

Abstract
Extensive necrosis of ischemic cardiomyocytes in the infarcted myocardium activates the innate immune response triggering an intense inflammatory reaction. Release of danger signals from dying cells and damaged matrix activates the complement cascade and stimulates Toll-Like Receptor (TLR)/Interleukin (IL)-1 signaling, resulting in activation of the Nuclear Factor (NF)-κB system and induction of chemokines, cytokines and adhesion molecules. Subsequent infiltration of the infarct with neutrophils and mononuclear cells serves to clear the wound from dead cells and matrix debris, while stimulating reparative pathways. In addition to its role in repair of the infarcted heart and formation of a scar, the immune system is also involved in adverse remodeling of the infarcted ventricle. Overactive immune responses and defects in suppression, containment and resolution of the post-infarction inflammatory reaction accentuate dilative remodeling in experimental models and may be associated with chamber dilation, systolic dysfunction and heart failure in patients surviving a myocardial infarction. Interventions targeting the inflammatory response to attenuate adverse remodeling may hold promise in patients with myocardial infarction that exhibit accentuated, prolonged, or dysregulated immune responses to the acute injury.
1. Introduction
Because the adult mammalian heart has negligible regenerative capacity, necrotic myocardial injury in myocardial infarction activates a reparative response that ultimately leads to replacement of dead cardiomyocytes with scar tissue1, Cardiac repair is dependent on mobilization of the immune system that serves to clear the wound from dead cells and matrix debris and to produce mediators that activate fibroblast growth and angiogenesis 2. However, activation of immune pathways to repair the injured heart does not have exclusively beneficial effects. Even in vulnerable species, the late manifestation of myocardial infarction well after the reproductive age does not impose any evolutionary pressures on the immune system. Thus, immune responses evolved to protect cutaneous and mucosal surfaces from bacterial contamination and may be inherently overactive for the sterile environment of the injured heart. In the myocardium, where normal function is dependent on optimal preservation of structure, even subtle accentuation of immunoinflammatory pathways may have catastrophic consequences.
Over the last 30 years a large amount of experimental work suggested that immune responses may extend injury and accentuate adverse remodeling in several distinct cardiac pathologic conditions 3, 4. Because sudden acute loss of a large number of cardiomyocytes in myocardial infarction is associated with an intense inflammatory reaction, the bulk of the evidence on the role of immune activation in cardiac injury is derived from investigations in models of ischemic heart disease. In the 1980s and 1990s extensive experimental evidence suggested that cytotoxic inflammatory cells infiltrating the infarcted myocardium may exacerbate ischemic injury causing death of viable cardiomyocytes 5. Unfortunately, translation of this concept in the clinical context was unsuccessful, as clinical trials examining the effects of targeted anti-inflammatory approaches in patients with myocardial infarction did not improve outcome and failed to reduce the size of the infarct 6, 7. In recent years, studies in genetically targeted mice led to the development of a new paradigm, suggesting that inflammatory signals may not directly extend ischemic cardiomyocyte injury, but may play an important role in development of dilative remodeling and heart failure 3. Our current review manuscript discusses the involvement of immune cells and pathways in cardiac repair and remodeling of the injured heart and attempts to identify promising therapeutic targets, through the lessons we have learned both from our experimental work and through the translational failures of the past. We highlight the importance of endogenous STOP signals that negatively regulate the inflammatory reaction, orchestrating the transition from inflammation to repair.
2. The immune system in post-infarction remodeling
2.1. Repair and remodeling of the infarcted heart: an overview of the cellular events
Activation of the immune system is a hallmark of myocardial infarction. After 15-20 minutes of severe ischemia cardiomyocytes exhibit irreversible alterations that ultimately result in their death. Subendocardial cardiomyocytes are more susceptible to ischemic injury; thus, a “wavefront” of necrosis is noted that extends from the subendocardial area to the subepicardium 8. Ischemia and necrotic cardiomyocytes trigger a reparative process that can be divided into three distinct but overlapping phases. During the inflammatory phase, activation of innate immune signals results in induction of chemokines and cytokines leading to infiltration of the infarct with neutrophils and pro-inflammatory monocytes that clear the infarct from dead cells and matrix debris. Apoptotic death of neutrophils marks the end of the inflammatory phase and may activate signals that inhibit inflammation and induce resolution of the leukocyte infiltrate. The proliferative phase follows, as reparative monocytes are recruited in the infarct and macrophages may acquire an angiogenic or fibrogenic phenotype that promotes differentiation and growth of myofibroblasts and endothelial cells. Activated myofibroblasts contribute to wound contraction and produce structural matrix proteins forming a scar. Maturation of the infarct follows and is characterized by progressive apoptotic death of the cellular elements and by collagen cross-linking. As the infarct heals, the ventricle dilates, while non-infarcted segments become hypertrophic and exhibit increased interstitial fibrosis. The extent of adverse remodeling is dependent not only on the size of the infarct, but also on the qualitative characteristics of the wound.
2.2. Initiation of the immune response in the infarcted myocardium
Acute sudden death of cardiomyocytes in the infarcted heart rapidly activates the immune system triggering an intense, but transient, inflammatory reaction. In the infarcted myocardium, cellular necrosis and matrix fragmentation generate alarm signals that activate the innate immune response; these molecules are known as danger-associated molecular patterns (DAMPs) 9. The nuclear chromatin-binding protein High-mobility group box-1 (HMGB1) plays a crucial role in initiation of the inflammatory response through actions that may involve Toll-Like Receptor (TLR) and RAGE signaling 10. Heat shock proteins (HSPs), ATP and matrix fragments (including low molecular weight hyaluronan and fibronectin fragments) also serve as “danger signals” activating the immune response 9, 11, 12, 13.
The immune response following myocardial infarction involves signaling through the TLRs, a family of transmembrane receptors that activate downstream pro-inflammatory cascades. TLRs recognize conserved motifs in pathogens, thus contributing to protection from infection, but are also activated by DAMPs, initiating immune responses that clear injured tissues from dead cells and matrix debris and activate reparative signals. 13 mammalian TLRs have been identified; in the infarcted myocardium, loss-of function approaches have identified TLR2 and TLR4, both localized in the cell surface, as important mediators of the inflammatory reaction. Impaired TLR4 signaling attenuated the inflammatory reaction following myocardial infarction and reduced adverse remodeling; complete loss of TLR4 was also associated with a reduction in the size of the infarct 14, 15. TLR2 signaling has also been reported to mediate inflammatory cardiomyocyte injury following coronary ischemia and reperfusion 16. DAMPs also activate the complement system, another essential cascade in transducing the immune response in the infarcted heart. Hill and Ward demonstrated C3 cleavage in the infarcted myocardium and documented a role for the complement system in leukocyte infiltration 17. Moreover, complement inhibition consistently attenuated leukocyte recruitment following myocardial infarction 18 highlighting the critical role of the complement cascade in triggering inflammation in the ischemic myocardium.
Generation of reactive oxygen species (ROS) also activates immune cells in the infarcted heart. ROS promote leukocyte chemotaxis 19 by activating complement, by stimulating expression of P-selectin and by upregulating cytokine and chemokine synthesis through activation of the Nuclear Factor (NF)-κB system 20. Although reactive oxygen generation is important for cardiac repair following infarction, overactive ROS-mediated signaling has injurious potential by promoting cell apoptosis and by inducing degradation of the extracellular matrix. The normal heart possesses substantial ability to counterbalance the generation of ROS through inhibitory enzymatic pathways (such as catalase, glutathione peroxidase and the superoxide dismutases) and through the actions of intracellular antioxidants. Following myocardial infarction, the antioxidant defenses are overwhelmed, resulting in net generation of free radicals that may suppress myocardial function and activate immune cells inducing inflammatory injury. Because of the pleiotropic effects of ROS, both beneficial and detrimental actions have been reported after administration of free radical scavengers in myocardial infarction.
2.3. Cytokine and chemokine signaling in the infarcted heart
TLR-mediated, complement-activated and ROS-induced pathways in the infarcted myocardium converge on activation of NF-κB that drives induction of adhesion molecules in endothelial cells and production of pro-inflammatory cytokines and chemokines by fibroblasts, leukocytes and vascular cells 21, 22. As the prototypical pro-inflammatory cytokine, Interleukin (IL)-1 mediates synthesis of chemotactic mediators in the infarct and stimulates leukocyte recruitment 23, 24. Processing and generation of active IL-1β in inflamed tissues requires formation of specialized molecular platforms, called “inflammasomes” 25. Two recent investigations have demonstrated activation of the inflammasome in the infarcted myocardium 26, 27. Inflammasome activation in the infarct is localized in leukocytes, resident fibroblasts and border zone cardiomyocytes 26, 27 and drives IL-1-mediated inflammatory cell infiltration and cytokine synthesis. ROS production, ATP and potassium efflux may play a role in inflammasome activation in infarct cells.
The pro-inflammatory cytokine Tumor Necrosis Factor (TNF)-α is also released in the infarcted myocardium and may play a role in stimulating cytokine synthesis by mononuclear cells infiltrating the infarct 28. In addition to its pro-inflammatory actions TNF-α may also exert cytoprotective effects 29 and may regulate post-infarction cardiac remodeling through divergent actions involving TNFR1 and TNFR2 receptors 30. TNFR1 signaling stimulates cardiomyocyte apoptosis and enhances inflammatory activity in the infarcted heart, whereas TNFR2 reduces TNF-induced NF-κB activation and attenuates cardiac injury 30.
Members of the IL-6 family of cytokines (including IL-6, cardiotrophin-1, oncostatin-M and leukemia inhibitory factor) are also consistently upregulated in experimental models of myocardial infarction and may modulate the inflammatory and reparative response, signaling through receptors that share the transmembrane glycoprotein (gp)130 31. Genetic loss of IL-6 did not affect infarct size, ventricular function and cardiac remodeling in non-reperfused infarcts32; in the absence of IL-6 other gp130 cytokines may act in a compensatory manner, activating JAK/STAT signaling and maintaining STAT3 phosphorylation. On the other hand, experiments targeting IL-6 through administration of an anti-IL-6 receptor antibody attenuated adverse remodeling33. Timely activation and suppression of gp130 signaling may play an important role in regulation of post-infarction remodeling. In an experimental model of myocardial infarction, failure to suppress gp130/STAT3 signaling resulted in prolonged and accentuated inflammation predisposing to cardiac rupture 34.
Several members of the chemokine family are also prominently involved in the post-infarction inflammatory response 35 and provide key directional signals for recruitment of leukocyte subpopulations into the infarct. CC chemokines that serve as mononuclear cell chemoattractants, ELR+ CXC chemokines that function to attract neutrophils in sites of injury, and ELR-negative CXC chemokines that recruit lymphocytes and may exert antifibrotic and angiostatic actions are rapidly, but transiently, upregulated in the infarcted myocardium. The chemotactic actions of chemokines are dependent on their immobilization to glycosaminoglycans on the endothelial surface, or on the extracellular matrix; as leukocytes are captured by activated endothelial cells and roll on the endothelial surface (through selectin-mediated actions) they “sense” the bound chemokines, and exhibit integrin activation, thus engaging in firm adhesive interactions with vascular endothelial cells (Figure 1). Various leukocyte subpopulations exhibit distinct chemokine receptor profiles and interact with different chemokines; thus selective activation of chemokine/chemokine receptor pairs orchestrates recruitment of leukocyte subpopulations with distinct properties 36, 37, 38. Activated leukocyte β2 integrins interact with endothelial Intercellular Adhesion Molecule (ICAM)-1 resulting in firm adhesion of leukocytes to the endothelial layer. Transmigration of activated leukocyte follows, and is dependent on several adhesion molecules, including ICAM-1, members of the Junctional Adhesion Molecule (JAM) family and Vascular-endothelial (VE)-cadherin 39, 40, 41.
Figure 1.
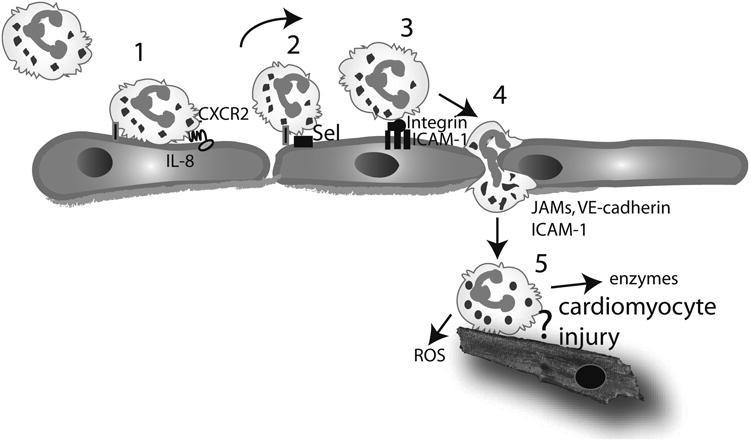
Neutrophil infiltration in the infarcted myocardium and the concept of leukocyte-mediated cardiomyocyte injury. Neutrophil extravasation in the infarcted heart occurs in a series of sequential steps. Circulating neutrophils are captured by activated endothelial cells (1) and roll on the endothelial surface (2) through selectin-mediated interactions. Captured neutrophils can “sense” chemokines immobilized in the endothelial surface (such as IL-8). Interactions between the chemokines and their corresponding chemokine receptors activate integrins on the surface of the neutrophils. Interactions between leukocyte b2 integrins and endothelial adhesion molecules (such as ICAM-1) result in firm adhesion of the neutrophils (3). Subsequently the neutrophils transmigrate across the endothelial layer through interactions that involve adhesion molecules, including VE-cadherin, ICAM-1 and members of the JAM family (4). In the infarcted tissue, neutrophils release reactive oxygen species (ROS) and proteolytic enzymes, playing a role in clearance of the infarct from dead cells and matrix debris (5). Experimental evidence has suggested that neutrophils may adhere to viable cardiomyocytes and exert cytotoxic effects; however, this concept has been challenged by recent studies using genetically targeted animals.
2.4. The cellular effectors of the post-infarction inflammatory reaction
Both resident and bone marrow-derived cells participate in the post-infarction inflammatory reaction; understanding the relative contribution of inflammatory activation of various cell types is limited by the absence of studies using cell-specific loss-of-function approaches in vivo.
2.4.1. Cardiomyocytes
Necrotic cardiomyocytes release a variety of danger signals, thus actively contributing to activation of innate immune cascades. Moreover, border zone cardiomyocytes may exhibit activation of the inflammasome, thus serving as a source of bioactive IL-1β.
2.4.2. Platelets
In addition to their hemostatic role, platelets modulate inflammatory responses and may link tissue injury with the activation of reparative pathways 42. In the healing infarct activated platelets aggregate in the site of injury and may contribute to the formation of a fibrin-based provisional matrix, necessary to support influx of hematopoietic cells and migration of reparative mesenchymal cells 43, 13. Platelets may also release chemokines, cytokines and growth factors in the infarcted tissue; however, their relative significance in the post-infarction inflammatory reaction remains unknown.
2.4.3. The neutrophils
Activation of the complement cascade, ROS generation, and ELR+ CXC chemokines have been implicated in recruitment of neutrophils in the infarcted myocardium. By generating ROS and through the release of proteolytic enzymes, neutrophils infiltrating the infarcted myocardium contribute to the clearance of the infarct from dead cells and matrix debris. In addition, neutrophils are capable of secreting pro-inflammatory mediators that may amplify recruitment of mononuclear cells. Experimental evidence, derived primarily through neutrophil depletion strategies, suggested that neutrophils may directly injure ischemic, but viable, cardiomyocytes increasing the size of the infarct through adhesive interactions involving neutrophil integrin activation and cardiomyocyte ICAM-1 expression 44. However, the in vivo significance of neutrophil-induced cardiomyocyte injury has been challenged by loss-of-function experiments in genetically targeted mice. ICAM-1 null mice had no significant difference in infarct size and scar formation after 1-3 weeks of reperfusion 45. Moreover, animals with a combined deficiency in both ICAM-1 and P-selectin and animals with disrupted IL-1 signaling showed no reduction in infarct size following ischemia and reperfusion despite a marked impairment in neutrophil trafficking 46, 23.
2.4.4. Monocytes and macrophages
Monocyte subpopulations are sequentially recruited in the infarcted myocardium and regulate inflammatory and reparative cascades. Infiltrating monocytes are primarily derived from the circulating blood; however, the contribution of alternative sources, such as the spleen is increasingly recognized 47, 48. During the first few hours after reperfusion, complement activation and induction of the CC chemokine CCL2/monocyte chemoattractant protein (MCP)-1 play an important role in recruitment of pro-inflammatory monocytes 49, 36; these cells exhibit potent pro-inflammatory and phagocytotic properties. At a later stage, recruitment of reparative mononuclear cell subsets may play an important role in activation of fibroblasts and endothelial cells, promoting formation of a scar. Understanding of the time course, phenotype and mechanism of recruitment of specific monocyte subsets in the healing infarct is limited.
In response to the dynamic alterations in cytokine and growth factor expression in the infarct, infiltrating monocytes undergo phenotypic changes. Maturation of monocytes into mature macrophages involves the local upregulation of growth factors, such as Macrophage-Colony Stimulating Factor (M-CSF) 50. Considering the remarkable potential of macrophages to acquire unique phenotypic characteristics in response to microenvironmental cues, differentiation of several distinct subpopulations of macrophages is likely to orchestrate the inflammatory and reparative response. Thus, macrophage subsets may play multiple roles in the healing infarct. First, they phagocytose dead cells and matrix debris and clear the infarct from apoptotic neutrophils and cardiomyocytes. Second, they may serve as a source of cytokines and growth factors regulating inflammatory activity, fibroblast growth and activation, and neovessel formation and maturation. Third, they may contribute to extracellular matrix remodeling by producing matrix metalloproteinases (MMPs) and their inhibitors.
2.4.5. The mast cells
In large animals, the heart contains a significant number of resident mast cells predominantly located in close proximity to vessels; the population of cardiac mast cells is smaller in mice, leading perhaps to underestimation of their role in heart disease when murine models of cardiac injury are used 51. In a canine model of experimental infarction, resident cardiac mast cells in the ischemic territory showed rapid and impressive degranulation, releasing large amounts of preformed pro-inflammatory mediators, including histamine and TNF-α 28.
2.4.6. The fibroblasts
Fibroblasts are the most abundant non-cardiomyocytes in the adult mammalian heart 52 and are strategically located in the interstitial and perivascular space. In the infarcted heart resident cardiac fibroblasts undergo inflammatory activation and activate the inflammasome platform 27, thus serving as a source of bioactive IL-1β. Activated fibroblasts may also secrete chemokines; however, their relative contribution to the activation and progression of the post-infarction inflammatory reaction remains unknown.
2.4.7. Endothelial cells
Endothelial cells actively participate in the post-infarction inflammatory reaction. Cytokine release in the infarct zone induces adhesion molecule expression on the endothelial cell surface, thus promoting adhesive interactions with circulating leukocytes. Endothelial cells are also a major source of pro-inflammatory chemokines (such as MCP-1 and IP-10) 53, 54 in the infarcted myocardium.
3. Resolution of the inflammatory response and activation of fibrogenic and angiogenic pathways
Optimal tissue repair is dependent on activation of endogenous signals that restrain the inflammatory reaction and stimulate reparative cascades. These STOP signals are particularly important in repair of the injured heart. In the myocardium, normal function is dependent on optimal preservation of structure; even subtle alterations in cardiac morphology may induce severe dysfunction. Thus, in the injured heart unrestrained, prolonged or extended inflammatory activity could have catastrophic consequences, leading to extensive matrix degradation, adverse dilative remodeling and progressive dysfunction 3. Because myocardial infarction activates several distinct inflammatory cascades in all cell types involved in cardiac repair, multiple inhibitory signals may co-operate to restrain and suppress inflammatory signaling. Recent studies have identified specific cellular processes (Figure 2) and STOP signals involved in negative regulation of the post-infarction inflammatory reaction; in their absence, remodeling of the infarcted heart is accentuated.
Figure 2.
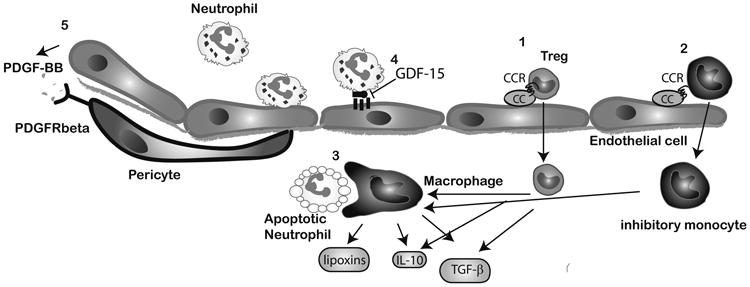
Cellular events associated with negative regulation of the post-infarction inflammatory response. Recruitment of inhibitory mononuclear cell subpopulations in the infarcted myocardium, such as Tregs (1) and inhibitory monocytes (2) may require activation of specific chemokine/chemokine receptor interactions. Macrophages acquire an anti-inflammatory phenotype and secrete inhibitory mediators upon phagocytosis of apoptotic neutrophils (3). Endothelial cells may participate in suppression of the inflammatory response (4) by producing mediators that inhibit adhesive interactions with leukocytes (such as GDF-15). Acquisition of a pericyte coat by infarct neovessels (5) may also suppress inflammatory activity and is mediated through PDGFR-b/PDGF-BB interactions.
3.1. The cellular effectors of resolution of inflammation
3.1.1. Clearance of apoptotic neutrophils and removal of matrix debris
Neutrophils participate in resolution of the inflammatory response through their death. Neutrophils infiltrating injured tissues are programmed to undergo apoptosis 55; ingestion of apoptotic neutrophils by macrophages exerts potent anti-inflammatory and immunosuppressive effects 56, mediated through macrophage-derived secretion of TGF-β, IL-10 and lipid-derived pro-resolving mediators (such as lipoxins). Clearance of apoptotic cells by macrophages may function as an important endogenous inhibitory mechanism; however, its role in resolution of inflammation following myocardial infarction has not been investigated.
Removal of matrix fragments generated in the injured heart also appears to play an essential role in restraining the post-infarction inflammatory response 13. Evidence from our laboratory suggested that clearance of low molecular weight hyaluronan fragments generates important inhibitory signals in the infarcted heart 12. In normal hearts, hyaluronan exists as a high molecular weight polymer; however following myocardial infarction hyaluronan undergoes degradation that may result in accumulation of low molecular weight species in the area of injury. Hyaluronan fragments activate endothelial cells and macrophages inducing synthesis of inflammatory cytokines and chemokines 57. Hyaluronan modulates the inflammatory response through interactions involving CD44, a ubiquitously distributed transmembrane adhesion molecule 58. CD44 null mice exhibit enhanced and prolonged post-infarction inflammation followed by reduced myofibroblast infiltration. Isolated CD44 -/- cardiac fibroblasts had decreased proliferative activity upon stimulation with serum and exhibited attenuated TGF-β-induced collagen synthesis when compared with wildtype cells. Accentuated inflammation and impaired fibroblast activation in CD44 -/- mice resulted in enhanced dilative remodeling of the infarcted ventricle, without affecting the size of the infarct. Thus, the effects of CD44 signaling in the infarcted myocardium may extend beyond its role in activating an anti-inflammatory cascade and may also involve regulation of fibrogenic responses.
3.1.2. Regulatory T cells (Tregs) and inhibitory monocytes/macrophages
Mononuclear cell subpopulations with inhibitory properties are ideally suited as negative regulators of the post-infarction inflammatory reaction. The sequential recruitment of inflammatory and reparative monocytes in the healing infarct 36 suggests that early activation of MCP-1/CCR2-dependent chemotaxis may be followed by late activation of chemotactic pathways that specifically attract inhibitory cells. The chemokine/chemokine receptor pairs that may be involved in recruitment of inhibitory monocytes remain poorly understood. Differentiated macrophages may also play an important role in suppression of the post-infarction inflammatory reaction. In the dynamic and complex microenvironment of the infarct, cytokine stimulation may drive differentiation of macrophage subpopulations with inhibitory properties that may serve as key effectors in negative regulation of the inflammatory reaction.
Inhibitory lymphocytes are also recruited in the infarcted myocardium and may play an important role in restraining the post-infarction inflammatory response. Regulatory T cells (Tregs) infiltrate the healing infarct and may suppress macrophage and lymphocyte inflammatory activity through contact-dependent interactions and by secreting soluble inhibitory mediators, such as IL-10 and TGF-β 37. Interactions involving the chemokine receptor CCR5 are crucial for recruitment of Tregs in the infarct and play an important role in negative regulation of the inflammatory reaction 37.
3.1.3. Vascular maturation and the role of the pericytes
Healing of the infarcted heart is associated with intense angiogenesis, as a rich microvascular network is formed to supply the wound with nutrients and oxygen, Rapid induction of angiogenic growth factors results in formation of a network of hyperpermeable neovessels 53 that may lack a pericyte coat 59. As the infarct vasculature matures, some infarct neovessels are coated with pericytes, whereas uncoated vessels regress 60. Mature coated vessels are protected from regression and exhibit decreased inflammatory activity and reduced angiogenic potential contributing to stabilization of the scar. Our experiments have demonstrated that acquisition of a muscular coat by infarct neovessels requires activation of Platelet Derived Growth Factor (PDGF)-BB/PDGF-Rβ interactions 61.
3.2. Molecular signals involved in suppression and resolution of inflammation
Suppression and resolution of the post-infarction inflammatory reaction involves activation of a complex network of endogenous STOP signals that may co-operate to inhibit inflammatory activity in all cell types involved in cardiac repair. Secretion of soluble inhibitory mediators (such as IL-10 and TGF-β), activation of endogenous cascades that inhibit innate immune response, increased expression of decoy receptors and deposition of matricellular proteins that modulate inflammatory signaling may play an important role in termination and resolution of the post-infarction inflammatory reaction.
3.2.1. IL-10
IL-10 expression shows a late and prolonged time course in the infarcted myocardium and is localized in T cells and in a subset of macrophages 62. Associative in vivo studies and in vitro experiments by our laboratory suggested that IL-10 may suppress pro-inflammatory cytokine synthesis and may contribute to stabilization of the matrix by inducing synthesis of protease inhibitors, such as Tissue Inhibitor of Metalloproteinases (TIMP)-1, by macrophages. Experiments in IL-10 null mice have produced contradictory results on the role of IL-10 in negative regulation of the immune response following myocardial infarction. Yang and coworkers suggested that IL-10 -/- mice had markedly increased mortality and exhibited an enhanced inflammatory response following reperfused myocardial infarction 63. In contrast, our laboratory found no effects of IL-10 disruption on survival of mice undergoing reperfused infarction protocols and showed only subtle differences between IL-10 null and wildtype animals 64. Although IL-10 -/- mice had higher peak myocardial TNF-α and MCP-1 mRNA levels following infarction than wildtype animals, both groups had timely repression of pro-inflammatory cytokine and chemokine mRNA synthesis, exhibited a similar time course of resolution of the neutrophil infiltrate and had comparable adverse remodeling 64. Our findings suggested that IL-10 signaling plays a non-critical role in suppression of inflammatory mediators, resolution of the inflammatory response, fibrosis and matrix metabolism following myocardial infarction.
3.2.2. TGF-β
TGF-β, a highly pleiotropic and multifunctional mediator with context-dependent actions, may play an important role in suppression of the immune response following infarction, orchestrating the transition from inflammation to fibrous tissue deposition. TGF-β suppresses cytokine and chemokine synthesis by macrophages and endothelial cells, reduces adhesion molecule expression and promotes Treg differentiation 65. On the other hand, TGF-β activates fibroblasts, inducing myofibroblast transdifferentiation, stimulating synthesis of extracellular matrix proteins (including collagens, fibronectin, matricellular proteins and proteoglycans) 66, and suppressing matrix degradation by inducing expression of protease inhibitors (such as Plasminogen Activator Inhibitor (PAI)-1 and TIMP-1) 67. In healing infarcts, TGF-β may serve as a “master switch” that links repression of the inflammatory reaction with activation of a fibrogenic program. Unfortunately, the complex biology of TGF-β activation, its pleiotropic and context-dependent actions, and its multiple cellular targets have hampered our efforts to understand its role in modulating the immune response in the infarcted heart. A crucial role for TGF-β in resolution of post-infarction inflammation was suggested inhibition studies through injection of an adenovirus harboring soluble TGF-β type II receptor in the hindlimb muscles. Late TGF-β inhibition attenuated left ventricular remodeling by reducing cardiac fibrosis 68, 69; however, early TGF-β inhibition significantly increased mortality and exacerbated left ventricular dilation enhancing cytokine synthesis. These findings suggested that TGF-β signaling may play an important role in suppression of inflammatory mediator synthesis following myocardial infarction 68.
TGF-β exerts its cellular effects by binding to the constitutively active type II receptor (TβRII); subsequently the complex transphosphorylates the cytoplasmic domain of the type I receptor (TβRI). Activation of TβRI propagates downstream intracellular signals, through the Smad proteins, essential components of the TGF-β signaling pathway 70, but also activates Smad-independent pathways. Our experiments demonstrated that the Smad3 cascade is responsible for the fibrogenic, but not for the anti-inflammatory actions of TGF-β in the infarcted myocardium 71, 72.
3.2.3. Growth differentiation factor (GDF)-15
GDF-15, a member of the TGF-β superfamily, has been recently identified as essential inhibitory molecule that suppresses chemokine-triggered conformational integrin activation in neutrophils, thus limiting activation of inflammatory leukocytes and adhesion to the endothelium 73. Loss of the anti-inflammatory actions of GDF-15 results in an increased incidence of cardiac rupture following myocardial infarction 73.
3.2.4. Expression of decoy receptors as a mechanism terminating inflammation
A growing body of evidence suggests that certain cytokine and chemokine receptors may bind to their ligands with high affinity without transducing a signal, thus serving as a molecular trap that removes the cytokine or chemokine from the circulation. Such decoy receptors have been identified for several members of the cytokine and chemokine family; however, their role in regulating the post-infarction inflammatory response remains unknown 74. The balance between surface expression of signaling and decoy receptors may determine cellular responsiveness to pro-inflammatory cytokines and chemokines. A recent investigation demonstrated that the decoy chemokine receptor D6 is expressed in healing infarcts and protects the heart from excessive inflammation and adverse remodeling 75.
3.2.5. Activation of intracellular pathways that inhibit the innate immune response
Because unrestrained innate immune responses could have catastrophic effects on tissue structure, multiple endogenous inhibitory signals have evolved to limit and contain activation of TLR/IL-1 signaling 76. Interleukin Receptor-Associated Protein (IRAK)-M is a member of the IRAK family that, unlike IRAK-1 and IRAK-4, does not transduce pro-inflammatory signals, but is predominantly expressed in macrophages and limits TLR/IRAK-1 dependent activation (Figure 3). Recent experiments from our laboratory demonstrated that IRAK-M is upregulated in both macrophages and fibroblasts in the infarcted heart and is critically involved in negative regulation of the innate immune response. IRAK-M loss results in accentuated dilative remodeling, associated with increased inflammation and enhanced MMP activation 77. Although macrophage IRAK-M had anti-inflammatory actions, IRAK-M upregulation in cardiac fibroblasts does not affect their inflammatory capacity but restrains expression and activation of MMPs promoting a matrix-preserving phenotype 77.
Figure 3.
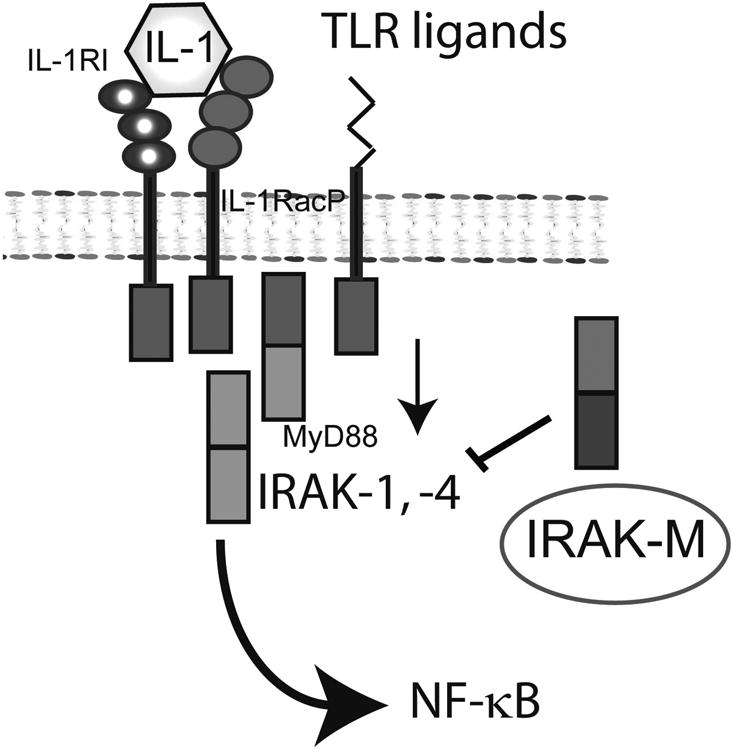
Negative regulation of IL-1/TLR responses plays an important role in restraining the post-infarction inflammatory reaction. IRAK-M is expressed in infarct macrophages and fibroblasts and limits IL-1/TLR-mediated NF-κB activation.
3.2.6. Upregulation of matricellular proteins in the infarct border zone may inhibit the inflammatory reaction
In addition to its structural role, the cardiac extracellular matrix is also capable of modulating cellular phenotype and function; components of the matrix activate and transduce key signals in inflammation and tissue repair. Following infarction, the cardiac extracellular matrix undergoes dynamic changes that drive the inflammatory and reparative response. During the first few hours after infarction generation of matrix fragments stimulates inflammatory mediator synthesis 13. Formation of a provisional matrix network comprised of fibrin and fibronectin provides a conduit for migrating cells facilitating the reparative process 43, 78. The proliferative phase of infarct healing is associated with marked induction and deposition of matricellular proteins, a family of macromolecules that do not play a structural role, but bind to the matrix and modulate cell-cell and cell-matrix interactions 79. Several members of the matricellular protein family modulate the post-infarction inflammatory reaction. Thrombospondin (TSP) -1, a potent angiostatic protein with a crucial role in TGF-β activation is selectively upregulated in the infarct border zone and suppresses the inflammatory reaction, serving as a barrier that prevents extension of inflammatory activity into the viable non-infarcted myocardium 80. Other matricellular proteins, such as osteonectin, osteopontin, tenascin-C and periostin are also induced in the infarcted heart and regulate the reparative response through direct effects on fibroblasts and inflammatory cells and by modulating growth factor signaling 81.
4. Targeting the immune response in the remodeling infarcted heart
4.1. The concept of inflammatory cardiomyocyte injury
Observations documenting abundant infiltration of the infarct border zone with inflammatory leukocytes, and in vitro studies suggesting that infiltrating neutrophils may cause cytotoxic cardiomyocyte injury supported the notion that the post-infarction inflammatory reaction may extend the size of the infarct following myocardial ischemia and reperfusion. Early attempts for broad inhibition of the inflammatory reaction in patients with myocardial infarction using systemic corticosteroids had, in some cases 82, catastrophic effects, presumed due to impairment of the reparative process. These effects highlighted the dual function of the immune system in myocardial injury and repair. In the following decades, the explosive growth in the biology of inflammation fueled experimental studies targeting specific immune pathways in large animal models of reperfused myocardial infarction. Extensive experimental evidence demonstrated that antibody neutralization of adhesion molecules, chemokines and cytokines protected the ischemic and reperfused myocardium, reducing the size of the infarct by 40-50% 83. These findings generated considerable enthusiasm regarding the potential effectiveness of targeted anti-inflammatory approaches in patients with acute myocardial infarction. Unfortunately, clinical trials failed to translate the concept into a successful therapeutic approach. Despite the success of anti-integrin approaches in reducing infarct size in experimental models, clinical trials showed no effects of early CD11b/CD18 integrin inhibition in human patients with ST elevation myocardial infarction 84, 6. Moreover, inhibition of the complement cascade, an essential component of the innate immune response, did not improve outcome in patients undergoing a percutaneous coronary intervention for acute myocardial infarction 7.
Why did early inhibition of inflammatory signals fail to reduce ischemic injury in patients with acute myocardial infarction, despite extensive experimental support? Perhaps, the enthusiasm generated by the concept and by an impressive volume of promising experimental evidence may have been misleading. A bias against negative experimental studies may have resulted in selective publication of studies showing positive results that may not have adequately reflected the collective experience of the research community. Over the last decade, studies using genetically manipulated animals have challenged the concept of inflammatory cardiomyocyte injury. Mice with disruption of key pro-inflammatory pathways (such as the P-selectin/ICAM-1 double knockout mice 46, the IL-1 type 1 receptor null mice 23 and the MCP-1 -/- animals 49) had no reduction in infarct size, despite a marked attenuation of inflammatory activity. Thus, the significance of inflammation-related extension of ischemic cardiomyocyte death may have been overstated.
Moreover, human patient populations with myocardial infarction are highly heterogeneous; for this reason use of animal models to derive direct translational conclusions is challenging and problematic. Animal models are extremely useful in studies designed to obtain mechanistic insights into the role of specific molecular signals and cellular responses. To fulfill this goal, animal studies are designed to eliminate variability so that the role of a specific signal or mediator in the pathophysiologic process of interest can be dissected. Patients with myocardial infarction on the other hand, exhibit variations in a range of factors that may affect outcome. The presence of comorbid conditions (such as diabetes, hypertension or hyperlipidemia), advanced age, gender-specific effects, differences in genetic background, and in the pattern and presentation of coronary disease, and therapy with various pharmacologic agents have profound effects on the inflammatory and reparative response to myocardial infarction; the complexity of these variations cannot be recapitulated by any animal model. An example of the effects of one of these factors is provided by our studies on the effects of aging on the post-infarction inflammatory reaction. We found that senescent mice develop worse adverse remodeling following myocardial infarction, but exhibit suppressed (and somewhat prolonged) inflammatory activation 85. Conclusions on the efficacy of targeted anti-inflammatory strategies were based exclusively on investigations in young animals (which exhibit robust and transient immune responses to tissue injury). These findings may not be directly relevant to the aged human population presenting with an acute coronary event. Thus, in order to interpret findings from experimental studies we need to recognize that a well-executed animal study should enhance our knowledge on the pathophysiologic mechanisms of disease. These insights may provide us with the necessary information in order to develop and design a therapeutic approach. However, the findings do not have direct predictive value regarding success in the clinical context.
4.2. Targeted anti-inflammatory approaches may prevent adverse post-infarction remodeling: the case for personalized approaches based on pathophysiologic insights
Inflammatory injury may not accentuate ischemic cardiomyocyte death; however, a growing body of evidence suggests that inflammatory mediators play an important role in the pathogenesis of dilative remodeling following myocardial infarction. In mouse models, loss of IL-1 or MCP-1 markedly reduced dilative remodeling without affecting the size of the infarct 23, 49. Moreover, animals with defects in pathways involved in resolution of inflammation 77 exhibited enhanced chamber dilation highlighting the importance of intact endogenous STOP signals in protection from dilative remodeling. Inflammation-induced adverse remodeling of the infarcted heart may be due to activation of several distinct detrimental processes. First, cytokine-mediated induction and activation of MMPs and subsequent loss of extracellular matrix proteins may result in reduced tensile strength of the infarct, cardiomyocyte slippage and loss of matrix-derived signals that may support cardiomyocyte function. Second, defective containment of the post-infarction inflammatory reaction may stimulate apoptotic pathways in viable cardiomyocytes of the infarct border zone. Third, extension of pro-inflammatory signaling in non-infarcted areas may trigger fibrosis exacerbating dysfunction.
Targeted anti-inflammatory strategies to inhibit IL-1 or MCP-1 signaling may be promising strategies to attenuate post-infarction remodeling. Two pilot studies examining the effects of IL-1 inhibition with IL-1 receptor antagonist (anakinra) 86, 87 in patients with clinically stable ST elevation myocardial infarction suggested promising beneficial effects. Treatment with anakinra suppressed the post-infarction inflammatory response and was associated with a lower incidence of heart failure 87.
However, in order to successfully translate this concept to the clinic, we need to recognize and exploit the pathophysiologic complexity of post-infarction remodeling. Patients surviving an acute infarction have very different remodeling responses that may depend on their comorbidities, genetic factors, age, or their unique pattern of coronary disease; these responses are to a significant extent independent of the size of the infarct. Some patients with myocardial infarction exhibit accelerated ventricular dilation accompanied by progression to systolic dysfunction and heart failure; others develop early and prominent fibrotic changes associated with concentric hypertrophy and diastolic dysfunction. The subset of patients with dilative remodeling may have prolonged or exaggerated myocardial inflammation following infarction, possibly due to defective STOP signals and impaired negative regulation of the inflammatory reaction. These patients may benefit from therapeutic strategies targeting specific pro-inflammatory cascades. In contrast, in patients with prominent fibrosis and diastolic dysfunction (such as individuals with diabetes) 88 inflammatory activity may be temporally and spatially restrained and an overactive TGF-β response may be responsible for the fibrotic alterations. This subpopulation of patients may benefit from therapies targeting TGF-β/Smad signaling 89. Biomarker-based approaches may prove extremely valuable in identifying patient subpopulations with specific pathophysiologic disturbances. Patients with a prolonged increase in circulating chemokine levels had worse prognosis following myocardial infarction 90; this may be due to the development of dilative remodeling in response to an overactive inflammatory reaction. These patients may benefit from targeted inhibition of inflammation. On the other hand, elevated levels of biomarkers indicating increased matrix protein synthesis 91 may contribute to the identification of patients with more intense fibrotic changes. Development of reliable biomarker-based approaches to identify subgroups of patients with specific pathophysiologic responses may prove extremely valuable for effective targeting of post-infarction cardiac remodeling 92.
Acknowledgments
Supported by NIH grants R01 HL76246 and R01 HL85440
References
- 1.Frangogiannis NG. The mechanistic basis of infarct healing. Antioxid Redox Signal. 2006 Nov-Dec;8(11-12):1907–1939. doi: 10.1089/ars.2006.8.1907. [DOI] [PubMed] [Google Scholar]
- 2.Frangogiannis NG. The immune system and cardiac repair. Pharmacol Res. 2008 Aug;58(2):88–111. doi: 10.1016/j.phrs.2008.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Frangogiannis NG. Regulation of the inflammatory response in cardiac repair. Circ Res. 2012 Jan 6;110(1):159–173. doi: 10.1161/CIRCRESAHA.111.243162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Frantz S, Bauersachs J, Ertl G. Post-infarct remodelling: contribution of wound healing and inflammation. Cardiovasc Res. 2009 Feb 15;81(3):474–481. doi: 10.1093/cvr/cvn292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Entman ML, Michael L, Rossen RD, Dreyer WJ, Anderson DC, Taylor AA, Smith CW. Inflammation in the course of early myocardial ischemia. Faseb J. 1991 Aug;5(11):2529–2537. doi: 10.1096/fasebj.5.11.1868978. [DOI] [PubMed] [Google Scholar]
- 6.Faxon DP, Gibbons RJ, Chronos NA, Gurbel PA, Sheehan F. The effect of blockade of the CD11/CD18 integrin receptor on infarct size in patients with acute myocardial infarction treated with direct angioplasty: the results of the HALT-MI study. J Am Coll Cardiol. 2002 Oct 2;40(7):1199–1204. doi: 10.1016/s0735-1097(02)02136-8. [DOI] [PubMed] [Google Scholar]
- 7.Armstrong PW, Granger CB, Adams PX, Hamm C, Holmes D, Jr, O'Neill WW, Todaro TG, Vahanian A, Van de Werf F. Pexelizumab for acute ST-elevation myocardial infarction in patients undergoing primary percutaneous coronary intervention: a randomized controlled trial. Jama. 2007 Jan 3;297(1):43–51. doi: 10.1001/jama.297.1.43. [DOI] [PubMed] [Google Scholar]
- 8.Reimer KA, Lowe JE, Rasmussen MM, Jennings RB. The wavefront phenomenon of ischemic cell death. 1. Myocardial infarct size vs duration of coronary occlusion in dogs. Circulation. 1977 Nov;56(5):786–794. doi: 10.1161/01.cir.56.5.786. [DOI] [PubMed] [Google Scholar]
- 9.Timmers L, Pasterkamp G, de Hoog VC, Arslan F, Appelman Y, de Kleijn DP. The innate immune response in reperfused myocardium. Cardiovasc Res. 2012 May 1;94(2):276–283. doi: 10.1093/cvr/cvs018. [DOI] [PubMed] [Google Scholar]
- 10.Andrassy M, Volz HC, Igwe JC, Funke B, Eichberger SN, Kaya Z, Buss S, Autschbach F, Pleger ST, Lukic IK, Bea F, Hardt SE, Humpert PM, Bianchi ME, Mairbaurl H, Nawroth PP, Remppis A, Katus HA, Bierhaus A. High-mobility group box-1 in ischemia-reperfusion injury of the heart. Circulation. 2008 Jun 24;117(25):3216–3226. doi: 10.1161/CIRCULATIONAHA.108.769331. [DOI] [PubMed] [Google Scholar]
- 11.Arslan F, de Kleijn DP, Pasterkamp G. Innate immune signaling in cardiac ischemia. Nat Rev Cardiol. 2011 May;8(5):292–300. doi: 10.1038/nrcardio.2011.38. [DOI] [PubMed] [Google Scholar]
- 12.Huebener P, Abou-Khamis T, Zymek P, Bujak M, Ying X, Chatila K, Haudek S, Thakker G, Frangogiannis NG. CD44 Is Critically Involved in Infarct Healing by Regulating the Inflammatory and Fibrotic Response. J Immunol. 2008 Feb 15;180(4):2625–2633. doi: 10.4049/jimmunol.180.4.2625. [DOI] [PubMed] [Google Scholar]
- 13.Dobaczewski M, Gonzalez-Quesada C, Frangogiannis NG. The extracellular matrix as a modulator of the inflammatory and reparative response following myocardial infarction. J Mol Cell Cardiol. 2010 Mar;48(3):504–511. doi: 10.1016/j.yjmcc.2009.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Oyama J, Blais C, Jr, Liu X, Pu M, Kobzik L, Kelly RA, Bourcier T. Reduced myocardial ischemia-reperfusion injury in toll-like receptor 4-deficient mice. Circulation. 2004 Feb 17;109(6):784–789. doi: 10.1161/01.CIR.0000112575.66565.84. [DOI] [PubMed] [Google Scholar]
- 15.Timmers L, Sluijter JP, van Keulen JK, Hoefer IE, Nederhoff MG, Goumans MJ, Doevendans PA, van Echteld CJ, Joles JA, Quax PH, Piek JJ, Pasterkamp G, de Kleijn DP. Toll-like receptor 4 mediates maladaptive left ventricular remodeling and impairs cardiac function after myocardial infarction. Circ Res. 2008 Feb 1;102(2):257–264. doi: 10.1161/CIRCRESAHA.107.158220. [DOI] [PubMed] [Google Scholar]
- 16.Arslan F, Smeets MB, O'Neill LA, Keogh B, McGuirk P, Timmers L, Tersteeg C, Hoefer IE, Doevendans PA, Pasterkamp G, de Kleijn DP. Myocardial ischemia/reperfusion injury is mediated by leukocytic toll-like receptor-2 and reduced by systemic administration of a novel anti-toll-like receptor-2 antibody. Circulation. 2010 Jan 5;121(1):80–90. doi: 10.1161/CIRCULATIONAHA.109.880187. [DOI] [PubMed] [Google Scholar]
- 17.Hill JH, Ward PA. The phlogistic role of C3 leukotactic fragments in myocardial infarcts of rats. J Exp Med. 1971 Apr 1;133(4):885–900. doi: 10.1084/jem.133.4.885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Weisman HF, Bartow T, Leppo MK, Marsh HC, Jr, Carson GR, Concino MF, Boyle MP, Roux KH, Weisfeldt ML, Fearon DT. Soluble human complement receptor type 1: in vivo inhibitor of complement suppressing post-ischemic myocardial inflammation and necrosis. Science. 1990 Jul 13;249(4965):146–151. doi: 10.1126/science.2371562. [DOI] [PubMed] [Google Scholar]
- 19.Granger DN. Role of xanthine oxidase and granulocytes in ischemia-reperfusion injury. Am J Physiol. 1988 Dec;255(6 Pt 2):H1269–1275. doi: 10.1152/ajpheart.1988.255.6.H1269. [DOI] [PubMed] [Google Scholar]
- 20.Hensley K, Robinson KA, Gabbita SP, Salsman S, Floyd RA. Reactive oxygen species, cell signaling, and cell injury. Free Radic Biol Med. 2000 May 15;28(10):1456–1462. doi: 10.1016/s0891-5849(00)00252-5. [DOI] [PubMed] [Google Scholar]
- 21.Gordon JW, Shaw JA, Kirshenbaum LA. Multiple facets of NF-kappaB in the heart: to be or not to NF-kappaB. Circ Res. 2011 Apr 29;108(9):1122–1132. doi: 10.1161/CIRCRESAHA.110.226928. [DOI] [PubMed] [Google Scholar]
- 22.Nian M, Lee P, Khaper N, Liu P. Inflammatory cytokines and postmyocardial infarction remodeling. Circ Res. 2004 Jun 25;94(12):1543–1553. doi: 10.1161/01.RES.0000130526.20854.fa. [DOI] [PubMed] [Google Scholar]
- 23.Bujak M, Dobaczewski M, Chatila K, Mendoza LH, Li N, Reddy A, Frangogiannis NG. Interleukin-1 receptor type I signaling critically regulates infarct healing and cardiac remodeling. Am J Pathol. 2008 Jul;173(1):57–67. doi: 10.2353/ajpath.2008.070974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bujak M, Frangogiannis NG. The role of IL-1 in the pathogenesis of heart disease. Arch Immunol Ther Exp (Warsz) 2009 May-Jun;57(3):165–176. doi: 10.1007/s00005-009-0024-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schroder K, Tschopp J. The inflammasomes. Cell. 2010 Mar 19;140(6):821–832. doi: 10.1016/j.cell.2010.01.040. [DOI] [PubMed] [Google Scholar]
- 26.Mezzaroma E, Toldo S, Farkas D, Seropian IM, Van Tassell BW, Salloum FN, Kannan HR, Menna AC, Voelkel NF, Abbate A. The inflammasome promotes adverse cardiac remodeling following acute myocardial infarction in the mouse. Proc Natl Acad Sci U S A. 2011 Dec 6;108(49):19725–19730. doi: 10.1073/pnas.1108586108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kawaguchi M, Takahashi M, Hata T, Kashima Y, Usui F, Morimoto H, Izawa A, Takahashi Y, Masumoto J, Koyama J, Hongo M, Noda T, Nakayama J, Sagara J, Taniguchi S, Ikeda U. Inflammasome activation of cardiac fibroblasts is essential for myocardial ischemia/reperfusion injury. Circulation. 2011 Feb 15;123(6):594–604. doi: 10.1161/CIRCULATIONAHA.110.982777. [DOI] [PubMed] [Google Scholar]
- 28.Frangogiannis NG, Lindsey ML, Michael LH, Youker KA, Bressler RB, Mendoza LH, Spengler RN, Smith CW, Entman ML. Resident cardiac mast cells degranulate and release preformed TNFalpha, initiating the cytokine cascade in experimental canine myocardial ischemia/reperfusion. Circulation. 1998 Aug 18;98(7):699–710. doi: 10.1161/01.cir.98.7.699. [DOI] [PubMed] [Google Scholar]
- 29.Kurrelmeyer KM, Michael LH, Baumgarten G, Taffet GE, Peschon JJ, Sivasubramanian N, Entman ML, Mann DL. Endogenous tumor necrosis factor protects the adult cardiac myocyte against ischemic-induced apoptosis in a murine model of acute myocardial infarction. Proc Natl Acad Sci U S A. 2000 May 9;97(10):5456–5461. doi: 10.1073/pnas.070036297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hamid T, Gu Y, Ortines RV, Bhattacharya C, Wang G, Xuan YT, Prabhu SD. Divergent tumor necrosis factor receptor-related remodeling responses in heart failure: role of nuclear factor-kappaB and inflammatory activation. Circulation. 2009 Mar 17;119(10):1386–1397. doi: 10.1161/CIRCULATIONAHA.108.802918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fischer P, Hilfiker-Kleiner D. Role of gp130-mediated signalling pathways in the heart and its impact on potential therapeutic aspects. Br J Pharmacol. 2008 Mar;153(1):S414–427. doi: 10.1038/bjp.2008.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fuchs M, Hilfiker A, Kaminski K, Hilfiker-Kleiner D, Guener Z, Klein G, Podewski E, Schieffer B, Rose-John S, Drexler H. Role of interleukin-6 for LV remodeling and survival after experimental myocardial infarction. Faseb J. 2003 Nov;17(14):2118–2120. doi: 10.1096/fj.03-0331fje. [DOI] [PubMed] [Google Scholar]
- 33.Kobara M, Noda K, Kitamura M, Okamoto A, Shiraishi T, Toba H, Matsubara H, Nakata T. Antibody against interleukin-6 receptor attenuates left ventricular remodelling after myocardial infarction in mice. Cardiovasc Res. 2010 Aug 1;87(3):424–430. doi: 10.1093/cvr/cvq078. [DOI] [PubMed] [Google Scholar]
- 34.Hilfiker-Kleiner D, Shukla P, Klein G, Schaefer A, Stapel B, Hoch M, Muller W, Scherr M, Theilmeier G, Ernst M, Hilfiker A, Drexler H. Continuous glycoprotein-130-mediated signal transducer and activator of transcription-3 activation promotes inflammation, left ventricular rupture, and adverse outcome in subacute myocardial infarction. Circulation. 2010 Jul 13;122(2):145–155. doi: 10.1161/CIRCULATIONAHA.109.933127. [DOI] [PubMed] [Google Scholar]
- 35.Frangogiannis NG. Chemokines in ischemia and reperfusion. Thromb Haemost. 2007 May;97(5):738–747. [PubMed] [Google Scholar]
- 36.Nahrendorf M, Swirski FK, Aikawa E, Stangenberg L, Wurdinger T, Figueiredo JL, Libby P, Weissleder R, Pittet MJ. The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J Exp Med. 2007 Nov 26;204(12):3037–3047. doi: 10.1084/jem.20070885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dobaczewski M, Xia Y, Bujak M, Gonzalez-Quesada C, Frangogiannis NG. CCR5 signaling suppresses inflammation and reduces adverse remodeling of the infarcted heart, mediating recruitment of regulatory T cells. Am J Pathol. 2010 May;176(5):2177–2187. doi: 10.2353/ajpath.2010.090759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Proudfoot AE, Handel TM, Johnson Z, Lau EK, LiWang P, Clark-Lewis I, Borlat F, Wells TN, Kosco-Vilbois MH. Glycosaminoglycan binding and oligomerization are essential for the in vivo activity of certain chemokines. Proc Natl Acad Sci U S A. 2003 Feb 18;100(4):1885–1890. doi: 10.1073/pnas.0334864100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Williams MR, Azcutia V, Newton G, Alcaide P, Luscinskas FW. Emerging mechanisms of neutrophil recruitment across endothelium. Trends Immunol. 2011 Oct;32(10):461–469. doi: 10.1016/j.it.2011.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Woodfin A, Voisin MB, Beyrau M, Colom B, Caille D, Diapouli FM, Nash GB, Chavakis T, Albelda SM, Rainger GE, Meda P, Imhof BA, Nourshargh S. The junctional adhesion molecule JAM-C regulates polarized transendothelial migration of neutrophils in vivo. Nat Immunol. 2011 Aug;12(8):761–769. doi: 10.1038/ni.2062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Corada M, Chimenti S, Cera MR, Vinci M, Salio M, Fiordaliso F, De Angelis N, Villa A, Bossi M, Staszewsky LI, Vecchi A, Parazzoli D, Motoike T, Latini R, Dejana E. Junctional adhesion molecule-A-deficient polymorphonuclear cells show reduced diapedesis in peritonitis and heart ischemia-reperfusion injury. Proc Natl Acad Sci U S A. 2005 Jul 26;102(30):10634–10639. doi: 10.1073/pnas.0500147102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.von Hundelshausen P, Weber C. Platelets as immune cells: bridging inflammation and cardiovascular disease. Circ Res. 2007 Jan 5;100(1):27–40. doi: 10.1161/01.RES.0000252802.25497.b7. [DOI] [PubMed] [Google Scholar]
- 43.Dobaczewski M, Bujak M, Zymek P, Ren G, Entman ML, Frangogiannis NG. Extracellular matrix remodeling in canine and mouse myocardial infarcts. Cell Tissue Res. 2006 Jun;324(3):475–488. doi: 10.1007/s00441-005-0144-6. [DOI] [PubMed] [Google Scholar]
- 44.Albelda SM, Smith CW, Ward PA. Adhesion molecules and inflammatory injury. Faseb J. 1994 May;8(8):504–512. [PubMed] [Google Scholar]
- 45.Metzler B, Mair J, Lercher A, Schaber C, Hintringer F, Pachinger O, Xu Q. Mouse model of myocardial remodelling after ischemia: role of intercellular adhesion molecule-1. Cardiovasc Res. 2001 Feb 1;49(2):399–407. doi: 10.1016/s0008-6363(00)00261-3. [DOI] [PubMed] [Google Scholar]
- 46.Briaud SA, Ding ZM, Michael LH, Entman ML, Daniel S, Ballantyne CM. Leukocyte trafficking and myocardial reperfusion injury in ICAM-1/P-selectin-knockout mice. Am J Physiol Heart Circ Physiol. 2001 Jan;280(1):H60–67. doi: 10.1152/ajpheart.2001.280.1.H60. [DOI] [PubMed] [Google Scholar]
- 47.Swirski FK, Nahrendorf M, Etzrodt M, Wildgruber M, Cortez-Retamozo V, Panizzi P, Figueiredo JL, Kohler RH, Chudnovskiy A, Waterman P, Aikawa E, Mempel TR, Libby P, Weissleder R, Pittet MJ. Identification of splenic reservoir monocytes and their deployment to inflammatory sites. Science. 2009 Jul 31;325(5940):612–616. doi: 10.1126/science.1175202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jung K, Kim P, Leuschner F, Gorbatov R, Kim JK, Ueno T, Nahrendorf M, Yun SH. Endoscopic time-lapse imaging of immune cells in infarcted mouse hearts. Circ Res. 2013 Mar 15;112(6):891–899. doi: 10.1161/CIRCRESAHA.111.300484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dewald O, Zymek P, Winkelmann K, Koerting A, Ren G, Abou-Khamis T, Michael LH, Rollins BJ, Entman ML, Frangogiannis NG. CCL2/Monocyte Chemoattractant Protein-1 regulates inflammatory responses critical to healing myocardial infarcts. Circ Res. 2005 Apr 29;96(8):881–889. doi: 10.1161/01.RES.0000163017.13772.3a. [DOI] [PubMed] [Google Scholar]
- 50.Frangogiannis NG, Mendoza LH, Ren G, Akrivakis S, Jackson PL, Michael LH, Smith CW, Entman ML. MCSF expression is induced in healing myocardial infarcts and may regulate monocyte and endothelial cell phenotype. Am J Physiol Heart Circ Physiol. 2003 Aug;285(2):H483–492. doi: 10.1152/ajpheart.01016.2002. [DOI] [PubMed] [Google Scholar]
- 51.Gersch C, Dewald O, Zoerlein M, Michael LH, Entman ML, Frangogiannis NG. Mast cells and macrophages in normal C57/BL/6 mice. Histochem Cell Biol. 2002 Jul;118(1):41–49. doi: 10.1007/s00418-002-0425-z. [DOI] [PubMed] [Google Scholar]
- 52.Souders CA, Bowers SL, Baudino TA. Cardiac fibroblast: the renaissance cell. Circ Res. 2009 Dec 4;105(12):1164–1176. doi: 10.1161/CIRCRESAHA.109.209809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Frangogiannis NG, Mendoza LH, Lewallen M, Michael LH, Smith CW, Entman ML. Induction and suppression of interferon-inducible protein 10 in reperfused myocardial infarcts may regulate angiogenesis. FASEB J. 2001 Jun;15(8):1428–1430. doi: 10.1096/fj.00-0745fje. [DOI] [PubMed] [Google Scholar]
- 54.Bujak M, Dobaczewski M, Gonzalez-Quesada C, Xia Y, Leucker T, Zymek P, Veeranna V, Tager AM, Luster AD, Frangogiannis NG. Induction of the CXC chemokine interferon-gamma-inducible protein 10 regulates the reparative response following myocardial infarction. Circ Res. 2009 Nov 6;105(10):973–983. doi: 10.1161/CIRCRESAHA.109.199471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Haslett C. Granulocyte apoptosis and its role in the resolution and control of lung inflammation. Am J Respir Crit Care Med. 1999 Nov;160(5 Pt 2):S5–11. doi: 10.1164/ajrccm.160.supplement_1.4. [DOI] [PubMed] [Google Scholar]
- 56.Savill J, Dransfield I, Gregory C, Haslett C. A blast from the past: clearance of apoptotic cells regulates immune responses. Nat Rev Immunol. 2002 Dec;2(12):965–975. doi: 10.1038/nri957. [DOI] [PubMed] [Google Scholar]
- 57.Taylor KR, Trowbridge JM, Rudisill JA, Termeer CC, Simon JC, Gallo RL. Hyaluronan fragments stimulate endothelial recognition of injury through TLR4. J Biol Chem. 2004 Apr 23;279(17):17079–17084. doi: 10.1074/jbc.M310859200. [DOI] [PubMed] [Google Scholar]
- 58.Ponta H, Sherman L, Herrlich PA. CD44: from adhesion molecules to signalling regulators. Nat Rev Mol Cell Biol. 2003 Jan;4(1):33–45. doi: 10.1038/nrm1004. [DOI] [PubMed] [Google Scholar]
- 59.Ren G, Michael LH, Entman ML, Frangogiannis NG. Morphological characteristics of the microvasculature in healing myocardial infarcts. J Histochem Cytochem. 2002 Jan;50(1):71–79. doi: 10.1177/002215540205000108. [DOI] [PubMed] [Google Scholar]
- 60.Dobaczewski M, Akrivakis S, Nasser K, Michael LH, Entman ML, Frangogiannis NG. Vascular mural cells in healing canine myocardial infarcts. J Histochem Cytochem. 2004 Aug;52(8):1019–1029. doi: 10.1369/jhc.3A6210.2004. [DOI] [PubMed] [Google Scholar]
- 61.Zymek P, Bujak M, Chatila K, Cieslak A, Thakker G, Entman ML, Frangogiannis NG. The role of platelet-derived growth factor signaling in healing myocardial infarcts. J Am Coll Cardiol. 2006 Dec 5;48(11):2315–2323. doi: 10.1016/j.jacc.2006.07.060. [DOI] [PubMed] [Google Scholar]
- 62.Frangogiannis NG, Mendoza LH, Lindsey ML, Ballantyne CM, Michael LH, Smith CW, Entman ML. IL-10 is induced in the reperfused myocardium and may modulate the reaction to injury. J Immunol. 2000 Sep 1;165(5):2798–2808. doi: 10.4049/jimmunol.165.5.2798. [DOI] [PubMed] [Google Scholar]
- 63.Yang Z, Zingarelli B, Szabo C. Crucial role of endogenous interleukin-10 production in myocardial ischemia/reperfusion injury. Circulation. 2000 Mar 7;101(9):1019–1026. doi: 10.1161/01.cir.101.9.1019. [DOI] [PubMed] [Google Scholar]
- 64.Zymek P, Nah DY, Bujak M, Ren G, Koerting A, Leucker T, Huebener P, Taffet G, Entman M, Frangogiannis NG. Interleukin-10 is not a critical regulator of infarct healing and left ventricular remodeling. Cardiovasc Res. 2007 May 1;74(2):313–322. doi: 10.1016/j.cardiores.2006.11.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bujak M, Frangogiannis NG. The role of TGF-beta signaling in myocardial infarction and cardiac remodeling. Cardiovasc Res. 2007 May 1;74(2):184–195. doi: 10.1016/j.cardiores.2006.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bassols A, Massague J. Transforming growth factor beta regulates the expression and structure of extracellular matrix chondroitin/dermatan sulfate proteoglycans. J Biol Chem. 1988 Feb 25;263(6):3039–3045. [PubMed] [Google Scholar]
- 67.Laiho M, Saksela O, Andreasen PA, Keski-Oja J. Enhanced production and extracellular deposition of the endothelial-type plasminogen activator inhibitor in cultured human lung fibroblasts by transforming growth factor-beta. J Cell Biol. 1986 Dec;103(6 Pt 1):2403–2410. doi: 10.1083/jcb.103.6.2403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ikeuchi M, Tsutsui H, Shiomi T, Matsusaka H, Matsushima S, Wen J, Kubota T, Takeshita A. Inhibition of TGF-beta signaling exacerbates early cardiac dysfunction but prevents late remodeling after infarction. Cardiovasc Res. 2004 Dec 1;64(3):526–535. doi: 10.1016/j.cardiores.2004.07.017. [DOI] [PubMed] [Google Scholar]
- 69.Okada H, Takemura G, Kosai K, Li Y, Takahashi T, Esaki M, Yuge K, Miyata S, Maruyama R, Mikami A, Minatoguchi S, Fujiwara T, Fujiwara H. Postinfarction gene therapy against transforming growth factor-beta signal modulates infarct tissue dynamics and attenuates left ventricular remodeling and heart failure. Circulation. 2005 May 17;111(19):2430–2437. doi: 10.1161/01.CIR.0000165066.71481.8E. [DOI] [PubMed] [Google Scholar]
- 70.Shi Y, Massague J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell. 2003 Jun 13;113(6):685–700. doi: 10.1016/s0092-8674(03)00432-x. [DOI] [PubMed] [Google Scholar]
- 71.Bujak M, Ren G, Kweon HJ, Dobaczewski M, Reddy A, Taffet G, Wang XF, Frangogiannis NG. Essential Role of Smad3 in Infarct Healing and in the Pathogenesis of Cardiac Remodeling. Circulation. 2007 Oct;22:116, 2127–2138. doi: 10.1161/CIRCULATIONAHA.107.704197. [DOI] [PubMed] [Google Scholar]
- 72.Dobaczewski M, Bujak M, Li N, Gonzalez-Quesada C, Mendoza LH, Wang XF, Frangogiannis NG. Smad3 signaling critically regulates fibroblast phenotype and function in healing myocardial infarction. Circ Res. 2010 Aug 6;107(3):418–428. doi: 10.1161/CIRCRESAHA.109.216101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kempf T, Zarbock A, Widera C, Butz S, Stadtmann A, Rossaint J, Bolomini-Vittori M, Korf-Klingebiel M, Napp LC, Hansen B, Kanwischer A, Bavendiek U, Beutel G, Hapke M, Sauer MG, Laudanna C, Hogg N, Vestweber D, Wollert KC. GDF-15 is an inhibitor of leukocyte integrin activation required for survival after myocardial infarction in mice. Nat Med. 2011 May;17(5):581–588. doi: 10.1038/nm.2354. [DOI] [PubMed] [Google Scholar]
- 74.Mantovani A, Locati M, Vecchi A, Sozzani S, Allavena P. Decoy receptors: a strategy to regulate inflammatory cytokines and chemokines. Trends Immunol. 2001 Jun;22(6):328–336. doi: 10.1016/s1471-4906(01)01941-x. [DOI] [PubMed] [Google Scholar]
- 75.Cochain C, Auvynet C, Poupel L, Vilar J, Dumeau E, Richart A, Recalde A, Zouggari Y, Yin KY, Bruneval P, Renault G, Marchiol C, Bonnin P, Levy B, Bonecchi R, Locati M, Combadiere C, Silvestre JS. The chemokine decoy receptor D6 prevents excessive inflammation and adverse ventricular remodeling after myocardial infarction. Arterioscler Thromb Vasc Biol. 2012 Sep;32(9):2206–2213. doi: 10.1161/ATVBAHA.112.254409. [DOI] [PubMed] [Google Scholar]
- 76.Kondo T, Kawai T, Akira S. Dissecting negative regulation of Toll-like receptor signaling. Trends Immunol. 2012 Sep;33(9):449–458. doi: 10.1016/j.it.2012.05.002. [DOI] [PubMed] [Google Scholar]
- 77.Chen W, Saxena A, Li N, Sun J, Gupta A, Lee DW, Tian Q, Dobaczewski M, Frangogiannis NG. Endogenous IRAK-M attenuates postinfarction remodeling through effects on macrophages and fibroblasts. Arterioscler Thromb Vasc Biol. 2012 Nov;32(11):2598–2608. doi: 10.1161/ATVBAHA.112.300310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Dobaczewski M, de Haan JJ, Frangogiannis NG. The extracellular matrix modulates fibroblast phenotype and function in the infarcted myocardium. J Cardiovasc Transl Res. 2012 Dec;5(6):837–847. doi: 10.1007/s12265-012-9406-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Frangogiannis NG. Matricellular proteins in cardiac adaptation and disease. Physiol Rev. 2012 Apr;92(2):635–688. doi: 10.1152/physrev.00008.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Frangogiannis NG, Ren G, Dewald O, Zymek P, Haudek S, Koerting A, Winkelmann K, Michael LH, Lawler J, Entman ML. The critical role of endogenous Thrombospondin (TSP)-1 in preventing expansion of healing myocardial infarcts. Circulation. 2005 Jun 7;111(22):2935–2942. doi: 10.1161/CIRCULATIONAHA.104.510354. [DOI] [PubMed] [Google Scholar]
- 81.Schellings MW, Vanhoutte D, Swinnen M, Cleutjens JP, Debets J, van Leeuwen RE, d'Hooge J, Van de Werf F, Carmeliet P, Pinto YM, Sage EH, Heymans S. Absence of SPARC results in increased cardiac rupture and dysfunction after acute myocardial infarction. J Exp Med. 2009 Jan 16;206(1):113–123. doi: 10.1084/jem.20081244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Roberts R, DeMello V, Sobel BE. Deleterious effects of methylprednisolone in patients with myocardial infarction. Circulation. 1976 Mar;53(3 Suppl):I204–206. [PubMed] [Google Scholar]
- 83.Simpson PJ, Todd RF, 3rd, Fantone JC, Mickelson JK, Griffin JD, Lucchesi BR. Reduction of experimental canine myocardial reperfusion injury by a monoclonal antibody (anti-Mo1, anti-CD11b) that inhibits leukocyte adhesion. J Clin Invest. 1988 Feb;81(2):624–629. doi: 10.1172/JCI113364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Baran KW, Nguyen M, McKendall GR, Lambrew CT, Dykstra G, Palmeri ST, Gibbons RJ, Borzak S, Sobel BE, Gourlay SG, Rundle AC, Gibson CM, Barron HV. Double-blind, randomized trial of an anti-CD18 antibody in conjunction with recombinant tissue plasminogen activator for acute myocardial infarction: limitation of myocardial infarction following thrombolysis in acute myocardial infarction (LIMIT AMI) study. Circulation. 2001 Dec 4;104(23):2778–2783. doi: 10.1161/hc4801.100236. [DOI] [PubMed] [Google Scholar]
- 85.Bujak M, Kweon HJ, Chatila K, Li N, Taffet G, Frangogiannis NG. Aging-related defects are associated with adverse cardiac remodeling in a mouse model of reperfused myocardial infarction. J Am Coll Cardiol. 2008 Apr 8;51(14):1384–1392. doi: 10.1016/j.jacc.2008.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Abbate A, Kontos MC, Grizzard JD, Biondi-Zoccai GG, Van Tassell BW, Robati R, Roach LM, Arena RA, Roberts CS, Varma A, Gelwix CC, Salloum FN, Hastillo A, Dinarello CA, Vetrovec GW. Interleukin-1 blockade with anakinra to prevent adverse cardiac remodeling after acute myocardial infarction (Virginia Commonwealth University Anakinra Remodeling Trial [VCU-ART] Pilot study) Am J Cardiol. 2010 May 15;105(10):1371–1377 e1371. doi: 10.1016/j.amjcard.2009.12.059. [DOI] [PubMed] [Google Scholar]
- 87.Abbate A, Van Tassell BW, Biondi-Zoccai G, Kontos MC, Grizzard JD, Spillman DW, Oddi C, Roberts CS, Melchior RD, Mueller GH, Abouzaki NA, Rengel LR, Varma A, Gambill ML, Falcao RA, Voelkel NF, Dinarello CA, Vetrovec GW. Effects of interleukin-1 blockade with anakinra on adverse cardiac remodeling and heart failure after acute myocardial infarction [from the Virginia Commonwealth University-Anakinra Remodeling Trial (2) (VCU-ART2) pilot study] Am J Cardiol. 2013 May 15;111(10):1394–1400. doi: 10.1016/j.amjcard.2013.01.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Carrabba N, Valenti R, Parodi G, Santoro GM, Antoniucci D. Left ventricular remodeling and heart failure in diabetic patients treated with primary angioplasty for acute myocardial infarction. Circulation. 2004 Oct 5;110(14):1974–1979. doi: 10.1161/01.CIR.0000143376.64970.4A. [DOI] [PubMed] [Google Scholar]
- 89.Dobaczewski M, Chen W, Frangogiannis NG. Transforming growth factor (TGF)-beta signaling in cardiac remodeling. J Mol Cell Cardiol. 2011 Oct;51(4):600–606. doi: 10.1016/j.yjmcc.2010.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.de Lemos JA, Morrow DA, Blazing MA, Jarolim P, Wiviott SD, Sabatine MS, Califf RM, Braunwald E. Serial measurement of monocyte chemoattractant protein-1 after acute coronary syndromes: results from the A to Z trial. J Am Coll Cardiol. 2007 Nov 27;50(22):2117–2124. doi: 10.1016/j.jacc.2007.06.057. [DOI] [PubMed] [Google Scholar]
- 91.Lopez B, Gonzalez A, Diez J. Circulating biomarkers of collagen metabolism in cardiac diseases. Circulation. 2010 Apr 13;121(14):1645–1654. doi: 10.1161/CIRCULATIONAHA.109.912774. [DOI] [PubMed] [Google Scholar]
- 92.Frangogiannis NG. Biomarkers: hopes and challenges in the path from discovery to clinical practice. Transl Res. 2012 Apr;159(4):197–204. doi: 10.1016/j.trsl.2012.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]