

Abstract
Congenital muscular dystrophy type Ullrich (UCMD) is a severe disorder of early childhood onset for which currently there is no effective treatment. UCMD commonly is caused by dominant-negative mutations in the genes coding for collagen type VI, a major microfibrillar component of the extracellular matrix surrounding the muscle fibers. To explore RNA interference (RNAi) as a potential therapy for UCMD, we designed a series of small interfering RNA (siRNA) oligos that specifically target the most common mutations resulting in skipping of exon 16 in the COL6A3 gene and tested them in UCMD-derived dermal fibroblasts. Transcript analysis by semiquantitative and quantitative reverse transcriptase PCR showed that two of these siRNAs were the most allele-specific, i.e., they efficiently knocked down the expression from the mutant allele, without affecting the normal allele. In HEK293T cells, these siRNAs selectively suppressed protein expression from a reporter construct carrying the mutation, with no or minimal suppression of the wild-type (WT) construct, suggesting that collagen VI protein levels are as also reduced in an allele-specific manner. Furthermore, we found that treating UCMD fibroblasts with these siRNAs considerably improved the quantity and quality of the collagen VI matrix, as assessed by confocal microscopy. Our current study establishes RNAi as a promising molecular approach for treating dominant COL6-related dystrophies.
Keywords: allele-specific siRNA, collagen type VI, dominant negative, Ullrich congenital muscular dystrophy
Introduction
Congenital muscular dystrophy type Ullrich (UCMD, MIM254090) is caused by mutations in one of three collagen VI genes (COL6A1 (MIM*120220), COL6A2 (MIM*120240), and COL6A3 (MIM*120250)).1,2,3 UCMD is a severe disorder of early childhood onset, characterized by muscle weakness, distal joint hypermobility, progressive joint contractures, skin changes, and progressive respiratory failure,4 eventually requiring ventilatory support.5 Currently, there are no primary or pharmacological treatment options available for individuals affected with this disease.
Collagen VI belongs to the class of nonfibrillar collagens forming a network of beaded microfibrils in the extracellular matrix.6,7 The major known collagen VI heterotrimer is composed of the α1(VI), α2(VI), and α3(VI) chains encoded respectively by the COL6A1, COL6A2, and COL6A3 genes.8 All three chains have relatively short triple helical domains of 335–336 amino acids each, which include cysteine residues important for higher order assembly at their N-terminal end.9 Collagen VI undergoes a sequential assembly process,10 where the first three chains associate in equal stoichiometry to form heterotrimeric monomers.10 Two monomers then associate in an antiparallel arrangement to form a dimer, stabilized by disulfide bonds. Finally, tetramers are generated by the parallel alignment of two dimers, and are secreted into the extracellular space, where they associate end-to-end to form a beaded microfibrillar network.9,10,11,12,13
The UCMD phenotype can result from either inherited loss-of-function recessive mutations,1,2 or from de novo dominant mutations,3,14,15 the latter accounting for more than 50% of UCMD cases.14,15,16,17,18 Bethlem myopathy is a milder allelic disorder, mostly caused by dominantly acting mutations,16,19,20,21,22 making dominant mutations the most common mutational mechanism in the COL6 genes. In-frame deletions in the N-terminal part of the triple helical domains of either of the three COL6 genes are a frequent dominant mechanism causing UCMD, as we and others have reported.3,14,15 In particular, in-frame skipping of exon 16 (caused by various genomic mutations) in the COL6A3 mRNA is the single most common mutation of this type.14,15 The resulting mutant α3(VI) chain contains a deletion of 18 amino acids near the aminoterminus of triple helical domain, but preserves the cysteine residue critical for higher order assembly.11,14,15 This mutation is presumed to exert a strong dominant-negative effect,3,14 as most tetramers will incorporate at least one mutant chain. Consequently, normal localization of collagen VI to the basement membrane surrounding muscle fibers is lost.3,15,23 In contrast, haploinsufficiency for the collagen VI genes is not associated with a clinical phenotype.1,15,24
In this study, we investigated the potential of RNA interference (RNAi) as a molecular therapeutic approach for dominant UCMD. Introduction of small interfering RNA (siRNA) into mammalian cells triggers RNAi, an endogenous intracellular pathway aimed at regulating gene expression and in which the RNA-induced silencing complex plays a central role.25,26 Using siRNA, we selectively suppressed expression of transcripts carrying the most frequent exon-skipping mutation causing UCMD – skipping of exon 16 in COL6A3 – in UCMD-derived fibroblasts, without affecting the wild-type (WT) transcripts. This selective knockdown subsequently reduced intracellular retention and increased the abundance and quality of the collagen VI extracellular matrix produced by the treated cells.
Results
siRNAs designed to bind the exon15/17 junction of COL6A3 silence the mutant mRNA with variable allele specificity
Skipping of exon 16 (henceforth designated as Δ16) of COL6A3 creates a novel junction between exons 15 and 17 in the mRNA transcripts arising from the mutant allele. This defines a unique target sequence for siRNA binding (Figure 1a). We designed a set of 11 siRNA oligos that sequentially tile across this mutant exon 15/17 junction (Figure 1b). The siRNA duplexes were stabilized by the introduction of two dT overhangs at the 3′ end of each 19-mer.25 To maximize entry of the guide (antisense) rather than the nonactive passenger strand into the RNA-induced silencing complex, a mismatch was also introduced at position 1 (5′ end) of the guide strand (Figure 1b).27
Figure 1.

Design of small interfering RNA (siRNA) oligos to target the mutant COL6A3 mRNA transcripts skipping exon 16 (Δ16). (a) Schematic diagram representing the hypothesis that siRNAs designed to target the COL6A3 exon15/17 junction, when loaded into the RNA-induced silencing complex, would specifically recognize and bind the mutant mRNA (Δ16), thus promoting its cleavage and clearance from the cytoplasm. In contrast, these oligos would not bind to the normal mRNA, because of insufficient base-pairing recognition. (b) Sequences of the 11 siRNA oligos targeting the exon 15/17 junction. Each oligo includes a mismatch at position 19 of the passenger (upper) strand (underlined). The seed region on the guide (lower) strand is also underlined.
Fibroblasts of muscle and dermal origins strongly express collagen VI,28 so that patient-derived dermal fibroblasts could be used in this study. To test their efficacy and allele specificity, the 11 siRNA oligos were transfected into cultured dermal fibroblasts derived from 2 UCMD patients with Δ16 mutations in COL6A3 (UCMD1 and UCMD2; Supplementary Figure S1), and 48 hours later, the total RNA was isolated from these cells. The mutant (Δ16) and the WT mRNA fragments were easily distinguished by their size difference (54 bp) upon gel electrophoresis following an unsaturated (26 cycles) reverse transcription PCR (RT-PCR; Figure 2a,b). Allele-specific knockdown is reported here as the ratio of intensity of the mutant over the WT fragments (Δ16/WT), so that 1 would designate no differential knockdown, while 0.5 and 0.25 would signify a 50 and 75% knockdown of the mutant relative to the WT allele, respectively (Figure 2a,b). With the exception of siRNA-8 and siRNA-9, all siRNA oligos achieved knockdown of the mutant compared to the WT transcripts, relative to a scrambled control (Figure 2a,b). siRNA-1 and siRNA-2 were the most potent, showing up to 81% of specific knockdown of the mutant allele at 48 hours (Figure 2a,b). In a dose–response experiment, siRNA-1 and siRNA-2 were found to be efficient at all concentrations tested, as shown by the decreased Δ16/WT ratio; however their allele specificity was optimal between 5 and 50 nmol/l concentrations (Supplementary Figure S2). At 1 nmol/l, the reduced allele specificity may be due to the only modest knockdown of the mutant allele; whereas at 100 nmol/l, it may be a consequence of the concomitant knockdown of the WT allele, in addition to the mutant, as both RT-PCR fragments were reduced in intensity on gel (Supplementary Figure S2a).
Figure 2.

In vitro silencing of mutant COL6A3 mRNA transcripts (Δ16) is achieved by allele-specific small interfering RNA (siRNA) oligos in two congenital muscular dystrophy type Ullrich (UCMD) fibroblast lines. (a,b) Agarose gel representing a single transfection (48 hours) of each siRNA (including a scrambled control – Scr) into two UCMD fibroblast cell lines (UCMD1, a; UCMD2, b). Semiquantitative RT-PCR was performed using primers in exons 14 and 20, and was ended after 26 cycles, to prevent saturation. The lower band on the gels represents the mutant transcript (Δ16). The intensity of each fragment was quantified using ImageJ, and the allele-specific knockdown was reported as the intensity ratio of the mutant over the wild-type (Δ16/WT) fragments (graphs shown below each gel image). Each bar on the graphs represents the average of three single transfections (amplified three times each) ± standard error of the mean (SEM). (c) Schematic of the COL6A3 mRNA from the wild-type (WT) and the mutant (Δ16) alleles, showing the position of the primers used for the quantitative RT-PCR (qRT-PCR) experiment. (d,e) The relative expression of only the WT COL6A3 transcripts (that include exon 16, primers placed in exons 15 and 16, upper panels) or of the total COL6A3 transcripts (primers placed in exons 9 and 10, lower panels) was measured by qRT-PCR, using β-actin (ACTB) and phosphoglycerate kinase 1 (PGK1) as endogenous controls, in fibroblasts from (d) UCMD1 and (e) UCMD2 following transfection of siRNA oligos. Graphs depict the 2−ΔCt data. Each bar is the average of three single transfections (each amplified in triplicate) ± SEM. *P ≤ 0.05, one-tailed Mann–Whitney. For UCMD2, comparison of scrambled- versus siRNA-11 treatments could not be completed (n = 2 for these datapoints).
The seed regions of both siRNA-1 and siRNA-2 lie in exon 17 of the COL6A3 mRNA, and thus in regions shared by both the WT and mutant transcripts. To carefully assess whether these siRNAs can incidentally knockdown the WT COL6A3 mRNA and thus compromise allele specificity, we used a quantitative RT-PCR (qRT-PCR) assay to specifically amplify WT transcripts (using primers located in exons 15 and 16, thus only amplifying transcripts that include exon 16; Figure 2c), and measure their level of expression following siRNA treatment (Figure 2d,e). A qRT-PCR assay amplifying all transcripts (using primers located in exons 9 and 10, thus amplifying both WT and mutant alleles; Figure 2c) was used for comparison (Figure 2d,e). In UCMD1 and UCMD2 cell lines, the specific expression of the WT COL6A3 allele was not decreased 48 hours after siRNA treatment (one-tailed Mann–Whitney t-test, siRNA-Scr versus siRNA-1 or siRNA-2, P > 0.05), suggesting that siRNA-1 and siRNA-2 specifically downregulate their intended target – mutant COL6A3 transcripts skipping exon 16 – without significant effect on the normal transcripts. In contrast, the relative expression of both the WT and the mutant transcripts were reduced after siRNA-11 treatment, whereas treatment with siRNA-9 had no effect on their expression levels (Figure 2d,e). Because of their efficacy and allele specificity, siRNA-1 and siRNA-2 were chosen for further characterization.
siRNA-1 and siRNA-2 suppress protein expression from a mutant, but not from a WT, α3(VI) triple helical domain reporter construct
Partial complementarity of a siRNA or miRNA to the targeted mRNA can lead to inhibition of translation without cleavage of the mRNA transcript.29 To confirm the ability of siRNA-1 and siRNA-2 to suppress protein expression only from the mutant allele, we generated green fluorescent protein (GFP)-fused reporter constructs for both the WT and the mutant (Δ16) triple helical domains (Figure 3a). Each plasmid (COL6A3THWT-GFP and COL6A3THΔ16-GFP) was transfected into HEK 293T cells, along with either one of the siRNAs (siRNA-1, siRNA-2, or siRNA-Scr). The effect of the siRNA oligos on the expression of the reporter constructs was examined 48 hours after cotransfections by detecting GFP signal in live cell imaging (Figure 3b). We found that compared to treatment with the scrambled siRNA, treatment with allele-specific siRNAs led to suppression of the fluorescence signal from COL6A3THΔ16-GFP transfected cells (Figure 3b, lower panel). In contrast, the allele-specific siRNAs did not substantially suppress fluorescence signal from COL6A3THWT-GFP transfected cells (Figure 3b, upper panel). GFP immunoblotting on the cell lysates confirmed that the allele-specific siRNAs preferentially suppressed COL6A3THΔ16-GFP expression, with no or only minimal suppression from the COL6A3THWT-GFP allele (Figure 3c,d).
Figure 3.

Small interfering RNA (siRNA)-mediated allele-specific reduction of protein expression from reporter constructs. (a) Schematic representation of the two constructs used in the experiments. (b) Representative live images of HEK293T cells at 48 hours posttransfection of a reporter construct (Col6A3THWT-GFP or Col6A3THΔ16-GFP) together with either one of the following siRNA oligos: siRNA-1, siRNA-2, or a scrambled control (siRNA-Scr). (c) Western-blot analysis of HEK293T cells in b. Cells were lysed 48 hours after cotransfections, and membrane was probed with polyclonal anti-GFP antibody. Tubulin is shown as a loading control. (d) Quantification of Western-blot GFP expression, normalized by tubulin, measured from three independent cotransfection experiments. *P ≤ 0.05, one-tailed Mann–Whitney.
Allele-specific silencing of the dominant-negative transcript results in reduced intracellular retention and increased extracellular matrix deposition of collagen VI in dermal fibroblast cultures
Previous work by us3,15,30 as well as by others14 has demonstrated that N-terminal triple helical in-frame deletions in COL6A1 and COL6A2 can produce mutated chains that incorporate along with WT chains into tetramers, which are then partially retained inside the cell, but are also secreted and yet incapable of forming a normal matrix as a result of the dominant-negative effect of the mutation. Similarly, fibroblasts from patients with exon 16-skipping mutations in COL6A3 also show intracellular retention of collagen VI and a reduced and disorganized matrix in culture. However, because of the large size of the α3(VI) chain, the presence of numerous alternative-splicing events and of heavily-glycosylated epitopes, immunoblot analyses cannot conclusively discriminate between the mutant and the WT chains, and cannot conclusively demonstrate that the mutant α3(VI) chain is secreted as a result of its assembly with the WT chain.
To first specify the compartment in which the intracellular collagen VI was retained, we used colocalization studies, and found that it was mainly localized with the ER marker HSP47 (Supplementary Figure S3a), but not with the Golgi marker giantin, nor with the lysosomal marker H4B4 (Lamp2) (Supplementary Figure S3b,c). We also tested whether the canonical ER stress pathway or the unfolded protein response were engaged by the retained mutant collagen VI; however, we found no evidence of activation of several markers (Ero1, Calnexin, XBP1, IRE1, S2P, CHOP, HSP47, BIP, and PDI) by immunoblot or RT-PCR (data not shown). It remains possible that there are alternative pathways of the mediation of ER stress that are not covered by the markers used here.
We next hypothesized that if skipping of exon 16 of COL6A3 does exert a dominant-negative interaction, elimination of the protein product generated from the mutant allele should remove this interference with the WT protein, thereby allowing for proper collagen VI assembly and significantly improved matrix deposition. We thus tested whether treatment with allele-specific siRNA oligos, in addition to effectively knocking-down the mutant transcripts, was also able to rescue the secretion and deposition of a collagen VI matrix in patient-derived fibroblast cells. Taking into consideration the relatively long half-life time of collagens in general,31 and accounting for the time it would take to completely deposit a matrix in culture, we performed three to five consecutive transfections (every 2–3 days) of the allele-specific siRNAs (5 nmol/l) in UCMD fibroblast cultures, using scrambled siRNA oligo (siRNA-Scr) as a control. Medium was then supplemented with L-ascorbic acid and cells were allowed an additional 3 to 5 days in culture, for final optimization of collagen synthesis and secretion.
We studied the intracellular retention of collagen VI by using immunofluorescence on permeabilized cells, and showed that the retention of collagen VI was markedly reduced after treatment with siRNA-1 and siRNA-2, compared to a control siRNA, for UCMD1 and UCMD3 (Figure 4a). The intracellular fraction of collagen VI was also assessed by carefully washing the trypsinized cells before the lysis, to remove all extracellular matrix. The immunoblot further confirmed the reduction of intracellular collagen VI following siRNA-2 treatment (Figure 4b). In a separate set of experiments, we assessed the impact of the siRNA oligos on the quantity of deposited matrix, using a confocal microscopy-based volumetric analysis of collagen VI matrix. In a normal control fibroblast culture (unaffected), we found that neither siRNA-1 nor siRNA-2 had an effect on the deposition of collagen VI extracellular matrix, which appeared similar to the control (siRNA-Scr) culture (Figure 5a, upper panel). Upon quantification, there was no significant difference in the collagen VI volume between cultures treated with allele-specific siRNAs versus scrambled siRNA in normal cultures (Kruskall–Wallis and Dunn's posttest: P > 0.05 for siRNA-1/-2 versus siRNA-Scr; Figure 5b). In contrast, in two UCMD mutant fibroblast cultures, allele-specific siRNA treatments led to a significant increase in the volume of collagen VI deposited in the matrix (siRNA-1, P < 0.05, and siRNA-2, P < 0.05 for UCMD1; siRNA-1, P < 0.05 for UCMD3; Figure 5a,b). Moreover, treatments with siRNA-1 and siRNA-2 improved the colocalization of collagen VI with fibronectin, another component of the fibroblast extracellular matrix (Figure 6a).32 Notably, siRNA treatments also modified the appearance of the collagen VI microfibrils formed in the matrix from a speckled appearance to a solid and linear aspect, indicating a more normal assembly (Figure 6b–d). Thus, treatment with the mutant allele-specific siRNA oligos appeared to improve both the quantity and the quality of the matrix, while at the same time decreasing intracellular retention, suggesting that with the treatment the WT isoform is freed up to be secreted, and to be deposited into the matrix.
Figure 4.

Reduced intracellular retention of collagen VI following small interfering RNA (siRNA) treatment in cultured congenital muscular dystrophy type Ullrich (UCMD) fibroblasts. (a) Immunofluorescence staining of collagen VI on permeabilized cells following three consecutive treatments with siRNA-1, siRNA-2, or siRNA-Scr (scrambled control), on two UCMD cell lines (UCMD1 and 3). (b) Immunofluorescence staining of collagen VI on permeabilized cells following three consecutive treatments with siRNA-2 or control siRNA-Scr, and corresponding immunoblot from these cell lysates (right panel) showing the intracellular fraction of collagen VI. Cells were washed three times before collected and lysed, to subtract the collagen secreted in the medium.
Figure 5.
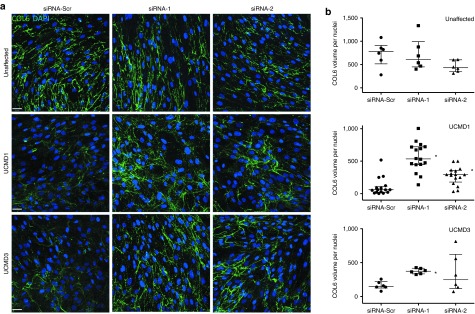
Allele-specific silencing of the exon 16-skipped mRNA increases extracellular collagen VI matrix deposition in cultured congenital muscular dystrophy type Ullrich (UCMD) fibroblasts. (a) Representative images of immunofluorescence staining of collagen VI matrix, as secreted and deposited by one normal (Unaffected) and two UCMD fibroblast cell lines (UCMD1 and UCMD3). Four consecutive small interfering RNA (siRNA) treatments (once every 48h) were performed with either siRNA-1, siRNA-2, or the scrambled control siRNA-Scr. Following the last treatment, culture medium was supplemented with L-ascorbic acid, and cells were maintained in culture 3 to 5 days to allow for collagen VI secretion and matrix deposition. Stacks of confocal microscopy images were acquired (section thickness of 0.5 μm, one image every 0.5 μm), and are presented as a merge. Scale bar = 38 μm. (b) Total collagen VI volume in the matrix was quantified in each image (between 6 and 10 images per experiment, where each experiment was performed either once or in duplicate) by measuring the sum of the volume of all objects displaying a longest axis greater than 15 μm, using Volocity. Total volume was normalized by the number of nuclei in each image, and plotted as dot-plot. Bars indicate the median and interquartile range. Kruskall–Wallis and Dunn's tests were applied. *P < 0.05.
Figure 6.

Allele-specific silencing of the exon-16 skipped mRNA improves the quality of the newly formed collagen VI matrix. (a) Immunofluorescence staining of collagen VI (green), co-stained with fibronectin (red) as secreted and deposited in scrambled small interfering RNA (siRNA)-treated samples (one patient, UCMD1, and unaffected cell line), or in siRNA-1 and siRNA-2–treated samples (UCMD1). Four consecutive siRNA treatments (once every 48 hours) were performed with either siRNA-1, siRNA-2, or the scrambled control siRNA-Scr. Following the last treatment, culture medium was supplemented with L-ascorbic acid, and cells were allowed 3 days for collagen VI to be secreted and deposited into the matrix. Images were taken on a confocal microscopy system. Scale bar = 12 μm. (b) Insets from images in a. (c,d) Qualitative assessment of collagen VI microfibrils. Five independent observers blinded to the treatment condition scored 85 different collagen VI microfibril images, selected from four different conditions (UCMD1 cells treated with siRNA-1, siRNA-2, or control siRNA-Scr, and cells from an unaffected individual treated with control siRNA-Scr). The aspect of the microfibrils was categorized as either “discontinuous/speckled”, “partially continuous”, or “continuous/solid”. (c) Examples of collagen VI microfibril images that were unambiguously scored as “discontinuous/speckled”, “partially continuous”, or “continuous/solid”. (d) Histogram of combined scoring results depicting the fraction of images that were scored as either “discontinuous/speckled”, “partially continuous”, or “continuous/solid” for each treatment condition.
Discussion
In this study, we tested an RNAi-based therapeutic targeting a dominant-negative exon-skipping mutation as a potential treatment for UCMD. Dominant-negatively acting mutations are frequent in the collagen VI genes, in particular in-frame exon skipping or glycine substitutions at the Gly-X-Y motifs located at the N-terminal end of the triple helical domain.4,14,15,16,33 In this location, mutations are “assembly-competent” explaining their strong dominant-negative effect.3 Molecular treatment design for this type of mutation is challenging, but the development of RNA-based strategies, such as RNAi to abolish the synthesis of the dominant-negative interfering protein product emerges as an appropriate therapeutic avenue. For this approach to be successful, it is important to ascertain (i) the functional consequence of haploinsufficiency, (ii) the appropriate cell type to which the compounds need to be delivered, and (iii) allele specificity of the therapeutic compounds. We as well as others have provided strong evidence that haploinsufficiency for any of the COL6 genes does not cause a phenotype, from the observation that parents of patients carrying loss-of-function UCMD mutations (either premature stop mutations or large genomic deletions, even spanning two of the collagen VI genes) can show a reduced collagen VI production, and yet are unaffected.1,2,24 We have also previously shown that the cellular source for collagen VI and hence the cell to be targeted in mature muscle is the interstitial fibroblast,28 making fibroblasts an appropriate cellular model for this study. Our current study now demonstrates that siRNA oligos can act as potent allele-specific probes to target the common exon 16-skipping mutation in COL6A3, and that they can alleviate several of the biochemical consequences of this mutation.
Two approaches were employed in our experiments to assess the mRNA transcript allele specificity of the siRNA oligos: unsaturated RT-PCR to compare the relative expression of both the mutant and the normal allele, and quantitative RT-PCR to measure the abundance of the normal transcripts (i.e., including exon 16) (Figure 2). Out of the systematic series of 11 oligos we used and which cover the mutant exon 15/17 junction, two (siRNA-1 and siRNA-2) displayed considerable allele specificity on both analyses. The high efficacy and allele specificity of siRNA-1 and siRNA-2 was further supported by the observation that they more specifically suppressed protein synthesis from a mutant (Δ16) compared to a WT reporter construct in cotransfection experiments (Figure 3), suggesting that there are also no adverse effects of the siRNA on WT translation. The mild decrease of the WT protein expression observed in cotransfection experiments for siRNA-1 (Figure 3d) could be the consequence of a higher siRNA:RNA ratio in this experimental cell model. In cultured fibroblasts, the two oligos maintained allele specificity up to a concentration that was 10-fold higher than the most effective concentration of 5 nmol/l, indicating a broad therapeutic range, i.e., the range in which the oligos are effective and yet specific, with a loss of specificity only occurring at 20-fold higher concentration (100 nmol/l) (Supplementary Figure S2). Although a mild degree of knockdown of the WT transcript in conjunction with significant knockdown of the mutant allele may not cause major problems, reducing the allele-specificity would certainly result in the treatment to lose efficacy. It remains to be determined at precisely which ratio of the mutant to WT allele improvement would still occur; however, clinical observation suggests that merely reducing the amount of mutant versus WT allele would still be clinically beneficial. This notion is supported by our observation of individuals with a mild Bethlem phenotype caused by somatic cellular mosaicism for a dominant-negative severe UCMD mutation, where decreased expression of the mutant in favor of the WT allele is the basis for the milder then expected phenotype (Donkervoort S., Hu Y., et al. unpublished data). Thus, even if partly complete, allele-specific knockdown would still modify the severe UCMD phenotype toward a milder Bethlem-like phenotype.
In-frame skipping of exon 16 of COL6A3 gene results in the deletion of 18 triple helical amino acid residues, while the critical cysteine residue thought to be important for monomer and dimer formation is retained,14,15 thus presumably allowing for mutant chain incorporation into the dimer and likely tetramer (Figure 7). As a consequence, mutant-containing tetramers may be secreted but not assembled into a normal matrix (Figure 7).14,15,30 Our current data now serve as proof-of-principle that this particular type of mutation does in fact act in a dominant-negative way in that we show that treatment with siRNA alleviates the collagen VI intracellular retention (Figure 4), and restores the secretion and deposition of a recognizable collagen VI matrix (Figures 5 and 6). Thus, depletion of the mutant chain appears to free up the normal chain to assemble a matrix.
Figure 7.

Correction of the dominant-negative impact on collagen VI assembly of the COL6A3 Δ16 mutation by small interfering RNA (siRNA) treatment. Schematic representation of the collagen VI assembly process (a) in normal conditions, and (b) in the presence of the mutated α3(VI) chain, resulting from the skipping of exon 16 in COL6A3. In a, the three chains – α1(VI), α2(VI), and α3(VI) – are incorporated with equal stoichiometry to form a monomer, followed by a stepwise process to generate normal functioning tetramers for secretion. In b, two different α3(VI) chains, encoded by the normal and the mutated allele, are produced and are allowed to incorporate in the monomer. This results in half of the monomers carrying a mutated chain (ratio of 1:1 of the normal versus the mutant monomer). The mutated monomer can also form dimers, such that about 3 out of 4 dimers will contain at least one mutated chain (ratio of 1:3 of the normal versus mutant dimers). Finally, 15 out of 16 tetramers should retain at least one mutated chain (ratio of 1:15), which are partially retained inside the cell or are secreted and incapable of forming a normal matrix. This explains the strong dominant-negative effect of this mutation on the collagen VI assembly. Representation of the consequence of the siRNA treatment on the collagen VI assembly if knockdown (KD) of the mutated allele is of 100% (c), or of 75% (d). In c, the mutated α3(VI) is not produced, because of the siRNA KD of the mutant mRNA. Thus all tetramers formed are fully functional and can be secreted outside the cell. However, if expression from the normal allele is not overregulated, the total amount of tetramers formed should be half of what a normal cell produces. Nevertheless, since haploinsufficiency of collagen VI does not cause a phenotype, this situation corresponds to the ideal therapeutic scenario. In d, even if the KD is only of about 75 %, which was about the KD observed in vitro for siRNA-1 and siRNA-2, the downstream effect is a net increase in the ratio of functional tetramers (10:15, compared to 1:15 without siRNA treatment).
In conclusion, our results contribute to establish allele-specific RNAi as a promising molecular approach for the treatment of dominant collagen VI-related disorders. While an epidemiological study in Northern UK found their prevalence to be of 0.9/100,000 in this geographical region,34 the broader prevalence of the COL6-related disorders is still insufficiently known. However, they have been found in various ethnic backgrounds and in fact represent the most common subgroup of congenital muscular dystrophies in UK,35 and the second most common congenital muscular dystrophies subgroup in Australia and in Japan.17,36 As dominant-negative mutations account for 50 to 75% of UCMD cases16,17,18 and nearly 100% of BM cases,16,22 the allele-specific antisense approach we tested here can eventually be adapted and applied to a significant portion of patients affected with collagen VI mutations. Recently, an antisense oligonucleotide (i.e., not an siRNA) targeting a polymorphism in cis of an in-frame exon-skipping COL6A2 mutation has been shown to promote exon skipping and out-of-framing for specifically the mRNA transcripts carrying the mutation.37 Similarly, allele-specific knockdown of COL3A1, COL1A2, and COL7A1 mutations (including glycine missense mutations) associated with other types of collagen-related disorders has been accomplished by siRNA in vitro,38,39,40 and corrected for the mutation adverse effects,40 providing assurance about the broader applicability of this approach to dominant mutations in collagens. The hurdles of using siRNA for in vivo application in humans are their poor stability and tissue delivery26,41,42; however, recent advances in siRNA pharmaceuticals successfully improved the physical and functional properties of these compounds by using a variety of chemical modifications.41,42 An attractive aspect of oligo-based approach in general is the reversible nature of the effects as it provides an additional safety margin. The next step will be to bring the siRNA tested in this study into preclinical studies, using an appropriate animal model to determine their in vivo efficacy and allele specificity and the optimal delivery method to reach the interstitial fibroblasts. We are in the process of characterizing a mouse model of the deletion of exon 16 in the Col6a3 as a model of the human UCMD mutation studied here (unpublished data). This model will be of great value to show the in vivo potential of RNAi to treat dominant mutations in the collagen VI-related disorders.
Materials and methods
UCMD patient samples. Four patients (UCMD1 to UCMD4) with mutations leading to in-frame skipping of COL6A3 exon 16 and a clinical diagnosis of UCMD were selected (Supplementary Figure S1). The genetic and clinical studies were performed in accordance with an institutional review board-approved protocol and after informed consents were obtained. For all patients we showed the presence of both the WT and the exon 16-deleted transcripts by an RT-PCR analysis performed on RNA isolated from primary fibroblast cultures. The in-frame skipping of exon 16 was confirmed by sequencing of the amplified cDNA product. In all patients, a de novo genomic splice site mutation was demonstrated by genomic sequencing. Dermal fibroblast cultures were established from skin biopsies of each UCMD patient and an unaffected individual.
siRNA oligos. The siRNA oligos were synthesized by Invitrogen/Life Technologies (Carlsbad, CA) and annealed following Invitrogen's protocol. A scrambled oligo with similar CG content (Stealth RNAi negative control, Invitrogen) was used as a control.
Reporter constructs. To create constructs expressing WT and mutant α3(VI) triple helical domains fused to a GFP reporter, the WT and mutant triple helical domains were RT-PCR amplified from RNA isolated from patient-derived dermal fibroblasts culture. Primers used for RT-PCR were 5′-aacttggagcggctaatgc-3′ and 5′-tcaagaccacatctcgcatc-3′. Wild-type and mutant PCR products were gel separated, purified, and cloned into pCDNA3.1/CT GFP TOPO (Invitrogen). Both constructs pcDNA3.1/COL6A3THWT-GFP and pcDNA3.1/COL6A3THDel-GFP were verified by sequencing.
Cell culture and transfection. Primary dermal fibroblasts and HEK 293T cells were cultured in Dulbecco's modified Eagle medium supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin at 37 °C in 5% CO2. Lipofectamine 2000 or RNAiMax (Invitrogen) was used to transfect primary dermal fibroblasts and HEK 293T cells according to the manufacturer's reverse transfection protocol. Briefly, siRNAs were diluted in OPTI-MEM with Lipofectamine, and transfected using 12- or 6-well plates. siRNA final concentration was 5 nmol/l unless otherwise indicated. Dulbecco's modified Eagle medium supplemented with 10% FBS, but without antibiotics, was added to each well. In the case of sequential transfections, fibroblasts were replated and retransfected 48 hours after the previous transfection. For reporter construct transfection experiments, HEK 293T cells were cotransfected with 12.5 nmol/l siRNAs and 1 µg of the WT or mutant reporter construct DNA.
RT-PCR and gel analysis. Forty-eight to 72 hours after siRNA transfection, total RNA was isolated from fibroblasts using the miniRNeasy Kit (Qiagen Sciences, Germantown, MD). Reverse transcription reactions were performed with 0.3 to 0.5 µg of RNA and random primers, using the TaqMan Reverse Transcription Kit or the SuperScript III Reverse Transcriptase (Life Technologies), following manufacturers' protocols. To assess the silencing of the mutant allele, a nonsaturating PCR protocol was developed to distinguish the mutant from the WT allele by molecular weight separation on a gel. PCRs were performed with 1 µl of cDNA in a total volume of 20 µl, using Taq Advantage 2 (Clontech, Mountain View, CA) and the following primers: 5′-atgtatgacaggcccctg-3′, 5′-tcgaatcccaacatctcct-3′. The amplification cycling was as followed: a first denaturation at 95 °C for 1 minute, followed by 5 cycles of 95 °C for 30 seconds, and 68 °C for 30 seconds, then 5 cycles of 95 °C for 30 seconds, 64 °C for 30 seconds, and 68 °C for 30 seconds, and then 15 or 16 cycles of 95 °C for 30 seconds, 55 °C for 30 seconds, and 68 °C for 30 seconds, for a total of 25 or 26 cycles. A final extension stage of 1 minute at 68 °C was performed. PCR products were run on a 2 to 3% agarose gel, and images were obtained on a Molecular Imager Gel Documentary System Gel Doc XR (Bio-Rad Laboratories, Hercules, CA). The intensity of each PCR band was quantified using ImageJ software, version 1.6 (Research Services Branch, National Institute of Mental Health, Bethesda, MD). Briefly, images were converted to 8-bit, and background was subtracted using a rolling ball radius of 20 pixels. Peaks representing each band were obtained using the Gel Analysis tool, and measures of the area under each peak were used for calculating the ratio of the mutant over the WT allele. Alternatively, the integrated density of each band was calculated with the Wand tool after establishing a threshold to eliminate the background, and these measures were used to calculate the ratio.
Quantitative PCR (qPCR). The FastStart Universal Probe Master (ROX) (Roche Applied Science, Penzberg, Germany) was used to perform the qPCR reactions. Two microliters of diluted cDNA (dilution of 1/10 in RNAse-free water) was used in a total volume of 10 µl for each qPCR reaction. Primers were added to a final concentration of either 500 or 700 nmol/l each, and the probe to a final concentration of 250 or 400 nmol/l. The primers/probe combinations were designed using the Universal ProbeLibrary Assay Design Center (Roche Applied Science) and were as follows: COL6A3 (WT allele) 5′-ggttccctgcaagtgctc-3′, 5′-accaggatagcctcggtagc-3′, and probe number 80 (Universal ProbeLibrary, Roche Applied Science); COL6A3 (exons 9–10) 5′-ccagaagctcttagccagca-3′, 5′- tgtctgcagcatcactctca-3′, and probe number 10; ACTB 5′ ccaaccgcgagaagatga 3′, 5′ ccagaggcgtacagggatag3′, and probe number 64; PGK1 5′ cagctgctgggtctgtcat 3′, 5′gctggctcggctttaacc 3′, and probe 67. Reactions were run in triplicate on a 7900HT instrument (Applied Biosystems/Life Technologies, Carlsbad, CA), using the default amplification settings, but 57 °C as the annealing/extension temperature. Relative expression was determined with the 2−ΔCt method normalized by ACTB and PGK1.
Live fluorescent imaging and Western blotting. Forty-eight hours after cotransfection of siRNA and reporter plasmid DNA into HEK 293E cells, the fluorescence signal emanating from the WT or mutant α3(VI) collagen domain-GFP fusion protein was observed and recorded with a Hamamatsu digital camera (Hamamatsu, Japan) mounted on an inverted microscope (Nikon TE300, Nikon Instruments, Tokyo, Japan). HEK293 cells were harvested and lysates were processed for immunoblotting with polyclonal anti-GFP antibody (Molecular Probes, Eugene, OR). Quantification of protein expression was done from the Western blot images using the integrated density measure tool of ImageJ software. GFP expression was normalized to tubulin. For immunobloting assessing intracellular retention of collagen VI in fibroblasts, cells were trypsinized, and washed at the time of harvesting. Cells were then lysed using lysis buffer (Cell Signaling Technology, Danvers, MA) containing protease inhibitors (Roche Applied Science). Cell lysates were then proceeded to immunoblot with anti-COL6A2 rabbit polyclonal antibody (F225, kindly provided by M.L. Chu).
Immunofluorescence staining. After three to five consecutive siRNA transfections, primary fibroblasts were plated and transfected on glass chamber slides (BD Biosciences, San Jose, CA) to a confluence of 40–50%. After 24 hours, the medium was replaced with Dulbecco's modified Eagle medium supplemented with 10% FBS and 50 µg/ml ascorbic acid for an additional 3 to 5 days. Every 48 hours, a supplement of ascorbic acid was added without changing the medium, for a final concentration of 50 µg/ml. At the time of harvesting, cultures were fixed with 4% PFA for 10 minutes, rinsed twice in phosphate-buffered saline (PBS), and kept in PBS before to proceed. For immunofluorescence staining, slides were blocked with 10% FBS in PBS for 1 hour, and incubated with a combination of anti-collagen type VI antibody (MAB1944; diluted 1:2,000, Chemicon International, Temecula, CA) and anti-fibronectin antibody (F3648; diluted 1:800, Sigma-Aldrich, St Louis, MO) in blocking buffer. Slides were incubated with the primary antibodies for 1 hour at RT or overnight at 4 °C, and washed twice in PBS for 5 minutes. Secondary antibodies (goat anti-mouse-488 and goat anti-rabbit-568; Life Technologies) were diluted 1:500 each in blocking buffer and applied on the section for 1 hour at RT. After two washes in PBS, sections were stained with 4',6-diamidino-2-phenylindole (Cell Biolabs, San Diego, CA) diluted 1:1,000 in PBS for 2 minutes or with Hoescht 33342 (Life Technologies) at 5 µg/ml in PBS for 5 minutes, and washed in PBS, and distillated water. Slides were mounted with Fluoromount (SouthernBiotech, Birmingham, AL). For staining of intracellular collagen VI staining, the same protocol was applied, but triton was added in the blocking buffer to a final concentration of 0.1%.
Confocal imaging and analysis. For matrix quantification, stack images of ×400 magnification were acquired on a TCS SP5 II system (Leica Microsystems, Wetzlar, Germany). Pinhole was adjusted to obtain a section thickness of 0.508 µm, and z-stacks were acquired using 0.5 µm-sized steps. Image sequences were imported into Volocity 3D Image Analysis Software, version 6.1 (PerkinElmer, Waltham, MA). To quantify the signal emanating from the collagen VI matrix, the Find Objects tool was threshold to eliminate background pixels (by using a pixel intensity threshold of at least 4), and to find objects of long axis (by using a longest axis threshold of at least 15 µm). The sum of the volume (µm3) in each image was used as the measure of the total volume occupied by the collagen VI matrix. The number of nuclei in each image was used to normalize. To select images for the qualitative scoring of collagen VI microfibril appearance, at least six fields of confocal images were captured for each treatment condition as described above, then three to four representative areas of 45-μm length were selected in each field (UCMD1: n = 19 for siRNA-Scr, n = 23 for siRNA-1, n = 19 for siRNA-2; unaffected: n = 24 for siRNA-Scr). The resulting 85 images were deidentified and scrambled and then scored by 5 independent observers blinded to the treatment conditions. The combined scores were expressed as the fraction of images called “discontinuous/speckled”, “partially continuous”, or “continuous/solid” as determined for each treatment group.
Statistics. Graphics and statistical analyses were performed with GraphPad Prism version 5.00 (GraphPad Software, San Diego, CA). Data in histograms are presented as mean ± standard error of the mean. For quantitative RT-PCR and immunoblot GFP expression, one-tailed Mann–Whitney t-test was performed. For collagen VI volume in extracellular matrix, Kruskall–Wallis test was used and Dunnet's test was used for multiple comparison tests. A significance level of 0.05 was used.
SUPPLEMENTARY MATERIAL Figure S1. Genetic and histological description of the UCMD patients used in this study. Figure S2. Allele specificity of siRNA-1 and siRNA-2 at concentration varying between 1 nM and 100 nM. Figure S3. The intracellular retained collagen VI in UCMD fibroblasts is mainly located in the endoplasmic reticulum (ER), but does not activate the unfolded protein response (UPR).
Acknowledgments
We thank Ying Hu for technical assistance, as well as Sungyoung Auh, Ami Mankodi, Carlo Rinaldi, and Laura Bott for critical review of the manuscript and helpful discussions. We thank Mon-Li Chu for sharing antibodies. V.B. is the recipient of a Canadian Institutes of Health Research fellowship. This study was initially supported by a grant from the Muscular Dystrophy Association (USA) to C.G.B., and then by funds of the NINDS intramural research program to C.G.B. The authors declared no conflict of interest.
Supplementary material
Genetic and histological description of the UCMD patients used in this study.
Allele specificity of siRNA-1 and siRNA-2 at concentration varying between 1 nM and 100 nM.
The intracellular retained collagen VI in UCMD fibroblasts is mainly located in the endoplasmic reticulum (ER), but does not activate the unfolded protein response (UPR).
References
- Camacho Vanegas O, Bertini E, Zhang RZ, Petrini S, Minosse C, Sabatelli P, et al. Ullrich scleroatonic muscular dystrophy is caused by recessive mutations in collagen type VI. Proc Natl Acad Sci USA. 2001;98:7516–7521. doi: 10.1073/pnas.121027598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demir E, Sabatelli P, Allamand V, Ferreiro A, Moghadaszadeh B, Makrelouf M, et al. Mutations in COL6A3 cause severe and mild phenotypes of Ullrich congenital muscular dystrophy. Am J Hum Genet. 2002;70:1446–1458. doi: 10.1086/340608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan TC, Zhang RZ, Sudano DG, Marie SK, Bönnemann CG, Chu ML. New molecular mechanism for Ullrich congenital muscular dystrophy: a heterozygous in-frame deletion in the COL6A1 gene causes a severe phenotype. Am J Hum Genet. 2003;73:355–369. doi: 10.1086/377107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bönnemann CG. The collagen VI-related myopathies: muscle meets its matrix. Nat Rev Neurol. 2011;7:379–390. doi: 10.1038/nrneurol.2011.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadeau A, Kinali M, Main M, Jimenez-Mallebrera C, Aloysius A, Clement E, et al. Natural history of Ullrich congenital muscular dystrophy. Neurology. 2009;73:25–31. doi: 10.1212/WNL.0b013e3181aae851. [DOI] [PubMed] [Google Scholar]
- Engel J, Furthmayr H, Odermatt E, von der Mark H, Aumailley M, Fleischmajer R, et al. Structure and macromolecular organization of type VI collagen. Ann N Y Acad Sci. 1985;460:25–37. doi: 10.1111/j.1749-6632.1985.tb51154.x. [DOI] [PubMed] [Google Scholar]
- Chu ML, Pan TC, Conway D, Saitta B, Stokes D, Kuo HJ, et al. The structure of type VI collagen. Ann N Y Acad Sci. 1990;580:55–63. doi: 10.1111/j.1749-6632.1990.tb17917.x. [DOI] [PubMed] [Google Scholar]
- Weil D, Mattei MG, Passage E, N'Guyen VC, Pribula-Conway D, Mann K, et al. Cloning and chromosomal localization of human genes encoding the three chains of type VI collagen. Am J Hum Genet. 1988;42:435–445. [PMC free article] [PubMed] [Google Scholar]
- Chu ML, Conway D, Pan TC, Baldwin C, Mann K, Deutzmann R, et al. Amino acid sequence of the triple-helical domain of human collagen type VI. J Biol Chem. 1988;263:18601–18606. [PubMed] [Google Scholar]
- Engvall E, Hessle H, Klier G. Molecular assembly, secretion, and matrix deposition of type VI collagen. J Cell Biol. 1986;102:703–710. doi: 10.1083/jcb.102.3.703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furthmayr H, Wiedemann H, Timpl R, Odermatt E, Engel J. Electron-microscopical approach to a structural model of intima collagen. Biochem J. 1983;211:303–311. doi: 10.1042/bj2110303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colombatti A, Mucignat MT, Bonaldo P. Secretion and matrix assembly of recombinant type VI collagen. J Biol Chem. 1995;270:13105–13111. doi: 10.1074/jbc.270.22.13105. [DOI] [PubMed] [Google Scholar]
- Lamandé SR, Sigalas E, Pan TC, Chu ML, Dziadek M, Timpl R, et al. The role of the alpha3(VI) chain in collagen VI assembly. Expression of an alpha3(VI) chain lacking N-terminal modules N10-N7 restores collagen VI assembly, secretion, and matrix deposition in an alpha3(VI)-deficient cell line. J Biol Chem. 1998;273:7423–7430. doi: 10.1074/jbc.273.13.7423. [DOI] [PubMed] [Google Scholar]
- Baker NL, Mörgelin M, Peat R, Goemans N, North KN, Bateman JF, et al. Dominant collagen VI mutations are a common cause of Ullrich congenital muscular dystrophy. Hum Mol Genet. 2005;14:279–293. doi: 10.1093/hmg/ddi025. [DOI] [PubMed] [Google Scholar]
- Lampe AK, Zou Y, Sudano D, O'Brien KK, Hicks D, Laval SH, et al. Exon skipping mutations in collagen VI are common and are predictive for severity and inheritance. Hum Mutat. 2008;29:809–822. doi: 10.1002/humu.20704. [DOI] [PubMed] [Google Scholar]
- Allamand V, Briñas L, Richard P, Stojkovic T, Quijano-Roy S, Bonne G. ColVI myopathies: where do we stand, where do we go. Skelet Muscle. 2011;1:30. doi: 10.1186/2044-5040-1-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peat RA, Smith JM, Compton AG, Baker NL, Pace RA, Burkin DJ, et al. Diagnosis and etiology of congenital muscular dystrophy. Neurology. 2008;71:312–321. doi: 10.1212/01.wnl.0000284605.27654.5a. [DOI] [PubMed] [Google Scholar]
- Briñas L, Richard P, Quijano-Roy S, Gartioux C, Ledeuil C, Lacène E, et al. Early onset collagen VI myopathies: Genetic and clinical correlations. Ann Neurol. 2010;68:511–520. doi: 10.1002/ana.22087. [DOI] [PubMed] [Google Scholar]
- Pan TC, Zhang RZ, Pericak-Vance MA, Tandan R, Fries T, Stajich JM, et al. Missense mutation in a von Willebrand factor type A domain of the alpha 3(VI) collagen gene (COL6A3) in a family with Bethlem myopathy. Hum Mol Genet. 1998;7:807–812. doi: 10.1093/hmg/7.5.807. [DOI] [PubMed] [Google Scholar]
- Jöbsis GJ, Keizers H, Vreijling JP, de Visser M, Speer MC, Wolterman RA, et al. Type VI collagen mutations in Bethlem myopathy, an autosomal dominant myopathy with contractures. Nat Genet. 1996;14:113–115. doi: 10.1038/ng0996-113. [DOI] [PubMed] [Google Scholar]
- Pepe G, Giusti B, Bertini E, Brunelli T, Saitta B, Comeglio P, et al. A heterozygous splice site mutation in COL6A1 leading to an in-frame deletion of the alpha1(VI) collagen chain in an italian family affected by bethlem myopathy. Biochem Biophys Res Commun. 1999;258:802–807. doi: 10.1006/bbrc.1999.0680. [DOI] [PubMed] [Google Scholar]
- Hicks D, Lampe AK, Barresi R, Charlton R, Fiorillo C, Bonnemann CG, et al. A refined diagnostic algorithm for Bethlem myopathy. Neurology. 2008;70:1192–1199. doi: 10.1212/01.wnl.0000307749.66438.6d. [DOI] [PubMed] [Google Scholar]
- Ishikawa H, Sugie K, Murayama K, Awaya A, Suzuki Y, Noguchi S, et al. Ullrich disease due to deficiency of collagen VI in the sarcolemma. Neurology. 2004;62:620–623. doi: 10.1212/01.wnl.0000113023.84421.00. [DOI] [PubMed] [Google Scholar]
- Foley AR, Hu Y, Zou Y, Yang M, Medne L, Leach M, et al. Large genomic deletions: a novel cause of Ullrich congenital muscular dystrophy. Ann Neurol. 2011;69:206–211. doi: 10.1002/ana.22283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elbashir SM, Harborth J, Lendeckel W, Yalcin A, Weber K, Tuschl T. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature. 2001;411:494–498. doi: 10.1038/35078107. [DOI] [PubMed] [Google Scholar]
- Burnett JC, Rossi JJ. RNA-based therapeutics: current progress and future prospects. Chem Biol. 2012;19:60–71. doi: 10.1016/j.chembiol.2011.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz DS, Hutvágner G, Du T, Xu Z, Aronin N, Zamore PD. Asymmetry in the assembly of the RNAi enzyme complex. Cell. 2003;115:199–208. doi: 10.1016/s0092-8674(03)00759-1. [DOI] [PubMed] [Google Scholar]
- Zou Y, Zhang RZ, Sabatelli P, Chu ML, Bönnemann CG. Muscle interstitial fibroblasts are the main source of collagen VI synthesis in skeletal muscle: implications for congenital muscular dystrophy types Ullrich and Bethlem. J Neuropathol Exp Neurol. 2008;67:144–154. doi: 10.1097/nen.0b013e3181634ef7. [DOI] [PubMed] [Google Scholar]
- Carthew RW, Sontheimer EJ. Origins and Mechanisms of miRNAs and siRNAs. Cell. 2009;136:642–655. doi: 10.1016/j.cell.2009.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimenez-Mallebrera C, Maioli MA, Kim J, Brown SC, Feng L, Lampe AK, et al. A comparative analysis of collagen VI production in muscle, skin and fibroblasts from 14 Ullrich congenital muscular dystrophy patients with dominant and recessive COL6A mutations. Neuromuscul Disord. 2006;16:571–582. doi: 10.1016/j.nmd.2006.07.015. [DOI] [PubMed] [Google Scholar]
- Kern P, Menasche M, Robert L. Relative rates of biosynthesis of collagen type I, type V and type VI in calf cornea. Biochem J. 1991;274 (Pt 2):615–617. doi: 10.1042/bj2740615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabatelli P, Bonaldo P, Lattanzi G, Braghetta P, Bergamin N, Capanni C, et al. Collagen VI deficiency affects the organization of fibronectin in the extracellular matrix of cultured fibroblasts. Matrix Biol. 2001;20:475–486. doi: 10.1016/s0945-053x(01)00160-3. [DOI] [PubMed] [Google Scholar]
- Butterfield RJ, Foley AR, Dastgir J, Asman S, Dunn DM, Zou Y, et al. Position of glycine substitutions in the triple helix of COL6A1, COL6A2, and COL6A3 is correlated with severity and mode of inheritance in collagen VI myopathies. Hum Mutat. 2013;34:1558–1567. doi: 10.1002/humu.22429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norwood FL, Harling C, Chinnery PF, Eagle M, Bushby K, Straub V. Prevalence of genetic muscle disease in Northern England: in-depth analysis of a muscle clinic population. Brain. 2009;132 Pt 11:3175–3186. doi: 10.1093/brain/awp236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clement EM, Feng L, Mein R, Sewry CA, Robb SA, Manzur AY, et al. Relative frequency of congenital muscular dystrophy subtypes: analysis of the UK diagnostic service 2001-2008. Neuromuscul Disord. 2012;22:522–527. doi: 10.1016/j.nmd.2012.01.010. [DOI] [PubMed] [Google Scholar]
- Kawahara G, Ogawa M, Okada M, Malicdan MC, Goto Y, Hayashi YK, et al. Diminished binding of mutated collagen VI to the extracellular matrix surrounding myocytes. Muscle Nerve. 2008;38:1192–1195. doi: 10.1002/mus.21030. [DOI] [PubMed] [Google Scholar]
- Gualandi F, Manzati E, Sabatelli P, Passarelli C, Bovolenta M, Pellegrini C, et al. Antisense-induced messenger depletion corrects a COL6A2 dominant mutation in Ullrich myopathy. Hum Gene Ther. 2012;23:1313–1318. doi: 10.1089/hum.2012.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pendaries V, Gasc G, Titeux M, Tonasso L, Mejía JE, Hovnanian A. siRNA-mediated allele-specific inhibition of mutant type VII collagen in dominant dystrophic epidermolysis bullosa. J Invest Dermatol. 2012;132:1741–1743. doi: 10.1038/jid.2012.11. [DOI] [PubMed] [Google Scholar]
- Lindahl K, Rubin CJ, Kindmark A, Ljunggren O. Allele dependent silencing of COL1A2 using small interfering RNAs. Int J Med Sci. 2008;5:361–365. doi: 10.7150/ijms.5.361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller GA, Hansen U, Xu Z, Griswold B, Talan MI, McDonnell NB, et al. Allele-specific siRNA knockdown as a personalized treatment strategy for vascular Ehlers-Danlos syndrome in human fibroblasts. FASEB J. 2012;26:668–677. doi: 10.1096/fj.11-182162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rettig GR, Behlke MA. Progress toward in vivo use of siRNAs-II. Mol Ther. 2012;20:483–512. doi: 10.1038/mt.2011.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deleavey GF, Damha MJ. Designing chemically modified oligonucleotides for targeted gene silencing. Chem Biol. 2012;19:937–954. doi: 10.1016/j.chembiol.2012.07.011. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Genetic and histological description of the UCMD patients used in this study.
Allele specificity of siRNA-1 and siRNA-2 at concentration varying between 1 nM and 100 nM.
The intracellular retained collagen VI in UCMD fibroblasts is mainly located in the endoplasmic reticulum (ER), but does not activate the unfolded protein response (UPR).