

Abstract
Vitamin E is likely the most important antioxidant in the human diet and α-tocopherol is the most active isomer. α-Tocopherol exhibits anti-oxidative capacity in vitro, and inhibits oxidation of LDL. Beside this, α-tocopherol shows anti-inflammatory activity and modulates expression of proteins involved in uptake, transport and degradation of tocopherols, as well as the uptake, storage and export of lipids such as cholesterol. Despite promising anti-atherogenic features in vitro, vitamin E failed to be atheroprotective in clinical trials in humans. Recent studies highlight the importance of long-chain metabolites of α-tocopherol, which are formed as catabolic intermediate products in the liver and occur in human plasma. These metabolites modulate inflammatory processes and macrophage foam cell formation via mechanisms different than that of their metabolic precursor α-tocopherol and at lower concentrations. Here we summarize the controversial role of vitamin E as a preventive agent against atherosclerosis and point the attention to recent findings that highlight a role of these long-chain metabolites of vitamin E as a proposed new class of regulatory metabolites. We speculate that the metabolites contribute to physiological as well as pathophysiological processes.
Keywords: α-Tocopherol, α-Tocopherol long-chain metabolites, α-13'-COOH, Atherosclerosis, Macrophage foam cells
Highlights
-
•
Macrophage recruitment, proliferation and transformation into foam cells trigger atherosclerosis.
-
•
α-Tocopherol acts as an anti-atherogenic agent in vitro but not in vivo.
-
•
α-Tocopherol and α-tocopherol long-chain metabolites modulate macrophage foam cell formation via different pathways.
-
•
α-Tocopherol long-chain metabolites modulate cellular processes at lower concentrations than α-tocopherol.
-
•
α-Tocopherol long-chain metabolites occur in human plasma and may represent a new class of regulatory metabolites.
Nomenclature
- α-13'-OH
α-13'-(6-hydroxy-2,5,7,8,-tetramethylchroman-2-yl)-2,6,10-trimethyl-tridecanol
- α-13'-COOH
α-13'-(6-hydroxy-2,5,7,8,-tetramethylchroman-2-yl)-2,6,10-trimethyl-tridecanoic acid
- α-CEHC
α-carboxyethyl-hydroxychroman
- α-LCM
α-tocopherol long-chain metabolites
- α-SCM
α-tocopherol short-chain metabolites
- α-TOH
α-tocopherol
- COX
cyclooxygenase
- CYP3A4
cytochrome P450, subfamily IIIA, polypeptide 4
- CYP4F2
cytochrome P450, subfamily IVF, polypeptide 2
- LDL
low density lipoprotein
- oxLDL
oxidized low density lipoprotein
Introduction
Atherosclerosis is a progressive inflammatory disease characterized by excessive deposition of cholesterol in the arterial wall. Despite intensive therapeutic treatment opportunities the atherosclerotic complications are still the leading cause of death in Western industrialized countries.
Leonardo da Vinci (1452–1519) was probably the first who described the macroscopic changes of atherosclerosis, when he illustrated the lesions in arteries obtained from an elderly man at autopsy. His visionary idea was that the pathological thickening of the arterial wall was due to ‘excessive nourishment’ from the blood. Many decades later da Vinci׳s observation was studied in more detail by Carl von Rokitansky (1852) and Rudolf L. K. Virchow (1821–1902). In 1856 Virchow proposed that injury of the endothelium may initiate the disease process of atherosclerosis. Based on this idea, Russell Ross (1929–1999) and John A. Glomset came up in 1973 with the ‘response-to-injury’ hypothesis which is still generally accepted today in the form of the more generalized concept of endothelial dysfunction as the initial cause of atherosclerosis.
The pioneering work of Virchow and Nikolai N. Anitschkow (1885–1964) provided first evidence for the importance of the deposition of lipids from the blood, in particular cholesterol, in the arterial wall. Their findings formed the basis for the lipid hypothesis which connects plasma cholesterol levels to the development of the disease. In 1951, G. Lyman Duff (1904–1956) and Gardner C. McMillan (1918–2004) formulated the lipid hypothesis in its modern form, which is, despite controversial discussions, still widely accepted today. Since the discovery of the importance of the cholesterol contained in low-density lipoprotein (LDL2) particles for the pathogenesis of atherosclerosis, the concept of endothelial dysfunction has become tightly linked to the lipid hypothesis.
Almost 30 years ago the concept originated from work by Daniel Steinberg and Joseph L. Witztum that oxidative stress and the oxidation of LDL particles might contribute to atherosclerosis. The idea came up from the observation that the incubation of macrophages with oxidized LDL (oxLDL3) but not with native LDL led to the intracellular accumulation of cholesteryl esters. The idea that oxidative stress is involved in atherogenesis gained much attention and created tremendous excitement to look for oxLDL in vivo as well as for different kinds of oxidized lipid species within the particle. Since oxLDL appears in human plasma as well as within the arterial wall it was even a small step to the idea that supplementation with antioxidants may prevent atherosclerosis by inhibiting the formation of oxLDL. This hypothesis appeared to be on solid ground due to epidemiological evidence and the success in several animal studies using a variety of antioxidants. The euphoria of initial success led to clinical trials to validate the hypothesis and natural antioxidants were of particular interest as the expectation was that these natural compounds would have less undesirable effects. Accordingly a number of clinical trials were performed using, for example, vitamin E, which surprisingly have not been overwhelmingly supportive of the hypothesis. An overview on the controversial findings for vitamin E obtained from clinical trials is given in Fig. 1.
Fig. 1.
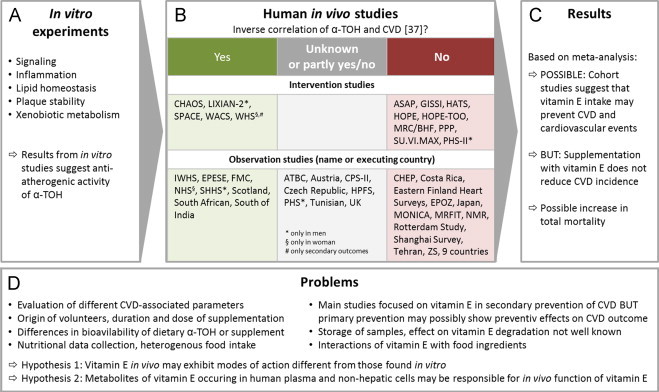
Effects of α-TOH in vivo. Many in vitro investigations (A) focused on the identification of α-TOH-regulated signaling pathways and its effects on inflammation, lipid homeostasis, atherosclerotic plaque stability as well as xenobiotic metabolism as key processes. Taken together most of the in vitro studies implicated that vitamin E, and particularly α-TOH, may be used to prevent or cure cardiovascular disease (CVD) and related diseases, such as atherosclerosis. Based on very promising studies in vitro and with animals several large-scaled human intervention trials were initiated and followed up over years. Unfortunately, the trials revealed controversial results and failed to demonstrate clear invers relations or positive effects of α-TOH supplementation with respect to the prevention of cardiovascular complications [21] (B). Further, α-TOH serum levels did not correlate with cardiovascular outcomes in different cohorts. Although some studies reported promising findings, such as the ‘Nurses’ Health Study (NHS) [22] including 87,000 volunteers in which vitamin E supplementation was associated with a lower risk of major coronary disease, other large-scale studies, such as the Heart Outcomes Prevention Evaluation (HOPE) study [23], the SU.VI.MAX study [24], the Gruppo Italiano per lo Studio della Sopravvivenza nell’Infarto Miocardico (GISSI) study [25] did not confirm that vitamin E intake correlates negatively with cardiovascular outcomes. Other studies revealed also contrary results depending on the time of follow-up and the cardiovascular parameter investigated or they showed unclear results (for example, the Alpha-Tocopherol, Beta-Carotene Cancer Prevention Study (ATBC) [26], [27], [28], [29], [30], the Physicians׳ Health Study [31], the Health Professionals Follow-up Study (HPFS) [32], [33]). In a recent meta-analysis Ye et al. also found no significant inverse correlation for CVD and cardiovascular mortality under vitamin E [34]. Reasons for the poor outcomes of clinical trials are hardly to define. Beside a general failure of vitamin E, other reasons may explain the lack of any cardio-preventive effect. Likely the selection of volunteers, the sizes of cohorts, doses and duration of supplementation or rather the application form of vitamin E with respect to its bioavailability, the food questionnaires and variability of food intake may explain the findings. Furthermore, it cannot be excluded that the interaction of vitamin E with other food ingredients contributes to the lack of its cardio-preventive activity. From a technical point of view, it cannot be excluded that vitamin E in stored sample is chemically modified or degraded so that frozen biological samples are not completely comparable to fresh samples. Another important point refers to the fact that most of the intervention trials concentrated on secondary prevention in patients with already existing CVD. It has been suggested that vitamin E supplementation may be more effectively for inhibiting the early stages of atherosclerosis [35] and should be considered for primary prevention, as recently supported by Meydani et al. emphasizing the beneficial effects of long-term vitamin E supplementation in Ldlr−/− mice under healthy life-style conditions, such as low fat diet [36]. However, this hypothesis has not yet been confirmed. In summary (C), there is no clear evidence that supplementation with vitamin E correlates inversely with CVD incidence. Meta-analyses of observation studies suggest that vitamin E intake may prevent CVD and cardiovascular events [37], [38], [39]. Knekt et al. performed a pooled analysis of observation studies with dietary vitamin E intake and supplementation in separate arms and found a significant inverse correlation of intake and CVD events only in the supplementation group [40]. Apart from that meta-analysis of intervention studies provide evidence that supplementation with vitamin E does not reduce CVD incidence [38], [41], [42], [43]. There are several drawbacks of meta-analysis that should be considered while interpreting these results, such as combination of heterogeneous data sets (regarding quality, statistics and focus within the topic), publication bias as well as criteria for inclusion and exclusion of the meta-analysis. However, it cannot yet be excluded that vitamin E intake is protective at least in some groups of humans against CVD as primary prevention. It is also important to remind that Miller et al. focused in their meta-analysis on some intervention studies, which provided evidence for an increase in all-cause mortality after supplementation with high doses of vitamin E [44]. These findings in humans raise the question whether α-TOH in vivo exhibits modes of action different from those found in vitro. Possible explanations for the inability of vitamin E to prevent CVD and its complications in clinical trials in humans have been outlined above and are summarized in (D). Abbreviations and references: Alpha-Tocopherol, Beta-Carotene Cancer Prevention (ATBC) [26], [27], [28], [29], [30], Antioxidant Supplementation in Atherosclerosis Prevention (ASAP) [45], Austria [46], Cambridge Heart Antioxidant Study (CHAOS) [47], Cancer Prevention Study II (CPS-II) [48], Costa Rica [49], Czech Republic [50], Eastern Finland Heart Surveys [51], Epidemiologic Study of Cardiovascular Risk Indicators (EPOZ Study) [52], Established Populations for Epidemiologic Studies of the Elderly (EPESE) [53], Finnish Mobile Clinic Examination Survey (FMC) [54], Gruppo Italiano per lo Studio della Sopravvivenza nell’Infarto miocardico (GISSI) [25], HDL-Atherosclerosis Treatment Study (HATS) [55], Health Check-up Program (CHEP) [56], Heart Outcomes Prevention Evaluation Study (HOPE) [23], HOPE-The Ongoing Outcomes [HOPE-TOO] [57], Iowa Women׳s Health Study (IWHS) [58], Japan [59], Lixian dysplasia trial (LIXIAN-2) [60], [61], Medical Research Council/British Heart Foundation (MRC/BHF) [62], Multinational MONItoring of trends and determinants in CArdiovascular disease (MONICA) [63], Noninstitutionalized Massachusetts Residents (NMR) [64], Physicians׳ Health Study (PHS) [31], Physicians׳ Health Study II (PHS-II) [65], Rotterdam Study [66], Scotland and UK [67], Scottish Heart Health Study (SHHS) [68], Secondary prevention with antioxidants of cardiovascular disease in endstage renal disease (SPACE) [69], Shanghai Survey [70], South African [71], South of India [72], Supplémentation en Vitamines et Minéraux Antioxydants (SU.VI.MAX) [24], Tehran [73], The Health Professionals Follow-up Study (HPFS) [32], [33], The Multiple Risk Factor Intervention Trial (MRFIT) [74], The Nurses׳ Health Study (NHS) [22], The Primary Prevention Project (PPP) [75], The Zutphen Study (ZS) [76], Tunisian [77], UK [78], Women׳s Antioxidant Cardiovascular Study (WACS) [79], Women׳s Health Study (WHS) [80], 9 countries [81]. The name of countries/cities refers to the countries/cities in which the studies have been performed, if no name for the studies is available.
In this review, we want to summarize the controversial role of vitamin E as a preventive agent against atherosclerosis and to point the attention to recent findings by our group that highlight a role of long-chain metabolites of vitamin E as a proposed new class of regulatory metabolites and to their potential contribution to atherosclerotic processes.
Pathogenesis of atherosclerosis
The endothelium covering the arterial walls comprises a physiological and selective barrier, the so-called intima, between blood and the inner layer of the arterial wall. This so-called media is comprised by contractile smooth muscle cells. Pathophysiological stimuli cause endothelial dysfunction triggering inflammatory processes in the vascular wall which result under chronic conditions in extensive morphological changes characterized by intimal thickening, deposition of cholesterol and fibrotic material, loss of elasticity, reduction of vascular lumen, and widening of the vessel diameter [1]. Endothelial dysfunction is thought to be caused by exogenous stimuli, such as environmental factors (e.g., toxicants such as dioxins, PCBs, and pesticides), unhealthy lifestyle (e.g., smoking and physical inactivity) and dietary habits (e.g., high intake of saturated fat). The impact of exogenous factors depends on endogenous local and systemic conditions. Local factors are vessel-associated junctions, bifurcations and curvatures which are responsible for increased shear stress caused by turbulences of the blood stream in these areas, which are thus predestinated for the formation of atherosclerotic lesions [2]. Pro-atherogenic systemic factors are determined either genetically or pathophysiologically, for example, in case of increased LDL and triglyceride plasma levels [3], [4] as well as inflammatory conditions [5]. The process of atherosclerosis is outlined and explained in more detail in Fig. 2.
Fig. 2.
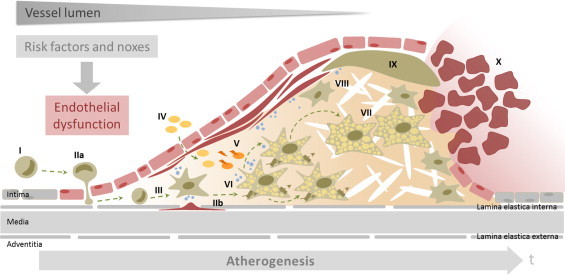
Key events of atherogenesis. Atherosclerosis is a complex and progressive inflammatory disease characterized by extensive morphological changes of the vascular wall. The arterial wall is composed of the intima, formed by endothelial cells (EC), the media, comprised of smooth muscle cells (SMC), and the adventitia, made of fibroblasts. The intermediate layers of the arterial vessel are the lamina elastica interna, connecting the intima with the media, and the lamina elastica externa, the connection between the media and the adventitia. Key changes of the vascular wall during the progression of atherosclerosis are intimal thickening, extensive extracellular deposition of cholesterol and fibrotic material, loss of elasticity, reduction of the vascular lumen and widening of the arterial diameter [1]. Endothelial dysfunction is caused by exogenous and endogenous noxes and is the initial event in atherosclerosis. The dysfunction of the endothelium is accompanied by up-regulation of adhesion molecules in EC, which promote attachment and recruitment of T-lymphocytes and monocytes from the blood (I) and initiate immigration of monocytes into the subendothelial area of the vessel wall (IIa). Migration of monocytes, T-lymphocytes and SMC from the media (IIb) finally results in intimal thickening and fibrosis. Following migration of monocytes through the intima, differentiation of these cells into macrophages occurs (III). Macrophages as the phagocytic cells of the immune system play a pivotal role in the progression of atherosclerosis. They actively engage by taking up lipids from oxLDL in a non-controlled manner, by storing large amounts of cholesteryl esters, and by mobilizing cholesterol for reverse cholesterol transport [82], [83]. Macrophages also orchestrate the inflammatory process, are responsible for the immigration of SMC from the media by releasing chemotactic molecules and proteases, and modulate the fibrotic process. LDL particles diffuse from the blood into the subendothelial space as a consequence of the loss of the endothelial barrier during endothelial dysfunction (IV). In the arterial wall the lipids and LDL are subjected to oxidation and enzymatic modification. The resulting oxLDL is taken up by macrophages (V) via scavenger receptors and phagocytosis in uncontrolled fashion [84]. Recent studies have also highlighted the intra-plaque proliferation of macrophages in the lipid-rich stage of atherosclerotic plaque development (VI) [85]. Accumulation and uptake of oxLDL by macrophages triggers the secretion of chemotactic molecules [86], which promote the migration of SMC from the media into the subendothelial tissue. The intimal SMC lose their ability to contract, proliferate and synthesize extracellular matrix which results in fibrosis as part of the plaque development. The deposition of the extracellular matrix leads to further accumulation of oxLDL and lipids, in particular cholesterol and cholesteryl esters. The uptake of oxLDL via scavenger receptors is not subjected to negative feedback regulation, thus resulting in excessive intracellular lipid accumulation and formation of macrophage foam cells (VII) [87], as well as the release of chemotactic mediators. During further progression of atherosclerosis, fatty streaks are formed and the thickening of the vascular wall progresses through the ongoing deposition of extracellular lipids and proliferation of intimal SMC, and the accompanied synthesis of extracellular matrix proteins. Over many years these processes together form the characteristic necrotic lipid core covered and stabilized by a fibrotic cap (VIII). The stability of the atherosclerotic plaque is defined by the amount of accumulated lipids and also by the amount and quality of the extracellular matrix of the fibrotic cap; the progressing accumulation of lipids is often accompanied by reduced stability of the fibrotic cap (IX). Weakening of the cap may finally result in rupturing of the plaque, particularly in areas rich in macrophages as these cells produce a broad range of proteases that degrade the extracellular matrix of the fibrotic cap [88], [89]. Plaque rupture causes thrombus formation (X) via the activation of the coagulation cascade [87]. Thrombi may occlude the artery at the site of plaque rupture or may flow through the blood stream and occlude downstream arteries that have a smaller lumen. In some cases, thrombi at the plaque site are reorganized and integrated into the plaque. This finally leads to the formation of the so-called complicated plaque (not shown).
A key event of atherogenesis is the loss of the selective endothelial barrier by endothelial dysfunction which allows, for example, LDL to enter the arterial wall. Once inside the vessel wall, LDL particles become prone to oxidation. The oxidized particles cause damage to the tissue thus triggering a cascade of immune and inflammatory responses. In addition, macrophages, the phagocytic cells of the immune system, are recruited to the affected tissue sites to clear the oxLDL particles. As a consequence oxidized lipids and particularly cholesterol accumulate within the macrophages as these cells are not able to process the oxLDL completely. This causes transformation of the cells into so-called foam cells and ultimately cell death as the excessive accumulation of intracellular lipids is cytotoxic. Death of macrophage foam cells results over time in the extracellular deposition of cholesterol in the arterial wall and the formation of an atheroma. The process of atherosclerosis is outlined and explained in more detail in Fig. 2.
Thus, vitamin E was considered as an anti-atherogenic agent for a long time as prevention of LDL oxidation by providing increased levels of antioxidants would prevent the formation of macrophage foam cells and atheroma, and would dampen the immune and inflammatory response.
Effects of α-Tocopherol on atherogenic processes
Vitamin E is likely the most important lipid antioxidant in the human diet. The term vitamin E comprises a group of eight abundant isomers (α-, β-, γ-, δ-tocopherol and -tocotrienol), that differ by their methylation patterns of the hydroxychromanol ring and saturation of the side-chain. Many in vitro studies have been performed with α-tocopherol (α-TOH4) which is the most active isomer within the group of vitamin E [6]. α-Tocopherol exhibits anti-oxidative capacity in vitro [7], and it has been shown to particularly inhibit, for example, the oxidation of LDL. Beside this, α-TOH shows anti-inflammatory features by, for example, inhibiting cyclooxygenase (COX5) 2. Next to its anti-inflammatory and anti-oxidative properties, the vitamin E isomers may have a variety of further independent properties, namely the modulation of gene expression, particularly that of genes encoding proteins involved in signaling but also the uptake, transport and degradation of tocopherols, as well as the uptake of lipoproteins and the storage and export of lipids such as cholesterol. The in vitro and ex vivo effects of α-tocopherol on cellular processes are depicted in Fig. 3.
Fig. 3.
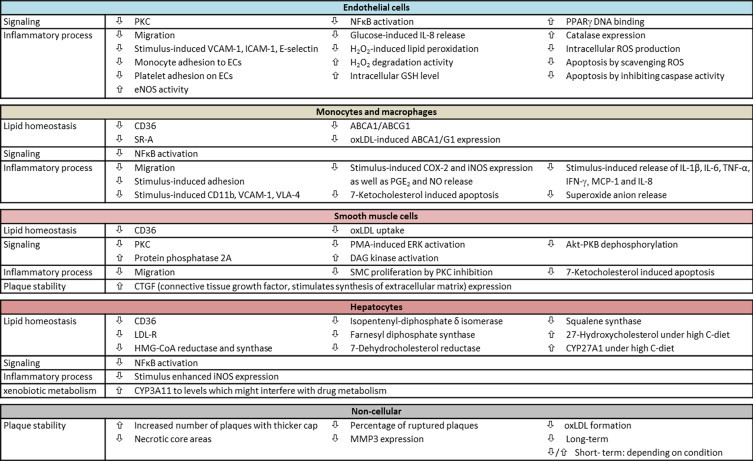
Atherosclerosis-relevant in vitro and ex vivo effects of α-tocopherol. Due to its function as an antioxidant, vitamin E was considered to interfere with key events in atherogenesis. To gain better insights into the contribution of vitamin E to the molecular processes underlying the hallmarks of atherosclerosis much effort was spend on in vitro and ex vivo experiments as well as studies involving animal models. As the complex pathogenesis of atherosclerosis involves several different cell types, the figure is divided according to the cells of interest (EC, SMC, monocytes and macrophages, hepatocytes) as well as non-cellular, plaque-specific processes and categories such as lipid homeostasis, signaling, inflammation, plaque stability and xenobiotic metabolism which are used to reflect the hallmarks of atherosclerosis. Endothelial cells surface the arterial wall and their dysfunction is the initial step of atherogenesis. α-Tocopherol reduces the stimulus-induced expression of adhesion molecules, such as vascular cell adhesion molecule-1 (VCAM-1), intercellular adhesion molecule 1 (ICAM-1) and E-selectin [90], [91], [92], and decreases thereby the adhesion [92] and immigration of leukocytes [93] onto the endothelium or into the arterial wall, respectively. α-Tocopherol makes it easier for EC to deal with oxidative stress, such as H2O2-induced lipid peroxidation [7], due to higher catalase expression [94], increased H2O2 degradation activity, and higher intracellular GSH levels [13]. Thus, apoptosis of EC induced by oxidative stress is reduced [95]. Beside reducing the release of inflammatory cytokines, such as interleukin (IL) 8 [96], in response to external stimuli, α-TOH interferes with cellular signaling by inhibiting, for example, PKC [97], activate nuclear factor κB (NFκB) [98] or binding of peroxisome proliferator-activated receptor γ (PPARγ) to its regulatory DNA elements [94]. Lipid homeostasis of SMC is also affected by α-TOH, for example by down-regulation of the scavenger receptor CD36 leading to reduced uptake of oxLDL [99]. α-Tocopherol inhibits migration [100] and proliferation of SMC by inhibiting PKC [101], and prevents 7-ketocholesterol-induced apoptosis [102]. Plaque stability is modulated by connective tissue growth factor (CTGF), which stimulates the synthesis of extracellular matrix. This factor is induced by α-TOH in SMC [103], suggesting that α-TOH contributes to plaque stability by inducing fibrotic processes. Several signaling cascades in SMC are also modulated by α-TOH, namely PKC [104], [105], extracellular signal-regulated kinase (ERK) [106], protein kinase B (PK/AKT) and protein phosphatase 2A [107]. Monocytes migrate in the arterial wall, differentiate into macrophages and transform to foam cells under atherogenic conditions. α-Tocopherol reduces adhesion and migration of leukocytes by down-regulating expression of CD11b [93] and very late antigen-4 (VLA-4) [92], [108]. Macrophage foam cell formation is prevented by reducing the expression of scavenger receptors CD36 [109], [110] and A [111]. On the other hand, foam cell formation is triggered by the down-regulation of the lipid exporters ATP-binding cassette transporter (ABC) A1 and G1 [110]. Furthermore, the inflammatory response of macrophages to stimuli such as lipopolysaccharide (LPS) is dampened. The diminished induction of COX2 and inducible nitric oxide synthase (iNOS) by α-TOH results in a reduced release of prostaglandin E2 (PGE2) and nitric oxide [112], [113]. The release of pro-inflammatory cytokines such as IL-1b, IL-6, tumor necrosis factor α (TNFα), and interferon γ (IFNγ) is also reduced [113]. Similar to SMC, 7-ketocholesterol-induced apoptosis is reduced by α-TOH in macrophages [114], [115]. Signaling pathways, such as NFκB [108] and oxLDL or lipid-free high density lipoprotein induced liver X receptor α (LXRα) activity, are also inhibited by α-TOH [110]. The liver is the major organ for cholesterol biosynthesis and metabolism of lipoproteins, xenobiotics and α-TOH. In liver cells α-TOH down-regulates expression of CD36 [116], [117] and the LDL receptor [118], as well as expression of enzymes involved in cholesterol biosynthesis, such as HMG-CoA reductase and HMG-CoA synthase [118]. In contrast, expression of cytochrome P450 subfamily 27A polypeptide 1 (CYP27A1), and thus synthesis of 27-hydroxycholesterol, is induced by α-TOH [119]. Similar to the situation in macrophages, activation of NFκB and expression of iNOS in response to stimuli is reduced by α-TOH [120]. α-Tocopherol also induces CYP3A11 to levels which might interfere with drug metabolism [121]. In addition to the effects of α-TOH on the different cell types involved in atherogenesis, α-TOH improves plaque stability in hyperlipidemic rabbits. Treatment of the animals with α-TOH increased the number of plaques with thicker, stabilizing fibrotic caps and reduced necrotic lipid core areas as well as reduced number of ruptured plaques [122]. Formation of oxLDL was also blocked by α-TOH [123]. Several studies using animals such as hypercholesterolemic rabbits or mice suggest that vitamin E inhibits atherogenesis in early [124] or advanced stages by its antioxidant capacity [125] or gene regulatory potential via signal transduction cascades and on adhesion molecules [126], [127]. While high dose supplementation of vitamin E can improve myocardial tolerance to ischemia and reperfusion [128], Keaney et al. found that low dose α-TOH improves and high dose worsens endothelial vasodilatory function in cholesterol-fed rabbits [129]. Under extreme conditions, such as pronounced elevation in systemic oxidative stress due to hyperlipidaemia and obesity, vitamin E seems to be not cardioprotective [130].
α-Tocopherol metabolites and their bioactivity
Metabolic degradation of α-TOH takes place almost exclusively in the liver. Beside metabolites resulting from oxidation of the chroman moiety, hepatic metabolism of α-TOH involves CYP3A46-dependent ω-hydroxylation and α-oxidation, which results in the formation of the α-tocopherol long-chain metabolites (α-LCM7) α-13'-OH8 (13'-(6-hydroxy-2,5,7,8,-tetramethylchroman-2-yl)-2,6,10-trimethyl-tridecanol) and α-13'-COOH9 (13'-(6-hydroxy-2,5,7,8,-tetramethylchroman-2-yl)-2,6,10-trimethyl-tridecanoic acid), and further steps of β-oxidation, which results in the formation of middle- and short-chain metabolites (α-SCM10) with the catabolic end-product α-carboxyethyl-hydroxychroman (α-CEHC11), respectively [8], [9], [10]. The short-chain metabolites are excreted via urine and are often used as a marker for α-TOH supply [11]. Other tocopherols, such as γ- and δ-tocopherol, are almost quantitatively degraded and excreted via the urine as the corresponding γ- and δ-CEHCs. The hepatic metabolism of α-TOH is illustrated in Fig. 4.
Fig. 4.
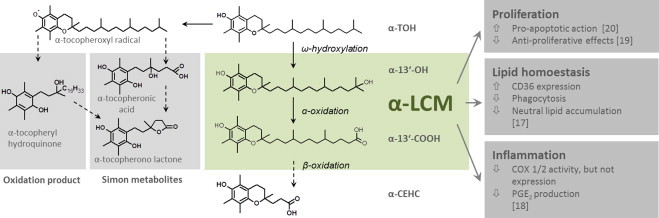
α-Tocopherol metabolism and in vitro characteristics of its liver-derived long-chain metabolites. Due to the antioxidant capacity of vitamin E, early studies on its metabolism have concentrated on metabolites resulting from oxidation of the chroman moiety. The major oxidation product in the liver was described as α-tocopheryl quinone. This metabolite derives from the reaction of the tocopheroxyl radical with a peroxyl radical; it can be reduced to α-tocopheryl hydroquinone by NAD(P)H-dependent microsomal and mitochondrial enzymes. For many years, the so-called Simon metabolites, α-tocopheronic acid and its lactone, were the only known urinary α-TOH metabolites. The Simon metabolites are characterized by the opened chroman ring. Opening of the chroman ring starts with the formation of an α-tocopheroxyl radical when α-TOH has exerted its antioxidant activity. The Simon metabolites were therefore considered as urinary indicators that α-TOH had reacted as an antioxidant [131]. Today some researcher rise the question whether Simon metabolites are artefacts produced during sample preparation as α-CEHC is easily converted to α-tocopheronolactone by oxygenation [10], [132]. α-Tocopherol is physiologically catabolized in the liver via the xenobiotic detoxification system involving CYP3A4 [8] and CYP4F2[133]. Cytochrome-dependent ω-hydroxylation results in the formation of the long-chain alcohol derivate α-13'-OH, 13'-(6-hydroxy-2,5,7,8,-tetramethylchroman-2-yl)-2,6,10-trimethyl-tridecanol. Subsequent α-oxidation leads to α-13'-COOH, 13'-(6-hydroxy-2,5,7,8,-tetramethylchroman-2-yl)-2,6,10-trimethyl-tridecanoic acid. The following β-oxidation steps in peroxisomes and mitochondria form the α-SCM, α-carboxyethyl-hydroxychroman (α-CEHC) [6]. This end-product of α-TOH metabolism can be conjugated and is excreted via urine [134]. The intact chroman structure indicates that α-CEHC is derived from α-TOH that has not reacted as an antioxidant. As α-CEHC excretion increases when certain plasma levels of RRR-α-TOH are exceeded, excretion of α-CEHC is considered as an indicator of adequate or excessive α-TOH supply. Although hepatic metabolism of α-TOH and the formation of the metabolic long- and short-chain intermediates are known for several years [8], [9], [10], the physiological function of the α-LCM α-13'-OH and α-13'-COOH is still unknown. Due to a lack of the pure compounds α-13'-OH and α-13'-COOH, only a few studies on the function of these α-LCM have been performed. Work so far focused on anti-proliferative effects, modulation of inflammatory processes and modulation of lipid homeostasis. Our group described anti-proliferative effects of the α-LCM due to pro-apoptotic action [20]. In HepG2 cells, the α-LCM induced cleavage of caspases 3, 7 and 9 as well as PARP-1 and induced mitochondrial dysfunction as characterized by reduced mitochondrial membrane potential and induced intra-mitochondrial ROS formation. The anti-proliferative effect of α-13'-COOH was shown also in murine glioma C6 cancer cells [19]. Others have reported that the α-LCM interfere with inflammatory processes by modulating activity of COX1 and COX2 and consequently by blocking production of PGE2[18]. Recent work by our group focused on the effects of the α-LCM on macrophage foam cell formation [17]. We have shown that α-LCM induce expression of the scavenger receptor CD36, the major receptor responsible for oxLDL uptake, in human macrophages, in contrast to the inhibiting actions of α-TOH [17]. Despite up-regulation of CD36, uptake of oxLDL and oxLDL-induced lipid accumulation was reduced in human macrophages, similar to the effects of α-TOH on oxLDL-mediated foam cell formation. An important finding of this recent study was that the metabolite α-13'-COOH was detected in serum providing for the first time evidence for the bioavailability of the α-LCM outside the liver. Another key finding of the study was that bioactivity of the α-LCM occur at lower concentrations and with mechanisms distinct from those of α-TOH. Taken together, these recent studies provide evidence for a role of the α-LCM as signaling molecules derived metabolically from α-TOH.
Regulatory activity is not restricted to α-TOH as its short-chain metabolite α-CEHC also exhibits bioactivity. It has been shown that α-CEHC is anti-proliferative [12], anti-inflammatory [13], and anti-oxidative [14], and inhibits oxLDL formation [15] and protein kinase C (PKC12) signaling [16]. Recently, researchers focused also on investigating the cellular effects of the α-LCM as α-13'-COOH was detected in human serum, a finding providing clear evidence for its systemic bioavailability. Until now, only a few cellular effects of the α-LCM have been described, such as pro-apoptotic, anti-proliferative and anti-inflammatory features [17], [18], [19], [20], which are highlighted in Fig. 4. A recent study by our group showed that α-LCM also affect macrophage foam cell formation by regulating uptake of oxLDL by macrophages via down-regulation of its phagocytic uptake (Fig. 4) [17]. A key finding of our study was that bioactivity of the α-LCMs occurs at much lower concentrations and with mechanisms distinct from those of their metabolic precursor α-TOH.
Perspective
The findings obtained from clinical trials with humans raise the question whether vitamin E in vivo exhibits modes of action different from those found in vitro. Recent studies shed new light on mechanistic aspects of α-TOH function, which appear to be complicated by α-LCM circulating in the blood. We speculate that the α-LCM represent a new class of regulatory metabolites and propose that unraveling the molecular modes of action of the α-LCM and identifying the key players involved in their signaling may provide new fundamental insights into the biology and mode of function of vitamin E. Further studies are therefore required to elucidate the physiological role of the α-LCM and their contribution to disease processes, such as atherosclerosis. We also hypothesize that the discrepancy between the results obtained in vitro and in vivo in humans may be due to the physiologic metabolism of α-TOH and the formation of α-LCM in the liver and their release into circulation.
Sources and Funding
Research of SL is funded by Grants from the Forschungskreis der Ernährungsindustrie (FEI) as part of the Arbeitsgemeinschaft industrieller Forschungsvereinigungen (AiF) and the Deutsche Forschungsgemeinschaft (DFG).
Disclosures
None.
Footnotes
LDL, low density lipoprotein.
oxLDL, oxidized low density lipoprotein.
α-TOH, α-tocopherol.
COX, cyclooxygenase.
CYP3A4, cytochrome P450, subfamily IIIA, polypeptide 4.
α-LCM, α-tocopherol long-chain metabolite(s).
α-13'-OH, α-13'-(6-hydroxy-2,5,7,8,-tetramethylchroman-2-yl)-2,6,10-trimethyl-tridecanol.
α-13'-COOH, α-13'-(6-hydroxy-2,5,7,8,-tetramethylchroman-2-yl)-2,6,10-trimethyl-tridecanoic acid.
α-SCM, α-tocopherol short-chain metabolite(s).
α-CEHC, α-carboxyethyl-hydroxychroman.
PKC, protein kinase C.
Contributor Information
Maria Wallert, Email: maria.wallert@uni-jena.de.
Lisa Schmölz, Email: lisa.schmoelz@uni-jena.de.
Francesco Galli, Email: f.galli@unipg.it.
Marc Birringer, Email: marc.birringer@he.hs-fulda.de.
Stefan Lorkowski, Email: stefan.lorkowski@uni-jena.de.
References
- 1.Cullen P., Rauterberg J., Lorkowski S. The pathogenesis of atherosclerosis. Handb. Exp. Pharmacol. 2005;170:3–70. doi: 10.1007/3-540-27661-0_1. [DOI] [PubMed] [Google Scholar]
- 2.Monajemi H., Arkenbout E.K., Pannekoek H. Gene expression in atherogenesis. Thromb. Haemost. 2001;86:404–412. [PubMed] [Google Scholar]
- 3.Jakubowski H. The pathophysiological hypothesis of homocysteine thiolactone-mediated vascular disease. J. Physiol. Pharmacol. 2008;59(Suppl 9):155–167. [PubMed] [Google Scholar]
- 4.Raal F.J. Pathogenesis and management of the dyslipidemia of the metabolic syndrome. Metab. Syndr. Relat. Disord. 2009;7:83–88. doi: 10.1089/met.2008.0079. [DOI] [PubMed] [Google Scholar]
- 5.Kullo I.J., Gau G.T., Tajik A.J. Novel risk factors for atherosclerosis. Mayo Clin. Proc. 2000;75:369–380. doi: 10.4065/75.4.369. [DOI] [PubMed] [Google Scholar]
- 6.Mustacich D.J., Leonard S.W., Patel N.K., Traber M.G. Alpha-tocopherol beta-oxidation localized to rat liver mitochondria. Free Radic. Biol. Med. 2010;48:73–81. doi: 10.1016/j.freeradbiomed.2009.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.van Dam B., van Hinsbergh, Victor W.M., Stehouwer, Coen D.A., Versteilen A., Dekker H., Buytenhek R., Princen H.M., Schalkwijk C.G. Vol. 57. 2003. Vitamin E inhibits lipid peroxidation-induced adhesion molecule expression in endothelial cells and decreases soluble cell adhesion molecules in healthy subjects; pp. 563–571. (Cardiovasc. Res.). [DOI] [PubMed] [Google Scholar]
- 8.Birringer M., Drogan D., Brigelius-Flohé R. Tocopherols are metabolized in HepG2 cells by side chain omega-oxidation and consecutive beta-oxidation. Free Radic. Biol. Med. 2001;31:226–232. doi: 10.1016/s0891-5849(01)00574-3. [DOI] [PubMed] [Google Scholar]
- 9.Zhao Y., Lee M.-J., Cheung C., Ju J.-H., Chen Y.-K., Liu B., Hu L.-Q., Yang C.S. Analysis of multiple metabolites of tocopherols and tocotrienols in mice and humans. J. Agric. Food Chem. 2010;58:4844–4852. doi: 10.1021/jf904464u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schultz M., Leist M., Petrzika M., Gassmann B., Brigelius-Flohé R. Novel urinary metabolite of alpha-tocopherol, 2,5,7,8-tetramethyl-2(2'-carboxyethyl)-6-hydroxychroman, as an indicator of an adequate vitamin E supply? Am. J. Clin. Nutr. 1995;62:1527S–1534S. doi: 10.1093/ajcn/62.6.1527S. [DOI] [PubMed] [Google Scholar]
- 11.Lebold K.M., Ang A., Traber M.G., Arab L. Urinary α-carboxyethyl hydroxychroman can be used as a predictor of α-tocopherol adequacy, as demonstrated in the energetics study. Am. J. Clin. Nutr. 2012;96:801–809. doi: 10.3945/ajcn.112.038620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Galli F., Stabile A.M., Betti M., Conte C., Pistilli A., Rende M., Floridi A., Azzi A. The effect of α- and γ-tocopherol and their carboxyethyl hydroxychroman metabolites on prostate cancer cell proliferation. Arch. Biochem. Biophys. 2004;423:97–102. doi: 10.1016/j.abb.2003.11.014. [DOI] [PubMed] [Google Scholar]
- 13.Grammas P., Hamdheydari L., Benaksas E.J., Mou S., Pye Q.N., Wechter W.J., Floyd R.A., Stewart C., Hensley K. Anti-inflammatory effects of tocopherol metabolites. Biochem. Biophys. Res. Commun. 2004;319:1047–1052. doi: 10.1016/j.bbrc.2004.05.082. [DOI] [PubMed] [Google Scholar]
- 14.Betancor-Fernandez A., Sies H., Stahl W., Polidori M.C. In vitro antioxidant activity of 2,5,7,8-tetramethyl-2-(2'-carboxyethyl)-6-hydroxychroman (alpha-CEHC), a vitamin E metabolite. Free Radic. Res. 2002;36:915–921. doi: 10.1080/1071576021000005357. [DOI] [PubMed] [Google Scholar]
- 15.Uto-Kondo H., Kiyose C., Ohmori R., Saito H., Taguchi C., Kishimoto Y., Machida N., Hasegawa M., Yoshioka E., Saita E., Hirata Y., Igarashi O., Kondo K. The coantioxidative effects of carboxyethyl-6-hydroxychromans and alpha-Tocopherol. J. Nutr. Sci. Vitaminol. 2007;53:301–305. doi: 10.3177/jnsv.53.301. [DOI] [PubMed] [Google Scholar]
- 16.Varga Z., Kosaras E., Komodi E., Katko M., Karpati I., Balla J., Paragh G., Aisa M.C., Galli F. Effects of tocopherols and 2,2'-carboxyethyl hydroxychromans on phorbol-ester-stimulated neutrophils. J. Nutr. Biochem. 2008;19:320–327. doi: 10.1016/j.jnutbio.2007.05.002. [DOI] [PubMed] [Google Scholar]
- 17.Wallert M., Mosig S., Rennert K., Funke H., Ristow M., Pellegrino R.M., Cruciani G., Galli F., Lorkowski S., Birringer M. Long-chain metabolites of α-tocopherol occur in human serum and inhibit macrophage foam cell formation in vitro. Free Radic. Biol. Med. 2013;68:43–51. doi: 10.1016/j.freeradbiomed.2013.11.009. [DOI] [PubMed] [Google Scholar]
- 18.Jiang Q., Yin X., Lill M.A., Danielson M.L., Freiser H., Huang J. Long-chain carboxychromanols, metabolites of vitamin E, are potent inhibitors of cyclooxygenases. Proc. Natl. Acad. Sci. USA. 2008;105:20464–20469. doi: 10.1073/pnas.0810962106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mazzini F., Betti M., Netscher T., Galli F., Salvadori P. Configuration of the vitamin E analogue garcinoic acid extracted from garcinia kola seeds. Chirality. 2009;21:519–524. doi: 10.1002/chir.20630. [DOI] [PubMed] [Google Scholar]
- 20.Birringer M., Lington D., Vertuani S., Manfredini S., Scharlau D., Glei M., Ristow M. Proapoptotic effects of long-chain vitamin E metabolites in HepG2 cells are mediated by oxidative stress. Free Radic. Biol. Med. 2010;49:1315–1322. doi: 10.1016/j.freeradbiomed.2010.07.024. [DOI] [PubMed] [Google Scholar]
- 21.Rodrigo R., Guichard C., Charles R. Clinical pharmacology and therapeutic use of antioxidant vitamins. Fundam. Clin. Pharmacol. 2007;21:111–127. doi: 10.1111/j.1472-8206.2006.00466.x. [DOI] [PubMed] [Google Scholar]
- 22.Stampfer M.J., Hennekens C.H., Manson J.E., Colditz G.A., Rosner B., Willett W.C. Vitamin E consumption and the risk of coronary disease in women. N. Engl. J. Med. 1993;328:1444–1449. doi: 10.1056/NEJM199305203282003. [DOI] [PubMed] [Google Scholar]
- 23.Yusuf S., Dagenais G., Pogue J., Bosch J., Sleight P. Vitamin E supplementation and cardiovascular events in high-risk patients. The heart outcomes prevention evaluation study investigators. N. Engl. J. Med. 2000;342:154–160. doi: 10.1056/NEJM200001203420302. [DOI] [PubMed] [Google Scholar]
- 24.Hercberg S., Galan P., Preziosi P., Bertrais S., Mennen L., Malvy D., Roussel A.-M., Favier A., Briançon S. The SU.VI.MAX study: a randomized, placebo-controlled trial of the health effects of antioxidant vitamins and minerals. Arch. Intern. Med. 2004;164:2335–2342. doi: 10.1001/archinte.164.21.2335. [DOI] [PubMed] [Google Scholar]
- 25.Gruppo Italiano per lo Studio della, Dietary supplementation with n-3 polyunsaturated fatty acids and vitamin E after myocardial infarction: results of the GISSI-Prevenzione trial. Sopravvivenza nell’Infarto miocardico, Lancet 354, 1999, pp. 447–455. [PubMed]
- 26.Wright M.E., Lawson K.A., Weinstein S.J., Pietinen P., Taylor P.R., Virtamo J., Albanes D. Higher baseline serum concentrations of vitamin E are associated with lower total and cause-specific mortality in the alpha-tocopherol, beta-carotene cancer prevention study. Am. J. Clin. Nutr. 2006;84:1200–1207. doi: 10.1093/ajcn/84.5.1200. [DOI] [PubMed] [Google Scholar]
- 27.Rapola J.M., Virtamo J., Ripatti S., Huttunen J.K., Albanes D., Taylor P.R., Heinonen O.P. Randomised trial of alpha-tocopherol and beta-carotene supplements on incidence of major coronary events in men with previous myocardial infarction. Lancet. 1997;349:1715–1720. doi: 10.1016/S0140-6736(97)01234-8. [DOI] [PubMed] [Google Scholar]
- 28.Virtamo J., Rapola J.M., Ripatti S., Heinonen O.P., Taylor P.R., Albanes D., Huttunen J.K. Effect of vitamin E and beta carotene on the incidence of primary nonfatal myocardial infarction and fatal coronary heart disease. Arch. Intern. Med. 1998;158:668–675. doi: 10.1001/archinte.158.6.668. [DOI] [PubMed] [Google Scholar]
- 29.Leppälä J.M., Virtamo J., Fogelholm R., Huttunen J.K., Albanes D., Taylor P.R., Heinonen O.P. Controlled trial of alpha-tocopherol and beta-carotene supplements on stroke incidence and mortality in male smokers. Arterioscler. Thromb. Vasc. Biol. 2000;20:230–235. doi: 10.1161/01.atv.20.1.230. [DOI] [PubMed] [Google Scholar]
- 30.Hirvonen T., Virtamo J., Korhonen P., Albanes D., Pietinen P. Intake of flavonoids, carotenoids, vitamins C and E, and risk of stroke in male smokers. Stroke. 2000;31:2301–2306. doi: 10.1161/01.str.31.10.2301. [DOI] [PubMed] [Google Scholar]
- 31.Muntwyler J., Hennekens C.H., Manson J.E., Buring J.E., Gaziano J.M. Vitamin supplement use in a low-risk population of US male physicians and subsequent cardiovascular mortality. Arch. Intern. Med. 2002;162:1472–1476. doi: 10.1001/archinte.162.13.1472. [DOI] [PubMed] [Google Scholar]
- 32.Rimm E.B., Stampfer M.J., Ascherio A., Giovannucci E., Colditz G.A., Willett W.C. Vitamin E consumption and the risk of coronary heart disease in men. N. Engl. J. Med. 1993;328:1450–1456. doi: 10.1056/NEJM199305203282004. [DOI] [PubMed] [Google Scholar]
- 33.Ascherio A., Rimm E.B., Hernán M.A., Giovannucci E., Kawachi I., Stampfer M.J., Willett W.C. Relation of consumption of vitamin E, vitamin C, and carotenoids to risk for stroke among men in the United States. Ann. Intern. Med. 1999;130:963–970. doi: 10.7326/0003-4819-130-12-199906150-00003. [DOI] [PubMed] [Google Scholar]
- 34.Frei B., Gaziano J.M. Content of antioxidants, preformed lipid hydroperoxides, and cholesterol as predictors of the susceptibility of human LDL to metal ion-dependent and -independent oxidation. J. Lipid Res. 1993;34:2135–2145. [PubMed] [Google Scholar]
- 35.Vivekananthan D.P., Penn M.S., Sapp S.K., Hsu A., Topol E.J. Use of antioxidant vitamins for the prevention of cardiovascular disease: meta-analysis of randomised trials. Lancet. 2003;361:2017–2023. doi: 10.1016/S0140-6736(03)13637-9. [DOI] [PubMed] [Google Scholar]
- 36.Meydani M., Kwan P., Band M., Knight A., Guo W., Goutis J., Ordovas J. Long-term vitamin E supplementation reduces atherosclerosis and mortality in Ldlr−/−mice, but not when fed Western style diet. Atherosclerosis. 2014;233:196–205. doi: 10.1016/j.atherosclerosis.2013.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cordero Z., Drogan D., Weikert C., Boeing H. Vitamin E and risk of cardiovascular diseases: a review of epidemiologic and clinical trial studies. Crit. Rev. Food Sci. Nutr. 2010;50:420–440. doi: 10.1080/10408390802304230. [DOI] [PubMed] [Google Scholar]
- 38.Asplund K. Antioxidant vitamins in the prevention of cardiovascular disease: a systematic review. J. Intern. Med. 2002;251:372–392. doi: 10.1046/j.1365-2796.2002.00973.x. [DOI] [PubMed] [Google Scholar]
- 39.Marchioli R. Antioxidant vitamins and prevention of cardiovascular disease: laboratory, epidemiological and clinical trial data. Pharmacol. Res. 1999;40:227–238. doi: 10.1006/phrs.1999.0480. [DOI] [PubMed] [Google Scholar]
- 40.Knekt P., Ritz J., Pereira M.A., O’Reilly E.J., Augustsson K., Fraser G.E., Goldbourt U., Heitmann B.L., Hallmans G., Liu S., Pietinen P., Spiegelman D., Stevens J., Virtamo J., Willett W.C., Rimm E.B., Ascherio A. Antioxidant vitamins and coronary heart disease risk: a pooled analysis of 9 cohorts. Am. J. Clin. Nutr. 2004;80:1508–1520. doi: 10.1093/ajcn/80.6.1508. [DOI] [PubMed] [Google Scholar]
- 41.Eidelman R.S., Hollar D., Hebert P.R., Lamas G.A., Hennekens C.H. Randomized trials of vitamin E in the treatment and prevention of cardiovascular disease. Arch. Intern. Med. 2004;164:1552–1556. doi: 10.1001/archinte.164.14.1552. [DOI] [PubMed] [Google Scholar]
- 42.Shekelle P.G., Morton S.C., Jungvig L.K., Udani J., Spar M., Tu W., Suttorp M.J., Coulter I., Newberry S.J., Hardy M. Effect of supplemental vitamin E for the prevention and treatment of cardiovascular disease. J. Gen. Intern. Med. 2004;19:380–389. doi: 10.1111/j.1525-1497.2004.30090.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Law M.R., Morris J.K. By how much does fruit and vegetable consumption reduce the risk of ischaemic heart disease? Eur. J. Clin. Nutr. 1998;52:549–556. doi: 10.1038/sj.ejcn.1600603. [DOI] [PubMed] [Google Scholar]
- 44.Miller E.R., Pastor-Barriuso R., Dalal D., Riemersma R.A., Appel L.J., Guallar E. Meta-analysis: high-dosage vitamin E supplementation may increase all-cause mortality. Ann. Intern. Med. 2005;142:37–46. doi: 10.7326/0003-4819-142-1-200501040-00110. [DOI] [PubMed] [Google Scholar]
- 45.Salonen J.T., Nyyssönen K., Salonen R., Lakka H.M., Kaikkonen J., Porkkala-Sarataho E., Voutilainen S., Lakka T.A., Rissanen T., Leskinen L., Tuomainen T.P., Valkonen V.P., Ristonmaa U., Poulsen H.E. Antioxidant Supplementation in Atherosclerosis Prevention (ASAP) study: a randomized trial of the effect of vitamins E and C on 3-year progression of carotid atherosclerosis. J. Intern. Med. 2000;248:377–386. doi: 10.1046/j.1365-2796.2000.00752.x. [DOI] [PubMed] [Google Scholar]
- 46.Kostner K., Hornykewycz S., Yang P., Neunteufl T., Glogar D., Weidinger F., Maurer G., Huber K. Is oxidative stress causally linked to unstable angina pectoris? A study in 100 CAD patients and matched controls. Cardiovasc. Res. 1997;36:330–336. doi: 10.1016/s0008-6363(97)00185-5. [DOI] [PubMed] [Google Scholar]
- 47.Stephens N.G., Parsons A., Schofield P.M., Kelly F., Cheeseman K., Mitchinson M.J. Randomised controlled trial of vitamin E in patients with coronary disease: Cambridge Heart Antioxidant Study (CHAOS) Lancet. 1996;347:781–786. doi: 10.1016/s0140-6736(96)90866-1. [DOI] [PubMed] [Google Scholar]
- 48.Watkins M.L., Erickson J.D., Thun M.J., Mulinare J., Heath C.W. Multivitamin use and mortality in a large prospective study. Am. J. Epidemiol. 2000;152:149–162. doi: 10.1093/aje/152.2.149. [DOI] [PubMed] [Google Scholar]
- 49.El-Sohemy A., Baylin A., Spiegelman D., Ascherio A., Campos H. Dietary and adipose tissue gamma-tocopherol and risk of myocardial infarction. Epidemiology. 2002;13:216–223. doi: 10.1097/00001648-200203000-00018. [DOI] [PubMed] [Google Scholar]
- 50.Bobák M., Brunner E., Miller N.J., Skodová Z., Marmot M. Could antioxidants play a role in high rates of coronary heart disease in the Czech Republic? Eur. J. Clin. Nutr. 1998;52:632–636. doi: 10.1038/sj.ejcn.1600616. [DOI] [PubMed] [Google Scholar]
- 51.Salonen J.T., Salonen R., Penttilä I., Herranen J., Jauhiainen M., Kantola M., Lappeteläinen R., Mäenpää P.H., Alfthan G., Puska P. Serum fatty acids, apolipoproteins, selenium and vitamin antioxidants and the risk of death from coronary artery disease. Am. J. Cardiol. 1985;56:226–231. doi: 10.1016/0002-9149(85)90839-2. [DOI] [PubMed] [Google Scholar]
- 52.Kok F.J., de Bruijn A.M., Vermeeren R., Hofman A., van Laar A., de Bruin M., Hermus R.J., Valkenburg H.A. Serum selenium, vitamin antioxidants, and cardiovascular mortality: a 9-year follow-up study in the Netherlands. Am. J. Clin. Nutr. 1987;45:462–468. doi: 10.1093/ajcn/45.2.462. [DOI] [PubMed] [Google Scholar]
- 53.Losonczy K.G., Harris T.B., Havlik R.J. Vitamin E and vitamin C supplement use and risk of all-cause and coronary heart disease mortality in older persons: the established populations for epidemiologic studies of the elderly. Am. J. Clin. Nutr. 1996;64:190–196. doi: 10.1093/ajcn/64.2.190. [DOI] [PubMed] [Google Scholar]
- 54.Knekt P., Reunanen A., Järvinen R., Seppänen R., Heliövaara M., Aromaa A. Antioxidant vitamin intake and coronary mortality in a longitudinal population study. Am. J. Epidemiol. 1994;139:1180–1189. doi: 10.1093/oxfordjournals.aje.a116964. [DOI] [PubMed] [Google Scholar]
- 55.Brown B.G., Zhao X.Q., Chait A., Fisher L.D., Cheung M.C., Morse J.S., Dowdy A.A., Marino E.K., Bolson E.L., Alaupovic P., Frohlich J., Albers J.J. Simvastatin and niacin, antioxidant vitamins, or the combination for the prevention of coronary disease. N. Engl. J. Med. 2001;345:1583–1592. doi: 10.1056/NEJMoa011090. [DOI] [PubMed] [Google Scholar]
- 56.Ito Y., Suzuki K., Ishii J., Hishida H., Tamakoshi A., Hamajima N., Aoki K. A population-based follow-up study on mortality from cancer or cardiovascular disease and serum carotenoids, retinol and tocopherols in Japanese inhabitants. Asian Pac. J. Cancer Prev. 2006;7:533–546. [PubMed] [Google Scholar]
- 57.Lonn E., Bosch J., Yusuf S., Sheridan P., Pogue J., Arnold J., Malcolm O., Ross C., Arnold A., Sleight P., Probstfield J., Dagenais G.R. Effects of long-term vitamin E supplementation on cardiovascular events and cancer: a randomized controlled trial. JAMA. 2005;293:1338–1347. doi: 10.1001/jama.293.11.1338. [DOI] [PubMed] [Google Scholar]
- 58.Kushi L.H., Folsom A.R., Prineas R.J., Mink P.J., Wu Y., Bostick R.M. Dietary antioxidant vitamins and death from coronary heart disease in postmenopausal women. N. Engl. J. Med. 1996;334:1156–1162. doi: 10.1056/NEJM199605023341803. [DOI] [PubMed] [Google Scholar]
- 59.Nojiri S., Daida H., Mokuno H., Iwama Y., Mae K., Ushio F., Ueki T. Association of serum antioxidant capacity with coronary artery disease in middle-aged men. Jpn. Heart J. 2001;42:677–690. doi: 10.1536/jhj.42.677. [DOI] [PubMed] [Google Scholar]
- 60.Mark S.D., Wang W., Fraumeni J.F., Li J.Y., Taylor P.R., Wang G.Q., Guo W., Dawsey S.M., Li B., Blot W.J. Lowered risks of hypertension and cerebrovascular disease after vitamin/mineral supplementation: the Linxian Nutrition Intervention Trial. Am. J. Epidemiol. 1996;143:658–664. doi: 10.1093/oxfordjournals.aje.a008798. [DOI] [PubMed] [Google Scholar]
- 61.Blot W.J., Li J.Y., Taylor P.R., Guo W., Dawsey S.M., Li B. The Linxian trials: mortality rates by vitamin-mineral intervention group. Am. J. Clin. Nutr. 1995;62:1424S–1426S. doi: 10.1093/ajcn/62.6.1424S. [DOI] [PubMed] [Google Scholar]
- 62.Heart Protection Study Collaborative Group MRC/BHF Heart Protection Study of antioxidant vitamin supplementation in 20 536 high-risk individuals: a randomised placebo-controlled trial//MRC/BHF Heart Protection Study of antioxidant vitamin supplementation in 20,536 high-risk individuals: a randomised placebo-controlled trial. Lancet. 2002;360:23–33. [Google Scholar]
- 63.Hense H.W., Stender M., Bors W., Keil U. Lack of an association between serum vitamin E and myocardial infarction in a population with high vitamin E levels. Atherosclerosis. 1993;103:21–28. doi: 10.1016/0021-9150(93)90036-t. [DOI] [PubMed] [Google Scholar]
- 64.Sahyoun N.R., Jacques P.F., Russell R.M. Carotenoids, vitamins C and E, and mortality in an elderly population. Am. J. Epidemiol. 1996;144:501–511. doi: 10.1093/oxfordjournals.aje.a008957. [DOI] [PubMed] [Google Scholar]
- 65.Sesso H.D., Buring J.E., Christen W.G., Kurth T., Belanger C., MacFadyen J., Bubes V., Manson J.E., Glynn R.J., Gaziano J.M. Vitamins E and C in the prevention of cardiovascular disease in men: the Physicians׳ Health Study II randomized controlled trial. JAMA. 2008;300:2123–2133. doi: 10.1001/jama.2008.600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Klipstein-Grobusch K., Geleijnse J.M., den Breeijen J.H., Boeing H., Hofman A., Grobbee D.E., Witteman J.C. Dietary antioxidants and risk of myocardial infarction in the elderly: the Rotterdam Study. Am. J. Clin. Nutr. 1999;69:261–266. doi: 10.1093/ajcn/69.2.261. [DOI] [PubMed] [Google Scholar]
- 67.Riemersma R.A., Wood D.A., Macintyre C.C., Elton R.A., Gey K.F., Oliver M.F. Risk of angina pectoris and plasma concentrations of vitamins A, C, and E and carotene. Lancet. 1991;337:1–5. doi: 10.1016/0140-6736(91)93327-6. [DOI] [PubMed] [Google Scholar]
- 68.Todd S., Woodward M., Tunstall-Pedoe H., Bolton-Smith C. Dietary antioxidant vitamins and fiber in the etiology of cardiovascular disease and all-causes mortality: results from the Scottish Heart Health Study. Am. J. Epidemiol. 1999;150:1073–1080. doi: 10.1093/oxfordjournals.aje.a009931. [DOI] [PubMed] [Google Scholar]
- 69.Boaz M., Smetana S., Weinstein T., Matas Z., Gafter U., Iaina A., Knecht A., Weissgarten Y., Brunner D., Fainaru M., Green M.S. Secondary prevention with antioxidants of cardiovascular disease in endstage renal disease (SPACE): randomised placebo-controlled trial. Lancet. 2000;356:1213–1218. doi: 10.1016/s0140-6736(00)02783-5. [DOI] [PubMed] [Google Scholar]
- 70.Ross R.K., Yuan J.M., Henderson B.E., Park J., Gao Y.T., Yu M.C. Prospective evaluation of dietary and other predictors of fatal stroke in Shanghai, China. Circulation. 1997;96:50–55. doi: 10.1161/01.cir.96.1.50. [DOI] [PubMed] [Google Scholar]
- 71.Delport R., Ubbink J.B., Human J.A., Becker P.J., Myburgh D.P., Vermaak W.J. Antioxidant vitamins and coronary artery disease risk in South African males. Clin. Chim. Acta. 1998;278:55–60. doi: 10.1016/s0009-8981(98)00131-4. [DOI] [PubMed] [Google Scholar]
- 72.Rajasekhar D., Srinivasa Rao P.V.L.N., Latheef S.A.A., Saibaba K.S.S., Subramanyam G. Association of serum antioxidants and risk of coronary heart disease in South Indian population. Indian J. Med. Sci. 2004;58:465–471. [PubMed] [Google Scholar]
- 73.Meraji S., Abuja P.M., Hayn M., Kostner G.M., Morris R., Oraii S., Tatzber F., Wonisch W., Zechner R., Gey K.F. Relationship between classic risk factors, plasma antioxidants and indicators of oxidant stress in angina pectoris (AP) in Tehran. Atherosclerosis. 2000;150:403–412. doi: 10.1016/s0021-9150(99)00394-9. [DOI] [PubMed] [Google Scholar]
- 74.Evans R.W., Shaten B.J., Day B.W., Kuller L.H. Prospective association between lipid soluble antioxidants and coronary heart disease in men. The Multiple Risk Factor Intervention Trial. Am. J. Epidemiol. 1998;147:180–186. doi: 10.1093/oxfordjournals.aje.a009432. [DOI] [PubMed] [Google Scholar]
- 75.de Gaetano G. Low-dose aspirin and vitamin E in people at cardiovascular risk: a randomised trial in general practice. Collaborative Group of the Primary Prevention Project. Lancet. 2001;357:89–95. doi: 10.1016/s0140-6736(00)03539-x. [DOI] [PubMed] [Google Scholar]
- 76.Keli S.O., Hertog M.G., Feskens E.J., Kromhout D. Dietary flavonoids, antioxidant vitamins, and incidence of stroke: the Zutphen study. Arch. Intern. Med. 1996;156:637–642. [PubMed] [Google Scholar]
- 77.Feki M., Souissi M., Mokhtar E., Hsairi M., Kaabachi N., Antebi H., Alcindor L.G., Mechmeche R., Mebazaa A. Vitamin E and coronary heart disease in Tunisians. Clin. Chem. 2000;46:1401–1405. [PubMed] [Google Scholar]
- 78.Ferns G., Williams J., Forster L., Tull S., Starkey B., Gershlick A. Cholesterol standardized plasma vitamin E levels are reduced in patients with severe angina pectoris. Int. J. Exp. Pathol. 2000;81:57–62. doi: 10.1111/j.1365-2613.2000.00141.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Cook N.R., Albert C.M., Gaziano J.M., Zaharris E., MacFadyen J., Danielson E., Buring J.E., Manson J.E. A randomized factorial trial of vitamins C and E and beta carotene in the secondary prevention of cardiovascular events in women: results from the Women׳s Antioxidant Cardiovascular Study. Arch. Intern. Med. 2007;167:1610–1618. doi: 10.1001/archinte.167.15.1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lee I.-M., Cook N.R., Gaziano J.M., Gordon D., Ridker P.M., Manson J.E., Hennekens C.H., Buring J.E. Vitamin E in the primary prevention of cardiovascular disease and cancer: the Women׳s Health Study: a randomized controlled trial. JAMA. 2005;294:56–65. doi: 10.1001/jama.294.1.56. [DOI] [PubMed] [Google Scholar]
- 81.Kardinaal A.F., Kok F.J., Ringstad J., Gomez-Aracena J., Mazaev V.P., Kohlmeier L., Martin B.C., Aro A., Kark J.D., Delgado-Rodriguez M. Antioxidants in adipose tissue and risk of myocardial infarction: the EURAMIC Study. Lancet. 1993;342:1379–1384. doi: 10.1016/0140-6736(93)92751-e. [DOI] [PubMed] [Google Scholar]
- 82.Goldstein J.L., Ho Y.K., Basu S.K., Brown M.S. Binding site on macrophages that mediates uptake and degradation of acetylated low density lipoprotein, producing massive cholesterol deposition. Proc. Natl. Acad. Sci. USA. 1979;76:333–337. doi: 10.1073/pnas.76.1.333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Schwartz C.J., Valente A.J., Sprague E.A., Kelley J.L., Nerem R.M. The pathogenesis of atherosclerosis: an overview. Clin. Cardiol. 1991;14:I1–16. doi: 10.1002/clc.4960141302. [DOI] [PubMed] [Google Scholar]
- 84.Erwig L.-P., Henson P.M. Clearance of apoptotic cells by phagocytes. Cell Death Differ. 2008;15:243–250. doi: 10.1038/sj.cdd.4402184. [DOI] [PubMed] [Google Scholar]
- 85.Robbins C.S., Hilgendorf I., Weber G.F., Theurl I., Iwamoto Y., Figueiredo J.-L., Gorbatov R., Sukhova G.K., Gerhardt L.M., Louisa M.S., Smyth D., Zavitz Caleb C.J., Shikatani E.A., Parsons M., van Rooijen N., Lin H.Y., Husain M., Libby P., Nahrendorf M., Weissleder R., Swirski F.K. Local proliferation dominates lesional macrophage accumulation in atherosclerosis. Nat. Med. 2013;19:1166–1172. doi: 10.1038/nm.3258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Cushing S.D., Berliner J.A., Valente A.J., Territo M.C., Navab M., Parhami F., Gerrity R., Schwartz C.J., Fogelman A.M. Minimally modified low density lipoprotein induces monocyte chemotactic protein 1 in human endothelial cells and smooth muscle cells. Proc. Natl. Acad. Sci. USA. 1990;87:5134–5138. doi: 10.1073/pnas.87.13.5134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Collot-Teixeira S., Martin J., McDermott-Roe C., Poston R., McGregor J.L. CD36 and macrophages in atherosclerosis. Cardiovasc. Res. 2007;75:468–477. doi: 10.1016/j.cardiores.2007.03.010. [DOI] [PubMed] [Google Scholar]
- 88.Newby A.C. Metalloproteinase expression in monocytes and macrophages and its relationship to atherosclerotic plaque instability. Arterioscler. Thromb. Vasc. Biol. 2008;28:2108–2114. doi: 10.1161/ATVBAHA.108.173898. [DOI] [PubMed] [Google Scholar]
- 89.Bäck M., Ketelhuth D.F., Agewall S. Matrix metalloproteinases in atherothrombosis. Prog. Cardiovas. Dis. 2010;52:410–428. doi: 10.1016/j.pcad.2009.12.002. [DOI] [PubMed] [Google Scholar]
- 90.Catalán U., Fernández-Castillejo S., Pons L., Heras M., Aragonés G., Anglès N., Morelló J.-R., Solà R. Alpha-tocopherol and BAY 11-7082 reduce vascular cell adhesion molecule in human aortic endothelial cells. J. Vasc. Res. 2012;49:319–328. doi: 10.1159/000337466. [DOI] [PubMed] [Google Scholar]
- 91.Wu D., Koga T., Martin K.R., Meydani M. Effect of vitamin E on human aortic endothelial cell production of chemokines and adhesion to monocytes. Atherosclerosis. 1999;147:297–307. doi: 10.1016/s0021-9150(99)00199-9. [DOI] [PubMed] [Google Scholar]
- 92.Noguchi N., Hanyu R., Nonaka A., Okimoto Y., Kodama T. Inhibition of THP-1 cell adhesion to endothelial cells by alpha-tocopherol and alpha-tocotrienol is dependent on intracellular concentration of the antioxidants. Free Radic. Biol. Med. 2003;34:1614–1620. doi: 10.1016/s0891-5849(03)00216-8. [DOI] [PubMed] [Google Scholar]
- 93.Berdnikovs S., Abdala-Valencia H., McCary C., Somand M., Cole R., Garcia A., Bryce P., Cook-Mills J.M. Isoforms of vitamin E have opposing immunoregulatory functions during inflammation by regulating leukocyte recruitment. J. Immunol. 2009;182:4395–4405. doi: 10.4049/jimmunol.0803659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Nakamura Y.K., Omaye S.T. Alpha-tocopherol modulates human umbilical vein endothelial cell expression of Cu/Zn superoxide dismutase and catalase and lipid peroxidation. Nutr. Res. 2008;28:671–680. doi: 10.1016/j.nutres.2008.07.002. [DOI] [PubMed] [Google Scholar]
- 95.Uemura M., Manabe H., Yoshida N., Fujita N., Ochiai J., Matsumoto N., Takagi T., Naito Y., Yoshikawa T. Alpha-tocopherol prevents apoptosis of vascular endothelial cells via a mechanism exceeding that of mere antioxidation. Eur. J. Pharmacol. 2002;456:29–37. doi: 10.1016/s0014-2999(02)02639-0. [DOI] [PubMed] [Google Scholar]
- 96.Nobata Y., Urakaze M., Temaru R., Sato A., Nakamura N., Yamazaki K., Kishida M., Takata M., Kobayashi M. alpha-Tocopherol Inhibits IL-8 synthesis induced by thrombin and high glucose in endothelial cells. Horm. Metab. Res. 2002;34:49–54. doi: 10.1055/s-2002-20523. [DOI] [PubMed] [Google Scholar]
- 97.McCary C.A., Yoon Y., Panagabko C., Cho W., Atkinson J., Cook-Mills J.M. Vitamin E isoforms directly bind PKCα and differentially regulate activation of PKCα. Biochem. J. 2012;441:189–198. doi: 10.1042/BJ20111318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Li D., Saldeen T., Mehta J.L. Effects of alpha-tocopherol on ox-LDL-mediated degradation of IkappaB and apoptosis in cultured human coronary artery endothelial cells. J. Cardiovasc. Pharmacol. 2000;36:297–301. doi: 10.1097/00005344-200009000-00003. [DOI] [PubMed] [Google Scholar]
- 99.Ricciarelli R., Zingg J.M., Azzi A. Vitamin E reduces the uptake of oxidized LDL by inhibiting CD36 scavenger receptor expression in cultured aortic smooth muscle cells. Circulation. 2000;102:82–87. doi: 10.1161/01.cir.102.1.82. [DOI] [PubMed] [Google Scholar]
- 100.Yasunari K., Kohno M., Kano H., Yokokawa K., Minami M., Yoshikawa J. Antioxidants improve impaired insulin-mediated glucose uptake and prevent migration and proliferation of cultured rabbit coronary smooth muscle cells induced by high glucose. Circulation. 1999;99:1370–1378. doi: 10.1161/01.cir.99.10.1370. [DOI] [PubMed] [Google Scholar]
- 101.Boscoboinik D., Szewczyk A., Azzi A. Alpha-tocopherol (vitamin E) regulates vascular smooth muscle cell proliferation and protein kinase C activity. Arch. Biochem. Biophys. 1991;286:264–269. doi: 10.1016/0003-9861(91)90039-l. [DOI] [PubMed] [Google Scholar]
- 102.Royer M.-C., Lemaire-Ewing S., Desrumaux C., Monier S., Pais de Barros, Jean-Paul, Athias A., Néel D., Lagrost L. 7-ketocholesterol incorporation into sphingolipid/cholesterol-enriched (lipid raft) domains is impaired by vitamin E: a specific role for alpha-tocopherol with consequences on cell death. J. Biol. Chem. 2009;284:15826–15834. doi: 10.1074/jbc.M808641200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Villacorta L., Graça-Souza A.V., Ricciarelli R., Zingg J.-M., Azzi A. Alpha-tocopherol induces expression of connective tissue growth factor and antagonizes tumor necrosis factor-alpha-mediated downregulation in human smooth muscle cells. Circ. Res. 2003;92:104–110. doi: 10.1161/01.res.0000049103.38175.1b. [DOI] [PubMed] [Google Scholar]
- 104.Ricciarelli R., Tasinato A., Clément S., Ozer N.K., Boscoboinik D., Azzi A. alpha-Tocopherol specifically inactivates cellular protein kinase C alpha by changing its phosphorylation state. Biochem. J. 1998;334(Pt 1):243–249. doi: 10.1042/bj3340243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Azzi A., Boscoboinik D., Marilley D., Ozer N.K., Stäuble B., Tasinato A. Vitamin E: a sensor and an information transducer of the cell oxidation state. Am. J. Clin. Nutr. 1995;62:1337S–1346S. doi: 10.1093/ajcn/62.6.1337S. [DOI] [PubMed] [Google Scholar]
- 106.Clément S.A., Tan C.C., Guo J., Kitta K., Suzuki Y.J. Roles of protein kinase C and alpha-tocopherol in regulation of signal transduction for GATA-4 phosphorylation in HL-1 cardiac muscle cells. Free Radic. Biol. Med. 2002;32:341–349. doi: 10.1016/s0891-5849(01)00802-4. [DOI] [PubMed] [Google Scholar]
- 107.Azzi A., Boscoboinik D., Clément S., Marilley D., Ozer N.K., Ricciarelli R., Tasinato A. Alpha-tocopherol as a modulator of smooth muscle cell proliferation. Prostaglandins Leukot. Essent. Fatty Acids. 1997;57:507–514. doi: 10.1016/s0952-3278(97)90436-1. [DOI] [PubMed] [Google Scholar]
- 108.Islam K.N., Devaraj S., Jialal I. alpha-Tocopherol enrichment of monocytes decreases agonist-induced adhesion to human endothelial cells. Circulation. 1998;98:2255–2261. doi: 10.1161/01.cir.98.21.2255. [DOI] [PubMed] [Google Scholar]
- 109.Devaraj S., Hugou I., Jialal I. Alpha-tocopherol decreases CD36 expression in human monocyte-derived macrophages. J. Lipid Res. 2001;42:521–527. [PubMed] [Google Scholar]
- 110.Rode S., Rubic T., Lorenz R.L. alpha-Tocopherol disturbs macrophage LXRalpha regulation of ABCA1/G1 and cholesterol handling. Biochem. Biophys. Res. Commun. 2008;369:868–872. doi: 10.1016/j.bbrc.2008.02.132. [DOI] [PubMed] [Google Scholar]
- 111.Teupser D., Thiery J., Seidel D. Alpha-tocopherol down-regulates scavenger receptor activity in macrophages. Atherosclerosis. 1999;144:109–115. doi: 10.1016/s0021-9150(99)00040-4. [DOI] [PubMed] [Google Scholar]
- 112.Kim S., Lee E.-H., Kim S.H., Lee S., Lim S.-J. Comparison of three tocopherol analogs as an inhibitor of production of proinflammatory mediators in macrophages. J. Pharmacol. Sci. 2012;118:237–244. doi: 10.1254/jphs.11152fp. [DOI] [PubMed] [Google Scholar]
- 113.Ng L.-T., Ko H.-J. Comparative effects of tocotrienol-rich fraction, α-tocopherol and α-tocopheryl acetate on inflammatory mediators and nuclear factor kappa B expression in mouse peritoneal macrophages. Food Chem. 2012;134:920–925. doi: 10.1016/j.foodchem.2012.02.206. [DOI] [PubMed] [Google Scholar]
- 114.Lyons N.M., Woods J.A., O’Brien N.M. alpha-Tocopherol, but not gamma-tocopherol inhibits 7 beta-hydroxycholesterol-induced apoptosis in human U937 cells. Free Radic. Res. 2001;35:329–339. doi: 10.1080/10715760100300861. [DOI] [PubMed] [Google Scholar]
- 115.Vejux A., Guyot S., Montange T., Riedinger J.-M., Kahn E., Lizard G. Phospholipidosis and down-regulation of the PI3-K/PDK-1/Akt signalling pathway are vitamin E inhibitable events associated with 7-ketocholesterol-induced apoptosis. J. Nutr. Biochem. 2009;20:45–61. doi: 10.1016/j.jnutbio.2007.12.001. [DOI] [PubMed] [Google Scholar]
- 116.Rimbach G., Fischer A., Stoecklin E., Barella L. Modulation of hepatic gene expression by alpha-tocopherol in cultured cells and in vivo. Ann. N. Y. Acad. Sci. 2004;1031:102–108. doi: 10.1196/annals.1331.011. [DOI] [PubMed] [Google Scholar]
- 117.Barella L., Muller P.Y., Schlachter M., Hunziker W., Stöcklin E., Spitzer V., Meier N., de Pascual-Teresa S., Minihane A.-M., Rimbach G. Identification of hepatic molecular mechanisms of action of alpha-tocopherol using global gene expression profile analysis in rats. Biochim. Biophys. Acta. 1689;2004:66–74. doi: 10.1016/j.bbadis.2004.02.002. [DOI] [PubMed] [Google Scholar]
- 118.Valastyan S., Thakur V., Johnson A., Kumar K., Manor D. Novel transcriptional activities of vitamin E: inhibition of cholesterol biosynthesis. Biochemistry. 2008;47:744–752. doi: 10.1021/bi701432q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Poirier J., Cockell K.A., Ratnayake W.M. Nimal, Scoggan K.A., Hidiroglou N., Gagnon C., Rocheleau H., Gruber H., Griffin P., Madère R., Trick K., Kubow S. Antioxidant supplements improve profiles of hepatic oxysterols and plasma lipids in butter-fed hamsters. Nutr. Metab. Insights. 2010;3:1–14. doi: 10.4137/NMI.S3911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.González R., Collado J.A., Nell S., Briceño J., Tamayo M.J., Fraga E., Bernardos A., López-Cillero P., Pascussi J.M., Rufián S., Vilarem M.-J., De la Mata Manuel, Brigelius-Flohe R., Maurel P., Muntané J. Cytoprotective properties of alpha-tocopherol are related to gene regulation in cultured D-galactosamine-treated human hepatocytes. Free Radic. Biol. Med. 2007;43:1439–1452. doi: 10.1016/j.freeradbiomed.2007.07.023. [DOI] [PubMed] [Google Scholar]
- 121.Kluth D., Landes N., Pfluger P., Müller-Schmehl K., Weiss K., Bumke-Vogt C., Ristow M., Brigelius-Flohé R. Modulation of Cyp3a11 mRNA expression by alpha-tocopherol but not gamma-tocotrienol in mice. Free Radic. Biol. Med. 2005;38:507–514. doi: 10.1016/j.freeradbiomed.2004.11.010. [DOI] [PubMed] [Google Scholar]
- 122.Li W., Hellsten A., Jacobsson L.S., Blomqvist H.M., Olsson A.G., Yuan X.-M. Alpha-tocopherol and astaxanthin decrease macrophage infiltration, apoptosis and vulnerability in atheroma of hyperlipidaemic rabbits. J. Mol. Cell. Cardiol. 2004;37:969–978. doi: 10.1016/j.yjmcc.2004.07.009. [DOI] [PubMed] [Google Scholar]
- 123.Satchell L., Leake D.S. Oxidation of low-density lipoprotein by iron at lysosomal pH: implications for atherosclerosis. Biochemistry. 2012;51:3767–3775. doi: 10.1021/bi2017975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Williams R.J., Motteram J.M., Sharp C.H., Gallagher P.J. Dietary vitamin E and the attenuation of early lesion development in modified Watanabe rabbits. Atherosclerosis. 1992;94:153–159. doi: 10.1016/0021-9150(92)90240-h. [DOI] [PubMed] [Google Scholar]
- 125.Praticò D., Tangirala R.K., Rader D.J., Rokach J., FitzGerald G.A. Vitamin E suppresses isoprostane generation in vivo and reduces atherosclerosis in ApoE-deficient mice. Nat. Med. 1998;4:1189–1192. doi: 10.1038/2685. [DOI] [PubMed] [Google Scholar]
- 126.Ozer N.K., Sirikçi O., Taha S., San T., Moser U., Azzi A. Effect of vitamin E and probucol on dietary cholesterol-induced atherosclerosis in rabbits. Free Radic. Biol. Med. 1998;24:226–233. doi: 10.1016/s0891-5849(97)00136-6. [DOI] [PubMed] [Google Scholar]
- 127.Koga T., Kwan P., Zubik L., Ameho C., Smith D., Meydani M. Vitamin E supplementation suppresses macrophage accumulation and endothelial cell expression of adhesion molecules in the aorta of hypercholesterolemic rabbits. Atherosclerosis. 2004;176:265–272. doi: 10.1016/j.atherosclerosis.2004.05.034. [DOI] [PubMed] [Google Scholar]
- 128.Axford-Gately R.A., Wilson G.J. Myocardial infarct size reduction by single high dose or repeated low dose vitamin E supplementation in rabbits. Can. J. Cardiol. 1993;9:94–98. [PubMed] [Google Scholar]
- 129.Keaney J.F., Gaziano J.M., Xu A., Frei B., Curran-Celentano J., Shwaery G.T., Loscalzo J., Vita J.A. Low-dose alpha-tocopherol improves and high-dose alpha-tocopherol worsens endothelial vasodilator function in cholesterol-fed rabbits. J. Clin. Invest. 1994;93:844–851. doi: 10.1172/JCI117039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Hasty A.H., Gruen M.L., Terry E.S., Surmi B.K., Atkinson R.D., Gao L., Morrow J.D. Effects of vitamin E on oxidative stress and atherosclerosis in an obese hyperlipidemic mouse model. J. Nutr. Biochem. 2007;18:127–133. doi: 10.1016/j.jnutbio.2006.03.012. [DOI] [PubMed] [Google Scholar]
- 131.Brigelius-Flohé R., Traber M.G. Vitamin E: function and metabolism. FEBS J. 1999;13:1145–1155. [PubMed] [Google Scholar]
- 132.Brigelius-Flohé R., Kelly F.J., Salonen J.T., Neuzil J., Zingg J.-M., Azzi A. The European perspective on vitamin E: current knowledge and future research. Am. J. Clin. Nutr. 2002;76:703–716. doi: 10.1093/ajcn/76.4.703. [DOI] [PubMed] [Google Scholar]
- 133.Sontag T.J., Parker R.S. Cytochrome P450 omega-hydroxylase pathway of tocopherol catabolism. Novel mechanism of regulation of vitamin E status. J. Biol. Chem. 2002;277:25290–25296. doi: 10.1074/jbc.M201466200. [DOI] [PubMed] [Google Scholar]
- 134.Traber M.G. Mechanisms for the prevention of vitamin E excess. J. Lipid Res. 2013;54:2295–2306. doi: 10.1194/jlr.R032946. [DOI] [PMC free article] [PubMed] [Google Scholar]