.Bhende PM, Seaman WT, Delecluse HJ, Kenney SC. BZLF1 activation of the methylated form of the BRLF1 immediate-early promoter is regulated by BZLF1 residue 186. J Virol. 2005;79:7338–7348. doi: 10.1128/JVI.79.12.7338-7348.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43*.Ramasubramanyan S, Kanhere A, Osborn K, Flower K, Jenner RG, Sinclair AJ. Genome-Wide Analyses of Zta Binding to the Epstein-Barr Virus Genome Reveals Interactions in both Early and Late Lytic Cycles and an Epigenetic Switch Leading to an Altered Binding Profile. J Virol. 2012;86:12494–12502. doi: 10.1128/JVI.01705-12. Demonstration of the role of Zta recognition of methylated cytosine in viral reactivation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Alazard N, Gruffat H, Hiriart E, Sergeant A, Manet E. Differential hyperacetylation of histones H3 and H4 upon promoter-specific recruitment of EBNA2 in Epstein-Barr virus chromatin. J Virol. 2003;77:8166–8172. doi: 10.1128/JVI.77.14.8166-8172.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Deng Z, Chen CJ, Chamberlin M, Lu F, Blobel GA, Speicher D, Cirillo LA, Zaret KS, Lieberman PM. The CBP bromodomain and nucleosome targeting are required for Zta-directed nucleosome acetylation and transcription activation. Mol Cell Biol. 2003;23:2633–2644. doi: 10.1128/MCB.23.8.2633-2644.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Miller G, El-Guindy A, Countryman J, Ye J, Gradoville L. Lytic cycle switches of oncogenic human gammaherpesviruses(1) Adv Cancer Res. 2007;97:81–109. doi: 10.1016/S0065-230X(06)97004-3. [DOI] [PubMed] [Google Scholar]
- 47.Lu F, Zhou J, Wiedmer A, Madden K, Yuan Y, Lieberman PM. Chromatin remodeling of the Kaposi’s sarcoma-associated herpesvirus ORF50 promoter correlates with reactivation from latency. J Virol. 2003;77:11425–11435. doi: 10.1128/JVI.77.21.11425-11435.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Day L, Chau CM, Nebozhyn M, Rennenkamp AJ, Showe M, Lieberman PM. Chromatin Profiling Of Epstein-Barr Virus Latency Control Region. J Virol. 2007 doi: 10.1128/JVI.02172-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49**.Arvey A, Tempera I, Tsai K, Chen HS, Tikhmyanova N, Klichinsky M, Leslie C, Lieberman PM. An atlas of the epstein-barr virus transcriptome and epigenome reveals host-virus regulatory interactions. Cell Host Microbe. 2012;12:233–245. doi: 10.1016/j.chom.2012.06.008. Analysis of ENCODE data sets for epigenetic features of EBV episomes during latency in LCLs. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50**.Toth Z, Maglinte DT, Lee SH, Lee HR, Wong LY, Brulois KF, Lee S, Buckley JD, Laird PW, Marquez VE, et al. Epigenetic analysis of KSHV latent and lytic genomes. PLoS Pathog. 2010;6:e1001013. doi: 10.1371/journal.ppat.1001013. Genome wide analysis of KSHV histone modification patterns revealed bivalent modifications of immediate early regulatory regions. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bernstein BE, Mikkelsen TS, Xie X, Kamal M, Huebert DJ, Cuff J, Fry B, Meissner A, Wernig M, Plath K, et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell. 2006;125:315–326. doi: 10.1016/j.cell.2006.02.041. [DOI] [PubMed] [Google Scholar]
- 52.Phillips JE, Corces VG. CTCF: master weaver of the genome. Cell. 2009;137:1194–1211. doi: 10.1016/j.cell.2009.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ohlsson R, Lobanenkov V, Klenova E. Does CTCF mediate between nuclear organization and gene expression? Bioessays. 2010;32:37–50. doi: 10.1002/bies.200900118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54*.Tempera I, Wiedmer A, Dheekollu J, Lieberman PM. CTCF prevents the epigenetic drift of EBV latency promoter Qp. PLoS Pathog. 2010;6 doi: 10.1371/journal.ppat.1001048. Genetic demonstration of the the role of chromatin boundary factor CTCF in protection of EBV latency promoter Qp. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Stedman W, Kang H, Lin S, Kissil JL, Bartolomei MS, Lieberman PM. Cohesins localize with CTCF at the KSHV latency control region and at cellular c-myc and H19/Igf2 insulators. EMBO J. 2008;27:654–666. doi: 10.1038/emboj.2008.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Avolio-Hunter TM, Lewis PN, Frappier L. Epstein-Barr nuclear antigen 1 binds and destabilizes nucleosomes at the viral origin of latent DNA replication. Nucleic Acids Res. 2001;29:3520–3528. doi: 10.1093/nar/29.17.3520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Avolio-Hunter TM, Frappier L. EBNA1 efficiently assembles on chromatin containing the Epstein-Barr virus latent origin of replication. Virology. 2003;315:398–408. doi: 10.1016/s0042-6822(03)00561-0. [DOI] [PubMed] [Google Scholar]
- 58.Dheekollu J, Lieberman PM. The replisome pausing factor Timeless is required for episomal maintenance of latent Epstein-Barr virus. J Virol. 2011;85:5853–5863. doi: 10.1128/JVI.02425-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dheekollu J, Chen HS, Kaye KM, Lieberman PM. Timeless-dependent DNA Replication-coupled Recombination Promote KSHV Episome Maintenance and Terminal Repeat Stability. J Virol. 2013 doi: 10.1128/JVI.02211-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60**.Tempera I, Klichinsky M, Lieberman PM. EBV latency types adopt alternative chromatin conformations. PLoS Pathog. 2011;7:e1002180. doi: 10.1371/journal.ppat.1002180. Identification of a DNA loop between the OriP enhancer and the active promoters at either Cp or Qp. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61*.Kang H, Wiedmer A, Yuan Y, Robertson E, Lieberman PM. Coordination of KSHV latent and lytic gene control by CTCF-cohesin mediated chromosome conformation. PLoS Pathog. 2011;7:e1002140. doi: 10.1371/journal.ppat.1002140. 3C methods to demonstrate that CTCF-cohesin mediate a DNA loop between the promoter regulatory regions of the latency transcript and the lytic immediate early genes. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nasmyth K, Haering CH. The structure and function of SMC and kleisin complexes. Annu Rev Biochem. 2005;74:595–648. doi: 10.1146/annurev.biochem.74.082803.133219. [DOI] [PubMed] [Google Scholar]
- 63.Losada A. Cohesin regulation: fashionable ways to wear a ring. Chromosoma. 2007;116:321–329. doi: 10.1007/s00412-007-0104-x. [DOI] [PubMed] [Google Scholar]
- 64.Dorsett D. Cohesin: genomic insights into controlling gene transcription and development. Curr Opin Genet Dev. 2011;21:199–206. doi: 10.1016/j.gde.2011.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chien R, Zeng W, Ball AR, Yokomori K. Cohesin: a critical chromatin organizer in mammalian gene regulation. Biochem Cell Biol. 2011;89:445–458. doi: 10.1139/o11-039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chen HS, Wikramasinghe P, Showe L, Lieberman PM. Cohesins repress Kaposi’s sarcoma-associated herpesvirus immediate early gene transcription during latency. J Virol. 2012;86:9454–9464. doi: 10.1128/JVI.00787-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Paschos K, Allday MJ. Epigenetic reprogramming of host genes in viral and microbial pathogenesis. Trends Microbiol. 2010;18:439–447. doi: 10.1016/j.tim.2010.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kirchmaier AL, Sugden B. Rep*: a viral element that can partially replace the origin of plasmid DNA synthesis of Epstein-Barr virus. J Virol. 1998;72:4657–4666. doi: 10.1128/jvi.72.6.4657-4666.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Dresang LR, Vereide DT, Sugden B. Identifying sites bound by Epstein-Barr virus nuclear antigen 1 (EBNA1) in the human genome: defining a position-weighted matrix to predict sites bound by EBNA1 in viral genomes. J Virol. 2009;83:2930–2940. doi: 10.1128/JVI.01974-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lu F, Wikramasinghe P, Norseen J, Tsai K, Wang P, Showe L, Davuluri RV, Lieberman PM. Genome-wide analysis of host-chromosome binding sites for Epstein-Barr Virus Nuclear Antigen 1 (EBNA1) Virol J. 2010;7:262. doi: 10.1186/1743-422X-7-262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ruf IK, Moghaddam A, Wang F, Sample J. Mechanisms that regulate Epstein-Barr virus EBNA-1 gene transcription during restricted latency are conserved among lymphocryptoviruses of Old World primates. J Virol. 1999;73:1980–1989. doi: 10.1128/jvi.73.3.1980-1989.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Altmann M, Pich D, Ruiss R, Wang J, Sugden B, Hammerschmidt W. Transcriptional activation by EBV nuclear antigen 1 is essential for the expression of EBV’s transforming genes. Proc Natl Acad Sci U S A. 2006;103:14188–14193. doi: 10.1073/pnas.0605985103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Gruhne B, Sompallae R, Marescotti D, Kamranvar SA, Gastaldello S, Masucci MG. The Epstein-Barr virus nuclear antigen-1 promotes genomic instability via induction of reactive oxygen species. Proc Natl Acad Sci U S A. 2009;106:2313–2318. doi: 10.1073/pnas.0810619106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wang S, Frappier L. Nucleosome assembly proteins bind to Epstein-Barr virus nuclear antigen 1 and affect its functions in DNA replication and transcriptional activation. J Virol. 2009;83:11704–11714. doi: 10.1128/JVI.00931-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tempera I, Deng Z, Atanasiu C, Chen CJ, D’Erme M, Lieberman PM. Regulation of Epstein-Barr virus OriP replication by poly(ADP-ribose) polymerase 1. J Virol. 2010;84:4988–4997. doi: 10.1128/JVI.02333-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Deng Z, Atanasiu C, Zhao K, Marmorstein R, Sbodio JI, Chi NW, Lieberman PM. Inhibition of Epstein-Barr virus OriP function by tankyrase, a telomere-associated poly-ADP ribose polymerase that binds and modifies EBNA1. J Virol. 2005;79:4640–4650. doi: 10.1128/JVI.79.8.4640-4650.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ballestas ME, Kaye KM. The latency-associated nuclear antigen, a multifunctional protein central to Kaposi’s sarcoma-associated herpesvirus latency. Future Microbiol. 2011;6:1399–1413. doi: 10.2217/fmb.11.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lu F, Tsai K, Chen HS, Wikramasinghe P, Davuluri RV, Showe L, Domsic J, Marmorstein R, Lieberman PM. Identification of Host-Chromosome Binding Sites and Candidate Gene Targets for KSHV LANA. J Virol. 2012 doi: 10.1128/JVI.07216-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Jin Y, He Z, Liang D, Zhang Q, Zhang H, Deng Q, Robertson ES, Lan K. LANA Carboxyl Terminal Amino Acids 1052 to 1082 Interact with RBP-Jkappa and are Responsible for LANA-Mediated RTA Repression. J Virol. 2012 doi: 10.1128/JVI.06788-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lu J, Verma SC, Cai Q, Robertson ES. The single RBP-Jkappa site within the LANA promoter is crucial for establishing Kaposi’s sarcoma-associated herpesvirus latency during primary infection. J Virol. 2011;85:6148–6161. doi: 10.1128/JVI.02608-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lan K, Kuppers DA, Robertson ES. Kaposi’s sarcoma-associated herpesvirus reactivation is regulated by interaction of latency-associated nuclear antigen with recombination signal sequence-binding protein Jkappa, the major downstream effector of the Notch signaling pathway. J Virol. 2005;79:3468–3478. doi: 10.1128/JVI.79.6.3468-3478.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82*.Lu J, Verma SC, Cai Q, Saha A, Dzeng RK, Robertson ES. The RBP-Jkappa binding sites within the RTA promoter regulate KSHV latent infection and cell proliferation. PLoS Pathog. 2012;8:e1002479. doi: 10.1371/journal.ppat.1002479. Genetic demonstration that RBP-jK binding sites regulate lytic immediate early promoters of KSHV. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Barbera AJ, Chodaparambil JV, Kelley-Clarke B, Joukov V, Walter JC, Luger K, Kaye KM. The nucleosomal surface as a docking station for Kaposi’s sarcoma herpesvirus LANA. Science. 2006;311:856–861. doi: 10.1126/science.1120541. [DOI] [PubMed] [Google Scholar]
- 84.Ottinger M, Christalla T, Nathan K, Brinkmann MM, Viejo-Borbolla A, Schulz TF. Kaposi’s sarcoma-associated herpesvirus LANA-1 interacts with the short variant of BRD4 and releases cells from a BRD4- and BRD2/RING3-induced G1 cell cycle arrest. J Virol. 2006;80:10772–10786. doi: 10.1128/JVI.00804-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85*.Krithivas A, Fujimuro M, Weidner M, Young DB, Hayward SD. Protein interactions targeting the latency-associated nuclear antigen of Kaposi’s sarcoma-associated herpesvirus to cell chromosomes. J Virol. 2002;76:11596–11604. doi: 10.1128/JVI.76.22.11596-11604.2002. Reveals a novel mechanism of chromatin control by KSHV latency proteins. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Sarek G, Jarviluoma A, Moore HM, Tojkander S, Vartia S, Biberfeld P, Laiho M, Ojala PM. Nucleophosmin phosphorylation by v-cyclin-CDK6 controls KSHV latency. PLoS Pathog. 2010;6:e1000818. doi: 10.1371/journal.ppat.1000818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Xiao B, Verma SC, Cai Q, Kaul R, Lu J, Saha A, Robertson ES. Bub1 and CENP-F can contribute to Kaposi’s sarcoma-associated herpesvirus genome persistence by targeting LANA to kinetochores. J Virol. 2010;84:9718–9732. doi: 10.1128/JVI.00713-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Shamay M, Liu J, Li R, Liao G, Shen L, Greenway M, Hu S, Zhu J, Zhi X, Ambinder RF, et al. A Protein Array Screen for KSHV LANA Interactors Links LANA to TIP60, PP2A activity and Telomere Shortening. J Virol. 2012 doi: 10.1128/JVI.00169-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Chen W, Hilton IB, Staudt MR, Burd CE, Dittmer DP. Distinct p53, p53:LANA, and LANA complexes in Kaposi’s Sarcoma--associated Herpesvirus Lymphomas. J Virol. 2010;84:3898–3908. doi: 10.1128/JVI.01321-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Purushothaman P, McDowell ME, McGuinness J, Salas R, Rumjahn SM, Verma SC. Kaposi’s sarcoma-associated herpesvirus-encoded LANA recruits topoisomerase IIbeta for latent DNA replication of the terminal repeats. J Virol. 2012;86:9983–9994. doi: 10.1128/JVI.00839-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lu F, Day L, Gao SJ, Lieberman PM. Acetylation of the latency-associated nuclear antigen regulates repression of Kaposi’s sarcoma-associated herpesvirus lytic transcription. J Virol. 2006;80:5273–5282. doi: 10.1128/JVI.02541-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Campbell M, Chang PC, Huerta S, Izumiya C, Davis R, Tepper CG, Kim KY, Shevchenko B, Wang DH, Jung JU, et al. Protein arginine methyltransferase 1-directed methylation of Kaposi sarcoma-associated herpesvirus latency-associated nuclear antigen. J Biol Chem. 2011;287:5806–5818. doi: 10.1074/jbc.M111.289496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Cheng F, Weidner-Glunde M, Varjosalo M, Rainio EM, Lehtonen A, Schulz TF, Koskinen PJ, Taipale J, Ojala PM. KSHV reactivation from latency requires Pim-1 and Pim-3 kinases to inactivate the latency-associated nuclear antigen LANA. PLoS Pathog. 2009;5:e1000324. doi: 10.1371/journal.ppat.1000324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Bajaj BG, Verma SC, Lan K, Cotter MA, Woodman ZL, Robertson ES. KSHV encoded LANA upregulates Pim-1 and is a substrate for its kinase activity. Virology. 2006;351:18–28. doi: 10.1016/j.virol.2006.03.037. [DOI] [PubMed] [Google Scholar]
- 95.Woodard C, Shamay M, Liao G, Zhu J, Ng AN, Li R, Newman R, Rho HS, Hu J, Wan J, et al. Phosphorylation of the Chromatin Binding Domain of KSHV LANA. PLoS Pathog. 2012;8:e1002972. doi: 10.1371/journal.ppat.1002972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Sivachandran N, Cao JY, Frappier L. Epstein-Barr virus nuclear antigen 1 Hijacks the host kinase CK2 to disrupt PML nuclear bodies. J Virol. 2010;84:11113–11123. doi: 10.1128/JVI.01183-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kang MS, Lee EK, Soni V, Lewis TA, Koehler AN, Srinivasan V, Kieff E. Roscovitine inhibits EBNA1 serine 393 phosphorylation, nuclear localization, transcription, and episome maintenance. J Virol. 2011;85:2859–2868. doi: 10.1128/JVI.01628-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Saridakis V, Sheng Y, Sarkari F, Holowaty MN, Shire K, Nguyen T, Zhang RG, Liao J, Lee W, Edwards AM, et al. Structure of the p53 binding domain of HAUSP/USP7 bound to Epstein-Barr nuclear antigen 1 implications for EBV-mediated immortalization. Mol Cell. 2005;18:25–36. doi: 10.1016/j.molcel.2005.02.029. [DOI] [PubMed] [Google Scholar]
- 99.Jager W, Santag S, Weidner-Glunde M, Gellermann E, Kati S, Pietrek M, Viejo-Borbolla A, Schulz TF. The ubiquitin-specific protease USP7 modulates the replication of Kaposi’s sarcoma-associated herpesvirus latent episomal DNA. J Virol. 2012;86:6745–6757. doi: 10.1128/JVI.06840-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Sarkari F, Wang X, Nguyen T, Frappier L. The herpesvirus associated ubiquitin specific protease, USP7, is a negative regulator of PML proteins and PML nuclear bodies. PLoS One. 2011;6:e16598. doi: 10.1371/journal.pone.0016598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Sears J, Ujihara M, Wong S, Ott C, Middeldorp J, Aiyar A. The amino terminus of Epstein-Barr Virus (EBV) nuclear antigen 1 contains AT hooks that facilitate the replication and partitioning of latent EBV genomes by tethering them to cellular chromosomes. J Virol. 2004;78:11487–11505. doi: 10.1128/JVI.78.21.11487-11505.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Nayyar VK, Shire K, Frappier L. Mitotic chromosome interactions of Epstein-Barr nuclear antigen 1 (EBNA1) and human EBNA1-binding protein 2 (EBP2) J Cell Sci. 2009;122:4341–4350. doi: 10.1242/jcs.060913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Lin A, Wang S, Nguyen T, Shire K, Frappier L. The EBNA1 protein of Epstein-Barr virus functionally interacts with Brd4. J Virol. 2008;82:12009–12019. doi: 10.1128/JVI.01680-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Viejo-Borbolla A, Ottinger M, Bruning E, Burger A, Konig R, Kati E, Sheldon JA, Schulz TF. Brd2/RING3 interacts with a chromatin-binding domain in the Kaposi’s Sarcoma-associated herpesvirus latency-associated nuclear antigen 1 (LANA-1) that is required for multiple functions of LANA-1. J Virol. 2005;79:13618–13629. doi: 10.1128/JVI.79.21.13618-13629.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Filippakopoulos P, Knapp S. The bromodomain interaction module. FEBS Lett. 2012;586:2692–2704. doi: 10.1016/j.febslet.2012.04.045. [DOI] [PubMed] [Google Scholar]
- 106.You J, Croyle JL, Nishimura A, Ozato K, Howley PM. Interaction of the bovine papillomavirus E2 protein with Brd4 tethers the viral DNA to host mitotic chromosomes. Cell. 2004;117:349–360. doi: 10.1016/s0092-8674(04)00402-7. [DOI] [PubMed] [Google Scholar]
- 107.Abbate EA, Voitenleitner C, Botchan MR. Structure of the papillomavirus DNA-tethering complex E2:Brd4 and a peptide that ablates HPV chromosomal association. Mol Cell. 2006;24:877–889. doi: 10.1016/j.molcel.2006.11.002. [DOI] [PubMed] [Google Scholar]
- 108.Wu SY, Lee AY, Hou SY, Kemper JK, Erdjument-Bromage H, Tempst P, Chiang CM. Brd4 links chromatin targeting to HPV transcriptional silencing. Genes Dev. 2006;20:2383–2396. doi: 10.1101/gad.1448206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.McPhillips MG, Oliveira JG, Spindler JE, Mitra R, McBride AA. Brd4 is required for e2-mediated transcriptional activation but not genome partitioning of all papillomaviruses. J Virol. 2006;80:9530–9543. doi: 10.1128/JVI.01105-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Wu SY, Chiang CM. The double bromodomain-containing chromatin adaptor Brd4 and transcriptional regulation. J Biol Chem. 2007;282:13141–13145. doi: 10.1074/jbc.R700001200. [DOI] [PubMed] [Google Scholar]
- 111.Devaiah BN, Singer DS. Two Faces of BRD4: Mitotic Bookmark and Transcriptional Lynchpin. Transcription. 2012;4 doi: 10.4161/trns.22542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Voigt P, Reinberg D. BRD4 jump-starts transcription after mitotic silencing. Genome Biol. 2011;12:133. doi: 10.1186/gb-2011-12-11-133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113**.Moore PS, Chang Y. Why do viruses cause cancer? Highlights of the first century of human tumour virology. Nat Rev Cancer. 2010;10:878–889. doi: 10.1038/nrc2961. Considers the role of abortive and aberrant infection in viral oncogenesis. [DOI] [PMC free article] [PubMed] [Google Scholar]