

Abstract
The Rieske/cytochrome b complexes, also known as cytochrome bc complexes, catalyze a unique oxidant-induced reduction reaction at their quinol oxidase sites (Qo), in which substrate hydroquinone reduces two distinct electron transfer chains, one through a series of high-potential electron carriers, the second through low-potential cytochrome b. This reaction is a critical step in energy storage by the Q-cycle. The semiquinone intermediate in this reaction can reduce O2 to produce deleterious superoxide. It is yet unknown how the enzyme controls this reaction, though numerous models are proposed. In previous work we trapped a Q-cycle semiquinone anion intermediate, termed SQo, in bacterial cyt bc1 by rapid freeze-quenching. In this work, we apply pulsed EPR techniques to determine the location and properties of SQo in the mitochondrial complex. In contrast to semiquinone intermediates in other enzymes, SQo is not thermodynamically stabilized, and may even be destabilized with respect to solution. It is trapped in the Qo at a site, which is distinct from previously described inhibitor-binding sites, yet sufficiently close to cytochrome bL to allow rapid electron transfer. The binding site and EPR analysis show that SQo is not stabilized by hydrogen bonds to proteins. The formation of SQo involves “stripping” of both substrate -OH protons during the initial oxidation step, as well as conformational changes of the semiquinone and Qo proteins. The resulting charged radical is kinetically trapped, rather than thermodynamically stabilized (as in most enzymatic semiquinone species), conserving redox energy to drive electron transfer to cytochrome bL, while minimizing certain Q-cycle bypass reactions including oxidation of pre-reduced cytochrome b and reduction of O2.
Keywords: Q Cycle, Complex III, bc Complex, Pulsed EPR, Paramagnetic Relaxation Enhancement
Introduction
The Rieske/b and related complexes, e.g., cytochrome (cyt) bc1 (Complex III, ubiquinol:cyt c oxidoreductase) are essential energy transduction components in respiration and photosynthesis of a wide range of organisms.[1] They translocate protons across a membrane against a concentration gradient, driven by oxidation of substrate hydroquinone (quinol, QH2). They act as a proton translocator running on the redox energy of the electron transport chain. Their enzyme mechanisms and their roles in oxidative stress are in dispute. Cyt bc1 catalyzes a Q-cycle, proposed by Mitchell and modified extensively,[1e, 2] with the remarkable feature that the two electrons carried by each QH2 enter two distinct electron transport chains leading from the quinol oxidase site, Qo, on the positive p-side of the membrane.[3] A ‘high-potential chain’ containing a Rieske Fe2S2 cluster acting as a tethered, mobile substrate and, typically, a c-type heme of cyt c1 on the p-side, relays the electron to a soluble cyt c. The Rieske Fe2S2 cluster pivots through an angle of about 60° to ferry electrons from the quinol substrate to the cyt c1. The ‘low-potential chain’ carries electrons across the membrane, building a transmembrane electrical field, from the Qo site, through a cyt b subunit with two hemes (cyt bL and cyt bH), to the quinone reductase site, Qi, on the negatively-charged n-side of the membrane. In all other enzymes utilizing ubiquinone, electrons are transferred to only one electron transport chain. The bifurcated electron transport is a distinctive feature that is central to the Q-cycle mechanism, and serves as an energy conserving strategy, increasing the ratio of protons translocated per electron transferred (H+/e) over what would otherwise be obtained through simple double reduction of the high-potential chain.
In most Q-cycle models,[4] Scheme 1, the first electron from QH2 enters the high-potential chain at the Rieske Fe2S2, which then reduces cyt c1. The one-electron oxidation of QH2 leaves a strongly-reducing semiquinone (SQ) intermediate, termed SQo, which passes the second electron to cyt bL in the low-potential chain. That second electron is then passed along to reduce cyt bH, which acts as electron donor to a quinone (Q) or SQ molecule bound within the Qi site. In contrast to SQo, the SQ formed at Qi is stabilized by the protein, raising its midpoint potential and facilitating reduction of Q by bH, but consequently making it harder for bH to reduce the SQ to QH2. Overall, the Q-cycle pumps four protons across the membrane, contributing to the transmembrane proton gradient that drives ATP synthesis. The Q-cycle involves a complex ballet of electron and proton transfers that takes four electrons and four protons from two QH2 at Qo, uses two electrons to regenerate one QH2 at Qi, while the other two travel the high-potential chain, and pumps the four protons out of one side of the membrane. Both electrons used to reduce Q in the Qi site come from the low-potential chain, requiring that the one-electron redox potential for SQo must be more reducing than the redox potentials of the Qi site. This requirement limits the extent to which SQo can be stabilized by the protein.
Scheme 1. Schematic representation of the Q-cycle.
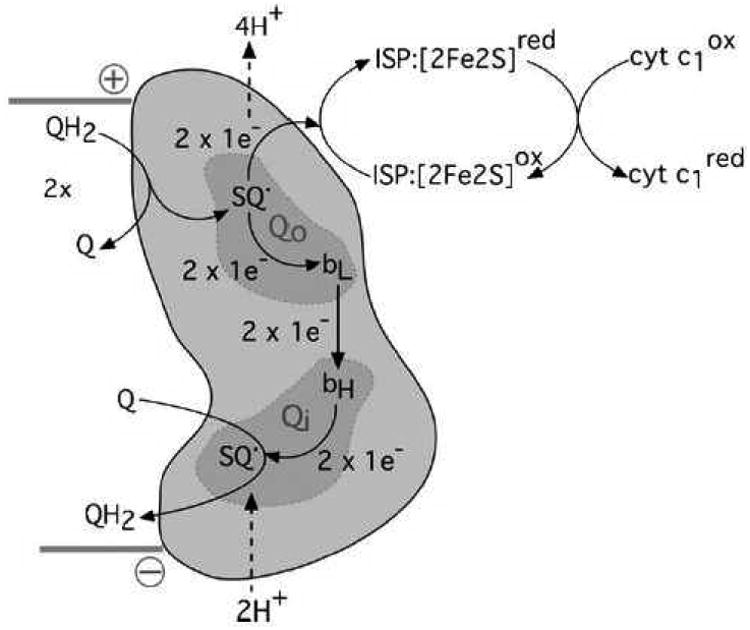
Cytochrome b is represented in orange cartoon form, as one half of the homodimeric bc1 complex. Quinol-oxidation and –reduction sites (Qo, Qi) are shaded. Proton movements are indicated by dashed arrows. Sequential oxidation of two molecules of quinol (QH2) at Qo results in the transfer of two electrons to the Rieske iron sulfur protein (ISP:[2Fe2S]) and the formation of one molecule of quinol (Q) at Qi following electron transfer through hemes bL and bH. The reduced Rieske protein is oxidised by cytochrome c1. The membrane orientation of cytochrome b is indicated by the solid gray bars, with the positive and negative sides of the membrane indicated accordingly.
Despite the availability of atomic structures of the bc1 complex from a variety of eukaryotic and prokaryotic organisms, there are no reported structures with substrate quinol or any intermediate species co-crystallised within the Qo site. There are, however, structures with inhibitors bound in the Qo site revealing it as a large, bifurcated volume within the bc1 complex, Fig. S1.[5] Qo-inhibitors show two modes of interaction with the Rieske protein. The ‘b-distal’ inhibitors, such as stigmatellin or famoxadone, form a strong hydrogen bond with a histidine ligand of the Fe2S2 cluster in the extrinsic domain of the Rieske protein, limiting its mobility and blocking its availability to substrate. Conversely, ‘b-proximal’ inhibitors, such as myxothiazol and MOA-stilbene, interact with a region of cytochrome b (the ef helix) in closer proximity to heme bL and do not affect Rieske mobility. The binding mode of stigmatellin is often used as a model for the initial encounter complex for the electron transfer reaction between QH2 and the Rieske protein. Given that the rate of electron transfer is strongly dependent on the distance between participating centers, it has been proposed that SQo moves within the Qo site, from the distal to the proximal region, in order to achieve the observed rate of bL reduction.[6]
The SQo presents a potential hazard to the cell because it has sufficiently low potential to generate superoxide in the presence of oxygen[7]. Thermodynamic or kinetic barriers are required to minimise superoxide-generating ‘bypass’ reactions within the Qo site during enzymatic turnover. In principle, superoxide production could be minimised by stabilization of the SQo intermediate, lowering its reactivity with oxygen but potentially creating a large steady-state occupancy. Enzyme-stabilized SQ intermediates are comparatively common in Nature (see Cape et al.[8] for discussion), but evidence for a stabilized SQo is lacking. Alternatively, destabilization of SQo would generate a highly-reactive intermediate with minimal superoxide production due to low SQo occupancy. Kinetic barriers to superoxide production by SQo might include simple steric exclusion of oxygen from Qo or otherwise entropically favoring non-bypass electron transfer reactions.
The SQo lies at the heart of several of the major questions about the Q-cycle. 1) What is the sequence of electron and proton transfer reactions in Qo during the Q-cycle? 2) Where is SQo formed and where does it travel within the Qo site? 3) How does SQo interact with the protein and how do those interactions favour the Q-cycle over side-reactions? The resolution of these questions requires experimental characterization of SQo.
Early attempts to detect the postulated SQo intermediate failed to find one that was sensitive to Qo site inhibitors[9] that block enzymatic activity. This missing intermediate prevented understanding of the strict use of two electron transport chains and the proton pumping, which are widely thought to involve motions of SQo within Qo and of the ISP subunit. Recent freeze-quench experiments[10] used different strategies to trap a SQ produced in the Qo site of cyt bc1 of Rhodobacter capsulatus. Both blocked the low-potential chain so it could accept only 1-2 electrons, before leaving a SQo stranded. Cape et al.[10a] rapidly mixed cyt bc1 with QH2 and froze it 10 ms later, long enough for 2-3 enzymatic turnovers to fill the low-potential chain and form SQo but not for SQo to react through bypass reactions[11] or escape from Qo. Electron paramagnetic resonance (EPR) measurements of the trapped SQ in these freeze-quenched samples showed it to be in or very near Qo. Zhang, et al.[10b] found consistent results in chromatophores of R. capsulatus mutants after flash excitation of the photosynthetic reaction centers to oxidize cyt bc1 complex followed by freezing.
Studies of the trapped SQ can probe reactions and interactions in Qo, but their interpretation depends on where that SQ is trapped and its chemical form, while their relevance to human or even eukaryotic pathophysiology is complicated by significant differences between bacterial and mitochondrial cyt bc1.[12] These considerations led us to apply rapid freeze-quench to mitochondrial cyt bc1 of the yeast Saccharomyces cerevisiae; to locate the trapped SQ using paramagnetic relaxation enhancement (PRE) of the SQ by the cyt bL and cyt c1 heme cofactors; and to characterize the trapped SQ and its interaction with the protein using pulsed EPR techniques.
We find that SQo is a radical anion contained within a predominantly hydrophobic region of Qo between the “b-distal” and “b-proximal” niches with minimal stabilizing hydrogen-bonding interactions with the surrounding protein. The lack of stabilization can both conserve energy and accelerate the Q-cycle reactions relative to side reactions.
Results
SQ is trapped in mitochondrial cyt bc1
The 77 K cw-EPR spectrum of freeze-quenched SQ is a single, nearly-symmetric line with g-factor ≈2.0044 and peak-to-peak line width of 1.16 mT (Fig. 1A), similar to SQ reported from R. capsulatus, g≈2.0054, 1.19 mT width[10a] and g≈2.0040, 1.17 mT width.[10b] Any contribution from SQ in the Qi site was prevented by using antimycin A (AA) to occupy Qi. SQ in the Qi site can also be ruled out because it has a distinctly narrower line width (∼0.8 mT).[13] The Qo site SQ signal largely disappears when slightly sub-stoichiometric stigmatellin (stig) blocks the Qo site, Fig. 1A; when UQH2 is absent; or in mutants lacking active Fe2S2.[10a] Fig. S3 shows the entire EPR spectrum with hemes and Fe2S2.
Figure 1.
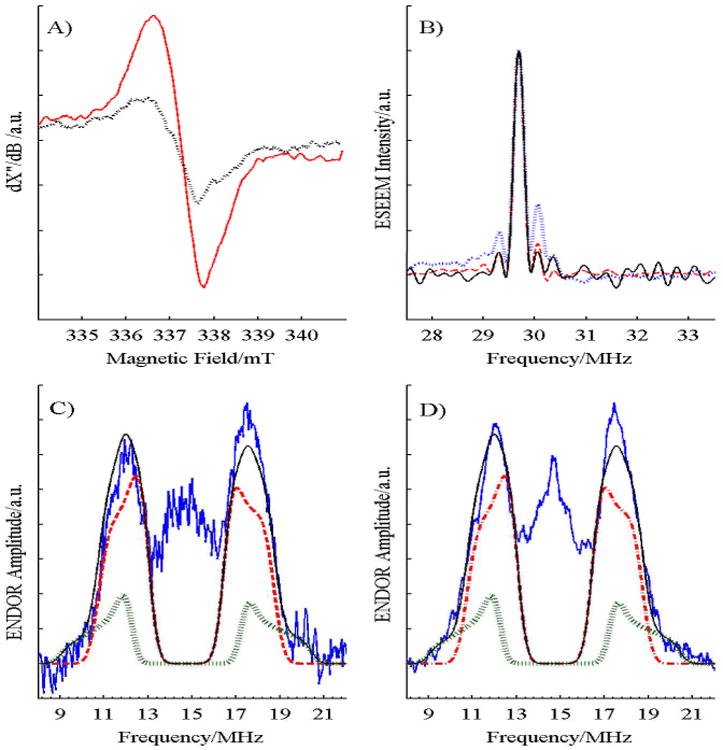
A) CW EPR spectrum of 15 μM mitochondrial cyt bc1 from S. cerevisiae, freeze-quench samples with 100 μM UQH2, 80 μM horse heart cyt c (to prevent complete reduction of the high-potential chain), 30 μM AA (to block the low-potential chain and keep Q out of Qi), with (Black, dotted) and without stig (Red, solid), at 6 mW of microwave power and a nominal temperature of 77 K. Slightly substoichiometric levels of stig were used to avoid interferences from an EPR-detectable radical in stig stock solutions. B) 4 pulse ESEEM spectra of SQ in mitochondrial cyt bc1 complex at 10 K with 1160 shots per phase step (Black, solid); and the UQ radical anion at 40 K in cyclohexanol (Red, dashed) and in CH3OH and CH2Cl2 (Blue, dotted) with 160 shots per phase step. There is no asymmetry near the peak at twice the Larmor frequency, 29.7 MHz, for SQ trapped in protein, indicating a lack of strong hydrogen bonds to the SQ. The spectra are normalized to the intensity of the main peak. Re-focused Mims ENDOR spectra at 40 K of SQo trapped in C) mitochondrial and D) bacterial cyt bc1 complex with τ = 60 ns to place the ‘blind spots’ near 7 and 23 MHz, well away from the observed features. The experimental spectra (Black) are well matched by the fit (Red) using 3 identical protons of the 5-methyl group, with hyperfine tensors of (8.0, 4.0, 3.5) MHz (Blue) and 1 proton of the 4-CH2 tail with (11.5, 5.5, 5.0) MHz (Green). The mitochondrial spectrum has 591 ‘stochastic’ shots and the bacterial has 464 shots per phase step taken during 8 scans.
Electron nuclear double resonance (ENDOR) measures the weak interactions between the unpaired electron spin and nearby nuclear spins, here protons that comprise the 1H nucleus. ENDOR provides an indication of the delocalization of the unpaired electron and its distribution in the SQ radical. The proton refocused Mims ENDOR spectrum of SQ in mitochondrial cyt bc1, Fig. 1C, consists of a peak centered at the proton Larmor frequency of ∼15 MHz from 1H protons in the methoxy groups and in the protein which have weak hyperfine couplings to the unpaired electron spin of the SQ. That peak is flanked by a pair of resolved peaks from protons with stronger hyperfine couplings. Its spectrum is very similar to that of SQ trapped in bacterial cyt bc1,[10a] Fig. 1D. The flanking peaks in both ENDOR spectra are assigned to the three protons of the 5-methyl group in the ubiquinone radical anion, while the weak tails extending 1.5 MHz beyond those peaks are assigned to a single methylene proton of the hydrocarbon tail. The entire spectrum lies well within the ‘blind spots’ of the Mims ENDOR measurement. The ENDOR spectra from the strongly-coupled protons of SQ in Qo of mitochondrial and bacterial cyt bc1 are well simulated, using the same hyperfine tensors: (Ax, Ay, Az) = (8.0, 4.0, 3.5) MHz and (11.5, 5.5, 5.0) MHz for the methyl and methylene protons respectively with a 3:1 stoichiometry in both the mitochondrial and the bacterial cyt bc1 (Figs. 1C-D). The two methylene protons of the phytyl chain are inequivalent with one large and one small coupling. The hyperfine coupling of the methylene proton depends on the dihedral angle between the plane of the SQ head group and the vector connecting C-H of the tail. The relatively large coupling indicates that the phytyl tail is twisted out of the plane of the radical macrocycle, giving the out-of-plane proton a strong coupling and the more in-plane proton a negligible coupling. The ENDOR results shows that the SQo trapped in both the bacterial and mitochondrial cyt bc1 are the same chemical species, in similar environments. The methyl hyperfine tensors lie in the range reported (see Table S1, Fig. S2 and their discussion in the Supporting Information) for UQ radical anion in proteins[14] and frozen solutions,[15] but about 50% smaller than for QH in cyt bo3 quinol oxidase where a controversial neutral SQ form is suggested.[15b, 16] There was no evidence of large hyperfine couplings from hydrogen-bonded protons as seen in other proteins with SQ radical anion,[14c] indicating an unusual trapping site.
SQ has no specific interactions with protein
Interactions of the SQ with protein or water were sought using the pulsed EPR technique known as ESEEM, which is highly effective at characterizing hydrogen bonds and coordination by nitrogens from proteins.[14c] Fig. 1B shows overlaid 4-pulse ESEEM spectra of various SQ samples at 15 K. Protons with small hyperfine anisotropy, e.g., 5-methyl, produce the strong, symmetric peak at 29.7 MHz while protons hydrogen bonded to the SQ carbonyl groups have a large anisotropy and are shifted 0.3-0.9 MHz to higher frequency, destroying the symmetry of the spectrum.[14c] The UQ radical anion in frozen methanol/dichloromethane has hydrogen bonds to the methanol and gives an asymmetric spectrum with extra intensity from 29.9-30.3 MHz. The less polar and more sterically-hindered cyclo-hexanol has less hydrogen bonding and less asymmetry in the same region. The spectrum of SQ in S. cerevisiae cyt bc1 is symmetric in that region, showing weak, or no, hydrogen bonding.
The 2- and 3-pulse ESEEM decays of SQ in cyt bc1 (Fig. 2) were carefully examined and totally lack the characteristic, low-frequency signals from coordination of SQ to protein nitrogens found in almost every reported SQ-containing protein.[14b, 14h, 15b, 16a] These two unexpected observations indicate that SQ does not interact with nitrogens from sidechains or the peptide backbone nor are there hydrogen bonds to the terminal oxygens of SQ.
Figure 2.
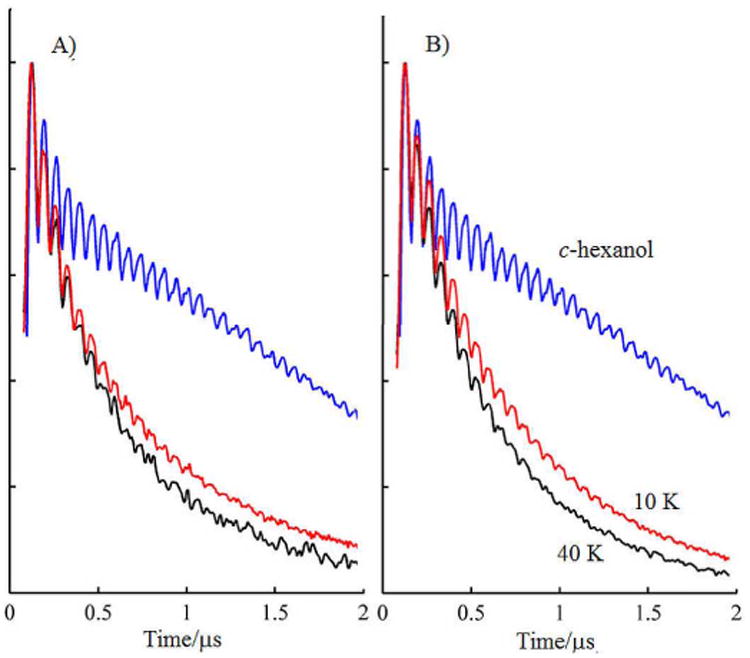
Comparison of the phase memory, TM, decay of the 2-pulse spin echo from SQo in A) mitochondrial and B) bacterial cyt bc1 at 10 K (red) and 40 K (black), with the UQ radical anion at 40 K in cyclohexanol (blue). For each spectrum, 256 points in τ separated by 8 ns were measured in the protein at a repetition rate of 163 Hz at 10 K with A) 640 or B) 128 shots per phase step, 245 Hz; at 40K with A) 480 or B) 320 shots per phase step and 490 Hz in cyclohexanol with 64 shots per phase step.
SQ is located in hydrophobic Qo pocket
The work of Prof. Bertini illustrated that nuclear spins located near paramagnetic centers in proteins were broadened, had faster spin relaxation rates and were shifted as a result of magnetic interactions with the electron spin of the paramagnetic center.[17] Similar effects occur for free radicals located in heme proteins. In particular, the electron transverse relaxation, TM, of SQ in the bacterial protein is strongly enhanced by the heme cofactors.[10a] This paramagnetic relaxation enhancement (PRE) was examined in detail in the mitochondrial cyt bc1 because PRE is useful for measuring distances between spins in proteins[18] including the Fe2S2-heme bL distance in cyt bc1 by Sarewicz et al.,[19] copper ion bound to cyt b6f,[20] cyt c bound to cyt c oxidase,[21] and spin-labeled metmyoglobin.[22]
The transverse relaxation time or TM of SQo had a significant PRE below ∼100 K (Figs. 2 and S4). The relaxation was distinctly non-exponential as noted for the Fe2S2 by cyt bL.[19] The temperature dependence of PRE was used to determine distance between the SQ and the nearby hemes, using theory developed for EPR by Zhidomirov and Salikhov.[23] The EPR signal of SQo is shifted by its dipolar interaction with the hemes. The spin relaxation of the heme causes the shift to vary with time. As the heme relaxation becomes faster, the TM relaxation of SQo initially speeds up until the heme spin-lattice relaxation rate matches the dipolar interaction and then slows down. Although all paramagnetic hemes in the protein contribute to the TM relaxation of SQo, The heme closest to the SQo will have the largest effect peaking at the lowest temperature. In most proteins the paramagnetic center nearest the free radical has a dominating effect.
The PRE could not be fit over its full temperature range based on interaction of SQ with only a single heme (or the Fe2S2) even allowing for multiple redox states of the protein. It was also clear that relaxation of the Fe2S2 is too slow to make a significant contribution to the observed PRE. The fitting was therefore extended to include interactions with two hemes. The freeze-quenched cyt bc1 sample contains a mixture of redox states, see Supporting Information, requiring four fitting parameters: the distances between SQ and each heme, and the fraction of each heme in its paramagnetic reduced state. The fractions of each of the two hemes in paramagnetic (and thus PRE-active) states were assumed to be independent and were varied independently in the fitting.
These fits with two hemes matched the measured relaxation to better than 3%. Examination of χ2 for the fit versus heme distances for the best-fitting heme redox states shows a well-defined global minimum at distances of 1.7 nm and 2.7 nm with 30% of each heme in its paramagnetic state. The 95% and 99% confidence limits, Fig. 3, for the distances are tight and consistent between samples. No interactions were seen between SQo and cyt bH, indicating that, as expected, heme bH was completely reduced during the first Qo site turnover and thus PRE-silent. However, cyt bL and/or cyt c1 could generate the observed PRE. Based on multiple measurements, the SQ trapped by rapid freeze-quench is localized 1.65-1.95 nm from the iron of one heme and 2.55-3.00 nm from that of another. These distances are large enough that the point dipole approximation is reasonable so that the distances correspond closely to the distance between the centers of SQ and the heme.
Figure 3.
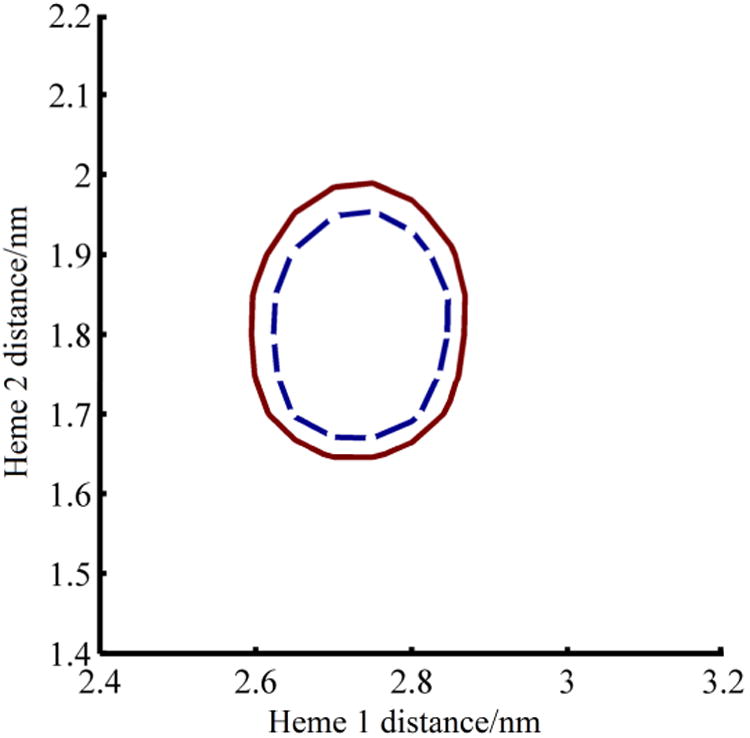
Plot of 95% (Blue, dashed) and 99% (Red, solid) confidence regions for one freeze-quenched sample as a function of distance from SQo to the two nearest hemes, when 30% of both hemes are paramagnetic. Heme 1 is the heme nearest SQo and heme 2 is the far heme. The confidence regions are calculated from the residuals and χ2 of fits to the experimental data. There is, for example, a 95% statistical likelihood that the distances to the hemes in the relaxation model lie within the blue curve.
To identify potential SQ trapping sites, a search was made for voids in a molecular model of yeast cyt bc1 (PDB: 3CX5) using intersecting spherical shell distance selections, consistent with the PRE distance ranges to the two nearest hemes. The only locations meeting these constraints are lobes of the Qo site, Figs. 4A-C and S1, distinct from the inhibitor binding sites known as the “distal” niche near Fe2S2 which binds stig and the “proximal” niche near cyt bL which binds myxothiazol or MOA-stilbene.
Figure 4.
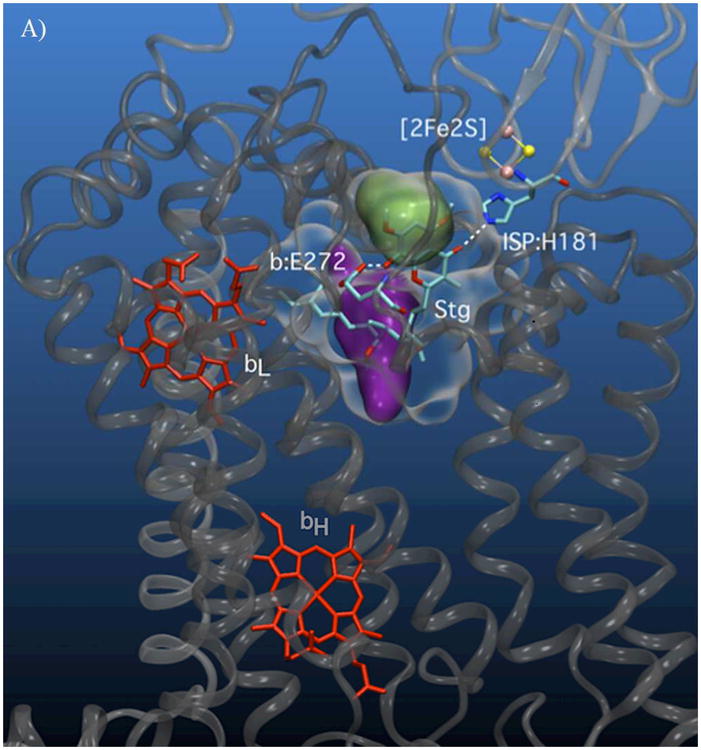
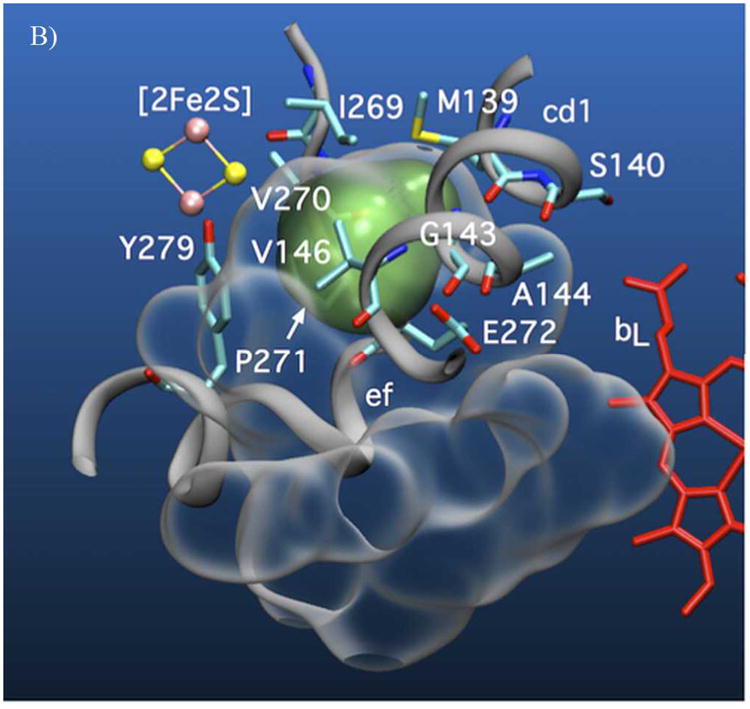
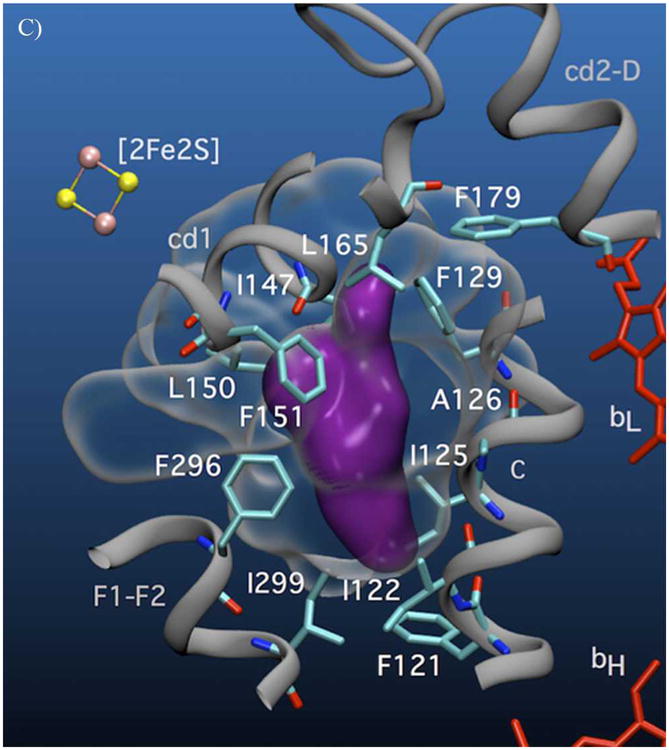
A) Volumetric representation of the SQ binding sites within yeast cyt bc1. The polypeptide backbones of cyt b and ISP from one functional monomeric unit of the yeast bc1 complex (3C×5.PDB[24]) are represented in dark and light grey respectively. The Qo void volume is represented as a glass surface. The Qo site volume overlapping the region defined by the bL(intramonomer)/c1(intramonomer) distance constraints (site I) is represented in green, and the bL(intramonomer)/bL(intermonomer) distance constraints (site II) is shown in purple. Also shown are hemes bL and bH (red) of cyt b and the Fe2S2 of the ISP (yellow/pink CPK). E272 (cyt b) and H181 (ISP Fe2S2 ligand) are represented in stick form. Qo-bound stigmatellin is labeled ‘Stg’. B) Cyt b residues in close contact with (< 4 Å) site I (green) and C) site II (purple) are shown in stick form.
The strongest contribution to the PRE for both lobes comes from cyt bL of the same monomer. A weaker, but significant, PRE contribution for one lobe comes from intermonomer cyt bL and for the other from the intramonomer cyt c1. The present data are unable to conclusively decide between these two lobes. Yet they share similar properties and place the SQ within the Qo site as the elusive SQo intermediate of the Q-cycle. The ENDOR and ESEEM results are therefore highly relevant to the Q-cycle mechanism and both sites are logical stages in the Q-cycle.
Discussion
Nature of SQo and its interaction with protein
To our knowledge, no crystal structures of cyt bc1 complexes have been solved with substrate QH2 or reaction intermediates bound in the Qo pocket. The reasonable default assumption is that QH2, at least initially, binds similarly to stig, which appears in the structures to be strongly hydrogen bonded to the imidazole of H181 of the ISP and the glutamyl of E272 of cyt b.[5] This QH2 binding configuration would require deprotonation of H181 which only occurs to appreciable extents for the oxidized Fe2S2 state. The spectroscopic characterization of the SQo reveals several remarkable features that we propose are related to its function.
The 2-, 3- and 4-pulse ESEEM spectra are highly unusual in their lack of detectable ESEEM from nitrogen or strong H-bonds, indicating that the interactions of trapped SQo are distinctly different from those of stig. This lack of strong interactions with the protein is unique. Previously-characterized UQ radicals show prominent ESEEM from amino acid side chain or peptide nitrogens and from hydrogen-bonded protons to carbonyl oxygens of the UQ.[14b, 14h, 15b, 16a]
This lack of H-bonding to the anionic SQo implies: 1) oxidation of QH2 occurs in a catalytic complex with both electron and H+ acceptors, removing one electron and two protons to form the SQ anion; and 2) formation of SQo is followed by conformational changes, moving SQ and/or protein components, to break or prevent hydrogen bonds.
SQ binding sites within Qo
The two possible SQo locations lie in lobes of the Qo site, Fig. 4A-C. These locations are consistent with our observations[10a] that: 1) the formation of SQo is abolished by binding of stig and 2) SQo is isolated from bulk solvent.
Both lobes place SQo nearest the intramonomer cyt bL heme, but they differ as to the next nearest heme. The lobe nearer the intramonomer cyt c1 provides a region of possible SQo locations that we call site I. It partly overlaps the chromone ring of bound stig (Figs. 4A-B, in green). The lobe nearer to intermonomer cyt bL, called site II, (Figs. 4A, C, in purple) is separate from the region occupied by the chromone ring of stig and is 1.3 nm from the Fe2S2 of Qo-docked ISP. Sites I and II are only 0.8 nm apart. More importantly, they have edge-to-edge separations of 1.2 and 1.35 nm respectively to heme bL. Using the Moser-Dutton approximation, these distances suggest a theoretical SQo/heme bL electron transfer rates of 1 & 106 s-1 for site I and 4 & 104 s-1 for site II, which are in reasonable agreement with those expected from turnover in the uninhibited cyt bc1.
In fact, these two locations need not be mutually exclusive and SQo may progress through both sites and may be trapped by freeze-quenching in one or both. Moreover, both sites are compatible with EPR analysis of Qo site occupancy in bacterial bc1 mutants where the SQ was proposed to move within Qo during catalysis.[25] The cyt bc1 provides at least two potential hydrogen-bonding groups in site I: H181 on the ISP and E272 on cyt b. To accommodate our data, H-bonding of SQo to both H181 and E272 would have to be blocked thermodynamically or sterically. Indeed, the ISP pivots around its flexible tether and adopts multiple positions to allow electron transfer from the ISP to cyt c1,[26] moving H181 and preventing H-bonding between H181 and SQo. Likewise, the E272 glutamyl sidechain is found in different orientations in different crystal structures.[12] In the stig-containing structure from yeast, the glutamyl carboxyl group makes a strong H-bond to the chromone hydroxyl,[27] whereas in the famoxadone-containing bovine structure, it has flipped by 150° rotation about its χ2 dihedral angle[28] and interacts with a potential proton channel within the cyt b protein. E272 is similarly rotated in the structure of the HHDBT-inhibited yeast bc1, a compound proposed to mimic a hydroxyquinone anion.[29] Motion of the two groups allows formation of an electron transfer complex analogous to the stig complex,[30] but also provides conformational changes to strip the two quinol protons, leaving the radical anion free to move through site I and into site II. By removing H-bonds from SQo these conformational changes would tend to trap the SQo in its deprotonated anionic state. Aside from peptide backbone amides, site II has no obvious H-bond donors or acceptors. Site II is formed from predominantly hydrophobic, conserved residues from regions of cyt b encompassing transmembrane helix C (residues 122-129), the cd1 helix (147-150) and the F1-F2 loop (296-299). The enthalpic cost of burying the anionic SQ in this non-polar niche (effectively a desolvation penalty) will likely be counterbalanced by entropic factors, i.e., the multiple possible conformations that can be accommodated within the site II volume. This effect could accommodate the anionic SQo in a relatively aprotic environment.
Reactions and Energetics of SQ in Qo
Previous freeze-quench[10a] and kinetics experiments[1e, 31] indicate a very low equilibrium constant for formation of SQo. Most SQ species found in enzymes, e.g. at the Qi site of bc1, the QB sites of bacterial reaction centers and photosystem II, the QH2 oxidation site of bacterial bo3 and aa3 quinol oxidases[32] etc., are bound considerably more tightly than Q or QH2, raising the stability constant (by five to ten orders of magnitude),[33] allowing reduction of Q or oxidation of QH2 to proceed by sequential, one-electron transitions. The high stability constant for these SQ species is critical for the so-called two-electron gating which connects the one-electron chemistry of cytochromes or Fe2S2 with the two-electron chemistry of Q/QH2.[4a] The redox properties of SQo more resemble those of the photosystem I phylloquinone, which also appears destabilized with respect to solution (i.e. with a more reducing Q/SQ.- redox potential) to allow electron transfer to the low potential Fx Fe4S4 cluster.[34] Similar anionic semiquinone destabilization has been observed at the QB site of the bacterial photosynthetic reaction center following mutation of arginine residues to non-polar isoleucines.[35] The small stability constant for SQo is consistent with its function in the Q-cycle since a stabilized SQ would lack sufficient reducing power to push electrons against the electrochemical gradient to the low-potential cyt b.[8] Thermodynamically, an unstable SQo should also be weakly bound in the Qo site since specific, tight binding through hydrogen bonds or salt bridges would stabilize the intermediate. The structural data presented here suggests that, because it is charged, SQo is kinetically trapped, rather than thermodynamically stabilized, via an “electrostatic cage” of hydrophobic residues of the surrounding Qo pocket, maintaining SQo reactivity yet preventing its rapid escape and subsequent loss of energy through disproportionation.[36] This same hydrophobic pocket would also act to prevent escape of any superoxide anion formed and would prevent influx of H+ to neutralize it and facilitate escape.
Conclusion
Our results lead us to propose that the bc1 complex traps SQo as an anionic, destabilized and non-hydrogen bonded intermediate in hydrophobic sites within Qo. The anionic nature of the SQ intermediate – a result of side chain-mediated ‘proton stripping’[37] in Qo – is critical for favoring the Q-cycle and slowing the rate of short-circuiting and other deleterious side reactions.[36] The first electron transfer is accompanied by proton transfer to ISP H181, and abstraction of the other proton by cyt b E272.[38] The deprotonated SQ anion is then trapped by conformational changes in SQo intermediates and protein components, consistent with several proposed models.[12, 26b, 39] The SQo is trapped because the surrounding low dielectric of the Qo site forms a hydrophobic “cage”,[40] but without thermodynamic stabilization.
The destabilized SQo not only conserves sufficient redox energy to reduce cyt bL, but should also suppress the rates of other potential Q-cycle bypass or short circuit reactions.[8, 36, 41] Reduction of a SQ anion to Q2- (without proton uptake) should be thermodynamically very unfavorable, slowing the oxidation of previously-reduced cyt bL, a potential Q-cycle bypass reaction[36]. Finally, as discussed in Cape et al.,[8] the low occupancy of an unstable SQo would also limit the diffusion-limited rate of O2 reduction, minimizing superoxide production.
Overall, we make the (counterintuitive) observation that Nature, contrary to most enzymes which specifically and tightly bind reactive intermediates, as in the Pauling paradigm,[42] guides the enigmatic Q-cycle reactions by forming an insulating “reaction chamber” that contains, but does not strongly interact with, the reactive intermediates of the Q-cycle. The chamber has the critical properties of preserving SQo reactivity while removing protons that could lead to deleterious side reactions
Experimental Section
Chemicals
Decylubiquinone, Co-Q10, AA and stig were obtained from Sigma (St. Louis, MO) and used without further purification. Samples of ubiquinone radical anion were prepared from decylubiquinone and Co-Q10 by air-oxidation in isopropanol/KOH or by photolysis at 77 K in cyclohexanol/KOH followed by annealing at 200 K. Decylubiquinol was prepared at each use by sodium borohydride reduction of decylubiquinone followed by quenching of excess sodium borohydride by HCl.
Biochemical Procedures
Yeast cyt bc1 complex was isolated from commercial baker's yeast using established protocols.[11a] The concentration of cyt bc1 was measured from dithionite reduced minus ferricyanide oxidized difference spectra at 562 and 575 nm, using an extinction coefficient of 28 mM-1 cm-1. Horse heart cytochrome c was purchased from Sigma (St. Louis, MO) and used without further purification.
Freeze-Quenched EPR Samples
Freeze-quenched samples were prepared by mixing anaerobically 15 μM cyt bc1 from S. cerevisiae, 100 μM UQH2, 80 μM horse heart cyt c to prevent complete reduction of the high-potential chain, and 30 μM AA, with a 10 ms delay between mixing and freezing in a freeze-quench apparatus (Model MPS-51; Biologic, Indianapolis, IN). Samples were subsequently stored and shipped at 77 K.
EPR Measurements
The CW X-band EPR measurements were carried out on a Bruker Elexsys-E560 spectrometer and ER4102ST resonator with a liquid nitrogen insertion dewar or ER4112HV helium flow cryostat. Pulsed EPR measurements were carried out on a Bruker Elexsys-E680 spectrometer with EN4118X-MD4 resonator, DICE ENDOR, a 500 W ENI A-500 RF amplifier and an ER4118CF cryostat with ITC503 controller. Echo-detected field-sweep EPR spectra were recorded by integrating the 2-pulse electron spin echo (ESE) generated by (π/2-τ-π) pulse sequence[43] with a 4-step phase cycle. The ESE decay was recorded by integrating the 2-pulse ESE and incrementing τ[43] with a 4-step phase cycle. Refocused Mims ENDOR was recorded by integrating the refocused stimulated echo that was altered by the RF pulses (π/2-τ-π/2-RF-π/2-τ‘-π pulse sequence with a 4-step phase cycle).[44] The 4-pulse ESEEM was measured with a (π/2-τ-π/2-T-π-T-π/2 pulse) sequence and a 16-step phase cycle with τ=158 ns and T incremented by 12 ns for 4096 points. The π/2 and π microwave pulses have nominal widths of 16 and 32 ns, respectively and the RF π pulses a width of 13 μs. The high reproducibility of normalized spectral intensity of Mims ENDOR spectra in our hands allows relative stoichiometry of protons in the spectrum to be determined in simulations using Easyspin.[45]
Theory
The decay of the two-pulse or primary echo was analyzed as a function of temperature using a custom MATLAB program to determine the dipolar interaction and the distance between the two spins based on the theory developed by Salikhov.23b] The program does a global least-squares fit to all measured decay curves with the distance to the heme and the fraction of that heme in its +3 state as adjustable parameters. The calculated PRE function is multiplied by the background decay of the primary echo from other causes and the ESEEM which were assumed to be independent of temperature over the experimental range. The average noise in each measurement was determined from the quadrature signal channel which contains noise but no signal. This allowed the χ2 to be calculated for the fits. All fits based on PRE from a single heme center had an inordinately large χ2 value as well as unacceptable residuals and led to the inclusion of a second heme. Because the spin-lattice relaxation of two hemes is uncorrelated, their effect on PRE can be multiplied together in the framework of Salikhov’s theory. Fits based on PRE from two centers with four adjustable parameters: distance from the radical to each heme and the fraction of each heme in its +3 state (assuming the probabilities were uncorrelated) dramatically improved the χ2 for the fits, greatly reduced the residuals and fit the experimental curves to within ∼3%.
Molecular Modeling of the SQ Distance Constraints
Q-Site Finder[46] was used to fill the void volume of the Qo site of the yeast bc1 atomic coordinates 3C×5.PDB[24] with methyl (-CH3) probe groups at a grid resolution of 0.09 nm. The resulting methyl-filled Qo void coordinates were appended to 3C×5 and written as a PDB file for visualization within VMD.[47] EPR distance constraints overlapping with probe groups within the Qo void volume were modeled in VMD using intersecting spherical shells centered on the appropriate heme-Fe atom coordinates, and displayed volumetrically using the ‘QuickSurf’ representation within VMD using a density isovalue and grid spacing of 0.5 and 0.2 respectively.
Supplementary Material
Acknowledgments
Research reported in this publication was supported by the National Institute of General Medical Sciences of the National Institute of Health under award number R01GM061904. The authors thank Dr. Jonathan Cape (Bend Research, Bend OR), Dr. Isaac Forquer (Oregon Health and Science University, Portland, Oregon) and Professor Mark Schumaker for interesting discussions and Dr. Alexander Maryasov (V. V. Voevodsky Institute of Chemical Kinetics and Combustion, Novosibirsk) for advice on PRE and Dr. Qiang Xu (Washington State University) for assistance in setting up the freeze quench apparatus.
Contributor Information
Dr. Preethi R. Vennam, Email: mkbowman@as.ua.edu.
Dr. Matthew D. Krzyaniak, Email: mkbowman@as.ua.edu.
Prof. Michael K. Bowman, Email: mkbowman@as.ua.edu.
References
- 1.a) Crofts AR. Annu Rev Physiol. 2004;66:689–733. doi: 10.1146/annurev.physiol.66.032102.150251. [DOI] [PubMed] [Google Scholar]; b) Berry EA, Guergova-Kuras M, Huang LS, Crofts AR. Annu Rev Biochem. 2000;69:1005–1075. doi: 10.1146/annurev.biochem.69.1.1005. [DOI] [PubMed] [Google Scholar]; c) Schutz M, Brugna M, Lebrun E, Baymann F, Huber R, Stetter KO, Hauska G, Toci R, Lemesle-Meunier D, Tron P, Schmidt C, Nitschke W. J Mol Biol. 2000;300:663–675. doi: 10.1006/jmbi.2000.3915. [DOI] [PubMed] [Google Scholar]; d) Cooley JW, Nitschke W, Kramer DM. In: The Purple Photosynthetic Bacteria. Hunter CN, Daldal F, Thurnauer MC, Beatty JT, editors. Springer; Dordrecht, The Netherlands: 2008. pp. 451–473. [Google Scholar]; e) Crofts AR, Wang ZG. Photosynthesis Res. 1989;22:69–87. doi: 10.1007/BF00114768. [DOI] [PubMed] [Google Scholar]
- 2.a) Mitchell P. FEBS Lett. 1975;59:137–139. doi: 10.1016/0014-5793(75)80359-0. [DOI] [PubMed] [Google Scholar]; b) Crofts AR, Shinkarev VP, Kolling DR, Hong S. J Biol Chem. 2003;278:36191–36201. doi: 10.1074/jbc.M305461200. [DOI] [PubMed] [Google Scholar]
- 3.a) Iwata S, Lee JW, Okada K, Lee JK, Iwata M, Rasmussen B, Link TA, Ramaswamy S, Jap BK. Science. 1998;281:64–71. doi: 10.1126/science.281.5373.64. [DOI] [PubMed] [Google Scholar]; b) Berry EA, Huang LS. FEBS Lett. 2003;555:13–20. doi: 10.1016/s0014-5793(03)01099-8. [DOI] [PubMed] [Google Scholar]; c) Esser L, Quinn B, Li YF, Zhang MQ, Elberry M, Yu L, Yu CA, Xia D. J Mol Biol. 2004;341:281–302. doi: 10.1016/j.jmb.2004.05.065. [DOI] [PubMed] [Google Scholar]; d) Hunte C, Palsdottir H, Trumpower BL. FEBS Lett. 2003;545:39–46. doi: 10.1016/s0014-5793(03)00391-0. [DOI] [PubMed] [Google Scholar]
- 4.a) Crofts AR, Holland JT, Victoria D, Kolling DR, Dikanov SA, Gilbreth R, Lhee S, Kuras R, Kuras MG. Biochim Biophys Acta. 2008;1777:1001–1019. doi: 10.1016/j.bbabio.2008.04.037. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Zhang H, Chobot SE, Osyczka A, Wraight CA, Dutton PL, Moser CC. J Bioenerg Biomembr. 2008;40:493–499. doi: 10.1007/s10863-008-9174-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Esser L, Quinn B, Li YF, Zhang MQ, Elberry M, Yu L, Yu CA, Xia D. J Mol Biol. 2004;341:281–302. doi: 10.1016/j.jmb.2004.05.065. [DOI] [PubMed] [Google Scholar]
- 6.Crofts AR, Barquera B, Gennis RB, Kuras R, Guergova-Kuras M, Berry EA. Biochemistry. 1999;38:15807–15826. doi: 10.1021/bi990962m. [DOI] [PubMed] [Google Scholar]
- 7.Muller FL, Roberts AG, Bowman MK, Kramer DM. Biochemistry. 2003;42:6493–6499. doi: 10.1021/bi0342160. [DOI] [PubMed] [Google Scholar]
- 8.Cape JL, Bowman MK, Kramer DM. Trends Plant Sci. 2006;11:46–55. doi: 10.1016/j.tplants.2005.11.007. [DOI] [PubMed] [Google Scholar]
- 9.Junemann S, Heathcote P, Rich PR. J Biol Chem. 1998;273:21603–21607. doi: 10.1074/jbc.273.34.21603. [DOI] [PubMed] [Google Scholar]
- 10.a) Cape JL, Bowman MK, Kramer DM. Proc Natl Acad Sci USA. 2007;104:7887–7892. doi: 10.1073/pnas.0702621104. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Zhang H, Osyczka A, Dutton PL, Moser CC. Biochim Biophys Acta. 2007;1767:883–887. doi: 10.1016/j.bbabio.2007.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.a) Kramer DM, Roberts AG, Muller F, Cape J, Bowman MK. Methods Enzymol. 2004;382:21–45. doi: 10.1016/S0076-6879(04)82002-0. [DOI] [PubMed] [Google Scholar]; b) Cape JL, Aidasani D, Kramer DM, Bowman MK. Biochemistry. 2009;48:10716–10723. doi: 10.1021/bi901205w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Berry EA, Huang LS. Biochim Biophys Acta. 2011;1807:1349–1363. doi: 10.1016/j.bbabio.2011.04.005. [DOI] [PubMed] [Google Scholar]
- 13.Kolling DRJ, Samoilova RI, Holland JT, Berry EA, Dikanov SA, Crofts AR. J Biol Chem. 2003;278:39747–39754. doi: 10.1074/jbc.M305913200. [DOI] [PubMed] [Google Scholar]
- 14.a) Schnegg A, Dubinskii AA, Fuchs MR, Grishin YA, Kirilina EP, Lubitz W, Plato M, Savitsky A, Mobius K. Appl Magn Reson. 2007;31:59–98. [Google Scholar]; b) Martin E, Samoilova RI, Narasimhulu KV, Wraight CA, Dikanov SA. J Am Chem Soc. 2010;132:11671–11677. doi: 10.1021/ja104134e. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Martin E, Samoilova RI, Narasimhulu KV, Lin TJ, O'Malley PJ, Wraight CA, Dikanov SA. J Am Chem Soc. 2011;133:5525–5537. doi: 10.1021/ja2001538. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Lubitz W, Feher G. Appl Magn Reson. 1999;17:1–48. [Google Scholar]; e) Rohrer M, MacMillan F, Prisner TF, Gardiner AT, Möbius K, Lubitz W. J Phys Chem B. 1998;102:4648–4657. [Google Scholar]; f) Isaacson RA, Abresch EC, Paddock ML, Feher G, Lendzian F, Lubitz W. Biophys J. 1998;74:A135–A135. doi: 10.1016/S0006-3495(95)79936-2. [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Lubitz W, Abresch EC, Debus RJ, Isaacson RA, Okamura MY, Feher G. Biochim Biophys Acta. 1985;808:464–469. doi: 10.1016/0005-2728(85)90155-0. [DOI] [PubMed] [Google Scholar]; h) Dikanov SA, Holland JT, Endeward B, Kolling DR, Samoilova RI, Prisner TF, Crofts AR. J Biol Chem. 2007;282:25831–25841. doi: 10.1074/jbc.M702333200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.a) MacMillan F, Lendzian F, Lubitz W. Magn Reson Chem. 1995;33:S81–S93. [Google Scholar]; b) MacMillan F, Kacprzak S, Hellwig P, Grimaldi S, Michel H, Kaupp M. Faraday Discuss. 2011;148:315. doi: 10.1039/c005149g. [DOI] [PubMed] [Google Scholar]
- 16.a) Yap LL, Samoilova RI, Gennis RB, Dikanov SA. J Biol Chem. 2007;282:8777–8785. doi: 10.1074/jbc.M611595200. [DOI] [PubMed] [Google Scholar]; b) Yap LL, Samoilova RI, Gennis RB, Dikanov SA. J Biol Chem. 2006;281:16879–16887. doi: 10.1074/jbc.M602544200. [DOI] [PubMed] [Google Scholar]; c) Grimaldi S, MacMillan F, Ostermann T, Ludwig B, Michel H, Prisner T. Biochemistry. 2001;40:1037–1043. doi: 10.1021/bi001641+. [DOI] [PubMed] [Google Scholar]
- 17.Bertini I, Turano P. In: Nuclear Magnetic Resonance of Paramagnetic Macromolecules. LaMar GN, editor. Kluwer Academic Publishers; Dordrecht: 1995. pp. 29–54. [Google Scholar]
- 18.a) Kulikov AV, Likhtenstein GI. Adv Mol Relax Int Proc. 1977;10:47–79. [Google Scholar]; b) Likhtenshtein GI. In: Distance Measurement in Biological Systems by EPR. Berliner LJ, Eaton GR, Eaton SS, editors. Kluwer Academic; New York: 2000. pp. 309–345. [Google Scholar]
- 19.Sarewicz M, Dutka M, Froncisz W, Osyczka A. Biochemistry. 2009;48:5708–5720. doi: 10.1021/bi900511b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rao BK, Tyryshkin AM, Roberts AG, Bowman MK, Kramer DM. Biochemistry. 2000;39:3285–3296. doi: 10.1021/bi991974a. [DOI] [PubMed] [Google Scholar]
- 21.Lyubenova S, Siddiqui MK, de Vries MJMP, Ludwig B, Prisner TF. J Phys Chem B. 2007;111:3839–3846. doi: 10.1021/jp065805t. [DOI] [PubMed] [Google Scholar]
- 22.Ulyanov D, Bowler BE, Eaton GR, Eaton SS. Biophys J. 2008;95:5306–5316. doi: 10.1529/biophysj.108.141887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.a) Zhidomirov GM, Salikhov KM. Sov Phys, JETP. 1969;29:1037–1040. [Google Scholar]; b) Salikhov KM, Dzuba SA, Raitsimring AM. J Magn Reson. 1981;42:255–276. [Google Scholar]
- 24.Solmaz SR, Hunte C. J Biol Chem. 2008;283:17542–17549. doi: 10.1074/jbc.M710126200. [DOI] [PubMed] [Google Scholar]
- 25.Crofts AR, Barquera B, Gennis RB, Kuras R, Guergova-Kuras M, Berry EA. Biochemistry. 2000;38:15807–15826. doi: 10.1021/bi990962m. [DOI] [PubMed] [Google Scholar]
- 26.a) Kim H, Xia D, Yu CA, Xia JZ, Kachurin AM, Zhang L, Yu L, Deisenhofer J. Proc Natl Acad Sci USA. 1998;95:8026–8033. doi: 10.1073/pnas.95.14.8026. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Cooley JW, Roberts AG, Bowman MK, Kramer DM, Daldal F. Biochemistry. 2004;43:2217–2227. doi: 10.1021/bi035938u. [DOI] [PubMed] [Google Scholar]
- 27.Hunte C, Koepke J, Lange C, Rossmanith T, Michel H. Structure. 2000;8:669–684. doi: 10.1016/s0969-2126(00)00152-0. [DOI] [PubMed] [Google Scholar]
- 28.Gao X, Wen X, Yu C, Esser L, Tsao S, Quinn B, Zhang L, Yu L, Xia D. Biochemistry. 2002;41:11692–11702. doi: 10.1021/bi026252p. [DOI] [PubMed] [Google Scholar]
- 29.Palsdottir H, Lojero CG, Trumpower BL, Hunte C. J Biol Chem. 2003;278:31303–31311. doi: 10.1074/jbc.M302195200. [DOI] [PubMed] [Google Scholar]
- 30.Rich PR. Biochim Biophys Acta. 2004;1658:165–171. doi: 10.1016/j.bbabio.2004.04.021. [DOI] [PubMed] [Google Scholar]
- 31.Zhu J, Egawa T, Yeh SR, Yu LD, Yu CA. Proc Natl Acad Sci USA. 2007;104:4864–4869. doi: 10.1073/pnas.0607812104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.a) Ingledew WJ, Ohnishi T, Salerno JC. Eur J Biochem. 1995;227:903–908. doi: 10.1111/j.1432-1033.1995.tb20217.x. [DOI] [PubMed] [Google Scholar]; b) Yi SM, Narasimhulu KV, Samoilova RI, Gennis RB, Dikanov SA. J Biol Chem. 2010;285:18241–18251. doi: 10.1074/jbc.M110.116186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.a) Hägerhäll C, Magnitsky S, Sled VD, Schröder I, Gunsalus RP, Cecchini G, Ohnishi T. J Biol Chem. 1999;274:26157–26164. doi: 10.1074/jbc.274.37.26157. [DOI] [PubMed] [Google Scholar]; b) Gunner MR, Madeo J, Zhu Z. J Bioenerg Biomembr. 2008;40:509–519. doi: 10.1007/s10863-008-9179-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shinkarev P, Vassiliev IR, Golbeck JH. Biophys J. 2000;78:363–372. doi: 10.1016/S0006-3495(00)76599-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cherepanov A, Bibikov SI, Bibikova MV, Bloch DA, Drachev LA, Gopta OA, Oesterhelt D, Semenov AY, Mulkidjanian AY. Biochim Biophys Acta. 2000;1459:10–34. doi: 10.1016/s0005-2728(00)00110-9. [DOI] [PubMed] [Google Scholar]
- 36.Muller F, Crofts AR, Kramer DM. Biochemistry. 2002;41:7866–7874. doi: 10.1021/bi025581e. [DOI] [PubMed] [Google Scholar]
- 37.Kramer M, Cape JL, Forquer I, Bowman MK. In: Photosynthesis: Fundamental Aspects to Global Perspectives. van der Est A, Bruce D, editors. Allen Publishing; Lawrence, KS: 2005. pp. 424–427. [Google Scholar]
- 38.Crofts R, Holland JT, Victoria D, Kolling DRJ, Dikanov SA, Gilbreth R, Lhee S, Kuras R, Kuras MG. Biochim Biophys Acta. 2008;1777:1001–1019. doi: 10.1016/j.bbabio.2008.04.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Crofts R, Guergova-Kuras M, Huang L, Kuras R, Zhang Z, Berry EA. Biochemistry. 1999;38:15791–15806. doi: 10.1021/bi990961u. [DOI] [PubMed] [Google Scholar]
- 40.Shinkarev VP. FEBS Lett. 2006;580:2534–2539. doi: 10.1016/j.febslet.2006.04.022. [DOI] [PubMed] [Google Scholar]
- 41.Rutherford AW, Osyczka A, Rappaport F. FEBS Lett. 2012;586:603–616. doi: 10.1016/j.febslet.2011.12.039. [DOI] [PubMed] [Google Scholar]
- 42.Pauling L. Nature. 1948;161:707–709. doi: 10.1038/161707a0. [DOI] [PubMed] [Google Scholar]
- 43.Bowman K. In: Electron Paramagnetic Resonance - A Practitioner's Toolkit. Brustalon M, Giamello E, editors. John Wiley & Sons; Hoboken: 2009. pp. 159–194. [Google Scholar]
- 44.a) Astashkin AV, Kawamori A, Kodera Y, Kuroiwa S, Akabori K. J Chem Phys. 1995;102:5583–5588. [Google Scholar]; b) Doan PE, Hoffman BM. Chem Phys Lett. 1997;269:208–214. [Google Scholar]
- 45.Stoll S, Schweiger A. J Magn Reson. 2006;178:42–55. doi: 10.1016/j.jmr.2005.08.013. [DOI] [PubMed] [Google Scholar]
- 46.Laurie AT, Jackson RM. Bioinformatics. 2005;21:1908–1916. doi: 10.1093/bioinformatics/bti315. [DOI] [PubMed] [Google Scholar]
- 47.Humphrey W, Dalke A, Schulten K. J Mol Graphics Model. 1996;14:33–38. 27–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.