

Bacteriophages hijack their host's transcription machinery, directing the host's RNA polymerase (RNAP) to switch promoter specificity from host genes to phage genes. To elucidate the structural basis of promoter specificity switching by the phage-encoded protein gp39, Tagami et al. solve the crystal structure of Thermus thermophilus RNAP holoenzyme bound to gp39. Biochemical analyses reveal a novel mechanism by which a phage-encoded protein switches the RNAP promoter specificity in favor of phage development without suppressing global transcription.
Keywords: bacteriophage, RNA polymerase, transcription factor, transcription control, X-ray crystallography
Abstract
Transcription of DNA to RNA by DNA-dependent RNA polymerase (RNAP) is the first step of gene expression and a major regulation point. Bacteriophages hijack their host's transcription machinery and direct it to serve their needs. The gp39 protein encoded by Thermus thermophilus phage P23-45 binds the host's RNAP and inhibits transcription initiation from its major “−10/−35” class promoters. Phage promoters belonging to the minor “extended −10” class are minimally inhibited. We report the crystal structure of the T. thermophilus RNAP holoenzyme complexed with gp39, which explains the mechanism for RNAP promoter specificity switching. gp39 simultaneously binds to the RNAP β-flap domain and the C-terminal domain of the σ subunit (region 4 of the σ subunit [σ4]), thus relocating the β-flap tip and σ4. The ∼45 Å displacement of σ4 is incompatible with its binding to the −35 promoter consensus element, thus accounting for the inhibition of transcription from −10/−35 class promoters. In contrast, this conformational change is compatible with the recognition of extended −10 class promoters. These results provide the structural bases for the conformational modulation of the host's RNAP promoter specificity to switch gene expression toward supporting phage development for gp39 and, potentially, other phage proteins, such as T4 AsiA.
Transcription of DNA to RNA is the first step of gene expression and is accomplished by DNA-dependent RNA polymerase (RNAP). The bacterial RNAP core enzyme is an ∼400-kDa protein complex with a crab claw-like shape consisting of five subunits, α2ββ′ω (Zhang et al. 1999). For transcription initiation, the RNAP core enzyme binds one of several promoter specificity σ subunits to form the holoenzyme α2ββ′ωσ (Murakami et al. 2002b; Vassylyev et al. 2002). Typically, the holoenzyme recognizes promoters through specific interactions between two DNA-binding domains in σ—σ2 and σ4 (regions 2 and 4 of the σ subunit)—and consensus promoter elements around the −10 and −35 positions, respectively, relative to the transcription start site (Fig. 1A; Campbell et al. 2002; Murakami et al. 2002a; Young et al. 2002; Feklistov and Darst 2011; Zhang et al. 2012). The interaction between σ and the −10/−35 promoter elements is essential for the subsequent formation of the open promoter complex in which the dsDNA is melted around positions −12 ∼ +1, and the template DNA strand is loaded into the RNAP nucleic acid-binding channel to initiate transcription.
Figure 1.

The structure of the RNAP holoenzyme•gp39 complex. (A) Schematic depictions of the −10/−35 and extended −10 promoters and the effect of gp39 on transcription. (B–D) Overall structure of the T. thermophilus RNAP holoenzyme•gp39 complex (holo•gp39) in three orientations. (E) Close-up view of the β-flap domain, σ factor, and gp39.
In addition to the major −10/−35 class promoters, there are the minor “extended −10” class promoters, which have the extended −10 element (the −10 element plus a TG motif located immediately upstream) but lack a discernible −35 promoter element (Fig. 1A; Mitchell et al. 2003). RNAP seems to select these promoters through extensive interactions between the σ2 domain and the extended −10 element and does not depend on the σ4 domain.
Transcription initiation is the major point of gene regulation by cellular factors. Bacteriophages employ their own proteins to appropriate the host's transcription system and direct it to serve their needs. Some of these proteins interact directly with the host's RNAP and switch its promoter specificity to modulate the entire transcriptome in favor of phage development (Nechaev and Severinov 2003). The σ4 domain and its binding site, the β-flap domain of the RNAP core enzyme, are among the preferred targets for phage transcription regulators (Dove et al. 2003; Lambert et al. 2004; Nechaev et al. 2004; Baxter et al. 2006; Yuan et al. 2009; Twist et al. 2011; Osmundson et al. 2012). These regulators directly bind to the interface of either σ–DNA, RNAP–σ, or RNAP–DNA and thus interfere with the RNAP–DNA interaction.
In Thermus thermophilus bacteriophage P23-45, the middle gene product, gp39, is a key factor that probably controls the switching of transcription from the host T. thermophilus to the phage genes (Minakhin et al. 2008; Berdygulova et al. 2011, 2012). This ∼16-kDa protein binds to the β-flap domain of the T. thermophilus RNAP holoenzyme and strongly inhibits transcription initiation from the −10/−35 class promoters. In contrast, gp39 has only a minor effect on transcription from the P23-45 middle/late promoters, which belong to the extended −10 class (Fig. 1A; Minakhin et al. 2008; Berdygulova et al. 2011). To elucidate the structural basis of the promoter specificity switching by the phage-encoded protein, we solved the crystal structure of the T. thermophilus RNAP holoenzyme bound to gp39 and performed structure-based biochemical analyses. Unexpectedly, our study revealed that gp39 switches the promoter specificity by modifying the σ4 orientation of RNAP without competitively dissociating the entire σ factor from RNAP or blocking σ4 binding to DNA.
Results
Structure determination
We determined the crystal structure of gp39 bound to the T. thermophilus RNAP holoenzyme (holo•gp39) at 3.6 Å resolution (Fig. 1B–E; Table 1). We also obtained single wavelength anomalous dispersion (SAD) data from crystals of the holoenzyme complex with selenomethionine (SeMet)-containing gp39 and precisely located the methionine residues in gp39 based on the Se anomalous peaks (Fig. 2A). Consistent with previous biochemical data (Berdygulova et al. 2012), the gp39 molecule primarily contacted the RNAP β-flap domain. Another gp39 molecule was present in the crystalline asymmetric unit, but its interaction with RNAP is probably functionally insignificant (Supplemental Fig. 1). Therefore, we focused on the first gp39 molecule, which directly binds the β flap. We also determined higher-resolution crystal structures of the gp39 variants bound to the β-flap domain fragment of RNAP (β residues 703–830) (Fig. 2B–D), which agree well with the interaction between the β flap and the first gp39 molecule in holo•gp39. For the σA subunit, models were built for the σ2 and σ4 domains and the σ3–4 linker but not for the remaining parts because of missing electron density. Significant conformational changes were observed in the RNAP structure, as described below (and in Supplemental Fig. 2).
Table 1.
Data collection and refinement statistics
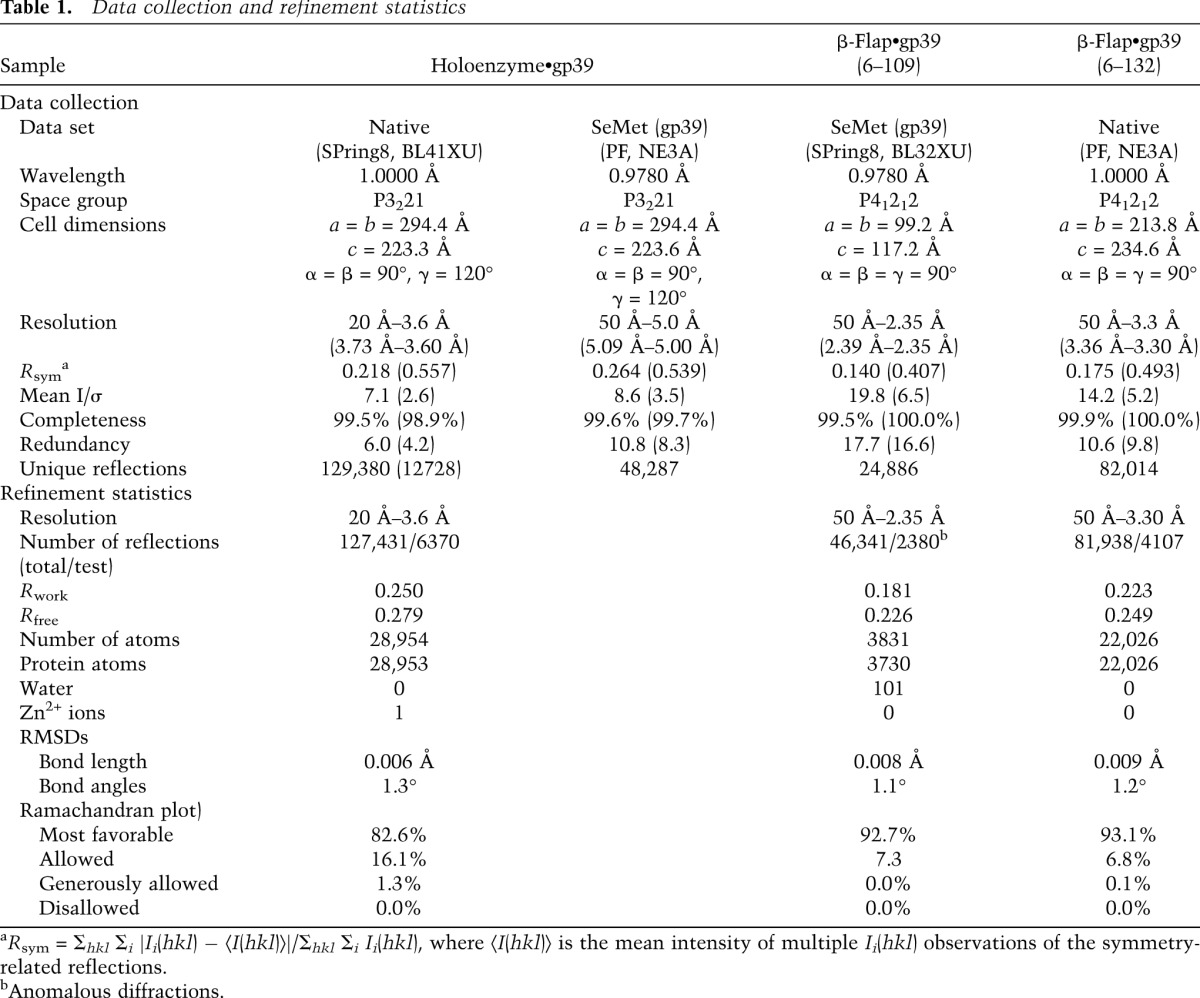
Figure 2.

The gp39 structure. (A) The holoenzyme-bound gp39. The N-terminal core domain (gp39 core) is colored blue, and the gp39 C-terminal tail is colored red. The anomalous difference map calculated with the Se SAD data is shown as a green mesh. (B) The structure of the complex between a truncated gp39 variant (residues 6–109) and the RNAP β-flap domain fragment (β residues 703–830). (C) The structure of the complex between another gp39 variant (residues 6–132) and the β-flap domain fragment, with the 2Fo − Fc electron density map contoured at 1.0σ. (D) The same structure as in B, overlaid with the 2Fo − Fc electron density map contoured at 1.0σ.
The gp39 structure and its binding mode to the RNAP β-flap domain
The gp39 structure consists of the gp39 core (residues 1–107) and the C-terminal tail (residues 119–141), which are connected by a linker (residues 108–118, mostly disordered) (Figs. 1D,E, 2A). The gp39 core comprises the central β sheet and the N-terminal α helix (Fig. 2) and binds primarily to the RNAP β-flap domain. A small protuberance from the RNAP flap β sheet (flap protuberance, residues 723–740) is the major gp39 core-binding site, and the interaction relies on shape complementarity and extensive hydrophobic/hydrophilic interactions (Fig. 3A–D). In gp39, Asp94, Asn95, Ile97, and His99 form a hydrogen-bonding network with Arg721, Thr723, and Asp725 in the flap (corresponding to Glu849, Thr851, and Asp853, respectively, in the Escherichia coli RNAP β subunit). The tip of the gp39 β sheet (residues 59–63) and the loop following the N-terminal helix (residues 16–21) wrap the loop of the β-flap domain (β737–740), where Arg61 in gp39 forms a salt bridge with Glu739 (E. coli Glu867) in the β flap. Bacterial two-hybrid experiments confirmed the gp39•β-flap interaction (Supplemental Fig. 3). The replacement of Asp725 in the β flap, at the middle of the interface with gp39, with a bulky Trp residue (β D725W) abolished the interaction. The reciprocal double substitution of Trp and Phe for Asp94 and Asn95, respectively, in gp39 (D94W/N95F), which would disrupt their interactions with Arg721, Thr723, and Asp725 in the β flap, also strongly impaired the interaction.
Figure 3.

The interaction between RNAP holoenzyme and gp39. (A) The gp39, β flap, and σ factor structures in the holo•gp39 complex. (B–D) Detailed views of the molecular interactions. (E) Analysis of gp39 interactions with Eco-tthflap RNAP (wild type or β D725W mutant). (Left panel) The Eco-tthflap RNAP holoenzymes containing E. coli σ70 were mixed with gp39 and resolved by native PAGE. (Right panel) The bands containing RNAP were excised from the gel and analyzed by denaturing SDS-PAGE. (Lane 3) The Eco-tthflap RNAP containing the D725W mutation migrated as a doublet on the native gel. Although the reason for the band separation remains unknown, the upper and lower band fractions both contain stoichiometric amounts of the RNAP subunits and lack gp39 (Supplemental Fig. 5). The D725W variant Eco-tthflap RNAP exhibits transcription activity comparable with that of the wild type (as shown in G). (F) Promoters used in this study. (P68M) Middle promoter of phage P23-45 belonging to the extended −10 class; (T5 N25) A model −10/−35 promoter. The positions of the −35, −10, and TG (extended −10) elements and the starting point of transcription (+1) are shown. (G) Effects of gp39 on the Eco-tthflap RNAP activity on −10/−35 (T5 N25) and extended −10 (P23-45 P68M) promoters. The position of the 3-nucleotide abortive transcription products is indicated.
To determine whether gp39 binding to the β-flap domain is functionally relevant, we analyzed the transcription inhibition of an E. coli RNAP variant on the −10/−35 and extended −10 class promoters (Fig. 3F). The wild-type E. coli RNAP is naturally resistant to gp39 due to the inability of gp39 to bind to the RNAP. However, the phage protein efficiently bound to the E. coli RNAP mutant bearing the T. thermophilus β flap instead of the E. coli β flap (hybrid Eco-tthflap) (Fig. 3E, lanes 2,2′). Similarly to T. thermophilus RNAP, gp39 inhibited transcription by the hybrid RNAP on the −10/−35 promoters but not on the extended −10 promoters of the phage (Fig. 3G). Equally efficient transcription inhibition by the hybrid E. coli RNAPs was observed with the E. coli σ70 and T. thermophilus σA subunits, demonstrating that σ does not contribute to the species specificity of gp39 action (Supplemental Fig. 4A). The β D725W substitution in the hybrid RNAP abolished gp39 binding (Fig. 3E, lanes 3,3′) and relieved the inhibition (Fig. 3G). Thus, the gp39-binding site in the β-flap domain is the major determinant for RNAP inhibition.
The gp39 C-terminal tail dramatically relocates σ4
In the holo•gp39 structure, the C-terminal tail of gp39 interacts with the gp39 core, the β-flap tip, and σ4 (Figs. 1E, 3A). The gp39 C-terminal helix (residues 122–132), the β-flap tip helix, and two σ4 helices (H1 and H4, residues 342–359 and 393–410, respectively) form the hydrophobic core of this tripartite interaction. The interface is formed between Tyr125, Leu128, Met129, Ala131, Met132, and Leu34 in gp39; Leu350, Leu354, and Ala357 in σ4 (corresponding to Leu540, Thr544, and Val547, respectively, in E. coli σ70); and Leu774, Ile777, and Phe778 in the β flap (corresponding to Leu902, Ile905, and Phe906, respectively, in E. coli β) (Fig. 3B). Importantly, the C-terminal tail of gp39 (6–132) was not observed in the structure bound to the β-flap domain fragment (Fig. 2C), demonstrating that the gp39 tail is fixed through its interaction with σ4. Bacterial two-hybrid data confirmed the weak interaction of gp39 with σ4 and revealed that this interaction is abolished by the deletion of the gp39 C-terminal helix (Supplemental Fig. 3). Thus, the mobile C-terminal helix of gp39 primarily interacts with σ4.
gp39 binding dramatically repositions σ4 and the β-flap tip helix in the holoenzyme (Fig. 4A). The C-terminal tail of gp39 plays a key role in this rearrangement. In the holoenzyme structure without gp39, the C-terminal H5 helix of σ4 forms hydrophobic interactions with the β-flap tip helix (Vassylyev et al. 2002). In the holo•gp39 complex, the gp39 C-terminal helix displaces σ4 H5 (Fig. 4B) and interacts with σ4 and the β-flap tip, causing σ4 to rotate together with the β-flap tip, while the gp39 core stays bound to the β-flap protuberance (β723–740). Consequently, σ4 and the β-flap tip are concomitantly rotated by ∼60° relative to the rest of the flap domain, thus shifting the position of σ4 by ∼45 Å. Although the displaced H5 helix of σ4 is not visible in the electron density, the relative positions between σ4 and the β-flap tip remain almost unchanged (Fig. 4B).
Figure 4.

The mechanism of transcription inhibition by gp39. (A,B) Superimposition of holo•gp39 and the free holoenzyme (Protein Data Bank [PDB] 1IW7; green) (Vassylyev et al. 2002). (A) The core modules of the two RNAP structures were superimposed by minimizing the root mean square deviation (RMSD) between the Cα atoms. (B) Superimposition of the σ4 domains of the two structures. (C) Transcription inhibition by wild-type gp39 (wt) and its truncated variant lacking the C-terminal tail (1–113) on a −10/−35 promoter (T5 N25). (D) Analysis of transcription inhibition by gp39 added either before (lane 2) or after (lane 3) the promoter DNA. Black triangles indicate order of addition. (E) Analysis of gp39 (wild type [wt] and 1–113) on promoter melting by potassium permanganate probing. Thymine positions in the melted promoter region (numbered relative to the transcription starting point) are indicated on the right.
The gp39 C-terminal tail is essential for transcription inhibition
To investigate the significance of the gp39 C-terminal tail, we examined the transcription inhibition activity of a mutant gp39 lacking the C-terminal tail (gp39 1–113). In vitro transcription experiments revealed that, in agreement with the structure, the deletion of the C-terminal tail strongly impairs the ability of gp39 to inhibit transcription (Fig. 4C) but only minimally affects gp39 binding to the RNAP core and holoenzymes (Supplemental Fig. 6). These data strongly suggested that σ4 displacement by the gp39 C-terminal tail is the major mechanism of transcription inhibition on promoters containing the −35 element.
Besides its activity as an initiation inhibitory factor, gp39 also acts as a potent transcription anti-terminator during transcription elongation (Berdygulova et al. 2012). However, the deletion of the gp39 C-terminal tail did not affect the gp39 anti-termination activity (Fig. 5). Thus, the anti-termination activity by gp39 does not require its C-terminal tail and is based on a mechanism distinct from that inhibiting initiation.
Figure 5.

Transcription anti-termination by gp39. (A) Transcription reactions were performed on a DNA fragment containing the T7A1 promoter followed by the tR2 terminator of phage λ, as described previously (Berdygulova et al. 2012). (B) Transcription anti-termination efficiencies by T. thermophilus RNAP with wild-type gp39 and its truncated variant lacking the C-terminal tail (1–113) were analyzed. The positions of the starting 21-mer RNA and terminated (tR2) and full-length run-off (RO) RNAs are indicated on the left.
gp39 prevents stable promoter complex formation
T. thermophilus RNAP forms highly unstable promoter complexes, which exist in rapid equilibrium with the free holoenzyme and DNA. To determine which step of transcription initiation is targeted by gp39, we performed order-of-addition experiments with the hybrid Eco-tthflap RNAP, which forms stable promoter complexes typical of E. coli RNAP (Supplemental Fig. 4B). gp39 inhibited Eco-tthflap RNAP transcription only when bound before the DNA and had no effect on preformed promoter complexes (Fig. 4D). Thus, the interaction of σ4 with the −35 promoter DNA element counteracts the action of gp39, likely by fixing the σ4/β-flap tip in the “proper” position.
If gp39 targets the interaction of σ4 with the −35 element, then it should inhibit an early stage in the promoter complex formation pathway. Indeed, potassium permanganate probing revealed that gp39 inhibited the open complex formation on a −10/−35 promoter (Fig. 4E, lanes 3,4), and the inhibition was strongly dependent on the gp39 C-terminal tail (Fig. 4E, lanes 5,6). On the other hand, gp39 exerted minimal effects on an extended −10 promoter (Supplemental Fig. 7C). To detect possible gp39-induced changes in the RNAP–promoter contacts, we performed DNase I and ExoIII footprinting analyses of promoter complexes in the absence and presence of gp39 (Supplemental Fig. 7). Eco-tthflap RNAP generated a clear footprint on the promoter DNA, corresponding to the open complex. In contrast, the footprint almost completely disappeared in the presence of gp39. Thus, gp39 inhibits an early step in the open complex formation pathway, probably by preventing the interaction of σ4 with the −35 element during the initial stage of promoter recognition.
Docking model with promoter DNA
To further examine the inhibition of promoter complex formation, we docked promoter DNA to holo•gp39 (Fig. 6A; Supplemental Fig. 8). The superimposition of the holo•gp39 and Thermus aquaticus closed promoter complex structures revealed that gp39 relocates the σ4 domain to the opposite side of the DNA, far away from the −35 promoter element. This clearly explains why gp39 prevents complex formation with the −10/−35 promoters. However, gp39 binding does not influence the σ region 2 (σ74–261, σ2), which is responsible for the −10 element recognition. Consequently, it is likely that gp39 does not interfere with σ2 binding to the −10 region, and thus gp39 allows transcription from promoters that depend on the extended −10 element. Although the superimposition suggested that a clash occurs between σ4 and DNA (region of −19 to approximately −26), the steric hindrance could be avoided by an ∼15° rotation of the DNA (Fig. 6B). In this new orientation, the promoter spacer region (region of −19 to approximately −32) is closer to the β′ zipper and zinc finger domains of the β′ subunit, which are critical for transcription from the extended −10 promoters (Yuzenkova et al. 2011). Therefore, the model reasonably explains why gp39 does not interfere with transcription from the extended −10 class promoters.
Figure 6.

Structural basis of promoter switching by gp39. (A) Superimposition of holo•gp39 and the T. aquaticus closed promoter complex (PDB 1L9Z) (Murakami et al. 2002a) on the clamp module and σ subunit. The latter structure shows only σ (moss green) and the promoter DNA (slate). The −10 and −35 elements are highlighted in yellow. (B) Model of an open promoter complex of holo•gp39 on DNA containing an extended −10 promoter. The DNA is reoriented by ∼15° and is located close to the β′ zipper and zinc finger domains (light green). The extended −10 element is highlighted in green, while the other parts of the DNA are colored as in A. (C) Diagram summarizing the mechanism by which gp39 switches the promoter specificity of RNAP. The left route shows transcription initiation from a −10/−35 promoter. The gp39-bound holoenzyme cannot form a stable promoter complex, and thus transcription is prevented. The right route shows transcription initiation from an extended −10 promoter, which is not affected by gp39.
Discussion
Many crystal structures of bacterial, archaeal, and eukaryotic RNAPs have been reported (Zhang et al. 1999; Cramer et al. 2001; Hirata et al. 2008; Spahr et al. 2009; Murakami 2013; Zuo et al. 2013). However, only a few structures of RNAP complexes with transcription factors are available (Murakami et al. 2002b; Vassylyev et al. 2002; Kostrewa et al. 2009; Wang et al. 2009; Liu et al. 2010; Tagami et al. 2010; Cheung and Cramer 2011). In this study, we solved the structure of the T. thermophilus RNAP holoenzyme (RNAP + σ) bound to the phage-encoded protein gp39. The structure revealed the complete molecular view of an external transcription factor modifying the holoenzyme conformation to switch the promoter specificity of the host's RNAP (Fig. 6C).
Transcription initiation may be inhibited by a few typical mechanisms. Transcription repressors directly bind DNA to prevent the RNAP from binding to a promoter (Pabo and Sauer 1984). Anti-σ factors bind to a σ factor and block its interaction with RNAP to prevent complete holoenzyme formation (Campbell et al. 2003; Lambert et al. 2004; Sorenson et al. 2004; Baxter et al. 2006). Some anti-σ factors also interfere with the interaction between σ and DNA by blocking the DNA-binding site of σ. The T7 gp2 protein binds and occludes the DNA-binding channel of RNAP (Camara et al. 2010; Nechaev and Severinov 2003). In contrast, the present study revealed that the phage protein gp39 functions via a novel, unanticipated mechanism by repositioning the promoter-binding determinant (σ4) without interacting with DNA, dissociating the host σ factor from RNAP, masking the DNA-binding determinants of σ, or occluding the RNAP channels. The repositioning prevents promoter complex formation on the −10/−35 promoters and thus shuts off most of the host's genes. However, it has a much smaller effect on the extended −10 promoters that drive the transcription of the middle and late genes of the phage (Berdygulova et al. 2011). The mechanism of transcription inhibition by gp39 does not involve significant conformational reorganization of σ4. Thus, the conformational modification of the RNAP holoenzyme by the small phage protein is sufficient to switch the entire gene transcription profile in the infected bacterial cells.
Similarly to gp39, the anti-σ factor AsiA from bacteriophage T4 inhibits transcription by E. coli RNAP from −10/−35 promoters but does not inhibit transcription from extended −10 promoters (Severinova et al. 1998). AsiA binds to the σ4 domain of the E. coli σ70 RNAP subunit to occlude the interface between σ4 and the β-flap tip. It also induces a significant conformational change in σ4 to deform the DNA-binding site (Lambert et al. 2004). On the other hand, AsiA also tightly binds to the β-flap tip, and this interaction is required for efficient transcription inhibition, suggesting that AsiA serves as a bridge between σ4 and the β-flap tip (Yuan et al. 2009). Therefore, the potential AsiA-induced relocation of σ4 could contribute to the transcription inhibition, as in the case of gp39. AsiA is not only a transcription inhibitor but also a coactivator protein required for T4 middle gene transcription dependent on the activator protein MotA. AsiA binds to both σ4 and MotA, and then MotA binds the “MotA box” DNA sequence around position −30. The potential relocation of σ4 by AsiA could facilitate the efficient interaction between σ4 and MotA to achieve the appropriation of σ by MotA/AsiA. The structure of the E. coli RNAP complex with AsiA will clarify this point. The repositioning of σ4 might be a widely used phage protein strategy to switch the host's gene expression.
Intriguingly, gp39 is a bifunctional regulator: Besides its initiation inhibitory activity, gp39 also has transcription anti-termination activity (Berdygulova et al. 2012). The deletion of the gp39 C-terminal tail, which is critical for inhibiting transcription initiation, does not affect the gp39 anti-termination activity (Fig. 5). Therefore, the gp39 C-terminal tail is dispensable for the anti-termination activity, consistent with the observation that the C-terminal tail is fixed only in the context of the σ-containing holoenzyme. The anti-termination activity thus requires the gp39 core to bind to the RNAP β-flap domain, a known modulator of transcription elongation and termination efficiency. Ongoing structural analyses of gp39-bound transcription elongation complexes should clarify the structural basis of this alternative activity of the bifunctional phage transcription regulator.
Materials and methods
Protein preparation
For the crystallization of RNAP, the RNAP holoenzyme was purified from T. thermophilus cells, as described previously (Vassylyev et al. 2002). For the biochemical analyses, T. thermophilus RNAP core and holoenzymes with the C-terminally 10His-tagged β′ subunit were purified from the T. thermophilus HB8 rpoC∷10H strain, as described (Sevostyanova et al. 2007). E. coli core RNAP was purchased from Epicentre. Hybrid E. coli RNAPs (Eco-tthflap and Eco-tthflapD725W) were created as follows. The E. coli β flap (residues 831–1060) was replaced with the T. thermophilus β flap (residues 702–833) by site-directed mutagenesis in the plasmid pIA545, bearing the E. coli rpoB gene. The D725W substitution (T. thermophilus numbering, corresponding to position 853 in the E. coli β subunit) was introduced into the β flap in the hybrid RNAP by site-directed mutagenesis. Both variants of the hybrid rpoB gene were recloned into the plasmid pIA679, which encodes all E. coli core RNAP subunits (the rpoA, rpoB, rpoC, and rpoZ genes), with a 6His tag appended to the C terminus of the β subunit. The resulting hybrid RNAPs were expressed in E. coli BL21(DE3) and purified as described previously (Borukhov and Goldfarb 1993; Pupov et al. 2010), using Polymin P precipitation followed by chromatography on Ni2+-equilibrated HiTrap chelating, HiTrap heparin, and Superose-6 columns (GE Healthcare). N-terminally 6His-tagged E. coli σ70 and T. thermophilus σA subunits were expressed in BL21(DE3) and purified as described previously (Borukhov and Goldfarb 1993; Sevostyanova et al. 2007).
For the preparation of gp39 and its variants, the genes encoding wild-type gp39 and the gp39 variant lacking 28 C-terminal amino acid residues (gp39 1–113) were cloned into the pET28a vector. To obtain gp39 variants with N-terminal cAMP-dependent protein kinase (PKA) sites, the corresponding genes were cloned into the appropriately modified pET16b vector. These gp39 variants were expressed and purified as described (Berdygulova et al. 2012). To obtain the SeMet derivative of wild-type gp39, the cells bearing the expression vector were cultured in M9 medium containing SeMet (Van Duyne et al. 1993). The protein was purified by chromatography on Ni-Sepharose FF, MonoQ, and Superdex 75 columns (GE Healthcare Biosciences). For the preparation of gp39 (6–109) and gp39 (6–132), the ORFs for these variants were cloned into the vector pET-47b, which allows expression of the variant proteins with a removable N-terminal 6His tag (Novagen). While gp39 (6–109) was expressed as a SeMet derivative, as described above, gp39 (6–132) was expressed as the native protein. The proteins were purified as described above, and the His tag was removed with HRV3C protease.
For the expression of the β-flap domain, its coding region (T. thermophilus rpoB 703–830) was cloned into the expression vector pGEX-6P, which allows expression of the protein as an N-terminal GST fusion (GE Healthcare). The protein was purified on a glutathione Sepharose FF column (GE Healthcare), with GST tag removal by HRV3C protease, followed by chromatography on HiTrap butyl FF and Superdex 75 columns (GE Healthcare).
Crystallization
For the cocrystallization of the T. thermophilus RNAP holoenzyme and the full-length gp39, the sample solution (5 μM holoenzyme + 25 μM full-length gp39) was mixed with the equivalent volume of the reservoir solution containing 45% tacsimate (pH 6.7). Crystallization was performed by the sitting-drop vapor diffusion technique at 20°C. Crystals with dimensions of 0.4 × 0.1 × 0.1 mm appeared in 3 wk.
For the cocrystallization of the β-flap domain and gp39 (6–109), the sample solution (50 μM β flap + 50 μM gp39 6–109) was mixed with the equivalent amount of the reservoir solution, containing 50 mM Tris-HCl buffer (pH 8.0), 12% polyethylene glycol (PEG) 8000, 8% 2-propanol, and 3% 1,6-hexanediol. Plate-like crystals appeared in a week.
For the cocrystallization of the β-flap domain and gp39 (6–132), the sample solution (50 μM β flap + 50 μM gp39 6–132) was mixed with the equivalent amount of the reservoir solution, containing 50 mM MES-NaOH buffer (pH 5.6), 6% PEG 8000, 200 mM KCl, and 10 mM MgCl2. Crystals with dimensions of 0.15 × 0.05 × 0.05 mm appeared in 3 mo.
Data collection and structure determination of holo•gp39
The X-ray diffraction data set for the native holo•gp39 crystal was obtained at beamline BL41XU of SPring-8 (Table 1). The SAD data set for the SeMet-containing crystal, in which the SeMet derivative of gp39 was complexed with the native holoenzyme, was collected at beamline NE3A of the Photon Factory. For the data collection, the crystallization solution containing 20% glycerol was used as the cryoprotectant. These data were processed with the HKL2000 software package (Otwinowski and Minor 1997).
The holo•gp39 crystal belongs to the space group P3221, with unit cell dimensions of a = b = 295 Å and c = 223 Å. The asymmetric unit contains one holoenzyme and two gp39 molecules. The structure was solved by molecular replacement with the program PHASER (McCoy et al. 2007) using the coordinates of the T. thermophilus holoenzyme structure (Protein Data Bank [PDB] 1IW7) (Vassylyev et al. 2002) as the search model. The holoenzyme structure was divided into ∼30 rigid bodies, and their positions were manually modified with the program COOT (Emsley et al. 2010) and refined with the program CNS (Brunger 2007). For the tip portion of the β′ nonconserved domain (NCD), the coordinates of that region of the T. thermophilus holoenzyme (PDB 3DXJ) (Mukhopadhyay et al. 2008) were used. After the placement of the RNAP model, extra electron density remained, corresponding to the gp39 molecules. One gp39 molecule was associated with the β-flap domain of RNAP, as seen in the β-flap•gp39 (6–109) structure. Therefore, we placed the model of β-flap•gp39 (6–109) within the electron density by superimposing the β-flap domains. This positioning of gp39 was confirmed to be correct by the identification of the Se anomalous peaks corresponding to Met38, Met129, and Met132 of gp39 in the anomalous difference map using the SAD data. We further built the model of the gp39 C-terminal helix in the 2Fo − Fc electron density map of the native crystal by using the positional information of Met129 and Met132 from the SAD data and secondary structure prediction with Phyre (Kelley and Sternberg 2009). Finally, we copied the coordinates of this gp39 molecule and manually placed them into the electron density for the second molecule (gp39_2). We observed an anomalous peak for Met38 of the second gp39 molecule but not for Met129 and Met132, probably because of the flexibility of the C-terminal helix. The structure was refined with the programs COOT, CNS, PHENIX, and Refmac5 (Vagin et al. 2004; Brunger 2007; Adams et al. 2010; Emsley et al. 2010; Winn et al. 2011). Restraints for structural refinement by Refmac5 were generated by ProSMART (Nicholls et al. 2012) using the structures of the T. thermophilus RNAP holoenzyme (PDB 2A6H) and β-flap•gp39 (6–109) as the reference models. The structure was refined at 3.6 Å to R and Rfree values of 0.258 and 0.282, respectively. Structural superimposition with other RNAP coordinates (Zhang et al. 1999, 2012; Cramer et al. 2001; Murakami et al. 2002a; Vassylyev et al. 2002, 2007; Tagami et al. 2010; Weixlbaumer et al. 2013) was accomplished with the CCP4 program suite (Winn et al. 2011).
Data collection and structure determination of β-flap•gp39 (6–109)
The cocrystals of β-flap•gp39 (6–109) consisted of the native protein of the β-flap domain and the SeMet derivative of gp39 (6–109). The crystals were immersed in the crystallization solution containing 35% glycerol as the cryoprotectant and were flash-cooled in liquid nitrogen. A SAD data set was collected at beamline BL32XU of SPring-8 (Table 1). The data were indexed, integrated, and scaled with the HKL2000 program (Otwinowski and Minor 1997). The space group is P41212, with unit cell dimensions of a = b = 99.2 Å and c = 117.2 Å. The asymmetric unit contains two pairs of β-flap•gp39 (6–109). Heavy-atom searches and phase calculations were performed by the MRSAD method using the program PHASER (McCoy et al. 2007) driven by PHENIX AutoSol (Adams et al. 2010). The coordinates of the β-flap domain in the T. thermophilus holoenzyme structure (PDB 1IW7) (Vassylyev et al. 2002) were used as the search model in the MRSAD method. The structure was refined at 2.35 Å to R and Rfree values of 0.191 and 0.238, respectively, by using COOT (Emsley et al. 2010) and PHENIX (Adams et al. 2010).
Data collection and structure determination of β-flap•gp39 (6–132)
The cocrystals of β-flap•gp39 (6–132) were immersed in the crystallization solution containing 35% glycerol as the cryoprotectant and were flash-cooled in liquid nitrogen. X-ray diffraction data were collected at beamline NE3A at the Photon Factory (Table 1). The data were indexed, integrated, and scaled with the HKL2000 software package (Otwinowski and Minor 1997). The crystal belongs to the space group P41212, with unit cell dimensions of a = b = 213.8 Å and c = 234.6 Å. The asymmetric unit contained 12 complexes. The structure was solved by molecular replacement with the program PHASER (McCoy et al. 2007) using the coordinates of the β-flap•gp39 (6–109) complex as the search model. The structure was refined at 3.3 Å to R and Rfree values of 0.223 and 0.249, respectively, by using COOT (Emsley et al. 2010) and PHENIX (Adams et al. 2010).
Ultracentrifugation analysis
To investigate the oligomeric state of gp39 in solution, a sedimentation equilibration experiment was performed using an analytical ultracentrifuge (Optima XL-I, Beckman Coulter). Analytical cells with a six-channel centerpiece were used, with each channel filled with 100 μL of sample or 110 μL of reference solution. The protein was dissolved in 10 mM Tris-HCl buffer (pH 8.0) containing 200 mM NaCl, and concentrations of 0.8, 0.4, and 0.2 mg/mL were examined. An eight-position rotor (An-50 Ti) was rotated at 20,000, 22,000, and 24,000 rpm (average g forces of ∼29,000, 35,000, and 42,000, respectively) at 20°C. Data were collected after 12, 14, and 16 h of revolution at each speed by observing the absorbance at 280 nm. Finally, the protein samples were completely sedimented at 40,000 rpm (106,000g) for 6 h to generate the baseline. Data were analyzed with the XL-A/XL-I data analysis software version 6.03 using a partial specific volume of 0.743 mL/g and a solvent density of 1.007 g/mL.
Native gel binding assay
Native gel binding assays with E. coli RNAP core and holoenzymes and wild-type or mutant gp39 were performed as described (Berdygulova et al. 2012). To compare the efficiencies of the interactions of wild-type and mutant gp39 with the E. coli RNAP core and holoenzymes, gp39 variants were radiolabeled with [γ-32P]-ATP at their PKA sites using the PKA enzyme (New England Biolabs) according to the manufacturer's protocol and were used in the native gel binding experiments under the same conditions.
Bacterial two-hybrid assay
Bacterial two-hybrid assays for analyses of gp39•RNAP interactions were performed using the E. coli strain FW102 OL2-62, containing the lacZ gene under the control of the test promoter placOR2-62 on an F′ episome, as described previously (Nickels 2009; Berdygulova et al. 2012). Plasmids containing gene fusions of gp39 (wild type, a variant with the 94W/95F double substitution, and a C-terminally truncated variant [residues 1–122]), the T. thermophilus β-flap (wild-type [residues 703–830] and a variant with the 725W substitution), and the σ4 domain (residues 337-423) were obtained by cloning the corresponding PCR fragments between the NotI and BamHI sites into the plasmids pBRαLN (encoding the N-terminal domain of the E. coli RNAP α subunit) and pACλCI32 (encoding the DNA-binding domain of the λCI protein). Empty vector plasmids were used as negative controls.
In vitro transcription
Abortive transcription initiation reactions were performed in transcription buffer containing 40 mM Tris-HCl (pH 7.9), 40 mM KCl, and 10 mM MgCl2, at 37°C for the E. coli and Eco-tthflap RNAPs and at 55°C for the T. thermophilus RNAP. The concentrations of the RNAPs and promoters were 0.1–1 and 0.05–0.5 μM, respectively. The gp39 concentrations in the transcription reactions were 0.03, 0.1, 0.3, 1, and 3 μM (Fig. 3G); 1.2 and 6 μM (Fig. 4C); and 5 μM (Fig. 4D). Transcription was initiated by the addition of dinucleotide primers and the [α-32P]-NTP, corresponding to the next promoter position, to preformed promoter complexes and was stopped after 5–10 min by the addition of an equal volume of urea–formamide loading buffer. RNA products were resolved in 20% polyacrylamide denaturing gels. Analyses of transcription anti-termination by gp39 were performed on a DNA fragment containing the T7A1 promoter followed by the tR2 terminator of phage λ, as described previously (Berdygulova et al. 2012).
Footprinting
For in vitro footprinting experiments, dsDNA promoter fragments were prepared from 100-nucleotide oligonucleotides corresponding to the −65/+35 promoter positions, as described (Mekler et al. 2011). In each case, either the nontemplate or template strand oligonucleotide was labeled with [γ-32P]-ATP at its 5′ end. The labeled promoter fragments were purified on Micro Bio-spin 6 columns (Bio-Rad) and used for the assays at 0.05-–0.1 μM concentrations.
DNase I footprinting
Promoter complexes were formed with 0.2 μM Eco-tthflap RNAP, 0.6 μM gp39, and 0.05 μM promoter DNA in 10 μL of transcription buffer. In most experiments, RNAP was preincubated with gp39 for 10 min at 37°C prior to the addition of DNA; in some reactions, DNA was added before gp39. The reactions were incubated for 10 min at 37°C followed by the addition of 0.2 U of DNase I (New England Biolabs). After 60 sec at 37°C, the reactions were stopped by the addition of 10 μg of calf thymus DNA in 10 μL of water followed by ethanol precipitation. The samples were dissolved in 8 μL of urea–formamide loading buffer and resolved in 7% polyacrylamide denaturing gels.
ExoIII footprinting
Promoter complexes were obtained as described above and treated with 0.5 U of ExoIII (New England Biolabs) for 60 sec at 37°C. The reactions were stopped by the addition of calf thymus DNA and processed as described above.
KMnO4 probing
The reactions were prepared as described above, except the concentrations of Eco-tthflap RNAP and gp39 were 0.1 μM and 0.06–0.3 μM, respectively. Promoter complexes were treated with 2 mM KMnO4 for 50 sec at 37°C. The reactions were then stopped by the addition of 30 mM β-mercaptoethanol followed by ethanol precipitation and treatment with 10% piperidine for 20 min at 95°C. The samples were treated with chloroform, ethanol-precipitated, dissolved in 8 μL of loading buffer, and resolved in 7% polyacrylamide denaturing gels.
Database deposition
The coordinates and structure–factor amplitudes have been deposited in PDB under accession numbers 3WOD (holo•gp39), 3WOE (β-flap•gp39 [6–109]), and 3WOF (β-flap•gp39 [6–132]).
Acknowledgments
We thank the beam line staffs of BL32XU and BL41XU at SPring-8 and NE3A at the Photon Factory for their assistance with data collection. We are grateful to I. Artsimovitch for providing plasmids pIA545 and pIA679 for our study. This work is based on experiments performed at SPring-8 with the approval of the Japan Synchrotron Radiation Research Institute (proposal nos. 2011B1382, 2012A1334, 2012B1392, and 2013A1149) and at the Photon Factory with the approval of the Photon Factory Program Advisory Committee (proposal no. 2011G554). This work was supported in part by a Japan Society for the Promotion of Science (JSPS) grant-in-aid for scientific research (to S.S. and S.Y.); a JSPS grant-in-aid for young scientists (to S.T.); the Targeted Proteins Research Program (TPRP) and the Ministry of Education, Culture, Sports, Science and Technology (MEXT) of Japan (to S.Y.); the National Institutes of Health (grant nos. R21 A1074769 to L.M. and R01 59295 to K.S.); the Russian Academy of Sciences Presidium Program in Molecular and Cellular Biology (to A.K. and K.S.); the Russian Foundation for Basic Research (grant nos. 12-04-33187 and 14-04-01696 to A.K.); and the Ministry of Education and Science of Russia (grants 8106 and 8475 and project 14.B25.31.0004 to A.K. and K.S.). S.T., S.S., L.M., A.K., K.S., and S.Y. designed the research. S.T. and S.S. performed the structural analyses. L.M., D.E., and A.K. performed the biochemical analyses. S.S., R.A., and M.S. performed the centrifugal analyses. S.T., S.S., L.M., D.E., A.K., K.S., and S.Y. interpreted the data and wrote the paper.
Footnotes
Supplemental material is available for this article.
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.233916.113.
Freely available online through the Genes & Development Open Access option.
References
- Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. 2010. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66: 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baxter K, Lee J, Minakhin L, Severinov K, Hinton DM 2006. Mutational analysis of σ70 region 4 needed for appropriation by the bacteriophage T4 transcription factors AsiA and MotA. J Mol Biol 363: 931–944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berdygulova Z, Westblade LF, Florens L, Koonin EV, Chait BT, Ramanculov E, Washburn MP, Darst SA, Severinov K, Minakhin L 2011. Temporal regulation of gene expression of the Thermus thermophilus bacteriophage P23-45. J Mol Biol 405: 125–142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berdygulova Z, Esyunina D, Miropolskaya N, Mukhamedyarov D, Kuznedelov K, Nickels BE, Severinov K, Kulbachinskiy A, Minakhin L 2012. A novel phage-encoded transcription antiterminator acts by suppressing bacterial RNA polymerase pausing. Nucleic Acids Res 40: 4052–4063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borukhov S, Goldfarb A 1993. Recombinant Escherichia coli RNA polymerase: Purification of individually overexpressed subunits and in vitro assembly. Protein Expr Purif 4: 503–511 [DOI] [PubMed] [Google Scholar]
- Brunger AT 2007. Version 1.2 of the crystallography and NMR system. Nat Protoc 2: 2728–2733 [DOI] [PubMed] [Google Scholar]
- Camara B, Liu M, Reynolds J, Shadrin A, Liu B, Kwok K, Simpson P, Weinzierl R, Severinov K, Cota E, et al. 2010. T7 phage protein Gp2 inhibits the Escherichia coli RNA polymerase by antagonizing stable DNA strand separation near the transcription start site. Proc Natl Acad Sci 107: 2247–2252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell EA, Muzzin O, Chlenov M, Sun JL, Olson CA, Weinman O, Trester-Zedlitz ML, Darst SA 2002. Structure of the bacterial RNA polymerase promoter specificity σ subunit. Mol Cell 9: 527–539 [DOI] [PubMed] [Google Scholar]
- Campbell EA, Tupy JL, Gruber TM, Wang S, Sharp MM, Gross CA, Darst SA 2003. Crystal structure of Escherichia coli σE with the cytoplasmic domain of its anti-σ RseA. Mol Cell 11: 1067–1078 [DOI] [PubMed] [Google Scholar]
- Cheung AC, Cramer P 2011. Structural basis of RNA polymerase II backtracking, arrest and reactivation. Nature 471: 249–253 [DOI] [PubMed] [Google Scholar]
- Cramer P, Bushnell DA, Kornberg RD 2001. Structural basis of transcription: RNA polymerase II at 2.8 Ångstrom resolution. Science 292: 1863–1876 [DOI] [PubMed] [Google Scholar]
- Dove SL, Darst SA, Hochschild A 2003. Region 4 of σ as a target for transcription regulation. Mol Microbiol 48: 863–874 [DOI] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, Cowtan K 2010. Features and development of Coot. Acta Crystallogr D Biol Crystallogr 66: 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feklistov A, Darst SA 2011. Structural basis for promoter –10 element recognition by the bacterial RNA polymerase σ subunit. Cell 147: 1257–1269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirata A, Klein BJ, Murakami KS 2008. The X-ray crystal structure of RNA polymerase from Archaea. Nature 451: 851–854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley LA, Sternberg MJ 2009. Protein structure prediction on the Web: A case study using the Phyre server. Nat Protoc 4: 363–371 [DOI] [PubMed] [Google Scholar]
- Kostrewa D, Zeller ME, Armache KJ, Seizl M, Leike K, Thomm M, Cramer P 2009. RNA polymerase II–TFIIB structure and mechanism of transcription initiation. Nature 462: 323–330 [DOI] [PubMed] [Google Scholar]
- Lambert LJ, Wei Y, Schirf V, Demeler B, Werner MH 2004. T4 AsiA blocks DNA recognition by remodeling σ70 region 4. EMBO J 23: 2952–2962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Bushnell DA, Wang D, Calero G, Kornberg RD 2010. Structure of an RNA polymerase II-TFIIB complex and the transcription initiation mechanism. Science 327: 206–209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ 2007. Phaser crystallographic software. J Appl Crystallogr 40: 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mekler V, Minakhin L, Sheppard C, Wigneshweraraj S, Severinov K 2011. Molecular mechanism of transcription inhibition by phage T7 gp2 protein. J Mol Biol 413: 1016–1027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minakhin L, Goel M, Berdygulova Z, Ramanculov E, Florens L, Glazko G, Karamychev VN, Slesarev AI, Kozyavkin SA, Khromov I, et al. 2008. Genome comparison and proteomic characterization of Thermus thermophilus bacteriophages P23-45 and P74-26: Siphoviruses with triplex-forming sequences and the longest known tails. J Mol Biol 378: 468–480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell JE, Zheng D, Busby SJ, Minchin SD 2003. Identification and analysis of ‘extended −10’ promoters in Escherichia coli. Nucleic Acids Res 31: 4689–4695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhopadhyay J, Das K, Ismail S, Koppstein D, Jang M, Hudson B, Sarafianos S, Tuske S, Patel J, Jansen R, et al. 2008. The RNA polymerase ‘switch region’ is a target for inhibitors. Cell 135: 295–307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami KS 2013. X-ray crystal structure of Escherichia coli RNA polymerase σ70 holoenzyme. J Biol Chem 288: 9126–9134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami KS, Masuda S, Campbell EA, Muzzin O, Darst SA 2002a. Structural basis of transcription initiation: An RNA polymerase holoenzyme-DNA complex. Science 296: 1285–1290 [DOI] [PubMed] [Google Scholar]
- Murakami KS, Masuda S, Darst SA 2002b. Structural basis of transcription initiation: RNA polymerase holoenzyme at 4 Å resolution. Science 296: 1280–1284 [DOI] [PubMed] [Google Scholar]
- Nechaev S, Severinov K 2003. Bacteriophage-induced modifications of host RNA polymerase. Annu Rev Microbiol 57: 301–322 [DOI] [PubMed] [Google Scholar]
- Nechaev S, Kamali-Moghaddam M, Andre E, Leonetti JP, Geiduschek EP 2004. The bacteriophage T4 late-transcription coactivator gp33 binds the flap domain of Escherichia coli RNA polymerase. Proc Natl Acad Sci 101: 17365–17370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholls RA, Long F, Murshudov GN 2012. Low-resolution refinement tools in REFMAC5. Acta Crystallogr D Biol Crystallogr 68: 404–417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickels BE 2009. Genetic assays to define and characterize protein-protein interactions involved in gene regulation. Methods 47: 53–62 [DOI] [PubMed] [Google Scholar]
- Osmundson J, Montero-Diez C, Westblade LF, Hochschild A, Darst SA 2012. Promoter-specific transcription inhibition in Staphylococcus aureus by a phage protein. Cell 151: 1005–1016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W 1997. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol 276: 307–326 [DOI] [PubMed] [Google Scholar]
- Pabo CO, Sauer RT 1984. Protein-DNA recognition. Annu Rev Biochem 53: 293–321 [DOI] [PubMed] [Google Scholar]
- Pupov D, Miropolskaya N, Sevostyanova A, Bass I, Artsimovitch I, Kulbachinskiy A 2010. Multiple roles of the RNA polymerase β′ SW2 region in transcription initiation, promoter escape, and RNA elongation. Nucleic Acids Res 38: 5784–5796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Severinova E, Severinov K, Darst SA 1998. Inhibition of Escherichia coli RNA polymerase by bacteriophage T4 AsiA. J Mol Biol 279: 9–18 [DOI] [PubMed] [Google Scholar]
- Sevostyanova A, Djordjevic M, Kuznedelov K, Naryshkina T, Gelfand MS, Severinov K, Minakhin L 2007. Temporal regulation of viral transcription during development of Thermus thermophilus bacteriophage φYS40. J Mol Biol 366: 420–435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorenson MK, Ray SS, Darst SA 2004. Crystal structure of the flagellar σ/anti-σ complex σ28/FlgM reveals an intact σ factor in an inactive conformation. Mol Cell 14: 127–138 [DOI] [PubMed] [Google Scholar]
- Spahr H, Calero G, Bushnell DA, Kornberg RD 2009. Schizosaccharomyces pombe RNA polymerase II at 3.6-Å resolution. Proc Natl Acad Sci 106: 9185–9190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tagami S, Sekine S, Kumarevel T, Hino N, Murayama Y, Kamegamori S, Yamamoto M, Sakamoto K, Yokoyama S 2010. Crystal structure of bacterial RNA polymerase bound with a transcription inhibitor protein. Nature 468: 978–982 [DOI] [PubMed] [Google Scholar]
- Twist KA, Campbell EA, Deighan P, Nechaev S, Jain V, Geiduschek EP, Hochschild A, Darst SA 2011. Crystal structure of the bacteriophage T4 late-transcription coactivator gp33 with the β-subunit flap domain of Escherichia coli RNA polymerase. Proc Natl Acad Sci 108: 19961–19966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vagin AA, Steiner RA, Lebedev AA, Potterton L, McNicholas S, Long F, Murshudov GN 2004. REFMAC5 dictionary: Organization of prior chemical knowledge and guidelines for its use. Acta Crystallogr D Biol Crystallogr 60: 2184–2195 [DOI] [PubMed] [Google Scholar]
- Van Duyne GD, Standaert RF, Karplus PA, Schreiber SL, Clardy J 1993. Atomic structures of the human immunophilin FKBP-12 complexes with FK506 and rapamycin. J Mol Biol 229: 105–124 [DOI] [PubMed] [Google Scholar]
- Vassylyev DG, Sekine S, Laptenko O, Lee J, Vassylyeva MN, Borukhov S, Yokoyama S 2002. Crystal structure of a bacterial RNA polymerase holoenzyme at 2.6 Å resolution. Nature 417: 712–719 [DOI] [PubMed] [Google Scholar]
- Vassylyev DG, Vassylyeva MN, Perederina A, Tahirov TH, Artsimovitch I 2007. Structural basis for transcription elongation by bacterial RNA polymerase. Nature 448: 157–162 [DOI] [PubMed] [Google Scholar]
- Wang D, Bushnell DA, Huang X, Westover KD, Levitt M, Kornberg RD 2009. Structural basis of transcription: Backtracked RNA polymerase II at 3.4 angstrom resolution. Science 324: 1203–1206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weixlbaumer A, Leon K, Landick R, Darst SA 2013. Structural basis of transcriptional pausing in bacteria. Cell 152: 431–441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AG, McCoy A, et al. 2011. Overview of the CCP4 suite and current developments. Acta Crystallogr D Biol Crystallogr 67: 235–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young BA, Gruber TM, Gross CA 2002. Views of transcription initiation. Cell 109: 417–420 [DOI] [PubMed] [Google Scholar]
- Yuan AH, Nickels BE, Hochschild A 2009. The bacteriophage T4 AsiA protein contacts the β-flap domain of RNA polymerase. Proc Natl Acad Sci 106: 6597–6602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuzenkova Y, Tadigotla VR, Severinov K, Zenkin N 2011. A new basal promoter element recognized by RNA polymerase core enzyme. EMBO J 30: 3766–3775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang G, Campbell EA, Minakhin L, Richter C, Severinov K, Darst SA 1999. Crystal structure of Thermus aquaticus core RNA polymerase at 3.3 Å resolution. Cell 98: 811–824 [DOI] [PubMed] [Google Scholar]
- Zhang Y, Feng Y, Chatterjee S, Tuske S, Ho MX, Arnold E, Ebright RH 2012. Structural basis of transcription initiation. Science 338: 1076–1080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo Y, Wang Y, Steitz TA 2013. The mechanism of E. coli RNA polymerase regulation by ppGpp is suggested by the structure of their complex. Mol Cell 50: 430–436 [DOI] [PMC free article] [PubMed] [Google Scholar]