

Abstract
Research on spermatogonia is hampered by complex architecture of the seminiferous tubule, poor viability of testicular tissue ex vivo and lack of physiologically relevant long-term culture systems. Therefore there is a need for an in vitro model that would enable long term survival and propagation of spermatogonia. We aimed at the most simplified approach to enable all different cell types within the seminiferous tubules to contribute to the creation of a niche for spermatogonia. In the present study we describe the establishment of a co-culture of mouse testicular cells that is based on proliferative and migratory activity of seminiferous tubule cells and does not involve separation, purification or differential plating of individual cell populations. The co-culture is composed of the constituents of testicular stem cell niche: Sertoli cells [identified by expression of Wilm's tumour antigen 1 (WT1) and secretion of glial cell line-derived neurotrophic factor, GDNF], peritubular myoid cells (expressing alpha smooth muscle actin, αSMA) and spermatogonia [expressing MAGE-B4, PLZF (promyelocytic leukaemia zinc finger), LIN28, Gpr125 (G protein-coupled receptor 125), CD9, c-Kit and Nanog], and can be maintained for at least five weeks. GDNF was found in the medium at a sufficient concentration to support proliferating spermatogonial stem cells (SSCs) that were able to start spermatogenic differentiation after transplantation to an experimentally sterile recipient testis. Gdnf mRNA levels were elevated by follicle-stimulating hormone (FSH) which shows that the Sertoli cells in the co-culture respond to physiological stimuli. After approximately 2–4 weeks of culture a spontaneous formation of cord-like structures was monitored. These structures can be more than 10 mm in length and branch. They are formed by peritubular myoid cells, Sertoli cells, fibroblasts and spermatogonia as assessed by gene expression profiling. In conclusion, we have managed to establish in vitro conditions that allow spontaneous reconstruction of testicular cellular microenvironments.
Introduction
Spermatogenic potential ultimately depends on a small population of spermatogonia called spermatogonial stem cells (SSCs). These cells are responsible for the life-long ability of sperm production in mammals and they are able to reconstitute spermatogenesis to recipients that are rendered experimentally sterile [1], [2]. Additionally, derivation of embryonic stem cell-like cells from SSCs has been demonstrated in a number of studies [3]–[5] and they hold thus a great promise for regenerative medicine by providing an ethically sustainable and immunologically competent source of pluripotent cells. A very small fraction of cells in SSC cultures behave like SSCs in vivo and are able to reconstitute spermatogenesis after transplantation [6]. Cells in SSC cultures are a heterogenous population and it is not clear how closely they resemble undifferentiated spermatogonia in vivo. Therefore their suitability to study germ cell biology can be justifiably questioned.
In the testis spermatogonia dwell on the basement membrane of the seminiferous epithelium. They are in physical contact with Sertoli cells and in close proximity of peritubular myoid cells. This is the physiologically relevant environment to study germ cell biology. Cell fate decisions of spermatogonia are thought to be directed by extracellular signals secreted by cells in their vicinity or carried to the testis by circulation. SSCs are maintained in the testis in specified areas, i.e. stem cell niches. There is a handful of molecular and anatomical cues about the location of testicular stem cell niche [7]–[11]. However, due to the complex architecture of the seminiferous tubules, in vivo studies on testis stem cell niche are limited and challenging. Moreover, there are not long-term organ cultures for adult testicular tissue to date. Fragments of rodent seminiferous tubules can be successfully cultured in defined conditions for up to 3–7 days [12]–[14], but already after a 16-hour culture a vast number of apoptotic cells can be observed [15] and the number increases rapidly within the next 2 days (Mäkelä J-A & Toppari J, unpublished data).
Recently, Sato and colleagues managed to produce functional male gametes in vitro using a neonatal mouse organ culture approach [16]–[17]. The same group demonstrated the progression of male germ cells at least up to meiotic prophase in in vitro reconstructed tubules of juvenile mouse testis cells [18]. Despite such progress in the field, their suitability to study dynamics of the adult stem cell niche is questionable. This is because adult SSCs are maintained in a quite distinct environment from that of the neonatal testis. In the adult testis somatic cells have seized proliferating and their secretory profile and mechanisms of regulation are different from the juvenile counterparts. Therefore new in vitro systems need to be developed to understand the regulation and function of adult testis stem cell niche.
The aim of the present study was to evaluate how well spermatogonia can be maintained in a seminiferous tubule cell-derived co-culture model. We hypothesized that co-culture of these cells would allow SSCs to survive and proliferate - something that SSCs cannot perform by themselves. Since the co-culture was maintained at 37°C and testosterone-producing Leydig cells did not contribute to it, we expected germ cells not to differentiate, the scope of the study being in early germ cell behaviour. We were able to support the survival and propagation of spermatogonia for at least 5 weeks and recorded relatively high levels of endogenous GDNF in the culture medium during that time. These data suggest that stem cell niche-like conditions had been reconstructed in vitro. Spermatogonia in the co-culture expressed a wide range of spermatogonial markers, including SSC markers PLZF (promyelocytic leukaemia zinc finger) [19], [20] and LIN28 [21], and undifferentiated spermatogonial marker Nanog [11] and they were able to colonize seminiferous tubules of busulfan-treated mice. Cells in the culture were also observed to give rise to secondary structures, i.e. spherical cell aggregates and cord-like structures. Therefore the present model might also be used to study early steps of development and regeneration of testis tissue in vitro.
Results
Establishment of the co-culture
Mouse testes were decapsulated and the seminiferous tubules were separated from each other and from the interstitial tissue in sterile conditions in DMEM/F12 supplemented with 15% (v/v) inactivated fetal calf serum (iFCS) (referred to as culture medium later in the text). The tubules were cut into small fragments and 50–100 approximately 1-mm-long segments of mouse seminiferous tubule were pipetted onto a culture dish in small volume of culture medium (100, 200 and 350 µl of suspension was plated onto 24-, 12-, and 6-wells plates, respectively). After 6–10 hours of incubation more culture medium was added (500, 1000 and 1500 µl was pipetted onto 24-, 12-, and 6-wells plates, respectively). Five to seven days later we could observe spreading of WT1 (Wilm's tumour antigen 1)- [22] and Vimentin-expressing Sertoli cells and alpha smooth muscle actin (αSMA)-expressing peritubular myoid cells [23] in the perimeter of seminiferous tubule fragments (Figure 1A–B; Figure S1 for positive and negative control stainings for testicular tissue sections). After 1–2 weeks of culture, the seminiferous tubule cells had spread greatly and formed a nearly confluent monolayer culture (Figure 1C). Immunocytochemical analysis revealed that many cells in the co-culture expressed MAGE-B4 (Figure 1D), a ubiquitous spermatogonial cell marker [24] (Figure S1), while only a few cells expressed A-single undifferentiated spermatogonial cell marker Nanog [11] (Figure 1E, S2). Utilising the same technique, we successfully established this kind of co-cultures from juvenile, pubertal, adult and elderly mice. Adult-derived co-cultures are used in analyses throughout this report if not otherwise stated.
Figure 1. Early stages in the establishment of seminiferous tubule-derived co-culture.

A) Five-day co-culture showing WT1 positive cells (red) in the perimeter of a seminiferous tubule fragment. B) WT1 (red), Vimentin (orange), Alpha smooth muscle actin (αSMA, green) immunocytochemical staining for 1-week co-culture. DAPI (white) stains the nuclei in A and B. DAPI dense areas are fragments of seminiferous tubule. C) Phase-contrast microscopy image of 1-week co-culture. D) MAGE-B4-expressing (red) cells in 1-week co-culture. E) An example of a rare Nanog-expressing cell in 2-week co-culture. DAPI stains the nuclei blue in D and E. Scale bar is 100 and 25 µm in A–D and E, respectively. Insets in A, B and D represent negative control stainings.
We followed the co-culture up to 8 weeks and performed a time-lapse imaging series by photographing specific areas in the co-culture every 1–3 days. During this period of time we observed partly dissociation of the seminiferous tubule fragments (during the first week), gain of confluence within the first 1–2 weeks and formation of cord-like structures and cell clusters during weeks 2–6 (Figure 2 and Video S1). Formation of clusters and cord-like structures was associated with dramatic and dynamic changes in the interaction between different cell populations in the co-culture, whereas during the first 1–2 weeks the cells were mainly found single-dwelling or in small populations within the co-culture. In the co-culture at least two different testicular cellular microenvironments were reconstructed: 1. testis stem cell niche-like conditions, and 2. in vitro milieu enabling cluster and cord-like structure formation by testicular somatic cells.
Figure 2. Eight-week follow-up of adult-derived seminiferous tubule cell co-culture.

Cells proliferated rapidly to form a confluent culture. First signs of cord-like structure formation were apparent after four weeks of culture in this case.
Maintenance and propagation of germ cells in the co-culture
To find out which germ cell populations are maintained in the prevailing conditions we analysed the steady state levels of CD9 (CD9 antigen) [25], c-Kit [26], Erm (Ets-related molecule) [27], [28], Gpr125 [29], Nanog, PLZF, Stra8 (Stimulated by retinoic acid gene 8) [30], [31] and Sycp3 (Synaptonemal complex protein 3) [32] mRNAs throughout 5 weeks of culture. Because the full complement of germ cells is present only in the adult testis, we used seminiferous tubule segments from adult mice to establish the co-cultures for this experiment. We observed that Gpr125, Erm and Stra8 mRNA levels slowly decreased with time until statistically significant differences were recorded at the late time points (Figure 3A). Transcript levels of c-Kit, Nanog and PLZF did not change in a statistically significant manner, whereas at 3 weeks the highest CD9 mRNA levels were recorded. Transcript for meiotic cell marker Sycp3 was not detectable after the first week (data not shown). These data suggest that spermatogonia survive in these culture conditions for many weeks, whereas more differentiated germ cells do not. This was also supported by propidium iodide (PI) staining of cell nuclei followed by flow cytometry showing that while haploid cells represented by far the greatest fraction in the beginning of the culture (Figure 3B) they virtually vanished after the first week. Most of the nuclei from then on were from cells in G0/G1 phase (Figure 3C–D). Loss of haploid cells is due to the 37°C temperature and perhaps also due to lack of testosterone: we were able to detect measurable but hypophysiological [33] levels of testosterone in 1–2 and 1-week cultures started from adult and juvenile mice, respectively (Table 1). Thereafter the levels fell below the sensitivity range of the used assay. Stimulation of 2–3-week cultures with an LH/hCG analogue did not increase the levels of testosterone in the medium (data not shown) proving the absence of Leydig cells in the co-cultures after 1–2 weeks.
Figure 3. Messenger RNAs of spermatogonial markers are expressed in the co-cultures until 5 weeks, whereas haploid cells are lost during the first week of culture.

A) Quantitative RT-PCR analysis of spermatogonial cell-associated markers during 1–5 weeks of culture. Steady state levels of CD9 mRNA were at their highest at three weeks. Erm, Gpr125 and Stra8 levels decreased towards the end of the culture. There were no statistically significant changes for c-Kit, Nanog and PLZF. Statistical significances are reported in comparison with the time point of the highest value. n = 3–6, SEM; a, p<0.01; b, p<0.05. Colour-coding in letters “a” and “b” refers to the colour of the lines. B–D) Analysis of haploid and diploid cells in co-cultures. Nuclei from co-cultures were stained with propidium iodide and analysed by flow cytometry: B) Starting material, C) 1-week co-culture and D) 2-week co-culture. Haploid cells (1C) were lost during the first week of culture. Relative fraction of G0/G1 cells (2C) increased rapidly, whereas the relative number of G1S/G2/M cells (4C) first decreased but later on stayed at a stable level. Histograms are representative of at least three parallel experiments.
Table 1. Testosterone concentration in the culture medium.
| 1-week (n = 3) | 2-week (n = 3) | 3–4-week (n = 6) | |
| Co-culture started from seminiferous tubules of adult mice | 1,79±0,24 nmol/l | 0,87±0,03 nmol/l | Measured values below the sensitivity range of the assay |
The values represent average ± standard deviation.
Spermatogonia expressing SSC-associated proteins PLZF, LIN28 and ubiquitous germ cell marker Ddx4 [34] were found to be proliferatively active as judged by the coexpression of proliferating cell antigens PH3 (phosphorylated Histone-3) and Ki-67 (Figure 4A–B, Figure S3). Generally, cells expressing spermatogonia-associated markers were usually located at areas of high cell density (Figure 1 and 4). These data suggest that at those areas the microenvironment was favourable for spermatogonial cell survival and propagation and provided testis stem cell niche-like conditions for SSCs.
Figure 4. Spermatogonial stem cells were identified in co-cultures by immunocytochemistry and transplantation assay, and these cells were supported by endogenous GDNF secretion.

A) Image of 1-week-old co-culture showing proliferating, phosphorylated Histone-3 (PH3) positive cells expressing spermatogonial stem cell marker PLZF. B) A high proportion of LIN28 positive cells also expressed PH3 after 10 days of culture. DAPI stains the nuclei of cells. C) Section of a wild-type SSC-depleted seminiferous tubule that has been colonized by transplanted SSCs. Samples were prepared five weeks after transplantation of GFP-expressing SSCs and show that spermatogenic differentiation of the transplanted SSCs had advanced to meiotic prophase. Arrows indicate pachytene spermatocytes. D) GDNF concentration in co-cultures. Analysis of GDNF levels in the culture medium showed that approximately 5–15 ng/ml were present in 1–5-week co-cultures.
After noting that the co-culture conditions were favourable for spermatogonia expressing SSC-associated markers, we wanted to study whether SSCs in the co-culture maintained their differentiation potential. For this purpose we established the co-cultures from adult GFP-expressing mice seminiferous tubules as described above. Then we treated adult wild-type male C57BL/6 mice with busulfan (40 mg/kg, i.p.) which results in the depletion of SSCs [35]. One month after the busulfan injection approximately 100 000 cells from 1-month-old co-cultures were transplanted via the rete testis according to a protocol described previously [36]. Five weeks later mice were sacrificed and seminiferous tubules were dissected and imaged under a fluorescence Zeiss SteREO Lumar V12 stereomicroscope with eGFP bandpass filter. The presence of GFP-positive pachytene spermatocytes inside seminiferous tubule segments indicated that the transplanted cells had started to undergo spermatogenic differentiation in vivo (Figure 4C). These data show that SSCs can be maintained in an undifferentiated state in the co-culture for at least a month. This was probably due to secretion of Sertoli cell-derived GDNF which has been shown to be crucial for SSC viability both in vivo and in vitro [37]. Using an ELISA-GDNF assay we detected 5–15 ng/ml of GDNF in the culture medium during 1–5 weeks of culture (Figure 4D), concentrations that are in the range of those that are commonly used in SSC cultures where GDNF is added to the culture medium as a supplement [6]. Addition of exogenous GDNF (10 ng/ml) to culture medium did not result in morphological alterations or dramatic changes in the number of spermatogonia in culture. At transcript level only the relative amount of CIP2A (cancerous inhibitor of PP2A) [38]–[39] mRNA was elevated (data not shown) by including exogenous GDNF in the culture medium.
To study if Sertoli cells in the co-culture respond to physiological stimuli, we treated 1-week-old co-cultures with recombinant human follicle-stimulating hormone (rhFSH; 10 ng/ml) for 6, 12, 24, 48 and 72 hours. The treatment increased the steady state levels of Gdnf mRNA 12, 24 and 72 hours after the treatment (Figure 5A). Moreover, FSH also stimulated the levels of Gdnf mRNA in the co-cultures that were established from juvenile (<10-day-old) mice (Figure 5B) albeit in a more acute fashion. FSH treatment also secondarily affected the steady state levels of some spermatogonia-associated mRNAs, most clearly those of Gfr1a and Stra8 (Figure S4).
Figure 5. Co-cultures responded to FSH.
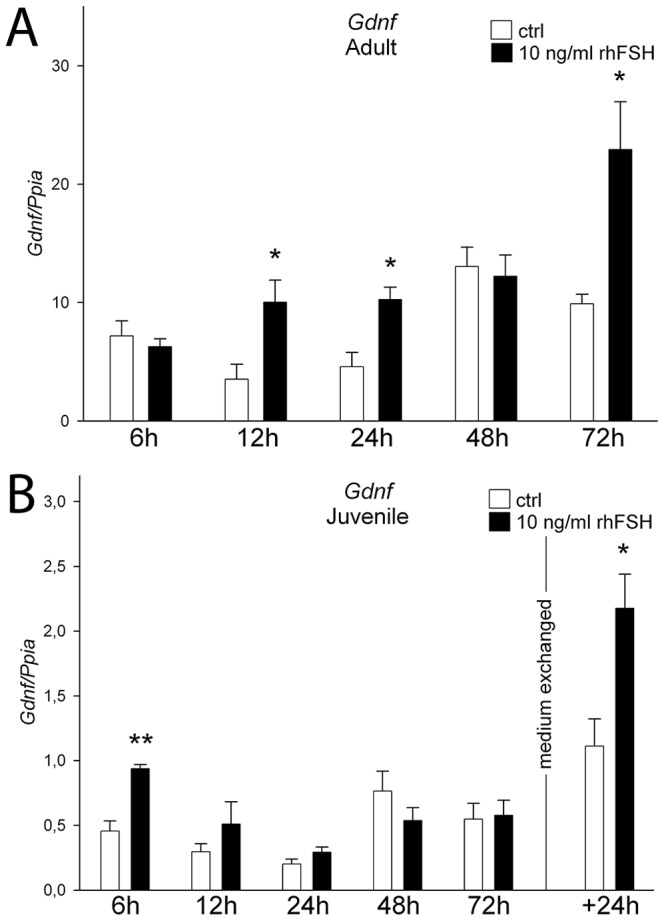
Recombinant human FSH treatment resulted in elevated Gdnf mRNA levels in 1-week co-cultures started from A) adult and B) juvenile seminiferous tubule fragments. In juvenile mouse-derived co-cultures FSH treatment was followed by an acute increase in Gdnf mRNA levels at 6 hours. Retreatment 72 hours after initial dose lead to increased levels 24 hours later. White bars, control; black bars, 10 ng/ml rhFSH; n = 3, SEM; *, p<0.05; **, p<0.01.
Formation of clusters and cord-like structures in vitro
After reaching certain density, cells in the co-culture formed a dynamic network that could tear and regrow over the course of hours (Video S2). These data suggest that the cells that contribute to it are contractile. In the mouse testis the only two cell types that have this capacity are peritubular myoid cells and endothelial cells both of which express αSMA (Figure S1). Usually towards the end of the second week of culture different cell populations started to form networks of cells (Figure 6). There were strain- and age-dependent variations, however, generally in the co-cultures started from C57BL/6 and juvenile mice seminiferous tubules this phenomenon was observed somewhat earlier than in NMRI or FVB/N and adult-derived co-cultures. Alpha smooth muscle actin positive cells were observed on the edge of a retreating frontier of cells (Figure 6A), whereas slender matrices of WT1-expressing Sertoli cells were often located at areas of lower cell density (Figure 6B). These areas were also recruited into clusters or cord-like structures later on. Spermatogonial cells (MAGE-B4) were found single-dwelling or in clusters that were associated with the tightening networks of cells (Figure 6A and 6C).
Figure 6. Peritubular myoid, Sertoli and spermatogonial cells are situated differently when secondary structure formation starts.
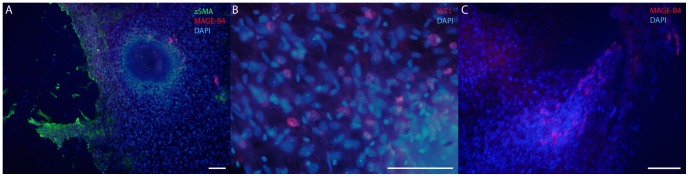
A) Alpha smooth muscle actin (green) positive cells were found at the frontier of a tightening cell network. B) Sertoli cells (WT1, red) were situated at areas of lower cell density, whereas C) spermatogonia (MAGE-B4, red) were found in clusters or single-dwelling at high cell density areas. There are MAGE-B4 positive cells (red) in A), as well. DAPI (blue) stains the nuclei. Scale bar 100 µm.
Depending on the origin of the seminiferous tubule fragments (strain and age of the used animals) the spontaneous formation of clusters and cord-like structures appeared between 1.5 to 4 weeks of culture. There were no exceptions to this although the number, shapes and sizes of the clusters and cord-like structures varied from plate to plate. The clusters (roughly 200–700 µm in diameter; Figure 7A–F) were formed by two alternative mechanisms: they were either remnants of the seminiferous tubules that later on gathered cells around them (Video S3), or they coalesced from a seemingly homogenous matrix of cells (Video S4). Cluster formation could be accelerated by exposing the co-cultures to angiotensin II, an inducer of peritubular myoid cell contraction [40] (Videos S5, S6). Based on qRT-PCR, melting curve analysis and gel electrophoresis these clusters were formed by Sertoli cells (4/10 were positive for WT1, 2/10 for Gdnf), fibroblasts [4/10 positive for Fsp1 (Fibroblast-specific protein-1)] [41], and peritubular myoid cells (9/10 positive for αSMA). This is in line with earlier findings showing that rodent Sertoli and peritubular cells form clusters of cells in a co-culture [42]. After 3–6 weeks of culture we occasionally observed spherical or slightly elongated clusters of cells also (in the range of 1–2 mm; Figure 7G–I, Figure S5). These were highly similar to the previously described protrusions formed during prolonged co-culture of juvenile rat Sertoli and peritubular myoid cells [43].
Figure 7. Collage of clusters.

Cluster formation started to take place in the co-cultures right after gain of confluency and thereafter throughout 8 weeks. A–F) Co-cultures invariably gave rise to clusters of approximately 200–700 µm in diameter. G–I) Occasionally we also observed bigger clusters that were partly detached from the underlying co-culture and held back by a stalk of cells. Scale bar 1 mm.
Co-cultures invariably gave rise to cord-like structures that could be up to 15 mm of length (Figure 8). Formation of cord-like structures took place through compaction of cellular networks suggesting an active role for peritubular myoid cells in the process, and clusters seemed to coordinate this process (Videos S7, S8). The role of the clusters in the orchestration of cord-like structure formation was also supported by the fact that no formation of cord-like structures was observed when we used single cell suspensions of testis homogenate to establish co-cultures. Quantitative RT-PCR and gel electrophoresis showed that transcripts of Sertoli cell (WT1), peritubular myoid cell (αSMA), fibroblast (Fsp1) and spermatogonial cell (c-Kit, CD9, Cip2a and Gfr1α) markers were present in these structures (data not shown).
Figure 8. Phase-contrast image of a cord-like structure.

First signs of cord-like structure formation were usually apparent after 3–4 weeks of culture. Cord-like structures sometimes branched and could reach a length of more than 10 mm.
When the cord-like structure formation had completed the co-cultures remained in a relatively stagnant state (see Video S1, for instance). As shown above, αSMA-expressing cells played a central role in the formation of clusters and cord-like structures and the relatively homogenous culture outside the cord-like structures and clusters had a relatively small fraction of αSMA, WT1 or spermatogonia-associated marker-expressing cells (data not shown). To answer which cells did not get recruited into secondary structures but throve in the culture conditions, we analysed how different testicular somatic cell populations acted during the course of culture by qRT-PCR (Figure 9). We used αSMA to represent peritubular myoid cells, WT1 for Sertoli cells, Fsp1 as a testicular fibroblast marker, and Cdh5 (Cadherin 5) [44], CD141 [45] and vWF (von Willebrand factor) [46] to represent endothelial cells. The levels of αSMA, Fsp1, Cdh5 and CD141 mRNAs were elevated shortly after the beginning of the cultures but all except for Fsp1 returned to control levels at later time points (Figure 9A–C). There were no statistically significant changes in the relative abundance of vWF mRNA (Figure 9C). WT1 levels plummeted right after the beginning of the culture suggesting that the number of Sertoli cells did not change much while other somatic cell populations actively proliferated in the co-culture (Figure 9A). This is understandable since Sertoli cells in the adult are thought to be terminally-differentiated and quiescent cells. Because Sertoli cells in the co-culture probably have to adopt features that developmentally only immature, juvenile-type Sertoli cells possess, we analysed whether immature Sertoli cell-associated markers are enriched over the course of culture. Indeed, markers of immature Sertoli cells such as Krt18 ((Cyto)keratin-18) [47] and Pdpn (podoplanin) [48], were elevated shortly after the beginning of the culture at mRNA (Figure 9D) and protein level (Figure S6). As expected, Krt18 mRNA levels in adult seminiferous tubules were undetectably low. These data suggest that adult-type mature Sertoli cells in the culture have to enter a dedifferentiation process to have an active role in secondary structure formation.
Figure 9. Quantitative RT-PCR analysis of somatic cell-specific marker expression in starting material and 1–5-week co-cultures.

A) Relative αSMA mRNA levels were increased in the beginning of the culture, whereas WT1 levels displayed the opposite. B) Fibroblast marker Fsp1 was found at a very high level from week 1 onwards. C) Endothelial cell marker mRNA levels were acutely increased (Cdh5 and CD141) or did not change (vWF) during the course of 5 weeks. D) Messenger-RNAs associated with juvenile, immature Sertoli cells (Krt18 and Pdpn) were found at their highest level in 1–2 week co-cultures. n = 3, SEM; *, p<0.05; **, p<0.01, ***, p<0.001.
Discussion
Due to the complexity of testicular architecture and function, novel in vitro approaches need to be developed to study the interaction of spermatogonia with testicular somatic cells. Even though SSCs can be maintained for extended periods of time ex vivo, co-culture with other stem cell niche contributors would provide the most physiologically relevant environment to study their biology. In the present study, a method for long-term co-culturing of spermatogonia with Sertoli and peritubular myoid cells was developed. Germ cells and somatic cells from small fragments of mouse seminiferous tubules proliferate in vitro and form a co-culture that can be maintained for at least five weeks. Since spermatogonia are able to survive and propagate for weeks and their life span is likely limited by Sertoli cells and not by the spermatogonia-intrinsic factors, we assume that stem cell niche-like conditions have been reconstructed in vitro. The advantages of this method are that no special isolation techniques and devices are required, no cell sorting is necessary, no exogenous growth factor stimulation is needed and it can be established in simple culture medium.
During the five-week culture expression of a wide range of spermatogonial markers was recorded. In adult-derived co-cultures great majority of differentiating cells was lost during the first week of culture. Taken together our results suggest that the conditions favour stem/progenitor spermatogonia but do not support differentiation of germ cells. Interestingly enough, a transcript for Nanog, a novel A-single spermatogonial marker [11], and rare Nanog-expressing cells were identified in the co-cultures. We also proved that the spermatogonia in our co-cultures remain able to enter spermatogenic differentiation if they are transplanted into in vivo settings. The presence of GFP-positive pachytene spermatocytes in mice depleted of germ cells clearly points at fully functional SSCs after a month of ex vivo co-culturing.
Sertoli cells secrete GDNF that is an important regulator of undifferentiated spermatogonia [37], [49]–[50], a factor that can be detected in our co-culture system at a comparable level as that used in SSC cultures in Sertoli cell-free conditions [6]. Additionally, it is highly likely that locally, i.e., in the interface between Sertoli cells and SSCs, the concentrations are even higher. Gdnf levels could be elevated by FSH, which shows that Sertoli cells were fully functional and responsive to physiological stimuli [51]. After the onset of spermatogenesis the dominant function of FSH on Sertoli cells shifts from driving their proliferation to nurturing germ cells via paracrine growth factor production [52]. Accordingly, FSH had a greater impact on one-week-old co-cultures established from adult than juvenile mice. Change in the paracrine milieu induced by FSH further activated SSCs as GDNF receptor Gfr1α mRNA expression was stimulated at the same time points. Our finding that FSH has a greater impact on the Stra8 level in cultured juvenile than in adult testicular cells probably stems from the developmental state of these cells. Premeiotic/meiotic transition of the most advanced germ cells takes place on day 8–10 in mice [53] and therefore a relatively greater number of spermatogonia are bound to enter meiosis in juvenile than in adult mouse testis.
Sertoli cells have a crucial role in the development of testis cords in the fetal gonad [54]. In the present study we noticed that the formation of cord-like structures was centered around cell clusters that often expressed Sertoli cell markers. It has been shown by a number of research groups that testicular single cell homogenates from a juvenile rat spontaneously give rise to cord-like structures in vitro [42]–[43], [55]–[58]. Recently, using early postnatal mouse testis cells, Yokonishi and colleagues were able to reconstruct well-organized tubular structures in vitro [18]. We show here that adult mouse seminiferous tubule cells have the same capacity and they spontaneously give rise to cord-like structures in vitro. Interestingly, formation of these structures was observed to be associated with stimulation of immature Sertoli cell markers (Keratin-18 and Podoplanin) suggesting that adult, mature Sertoli cells may have to dedifferentiate to be able to support the development of secondary structures in vitro. Schlatt and co-workers have demonstrated that only early steps of testis tissue regeneration can take place in vitro. A change in the environment that is gained by grafting spherical cell aggregates into an immunodeficient mouse is required to enable further morphogenesis [57]. However, fairly good level or organization can be achieved by optimizing the culture conditions [18].
We describe here a novel method to study the interaction of Sertoli cells, myoid cells and spermatogonia in vitro. Seminiferous tubule cells spontaneously gave rise to a co-culture that could be maintained for weeks. Based on relatively high levels of GDNF in the culture medium and a wide range of spermatogonia present in the culture during the first five weeks of culture, we propose that this model could be used to study the interaction of testis stem cell niche components in vitro. Formation of secondary structures takes place spontaneously and in a highly repetitive manner possibly enabling research on early steps of testis development and regeneration in vitro.
Materials and Methods
Ethics statement
Turku University Committee on the Ethics of Animal Experimentation approved all animal experiments (permission no. 2008-03959).
Establishing a two-dimensional co-culture of mouse seminiferous tubule cells
To establish a co-culture for mouse seminiferous tubule cells, we exploited a phenomenon described by Eddy and Kahri for rat seminiferous tubules [59]: spontaneous proliferation and migration of testicular cells out from small segments of seminiferous tubules in vitro. Male C57BL/6 and transgenic mice ubiquitously expressing eGFP [60]; purchased from The Jackson Laboratory (Bar Harbor, Maine, USA) and maintained in NMRI strain in Turku Center for Disease Modeling (TCDM, Turku, Finland)) were housed under environmentally controlled conditions (12 h light/12 h darkness; temperature, 21±1°C) at the Animal Centre of Turku University (Turku, Finland).
The mice were sacrificed by cervical dislocation under CO2 anaesthesia and the testes were collected. Under sterile conditions the testes were decapsulated and seminiferous tubules were dissected free from the interstitial tissue on a Petri dish in 15% inactivated fetal calf serum (iFCS; v/v) in DMEM/F12 (referred to hereafter as culture medium). Next the seminiferous tubules were cut into approximately 1-mm-long segments and 50–100 segments were moved onto cell culture plates with small amount of medium (100, 200 and 350 µl of suspension was plated onto 24-, 12-, and 6-wells plates, respectively). During 6–10 hours incubation [37°C, 5% (v/v) CO2, humidified atmosphere] the short fragments of seminiferous tubules attached on the bottom of the wells, and thereafter the amount of medium was increased. Culture medium was exchanged every two to three days. Originally we used seminiferous tubule segments from immature mice but noticed later that mice of different ages (from <10-day-old to 17-month-old) can be successfully used to establish the culture. The co-cultures were maintained for up to eight weeks as such and monitored by phase contrast microscopy, or samples were collected for experiments and analyses as described in the following sections.
Immunocytochemistry
For immunocytochemistry, the co-cultures were established on uncoated or laminin-coated (20 µg/ml; L2020, Sigma Chemical Co., St. Louis, Mo, USA) (for less than 20-day cultures) coverslips. After the culture the cells on a coverslip were fixed with cold 4% (v/v) PFA (paraformaldehyde) for 10 minutes and stored in cold PBS (phosphate-buffered saline, pH 7.4) or TBS (Tris-buffered saline, pH 7.55). Fixed cells were washed three times 5 min in PBS/TBS, and then permeabilized with 0.1% (v/v) Triton X-100 in PBS/TBS for 10 min followed by three washes with PBS/TBS for 5 min each. Before primary antibody treatment the specimens were incubated (1 h, RT) in 10% (v/v) normal serum (NS) or bovine serum albumin (BSA) in PBS/TBS. The specimens were then incubated with rabbit polyclonal anti-WT1 (1∶500; C-19, sc-846, Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA), polyclonal goat anti-Vimentin (1∶200; C-20, sc-7557, Santa Cruz), rabbit polyclonal anti-MAGE-B4 (1∶200; [24]), mouse monoclonal anti-α-smooth muscle actin (1∶500; A2547, Sigma), rabbit polyclonal anti-Nanog (1∶200; M-149, sc-33760, Santa Cruz), rabbit polyclonal antiphosphorylated-histone H3 (1∶500; #9701, Cell Signaling Technology, Danvers, MA, USA), goat polyclonal anti-PLZF (1∶500; AF2944, R&D Systems Inc., Minneapolis, MI, USA), goat polyclonal anti-LIN28 (1∶500; AF3757, R&D Systems), rabbit polyclonal anti-Ddx4 (1∶500; ab13840 Abcam, Cambridge, UK), mouse monoclonal anti-Ki-67 (1∶5000; M7240, DAKO, Glostrup, Denmark) or mouse monoclonal anti-Keratin-18 (1∶100; C-04, sc-51582, Santa Cruz) antibody overnight. The antibodies were diluted in 1% (v/v) NS/BSA in PBS/TBS. Negative controls lacked primary antibody.
After three washes with PBS/TBS 5 min each, the specimens were incubated with secondary antibody solution. The following secondary fluorescent antibodies were used: A11029, A11032, A11037, A11055, A11058, A21200, A31573 (diluted 1∶200 to 1∶2000 in 1% (v/v) NS/BSA in PBS/TBS; all purchased from Invitrogen, Carlsbad, CA, USA). After 1 hour of secondary antibody incubation (RT) the slides were washed three times with PBS/TBS and mounted with UltraCruz mounting medium (sc-24941, Santa Cruz Biotechnology Inc.). Images were captured using Olympus DP72 camera (Olympus Optical Co., Ltd, Tokyo, Japan) installed on Leica DMRBE microscope (Leica, Wetzlar, Germany), or Zeiss AxioImager M1 (Carl Zeiss MicroImaging GmbH, Heidelberg, Germany) coupled with a Zeiss AxioCam MR3 monochrome cooled-CCD camera and analyzed with Zen 2012 software. At least three parallel experiments were performed. For testis tissue sections, a protocol described previously was used [15] with slight modifications: for nuclear antigens the antigen retrieval step was performed in a pressure cooker (PickCell Laboratories, Amsterdam, the Netherlands).
FSH and GDNF treatments on one-week-old co-cultures
To study the effect of different factors on 1-week co-culture, they were treated with recombinant human FSH (Gonal F, Serono, Geneva, Switzerland) or recombinant mouse glial cell-line derived neurotrophic factor (PeproTech, London, UK). FSH and GDNF were added to 15% (v/v) iFCS in DMEM medium (final concentration 10 ng/ml for both) that was supplemented with MIX (1-methyl 3-isobutyl xanthine, 0.2 mmol/l; Aldrich Chemie, Steinheim, Germany) in the FSH experiment. Samples were collected 6, 12, 24, 48 and 72 hours after treatment by scraping off the cells, pelleting them by centrifugation (1200 rpm, RT, 5 min) and snap-freezing them in liquid nitrogen. Additionally, in the FSH experiment the medium was exchanged after 72 hours and samples were collected 24 hours later. Within three independent experiments each treatment was applied on three parallel samples.
Angiotensin II and LH/hCG analogue exposure to co-cultures
Angiotensin II has been shown to induce contraction of peritubular myoid cells in vitro [40]. We treated co-cultures at various time points with 100 nmol/l angiotensin II (A9525, Sigma) or vehicle (PBS) only and imaged and observed the co-culture by time-lapse imaging (one frame per 10 sec; see below for details).
LH/hCG analogue Pregnyl (Organon, Oss, the Netherlands) was administered in the culture medium of 2–3-week co-cultures at a final concentration of 10 ng/ml. Control samples got the vehicle only. Samples were collected 24 and 48 hours later (n = 4) and snap-frozen in liquid N2.
RNA isolation and cDNA synthesis
RNA was isolated from the original suspension that was to be plated (control sample), cell pellets collected 1–5 weeks after the initiation of the culture, or cell pellets collected at certain time points after different treatments specified in other paragraphs of Materials and Methods. Trisure reagent (Bioline, London, UK) was used for total RNA isolation according to the manufacturer's instructions. After isolation, RNA concentration was measured using a NanoDrop device (ND-1000; NanoDrop Technologies, Wilmington, DE, USA) and the RNA sample was run on agarose gel to confirm good quality of the isolated RNA (intact 28S and 18S ribosomal RNA bands). One microgram of RNA was processed further. Firstly, traces of contaminating genomic DNA were removed by treating the samples with DNase I (Invitrogen, Carlsbad, CA, USA). DyNAmo SYBR Green 2-step qRT-PCR Kit (Finnzymes, Espoo, Finland) was used for cDNA synthesis and 0.5 µg of template RNA was reverse-transcribed in a 20-µl-reaction with oligo(dT) primers while another 0.5 µg was used as a template in RT- reaction.
Whole transcriptome amplification
For samples of low RNA yield (clusters and cord-like structures) whole transcriptome amplification was performed using Qiagen Quantitect kit (207043; Qiagen, Hilden, Germany) according to the manufacturer's instructions. Complementary DNA obtained from the high yield reaction was diluted 1∶200 and used in qRT-PCR.
Real-time PCR
With the help of Primer 3 software (http://frodo.wi.mit.edu/) and mRNA sequence data available at NCBI and Ensembl databases, primers were designed to be located to different exonic sequences (Table 2). For Nanog the primers lie within one exon. However, RT-minus reactions that were run in parallel never gave a positive signal. Amplification of target cDNAs was done using CFX96 real-time PCR detection system device (Bio-Rad Laboratories Inc., Hercules, CA, USA) and the DyNAmo Flash SYBR green qPCR kit (F-415L; Finnzymes, Espoo, Finland) according to the manufacturers' instructions. Quantitative real-time PCR was performed under the following conditions: 95°C for 7 min followed by 40 cycles of 94°C for 1 s and 55–64°C (depending on the primer pair; see Table 2) for 15 s. Relative gene expression data was normalized to transcript levels of Ppia (cyclophilin A) and L19 (L19 ribosomal protein) using 2∧−ΔΔC(t) method [61]. PCR specificity was verified by gel electrophoresis and melting curve analysis, one band of the expected size and a single peak, respectively, were required.
Table 2. Primer design, annealing temperatures and PCR product lengths of the studied mRNAs.
| Gene | Accession number | Annealing temperature | Primers | Product length |
| αSMA | NM_007392 | 62°C | 5′-TGCTGTCCCTCTATGCCTCT-3′ 5′-GAAGGAATAGCCACGCTCAG-3′ | 184 bp |
| CD141 | NM_009378 | 55°C | 5′- TCTGCGAGCATTTTTGTGTC-3′ 5′-GCACTCTCCATCCACCAACT-3′ | 215 bp |
| CD9 | NM_007657 | 56°C | 5′-TGCAGTGCTTGCTATTGGAC-3′ 5′-GGCGAATATCACCAAGAGGA-3′ | 219 bp |
| Cdh5 | NM_009868 | 62°C | 5′-ATTGAGACAGACCCCAAACG-3′ 5′-TTCTGGTTTTCTGGCAGCTT-3′ | 238 bp |
| CIP2A | NM_172616 | 57°C | 5′-GCGCCATGTACTCAGTCAGA-3′ 5′-AGGAAGCAGAAGGGTCACAA-3′ | 234 bp |
| c-Kit | NM_021099 | 56°C | 5′-ATCCCGACTTTGTCAGATGG-3′ 5′-AAGGCCAACCAGGAAAAGTT-3′ | 192 bp |
| Erm | NM_023794 | 57°C | 5′-CAAGAGCCCCGAGATTACTG-3′ 5′-CTCGGGTACCACGCAAGTAT-3′ | 146 bp |
| Fsp1 | NM_011311 | 64°C | 5′-ACTCAGGCAAAGAGGGTGAC-3′ 5′-TGCAGGACAGGAAGACACAG-3′ | 188 bp |
| Gdnf | NM_010275 | 61°C | 5′-CGGACGGGACTCTAAGATGA-3′ 5′-CGTCATCAAACTGGTCAGGA-3′ | 209 bp |
| Gfr1α | NM_010279 | 63°C | 5′-TGTACTTCGTGCTGCCACTC-3′ 5′-GCTGAAGTTGGTTTCCTTGC-3′ | 166 bp |
| Gpr125 | XM_991709 | 59°C | 5′-GACCTGACGAACAACCGAAT-3′ 5′-CTGGTGTCTCGCACAGTGAT-3′ | 235 bp |
| Krt18 | NM_010664 | 55°C | 5′-CGAGGCACTCAAGGAAGAAC-3′ 5′-CTTGGTGGTGACAACTGTGG-3′ | 246 bp |
| L19 | NM_009078 | 55°C | 5′-GGACAGAGTCTTGATGATCTC-3′ 5′-CTGAAGGTCAAAGGGAATGTG-3′ | 195 bp |
| Nanog | NM_028016 | 64°C | 5′-CCAGTGGAGTATCCCAGCAT-3′ 5′-GAAGTTATGGAGCGGAGCAG-3′ | 236 bp |
| Pdpn | NM_010329 | 58°C | 5′-GCCAGTGTTGTTCTGGGTTT-3′ 5′-AGAGGTGCCTTGCCAGTAGA-3′ | 191 bp |
| PLZF | NM_001033324 | 59°C | 5′-AACGGTTCCTGGACAGTTG-3′ 5′-CCCACACAGCAGACAGAAGA-3′ | 172 bp |
| Ppia | NM_008907 | 63°C | 5′-CATCCTAAAGCATACAGGTCCTG-3′ 5′-TCCATGGCTTCCACAATGTT-3′ | 164 bp |
| Stra8 | NM_009292 | 57°C | 5′-ATGCAATGTTGCTGAAGTGC-3′ 5′-GGAAGCAGCCTTTCTCAATG-3′ | 161 bp |
| Sycp3 | NM_011517 | 60°C | 5′-AGCCAGTAACCAGAAAATTGAGC-3′ 5′-CCACTGCTGCAACACATTCATA-3′ | 106 bp |
| vWF | NM_011708 | 63°C | 5′-GGCAAGAGAATGAGCCTGTC-3′ 5′-AAGCCAAAGGTCTCACTGGA-3′ | 172 bp |
| WT1 | NM_144783 | 64°C | 5′-AGGTTTTCTCGCTCAGACCA-3′ 5′-GCTGAAGGGCTTTTCACTTG-3′ | 160 bp |
GDNF enzyme-linked immunosorbent assay (ELISA) and testosterone radioimmunoassay
Culture medium samples were collected on a weekly basis and stored at −80°C. GDNF concentration in the medium was determined using a mouse glial cell-line derived neurotrophic factor ELISA kit (CSB-EL009356MO, Cusabio Biotech Co., Ltd, Wuhan, China) according to the manufacturer's instructions. Testosterone levels in the culture media were measured using Spectria testosterone RIA (radio-immunoassay; Orion Diagnostica, Espoo, Finland).
Flow cytometry (FACS)
Cells were scraped off the bottom of the plates into a small volume of medium, and moved to 4 ml of prewarmed culture medium containing collagenase/dispase (0,5 mg/ml; Roche, Indianapolis, IN, USA), collagen IV (0,5 mg/ml; Worthington Biochemical, Freehold, NJ, USA), hyaluronidase (1 mg/ml; Sigma) and DNase I (0,04 mg/ml; Sigma). The samples were incubated at 37°C for 25 min in horizontal rotation. Thereafter cells were pelleted by centrifugation (1500 rpm, 5 min) and the supernatant was removed. The cells were resuspended in lysis buffer (PBS+2% Triton X-100) containing propidium iodide (5 µg/ml; P4864, Sigma) and incubated 15 min at +4°C in horizontal rotation. Thereafter the nuclei were pelleted and resuspended to 500 µl of PBS. The solution was passed through 100 µm cell strainer and analysed by FACS. Determination and analysis of the cell populations in the suspension were done using FACSCalibur flow cytometer, Cell Quest software (BD Biosciences, Mansfield, MA, USA) and Flowing-Software Version 2.5.0 (Turku Center for Biotechnology, Turku, Finland).
Transplantation of cells to rete testis
Adult C57BL/6 mice were rendered sterile by treating them with busulfan (40 mg/kg, i.p.). One-month after the injection approximately 100 000 cells isolated from 1-month-old adult ubiquitously GFP-expressing mouse seminiferous tubule fragment-derived cultures were transplanted via the rete testis according to a protocol described previously [36]. Single cell suspension for this purpose was prepared as described above for FACS. Five weeks post-operatively the testes were dissected and seminiferous tubules were imaged with a Zeiss SteREO Lumar V12 microscope with eGFP bandpass filter (Carl Zeiss MicroImaging, Thornwood, NY, USA).
Time-lapse imaging
During live-cell imaging the samples were maintained in an incubator at 37°C and 5% CO2, humidified atmosphere. Frames were captured at defined intervals (5 sec to 30 min) using Olympus DBH1 camera attached to a Olympus IX71 microscope with 10X phase-contrast objective. All images were acquired digitally using Cell R software (Olympus).
Statistical analysis
The results were analyzed for statistically significant differences using one-way analysis of variance, followed by Tukey's test for multiple comparisons of independent groups of samples. An independent samples t-test was used for statistical analyses of pair-wise comparisons. The p values less than 0.05 were considered statistically significant.
Supporting Information
Validation of the used antibodies. Formalin-fixed, paraffin-embedded adult mouse testis tissue sections were stained with the same antibodies that were also used for co-cultures. Positive control staining for A) αSMA (red), B) MAGE-B4 (red), C) WT1 (red) and D) Vimentin (red). DAPI stains the nuclei blue. Insets represent negative control stainings.
(TIF)
Nanog staining for ESCs. To validate the functionality of the used Nanog antibody, we stained mouse embryonic stem cells side-by-side with sc-33760 (top panel) and ab80892 (middle panel) (Abcam Inc., rabbit polyclonal anti-mouse Nanog antibody). The antibodies gave identical result. Negative control staining is in the bottom panel. M, murine feeder cell (mouse embryonic fibroblast). Scale bars: top and middle panel 10 µm, bottom panel 20 µm.
(TIF)
Proliferation of germ cells in co-cultures. Double-immunofluorescent staining for 1-week-old co-culture showed that some Ddx4 (green) positive germ cells also expressed proliferating cell antigen Ki-67 (red). DAPI stains nuclei blue. Scale bar 20 µm.
(TIF)
FSH treatment indirectly affected mRNA levels of spermatogonial markers. Steady state levels of Gfr1α mRNAs were acutely increased by FSH treatment in co-cultures established from A) adult and B) juvenile mouse seminiferous tubules. C) FSH did not consistently affect Stra8 levels in adult-derived co-cultures, D) whereas they were uniformly upregulated by the treatment in juvenile-derived co-cultures. FSH elevated E) c-Kit, F) Gpr125 and G) PLZF mRNA levels in 1-week adult-derived co-cultures. White bars, control; black bars, 10 ng/ml rhFSH; n = 3, SEM; *, p<0.05; **, p<0.01; *** p<0.001.
(TIF)
Haematoxylin-eosin-stained cross-section of a cluster that exhibits relatively high degree of bilateral symmetry. Clusters that had a diameter of 1–2 mm were only partially connected to the underlying co-culture and moved back and forth when the medium was exchanged. These structures were slightly disorganized but occasionally displayed relatively high level of symmetry.
(TIF)
Immunocytochemical staining for 4-week co-culture showing the presence of Vimentin (green) and Keratin-18 (red) positive cells side-by-side. Vimentin and Keratin-18 only colocalize (orange) at areas where the cells are in a physical contact. DAPI stains the nuclei of cells (blue).
(TIF)
Eight-week follow-up of co-culture. One frame per every 1–4 days.
(MOV)
Dynamic nature of confluent co-cultures. One frame per 20 minutes; total 45 hours.
(MOV)
Formation of a cluster by attraction of cells at a near distance to a seminiferous tubule remnant.
(MOV)
Formation of a cluster by coalescence of seemingly homogenous matrix of cells taking place in the lower right corner.
(MOV)
Angiotensin II-induced contraction of cells in the co-culture followed 5 minutes after the exposure. One frame per 5 seconds; total 5 min.
(MOV)
Vehicle-treated (PBS) control co-culture that is followed three minutes after addition of vehicle. One frame per 5 seconds; total 3 min.
(MOV)
Long-term follow-up of cord-like structure formation. One frame per every 1–4 days.
(MOV)
Short-term follow-up of cord-like structure formation. One frame per 12 minutes; total 29 hours.
(MOV)
Acknowledgments
We thank Matteo Da Ros, MSc, Sheyla Estefani Cisneros Montalvo, BSc, Ms Taina Kirjonen and Ms Erica Nyman for skilful technical assistance, Pia Rantakari, PhD, for help with transgenic animals, and Robin Hobbs, PhD for providing us with PH3, PLZF and LIN28 antibodies.
Funding Statement
This work was supported by grants from Sigrid Jusélius Foundation (http://www.sigridjuselius.fi), Emil Aaltonen's Foundation (http://www.emilaaltonen.fi), Finnish Cultural Foundation (www.skr.fi), The Finnish Medical Foundation (http://www.laaketieteensaatio.fi), The Academy of Finland (www.aka.fi) and Turku University Hospital. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Brinster R, Avarbock M (1994) Germline transmission of donor haplotype following spermatogonial transplantation. Proc Natl Acad Sci U S A 91: 11303–11307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Brinster R, Zimmermann J (1994) Spermatogenesis following male germ-cell transplantation. Proc Natl Acad Sci U S A 91: 11298–11302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kanatsu-Shinohara M, Inoue K, Lee J, Yoshimoto M, Ogonuki N, et al. (2004) Generation of pluripotent stem cells from neonatal mouse testis. Cell 119: 1001–1012. [DOI] [PubMed] [Google Scholar]
- 4. Guan K, Nayernia K, Maier L, Wagner S, Dressel R, et al. (2006) Pluripotency of spermatogonial stem cells from adult mouse testis. Nature 440: 1199–1203. [DOI] [PubMed] [Google Scholar]
- 5. Kossack N, Meneses J, Shefi S, Nguyen HN, Chavez S, et al. (2009) Isolation and characterization of pluripotent human spermatogonial stem cell-derived cells. Stem Cells 27: 138–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kanatsu-Shinohara M, Miki H, Inoue K, Ogonuki N, Toyokuni S, et al. (2005) Long-term culture of mouse male germline stem cells under serum-or feeder-free conditions. Biol Reprod 72: 985–991. [DOI] [PubMed] [Google Scholar]
- 7. Chiarini-Garcia H, Hornick JR, Griswold MD, Russell LD (2001) Distribution of type A spermatogonia in the mouse is not random. Biol Reprod 65: 1179–1185. [DOI] [PubMed] [Google Scholar]
- 8. Chiarini-Garcia H, Raymer AM, Russell LD (2003) Non-random distribution of spermatogonia in rats: evidence of niches in the seminiferous tubules. Reproduction 126: 669–680. [DOI] [PubMed] [Google Scholar]
- 9. Chiarini-Garcia H, Russell LD (2001) High-resolution light microscopic characterization of mouse spermatogonia. Biol Reprod 65: 1170–1178. [DOI] [PubMed] [Google Scholar]
- 10. Yoshida S, Sukeno M, Nabeshima Y (2007) A vasculature-associated niche for undifferentiated spermatogonia in the mouse testis. Science 317: 1722–1726. [DOI] [PubMed] [Google Scholar]
- 11. Ventelä S, Mäkelä JA, Kulmala J, Westermarck J, Toppari J (2012) Identification and regulation of a stage-specific stem cell niche enriched by nanog positive spermatogonial stem cells in the mouse testis. Stem Cells 30: 1008–1020. [DOI] [PubMed] [Google Scholar]
- 12. Toppari J, Eerola E, Parvinen M (1985) Flow cytometric DNA analysis of defined stages of rat seminiferous epithelial cycle during in vitro differentiation. J Androl 6: 325–333. [DOI] [PubMed] [Google Scholar]
- 13. Toppari J, Parvinen M (1985) In vitro differentiation of rat seminiferous tubular segments from defined stages of the epithelial cycle morphologic and immunolocalization analysis. J Androl 6: 334–343. [DOI] [PubMed] [Google Scholar]
- 14. Toppari J, Vihko KK, Räsänen KG, Eerola E, Parvinen M (1985) Regulation of stages VI and VIII of the rat seminiferous epithelial cycle in vitro. J Endocrinol 108: 417–422. [DOI] [PubMed] [Google Scholar]
- 15. Mäkelä JA, Saario V, Bourguiba-Hachemi S, Nurmio M, Jahnukainen K, et al. (2011) Hedgehog signalling promotes germ cell survival in the rat testis. Reproduction 142: 711–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sato T, Katagiri K, Gohbara A, Inoue K, Ogonuki N, et al. (2011) In vitro production of functional sperm in cultured neonatal mouse testes. Nature 471: 504–507. [DOI] [PubMed] [Google Scholar]
- 17. Sato T, Katagiri K, Kubota Y, Ogawa T (2013) In vitro sperm production from mouse spermatogonial stem cell lines using an organ culture method. Nat Protoc 8: 2098–2104. [DOI] [PubMed] [Google Scholar]
- 18. Yokonishi T, Sato T, Katagiri K, Komeya M, Kubota Y, et al. (2013) In Vitro Reconstruction of Mouse Seminiferous Tubules Supporting Germ Cell Differentiation. Biol Reprod 89: 1–6. [DOI] [PubMed] [Google Scholar]
- 19. Buaas F, Kirsh A, Sharma M, McLean D, Morris J, et al. (2004) Plzf is required in adult male germ cells for stem cell self-renewal. Nat Genet 36: 647–652. [DOI] [PubMed] [Google Scholar]
- 20. Costoya J, Hobbs R, Barna M, Cattoretti G, Manova K, et al. (2004) Essential role of Plzf in maintenance of spermatogonial stem cells. Nat Genet 36: 653–659. [DOI] [PubMed] [Google Scholar]
- 21. Zheng K, Wu X, Kaestner KH, Wang PJ (2009) The pluripotency factor LIN28 marks undifferentiated spermatogonia in mouse. BMC Dev Biol 9: 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Armstrong J, Pritchard-Jones K, Bickmore W, Hastie N, Bard J (1993) The expression of the wilms-tumor gene, wt1, in the developing mammalian embryo. Mech Dev 40: 85–97. [DOI] [PubMed] [Google Scholar]
- 23. Tung P, Fritz I (1990) Characterization of rat testicular peritubular myoid cells in culture - alpha-smooth muscle isoactin is a specific differentiation marker. Biol Reprod 42: 351–365. [DOI] [PubMed] [Google Scholar]
- 24. Österlund C, Töhönen V, Forslund KO, Nordqvist K (2000) Mage-b4, a novel melanoma antigen (MAGE) gene specifically expressed during germ cell differentiation. Cancer Res 15: 1054–1061. [PubMed] [Google Scholar]
- 25. Kanatsu-Shinohara M, Toyokuni S, Shinohara T (2004) CD9 is a surface marker on mouse and rat male germline stem cells. Biol Reprod 70: 70–75. [DOI] [PubMed] [Google Scholar]
- 26. Sorrentino V, Giorgi M, Geremia R, Besmer P, Rossi P (1991) Expression of the C-Kit Protooncogene in the Murine Male Germ-Cells. Oncogene 6: 149–151. [PubMed] [Google Scholar]
- 27. Chen C, Ouyang W, Grigura V, Zhou Q, Carnes K, et al. (2005) ERM is required for transcriptional control of the spermatogonial stem cell niche. Nature 436: 1030–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Oatley JM, Avarbock MR, Brinster RL (2007) Glial cell line-derived neurotrophic factor regulation of genes essential for self-renewal of mouse spermatogonial stem cells is dependent on src family kinase signaling. J Biol Chem 282: 25842–25851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Seandel M, James D, Shmelkov SV, Falciatori I, Kim J, et al. (2007) Generation of functional multipotent adult stem cells from GPR125(+) germline progenitors. Nature 449: 346–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Giuili G, Tomljenovic A, Labrecque N, Oulad-Abdelghani M, Rassoulzadegan M, et al. (2002) Murine spermatogonial stem cells: targeted transgene expression and purification in an active state. EMBO Rep 3: 753–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhou Q, Nie R, Li Y, Friel P, Mitchell D, et al. (2008) Expression of stimulated by retinoic acid gene 8 (Stra8) in spermatogenic cells induced by retinoic acid: An in vivo study in vitamin A-sufficient postnatal murine testes. Biol Reprod 79: 35–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. La Salle S, Sun F, Handel MA (2009) Isolation and short-term culture of mouse spermatocytes for analysis of meiosis. Methods Mol Biol 558: 279–297. [DOI] [PubMed] [Google Scholar]
- 33. Zhang FP, Pakarainen T, Poutanen M, Toppari J, Huhtaniemi I (2003) The low gonadotropin-independent constitutive production of testicular testosterone is sufficient to maintain spermatogenesis. Proc Natl Acad Sci U S A 100: 13692–13697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Castrillon DH, Quade BJ, Wang TY, Quigley C, Crum CP (2000) The human VASA gene is specifically expressed in the germ cell lineage. Proc Natl Acad Sci U S A 97: 9585–9590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bucci LR, Meistrich ML (1987) Effects of busulfan on murine spermatogenesis: cytotoxicity, sterility, sperm abnormalities, and dominant lethal mutations. Mutat Res 176: 259–268. [DOI] [PubMed] [Google Scholar]
- 36. Ogawa T, Aréchaga JM, Avarbock MR, Brinster RL (1997) Transplantation of testis germinal cells into mouse seminiferous tubules. Int J Dev Biol 41: 111–122. [PubMed] [Google Scholar]
- 37. Meng X, Lindahl M, Hyvonen M, Parvinen M, de Rooij D, et al. (2000) Regulation of cell fate decision of undifferentiated spermatogonia by GDNF. Science 287: 1489–1493. [DOI] [PubMed] [Google Scholar]
- 38. Junttila MR, Puustinen P, Niemela M, Ahola R, Arnold H, et al. (2007) CIP2A inhibits PP2A in human malignancies. Cell 130: 51–62. [DOI] [PubMed] [Google Scholar]
- 39. Ventelä S, Côme C, Mäkelä JA, Hobbs RM, Mannermaa L, et al. (2012) CIP2A promotes proliferation of spermatogonial progenitor cells and spermatogenesis in mice. PLoS One 7: e33209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Rossi F, Ferraresi A, Romagni P, Silvestroni L, Santiemma V (2002) Angiotensin II stimulates contraction and growth of testicular peritubular myoid cells in vitro. Endocrinology 143: 3096–3104. [DOI] [PubMed] [Google Scholar]
- 41. Strutz F, Okada H, Lo CW, Danoff T, Carone RL, et al. (1995) Identification and characterization of a fibroblast marker: FSP1. J Cell Biol 130: 393–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Skinner MK, Moses HL (1989) Transforming growth factor b gene expression and action in the seminiferous tubule: Peritubular cell-Sertoli interactions. Mol Endocrinol 3: 625–634. [DOI] [PubMed] [Google Scholar]
- 43. Tung PS, Fritz IB (1980) Interactions of sertoli cells with myoid cells in vitro. Biol Reprod 23: 207–217. [DOI] [PubMed] [Google Scholar]
- 44. Rahimi N, Kazlauskas A (1999) A role for Cadherin-5 in regulation of vascular endothelial growth factor receptor 2 activity in endothelial cells. Mol Biol Cell 10: 3401–3407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Maruyama I, Bell CE, Majerus PW (1985) Thrombomodulin is found on endothelium of arteries, veins, capillaries, and lymphatics, and on syncytiotrophoblast of human placenta. J Cell Biol 101: 363–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Yamamoto K, de Waard V, Fearns C, Loskutoff DJ (1998) Tissue distribution and regulation of murine von Willebrand factor gene expression in vivo. Blood 92: 2791–2801. [PubMed] [Google Scholar]
- 47. Steger K, Rey R, Kliesch S, Louis F, Schleicher G, et al. (1996) Immunohistochemical detection of immature Sertoli cell markers in testicular tissue of infertile adult men: a preliminary study. Int J Androl 19: 122–128. [DOI] [PubMed] [Google Scholar]
- 48. Sonne SB, Herlihy AS, Hoei-Hansen CE, Nielsen JE, Almstrup K, et al. (2006) Identity of M2A (D2-40) antigen and gp36 (Aggrus, T1A-2, podoplanin) in human developing testis, testicular carcinoma in situ and germ-cell tumours. Virchows Arch 449: 200–206. [DOI] [PubMed] [Google Scholar]
- 49. Kanatsu-Shinohara M, Ogonuki N, Inoue K, Miki H, Ogura A, et al. (2003) Long-term proliferation in culture and germline transmission of mouse male germline stem cells. Biol Reprod 69: 612–616. [DOI] [PubMed] [Google Scholar]
- 50. Kubota H, Avarbock M, Brinster R (2004) Growth factors essential for self-renewal and expansion of mouse spermatogonial stem cells. Proc Natl Acad Sci U S A 101: 16489–16494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Tadokoro Y, Yomogida K, Ohta H, Tohda A, Nishimune Y (2002) Homeostatic regulation of germinal stem cell proliferation by the GDNF/FSH pathway. Mech Dev 113: 29–39. [DOI] [PubMed] [Google Scholar]
- 52. Meachem S, Ruwanpura S, Ziolkowski J, Ague J, Skinner M, et al. (2005) Developmentally distinct in vivo effects of FSH on proliferation and apoptosis during testis maturation. J Endocrinol 186: 429–446. [DOI] [PubMed] [Google Scholar]
- 53. Bellve A, Cavicchia J, Millette C, Obrien D, Bhatnagar Y, et al. (1977) Spermatogenic cells of prepuberal mouse - isolation and morphological characterization. J Cell Biol 74: 68–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Griswold MD (1998) The central role of Sertoli cells in spermatogenesis. Semin Cell Dev Biol 9: 411–416. [DOI] [PubMed] [Google Scholar]
- 55. Zenzes MT, Engel W (1981) The capacity of testicular cells of the postnatal rat to reorganize into histotypic structures. Differentiation 20: 157–161. [DOI] [PubMed] [Google Scholar]
- 56. Hadley MA, Weeks BS, Kleinman HK, Dym M (1990) Laminin promotes formation of cord-like structures by Sertoli cells in vitro. Dev Biol 140: 318–327. [DOI] [PubMed] [Google Scholar]
- 57. Gassei K, Schlatt S, Ehmcke J (2006) De novo morphogenesis of seminiferous tubules from dissociated immature rat testicular cells in xenografts. J Androl 27: 611–618. [DOI] [PubMed] [Google Scholar]
- 58. Pan F, Chi L, Schlatt S (2013) Effects of nanostructures and mouse embryonic stem cells on in vitro morphogenesis of rat testicular cords. PLoS One 8: e60054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Eddy E, Kahri A (1976) Cell associations and surface features in cultures of juvenile rat seminiferous tubules. Anat Rec 185: 333–358. [DOI] [PubMed] [Google Scholar]
- 60. Okabe M, Ikawa M, Kominami K, Nakanishi T, Nishimune Y (1997) ‘Green mice’ as a source of ubiquitous green cells. FEBS Lett 407: 313–319. [DOI] [PubMed] [Google Scholar]
- 61. Livak K, Schmittgen T (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2∧(−Delta Delta C(t)) method. Methods 25: 402–408. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Validation of the used antibodies. Formalin-fixed, paraffin-embedded adult mouse testis tissue sections were stained with the same antibodies that were also used for co-cultures. Positive control staining for A) αSMA (red), B) MAGE-B4 (red), C) WT1 (red) and D) Vimentin (red). DAPI stains the nuclei blue. Insets represent negative control stainings.
(TIF)
Nanog staining for ESCs. To validate the functionality of the used Nanog antibody, we stained mouse embryonic stem cells side-by-side with sc-33760 (top panel) and ab80892 (middle panel) (Abcam Inc., rabbit polyclonal anti-mouse Nanog antibody). The antibodies gave identical result. Negative control staining is in the bottom panel. M, murine feeder cell (mouse embryonic fibroblast). Scale bars: top and middle panel 10 µm, bottom panel 20 µm.
(TIF)
Proliferation of germ cells in co-cultures. Double-immunofluorescent staining for 1-week-old co-culture showed that some Ddx4 (green) positive germ cells also expressed proliferating cell antigen Ki-67 (red). DAPI stains nuclei blue. Scale bar 20 µm.
(TIF)
FSH treatment indirectly affected mRNA levels of spermatogonial markers. Steady state levels of Gfr1α mRNAs were acutely increased by FSH treatment in co-cultures established from A) adult and B) juvenile mouse seminiferous tubules. C) FSH did not consistently affect Stra8 levels in adult-derived co-cultures, D) whereas they were uniformly upregulated by the treatment in juvenile-derived co-cultures. FSH elevated E) c-Kit, F) Gpr125 and G) PLZF mRNA levels in 1-week adult-derived co-cultures. White bars, control; black bars, 10 ng/ml rhFSH; n = 3, SEM; *, p<0.05; **, p<0.01; *** p<0.001.
(TIF)
Haematoxylin-eosin-stained cross-section of a cluster that exhibits relatively high degree of bilateral symmetry. Clusters that had a diameter of 1–2 mm were only partially connected to the underlying co-culture and moved back and forth when the medium was exchanged. These structures were slightly disorganized but occasionally displayed relatively high level of symmetry.
(TIF)
Immunocytochemical staining for 4-week co-culture showing the presence of Vimentin (green) and Keratin-18 (red) positive cells side-by-side. Vimentin and Keratin-18 only colocalize (orange) at areas where the cells are in a physical contact. DAPI stains the nuclei of cells (blue).
(TIF)
Eight-week follow-up of co-culture. One frame per every 1–4 days.
(MOV)
Dynamic nature of confluent co-cultures. One frame per 20 minutes; total 45 hours.
(MOV)
Formation of a cluster by attraction of cells at a near distance to a seminiferous tubule remnant.
(MOV)
Formation of a cluster by coalescence of seemingly homogenous matrix of cells taking place in the lower right corner.
(MOV)
Angiotensin II-induced contraction of cells in the co-culture followed 5 minutes after the exposure. One frame per 5 seconds; total 5 min.
(MOV)
Vehicle-treated (PBS) control co-culture that is followed three minutes after addition of vehicle. One frame per 5 seconds; total 3 min.
(MOV)
Long-term follow-up of cord-like structure formation. One frame per every 1–4 days.
(MOV)
Short-term follow-up of cord-like structure formation. One frame per 12 minutes; total 29 hours.
(MOV)