

Abstract
Type I diabetes is a disease caused by autoimmune destruction of the beta cells in the pancreas that leads to a deficiency in insulin production. The aim of this study was to evaluate the prophylactic potential of a prime-boost strategy involving bacille Calmette–Guérin (BCG) and the pVAXhsp65 vaccine (BCG/DNAhsp65) in diabetes induced by streptozotocin (STZ) in C57BL/6 mice and also in spontaneous type 1 diabetes in non-obese diabetic (NOD) mice. BCG/DNAhsp65 vaccination in NOD mice determined weight gain, protection against hyperglycaemia, decreased islet inflammation, higher levels of cytokine production by the spleen and a reduced number of regulatory T cells in the spleen compared with non-immunized NOD mice. In the STZ model, however, there was no significant difference in the clinical parameters. Although this vaccination strategy did not protect mice in the STZ model, it was very effective in NOD mice. This is the first report demonstrating that a prime-boost strategy could be explored as an immunomodulatory procedure in autoimmune diseases.
Keywords: BCG, hsp65, immunomodulation, NOD mice, type 1 diabetes
Introduction
Type 1 diabetes (T1D) is an autoimmune disease characterized by T cell-mediated destruction of the β cells in pancreatic islets. It affects the insulin production and leads to hyperglycaemia, polyuria and hypoinsulinaemia [1]. As a chronic condition, it may cause blindness, cardiovascular injury and harm in other systems at later stages [2].
A variety of animal models have been used extensively in diabetes research and, in this study, we used two of these models. The first was the one induced with multiple low doses of streptozotocin (MLD–STZ). STZ is a chemical substance with alkylation properties that interferes with glucose transportation. A single high-dose strategy results in severe toxicity and acute diabetes. Conversely, the multiple low-dose regimen, characterized by minimal β cell toxicity, results in autoantigen release and a possible break in self-tolerance [3]. The T cell dependence of this model is a debated topic, and needs further evaluation. What is well established is that diabetes in this model cannot be transferred reliably to syngenic recipients by transfer of splenocytes [4]. Non-obese diabetic (NOD) mice are an inbred strain derived from Jcl:ICR mice [5], which develop type 1 diabetes spontaneously. The infiltration in the islets starts around 4–5 weeks, when pockets of lymphocytes are first observed juxtaposed to the pancreatic islets of young NOD mice. As the animals grow older, these mononuclear cells migrate into the islets, and by the time hyperglycaemia occurs destructive insulitis is present. This model is very similar to the human disease. Disease onset, for example, is preceded by infiltration of pancreatic islets by mononuclear cells and is controlled by many quantitative trait loci, particularly major histocompatibility complex (MHC) class II genes. Diabetes in NOD mice is the most extensively studied model of autoimmune disease [6,7].
The discovery of regulatory T cells (Tregs) disclosed a new field to be explored in the control of autoimmune pathologies [8]. Heat shock proteins (hsps) are molecules up-regulated in conditions of stress that are highly conserved throughout evolution [9]. Although recent research implicates hsp60 as an autoantigen involved in type 1 diabetes pathogenesis [10], this protein also contributes to protection against autoimmune diseases. It has been described that microbial homologues of mammalian hsps could induce the recruitment of Tregs to inflamed tissues [9].
In this study, we investigated the possible protection against type 1 diabetes through a prime-boost vaccination strategy. This strategy consists in priming the system with the antigen administered in one vector and then boosting it with the same antigen, but through another vector [11]. Thus, we made use of two different vaccines containing mycobacterial hsp65: bacille Calmette–Guérin (BCG) and pVAXhsp65, a DNA vaccine. This association could, theoretically, be interesting because both vaccines have been already tested separately against diabetes and other autoimmune diseases and showed positive results [12–15]. We hypothesized that the prime-boost strategy could expand these beneficial effects.
Materials and methods
Animals
Female NOD mice and male C57BL/6 mice were obtained from the animal facility of State University of Campinas (UNICAMP, Campinas, São Paulo, Brazil). The animals were maintained on a 12-h light/dark cycle and given free access to autoclaved water and food. All animal procedures and experimental protocols were in accordance with the local Ethical Committee for Animal Research (CEEA – Protocol no. 212).
Study groups
NOD mice were distributed in three groups: non-immunized NOD mice (NOD); NOD mice immunized with BCG vaccine (BCG–NOD) and NOD mice immunized with the prime-boost BCG/pVAXhsp65 (BCG/DNAhps65–NOD). Diabetes type 1 in male C57BL/6 mice was induced with STZ and animals were allocated into four groups: non-immunized, non-diabetic mice (control); non-immunized diabetic mice (STZ), mice immunized with BCG (BCG-STZ) and mice immunized with the prime-boost BCG/pVAX-hsp65 (BCG/DNAhps65–STZ).
Genetic vaccine construction and purification
The vaccine pVAXhsp65 was derived from the pVAX vector (Invitrogen, Carlsbad, CA, USA), digested previously with BamHI and NotI (Gibco BRL, Gaithersburg, MD, USA) by inserting a 3·3 kb fragment corresponding to the Mycobacterium leprae hsp65 gene and the cytomegalovirus (CMV) intron A. DH5a Escherichia coli transformed with plasmid pVAX or the plasmid carrying the hsp65 gene (pVAXhsp65) were cultured in Luria-Bertani liquid medium (Gibco BRL) containing kanamycin (100 μg/ml). The plasmids were purified using the Concert High Purity Maxiprep System (Gibco BRL). Plasmid concentrations were determined by spectrophotometry at 260 and 280 nm by using the Gene Quant II apparatus (Pharmacia Biotech, Amersham, UK).
Immunizations with BCG and pVAXhsp65
BCG vaccine [50 μl containing around 105 colony-forming units (CFU)] was administered subcutaneously at the base of the tail when NOD mice were 7 weeks old and C57BL/6 mice were 4–6 weeks old. In the prime-boost group, animals were additionally injected with pVAXhsp65 (100 μg/100 μl) associated with 25% of saccharose by the intramuscular route (quadriceps muscle) 15 days after BCG immunization. NOD mice were monitored until their 29th week of life, whereas STZ groups were monitored for 21 days after diabetes induction. Body weight and blood glucose level were measured weekly and insulitis scores were measured only after euthanasia. In addition, in the NOD mice, cytokine production by spleen cells and the presence of Treg cells in the spleen were analysed.
Streptozotocin-induced diabetes and blood glucose level determination
In order to induce diabetes, male C57BL/6 mice were given intraperitoneal injections of STZ diluted in citrate buffer (40 mg/kg; Sigma-Aldrich, St Louis, MO, USA) for 5 consecutive days. Using this protocol, glycaemia was determined once before the first STZ dose and three times after the last dose. Non-fasted glucose concentration was determined in blood samples collected from the facial vein and measured using Prestige LX Smart System Test-strips (Home Diagnostic, Inc., Fort Lauderdale, FL, USA). NOD mice are known to develop hyperglycaemia around week 12 and, therefore, blood glucose concentration was measured from the 11th week onwards. Animals were considered diabetic when blood glucose levels were higher than 200 mg/dl during 2 consecutive weeks. If they reached more than 400 mg/dl, mice were injected daily with 50U of Humulin N (Eli Lilly and Company, Indianapolis, IN, USA).
Histopathological analysis
After euthanasia, pancreas were removed and fixed in phosphate-buffered formalin 10% (phosphate buffer pH = 7·2) for 24 h. The organs were conserved in alcohol 70% until histological processing and paraffin inclusion. Five-μm sections were cut and stained with haematoxylin and eosin (H&E). All islets on the slides were analysed and the following criteria were employed to determine insulitis score: 0 = intact islet; 1 = peri-insulitis; 2 = moderate insulitis (< 50% mononuclear infiltration); and 3 = severe insulitis (more than 50% mononuclear infiltration).
Cytokine production by spleen cells
Spleen cells were cultured in RPMI-1640 medium supplemented with 10% fetal bovine serum, 2 mM L-glutamine and 40 mg/l of gentamicin and then plated at 5 × 106 cells/ml in 48-well flat-bottomed culture plates (Nunc, Sigma-Aldrich) and stimulated with 10 μg/ml of recombinant heat shock protein 65-kDa (rhsp65). Cytokine levels were evaluated 48 h later by enzyme-linked immunosorbent assay (ELISA) in culture supernatants using interferon (IFN)-γ, interleukin (IL)-5 and IL-10 BD OptEIA Sets (Becton Dickinson, San Jose, CA, USA) and tumour necrosis factor (TNF)-α Duoset (R&D Systems, Minneapolis, MN, USA). The assays were performed according to the manufacturer's instructions.
Frequency of CD4+CD25+forkhead box protein 3 (FoxP3+) cells in the spleen
Spleen cells were collected, the red blood cells were lysed with Hanks's buffer containing NH4Cl and the remaining cells were adjusted to 2·5 × 106 cells/100 μl. These cells were incubated with 0·5 μg of fluorescein isothiocianate (FITC) anti-mouse CD4 (clone GK1·5) and 0·25 μg of allophycocyanin (APC) anti-mouse CD25 (clone PC61·5) for 20 min at room temperature. Staining for FoxP3 was then performed utilizing the phycoerythrin (PE) anti-mouse/rat FoxP3 Staining Set (eBioscience, San Diego, CA, USA), according to the manufacturer's instructions. After incubation, the cells were fixed in paraformaldehyde 1%. The cells were analysed by flow cytometry using FACSCalibur (Becton Dickinson) and BD CellQuest Pro software (Becton Dickinson, San Jose, CA).
Statistical analysis
Results are presented as mean ± standard error of the mean (s.e.m.). For diabetes incidence, the χ2 test was used. In all other cases, one-way analysis of variance (anova) was used for parameters with normal distribution and the Kruskal–Wallis test for parameters with non-normal distribution. Dunn's test was used when necessary. Significance level was P < 0·05. Statistical analysis was accomplished with SigmaStat for Windows version 3·5 (Systat Software Inc., Chicago, IL, USA).
Results
Effect of BCG/DNAhsp65 prime-boost in STZ-induced diabetes
Weight variation, glycaemia and the score of mononuclear infiltration in the pancreas were analysed in mice immunized with BCG alone or with prime-boost (BCG followed by pVAXhsp65) before diabetes induction with STZ. As shown in Fig. 1a, although all the groups gained weight, BCG–STZ and BCG/DNAhsp65–STZ exhibited a smaller variation (3 and 1%, respectively) in comparison to the control group (9%). BCG alone and BCG/DNAhsp5 prime-boost were not able to avoid blood glucose increase in any period and glycaemia in these groups was always similar to the STZ group (Fig. 1b); histopathological pancreas analysis revealed that the vaccines did not prevent insulitis either. As shown in Fig. 1c, BCG and BCG/DNAhsp65 reduced the percentage of intact islets (0 and 8%, respectively) in comparison to the STZ group (10%) and increased score 3 mononuclear infiltration (6 and 14%, respectively), also in comparison to the STZ group (2%).
Figure 1.
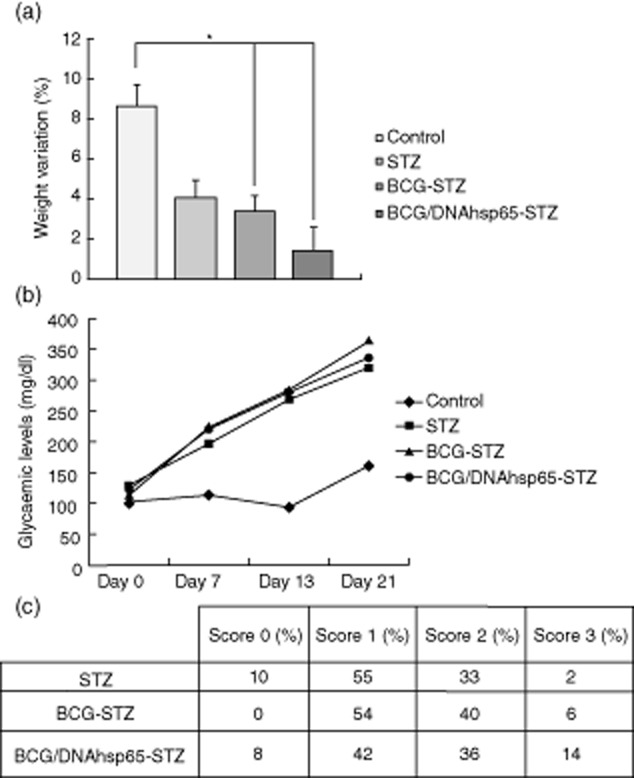
Effect of bacille Calmette–Guérin (BCG)/DNAhsp65 prime-boost in streptozotocin (STZ)-induced diabetes. (a) Weight variation and (b) blood glucose concentration were measured weekly in C57BL/c mice. The first measurement was performed before STZ inoculation and the other three were performed 7, 13 and 21 days after the last STZ dose. BCG and pVAXhsp65 were administered before diabetes induction. The histopathological analysis of the pancreatic islets (c) was performed 21 days after the last STZ administration in pancreas sections stained with haematoxylin and eosin (H&E) and all the islets were analysed. Results are expressed as mean ± standard error of the mean. *P < 0·05.
BCG/DNAhsp65 reduces diabetes clinical manifestation in NOD mice
Despite the negative results of the vaccination protocols in the MLD–STZ model, BCG alone and prime-boost BCG/DNAhps65 protected NOD mice against diabetes type 1 development. Seven-week-old NOD mice were immunized with BCG, and in the prime-boost group they also received a pVAXhsp65 dose 15 days later. Body weight and glycaemia were then measured until week 29. The weight variation from weeks 11–29 is shown in Fig. 2a. All the animals gained weight; however, the variation in BCG–NOD and BCG/DNAhsp65–NOD groups (20 and 21%, respectively) was significantly higher than in non-immunized NOD mice (13%). Weight gain was similar in the two immunized groups. The blood glucose variation during the experimental period can be observed in Fig. 2b. Blood glucose levels in the NOD group were always higher than 200 mg/dl from week 18 onwards. Both BCG–NOD and BCG/DNAhsp65–NOD groups had glycaemia measurements below the diabetic threshold; however, they were even lower in mice immunized with the prime-boost. Therefore, the vaccines protected mice against diabetes and data for the disease incidence are shown in Fig. 2c. In the non-immunized group, mice started to become diabetic by week 15. BCG alone was able to delay diabetes onset until week 24 and prime-boost BCG followed by pVAXhsp65 protected mice completely until week 29. Figure 2d shows the percentage of diabetic and non-diabetic mice per group, considering all animals. By week 29, 78% of all diabetic mice were in the non-immunized NOD group while the remaining 22% were in the BCG–NOD group; there were no diabetic mice in the BCG/DNAhsp65–NOD group. Thus, when analysing the non-diabetic mice, only 17% of all animals were in the NOD group, 38% were in the BCG–NOD group and almost half of them (45%) were in the BCG/DNAhsp65–NOD group.
Figure 2.
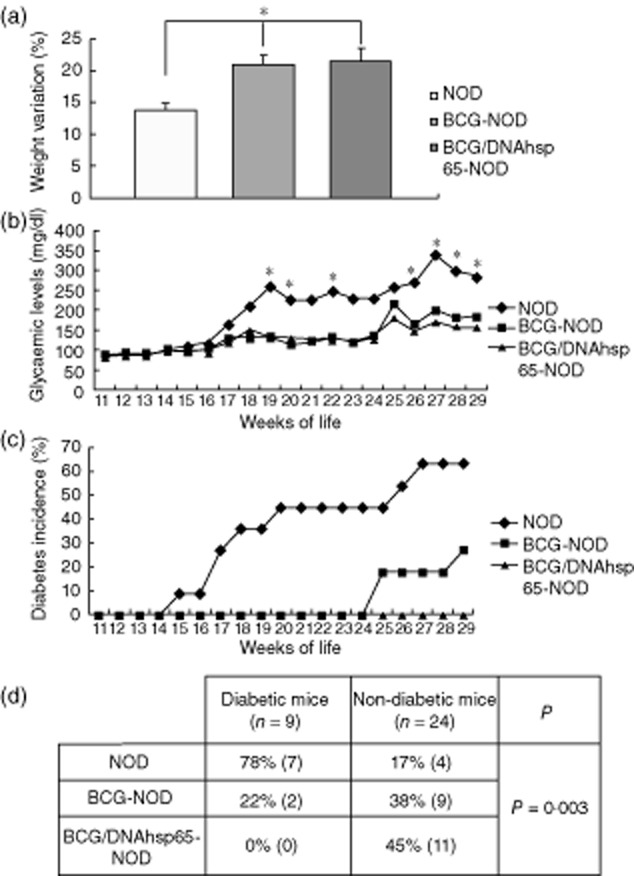
Effect of prime-boost bacille Calmette–Guérin (BCG)/DNAhsp65 in diabetes manifestations in non-obese diabetic (NOD) mice. Weight variation (a) and blood glucose (b) were measured once a week from weeks 11 to 29. BCG was administered on week 7; the prime-boost group was also vaccinated with pVAXhsp65 at week 9. Diabetes incidence through the weeks can be analysed in (c). Results are expressed as mean ± standard error of the mean. *P < 0·05 in comparison to the NOD group. The percentage of diabetic and non-diabetic mice at week 29 can be observed in (d) and P = 0·003.
Insulitis score is modulated by prime-boost BCG/DNAhsp65 in NOD mice
Examples of each one of the inflammatory scores found in the pancreas islets are shown in Fig. 3a: (i) presents a score 0, intact islet; (ii) shows a score 1 of infiltration, characterized by peri-insulitis; (iii) is a moderate infiltration defined as score 2 and (iv) shows an accentuated level of inflammatory infiltration, i.e. a score 3. Based on this score system, Fig. 3b illustrates the diversity of insulitis scores found in NOD mice. Although the three groups exhibit a similar percentage of islets on score 0, there is a descending pattern from score 1 to score 3 in BCG–NOD and BCG/DNAhsp65–NOD groups and the opposite occurs in the non-immunized NOD group. The major difference is observed in score 3; mice from the NOD group showed 50% of destructive islets while BCG alone and prime-boost BCG/DNAhsp65 groups showed only 20 and 17%, respectively.
Figure 3.
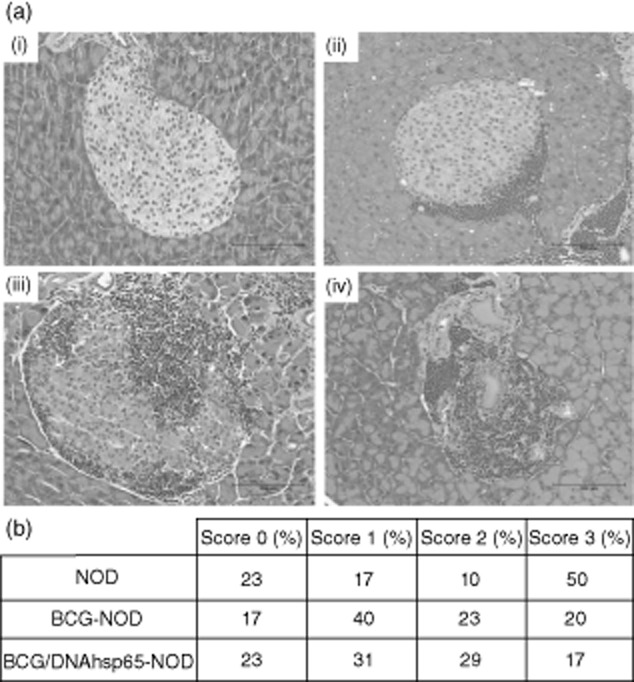
Effect of prime-boost bacille Calmette–Guérin (BCG)/DNAhsp65 in the insulitis score in non-obese diabetic (NOD) mice. The degrees of insulitis (a) were evaluated using a semi-quantitative scale: (i) 0 = intact islet; (ii) 1 = peri-insulitis; (iii) 2 = moderate insulitis (< 50% mononuclear infiltration); and (iv) 3 = severe insulitis (more than 50% mononuclear infiltration). The analysis of the severity of mononuclear infiltration in NOD immunized with the prime boost BCG/DNAhsp65 is shown in (c) based on the score described above. All islets of all pancreas sections were analysed.
Diabetes in C57BL/6 and NOD mice: comparison of clinical and histopathological parameters
Comparisons between clinical and histopathological data from induced (day 21) and spontaneous (week 29) diabetes are shown in Table 1. Previous vaccination in NOD mice, but not in the C57BL/6 strain, had blood glucose levels considered non-diabetic. This protection was more pronounced when NOD mice were immunized with the prime-boost procedure. Analysis of diabetes incidence revealed the same pattern, i.e. protection in spontaneous but not in induced disease and superior efficacy of the prime-boost strategy compared to BCG alone. Vaccination increased insulitis in STZ-induced diabetes but decreased this process in NOD mice.
Table 1.
Diabetes in C57BL/6 and non-obese diabetic (NOD) mice: comparison of clinical and histopathological parameters.
| C57BL/6: day 21 (15 animals per group) |
NOD: week 29 (11 animals per group) |
|||||
|---|---|---|---|---|---|---|
| STZ | BCG – STZ | BCG/DNAhsp65 – STZ | NOD | BCG – NOD | BCG/DNAhsp65 – NOD | |
| Glycaemic levels (mg/dl) | 322 | 366 | 338 | 287 | 186 | 159 |
| Diabetes incidence (%) | 100 | 100 | 100 | 64 | 27 | 0 |
| Insulitis score 3 (%) | 2 | 6 | 14 | 50 | 20 | 17 |
BCG: bacille Calmette–Guérin; STZ: streptozotocin.
Effect of vaccinations on cytokine production and presence of Treg cells
The cytokine profile in NOD mice was investigated based on their production by cultured spleen cells stimulated with rhsp65. Mice immunized with BCG alone and the prime-boost BCG/DNAhsp6 presented a significantly higher production of IFN-γ in comparison to non-immunized NOD mice (Fig. 4a). As shown in Fig. 4b, this increased production by mice immunized with BCG followed by pVAXhsp65 was also seen in TNF-α levels compared to the NOD group. The BCG/DNAhsp65 group showed a high production of IL-5 in comparison with the NOD and BCG–NOD groups, although there was no statistical difference (Fig. 4c). IL-10 levels seen in spleen cells stimulated with rhsp65 were similar among the groups; however, there was a small increase in the BCG/DNAhsp65–NOD group. CD4+CD25+FoxP3+ Treg cells were quantified in the spleen by using flow cytometry. As shown in Fig. 4e, the BCG-and BCG/DNAhsp65-immunized groups presented significantly lower percentages of Treg cells in the spleen than the non-immunized NOD mice.
Figure 4.
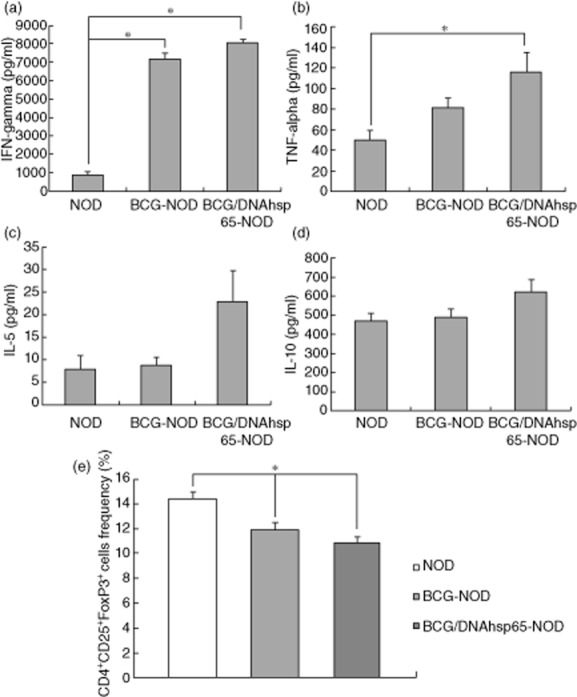
Effect of prime-boost bacille Calmette–Guérin (BCG)/DNAhsp65 in cytokine production and frequency of regulatory T cells (Treg) cells. The production of interferon (IFN)-γ (a), tumour necrosis factor (TNF)-α (b), interleukin (IL)-5 (c) and IL-10 (d) by spleen cells stimulated with rhsp65 in non-obese diabetic (NOD) mice were determined by enzyme-linked immunosorbent assay (ELISA). The effect of prime-boost BCG/DNAhsp65 in the frequency of Tregs in the spleen is represented at (e). Data were obtained by flow cytometry. Results are expressed as mean ± standard error of the mean. *P < 0·05.
Discussion
T1D is an autoimmune condition associated with T cell-mediated destruction of pancreatic beta-cells, resulting in loss of the ability to produce insulin [16]. As diabetes has no cure and the only available treatment consists in insulin administration, there is a great deal of interest to investigate immune-based interventions capable of protecting against the disease. Various studies have shown the potential of hsps to suppress immune responses in inflammatory diseases, such as rheumatoid arthritis, allergy and T1D [9,17]. In this scenario, we hypothesized that a prime-boost approach with administration of BCG (a M. bovis that naturally expresses the mycobaterial hsp65) followed by the vaccine pVAXhsp65 (DNA vaccine encoding the hsp65 gene from M. leprae) could protect mice against the development of type 1 diabetes. These vaccines have already been tested separately and showed promising results not only in T1D, but also in other autoimmune diseases as arthritis and experimental autoimmune encephalomyelitis [12–15,18,19]. Thus, we expected an additive or synergistic effect from combining BCG and pVAXhsp65.
Seven-week-old female NOD mice were primed with BCG vaccine and, 2 weeks later, they received the pVAXhps65 booster. A non-immunized group and a group immunized with BCG alone were used as experimental control groups. In this model, animals start to present mononuclear infiltration on the islets by the age of 4–5 weeks; however, clinical evidence for diabetes is only measurable around week 12 [4,7]. For this reason, body weight and glycaemia were evaluated from weeks 11–29. Weight gain was evaluated daily and indicated that all three groups gained weight; however, the immunized mice presented a significantly higher percentage of weight acquisition. Most relevant, the incidence of diabetes was also affected. While the hyperglycaemia in non-immunized mice began to be observed by week 15, in the BCG–NOD group it was delayed until week 24 and in NOD mice immunized with the prime-boost it was not detected during the whole protocol. Also, the percentage of diabetic mice was significantly higher in the NOD group compared to the BCG–NOD and BCG/DNAhsp65–NOD groups. These results suggest that although BCG alone is protective, the booster with pVAXhsp65 increased its potential to modulate the disease. We then analysed the insulitis score in the pancreas. Even though there was no difference in the score 0, BCG alone and BCG followed by pVAXhsp65 were able to reduce the percentage of destructive insulitis (score 3) in NOD mice. Comparisons of cytokine production indicated that there was significantly higher production of IFN-γ in both immunized groups and that the BCG/DNAhsp65–NOD group also exhibited higher levels of TNF-α in comparison to the non-immunized group. These cytokines, better known by their proinflammatory profile, could mediate one of the mechanisms by which both vaccine strategies protect mice against diabetes. Studies from [13] Qin et al. demonstrated that the co-operation of IFN-γ and TNF-α triggers the apoptosis of diabetogenic T cells through both Fas-FasL and TNF–TNFR1 pathways. IFN-γ is also known to induce MHC class II in various cell types. Thus, MHC class II presentation of hsp fragments in the absence of proper co-stimulation could boost regulatory T cell responses [20]. IL-5 and IL-10 levels were not statistically different among the groups; however, their production was slightly higher in the BCG/DNAhsp65–NOD group. To evaluate the possible contribution of Treg cells to this protective effect, we quantified these cells in the spleen. A decreased percentage of CD4+CD25+FoxP3+ cells in the immunized groups was detected in comparison to the NOD group. Hypothetically, these regulatory cells could have exited the spleen in these immunized groups and entered the pancreas to play their regulatory role on the inflammatory site. This possible explanation finds support in studies that show migration of Treg cells from lymphoid organs to the inflammatory site. For example [21], Tonkin and Haskins demonstrated that the migration of regulatory T cells from the peripheral lymphoid organs to the pancreas in the NOD mouse model resulted in reduced T helper type 1 (Th1) effector cells and their cytokines in the islets. We also suggest that these migrating Treg lymphocytes could be hsp-specific T cells. These cells exert their regulatory effect when exposed to hsp, which is a stress protein and could therefore be up-regulated at the inflammatory site [9].
Altogether, these results showed that the prime-boost procedure protected NOD mice against diabetes and that this strategy was even more effective than BCG alone, as suggested by diabetes incidence findings. Further investigation will allow us to determine if Treg cells are really located in the pancreas and if these cells are hsp-specific, as we are proposing.
Interestingly, the protective effects observed in NOD mice were not detected in the MLD–STZ model. This finding was unexpected and differ, to some extent, from what has been suggested by a few papers. There is only one report where the authors demonstrated that a BCG vaccine prevented insulitis in MLD–STZ diabetes in mice [12]. However, a direct comparison with the present work is hardly possible because distinct protocols, including mouse strain, timing and the BCG immunization route, were adopted. In addition, two other studies showed that vaccination with a heat shock protein (hsp65) was able to protect mice against diabetes induced by STZ [19,22]. Considering that the prime-boost strategy was able to decrease significantly the severity of insulitis and to avoid hyperglycaemia in NOD mice, we are tempted to attribute the observed failure to the STZ model itself. These two diabetes type 1 models present characteristics that could account for their distinct behaviour. The NOD mouse has been considered to be the model that resembles human type 1 diabetes most accurately in its genetic and immunopathogenic complexity [23,24]. For this reason it has been the preferred choice in investigating the role played by different T cell subsets in insulitis [25,26] and also to explore treatment strategies that target the autoimmune process [27,28]. The MLD–STZ is also considered a type 1 diabetes model in which the contribution of macrophages, Th subsets and Tc cells have been characterized [19,29,30. However, STZ can induce diabetes even in the absence of T and B cells, suggesting that it does not model the human pathology as closely as the disease developed by NOD mice [31]. This model is indicated preferentially to pursue therapies targeting cytokines and oxidants and also approaches to prevent beta cell death [28,32]. The need to use a toxic diabetogenic drug could also contribute to the inefficacy of BCG/pVAXhsp65 over the STZ model. The current view is that STZ determines strong immunosuppression associated with significant lymphopenia [33]. A direct effect of this drug over the immune system has been ascertained in vitro and in vivo [34,35]. This effect of STZ over the immune system could render it refractory to a potential immunomodulatory activity of the BCG/pVAXhsp65 association. The concurrence of the C57BL/6 strain background, especially the peculiarities associated with their Treg cell subset, can also be considered.
In conclusion, these results indicate that the prime-boost BCG/DNAhsp65 is able to protect NOD mice against type 1 diabetes, although a more detailed investigation will be necessary to clarify the immunological mechanisms. Our findings suggest that apoptosis of diabetogenic T cells and activity of Treg cells could be involved.
Acknowledgments
The authors would like to thank to Secretaria da Saúde do Estado de São Paulo for providing BCG. We are also thankful to Ana Paula Masson for her technical assistance. This study was supported by grants from Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP) and Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq).
Disclosures
None.
References
- 1.Cabrera SM, Rigby MR, Mirmir RG. Targeting regulatory T cells in the treatment of type 1 diabetes mellitus. Curr Mol Med. 2012;12:1261–1272. doi: 10.2174/156652412803833634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Maiese K, Shang YC, Chong ZZ, Hou J. Diabetes mellitus: channeling care through cellular discovery. Curr Neurovasc Res. 2010;7:59–64. doi: 10.2174/156720210790820217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wood SC, Rao TD, Frey AB. Multidose streptozotocin induction of diabetes in BALB/c By mice induces a T cell proliferation defect in thymocytes which is reversible by interleukin-4. Cell Immunol. 1999;192:1–12. doi: 10.1006/cimm.1998.1413. [DOI] [PubMed] [Google Scholar]
- 4.Rees DA, Alcolado JC. Animal models of diabetes mellitus. Diabet Med. 2005;22:359–370. doi: 10.1111/j.1464-5491.2005.01499.x. [DOI] [PubMed] [Google Scholar]
- 5.Aoki CA, Borchers AT, Ridgway WM, Keen CL, Ansari AA, Gershwin ME. NOD mice and autoimmunity. Autoimmun Rev. 2005;4:373–379. doi: 10.1016/j.autrev.2005.02.002. [DOI] [PubMed] [Google Scholar]
- 6.Yang Y, Santamaria P. Lessons on autoimmune diabetes from animal models. Clin Sci (Lond) 2006;110:627–639. doi: 10.1042/CS20050330. [DOI] [PubMed] [Google Scholar]
- 7.Driver JP, Serreze DV, Chen YG. Mouse models for the study of autoimmune type 1 diabetes: a NOD to similarities and differences to human disease. Semin Immunopathol. 2011;33:67–87. doi: 10.1007/s00281-010-0204-1. [DOI] [PubMed] [Google Scholar]
- 8.Jäger A, Kuchroo VK. Effector and regulatory T-cell subsets in autoimmunity and tissue inflammation. Scand J Immunol. 2010;72:173–184. doi: 10.1111/j.1365-3083.2010.02432.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.van Eden W, van der Zee R, Prakken B. Heat-shock proteins induce T-cell regulation of chronic inflammation. Nat Rev Immunol. 2005;5:318–330. doi: 10.1038/nri1593. [DOI] [PubMed] [Google Scholar]
- 10.Nicholas D, Odumosu O, Landridge WH. Autoantigen based vacines for type 1 diabetes. Discov Med. 2011;11:293–301. [PMC free article] [PubMed] [Google Scholar]
- 11.Woodland DL. Jump-starting the immune system: prime-boosting comes of age. Trends Immunol. 2004;25:98–104. doi: 10.1016/j.it.2003.11.009. [DOI] [PubMed] [Google Scholar]
- 12.Baik SH, Park IB, Choi KM, et al. BCG vaccine prevents insulitis in low dose streptozotocin-induced diabetic mice. Diabetes Res Clin Pract. 1999;46:91–97. doi: 10.1016/s0168-8227(99)00079-0. [DOI] [PubMed] [Google Scholar]
- 13.Qin HY, Chaturvedi P, Bhagirath S. In vivo apoptosis of diabetogenic T cells in NOD mice by IFN-γ/TNF-α. Int Immunol. 2004;16:1723–1732. doi: 10.1093/intimm/dxh173. [DOI] [PubMed] [Google Scholar]
- 14.Santos Junior RR, Sartori A, Bonato VL, et al. Immune modulation induced by tuberculosis DNA vaccine protects non-obese diabetic mice from diabetes progression. Clin Exp Immunol. 2007;149:570–578. doi: 10.1111/j.1365-2249.2007.03433.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zorzella-Pezavento SF, Chiuso-Minicucci F, França TG, et al. Immunization with pVAXhsp65 decreases inflammation and modulates immune response in experimental encephalomyelitis. Neuroimmunomodulation. 2010;17:287–297. doi: 10.1159/000292018. [DOI] [PubMed] [Google Scholar]
- 16.Chen W, Xie A, Chan L. Mechanistic basis of immunotherapies for type 1 diabetes mellitus. Transl Res. 2013;161:217–229. doi: 10.1016/j.trsl.2012.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wieten L, van der Zee R, Spiering R, et al. A novel heat-shock protein coinducer boosts stress protein Hsp70 to activate T cell regulation of inflammation in autoimmune arthritis. Arthritis Rheum. 2010;62:1026–1035. doi: 10.1002/art.27344. [DOI] [PubMed] [Google Scholar]
- 18.Quintana FJ, Carmi P, Mor F, Cohen IR. DNA fragments of the human 60-kDa heat shock protein (HSP60) vaccinate against adjuvant arthritis: identification of a regulatory HSP60 peptide. J Immunol. 2003;171:3533–3541. doi: 10.4049/jimmunol.171.7.3533. [DOI] [PubMed] [Google Scholar]
- 19.Santos Junior RR, Sartori A, Lima DS, et al. DNA vaccine containing the mycobacterial hsp65 gene prevented insulitis in MLD-STZ diabetes. J Immune Based Ther Vaccines. 2009;7:4. doi: 10.1186/1476-8518-7-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Borges TJ, Wieten L, van Herwijnen MJ, et al. The anti-inflammatory mechanisms of Hsp70. Front Immunol. 2012;3:95. doi: 10.3389/fimmu.2012.00095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tonkin DR, Haskins K. Regulatory T cells enter the pancreas during suppression of type 1 diabetes and inhibit effector T cells and macrophages in a TGF-β-dependent manner. Eur J Immunol. 2009;39:1313–1322. doi: 10.1002/eji.200838916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Szebeni A, Prohászka Z, Buzás E, Falus A, Kecskeméti V. Effects of vaccination with heat shock proteins on streptozotocin induced diabetes in histidine decarboxylase knockout mice. Inflamm Res. 2008;57:178–182. doi: 10.1007/s00011-007-7135-x. [DOI] [PubMed] [Google Scholar]
- 23.Thayer TC, Wilson SB, Mathews CE. Use of nonobese diabetic mice to understand human type 1 diabetes. Endocrinol Metab Clin North Am. 2010;39:541–561. doi: 10.1016/j.ecl.2010.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chaparro RJ, Dilorenzo TP. An update on the use of NOD mice to study autoimmune (Type 1) diabetes. Exp Rev Clin Immunol. 2010;6:939–955. doi: 10.1586/eci.10.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sgouroudis E, Piccirillo CA. Control of type 1 diabetes by CD4+Foxp3+ regulatory T cells: lessons from mouse models and implications for human disease. Diabetes Metab Res Rev. 2009;25:208–218. doi: 10.1002/dmrr.945. [DOI] [PubMed] [Google Scholar]
- 26.Shao S, He F, Yang Y, Yuan G, Zhang M, Yu X. Th17 cells in type 1 diabetes. Cell Immunol. 2012;280:16–21. doi: 10.1016/j.cellimm.2012.11.001. [DOI] [PubMed] [Google Scholar]
- 27.Fierabracci A. Peptide immunotherapies in Type 1 diabetes: lessons from animal models. Curr Med Chem. 2011;18:577–586. doi: 10.2174/092986711794480230. [DOI] [PubMed] [Google Scholar]
- 28.King AJ. The use of animal models in diabetes research. Br J Pharmacol. 2012;166:877–894. doi: 10.1111/j.1476-5381.2012.01911.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lukić ML, Stosić-Grujicić S, Shahin A. Effector mechanisms in low-dose streptozotocin-induced diabetes. Dev Immunol. 1998;6:119–128. doi: 10.1155/1998/92198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yaochite JN, Caliari-Oliveira C, Davanso MR, et al. Dynamic changes of the Th17/Tc17 and regulatory T cell populations interfere in the experimental autoimmune diabetes pathogenesis. Immunobiology. 2013;218:338–352. doi: 10.1016/j.imbio.2012.05.010. [DOI] [PubMed] [Google Scholar]
- 31.Reddy S, Wu D, Elliott RB. Low dose streptozotocin causes diabetes in severe combined immunodeficient (SCID) mice without immune cell infiltration of the pancreatic islets. Autoimmunity. 1995;20:83–92. doi: 10.3109/08916939509001931. [DOI] [PubMed] [Google Scholar]
- 32.Kawasaki E, Abiru N, Eguchi K. Prevention of type 1 diabetes: from the view point of beta cell damage. Diabetes Res Clin Pract. 2004;66:S27–32. doi: 10.1016/j.diabres.2003.09.015. [DOI] [PubMed] [Google Scholar]
- 33.Luo B, Chan WF, Lord SJ, et al. Diabetes induces rapid suppression of adaptive immunity followed by homeostatic T-cell proliferation. Scand J Immunol. 2007;65:22–31. doi: 10.1111/j.1365-3083.2006.01863.x. [DOI] [PubMed] [Google Scholar]
- 34.Gaulton GN, Schwartz JL, Eardley DD. Assessment of the diabetogenic drugs alloxan and streptozotocin as models for the study of immune defects in diabetic mice. Diabetologia. 1985;28:769–775. doi: 10.1007/BF00265026. [DOI] [PubMed] [Google Scholar]
- 35.Koulmanda M, Qipo A, Auchincloss H, Jr, Smith RN. Effects of streptozotocin on autoimmune diabetes in NOD mice. Clin Exp Immunol. 2003;134:210–216. doi: 10.1046/j.1365-2249.2003.02293.x. [DOI] [PMC free article] [PubMed] [Google Scholar]