

Abstract
After injury or during neurodegenerative disease in the central nervous system (CNS), the concentration of tumor necrosis factor alpha (TNFα) rises above normal during the inflammatory response. In vitro and in vivo, addition of exogenous TNFα to neurons has been shown to induce rapid plasma membrane-delivery of AMPA-type glutamate receptors (AMPARs) potentiating glutamatergic excitotoxicity. Thus the discovery of drug targets reducing excess TNFα-induced AMPAR surface expression may help protect neurons after injury. In this study, we investigate the neuroprotective role of the CB1 cannabinoid receptor using quantitative immunofluorescent and real-time video microscopy to measure the steady state plasma membrane AMPAR distribution and rate of AMPAR exocytosis after TNFα exposure in the presence or absence of CB1 agonists. The neuroprotective potential of CB1 activation with TNFα was measured in hippocampal neuron cultures challenged by an in vitro kainate (KA)-mediated model of Excitotoxic Neuroinflamatory Death (END). Here, we demonstrate that CB1 activation blocks the TNFα-induced increase in surface AMPARs and protects neurons from END. Thus, neuroprotective strategies which increase CB1 activity may help to reduce the END that occurs as a result of a majority of CNS insults.
Keywords: AMPA receptor, cannabinoid receptor, excitotoxicity, glutamate receptor, neuroprotection
1. Introduction
Control over normal neuronal glutamatergic signaling depends largely on the localization of AMPARs on synaptic and extrasynaptic plasma membrane (Malinow and Malenka, 2002). This balance can be disrupted during CNS injury or disease when the inflammatory response is initiated and the population of AMPARs on neuronal plasma membrane increases dramatically, potentiating END (reviewed in (Leonoudakis et al., 2004; Pickering et al., 2005). We previously characterized TNFα as a powerful glial-derived instigator of AMPAR trafficking to the plasma membrane (Beattie et al., 2002; Stellwagen et al., 2005). The presence of TNFα in the CNS is required for synaptic development (Stellwagen and Malenka, 2006), but the increased TNFα concentrations induced following infection or injury (Nemeth et al., 1997) contribute to END (Lock et al., 1999; Nagatsu et al., 2000; New et al., 1998; Perry et al., 2001; Shohami et al., 1999; Szelenyi, 2001). Several groups attribute this toxicity to long-term, translation-dependent apoptotic signaling pathways (Fontaine et al., 2002; Reimann-Philipp et al., 2001; Yang et al., 2002; Zhao et al., 2001; Zou and Crews, 2005). However, TNFα also induces a rapid (within 15 minutes) increase in surface expression of AMPARs (Beattie et al., 2002; Ogoshi et al., 2005; Stellwagen et al., 2005) which are calcium permeable and principally localized extrasynaptically (Leonoudakis et al., 2008). Additionally, acute neurotrauma increases surface expression of calcium permeable AMPARs composed of GluR1 subunits (Grooms et al., 2000; Grossman et al., 1999; Sanchez et al., 2001; Ying et al., 1998), and potentiates neuronal excitotoxicity (Feldmeyer et al., 1999; Oguro et al., 1999). Together, these studies suggest that TNFα-induced AMPAR surface expression may be a mechanism contributing to the early stages of END. Agents which decrease TNFα-induced AMPAR surface expression may reduce excitotoxicity in neurons and our previous studies suggest the use of cannabinoid receptor agonists may serve as a potential pharmacological intervention (Abood et al., 2001).
The cannabinoid receptor system is found in hippocampal neurons (Leterrier et al., 2006; Mackie, 2007; McDonald et al., 2007) and protects against excitotoxicity (Kim et al., 2006; Marsicano et al., 2003; Shen and Thayer, 1998, 1999). Activation of the G-protein coupled CB1 cannabinoid receptor initiates complex signaling cascades and leads to the hyperpolarization of neuronal membranes by increasing potassium and decreasing calcium conductance (Howlett, 2002). Our current results suggest a novel neuroprotective mechanism under the control of CB1, namely the reduction of excessive AMPAR surface expression caused by TNFα. We have used hippocampal culture systems to examine AMPAR surface expression and neuron survival after KA-induced excitotoxicity while exogenously applying TNFα. Our results demonstrate that CB1 activation prevents the TNFα-induced increase in surface GluR1-containing AMPARs and protects neurons from TNFα potentiated excitotoxic stress.
2. Materials and Methods
2.1. Preparation of hippocampal cultures
Mixed hippocampal neuron cultures were prepared from E18 rat pups as previously described (Beattie et al., 2002). Briefly, hippocampi of embryonic day 18 (E18) Sprague Dawley rats were removed, digested with papain, and dissociated by trituration. Neurons were plated in Neurobasal medium containing B27 supplement and Glutamax (Invitrogen) on coverslips or plastic plates coated with poly-D-lysine. After cell attachment (3–4 h), the medium was replaced. Three days later, an equal volume of Neurobasal medium containing N2 supplement and Glutamax was added. Cultures were fed weekly by replacing one-half of the medium with Neurobasal/N2/Glutamax. Astrocyte growth was inhibited by adding 5-fluoro-2′-deoxyuridine after 6 days in vitro (div). Cultures were used for experiments between weeks 3 and 4.
2.2. Immunofluorescence AMPAR surface localization
Neurons were fixed and processed as described (Leonoudakis et al., 2008). Before neuron treatment, the medium was changed to prewarmed artificial CSF (ACSF) (25 mM HEPES, pH 7.4, 125 mM NaCl, 2.5 mM KCl, 1 mM MgCl2, 2 mM CaCl2, 33 mM D-glucose with the following receptor antagonists: 0.5 μM tetrodotoxin, 3 μM strychnine, 20 μM bicuculline methiodide, and 20 μM D-APV to eliminate the contributions of spontaneous action potentials, inhibitory signaling, and NMDA receptor activation, respectively) for 30 minutes. The cannabinoid receptor agonists WIN55212-2 (0.5 μM, Sigma, St. Louis, MO), THC (0.5 μM, RTI), 2AG (0.5 μM, Tocris), and anandamide (0.5 μM, Tocris) were added 30 min before TNFα treatment. Recombinant rat TNFα (6 nM, R&D Systems) was then added to cells for an additional 15 minutes. Cannabinoid receptor antagonists (SR141716A, SR144528 and VCHSR1-used at 0.5 μM) were added 5 min before agonist application. Antagonists were provided by Drs. Herb Seltzman (RTI, Research Triangle Park, NC) and Patricia Reggio (UNC Greensboro). After the indicated times, neurons were fixed with 4% paraformaldehyde/4% sucrose in PBS for 10 min, washed with PBS, blocked with 3% BSA, 2% goat serum in PBS (blocking buffer) for 1 h, and incubated with rabbit anti-GluR1 (Calbiochem) or rabbit anti-CB1 (Sigma-Aldrich) antibodies (both against extracellular epitopes) and with goat anti-rabbit Alexafluor 555 and/or goat anti-mouse Alexafluor 488 (Invitrogen). For surface CB1 colocalization with synaptic markers, cells were permeabilized with 0.2% Triton X-100 and incubated with mouse monoclonal anti-PSD-95 (Affinity BioReagents, Inc.) or synaptophysin (Sigma-Aldrich) antibodies. Colocalization of PSD-95 or synaptophysin with CB1 was examined by imaging neurons with a single 0.5 μm thick plane at 1024×1024 pixel resolution with a Nikon C1 confocal microscope. Steady-state surface expression of AMPARs was determined as described by reports from our lab and others (Beattie et al., 2002; Gao et al., 2006; Leonoudakis et al., 2008; Ogoshi et al., 2005; Stellwagen et al., 2005). For individual experiments, images for all conditions were analyzed using identical acquisition parameters and untreated and treated cells from the same culture preparation were compared. Regions of equal size containing dendrites 30 μm away from the cell body were used for quantification. AMPAR surface expression level is represented as a ratio of surface AMPAR area divided by the total dendritic area in the visual field using MetaMorph software (Molecular Devices). Each experiment was repeated at least three times, and individual normalized cell values were averaged for each experimental condition.
2.3. Live imaging of superecliptic pHluorin-tagged GluR1 in hippocampal neurons
AMPAR exocytosis events were visualized by transfection of hippocampal neurons by AMAXA nucleofection with cDNA encoding superecliptic pHluorin (SEP)-tagged GluR1 imaged with live video microscopy as described except for the composition of the imaging media (Yudowski et al., 2007). Live neurons were imaged in 125 mM NaCl, 2.5 mM KCl, 2 mM CaCl2, 10 mM D-glucose, 25 mM HEPES, pH 7.4., 0.5 μM tetrodotoxin, 20 μM bicuculine methiodide, 20 μM CNQX, and 50 μM D-APV in the presence or absence of WIN (0.5 μM), TNFα (100ng/ml), or WIN+TNFα. Temperature of the imaging chamber was maintained at 37°C. The rate of AMPAR exocytosis events was measured in real time (100 msec time resolution). Sixty second videos of AMPAR exocytosis were collected from dendritic fields of over 100 separate hippocampal neurons exposed to four different conditions. For exocytotic rate analysis after WIN or WIN+TNFα treatment, neurons were pretreated with WIN for at least 15 minutes prior to examination. Then during WIN+TNFα treatment neurons were examined within a 15–45 minute window after this TNFα application which we have determined is the maximum AMPAR surface delivery time zone after TNFα application (Leonoudakis et al., 2008). Standard criteria were used to ensure that only healthy pyramidal hippocampal neurons transfected with comparable amounts of SEP-GluR1 were used for analysis (Leonoudakis et al., 2008; Yudowski et al., 2007). Newly inserted SEP-AMPARs were readily distinguished as pronounced “spikes” of increased fluorescence intensity in videos of 1 minute duration. Blinded quantitative analysis of these videos was performed using MetaMorph image analysis software (see supplementary materials).
2.4. Excitotoxicity assay
Propidium iodide (PI) is a cell-impermeant fluorescent biomarker that stains DNA. Healthy viable cells exclude PI from their cytoplasm, whereas dead and dying cells take it up (Aras et al., 2008). Lactose dehydrogenase (LDH) is a stable cytosolic enzyme released during cell lysis from dead or dying cells and was used as a quantitative measure of neuron toxicity and death (Glass et al., 2004; Patel et al., 1993). TNFα (6 nM, R&D Systems) was added 15 min before kainate (KA, 20 μM, Sigma), WIN (0.5 μM) was added 30 min before TNFα and incubated for 24 hours. Nuclei of dead cells and total cells were stained with (PI) (5 μg/ml, Sigma) and Hoechst (1 μg/ml, Molecular Probes). Cultures were then washed 3 times by PBS and fluorescence images were collected. Images were thresholded to only show the nucleus by Metamorph software. Nuclei were counted automatically by the software and puncta ≤ 3 pixels were excluded. Percentage of cell death was calculated as (PI nuclei number)/(Hoechst nuclei number) × 100%. Immediately prior to staining, media in the cultures was collected for LDH quantification following the CytoTox 96 (Promega) protocol. All quantitative measures are expressed as means ± SEM. Statistical significance was determined by either one way ANOVA followed by Bonferroni’s multiple comparison post test or unpaired t test. A value of p < 0.05 was considered significant.
3. Results
3.1. Expression of GluR1 and CB1 receptors on hippocampal neurons
In hippocampal neurons, GluR1-containing AMPARs co-localize with postsynaptic markers on somatic and dendritic plasma membranes (Beattie et al., 2002; Stellwagen et al., 2005) and CB1 has been detected in plasma membrane and cytosolic compartments of somato-dendritic regions as well as on axonal plasma membranes (Leterrier et al., 2006; McDonald et al., 2007). To confirm surface expression of CB1 in or near postsynaptic membrane in our hippocampal neuron cultures we have used immunofluorescence confocal microscopy methods which selectively detect surface receptors (see methods). We observed strong labeling of CB1 on axonal processes (Figure 1A and B, arrows) and colocalization of CB1 with PSD-95 staining on dendritic processes (Figure 1A, arrowheads). We also found CB1 is in close apposition to presynaptic synaptophysin staining (Figure 1B, arrowheads).
Figure 1. Surface CB1 is closely apposed to and colocalized with synaptic markers on dendrites of cultured hippocampal neurons.
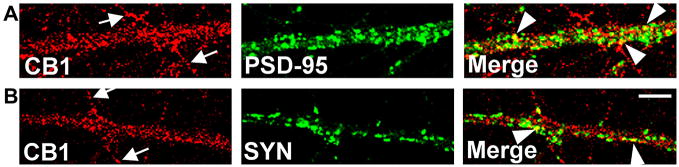
(A) The top three panels show confocal micrographs of the same field of view with either CB1, PSD-95, or CB1+PSD-95 (Merge) staining respectively. Co-staining of surface CB1 and internally localized PSD-95 demonstrates that cannabinoid receptors are expressed on dendrites of hippocampal neurons (arrow heads). Surface localization of CB1 on axonal processes which are devoid of PSD-95 is also shown (arrows) (B) The bottom three panels show another field of view with either CB1, the presynaptic marker synaptophysin (SYN), or CB1+SYN (Merge) staining respectively. Some CB1 receptors are colocalized with SYN (arrow heads), while others on the surface of axons are not overlapped with SYN (arrows). Scale bar, 5 μm.
3.2. Cannabinoid receptor agonists inhibit surface AMPAR increase induced by TNFα
We next addressed the hypothesis that activation of CB1 reduces AMPAR surface expression. We first tested whether the CB1/CB2 agonist WIN55212-2 (WIN) had short term effects (minutes) on the constitutive level of AMPAR surface expression. WIN alone for 45 minutes did not significantly change the surface expression of GluR1 compared to control (Fig 2A and B) suggesting that short term receptor activation does not change constitutive AMPAR surface expression. In agreement with previous studies, 15 minutes of TNFα treatment alone increased steady state surface expression of GluR1-containing AMPARs compared to control on hippocampal neurons (Leonoudakis et al., 2008), but pretreatment with WIN for 30 minutes prior to 15 minutes of WIN + TNFα treatment blocked this increase in surface expression (Fig. 2. A, B). These results demonstrating a WIN-induced block of TNFα-induced AMPAR surface delivery led us to examine another exogenous cannabinoid receptor agonist, THC, and two endogenous cannabinoid agonists (endocannabinoids) (2AG) and anandamide (ANA). These agonists also blocked the TNFα-induced surface expression of GluR1-containing AMPARs [THC (106±6.3%; * p<0.05; n ≥ 80), 2AG (108±3.5%; * p<0.05; n ≥ 100) and anandamide (109±3%; * p<0.05; n ≥ 100)].
Figure 2. Cannabinoid receptor agonists block the steady state increase in AMPAR surface expression and the increased rate of AMPAR exocytosis induced by TNFα.

(A) Immunofluorescence microscopy demonstrating that TNFα treatment (15 min.) increases GluR1 surface staining on dendrites of hippocampal neurons compared to control, untreated neurons. Pretreatment of cultures with WIN blocked the GluR1 surface increase induced by TNFα. WIN alone (45 min.) did not change surface expression of GluR1. Scale bar, 10 μm. (B) Immunofluorescent image quantification shows a significant increase in TNFα-induced surface GluR1 (128±3.4%; *** p<0.001) compared to control (100±2.6%). Pretreatment of cells with WIN for 30 minutes blocked the TNFα-induced GluR1 surface increase (100±3.2%; *** p<0.001). WIN alone (45 min.) did not have a significant effect on GluR1 surface expression (109±2.8%; p>0.05). n ≥ 100 neurons each condition from 5 independent experiments. (C) TNFα increases the rate of AMPAR exocytotic events on dendritic plasma membrane. Videos (1 minute duration and shown on-line in supplemental materials) of SEP-AMPAR exocytosis from over 100 hippocampal neurons were collected after either, 1) no treatment, 2) TNFα treatment, 3) WIN treatment, or 4) WIN+TNF treatment. TNFα treatment significantly increases the rate of AMPAR exocytosis compared to control (TNF: 237.4±30.4%, p<0.01; Control, n=23; TNF, n=41), while TNFα treatment in the presence of WIN did not change AMPAR exocytosis rates (WIN+TNF: 92.6±10.7%, p>0.05; WIN, n=16; WIN+TNF, n=40) compared to WIN treatment alone.
3.3. WIN inhibits the exocytosis increase of AMPAR induced by TNFα
The TNFα-induced increase in surface AMPARs is due to an increase in exocytosis of AMPARs from intracellular stores and is not the result of a decrease in surface receptor endocytosis (Stellwagen et al., 2005). Therefore, we hypothesized that WIN blocks the TNFα-induced increase of surface AMPARs by preventing an increase in the rate of AMPAR exocytosis in extrasynaptic plasma membrane. To test this hypothesis we used a quantitative video microscopy assay to measure the rate of AMPAR exocytosis events in real time (100 msec time resolution). Videos were taken after either 1) no treatment, 2) WIN treatment (between 15 and 45 minutes), or 3) TNFα treatment (between 15 and 45 minutes), or 4) WIN + TNFα treatment (between 15 and 45 minutes) (see supplementary materials). Figure 2C shows the graphic representation of the change in rate of exocytosis events (expressed as events/minute/μm2 of plasma membrane surface) between control vs. TNFα treatment and WIN vs. WIN + TNFα treatment. TNFα treatment in the absence of WIN doubles the rate of AMPAR exocytosis, while TNFα treatment does not increase the rate of exocytosis measured in WIN-pretreated neurons.
3.4. CB1, not CB2 inhibits the effects of TNFα on AMPAR expression
Since WIN and THC activate both CB1 and CB2, we pretreated neurons with specific CB1 or CB2 antagonists prior to CB1/CB2 agonist application to identify the CB receptor that might be responsible for preventing the TNFα-induced increase in AMPAR surface expression. As shown in Figure 3A, treatment with WIN blocked the TNFα-induced increase in cell surface AMPARs. This block was CB1-mediated, as it was reversed by the CB1-selective antagonist/inverse agonist SR141716A (SR1) (Hurst et al., 2006), but not the CB2-selective antagonist SR144528 (SR2) (Fig 3A). CB1 appeared to be constitutively active in this preparation, as treatment with SR1 enhanced the TNFα effect (Fig 3B). Therefore, we tested the effects of a neutral antagonist, VCHSR1 (VCH) (Hurst et al., 2006), and found that while VCH reversed the effects of WIN on AMPAR surface expression, it did not have intrinsic activity, as predicted for a neutral antagonist (Fig 3B).
Figure 3. The block of TNFα-induced increase of AMPAR surface expression requires CB1 but not CB2 activity.

(A) CB1 antagonists SR1 and VCH antagonized the effect of WIN on TNFα-induced increases in surface GluR1 (TNF, 128±3.4%; TNF+WIN, 100±3.2%; SR1+WIN+TNF, 150±4.9%; VCH+WIN+TNF, 135±6.7%; compared to TNF+WIN *** p<0.001). The CB2 antagonist SR2 did not (SR2+WIN+TNF, 109±4.5%). (B) Co-treatment of SR1 and TNF together induced a higher GluR1 increase than TNF alone (SR1+TNF, 164±5.4%; compared to TNF alone *** p<0.001). SR2 and VCH showed no effect on TNF-induced surface GluR1 expression (SR1, 108±4.4%; SR2+TNF (129±4.9%), VCH+TNF (133±6.9%). n ≥ 100 neurons each condition from 5 independent experiments.
3.5. CB1 activation mitigates the enhanced excitotoxic death caused by TNFα
Our results showing that CB1 activation prevents the rate of increase in TNFα-induced AMPAR exocytosis suggest that CB1 activation might protect neurons from TNFα-enhanced excitotoxic death. Thus, we challenged hippocampal neurons with TNFα (15 min pretreatment) and kainate [(KA): selective AMPA/kainate receptor agonist] and measured cell death 24 hrs later by propidium iodide (PI) uptake and Lactose dehydrogenase (LDH) release assays (Fig 4. A, B, and C). KA alone increased cell death compared to the control condition while TNFα alone did not show a change in cell death compared with control. Pretreatment of neurons with WIN prior to KA challenge did not protect neurons from cell death compared to the addition of KA alone as shown previously (Shen and Thayer, 1998). Pretreatment of the cultures with TNFα followed by the addition of KA significantly enhanced cell death compared with KA alone, suggesting that TNFα potentiates neuronal excitotoxicity. In contrast, preincubation with WIN prior to TNFα and KA addition significantly reduced the observed cell death compared to TNFα+KA co-application. Levels of cell death with KA alone or with WIN+TNFα+KA were not significantly different. Therefore, the potentiated KA excitotoxicity by TNFα is blocked by cannabinoid receptor activation.
Figure 4. Excitotoxicity is potentiated by TNFα and this potentiation blocked by cannabinoid receptor agonists.

(A) Nuclear staining of dead cells (red) by PI and total cells (blue) by Hoechst demonstrates kainate (KA) induced excitotoxicity. TNFα potentiated the cell death caused by KA (TNF+KA). Pretreatment with WIN blocked the potentiated cell death induced by TNFα (WIN+TNF+KA). Arrowheads indicate dead cells. Scale bar, 200 μm. (B) Quantification of image data in (A) demonstrate that KA induced more neuron death (28.4±1.2%, * p<0.05) than the control, untreated neurons (16.0±1.1%). Adding TNFα before KA potentiated cell death (43.6±4.2%, compared to KA ** p<0.01). Pretreatment of cells with WIN significantly blocked cell death enhancement by TNFα (31.2±2.0%, compared to TNF+KA ** p<0.01). WIN alone (17.5±2.6%) or TNFα alone (14.9±2.5%) did not show a significant change in cell death compared to control. [n=25 images for each condition from 5 cultures]. (C) The amount of LDH released from dead or dying neurons was measured to confirm that WIN blocks TNFα-induced potentiation of excitotoxicity. TNF (85.1%±7.1); WIN (101.2%±1.6); KA (161.2%±12.3, compared to control * p<0.05); TNF+KA (230.7%±28.1, compared to KA ** p<0.01); WIN+KA (154.6%±12.9, compared to KA p>0.05) and WIN+TNF+KA (153.9%±8.3, compared to TNF+KA ** p<0.01). n=21 wells of cells for each condition from 7 cultures.
4. Discussion
Our data is supportive of the hypothesis that activation of CB1 decreases TNFα-induced surface expression of AMPARs and protects against END in hippocampal cultures. CB1, AMPARs, and TNFα receptors are all expressed in dendrites in our cultured hippocampal neurons where intracellular signaling between CB1 and TNFα receptors may compete to regulate AMPAR trafficking (Marsicano et al., 2003) as suggested in studies of cortical pyramidal neurons (Hill et al., 2007; Kim et al., 2006) and spinal cord neurons (Salio et al., 2002). Our data showing a correlation of CB1 signaling with a block of the TNFα–induced increase in AMPAR surface trafficking will guide future studies that will seek to identify intracellular signaling mechanisms driving this potential receptor cross-talk.. Regardless of the precise mechanism, our results suggests a potential therapeutic benefit from CB1 activation in the vulnerable time-window immediately following CNS injury. Our studies show that pretreatment with CB1 agonists saves neurons in our excitotoxicity assay. How is this relevant to practical therapeutic intervention in neurotrauma patients since neurons lost in the “crush zone” of a spinal cord injury event, for example, would not likely be recovered by subsequent blocking of TNFα signaling?. The answer comes from studies showing an extensive amount of slowly-spreading neuron death (spanning several days after the injury event) around the initial injury zone which presents a significant opportunity for therapeutic intervention targeting TNFα signaling (Ferguson et al., 2008; Gensel et al., 2006; Mihai et al., 2008). If, as we hypothesize, CB1 activation protects neurons through a mechanism that regulates AMPAR trafficking, how might this occur? The modulation of AMPARs by other GPCRs has been previously demonstrated. D1 dopamine receptor activation increases AMPAR surface expression (Gao et al., 2006; Sun et al., 2005), whereas activation of the metabotropic glutamate receptor (mGluR) decreases surface AMPARs (Davidkova and Carroll, 2007; Snyder et al., 2001; Xiao et al., 2001). PKA activation followed by phosphorylation of AMPARs is a gate to AMPAR surface delivery (Oh et al., 2006), and TNFα stimulation of neurons has been shown to increase PKA activity (Zhang et al., 2002a; Zhang et al., 2002b). It is possible that activation of CB1 may counter this TNFα-induced AMPAR surface delivery through an intracellular signaling mechanism that decreases PKA activation (Di Marzo et al., 2004; Kim et al., 2006; Wade et al., 2004).
CB1 appeared to be constitutively active in our hippocampal neuron preparation, as treatment with the CB1 antagonist/inverse agonist SR1 enhanced the TNFα effect. Therefore, we tested the effects of a neutral antagonist, VCHSR1 (Hurst et al., 2006), and found that while VCHSR1 reversed the effects of WIN on AMPAR surface expression, it did not have intrinsic activity, as would be predicted for a neutral antagonist. These data support a role for CB1 signaling in the modulation of TNFα-induced AMPAR surface-expression.
We hypothesized that the observed block of the TNFα-induced increase in surface AMPARs by cannabinoid receptor activation was mediated by a reduction in the rate of exocytosis. We previously reported that TNFα delivers AMPARs preferentially to extrasynaptic plasma membrane (Leonoudakis et al., 2008) and we have recently developed a technique that allows for the real-time observation AMPAR exocytotic events (Yudowski et al., 2007). Our current results using this new live imaging technique demonstrate that during CB1 activation TNFα treatment is unable to increase the rate of AMPAR exocytosis.
CB1 activation does not confer neuroprotection through non-AMPAR trafficking effects in our excitotoxicity assays. Our previous work showed that neurons pretreated with the AMPAR/KA receptor-specific antagonist CNQX before exposure to kainate were completely protected from excitotoxicity, indicating that the cell death produced by kainate requires AMPAR/KA receptor activity (Leonoudakis et al., 2008). These are in agreement with previous in vitro hippocampal studies showing the AMPAR dependence of TNF-potentiated neuron death (Bernardino et al., 2005). Our previous work as well as others’ studies (Ogoshi et al., 2005; Stellwagen and Malenka, 2006) suggest that TNFα increases the surface delivery of Ca2+-permeable, GluR2-lacking AMPARs within 15 min of TNFα exposure. Ca2+-permeable AMPARs have been demonstrated to be major mediators of excitotoxic cell death (Carriedo et al., 1996; Kwak and Weiss, 2006; Oguro et al., 1999). If these Ca2+-permeable AMPARs are a significant component of the TNFα-induced surface AMPAR accumulation, then specifically blocking their activity, and thus Ca2+ influx through these channels, should reduce the extent of kainate-induced toxicity. Indeed, pretreatment of cultures with NASPM, a Ca2+-permeable AMPAR antagonist, followed by KA plus TNFα produced no additional potentiation than NASPM+ KA (Leonoudakis et al., 2008). Our experiments show that WIN blocks this TNFα-induced increase in GluR1-containing AMPARs. We also show that KA alone, WIN+KA, and WIN+KA+TNFα all induce a similar amount of cell death, whereas KA+TNFα results in significantly more cell death. This suggests that WIN selectively mitigates excitotoxicity caused by a TNFα-induced increase in AMPAR surface expression, and not through other neuron death mechanisms (ie- programmed apoptotic pathways). The model supported by the data is one where WIN interferes with TNF-induced AMPAR exocytosis to reduce TNFα-potentiated excitotoxicity. We have previously shown this TNFα-potentiated excitotoxicity to be a significant proportion of the total measured in vitro excitotoxicity (Leonoudakis et al., 2008).
Normally, the surface expression of AMPARs and their delivery to synapses is tightly regulated. This precise regulation of surface AMPAR number and location as well as the control over glutamate availability prevent excitotoxicity (Malinow and Malenka, 2002). However, after injury, seizures, or during neurodegenerative disease there is an increase in the concentration of extracellular glutamate along with an increased potential for excessive AMPAR surface delivery and excitotoxicity (reviewed in (Leonoudakis et al., 2004; Pickering et al., 2005). Our previous biochemical and microscopic data examining total surface AMPAR levels over a 1 h time course of specific TNFα receptor activation is supportive of a maximum increase in total surface accumulation of calcium permeable AMPARs (CP-AMPARs) at 15 min, which returns to baseline by 1 h (Leonoudakis et al., 2008). CP-AMPARs are important functionally because they have stronger current passing capabilities compared with GluR2-containing AMPARs and are Ca2+-permeable (Burnashev et al., 1992; Hollmann et al., 1991). When neurons are exposed to excess glutamate after neuronal damage, excessive surface expression of CP-AMPARs induced by increased TNFα levels may lead to the abnormal influx of Ca2+ and overactivation of AMPARs resulting in potentiated excitotoxicity (Bernardino et al., 2005; Chao and Hu, 1994; Gelbard et al., 1993; Hermann et al., 2001). These data reveal a critical window of excitotoxic vulnerability caused by TNFα. Inhibiting this activity through modulation of CP-AMPAR trafficking (e.g., by CB1 activation) may reduce excitotoxicity after acute trauma or during neurological disorders.
Cannabinoid receptors protect neurons from excitotoxic stress and the mechanisms of this protection are diverse (Kim et al., 2006; Marsicano et al., 2003; Shen and Thayer, 1998, 1999). One of these mechanisms is believed to be mediated by the retrograde release of endogenous cannabinoids from the postsynaptic neuron to CB1 receptors located on presynaptic neurons [reviewed in (Chevaleyre et al., 2006)] but a role for post-synaptic CB1 signaling cannot be ruled out.
Our results provide the basis for further in vitro studies that will address the specific mechanisms underlying the CB1-mediated block of TNFα-induced increase in AMPAR surface expression as well as animal studies that will test the in vivo neuroprotective potential of CB1 activation.
Supplementary Material
Acknowledgments
MEA is supported by grants from the NIH (DA09978, DA05274) and by The Forbes Norris ALS Center. EB is supported by grants from the NIH (MH067931 and NS038079) as well as by The Forbes Norris ALS Center, the California Pacific Medical Center Research Institute, and ALSA (starter grant 766). We thank Robert Kim and Sean Prasad for excellent technical assistance with the live imaging experiments. We thank Dr. Patricia Reggio (University of North Carolina, Greensboro) and Dr. Herb Seltzman (RTI) for providing VCHSR1 (DA039434).
Abbreviations
- 2AG
2-Arachidonoylglycerol
- AMPARs
α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors
- ANA
anandamide
- CB1
cannabinoid receptor 1
- END
Excitotoxic Neuroinflamatory Death
- GluR1
glutamate receptor 1
- GPCR
G-protein-coupled receptor
- KA
kainate
- LDH
lactose dehydrogenase
- SYN
synaptophysin
- THC
Δ9-tetrahydrocannabinol
- TNFα
Tumor necrosis factor α
- WIN
WIN55212-2
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abood ME, Rizvi G, Sallapudi N, McAllister SD. Activation of the CB(1) cannabinoid receptor protects cultured mouse spinal neurons against excitotoxicity. Neurosci Lett. 2001;309:197–201. doi: 10.1016/s0304-3940(01)02065-1. [DOI] [PubMed] [Google Scholar]
- Aras MA, Hartnett KA, Aizenman E. Assessment of cell viability in primary neuronal cultures. Curr Protoc Neurosci. 2008;Chapter 7(Unit 7):18. doi: 10.1002/0471142301.ns0718s44. [DOI] [PubMed] [Google Scholar]
- Beattie EC, Stellwagen D, Morishita W, Bresnahan JC, Ha BK, Von Zastrow M, Beattie MS, Malenka RC. Control of synaptic strength by glial TNFalpha. Science. 2002;295:2282–2285. doi: 10.1126/science.1067859. [DOI] [PubMed] [Google Scholar]
- Bernardino L, Xapelli S, Silva AP, Jakobsen B, Poulsen FR, Oliveira CR, Vezzani A, Malva JO, Zimmer J. Modulator effects of interleukin-1beta and tumor necrosis factor-alpha on AMPA-induced excitotoxicity in mouse organotypic hippocampal slice cultures. J Neurosci. 2005;25:6734–6744. doi: 10.1523/JNEUROSCI.1510-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burnashev N, Khodorova A, Jonas P, Helm PJ, Wisden W, Monyer H, Seeburg PH, Sakmann B. Calcium-permeable AMPA-kainate receptors in fusiform cerebellar glial cells. Science. 1992;256:1566–1570. doi: 10.1126/science.1317970. [DOI] [PubMed] [Google Scholar]
- Carriedo S, Yin H, Weiss J. Motor neurons are selectively vulnerable to AMPA/kainate receptor-mediated injury in vitro. J Neurosci. 1996;16:4069–4079. doi: 10.1523/JNEUROSCI.16-13-04069.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao CC, Hu S. Tumor necrosis factor-alpha potentiates glutamate neurotoxicity in human fetal brain cell cultures. Dev Neurosci. 1994;16:172–179. doi: 10.1159/000112104. [DOI] [PubMed] [Google Scholar]
- Chevaleyre V, Takahashi KA, Castillo PE. Endocannabinoid-mediated synaptic plasticity in the CNS. Annu Rev Neurosci. 2006;29:37–76. doi: 10.1146/annurev.neuro.29.051605.112834. [DOI] [PubMed] [Google Scholar]
- Davidkova G, Carroll RC. Characterization of the role of microtubule-associated protein 1B in metabotropic glutamate receptor-mediated endocytosis of AMPA receptors in hippocampus. J Neurosci. 2007;27:13273–13278. doi: 10.1523/JNEUROSCI.3334-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Marzo V, Bifulco M, De Petrocellis L. The endocannabinoid system and its therapeutic exploitation. Nat Rev Drug Discov. 2004;3:771–784. doi: 10.1038/nrd1495. [DOI] [PubMed] [Google Scholar]
- Feldmeyer D, Kask K, Brusa R, Kornau H, Kolhekar R, Rozov A, Burnashev N, Jensen V, Hvalby O, Sprengel R, Seeburg P. Neurological dysfunctions in mice expressing different levels of the Q/R site-unedited AMPAR subunit GluR-B. Nat Neurosci. 1999;2:57–64. doi: 10.1038/4561. [DOI] [PubMed] [Google Scholar]
- Ferguson AR, Christensen RN, Gensel JC, Miller BA, Sun F, Beattie EC, Bresnahan JC, Beattie MS. Cell death after spinal cord injury is exacerbated by rapid TNF alpha-induced trafficking of GluR2-lacking AMPARs to the plasma membrane. J Neurosci. 2008;28:11391–11400. doi: 10.1523/JNEUROSCI.3708-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontaine V, Mohand-Said S, Hanoteau N, Fuchs C, Pfizenmaier K, Eisel U. Neurodegenerative and neuroprotective effects of tumor Necrosis factor (TNF) in retinal ischemia: opposite roles of TNF receptor 1 and TNF receptor 2. J Neurosci. 2002;22:RC216. doi: 10.1523/JNEUROSCI.22-07-j0001.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao C, Sun X, Wolf ME. Activation of D1 dopamine receptors increases surface expression of AMPA receptors and facilitates their synaptic incorporation in cultured hippocampal neurons. J Neurochem. 2006;98:1664–1677. doi: 10.1111/j.1471-4159.2006.03999.x. [DOI] [PubMed] [Google Scholar]
- Gelbard HA, Dzenko KA, DiLoreto D, del Cerro C, del Cerro M, Epstein LG. Neurotoxic effects of tumor necrosis factor alpha in primary human neuronal cultures are mediated by activation of the glutamate AMPA receptor subtype: implications for AIDS neuropathogenesis. Dev Neurosci. 1993;15:417–422. doi: 10.1159/000111367. [DOI] [PubMed] [Google Scholar]
- Gensel JC, Tovar CA, Hamers FP, Deibert RJ, Beattie MS, Bresnahan JC. Behavioral and histological characterization of unilateral cervical spinal cord contusion injury in rats. J Neurotrauma. 2006;23:36–54. doi: 10.1089/neu.2006.23.36. [DOI] [PubMed] [Google Scholar]
- Glass TF, Reeves B, Sharp FR. The impact of excitotoxic blockade on the evolution of injury following combined mechanical and hypoxic insults in primary rat neuronal culture. Neurobiol Dis. 2004;17:378–384. doi: 10.1016/j.nbd.2004.07.017. [DOI] [PubMed] [Google Scholar]
- Grooms SY, Opitz T, Bennett MV, Zukin RS. Status epilepticus decreases glutamate receptor 2 mRNA and protein expression in hippocampal pyramidal cells before neuronal death. Proc Natl Acad Sci U S A. 2000;97:3631–3636. doi: 10.1073/pnas.050586497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossman SD, Wolfe BB, Yasuda RP, Wrathall JR. Alterations in AMPA receptor subunit expression after experimental spinal cord contusion injury. J Neurosci. 1999;19:5711–5720. doi: 10.1523/JNEUROSCI.19-14-05711.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermann GE, Rogers RC, Bresnahan JC, Beattie MS. Tumor necrosis factor-alpha induces cFOS and strongly potentiates glutamate-mediated cell death in the rat spinal cord. Neurobiol Dis. 2001;8:590–599. doi: 10.1006/nbdi.2001.0414. [DOI] [PubMed] [Google Scholar]
- Hill EL, Gallopin T, Ferezou I, Cauli B, Rossier J, Schweitzer P, Lambolez B. Functional CB1 receptors are broadly expressed in neocortical GABAergic and glutamatergic neurons. J Neurophysiol. 2007 doi: 10.1152/jn.00603.2006. [DOI] [PubMed] [Google Scholar]
- Hollmann M, Hartley M, Heinemann S. Ca2+ permeability of KA-AMPA--gated glutamate receptor channels depends on subunit composition. Science. 1991;252:851–853. doi: 10.1126/science.1709304. [DOI] [PubMed] [Google Scholar]
- Howlett AC. The cannabinoid receptors. Prostaglandins Other Lipid Mediat. 2002;68–69:619–631. doi: 10.1016/s0090-6980(02)00060-6. [DOI] [PubMed] [Google Scholar]
- Hurst D, Umejiego U, Lynch D, Seltzman H, Hyatt S, Roche M, McAllister S, Fleischer D, Kapur A, Abood M, Shi S, Jones J, Lewis D, Reggio P. Biarylpyrazole Inverse Agonists at the Cannabinoid CB1 Receptor: Importance of the C-3 Carboxamide Oxygen/Lysine3.28(192) Interaction. J Med Chem. 2006;49:5969–5987. doi: 10.1021/jm060446b. [DOI] [PubMed] [Google Scholar]
- Kim SH, Won SJ, Mao XO, Jin K, Greenberg DA. Molecular mechanisms of cannabinoid protection from neuronal excitotoxicity. Mol Pharmacol. 2006;69:691–696. doi: 10.1124/mol.105.016428. [DOI] [PubMed] [Google Scholar]
- Kwak S, Weiss JH. Calcium-permeable AMPA channels in neurodegenerative disease and ischemia. Curr Opin Neurobiol. 2006;16:281–287. doi: 10.1016/j.conb.2006.05.004. [DOI] [PubMed] [Google Scholar]
- Leonoudakis D, Braithwaite SP, Beattie MS, Beattie EC. TNFalpha-induced AMPA-receptor trafficking in CNS neurons; relevance to excitotoxicity? Neuron Glia Biol. 2004;1:263–273. doi: 10.1017/S1740925X05000608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonoudakis D, Zhao P, Beattie EC. Rapid tumor necrosis factor alpha-induced exocytosis of glutamate receptor 2-lacking AMPA receptors to extrasynaptic plasma membrane potentiates excitotoxicity. J Neurosci. 2008;28:2119–2130. doi: 10.1523/JNEUROSCI.5159-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leterrier C, Laine J, Darmon M, Boudin H, Rossier J, Lenkei Z. Constitutive activation drives compartment-selective endocytosis and axonal targeting of type 1 cannabinoid receptors. J Neurosci. 2006;26:3141–3153. doi: 10.1523/JNEUROSCI.5437-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lock C, Oksenberg J, Steinman L. The role of TNFalpha and lymphotoxin in demyelinating disease. Ann Rheum Dis. 1999;58(Suppl 1):I121–128. doi: 10.1136/ard.58.2008.i121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackie K. From active ingredients to the discovery of the targets: the cannabinoid receptors. Chem Biodivers. 2007;4:1693–1706. doi: 10.1002/cbdv.200790148. [DOI] [PubMed] [Google Scholar]
- Malinow R, Malenka RC. AMPA receptor trafficking and synaptic plasticity. Annu Rev Neurosci. 2002;25:103–126. doi: 10.1146/annurev.neuro.25.112701.142758. [DOI] [PubMed] [Google Scholar]
- Marsicano G, Goodenough S, Monory K, Hermann H, Eder M, Cannich A, Azad SC, Cascio MG, Gutierrez SO, van der Stelt M, Lopez-Rodriguez ML, Casanova E, Schutz G, Zieglgansberger W, Di Marzo V, Behl C, Lutz B. CB1 cannabinoid receptors and on-demand defense against excitotoxicity. Science. 2003;302:84–88. doi: 10.1126/science.1088208. [DOI] [PubMed] [Google Scholar]
- McDonald NA, Henstridge CM, Connolly CN, Irving AJ. An essential role for constitutive endocytosis, but not activity, in the axonal targeting of the CB1 cannabinoid receptor. Mol Pharmacol. 2007;71:976–984. doi: 10.1124/mol.106.029348. [DOI] [PubMed] [Google Scholar]
- Mihai G, Nout YS, Tovar CA, Miller BA, Schmalbrock P, Bresnahan JC, Beattie MS. Longitudinal comparison of two severities of unilateral cervical spinal cord injury using magnetic resonance imaging in rats. J Neurotrauma. 2008;25:1–18. doi: 10.1089/neu.2007.0338. [DOI] [PubMed] [Google Scholar]
- Nagatsu T, Mogi M, Ichinose H, Togari A. Cytokines in Parkinson’s disease. J Neural Transm. 2000;(Suppl):143–151. [PubMed] [Google Scholar]
- Nemeth ZH, Hasko G, Szabo C, Vizi ES. Amrinone and theophylline differentially regulate cytokine and nitric oxide production in endotoxemic mice. Shock. 1997;7:371–375. doi: 10.1097/00024382-199705000-00010. [DOI] [PubMed] [Google Scholar]
- New DR, Maggirwar SB, Epstein LG, Dewhurst S, Gelbard HA. HIV-1 Tat induces neuronal death via tumor necrosis factor-alpha and activation of non-N-methyl-D-aspartate receptors by a NFkappaB-independent mechanism. J Biol Chem. 1998;273:17852–17858. doi: 10.1074/jbc.273.28.17852. [DOI] [PubMed] [Google Scholar]
- Ogoshi F, Yin HZ, Kuppumbatti Y, Song B, Amindari S, Weiss JH. Tumor necrosis-factor-alpha (TNF-alpha) induces rapid insertion of Ca2+-permeable alpha-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate (AMPA)/kainate (Ca-A/K) channels in a subset of hippocampal pyramidal neurons. Exp Neurol. 2005;193:384–393. doi: 10.1016/j.expneurol.2004.12.026. [DOI] [PubMed] [Google Scholar]
- Oguro K, Oguro N, Kojima T, Grooms SY, Calderone A, Zheng X, Bennett MV, Zukin RS. Knockdown of AMPA receptor GluR2 expression causes delayed neurodegeneration and increases damage by sublethal ischemia in hippocampal CA1 and CA3 neurons. J Neurosci. 1999;19:9218–9227. doi: 10.1523/JNEUROSCI.19-21-09218.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh MC, Derkach VA, Guire ES, Soderling TR. Extrasynaptic membrane trafficking regulated by GluR1 serine 845 phosphorylation primes AMPA receptors for long-term potentiation. J Biol Chem. 2006;281:752–758. doi: 10.1074/jbc.M509677200. [DOI] [PubMed] [Google Scholar]
- Patel MN, Yim GK, Isom GE. N-methyl-D-aspartate receptors mediate cyanide-induced cytotoxicity in hippocampal cultures. Neurotoxicology. 1993;14:35–40. [PubMed] [Google Scholar]
- Perry RT, Collins JS, Wiener H, Acton R, Go RC. The role of TNF and its receptors in Alzheimer’s disease. Neurobiol Aging. 2001;22:873–883. doi: 10.1016/s0197-4580(01)00291-3. [DOI] [PubMed] [Google Scholar]
- Pickering M, Cumiskey D, O’Connor JJ. Actions of TNF-alpha on glutamatergic synaptic transmission in the central nervous system. Exp Physiol. 2005;90:663–670. doi: 10.1113/expphysiol.2005.030734. [DOI] [PubMed] [Google Scholar]
- Reimann-Philipp U, Ovase R, Weigel PH, Grammas P. Mechanisms of cell death in primary cortical neurons and PC12 cells. J Neurosci Res. 2001;64:654–660. doi: 10.1002/jnr.1119. [DOI] [PubMed] [Google Scholar]
- Salio C, Cottone E, Conrath M, Franzoni MF. CB1 cannabinoid receptors in amphibian spinal cord: relationships with some nociception markers. J Chem Neuroanat. 2002;24:153–162. doi: 10.1016/s0891-0618(02)00040-6. [DOI] [PubMed] [Google Scholar]
- Sanchez C, Rueda D, Segui B, Galve-Roperh I, Levade T, Guzman M. The CB(1) cannabinoid receptor of astrocytes is coupled to sphingomyelin hydrolysis through the adaptor protein fan. Mol Pharmacol. 2001;59:955–959. doi: 10.1124/mol.59.5.955. [DOI] [PubMed] [Google Scholar]
- Shen M, Thayer S. Cannabinoid Receptor Agonists Protect Cultured Rat Hippocampal Neurons from Excitotoxicity. Mol Pharmacol. 1998;54:459–462. doi: 10.1124/mol.54.3.459. [DOI] [PubMed] [Google Scholar]
- Shen M, Thayer S. Delta9-Tetrahydrocannabinol Acts as a Partial Agonist to Modulate Glutamatergic Synaptic Transmission between Rat Hippocampal Neurons in Culture. Mol Pharmacol. 1999;55:8–13. doi: 10.1124/mol.55.1.8. [DOI] [PubMed] [Google Scholar]
- Shohami E, Ginis I, Hallenbeck JM. Dual role of tumor necrosis factor alpha in brain injury. Cytokine Growth Factor Rev. 1999;10:119–130. doi: 10.1016/s1359-6101(99)00008-8. [DOI] [PubMed] [Google Scholar]
- Snyder EM, Philpot BD, Huber KM, Dong X, Fallon JR, Bear MF. Internalization of ionotropic glutamate receptors in response to mGluR activation. Nat Neurosci. 2001;4:1079–1085. doi: 10.1038/nn746. [DOI] [PubMed] [Google Scholar]
- Stellwagen D, Beattie EC, Seo JY, Malenka RC. Differential regulation of AMPA receptor and GABA receptor trafficking by tumor necrosis factor-alpha. J Neurosci. 2005;25:3219–3228. doi: 10.1523/JNEUROSCI.4486-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stellwagen D, Malenka RC. Synaptic scaling mediated by glial TNF-alpha. Nature. 2006;440:1054–1059. doi: 10.1038/nature04671. [DOI] [PubMed] [Google Scholar]
- Sun X, Zhao Y, Wolf ME. Dopamine receptor stimulation modulates AMPA receptor synaptic insertion in prefrontal cortex neurons. J Neurosci. 2005;25:7342–7351. doi: 10.1523/JNEUROSCI.4603-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szelenyi J. Cytokines and the central nervous system. Brain Res Bull. 2001;54:329–338. doi: 10.1016/s0361-9230(01)00428-2. [DOI] [PubMed] [Google Scholar]
- Wade MR, Tzavara ET, Nomikos GG. Cannabinoids reduce cAMP levels in the striatum of freely moving rats: an in vivo microdialysis study. Brain Res. 2004;1005:117–123. doi: 10.1016/j.brainres.2004.01.039. [DOI] [PubMed] [Google Scholar]
- Xiao MY, Zhou Q, Nicoll RA. Metabotropic glutamate receptor activation causes a rapid redistribution of AMPA receptors. Neuropharmacology. 2001;41:664–671. doi: 10.1016/s0028-3908(01)00134-4. [DOI] [PubMed] [Google Scholar]
- Yang L, Lindholm K, Konishi Y, Li R, Shen Y. Target depletion of distinct tumor necrosis factor receptor subtypes reveals hippocampal neuron death and survival through different signal transduction pathways. J Neurosci. 2002;22:3025–3032. doi: 10.1523/JNEUROSCI.22-08-03025.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying Z, Babb TL, Comair YG, Bushey M, Touhalisky K. Increased densities of AMPA GluR1 subunit proteins and presynaptic mossy fiber sprouting in the fascia dentata of human hippocampal epilepsy. Brain Res. 1998;798:239–246. doi: 10.1016/s0006-8993(98)00421-1. [DOI] [PubMed] [Google Scholar]
- Yudowski GA, Puthenveedu MA, Leonoudakis D, Panicker S, Thorn KS, Beattie EC, von Zastrow M. Real-time imaging of discrete exocytic events mediating surface delivery of AMPA receptors. J Neurosci. 2007;27:11112–11121. doi: 10.1523/JNEUROSCI.2465-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang HH, Halbleib M, Ahmad F, Manganiello VC, Greenberg AS. Tumor necrosis factor-alpha stimulates lipolysis in differentiated human adipocytes through activation of extracellular signal-related kinase and elevation of intracellular cAMP. Diabetes. 2002a;51:2929–2935. doi: 10.2337/diabetes.51.10.2929. [DOI] [PubMed] [Google Scholar]
- Zhang JM, Li H, Liu B, Brull SJ. Acute topical application of tumor necrosis factor alpha evokes protein kinase A-dependent responses in rat sensory neurons. J Neurophysiol. 2002b;88:1387–1392. doi: 10.1152/jn.2002.88.3.1387. [DOI] [PubMed] [Google Scholar]
- Zhao X, Bausano B, Pike BR, Newcomb-Fernandez JK, Wang KK, Shohami E, Ringger NC, DeFord SM, Anderson DK, Hayes RL. TNF-alpha stimulates caspase-3 activation and apoptotic cell death in primary septo-hippocampal cultures. J Neurosci Res. 2001;64:121–131. doi: 10.1002/jnr.1059. [DOI] [PubMed] [Google Scholar]
- Zou JY, Crews FT. TNF alpha potentiates glutamate neurotoxicity by inhibiting glutamate uptake in organotypic brain slice cultures: neuroprotection by NF kappa B inhibition. Brain Res. 2005;1034:11–24. doi: 10.1016/j.brainres.2004.11.014. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.