

Abstract
Self-assembling peptides play increasingly important roles in the development of novel materials and drug delivery vehicles. Understanding mechanisms governing the assembly of nanoarchitectures is essential for the generation of peptide-based nanodevices. We find that a cone-shaped derivative of the second transmembrane domain of CXCR4 receptor, x4-2-6 self-assembles into nanospheres, while a related cylindrical peptide, x4-2-9 forms fibrils. Stronger intermolecular interactions in nanospheres than in fibrils result in slow rates of particle disassembly and protection against proteolytic degradation.
Nanoscale technologies are changing the methods of diagnostics and treatment of human disease. Among known nanoscale devices, peptide-based nanoparticles represent an emerging group of therapeutic agents, drug delivery vehicles and tissue-engineering materials. The advantages of peptide-based nanostructures include biodegradability, versatility of design and ease of synthetic functionalization. Complex structures can be built out of many simple units with minimal manufacturing interference. Consequently, molecular design using short peptides as new materials is likely to play an increasingly important role in nanoscience, nanotechnology, nanobiotechnology and nanomedicine.
Peptides are capable of assembly into a variety of structures including spherical, fibrous, tubular and discoid shapes. For pharmaceutical applications, nanoparticles that are uniform in size and shape are desirable, especially if they are used for drug delivery where uniform loading with medication is important. Finite assemblies, such as spherical and discoid nanoparticles, may satisfy this requirement, while infinite assemblies do not. Conversely, infinite assemblies, such as fibrous and tubular peptide structures, may be employed for tissue engineering.
We recently discovered that peptides corresponding to transmembrane helices assemble into nanoparticles in aqueous solutions [1]. In the case of CXCR4 chemokine receptor, peptides derived from the second transmembrane helix, x4-2-6 forms uniform spherical nanoparticles that have intrinsic biological activity and inhibit HIV-1 cell entry as well as CXCR4-mediated metastasis in a tumor model of breast cancer. These peptides insert into the plasma membrane and act as antagonists of CXCR4 signaling by blocking ligand-induced conformational transitions of CXCR4 dimer and by disrupting CXCR4 hydrophobic core architecture [2, 3]. Surprisingly, a peptide just two amino acid residues shorter than x4-2-6, x4-2-9 assembles into nanofibers. The differences in the shape of nanoparticles formed by x4-2-6 and x4-2-9 peptides are confirmed by transmission electron microscopy and dynamic light scattering in solution [1].
In order to investigate molecular mechanisms that define the ability of these peptides to form such different molecular assemblies, we undertook structural and biophysical comparisons of the two peptides. We find that, although x4-2-6 and x4-2-9 have almost identical amino acid sequences, their monomeric structures of peptide regions differ significantly. While x4-2-1, the non-PEGylated form of x4-2-6, exhibits a conical topology, the x4-2-9 peptide has a cylindrical structure. It is known that conical lipids assemble into micelles, while cylindrical lipids form bilayers [4]. Our results suggest that peptide nanoassembly may follow similar rules to those governing aggregation of lipids. In addition, we address the question of differential stability of spherical and fibrous peptide assemblies against proteolytic degradation. Due to more extensive intermolecular interactions the spherical x4-2-6 nanoparticles are less susceptible to proteolysis than x4-2-9 fibers. Proteolysis in the bloodstream commonly prevents development of peptides into therapeutic agents. Therefore, knowledge of which peptide assembly is more efficient in abating proteolytic degradation is highly desirable.
1. Experimental procedures
1.1. Peptide synthesis
The peptides were synthesized by solid phase peptide synthesis on a 432A Applied Biosystems peptide synthesizer equipped with a conductivity monitoring unit utilizing Fmoc (N-(9-fluorenyl)methoxycarbonyl) amino acid derivatives as described previously [5].
1.2. Nuclear magnetic resonance (NMR) experiments
The 1H–13C heteronuclear single quantum correlation (HSQC) NMR experiments were performed on 600 and 800 MHz Bruker Avance spectrometers equipped with cryogenic probes. All experiments were carried out at 25 °C. The peptides were dissolved in dimethyl sulfoxide, DMSO-d6. The peptide resonance assignments were performed using homonuclear total correlation spectroscopy (TOCSY) and nuclear Overhauser effect spectroscopy (NOESY) experiments with mixing times of 70 ms and 500 ms, respectively. All the data were processed and analyzed using NMRPipe [6].
1.3. Peptide structure calculations
NOEs and dihedral angles were used for structural calculations. NOEs were classified as strong, medium or weak, corresponding to distance restraints of 1.8–2.5, 1.8–3.5, 1.8–6.0 and 1.8–7.0 Å (for methyl NOEs), respectively. The distance restraints were derived from cross-peak volumes calibrated against the Hε–Hζ interproton distance (2.45 Å) of Trp18. Dihedral angle restraints were acquired from the analysis of the chemical shift index for 13Cα and 13Cβ using TALOS software [7]. The initial 100 structures were generated using CNS 1.1 [8]. Twenty structures with the lowest energy, showing the least violation of restraints, were selected among the initial structures. The structural analysis was carried out using MOLMOL to identify violations of NOE and dihedral angle restraints and to superimpose structures and measure the root-mean-square deviation (RMSD) [9].
1.4. Determination of β-sheet formation in assemblies of x4-2-6 and x4-2-9
The environmentally sensitive fluorescence dye Thioflavin T (ThT) was used to determine the appearance of β-sheet structure upon assembly of x4-2-6 and x4-2-9. ThT was dissolved in hot phosphate buffered saline (PBS) to make 28.14 μM stock solutions. The 0.8 mg ml−1 solutions of x4-2-6 and x4-2-9 peptide were diluted with PBS to vary the concentration of nanoparticles. The peptide solutions were mixed with the ThT stock solution resulting in a final ThT concentration for all solutions was 13.69 μM determined by UV–vis. Fluorescence measurements were performed on a steady-state fluorescence spectroscopy instrument. The fluorescence emission and excitation spectra were recorded at 25 °C on a single-photon counting spectrofluorometer FluoroMax-2 from Jobin Yvon Horiba-SPEX (Edison, NJ) using a 1 mm path quartz cell. The excitation and emission monochromator slits were set to 7 and 10 nm bandwidth. The values of ThT fluorescence intensity were obtained by performing emission scans from 455 to 600 nm when excited at 440 nm and excitation scans from 350 to 470 nm for fixed emission at 482 nm. An extinction coefficient, EmM = 26.6, at 412 nm in PBS was used to determine the concentration of the dye in the final probe.
1.5. Peptide degradation by proteinase K
The 2 mg ml−1 x4-2-6 and x4-2-9 peptide nanoparticles were incubated with 0.4 mg ml−1 proteinase K at a molar ratio of 1:1000 (proteinase K:peptide nanoparticles) at 37 °C. The 50 μl aliquots were taken at various time points from 0 to 2 h and phenylmethanesulfonylfluoride (PMSF) was added to a final concentration of 5 mM to quench the proteinase K activity. All samples were stored on dry ice until further analysis by HPLC.
1.6. HPLC analysis
A Shimadzu Prominence HPLC system with a UV/vis photodiode array detector was used for analysis of peptide degradation. A Shimadzu C18 reverse-phase column (50 mm × 4.5 mm) was equilibrated with 0.1% of trifluoroacetic acid (TFA). The sample injection volume was 5 μl. The peptide was eluted with a linear gradient of 90% acetonitrile containing 0.1% TFA. The amount of non-degraded peptide was estimated by measuring the peak area at 280 nm.
1.7. Determination of critical aggregation concentration
To determine the critical aggregation concentration (CAC) a fluorescence probe, pyrene, was used. Pyrene was dissolved in 100% ethanol to make 2 mM stock solution because of its low solubility in water. The 4 mg ml−1 solutions of x4-2-6 and x4-2-9 peptide nanoparticles were diluted with PBS to vary the concentration of nanoparticles. The peptide solutions were mixed with the pyrene stock solution, resulting in a final pyrene concentration of 1 μM. The mixtures were incubated overnight at room temperature. Fluorescence measurements were performed on a PTI Quantamaster instrument. All experiments were carried out in quartz cuvettes using 1200 μl of the peptide and pyrene mixture. The excitation profile was scanned from 315 to 360 nm when the emission was set to 390 nm.
2. Results
The amino acid sequences of x4-2-1, x4-2-6 and x4-2-9 peptides shown in figure 1(A) are very similar. The two C-terminal aspartates are not present in the original CXCR4 sequence and are added to improve the inhibitory activity of the peptides. The x4-2-1 peptide does not have the polyethylene glycol (PEG) moiety at the C-terminal, whereas x4-2-6 and x4-2-9 possess 27 repeat units of PEG. The x4-2-6 and x4-2-9 peptides have identical primary structures, except that the two N-terminal leucines are lacking in the structure of x4-2-9.
Figure 1.
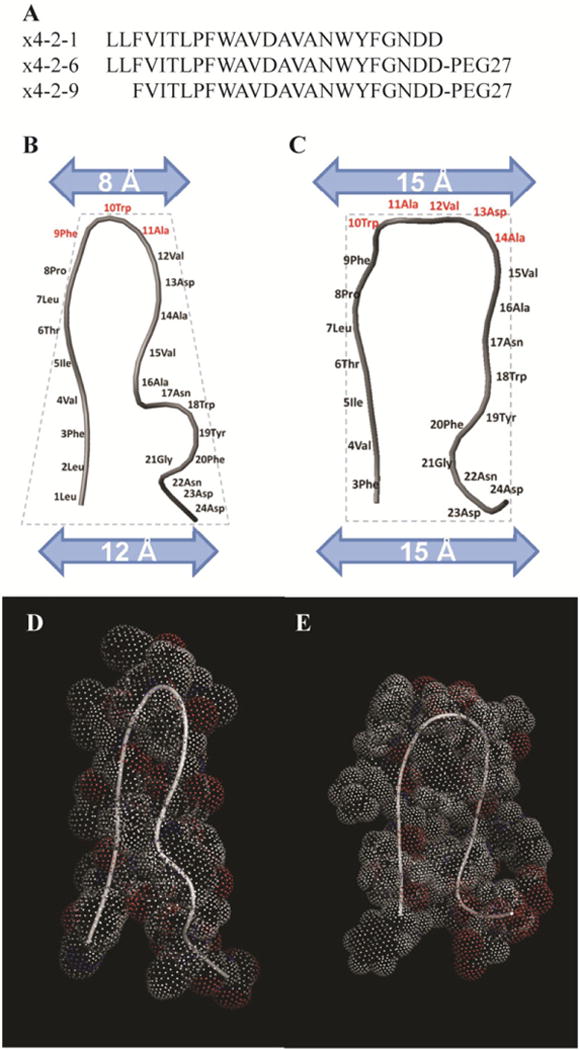
Structural characterization of x4-2-1, x4-2-6, and x4-2-9 peptides. (A) Amino acid sequences of x4-2-6 and x4-2-9 peptides differ by only two N-terminal residues. Comparison of average structures of x4-2-1 (B) and x4-2-9 (C) shows differences in the overall topology of peptide monomers. The width of the head and the base of the hairpin structures were measured. Residues involved in the head region are shown in dark gray. The dotted outline shows the conical shape of the x4-2-1 structure and the cylindrical shape of the x4-2-9 structure. The blue (light gray) bars represent the widths of the head and the base of x4-2-1 and x4-2-9 hairpins. The space filling models of average x4-2-1 and x4-2-9 structures are shown in (D) and (E), respectively.
We recently found that x4-2-1 and x4-2-6 peptides self-assemble into nanospheres 10 nm in diameter, while the truncated version of the peptide lacking two N-terminal Leu residues, x4-2-9, forms fibrils using transmission electron microscopy and dynamic light scattering [1]. To better understand the mechanisms defining the architecture of the self-assembled aggregates we studied structures of x4-2-1, x4-2-6 and x4-2-9 monomers in DMSO-d6 by NMR. DMSO is known to interfere with peptide aggregation without disrupting its tertiary structure [10]. DMSO is relevant to our studies because the assembly of x4-2-1, x-4-2-6 and x4-2-9 is initiated in a highly concentrated DMSO solution phase, since dissolving solid peptides directly in an aqueous buffer produces amorphous aggregates instead of ordered assemblies.
We initially solved the structure of x4-2-1 without the C-terminal PEG moiety that is present in x4-2-9. The PEG extension was added to prevent superaggregation in the aqueous solution. We recorded a 1H-13C HSQC spectrum of the x4-2-6 peptide and compared it to the 1H-13C HSQC spectrum of x4-2-1 (figure 2). The only changes in chemical shifts were observed in the vicinity of the PEG moiety. Since no significant changes in chemical shifts were observed in the rest of the peptide molecule, we assumed that the peptide structure with PEG is similar to the structure without PEG.
Figure 2.
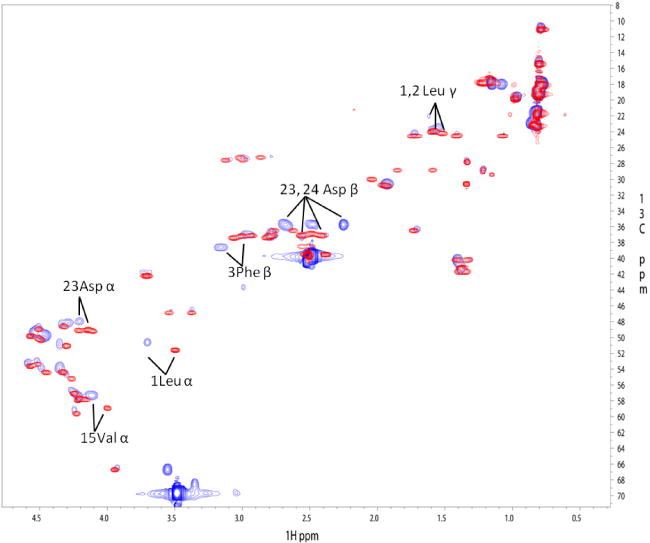
1H-13C HSQC spectra of x4-2-1 shown in red (dark gray) and x4-2-6 shown in blue (light gray). The labeled residues showing perturbations in chemical shifts are located in both the N- and C-termini of the peptide, which are in close proximity to the C-terminally attached PEG.
As we addressed the structure of x4-2-1 in a previous study, the structural topology of monomeric x4-2-1 is a hairpin with two tight helix-like turns in the C-terminus (figure 1(B)) [1]. Most of the residues in the hairpin structure (71%) exhibit a β-strand conformation, as shown by analysis of the distribution of ψ-dihedral angles in the Ramachandran plot (figure 3). The backbone and heavy atom root mean square deviation (RMSD) values calculated for residues 4– 20 are 0.37 ± 0.16 Å and 1.18 ± 0.28 Å, respectively. The amino acids responsible for stabilization of the hairpin structure include Val4, Thr6, Pro8, Asp13 and Ala14. The hydrogen bonds between two strands, such as the side chain of Thr6 to the amide backbone hydrogen of Val15 and the nitrogen of Val4 to the δ proton of Asn17, may be present. The hydrophobic interactions between Pro8 and Val12, and Val4 and Ala16, stabilize the hairpin-like structure. The negative charges provided by C-terminal aspartates further stabilize the hairpin conformation of x4-2-1 by interacting with the positively charged N-terminus. This structural arrangement allows close packing and subsequent stabilization of the N-terminal turns by the C-terminal strand. The hairpin conformation of the x4-2-1 structure and C-terminal turns define a conical overall shape of x4-2-1. This is consistent with our hypothesis that the conical shape of the x4-2-1 peptide may facilitate assembly into spherical nanoparticles.
Figure 3.
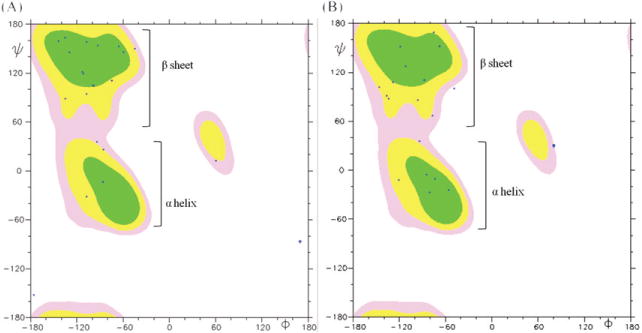
The Ramachandran plot of average conformations for 20 x4-2-1 and x4-2-9 structures determined by NMR. The MOLMOL 2K.1 software was used for analysis. Phi (ϕ) and psi (ψ) angles of most residues in both x4-2-1 (A) and x4-2-9 (B) structures are positioned in the β-sheet region, except those for residues involved in hairpin bending and the C-terminal helix. This plot shows that monomers of x4-2-1 and x4-2-9 adopt a β-hairpin conformation.
The x4-2-9 structures in DMSO-d6 were calculated with the RMSD of 0.53 ± 0.34 Å for backbone and 1.13 ± 0.39 Å for heavy atoms for 20 superimposed structures over residues Val4 to Phe20. The average structure of x4-2-9 is shown in figure 1(C). The PEG moiety was unstructured and therefore is not shown in figure 1. The residues involved in stabilization of the hairpin structure are Ile5, Thr6, Asn17 and Tyr19 through H-bonds. Helical turns are absent in the structure of x4-2-9. This observation reveals that the structural significance of Leu1 and Leu2 is to stabilize the helical turns in the C-terminus of x4-2-1. Without the stabilizing effect of the two leucine residues the N-terminal strand packs loosely next to the C-terminal strand, yielding a cylinder-like topology of x4-2-9. In addition, the number of amino acids in the β-strand conformation in x4-2-9 (50%) is significantly reduced, as compared to x4-2-1 (figure 3).
Further comparison of x4-2-1 and x4-2-9 structures revealed that five residues from Trp10 to Ala14 are involved in the head region of the x4-2-9 hairpin, whereas only three residues from Phe9 to Ala11 form the head of the x4-2-2 hairpin (figure 1). This implies that the x4-2-2 hairpin has a much narrower head (8 Å) than the base (12 Å). This feature may be responsible for the cone-like topology of this peptide. In contrast, both widths of the head and the base of x4-2-9 are approximately 15 Å, giving rise to the cylindrical shape of x4-2-9. This result is again consistent with our hypothesis that the cylinder-like peptides may assemble into fibrils, while conical peptides form spherical nanoparticles. The overall structures of x4-2-1 and x4-2-9 with the space-filling method support that peptides which have similar amino acid sequences show different structures as conical and cylinder-like shapes (figures 1(D) and (E)). Several long range NOE signals between two strands indicate the hairpin-like structure of x4-2-1 and x4-2-9. However, the set of NOEs in the x4-2-1 structure that support hairpin conformation was different from the set of those in the x4-2-9 structure. For instance, long range NOEs between Thr6 Hγ2, Ala14 Hα and Val15 Hα in the x4-2-1 NOESY spectrum were absent in the NOESY spectrum of x4-2-9. Based on the analysis of the NOESY spectrum of x4-2-9, Hα of Ile5 and Thr6 are close to Asn17 Hδ2 which were missing in the NOESY spectrum of x4-2-1 (figure 4). Therefore, the x4-2-9 peptide adopts a different conformation from the structure of x4-2-1. This observation allows us to conclude that the final morphology of nanoparticles may depend on the structure of the monomer.
Figure 4.
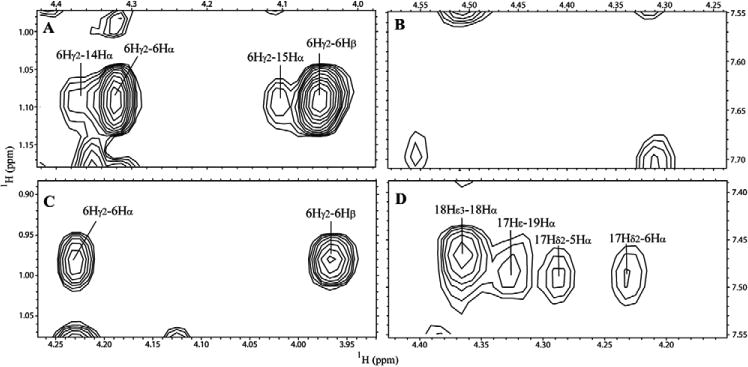
Partial NOESY spectra of x4-2-1 ((A) and (B)) and x4-2-9 ((C) and (D)) show that x4-2-1 and x4-2-9 do not share the same long range NOEs, supporting the hypothesis that the structures of these peptides are different.
To gain insight into the possible mechanism of peptide assembly we utilized a fluorescent dye Thioflavin-T (ThT) that is widely used for selectively staining and identifying β-sheet structures in amyloid fibrils both in vivo and in vitro [11]. The changes in fluorescent properties of ThT upon binding amyloids include a shift in its excitation spectrum and an increase in quantum yield [12]. ThT absorbs at 340 nm with an emission maximum at 445 nm. Upon binding to self-assembled x4-2-6 and x4-2-9 peptides a peak at 440 nm became dominant in excitation spectra with the fluorescent emission maximum shifting to 480 nm. This was accompanied by a strong enhancement of fluorescence (figure 5). Spectral changes of this type are broadly accepted as defining characteristics for the presence of β-sheets and thus suggest that the structural organization of self-assembled CXCR4 antagonists occurs via β-sheet formation from β-strands in the peptide hairpins. If the appearance of β-sheets marks the formation of nanostructures then the range of critical aggregation concentration (CAC) for the peptides may be determined by measuring absorbance of particle-bound ThT at 482 nm. Using this approach we found that CAC is much lower for the conical x4-2-6 peptide (between 0.004 and 0.01 mg ml−1) than for the cylindrical x4-2-9 peptide (between 0.035 and 0.08 mg ml−1).
Figure 5.
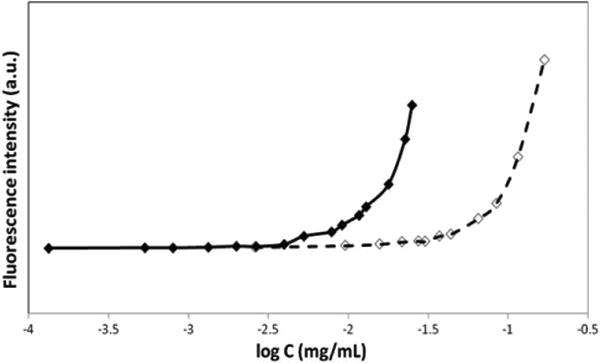
Peptide concentration dependence of ThT fluorescence intensity at 482 nm allowed us to determine that the critical aggregation concentration is much lower for x4-2-6 (filled diamonds) than for x4-2-9 (open diamonds).
Next, we wished to address the question of the significance of the shape of peptide assemblies. Since the x4-2-6 and x4-2- 9 peptides may have therapeutic value as inhibitors of CXCR4, it is important to determine if the type of peptide assembly correlates with the degree of protection against proteolysis by serum proteases. By studying x4-2-6 and x4-2-9 stability in bovine serum we were able to uncover significant differences in protection from proteolysis in these peptides [1]. However, since serum contains a mixture of proteases and the tested peptides do not have identical amino acid sequences, the possible differences in half-life values for the two peptides may also be interpreted in terms of preferential recognition of x4-2-6 and x4-2-9 by distinct proteases that differ in abundance in serum. To address this potential issue we performed a peptide degradation assay with 2 mg ml−1 of either x4-2-6 or x4-2-9 and purified proteinase K. The results of this experiment are shown in figure 6. The results of the peptide degradation assay demonstrate that x4-2-6 peptide within spherical nanospheres with the half-life time of 11.8 ± 1.8 min is significantly more protected against proteolysis by proteinase K than the x4-2-9 peptide within fibrils (2.9 ± 1.2 min).
Figure 6.
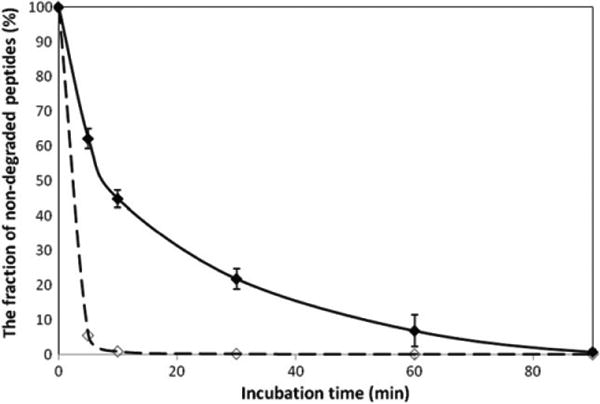
Time course of 2 mg ml−1 x4-2-6 (filled diamonds) and x4-2-9 (open diamonds) peptide degradation in PBS, pH 7.1 by proteinase K. The reaction was stopped after defined time intervals by the addition of 5 mM PMSF (proteinase K inhibitor) after incubation. All experiments were carried out at 37 °C and repeated three times for statistical analysis.
The reason for enhanced protection against proteolysis of peptides assembled into nanospheres versus fibrils may be (i) minimized surface exposure within nanospheres compared to fibrils and/or (ii) a difference in strength of intermolecular interactions in nanospheres and in fibrils leading to different rates of their disassembly. These explanations may not necessarily be mutually exclusive. Although the role of the difference in surface exposure in proteolytic degradation of peptides is challenging to demonstrate, the strength of intermolecular contacts in peptide assemblies may be readily assessed by studying the peptide CAC.
Pyrene is a small molecule that changes its fluorescence upon transitioning from the aqueous to the micellar phase. This property of pyrene has been extensively used to measure CAC of polymers [13, 14]. The CAC marks the onset of polymer aggregation when the first aggregates are observed. We incubated pyrene with varying concentrations of x4-2-6 and x4-2-9 and recorded pyrene excitation spectra at 390 nm emission. As the peptide concentration increased, we observed an increase in total fluorescence intensity and a redshift indicating that pyrene was incorporated into the hydrophobic core structure of nanoparticles. The CAC was determined using the fluorescence intensity ratio I337/I334 of pyrene from excitation spectra [15]. The results of our analysis are shown in figure 7. The error in measurements was estimated based on taking double points at several peptide concentrations. The measured CAC values of 0.02 mg ml−1 and 0.1 mg ml−1 for the x4-2-6 and x4-2-9 peptides, respectively, are in agreement with the CAC determination using ThT fluorescence. The CAC measurements suggest that stronger intermolecular interactions must exist within spherical nanoparticles than in fibrils. This observation implies that the spherical peptide nanoparticles disassemble at a slower rate than peptide fibrils, allowing enhanced protection against proteases.
Figure 7.
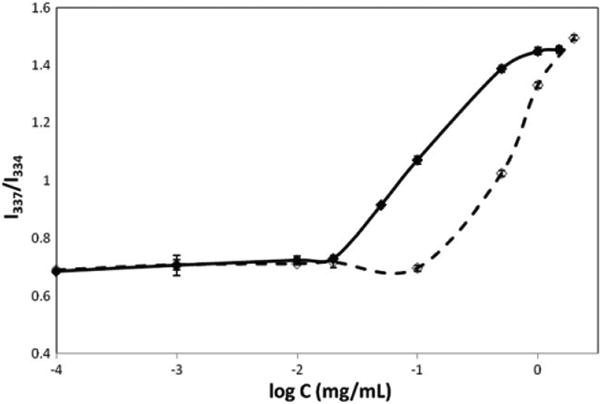
The plot of fluorescence intensity ratio (I337/I334) from pyrene excitation spectra as a function of concentration of x4-2-6 or x4-2-9 nanoparticles. The CAC values for x4-2-6 (filled diamonds) and x4-2-9 (open diamonds) were determined at the onset of peptide aggregation. The experiments were repeated for statistical analysis.
3. Discussion
There are a number of literature reports describing peptide-based spherical assemblies [16–18]. There is also abundant literature on peptide aggregation into amyloid fibrils [19]. These data suggest that common rules governing peptide assembly into higher-order structures may exist. In this publication we do not address the generality of mechanisms of peptide assembly. However, it is possible that the requirements for peptide monomeric structure delineated in this report apply to other peptides.
The mechanism of peptide aggregation into amyloid fibrils is still under investigation. However, it is already clear that it involves interactions of linear β-structures [20]. The β-strands and β-hairpins assembling into amyloid fibrils would satisfy the cylindrical shape requirement for fibril formation. The initial cones and cylinders may not directly assemble into macrostructures. Intermediate assembly states may be necessary for the final architectures. However, the clear differences in the structure of the monomeric building blocks may dictate the assembly pathways.
There have been many attempts to create diverse higher-order assemblies using inorganic and organic materials as building blocks [21–24]. However, the mechanism of self-assembly into nanostructures is not clearly understood. Self-assembly of inorganic molecules is currently better studied than self-assembly of organic materials, such as proteins and peptides. This is mostly due to the lack of well-developed techniques to study nanoassembly of proteins and peptides. The current knowledge of assembly mechanisms has been recently reviewed [25–27]. The consensus is that the shape of the nanostructures is mostly defined by the nature of intermolecular interactions between the monomer building blocks. The driving force for self-assembly events is largely due to desolvation, collapse and intermolecular association of hydrophobic molecular interfaces. It is possible that electrostatics and hydrogen bonding also contribute to the assembly process. This general conclusion seems to hold for both inorganic and organic materials. The importance of the overall topology of monomeric structures for definition of the shape of nanostructures has been recognized to a much lesser extent. The monomeric structure tends to be viewed as a way to create amphiphilic molecules by polarizing charged and hydrophobic surfaces rather than to limit the possibilities for intermolecular interactions through steric interference. A model reflecting this idea has been recently described for peptides assembling into rods, tubes and spheres [28]. A modification of this hypothesis suggests that conical amphiphilic peptides containing terminal bulky aromatic residues or amphiphilic tree-like peptides may also form spherical nanoparticles based on steric compatibility [29, 30].
We recently reported that peptides with almost identical amino acid sequences can assemble into different nanostructures [1]. In this paper, we continue structural characterization of peptides that form different shapes of nanoparticles. We also introduce new ways of CAC determination. In addition, the new proteolytic protection assay with proteinase K allows us to clearly uncover striking differences between the peptides in terms of their proteolytic stability.
By studying peptide molecules of almost identical amino acid composition but very different in their topology we propose that the three-dimensional structure of peptide monomeric components may function as a determinant of the final shape of the nanoassembly. Unlike in protein molecules that are much larger than peptides, small changes in the amino acid composition may have a drastic effect on the three-dimensional structure. For example, the x4-2-6 peptide that differs only by two N-terminal amino acids from the x4-2-9 peptide displays a cone-shaped conformation, while the x4-2-9 peptide is cylindrical in shape (figures 1(B) and (C)).
Based on the reports that the structure of nanoparticles depends on the shape of monomeric building blocks and based on our structural characterization of x4-2-1 and x4-2-9 peptides assembling into spherical nanoparticles and fibrils, we propose that the conical structures of monomeric components are required for spherical assemblies, while cylindrical shapes produce fibrils. This principle is schematically illustrated in figure 8. Although our data suggest that the PEG moiety does not significantly interfere with the monomeric structures of peptides in DMSO, undoubtedly it affects the structure of the final nanoassembly. Many literature reports testify to this assumption [31–33]. However, in this report by comparing the architectures of x4-2-6 and x4-2-9 we uncover the role of the structure of peptide monomers in the assembly mechanism. Since both peptides are PEGylated, the structure of the peptide part clearly dominates over the influence of PEG as the determining factor of the topology of the resulting nanoparticles.
Figure 8.
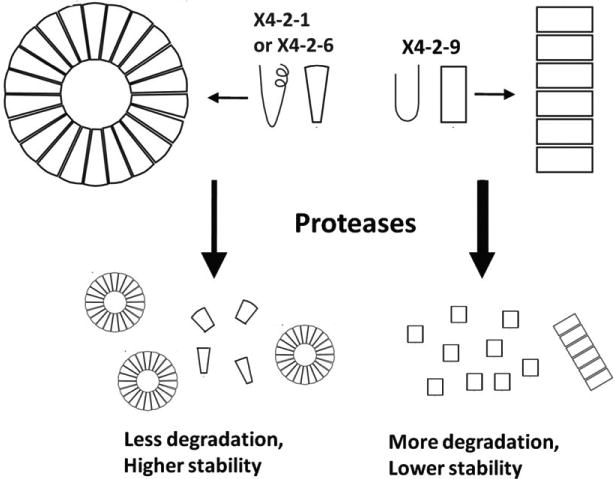
The model of nanoparticle assembly from the monomers of x4-2-6 or x4-2-9. The conical shape of x4-2-1 or x4-2-6 monomer leads to spherical nanoparticles whereas the cylindrical x4-2-9 monomer prefers to form fibrils. The nanospheres are protected from proteolysis more than peptide fibrils because of tighter intermolecular interactions in nanospheres.
The conical x4-2-6 peptide engages in more significant intermolecular interactions than the cylindrical x4-2-9 peptide as shown by differences in CAC (figure 7). These differences in intermolecular binding and perhaps differences in surface accessibility result in enhanced protection of peptides within spherical nanoparticles versus the peptides in fibrils against proteolytic degradation (figure 6).
Proteolytic stability of peptide molecules in the bloodstream is an important parameter in the development of their therapeutic applications. In addition, well-defined structures are important for drug delivery. The spherical shapes are necessary for encapsulation of drug molecules. Fibrils may also have value in tissue repair. Conversely, random aggregates may be neurotoxic. Various well-defined shapes of nanostructures will have their own applications in medicine. The studies reported here will contribute to the definition of requirements that may allow engineering of the shape of peptide-based nanostructures.
Acknowledgments
We are grateful for a National Cancer Institute grant 1R01CA135341-01A1 to VG and a Prostate Cancer Research Award PC08116, Congressionally Directed Medical Research Program, to NIT.
References
- 1.Tarasov SG, Gaponenko V, Howard OMZ, Chen Y, Oppenheim JJ, Dyba MA, Subramaniam S, Lee Y, Michejda CJ, Tarasova NI. Structural plasticity of a transmembrane peptide allows selfassembly into biologically active nanoparticles. Proc Natl Acad Sci USA. 2011;108:9798–803. doi: 10.1073/pnas.1014598108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Percherancier Y, Berchiche YA, Slight I, Volkmer-Engert R, Tamamura H, Fujii N, Bouvier M, Heveker N. Bioluminescence resonance energy transfer reveals ligand-induced conformational changes in CXCR4 homo- and heterodimers. J Biol Chem. 2005;280:9895–903. doi: 10.1074/jbc.M411151200. [DOI] [PubMed] [Google Scholar]
- 3.George SR, Ng GY, Lee SP, Fan T, Varghese G, Wang C, Deber CM, Seeman P, O'Dowd BF. Blockade of G protein-coupled receptors and the dopamine transporter by a transmembrane domain peptide: novel strategy for functional inhibition of membrane proteins in vivo. J Pharmacol Exp Ther. 2003;307:481–9. doi: 10.1124/jpet.103.053843. [DOI] [PubMed] [Google Scholar]
- 4.Brasseur R, De Meutter J, Goormaghtigh E, Ruysschaert JM. Mode of organization of galactolipids: a conformational analysis. Biochem Biophys Res Commun. 1983;115:666–72. doi: 10.1016/s0006-291x(83)80196-x. [DOI] [PubMed] [Google Scholar]
- 5.Tarasova NI, Rice WG, Michejda CJ. Inhibition of G-protein-coupled receptor function by disruption of transmembrane domain interactions. J Biol Chem. 1999;274:34911–5. doi: 10.1074/jbc.274.49.34911. [DOI] [PubMed] [Google Scholar]
- 6.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:277–93. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 7.Cornilescu G, Delaglio F, Bax A. Protein backbone angle restraints from searching a database for chemical shift and sequence homology. J Biomol NMR. 1999;13:289–302. doi: 10.1023/a:1008392405740. [DOI] [PubMed] [Google Scholar]
- 8.Brunger AT, et al. Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr. 1998;54:905–21. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 9.Koradi R, Billeter M, Wuthrich K. MOLMOL: a program for display and analysis of macromolecular structures. J Mol Graph. 1996;14:29–32. doi: 10.1016/0263-7855(96)00009-4. [DOI] [PubMed] [Google Scholar]
- 10.Toyama BH, Kelly MJ, Gross JD, Weissman JS. The structural basis of yeast prion strain variants. Nature. 2007;449:233–7. doi: 10.1038/nature06108. [DOI] [PubMed] [Google Scholar]
- 11.Wolfe LS, Calabrese MF, Nath A, Blaho DV, Miranker AD, Xiong Y. Protein-induced photophysical changes to the amyloid indicator dye thioflavin T. Proc Natl Acad Sci USA. 2010;107:16863–8. doi: 10.1073/pnas.1002867107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.LeVine H., III Thioflavine T interaction with synthetic Alzheimer's disease beta-amyloid peptides: detection of amyloid aggregation in solution. Protein Sci. 1993;2:404–10. doi: 10.1002/pro.5560020312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ferreira SA, Coutinho PJ, Gama FM. Self-assembled nanogel made of mannan: synthesis and characterization. Langmuir. 2010;26:11413–20. doi: 10.1021/la100903j. [DOI] [PubMed] [Google Scholar]
- 14.Park K, Kim K, Kwon IC, Kim SK, Lee S, Lee DY, Byun Y. Preparation and characterization of self-assembled nanoparticles of heparin-deoxycholic acid conjugates. Langmuir. 2004;20:11726–31. doi: 10.1021/la048646i. [DOI] [PubMed] [Google Scholar]
- 15.Wilhelm M, Zhao CL, Wang Y, Xu R, Winnik MA, Mura JL, Riess G, Croucher MD. Poly(styrene-ethylene oxide) block copolymer micelle formation in water: a fluorescence probe study. Macromolecules. 1991;24:1033–49. [Google Scholar]
- 16.Liu L, Guo K, Lu J, Venkatraman SS, Luo D, Ng KC, Ling EA, Moochhala S, Yang YY. Biologically active core/shell nanoparticles self-assembled from cholesterol-terminated PEG-TAT for drug delivery across the blood-brain barrier. Biomaterials. 2008;29:1509–17. doi: 10.1016/j.biomaterials.2007.11.014. [DOI] [PubMed] [Google Scholar]
- 17.Dreher MR, Simnick AJ, Fischer K, Smith RJ, Patel A, Schmidt M, Chilkoti A. Temperature triggered self-assembly of polypeptides into multivalent spherical micelles. J Am Chem Soc. 2008;130:687–94. doi: 10.1021/ja0764862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Choi H, Ahn JY, Sim SJ, Lee J. Glutamate decarboxylase-derived IDDM autoantigens displayed on self-assembled protein nanoparticles. Biochem Biophys Res Commun. 2005;327:604–8. doi: 10.1016/j.bbrc.2004.12.046. [DOI] [PubMed] [Google Scholar]
- 19.Hamley IW. Peptide fibrillization. Angew Chem Int Edn. 2007;46:8128–47. doi: 10.1002/anie.200700861. [DOI] [PubMed] [Google Scholar]
- 20.Sandberg A, et al. Stabilization of neurotoxic Alzheimer amyloid-beta oligomers by protein engineering. Proc Natl Acad Sci USA. 2010;107:15595–600. doi: 10.1073/pnas.1001740107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Du J, O'Reilly RK. Advances and challenges in smart and functional polymer vesicles. Soft Matter. 2009;5:3544–61. [Google Scholar]
- 22.Adler-Abramovich L, Aronov D, Beker P, Yevnin M, Stempler S, Buzhansky L, Rosenman G, Gazit E. Self-assembled arrays of peptide nanotubes by vapour deposition. Nature Nanotechnol. 2009;4:849–54. doi: 10.1038/nnano.2009.298. [DOI] [PubMed] [Google Scholar]
- 23.Reches M, Gazit E. Casting metal nanowires within discrete self-assembled peptide nanotubes. Science. 2003;300:625–7. doi: 10.1126/science.1082387. [DOI] [PubMed] [Google Scholar]
- 24.Kwon S, Jeon A, Yoo SH, Chung IS, Lee H. Unprecedented molecular architectures by the controlled self-assembly of a b-peptide foldamer. Angew Chem Int Ed. 2010;49:8232–6. doi: 10.1002/anie.201003302. [DOI] [PubMed] [Google Scholar]
- 25.Branco MC, Schneider JP. Self-assembling materials for the therapeutic delivery. Acta Biomater. 2009;5:817–31. doi: 10.1016/j.actbio.2008.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu L, Busuttil K, Zhang S, Yang Y, Wang C, Besenbacher F, Dong M. The role of self-assembling polypeptides in building nanomaterials. Phys Chem Chem Phys. 2011;13:17435–44. doi: 10.1039/c1cp21338e. [DOI] [PubMed] [Google Scholar]
- 27.Zhao X, Pan F, Xu H, Yassen M, Shan H, Hauser CAE, Zhang S, Lu JR. Molecular self-assembly and applications of designer peptide amphiphiles. Chem Soc Rev. 2010;39:3480–98. doi: 10.1039/b915923c. [DOI] [PubMed] [Google Scholar]
- 28.Reches M, Gazit E. Molecular self-assembly of peptide nanostructures: mechanism of association and potential uses. Curr Nanosci. 2006;2:105–11. [Google Scholar]
- 29.van Hell AJ, Costa CICA, Flesch FM, Sutter M, Jiskoot W, Crommelin DJA, Hennink WE, Mastrobattista E. Self-assembly of recombinant amphiphilic oligopeptides into vesicles. Biomacromolecules. 2007;8:2753–61. doi: 10.1021/bm0704267. [DOI] [PubMed] [Google Scholar]
- 30.Tian L, Hammond T. Comb-dendritic block copolymers as tree-shaped macromolecular amphiphiles for nanoparticle self-assembly. Chem Mater. 2006;18:3976–84. [Google Scholar]
- 31.Xiao RZ, Zeng ZW, Zhou GL, Wang JJ, Li FZ, Wang AM. Recent advances in PEG-PLA block copolymer nanoparticles. Int J Nanomed. 2010;5:1057–65. doi: 10.2147/IJN.S14912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dong Y, Feng S. Methoxy poly(ethylene glycol)-poly(lactide) (MPEG-PLA) nanoparticles for controlled drug delivery of anticancer drugs. Biomaterials. 2004;25:2843–9. doi: 10.1016/j.biomaterials.2003.09.055. [DOI] [PubMed] [Google Scholar]
- 33.Castelletto V, Hamley IW. self assembly of a model amphiphilic phenylalanine peptide/polyethylene glycol block copolymer in aqueous solution. Biophys Chem. 2009;141:169–74. doi: 10.1016/j.bpc.2009.01.008. [DOI] [PubMed] [Google Scholar]