

Abstract
Due to its cytotoxicity, free copper is chelated by protein side chains and does not exist in vivo. Several chaperones transport copper to various cell compartments, but none has been identified that traffic copper to the nucleus. Copper-64 decays by β+ and β- allowing for PET imaging and targeted radionuclide therapy of cancer. Because the delivery of 64Cu to the cell nucleus may enhance the therapeutic effect of copper radiopharmaceuticals, elucidation of the pathway(s) involved in transporting copper to the tumor cell nucleus is important for optimizing treatment. We identified Atox1 as one of the proteins that binds copper in the nucleus. Mouse embryonic fibroblast (MEF) cell lines, positive and negative for Atox1, were used to determine the role of Atox1 in 64Cu transport to the nucleus. MEF Atox1+/+ accumulated more 64Cu in the nucleus compared to Atox1-/- cells. HCT116 colorectal cancer cell lines expressing p53 (+/+) and non-expressing (-/-) were used to evaluate the role of this tumor suppressor protein in 64Cu transport. In cells treated with cisplatin (cisPt), the uptake of 64Cu in the nucleus of HCT116 p53+/+ was greater compared to p53-/- cells. Atox1 expression increased in p53 positive and negative HCT116 cells treated with cisPt; however, Atox1 localized to the nuclei of p53+/+ cells more than in the p53-/- cells. The data presented here indicate that Atox1 is involved in copper transport to the nucleus, and cisPt affects nuclear transport of 64Cu in HCT116 cells by up-regulating the expression and the nuclear localization of Atox1.
Keywords: Atox1, p53, copper-64, radiotherapy, copper chaperone
Introduction
Copper is a vital micronutrient and is essential for the activity of multiple enzymes in the cell, such as cytochrome C, superoxide dismutase and lysyl oxidase [1]. Due to its cytotoxicity, free copper essentially does not exist in vivo, as it is chelated through side chains on proteins [2]. Copper-64 (T1/2= 12.7 h, β+:0.655 MeV, 17.8%; β-: 0.573 MeV, 41%) is a β+ and β- emitter making it an ideal radionuclide for PET imaging and targeted radionuclide therapy. For example, copper radiopharmaceuticals have been widely investigated for PET imaging, both in animal models and humans [3]. Therapeutic efficacy of 64Cu radiopharmaceuticals in rat and hamster models was demonstrated with 64Cu-labeled somatostatin analogs and monoclonal antibody (mAb) conjugates, respectively [4, 5]. Moreover, enhanced therapeutic efficacy of an internalizing 64Cu-labeled mAb, 64Cu-BAT-2IT-IA3, in a tumor bearing hamster model was observed compared to published studies of 131I or 90Y- labeled mAbs in the same animal model [6]. Wang et al. [7] found the nuclear uptake of 64Cu from 64Cu-TETA-octreotide localized in the nucleus to a greater extent than 111In from 111In-DTPA-octreotide.
Copper binds to DNA and has been suggested to play an important role in DNA folding and repair [8]. Chui et al. [9, 10] found that treatment of isolated nuclei with unlabeled Cu2+ enhanced DNA crosslinking to nuclear matrix proteins upon irradiation, and that Cu2+ can cause additional damage to DNA and/or nuclear proteins by producing free radicals at copper binding sites. Because the delivery of 64Cu to the cell nucleus may enhance the therapeutic effect of β- and low energy electron-emitting tumor targeting copper radiopharmaceuticals, elucidating the pathway(s) involved in transporting copper to the nucleus is important for optimizing therapy.
Multiple studies have been performed to evaluate the cellular transport of copper. Copper enters cells through the hCtr1 (human copper transporter 1) protein and is delivered to different compartments [11]. Several copper binding proteins and chaperones have been identified including metallothionein, Cox 17, and Atox1, which are involved in copper storage, transport to the mitochondria, and transport to the Golgi apparatus [1, 11-13]. To date, there is no definitive evidence for a chaperone that transports copper to the cell nucleus.
Cisplatin (cisPt) is a potent chemotherapeutic agent. Multiple lines of evidence indicate that the mechanism of transport of cisPt into the cell and its distribution to different cell compartments involves copper transporters [14]. CisPt binds to the metal binding site of Atox1, as well as to Cu-loaded Atox1, without loss of copper [15, 16]. A relationship between nuclear transport of copper and cisPt may exist, but to our knowledge has not yet been reported. After nuclear transport, cisPt crosslinks DNA to interrupt transcription and cell replication resulting in cell death [17, 18].
The tumor suppressor protein, p53, plays a major role in the cell stress response. When activated, p53 accumulates in the nucleus to increase transcription and activation of proteins involved in DNA repair and/or apoptosis. For example, cisPt treatment of HCT116 colorectal cancer cells results in p53 activation leading to p38MAPK activation and resulting in apoptosis [19]. We previously demonstrated that HCT116 p53+/+ cells accumulate more copper in their nuclei than HCT116 p53-/- cells, suggesting a role for p53 in the transport of 64Cu to the nucleus [20].
The purpose of the current study is to elucidate the mechanism of copper transport into the nucleus. Here we present data suggesting that Atox1 is one of the proteins involved in the transport of copper to the nucleus, and p53 influences the nuclear copper transport by affecting the regulation of Atox1 expression. Our data also demonstrate that cisPt enhances the copper transport to the nucleus of HCT116 cells by up-regulating Atox1 and increasing its nuclear localization.
Materials and methods
Reagents
64Cu (t1/2 = 12.7 hours, β+; 17.8%, Eβ + max = 656 KeV, β-, 38.4%, Eβ- max = 573 KeV) was obtained from Washington University (St. Louis, MO) and University of Wisconsin (Madison, WI). All chemicals and solvents were purchased from Sigma-Aldrich Chemical Co. (St. Louis, MO), unless otherwise specified. Cell culture media were purchased form Invitrogen (Grand Island, NY). Cisplatin was purchased from Sigma-Aldrich Chemical Co. (St. Louis, MO). Aqueous solutions were prepared using ultrapure water (resistivity, 18 M). Mouse Anti-Human p53 and mouse anti-β-Actin were purchased from cell signaling (Danvers, MA). Mouse Anti-Human p53 (PAb240) and mouse Anti-Human Atox1 were purchased from Abcam (Cambridge, MA), and mouse Anti-Human TBP was purchased from Pierce (Rockford, IL).
Isolation and identification of copper binding partner from HCT116 cells
Cells were pre-treated with cisPt (40 μM) for 24 h and incubated with [64Cu]copper acetate (300 μCi) for another 24 h. Nuclear fractions were collected as previously described [7]. Nuclear pellets were sonicated for 30 sec and spun down to obtain the supernatants. Size-exclusion high performance liquid chromatography (HPLC) was used to separate the copper-binding partner from the supernatant. The sample was run on a Superose 12 HR 10/300 column (Amersham Biosciences, Piscataway, NJ) for 60 min and detected with an Ortec Model 661 radioactive detector (EG&G instruments). The mobile phase was 0.15 M NaCl and 20 mM HEPES (pH 7.3) with a flow rate of 0.5 mL/min. One sample per minute was collected and a Beckman 8000 (Beckman) gamma counter was used to count the radioactivity in the size-exclusion HPLC fractions. Fractions with high counts were collected and placed in the Speed-Vac (Thermo Scientific, Waltham, MA) to reduce the volume to approximately 20-30 μl. Samples were reconstituted in SDS-PAGE sample buffer and loaded onto Criterion XT 4-12% precast gels (Bio-Rad, Hercules, CA). The gels were then silver-stained and identified bands were isolated and subjected to trypsinization as previously described [21]. Fragments were analyzed by matrix-assisted laser desorption ionization (MALDI) mass spectrometry (Applied Biosystems, Grand Island, NY) at the NIH/NCRR Mass Spectrometry Facility at Washington University. Identified sequences were screened on the NCBI database.
Cell culture
Human Colorectal Tumor cells (HCT116) p53+/+ and p53-/- (provided by Dr. Bert Vogelstein, Johns Hopkins University) were cultured in McCoy's 5A medium supplemented with 10% FBS, penicillin (100 units/mL), streptomycin (100 μg/mL) l-glutamine (300 μg/mL), and immortalized Mouse Embryonic Fibroblast (MEF) Atox1+/+ and Atox1-/- cells (provided by Dr. Stephen Howell, UCSD, through Dr. Jonathan Gitlin) were cultured in Dulbecco's Modified Eagle Medium, supplemented with 10% FBS, penicillin (100 units/mL), streptomycin (100 μg/mL) l-glutamine (300 μg/mL) and sodium pyruvate (100 mg/mL), glucose (4.5 g/l). All tissue culture media and supplements were obtained from Invitrogen (Grand Island, NY).
Nuclear localization and internalization of [64Cu]copper acetate in HCT 116 and MEF cell lines
HCT116 and MEF cell nuclei were collected as previously described by Wang et al. [7] Experiments were performed in 6-well plates containing approximately 1.5 × 106 cells at the beginning of the time course. [64Cu]copper acetate (50 μCi) was added, and after incubation for 2, 6 and 24 h, the cells were pelleted, re-suspended in CSK buffer (1 mM EGTA, 2 mM MgCl2 and 10 mM PIPES, pH 6.8), and incubated on ice for 2 min. Cell lysates were centrifuged at 560G for 5 min at 4°C, and the supernatant was discarded. The nuclear pellet was re-suspended in 4 mL CSK buffer without Triton X-100, and centrifuged at 560G at 4°C for 5 min. The supernatant was discarded and the nuclear pellet was counted with a gamma counter. The yield of nuclei was determined by counting the initial cell number (stained with trypan blue) and the collected nuclei (stained with crystal violet) using a hemocytometer. The percentage in the cell nuclei was determined by the cpm in the pure nuclei divided by cpm internalized in whole cells, and was corrected for the yield of nuclei or to the protein amount. For the experiments with HCT116 cell lines, cells were pretreated with cisPt (40 μM) for 24 h. Fresh media was added prior to incubation with [64Cu]copper acetate. Cells were assayed for 64Cu internalization and nuclear localization two hours after the addition of [64Cu]copper acetate.
Western blot
Cells were harvested with trypsin, pelleted by centrifugation at 1500 rpm at 4°C and solubilized with RIPA lysis buffer (Pierce, Rockford, IL) containing protease inhibitors (Complete mini, Roche, Indianapolis, IN) for 20 minutes on ice. The cells were then centrifuged at 14,500 rpm for 15 min, the pellet was discarded and the supernatant was assayed for protein concentration using BCA assay (Pierce, Rockford, IL). Electrophoresis was performed under denaturating conditions with 20 to 35 μg protein per lane (100 μg protein per lane was loaded in nuclear fraction western blots) on 4-20% TGX precast gels (Bio-Rad, Hercules, CA) using a Bio-Rad Mini electrophoresis system. The proteins were transferred to a nitrocellulose membrane (transblot mini membranes, Bio-Rad, Hercules, CA) using a transblot turbo system (Bio-Rad, Hercules, CA). Blots were saturated in 5% dry non-fat milk in TBS-T buffer (tris buffered saline + Tween 20, Bio-Rad) for 30 minutes at room temperature (RT). The blots were incubated with primary antibodies (1 μg/mL) in 5% milk/TBS-T overnight at 4°C followed by secondary antibody horseradish peroxidase-conjugated (Bio-Rad, Hercules, CA) for 1 h at room temperature. The blots were exposed to the ECL chemiluminescence reagents (Immobilon western Millipore, Billerica, MA) and detected on x-ray films or using a gel imager (ChemiDoc MP from Bio-Rad, Hercules, CA). Western blot relative quantification was realized using Image Lab software from Bio-Rad by calculating the ratio of the integrated intensity of the band of the protein of interest over the integrated intensity of the band of β-actin.
siRNA knock-down of p53 in MEF cell lines
MEF cells were seeded in 6-well plates or on glass cover slips in 24-well plates to attain 20-30% confluence after 24 h. The transfection was done with p53 siRNA (Santa Cruz Biotechnology, Dallas, TX) using RNAiMAX lipofectamine reagent (Invitrogen, Grand Island, NY) according to the manufacturer's instructions (20 pmol siRNA). The transfection mixture was added to the cells, and after 5 h fresh medium was added. Cells were assayed 48 h after transfection.
siRNA knock-down of Atox1 in HCT116 cell lines
HCT116 p53+/+ and p53-/- cells were seeded in 6-well plates to attain 30-40% confluence after 24 h. The transfection was performed with Atox1 siRNA (Origene, Rockville, MD) using RNAiMAX lipofectamine reagent (Invitrogen, Grand Island, NY) according to the manufacturer's instructions (80 pmol siRNA). The transfection mixture was added to the cells, and after 5 h fresh medium was added. Cells were assayed 48 h after transfection.
Immunofluorescence and confocal microscopy
HCT116 p53+/+ and p53-/- cells or MEF Atox1+/+ cells were seeded on glass coverslips in a 24 well plate and left for adhesion overnight. Cells were then fixed with 4% paraformaldehyde (PFA) in PBS for 15 min at room temperature and permeabilized with 0.2% triton X100 in PBS for 5 min at room temperature. After washing with PBS, non-specific binding was saturated with 0.1%BSA for 30 min. Mouse Anti-Human Atox1 (Abcam, Cambridge, MA) (1/200 dilution in PBS/0.1%BSA) or goat Anti-Mouse HAH1 (Santa Cruz biotechnology, Dallas, TX) (1/200 dilution in PBS/0.1%BSA) and rabbit Anti-Human p53 antibody (cell signaling, Danvers, MA) (1/200 dilution in PBS/0.1%BSA) were added for one hour at RT followed by a secondary antibody, Anti-Rabbit-Alexa 488 and Anti-Mouse-Alexa 594 (Invitrogen, Grand Island, NY) or Anti-Goat-Alexa 564 (1/400 dilution in PBS/0.1%BSA) for one hour at RT. After washing in PBS, coverslips were mounted using ProLong anti-fading medium (Invitrogen, Grand Island, NY). Immunofluorescence staining was monitored using a Zeiss Apotome system equipped with a Zeiss HPO PL APO 40× or 63× oil immersion lens (numerical aperture 1.4-0.6). All comparable images were taken with the same exposure time. Fluorescence quantification was made using the AutoQuant X3 software, and the regions of interest (ROIs) were drawn around the cells, and the sum of the total voxel intensity was measured for each cell. A total of thirty-five cells from three different slides per cell line and per treatment conditions were compared. Colocalized images were generated using AutoQuant X3 software; the colocalization gate was the same for all images, and Menders and Pearson's correlation indices were greater than 0.9. Integrated density calculation was done using image J software.
Statistical analysis
All data are presented as mean ± standard deviation. Groups were compared using two-tailed student t-test. Values of p < 0.05 were considered statistically significant.
Results
Identification of the protein that binds 64Cu in the nucleus of HCT116 cells
In previous studies, Eiblmaier at al. [20] showed that HCT116 p53+/+ cells accumulate more copper in the nucleus than HCT116 p53-/- cells. To identify protein candidates that may bind copper in the nucleus, HCT116 p53+/+ and p53-/- cells were incubated with [64Cu]copper acetate (300 μCi) for 24 h. Cell nuclei were isolated and nuclear proteins were separated by size exclusion HPLC. Fractions were collected and radioactivity was measured by gamma counter. The fractions with high counts were analyzed by mass spectrometry and screened with the NCBI data base to identify their protein content. The results demonstrated that one of the major proteins in these HPLC fractions is the copper chaperone Atox1.
Internalization and nuclear localization of [64Cu]copper acetate in MEF cells
To evaluate the role of Atox1 in the transport of copper to the nucleus, Atox1+/+ and Atox1-/- MEF cells were incubated with [64Cu]copper acetate (50 μCi) over a 24 h time course to measure internalization and nuclear localization. After 2 h, the copper uptake in MEF Atox1+/+ was comparable to MEF Atox1-/- (5.5 ± 0.6 vs 4.9 ± 0.5 %ID/mg); however, at later times the MEF Atox1-/- cells accumulated significantly more 64Cu than Atox1+/+ cells (n=3, p<0.05). At 6 and 24 h, 64Cu uptake in Atox1+/+ cells was 6.8 ± 1.3 and 12.8 ± 0.6 %ID/mg, respectively, and Atox1-/- cells accumulated 10.6 ± 1.2 and 15.9 ± 1.0 %ID/mg, respectively (Figure 1a). This difference was similar to that previously reported [22, 23]. Although MEF Atox1-/- cells amassed more cell-associated 64Cu, after 2 h MEF Atox1+/+ cells have 4-fold more copper in the nucleus than MEF Atox1-/- cells (3.4 ± 0.5 vs 0.7 ± 0.1 %ID/nucleus). At 6 h the difference remained 4-fold (3.0 ± 0.4 vs 0.6 ± 0.1 %ID/nucleus for Atox1+/+ and Atox1-/- cells) and decreased to 2-fold by 24 h (2.9 ± 0.1 vs 1.5 ± 0.3 %ID/nucleus for Atox1+/+ and Atox1-/- cells) (Figure 1b). These results suggest that Atox1 plays a major role in the transport of copper to the nucleus.
Figure 1.
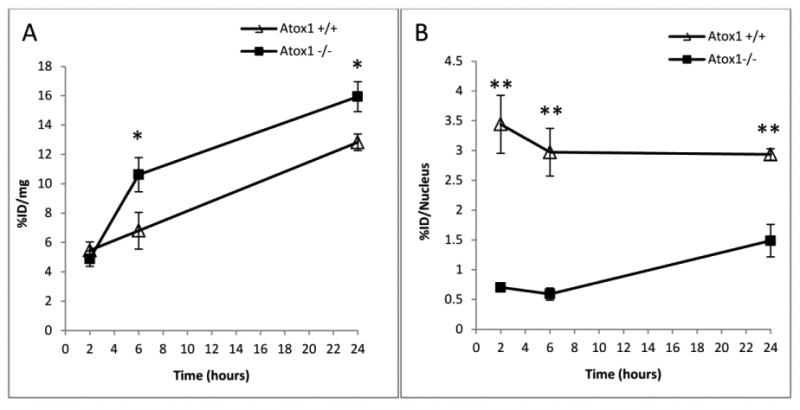
Internalization (a) and nuclear localization (b) of [64Cu]copper acetate in MEF cell lines positive (Δ) and negative ■ for Atox1 (n=3). Data are presented as %ID/mg and %ID/Nuclei. (*) p<0.05, (**) p<0.01. The data are representative of three experiments.
Atox1 expression in HCT116 cells
To investigate potential differences in Atox1 expression between HCT116 p53+/+ and p53-/-, Western blot analysis was performed. The Atox1 ratio (n=3) was normalized to β-actin, with values of 1.1 ± 0.1 for HCT116 p53+/+ vs 0.6 ± 0.1 for HCT116 p53-/-, demonstrating that HCT116 p53+/+ cells have approximately 2-fold more Atox1 than HCT116 p53-/- cells (Figure 2). These results suggest that p53 also plays a role in the regulation of Atox1 expression.
Figure 2.
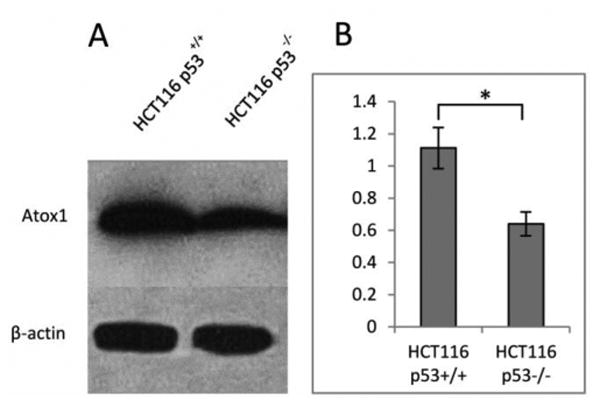
Atox1 level in HCT116 p53+/+ and p53-/- cell lines. Western blot analysis of lysates prepared from p53+/+ and p53-/- cell lines (a). Ratio of Atox1 to β-actin in p53+/+ and p53-/-cell lines (b). (*) p<0.05 (n=3). These results are representative of three independent experiments.
Effect of Atox1 knock-down (KD) on nuclear localization of [64Cu]copper acetate
To confirm the role of Atox1 in the transport of copper to the nucleus we evaluated the nuclear localization of 64Cu in HCT116 p53+/+ and p53-/- after Atox1 KD. Cell were transfected with Atox1 siRNA for 48 h, then incubated with [64Cu]copper acetate (50 μCi) for 2 h. The results show that nuclear localization of 64Cu decreases significantly after Atox1 KD in both HCT116 p53+/+ (Atox1 siRNA-treated: 0.75 ± 0.17 %ID/nucleus; non-treated: 0.26 ± 0.03 %ID/nucleus; p<0.05) and HCT116 p53-/- (Atox1 siRNA-treated: 0.63 ± 0.11 %ID/nucleus; non-treated: 0.32 ± 0.01 %ID/nucleus; p<0.05) (Figure 3a). These results are in strong correlation with the results obtained in MEF cells and confirm the importance of Atox1 in copper transport to the nuclei.
Figure 3.
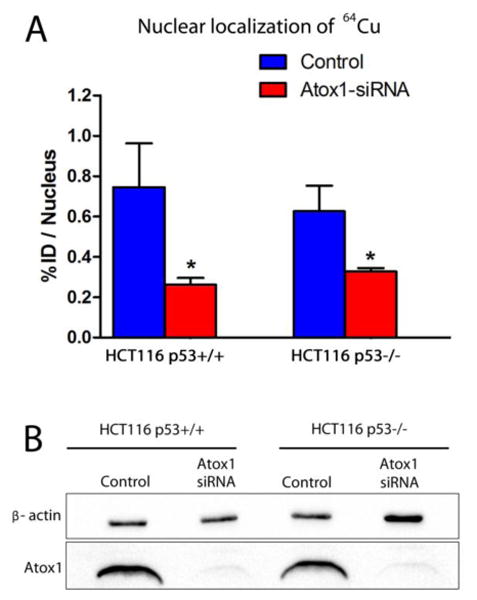
Effect of Atox1 knock-down on nuclear localization of [64Cu]copper acetate in HCT116 p53+/+ and p53-/- cells. Nuclear localization of [64Cu]copper acetate in HCT116 p53+/+ and p53-/- cell lines before and after Atox1 knock-down (n=3) (a). Western blot showing Atox1 knock-down in HCT116 p53+/+ and p53-/- cells (b). (*) p<0.05 (n=3).
Effect of p53 knock-down (KD) on Atox1 expression in MEF cells and on nuclear localization of [64Cu]copper acetate
MEF cells express high levels of p53 without any stress agents. In order to validate a relationship between p53 and Atox1, we knocked down p53 in MEF Atox1+/+ cells using siRNA. The expression of Atox1 was evaluated by immunofluorescence and the results show that the KD MEF cells overexpressed Atox1 (Figure 4a). We should note that evaluation of Atox1 level by western blot was not possible because the lack of western blot compatible anti-mouse Atox1 antibody. The nuclear localization of 64Cu in MEF Atox1+/+ and Atox1-/- cells after p53 KD was also evaluated, and higher nuclear localization of 64Cu was observed in MEF Atox1+/+ treated with p53-siRNA compared to the control (p53 siRNA-treated: 4.77 ± 0.7 %ID/nucleus; non-treated: 3.33 ± 0.3 %ID/nucleus; p=0.057) (Figure 4c), which is probably due to the overexpression of Atox1. MEF Atox1-/- cells did not show any difference in 64Cu nuclear localization after p53 KD.
Figure 4.
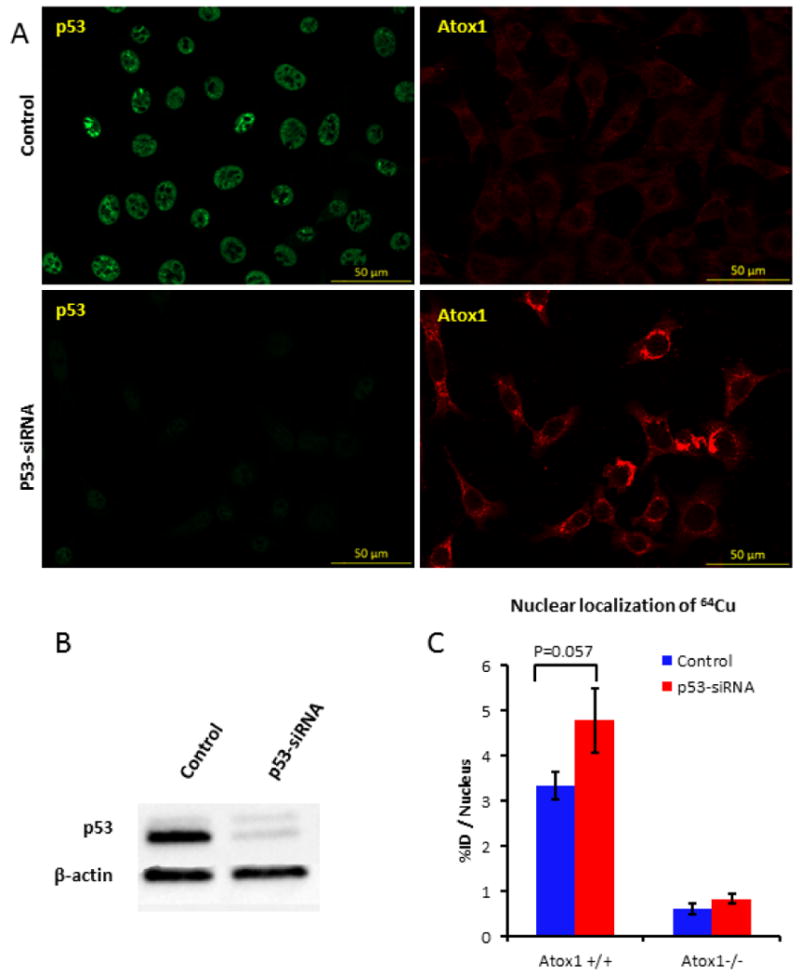
Effect of p53 knock-down on Atox1 expression in MEF Atox1+/+ cells. (a) Confocal images of MEF Atox1+/+ cells after KD of p53 using p53-siRNA. Cells were seeded on glass coverslips, treated with 20 pmol p53-siRNA for 48 h and fixed with 4% PFA. p53 (green) was detected with Mouse Anti-Human p53 antibody and Atox1 (red) was detected with Goat Anti-mouse HAH1 antibody. (b) Western blot showing p53 knock-down in MEF Atox1+/+ cells. (c) Nuclear localization of [64Cu]copper acetate in MEF Atox1+/+ and Atox1-/- cell lines before and after p53 knock-down (n=3). These results are representative of two independent experiments.
Effect of cisPt on internalization and nuclear localization of [64Cu]copper acetate in HCT116 cells
In order to investigate the influence of cisPt on copper transport in tumor cells, we studied the internalization and nuclear localization of [64Cu]copper acetate in HCT116 p53+/+ and p53-/- cells. Cells were pretreated with cisPt (40 μM) for 24 h before adding [64Cu]copper acetate (50 μCi). At 2 h post-incubation with 64Cu-acetate, HCT116 p53+/+ cells pretreated with cisPt internalized 2-fold less 64Cu than non-treated cells (cisPt-treated: 10.7 ± 0.6 %ID/mg, non-treated: 22.6 ± 0.7 %ID/mg), which is likely due to the down regulation of the copper transporter, hCtr1, by cisPt [24-26]. Interestingly, internalization of 64Cu in HCT116 p53-/- cells was not affected by cisPt treatment (cisPt-treated: 26.0 ± 1.1 %ID/mg; non-treated: 27.0 ± 0.7 %ID/mg) (Figure 5a). The pre-treatment with cisPt increased the nuclear localization of 64Cu in both p53+/+ and p53-/- HCT116 cells. In the p53+/+ cells pre-treated with cisPt, nuclear 64Cu is approximately 3.5-fold higher than in non-treated cells (26.5 ± 4.2 vs 7.4 ± 0.1 %ID/mg, respectively; p<0.001), whereas in HCT116 p53-/- cells pre-treated with cisPt, 64Cu in the nuclei is approximately 1.4-fold higher than in non-treated cells (9.7 ± 0.3 vs 7.1 ± 0.1 %ID/mg, respectively; p<0.05) (Figure 5b). A longer incubation time with [64Cu]copper acetate was not possible due to the high mortality of the cisPt pre-treated cells (>80%).
Figure 5.
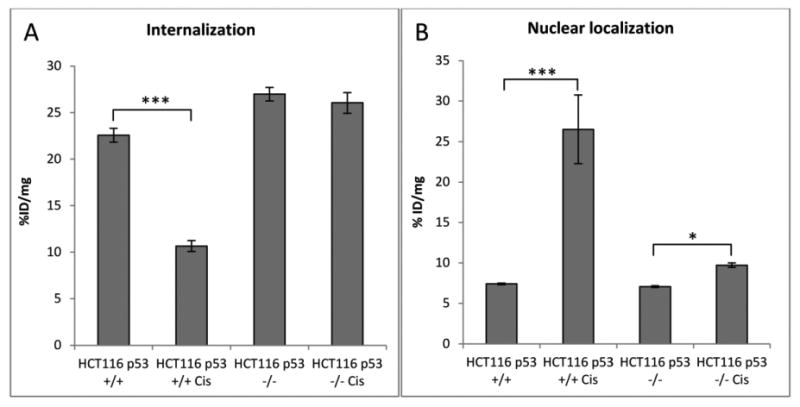
Internalization (a) and nuclear localization (b) of [64Cu]copper acetate in HCT116 cell lines positive and negative for p53 (n=3) in %ID/mg. Cells were pre-treated with cisPt for 24 h and [64Cu]copper acetate incubation time is 2 h. (*) p<0.05, (***) p<0.001. This experiment is representative of two experiments.
Effect of cisPt on the expression and localization of Atox1 in HCT116 cells
The data indicate that cisPt impacts internalization and the nuclear localization of 64Cu in HCT116 cells, with differing effects between p53+/+ and p53-/- cells. In addition, the data suggest that Atox1 plays an important role in the transport of 64Cu to the nucleus. Immunofluorescence studies were conducted to evaluate the effects of cisPt on the expression and localization of Atox1 in HCT116 p53+/+ and p53-/- cells. Confocal images demonstrate that cisPt induced Atox1 overexpression in HCT116 p53+/+ and p53-/- cells, and Atox1 localized to the nucleus (Figure 6a and d). Fluorescence quantification shows an approximately 2-fold increase of Atox1 in cells incubated with cisPt, with a small but significant difference between p53+/+ and p53-/- cell lines (Figure 6b and e). Western blot results correlate with the data obtained from immunofluorescence and confirms that cisPt increases the expression of Atox1 (Figure 6c and f). In addition, confocal images showed higher staining for Atox1 in the nuclei of HCT116 p53+/+ and p53-/- cells treated with cisPt compared to non-treated cells. Colocalization of Atox1 and DAPI staining of the nuclei clearly shows that Atox1 translocate to the nucleus in HCT116 p53+/+ and p53-/- cells treated with cisPt, with a higher colocalization in HCT116 p53+/+ cells (Figure 7a, b). These data correlate with results from Western blot analysis that also show an increase of nuclear Atox1 after treatment with cisPt, with a higher increase of Atox1 levels in HCT116 p53+/+ compared to p53-/- cells (Figure 7c).
Figure 6.
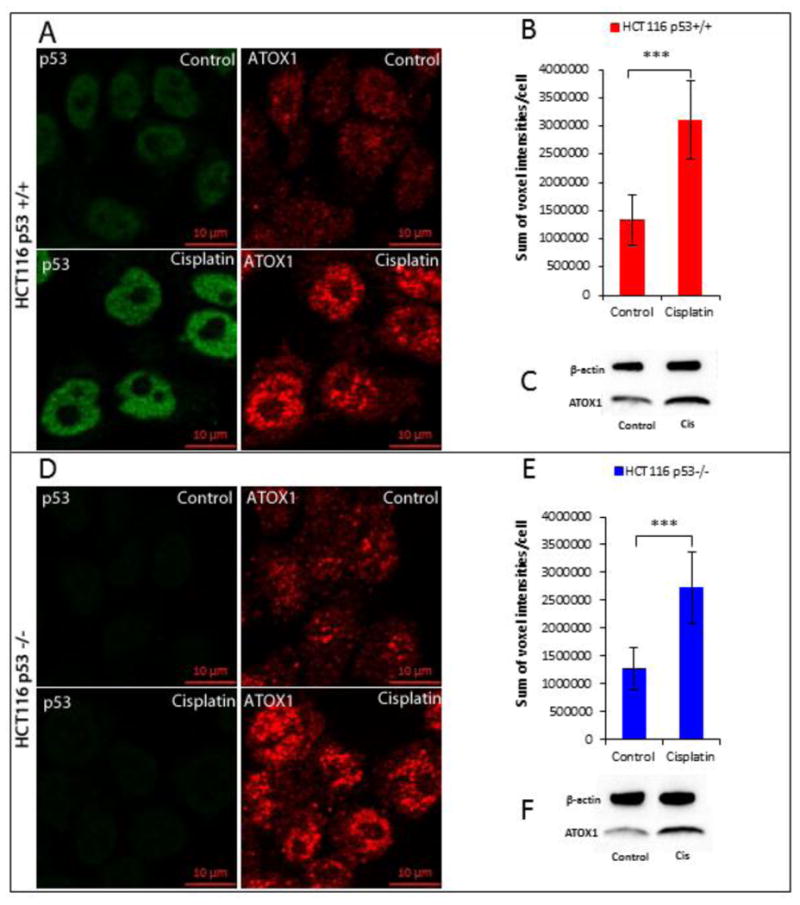
Effect of cisPt on Atox1 expression in HCT116 p53+/+ and p53-/- cell lines. Confocal images of HCT116 p53+/+ (a) and p53-/- (d) cells treated and non treated with cisPt; cells were seeded on glass coverslips, treated with 40 μM cisPt for 24h h then fixed with 4% PFA; p53 (green) was detected with Mouse Anti-Human p53 antibody and Atox1 (red) was detected with Mouse Anti-Human Atox1 antibody. Quantification of the fluorescence of Atox1 in HCT116 p53+/+ (b) and p53-/- (e) cells treated and non treated with cisPt. Western blot of Atox1 in HCT116 p53+/+ (c) and p53-/- (f) cells treated and non treated with cisPt. (*) p<0.05, (***) p<0.001.
Figure 7.
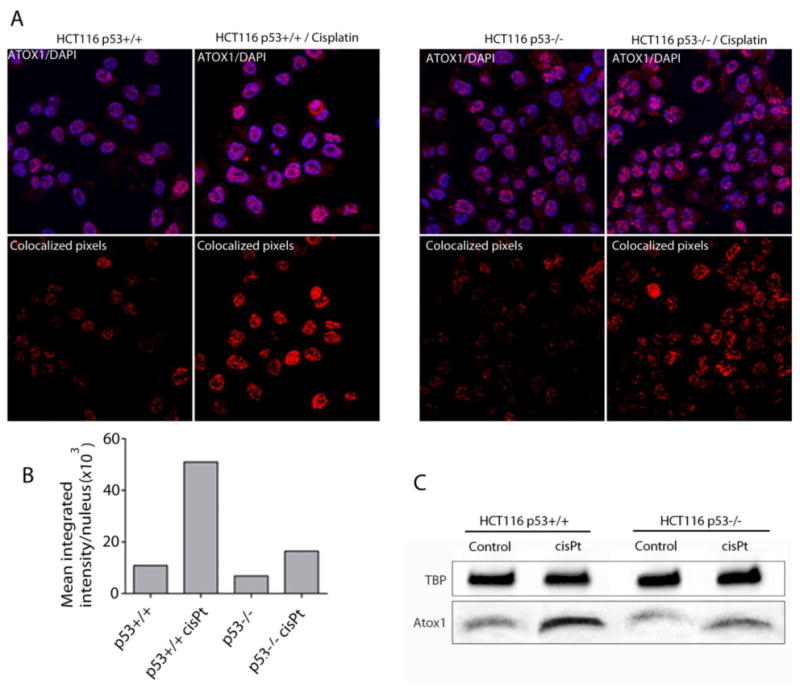
Nuclear localization of Atox1 in HCT116 p53+/+ and p53-/- cells, treated and non-treated with cisPt. Confocal images of Atox1 (red) and nuclear staining (blue) in HCT116 p53+/+ (a, upper left panel) and p53-/- (a, upper right panel) cells; Images show the colocalized pixels from the total Z-stack between Atox1 and DAPI staining in HCT116 p53+/+ (a, lower left panel) and p53-/- (a, lower right panel) cells treated and non treated with cisPt. (b) Pixel density analysis of the Atox1 and DAPI colocalized images. (c) Western blot showing Atox1 levels in HCT116 p53+/+ and p53-/- nuclei before and after 24 h treatment with cisPt.
Discussion
Copper homeostasis has been well studied, and significant progress has been made to identify the chaperones and proteins that bind and transport copper to different compartments of the cell; however, to date there is no clear evidence for chaperones that transfer copper to the cell nuclei. This study provides the first evidence that Atox1 is one of the proteins that plays a role in the transport of copper to the cell nuclei. After treatment of HCT116 cells with [64Cu]copper acetate, mass spectrometry analysis of the nuclear fractions containing the 64Cu showed that Atox1 is one of the proteins in these fractions. Our results show that MEF Atox1+/+ cells accumulate more copper in their nuclei compared to the cells that lack Atox1, suggesting a role of Atox1 in the transport and accumulation of copper in the cell nuclei. Knock-down of Atox1 in HCT116 p53+/+ and p53-/- cells decreased the nuclear localization in both cell lines, which confirm our observation in MEF cells that Atox1 plays a role in the transport of copper to the nucleus. Itoh et al. [27] identified Atox1 as a copper-dependent transcription factor involved in cell proliferation and showed that Atox1 undergoes nuclear translocation when activated by copper. These data by Itoh et al. are consistent with our results that indicate Atox1 transports copper to the cell nuclei. In addition, the data presented here show that although internalization of 64Cu was greater in Atox1 -/- cells, nuclear localization was significantly higher in the Atox1 +/+ cells (Figure 1), suggesting a role of Atox1 for binding copper in the cytoplasm via the CXXC motif and transporting it to the nucleus. It should be noted that our data contradict a report by McRae et al. [28], where quantitative synchrotron X-ray fluorescence data suggested that MEF Atox1-/- cells accumulate more copper in the nuclei compared to MEF Atox1+/+ cells. The discrepancy may be explained by the fact that the copper distribution in the cells and the analysis done by McRae et al. was performed on 2D fluorescent images and limited to only three cells, where the experiments described here were performed with large numbers of cells (>106 for each condition and/or treatment). Additionally, the relative amount of copper in cells was determined directly using a highly accurate tracer technique (64Cu), and all experiments were performed in triplicate.
Greater copper accumulation (internalization) over time was found in Atox1-/- compared to Atox1+/+ MEFs; however, the nuclear accumulation of copper was much higher in Atox1+/+ cells. Miyayama et al. [29] demonstrated that knocking-down Atox1 in metallothionein knock-out MEF cells increased the concentration of the copper influx pump, Ctr1, and copper chaperone Ccs, which is consistent with the higher accumulation over time of copper in Atox1-/- compared to Atox1+/+ cells. In addition, studies have shown that Atox1 is important for the efflux of copper out of cells [30].
We previously showed that p53 plays a role in copper transport to the nuclei of HCT116 cells [20]. As a crucial transcription factor, p53 affects many cellular processes and regulates the expression of several proteins [31]. Our results suggest that p53 may affect nuclear transport of copper by increasing the Atox1 expression level. Interestingly, the level of Atox1 in HCT116 p53 knock-out cells is less than in HCT116 p53+/+ cells (Figure 3). In contrast, p53 KD in MEF cells resulted in overexpression of Atox1. These conflicting results indicate that the effect of p53 on Atox1 expression is cell-line dependent and complex, with potential mediators also playing a role between p53 and Atox1 expression. Despite the differences in the effect of p53 on Atox1 expression observed between HCT116 and MEF cell lines, the correlation between cellular Atox1 levels and nuclear localization of 64Cu was consistent between both cell lines. Increased nuclear localization of copper was observed with higher Atox1 protein expression, suggesting that Atox1 plays a significant role in the transport copper to the cell nuclei.
There has been ongoing interest in targeted radionuclide therapy over the past few decades, as it may improve tumor kill with less toxicity compared to chemotherapeutic agents. In addition to decaying by positron emission for PET imaging, 64Cu decays by beta minus and Auger electron emission leading to tumor cell DNA damage that may enhance cancer therapy. Understanding the mechanism of transport of copper to tumor cell nuclei and how platinum chemotherapy affects this transport is of great importance for combining chemotherapy with targeted radiotherapy with 64Cu-based agents for cancer treatment.
It is known that cisPt binds to DNA and induces cell death. There is evidence that copper transporter Ctr1 contributes to the transport of cisPt into the cells [14, 32-34]. Here we studied the effect of cisPt on copper internalization and nuclear localization in HCT116 p53+/+ and p53-/- cells. Cells were pre-treated with cisPt for 24 h prior to incubation with 64Cu, and cisPt reduced the copper uptake in HCT116 p53+/+ cells, possibly due to the down-regulation of Ctr1 by cisPt. Copper internalization in HCT116 p53-/- cells was not affected by cisPt treatment possibly because the lack of p53 may disrupt the mechanism by which cisPt down-regulates Ctr1. However, cisPt significantly increased nuclear localization of 64Cu in p53+/+ cells, whereas in p53-/- there was only a modest but significant increase. Greater nuclear localization of copper after treatment with cisPt is likely due to the up-regulation of Atox1 by cisPt. Our data show that cisPt increases expression of Atox1 in both p53+/+ and p53-/- cells, but to a higher extent in p53+/+ cells (Figure 6). In addition, after treatment with cisPt, Atox1 colocalizes more in the nuclei of HCT116 p53+/+ cells compared to p53-/- cells as shown by immunofluorescence and Western blot (Figure 7). This finding is consistent with the nuclear localization results and also supports our hypothesis that Atox1 transports copper to nuclei. In addition, we observed a higher amount of nuclear copper in HCT116 p53+/+ compared to p53-/- cells. Studies have shown that apoptosis increases the nuclear envelope permeability [35, 36]. HCT116 p53+/+ are more sensitive to cisPt than p53-/- cells and undergo much more apoptosis and cell death, which may increase their nuclear permeability and explains the higher amount of copper and Atox1 in the nuclei of p53+/+ compared to p53-/- cells.
In conclusion, here we demonstrated that Atox1 plays a significant role in the transport of copper into the nucleus. CisPt up-regulates Atox1 and increases its localization to the nuclei. The nuclear localization of Atox1 correlates with nuclear localization of 64Cu. The data presented here provide insights into combining platinum drug therapy and 64Cu targeted radiotherapy for cancer. Increased nuclear localization of 64Cu after cisPt treatment may increase DNA damage to the cancer cells and enhance cell killing by 64Cu. Combining chemotherapy and targeted radiotherapy could also help reduce the dose of chemotherapy drugs, and thereby decrease their side effects, improving quality of life for cancer patients. The combination of 64Cu and cisPt may also improve the efficiency of treating resistant tumors that have a wild-type p53.
Supplementary Material
Acknowledgments
This research was supported by NIH/NCI 5R01CA064475 (CJA) and DOE DE-FG02-08ER64671 (Integrated Research Training Program of Excellence in Radiochemistry awarded to Suzanne Lapi supporting YG). The mass spectroscopy portion of the project was supported by grants from the National Center for Research Resources (5P41RR000954-35) and the National Institute of General Medical Sciences (8 P41 GM103422-35) from the National Institutes of Health.
Abbreviations
- Ab
antibody
- BSA
bovine serum albumin
- cisPt
Cisplatin
- DTPA
diethylenetriaminepentaacetic acid
- FBS
fetal bovine serum
- HCT116
human colorectal tumor cells 116
- hCtr1
human copper transporter 1
- HPLC
high performance liquid chromatography
- ICP-MS
inductively coupled plasma mass spectrometry
- Ir
Iridium
- KD
knock-down
- MEF
mouse embryonic fibroblasts
- OC
octreotide
- PET
positron emission tomography
- PBS
phosphate buffered solution
- PFA
paraformaldehyde
- QC
quality control
- RT
room temperature
- SOD
superoxide dismutase
- TBT-T
tris buffered salin-tween 20
- TBP
TATA binding protein
- TETA
1,4,8,11-tetraazacyclotetradecane-1,4,8,11-tetraacetic acid
References
- 1.Puig S, Thiele DJ. Current opinion in chemical biology. 2002;6:171–180. doi: 10.1016/s1367-5931(02)00298-3. [DOI] [PubMed] [Google Scholar]
- 2.Rae TD, Schmidt PJ, Pufahl RA, Culotta VC, O'Halloran TV. Science. 1999;284:805–808. doi: 10.1126/science.284.5415.805. [DOI] [PubMed] [Google Scholar]
- 3.Anderson CJ, Ferdani R. Cancer biotherapy & radiopharmaceuticals. 2009;24:379–393. doi: 10.1089/cbr.2009.0674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lewis MR, Wang M, Axworthy DB, Theodore LJ, Mallet RW, Fritzberg AR, Welch MJ, Anderson CJ. Journal of nuclear medicine : official publication. Society of Nuclear Medicine. 2003;44:1284–1292. [PubMed] [Google Scholar]
- 5.Anderson CJ, Pajeau TS, Edwards WB, Sherman EL, Rogers BE, Welch MJ. Journal of nuclear medicine : official publication. Society of Nuclear Medicine. 1995;36:2315–2325. [PubMed] [Google Scholar]
- 6.Connett JM, Anderson CJ, Guo LW, Schwarz SW, Zinn KR, Rogers BE, Siegel BA, Philpott GW, Welch MJ. Proceedings of the National Academy of Sciences of the United States of America. 1996;93:6814–6818. doi: 10.1073/pnas.93.13.6814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang M, Caruano AL, Lewis MR, Meyer LA, VanderWaal RP, Anderson CJ. Cancer research. 2003;63:6864–6869. [PubMed] [Google Scholar]
- 8.George AM, Sabovljev SA, Hart LE, Cramp WA, Harris G, Hornsey S. The British journal of cancer. 1987;(8):141–144. [PMC free article] [PubMed] [Google Scholar]
- 9.Chiu SM, Xue LY, Friedman LR, Oleinick NL. Radiation research. 1992;129:184–191. [PubMed] [Google Scholar]
- 10.Chiu SM, Xue LY, Friedman LR, Oleinick NL. Biochemistry. 1993;32:6214–6219. doi: 10.1021/bi00075a014. [DOI] [PubMed] [Google Scholar]
- 11.Abada P, Howell SB. Metal-based drugs. 2010;2010:317581. doi: 10.1155/2010/317581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Robinson NJ, Winge DR. Annual review of biochemistry. 2010;79:537–562. doi: 10.1146/annurev-biochem-030409-143539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Prohaska JR, Gybina AA. The Journal of nutrition. 2004;134:1003–1006. doi: 10.1093/jn/134.5.1003. [DOI] [PubMed] [Google Scholar]
- 14.Howell SB, Safaei R, Larson CA, Sailor MJ. Molecular pharmacology. 2010;77:887–894. doi: 10.1124/mol.109.063172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Palm ME, Weise CF, Lundin C, Wingsle G, Nygren Y, Bjorn E, Naredi P, Wolf-Watz M, Wittung-Stafshede P. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:6951–6956. doi: 10.1073/pnas.1012899108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Boal AK, Rosenzweig AC. Journal of the American Chemical Society. 2009;131:14196–14197. doi: 10.1021/ja906363t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rabik CA, Dolan ME. Cancer treatment reviews. 2007;33:9–23. doi: 10.1016/j.ctrv.2006.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fuertes MA, Alonso C, Perez JM. Chemical reviews. 2003;103:645–662. doi: 10.1021/cr020010d. [DOI] [PubMed] [Google Scholar]
- 19.Bragado P, Armesilla A, Silva A, Porras A. Apoptosis. 2007;12:1733–1742. doi: 10.1007/s10495-007-0082-8. [DOI] [PubMed] [Google Scholar]
- 20.Eiblmaier M, Meyer LA, Anderson CJ. Cancer biology & therapy. 2008;7:63–69. doi: 10.4161/cbt.7.1.5130. [DOI] [PubMed] [Google Scholar]
- 21.Shevchenko A, Wilm M, Vorm O, Mann M. Anal Chem. 1996;68:850–858. doi: 10.1021/ac950914h. [DOI] [PubMed] [Google Scholar]
- 22.Safaei R, Maktabi MH, Blair BG, Larson CA, Howell SB. Journal of inorganic biochemistry. 2009;103:333–341. doi: 10.1016/j.jinorgbio.2008.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hamza I, Faisst A, Prohaska J, Chen J, Gruss P, Gitlin JD. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:6848–6852. doi: 10.1073/pnas.111058498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Holzer AK, Katano K, Klomp LW, Howell SB. Clinical cancer research : an official journal of the American Association for Cancer Research. 2004;10:6744–6749. doi: 10.1158/1078-0432.CCR-04-0748. [DOI] [PubMed] [Google Scholar]
- 25.Petris MJ, Smith K, Lee J, Thiele DJ. The Journal of biological chemistry. 2003;278:9639–9646. doi: 10.1074/jbc.M209455200. [DOI] [PubMed] [Google Scholar]
- 26.Jandial DD, Farshchi-Heydari S, Larson CA, Elliott GI, Wrasidlo WJ, Howell SB. Clinical cancer research : an official journal of the American Association for Cancer Research. 2009;15:553–560. doi: 10.1158/1078-0432.CCR-08-2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Itoh S, Kim HW, Nakagawa O, Ozumi K, Lessner SM, Aoki H, Akram K, McKinney RD, Ushio-Fukai M, Fukai T. The Journal of biological chemistry. 2008;283:9157–9167. doi: 10.1074/jbc.M709463200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McRae R, Lai B, Fahrni CJ. Journal of biological inorganic chemistry : JBIC : a publication of the Society of Biological Inorganic Chemistry. 2010;15:99–105. doi: 10.1007/s00775-009-0598-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Miyayama T, Suzuki KT, Ogra Y. Toxicology and applied pharmacology. 2009;237:205–213. doi: 10.1016/j.taap.2009.03.024. [DOI] [PubMed] [Google Scholar]
- 30.Hamza I, Prohaska J, Gitlin JD. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:1215–1220. doi: 10.1073/pnas.0336230100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Beckerman R, Prives C. Cold Spring Harb Perspect Biol. 2010;2:a000935. doi: 10.1101/cshperspect.a000935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Holzer AK, Manorek GH, Howell SB. Molecular pharmacology. 2006;70:1390–1394. doi: 10.1124/mol.106.022624. [DOI] [PubMed] [Google Scholar]
- 33.Holzer AK, Samimi G, Katano K, Naerdemann W, Lin X, Safaei R, Howell SB. Molecular pharmacology. 2004;66:817–823. doi: 10.1124/mol.104.001198. [DOI] [PubMed] [Google Scholar]
- 34.Larson CA, Blair BG, Safaei R, Howell SB. Molecular pharmacology. 2009;75:324–330. doi: 10.1124/mol.108.052381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mason DA, Shulga N, Undavai S, Ferrando-May E, Rexach MF, Goldfarb DS. FEMS yeast research. 2005;5:1237–1251. doi: 10.1016/j.femsyr.2005.07.008. [DOI] [PubMed] [Google Scholar]
- 36.Ferrando-May E, Cordes V, Biller-Ckovric I, Mirkovic J, Gorlich D, Nicotera P. Cell death and differentiation. 2001;8:495–505. doi: 10.1038/sj.cdd.4400837. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.