

Abstract
Objective
Myeloperoxidase-enriched monocytes play important roles in inflammatory disease such as atherosclerosis. We previously demonstrated α-chlorofatty aldehydes (α-ClFALD) are produced as a result of plasmalogen targeting by myeloperoxidase-derived hypochlorous acid (HOCl) in activated monocytes. Here we show α-chlorofatty acid (α-ClFA), a stable metabolite of α-ClFALD, accumulates in activated monocytes and delineate the molecular effects of α-ClFA on monocytes/macrophages.
Approach and Results
Liquid chromatography-mass spectrometry revealed that α-ClFA is elevated 5-fold in PMA-stimulated human monocytes rising to ~ 20 μM, compared to unstimulated cells. Using human THP-1 monocytes and RAW 264.7 cells as in vitro models, we tested the hypothesis that α-ClFA is a cell death mediator, which could potentially participate in pathophysiological roles of monocytes in diseases such as atherosclerosis. Indeed, 2-ClHA, the 16 carbon molecular species of α-ClFA, caused significant apoptosis of primary monocytes. Similarly, 2-ClHA also caused apoptosis in THP-1 human monocytes and RAW 264.7 mouse macrophages as determined by annexin V-PI staining and TUNEL staining, respectively. 2-ClHA treatment also increased caspase-3 activity and poly (ADP-ribose) polymerase (PARP) cleavage in THP-1 cells. 2-ClHA likely elicits apoptosis by increasing both reactive oxygen species (ROS) production and endoplasmic reticulum (ER) stress since antioxidants and CCAAT/enhancer-binding protein homologous protein (CHOP) block such induced cell apoptosis.
Conclusion
The stable chlorinated lipid, α-ClFA accumulates in activated primary human monocytes, and elicits monocyte apoptosis through increased ROS production and endoplasmic reticulum stress, providing a new insight of chlorinated lipids and monocytes in inflammatory disease.
Keywords: Chlorinated lipids, myeloperoxidase, apoptosis, macrophages, monocytes
Myeloperoxidase (MPO)-enriched phagocytes are important cellular mediators in many inflammatory diseases including atherosclerosis.1–4 In response to inflammation, monocytes are recruited to the sites of inflammation and differentiate into macrophages to elicit immune response.5 Apoptosis of monocytes/macrophages is a component of the pathophysiologic sequelae of atherosclerosis.6 Oxidized lipid species have been suggested to be mediators of apoptosis and the progression of atherosclerosis.7, 8 However the role of chlorinated lipids, produced as a result of MPO activity, as mediators of apoptosis remain to be explored. Chlorinated lipids are initially produced by the targeting of the vinyl ether bond linking an sn-1 aliphatic group to the glycerol backbone of plasmalogens, resulting in the formation of two products, lysophospholipid and α-chlorofatty aldehydes (α-ClFALD).2, 9, 10 It should be noted that plasmalogens are a major lipid subclass found in many mammalian cell types, including endothelial cells, neutrophils, monocytes, smooth muscle cells and cardiac myocytes. α-ClFALD, including 2-chlorohexadecanal (2-ClHDA) and 2-chlorooctadecanal (2-ClODA), are produced in activated neutrophils and monocytes.2, 9, 10 Furthermore, the concentration of α-ClFALD is elevated in human atherosclerotic lesions and infarcted myocardium.11, 12 In neutrophils α-ClFALD has been shown to be predominantly oxidized to α-chlorofatty acid (α-ClFA) while some metabolism of α-ClFALD involves reduction to α-chlorofatty alcohol (α-ClFOH).13, 14 α-ClFA is elevated in rats treated with lipopolysaccharide and can be further catabolized in hepatocytes through ω-oxidation and subsequent β-oxidation resulting in the production of 2-chloroadipic acid (2-ClAdA), which is excreted in the urine.15
While the α-ClFALD, 2-ClHDA, has been shown to be a potent neutrophil chemoattractant, inhibitor of endothelial nitric oxide synthase (eNOS), inducer of the expression of cyclooxygenase-2, and mediator of endothelial cell dysfunction;10, 16–18 the biological properties of the α-ClFA, 2-chlorohexadecanoic acid (2-ClHA), have been relatively unexplored. Accordingly, in the study herein we have revealed the accumulation of α-ClFA and α-ClFOH in activated human monocytes. Furthermore, using monocytic and macrophagic cell lines, as well as primary human monocytes, we have revealed that 2-ClHA causes significant apoptosis through mechanisms involving ROS production and ER stress.
MATERIALS AND METHODS
Materials and Methods are available in the online-only Data Supplement.
RESULTS
2-ClHA and 2-ClOA increase significantly in primary human monocytes upon PMA stimulation
Our previous study showed 2-ClHDA and 2-ClODA increases to levels of ~3–6 pmol/106 PMA-activated primary human monocytes. In these studies we tested whether endogenously-produced α-ClFALD is metabolized to α-ClFA and α-ClFOH. To determine the levels of both α-ClFA and α-ClFOH in human primary monocytes, the cells were stimulated with 300 nM PMA for 1h. Cellular lipids were extracted and quantitatively analyzed by LC-MS and gas chromatography-MS for α-ClFA and α-ClFOH, respectively. As expected, the total amount of 2-ClHA and 2-ClOA was elevated 5-fold (rising to ~ 20 μM) compared to unstimulated cells (Figure 1A). For α-ClFOH levels, only 2-chlorohexadecanol (2-ClHOH) was found to significantly increase compared to unstimulated cells, while 2-chlorooctadecanol was undetectable (Figure 1B). Notably, the α-ClFA amount is much more than α-ClFOH, indicating that α-ClFALD is mainly metabolized to α-ClFA instead of α-ClFOH in human monocytes.
Figure 1. Both α-ClFA and α-ClFOH accumulate in activated primary human monocytes.

Cells were treated with 300 nM PMA for 1h, and the cellular free α-ClFA and α-ClFOH were quantified by LC-MS and gas chromatography-MS, respectively. N=3.
2-ClHA induces apoptosis in primary human monocytes, THP-1 monocytes and RAW 264.7 macrophages
Human primary monocytes were treated with various concentrations of 2-ClHA for 3h and apoptosis was determined by TUNEL staining. 2-ClHA significantly increased apoptotic cell numbers compared to either blank or HA group (Figure 2A). Importantly, significant apoptosis was observed in primary human monocytes at physiological concentrations of 2-ClHA found in activated human monocytes. The pro-apoptotic effect of 2-ClHA was then tested on a human monocytic cell line, THP-1 cells. The cells were treated with 2-ClHA for 6h, and apoptosis was subsequently examined by Annexin V-PI double staining followed by flow cytometry. Consistent with the findings in primary human monocytes, 2-ClHA increased apoptosis in THP-1 cells compared to either blank or HA group (Figure 2B). Under all conditions of treatments of THP-1 cells with 2-ClHA, the intracellular 2-ClHA concentration did not exceed the treatment extracellular concentration (e.g., treatments for 18h with 10 μM and 50 μM 2-ClHA resulted in intracellular concentrations of 7.2 ± 1.4 and 45.6 ± 4.9 μM, respectively; and treatments for 1h with 10 μM and 50 μM 2-ClHA resulted in intracellular concentrations of 0.8 ± 0.1 and 14.2 ± 1.1 μM). 2-ClHA-elicited apoptosis was further confirmed in RAW 264.7 macrophages by TUNEL staining, which further demonstrated the disparate pro-apoptotic effect of 2-ClHA compared to HA (Figure SIA).
Figure 2. 2-ClHA induces apoptosis of primary human monocytes and THP-1 monocytes.

A. Primary human monocytes were treated with indicated concentrations of HA or 2-ClHA for 3h, and the apoptotic cells were determined under fluorescent microscopy after TUNNEL staining. B. THP-1 monocytes were treated with indicated concentrations of HA or 2-ClHA for 6h, and the apoptotic cells were counted by flow cytometry after Annexin V-PI double staining. N=3. *P<0.05 and **P<0.01 vs. Blank.
2-ClHA increases caspase-3 activity, PARP cleavage and CHOP expression
Caspase-3 activity was evaluated to further examine 2-ClHA-induced apoptosis. Indeed, 2-ClHA treatment resulted in increased caspase-3 activity in both THP-1 monocytes and RAW 264.7 macrophages (Figure 3A and SIB). Furthermore, PARP cleavage was determined to confirm 2-ClHA-mediated cell apoptosis. 2-ClHA induces a concentration and time-dependent PARP cleavage (Figure 3B and 3C). Additionally, CHOP, also known as GADD153, which is a pro-apoptotic transcription factor activated by ER stress, was evaluated. In THP-1 monocytes, CHOP was present at undetectable or very low levels in untreated or HA-treated groups, while 50 μM of 2-ClHA induced a significant expression of CHOP at both mRNA level and protein level, as measured by qRT-PCR and Western blots, respectively (Figure 3B, 3C and SII). CHOP expression at the mRNA level in RAW 264.7 macrophages was also significantly up-regulated by 2-ClHA as determined by qRT-PCR (Figure SII)
Figure 3. 2-ClHA increases caspase 3 activity and induces PARP cleavage and CHOP expression.

A. Caspase 3 activity in THP-1 monocytes treated with indicated concentrations of HA or 2-ClHA for 6h was determined as described in “Materials and Methods”. B. THP-1 monocytes were treated with indicated concentrations of 2-ClHA for 18h, and PARP cleavage and CHOP expression was determined by Western blots. C. THP-1 monocytes were treated with 50 μM of 2-ClHA for indicated time, and PARP cleavage and CHOP expression was determined by Western blots. Numbers under the blots represent the ratio of the intensity of the response to that of the maximum response in that blot with normalization to the β-actin loading control, and values are the average of ratios from 3 independent experiments. **P<0.01 vs. Blank.
2-ClHA initiates an ER stress response
Since the induction of CHOP expression indicated ER stress might be involved in 2-ClHA-induced apoptosis, we further interrogated this possibility. 2-ClHA induced XBP1 mRNA splicing in both THP-1 and RAW 264.7 cells as determined by RT-PCR (Figure 4A and SIII). Furthermore, 2-ClHA significantly increased the phosphorylation of eIF2α without changing total eIF2α expression (Figure 4B). In concert with eIF2α phosphorylation, the target gene of eIF2α, ATF4, was up-regulated. Additionally, GRP78, which serves as a gatekeeper to the activation of ER stress transducers, was increased in response to 2-ClHA treatment (Figure 4B). Other data showed that mRNA expression of these ER stress-associated proteins in THP-1 monocytes or RAW 264.7 cells was increased in response to 2-ClHA (Figure SII).
Figure 4. 2-ClHA induces ER stress response.
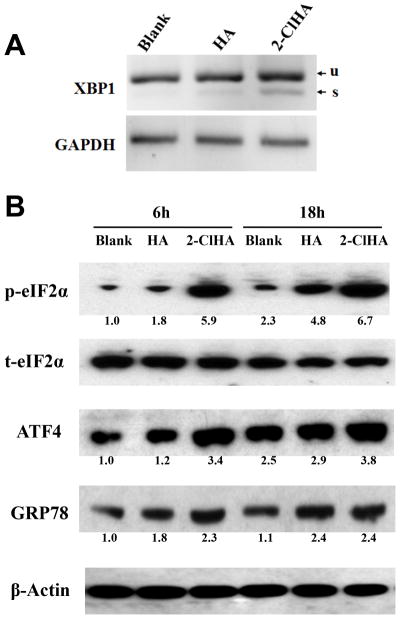
A. THP-1 cells were treated with HA or 2-ClHA for 18h, and XBP1 mRNA splicing was determined by qRT-PCR. u=unspliced; s=spliced. B. THP-1 cells were treated with 50 μM of 2-ClHA for 6h or 18h, the expression of phospho-eIF2α, total eIF2α, ATF4, and GRP78 was determined by Western blotting. Numbers under the blots represent the ratio of the intensity of the response to that of the response in the blank (control) condition, and values are the average of ratios from 3 independent experiments. Additionally, fold induction of phospho-eIF2α was normalized to total eIF2α, while that of other proteins was normalized to β-actin.
CHOP silencing attenuates 2-ClHA-induced apoptosis
To further assess ER stress involvement in 2-ClHA-induced apoptosis, the effector gene of ER stress, CHOP, was knocked down by its specific siRNA. Transfection of CHOP siRNA effectively reduced CHOP expression, while the control siRNA had no such effect (Figure 5A). CHOP silencing resulted in reduced PARP cleavage in response to 2-ClHA treatment (Figure 5A). Furthermore, CHOP silencing significantly decreased caspase-3 activity induced by 2-ClHA (Figure 5B).
Figure 5. Effect of CHOP siRNA on 2-ClHA-induced apoptosis.

THP-1 monocytes transfected with scrambled siRNA or CHOP siRNA were treated with 50 μM of 2-ClHA, then PARP cleavage as well as CHOP expression (18h treatment) was determined by Western blots (A) and caspase 3 activity (6h treatment) was measured as described in “Materials and Methods”. (B). Numbers under the blots represent the ratio of the intensity of the response to that of the maximum response in that blot with normalization to the β-actin loading control, and values are the average of ratios from 3 independent experiments. **P<0.01 between treatments that included the addition of 2-ClHA.
ROS is involved in 2-ClHA-induced apoptosis
A previous study has shown that hypochlorite-modified LDL-induced apoptosis is mediated by ROS in Jurkat T cells.19 Since LDL contains plasmalogens,20 it is likely that one component of hypochlorite-modified LDL is 2-ClHDA,9 which can be oxidized intracellularly to 2-ClHA. Accordingly, we investigated whether 2-ClHA alone is sufficient to induce ROS generation. Amplex Red was used to show that 2-ClHA treatments lead to H2O2 release from THP-1 cells (Figure 6A). Additionally, both N-acetylcysteine (NAC) and glutathione (GSH) ameliorated the production of H2O2 induced by 2-ClHA. Moreover, NAC and GSH attenuated 2-ClHA-induced caspase 3 activity, PARP cleavage and CHOP expression (Figures 6B & C). Thus, these results suggest that ROS might mediate 2-ClHA-induced ER stress.
Figure 6. ROS is involved in 2-ClHA-induced apoptosis.

THP-1 monocytes were pretreated or not with the antioxidants, NAC (1 mM) or GSH (1 mM) for 1h, and then with 50 μM of 2-ClHA for 3h (A), 6h (B) or 18h (C). Extracellular H2O2 levels (A) and caspase 3 activity (B) were determined as described in “Materials and Methods”. N=3. **P<0.01 vs Blank. ##P<0.01 vs 2-ClHA. PARP cleavage and CHOP expression (C) were determined by Western blotting. Numbers under the blots represent the ratio of the intensity of the response to that of the maximum response in that blot with normalization to the β-actin loading control, and values are the average of ratios from 3 independent experiments.
DISCUSSION
The present study shows for the first time that human monocyte activation results in α-ClFA and α-ClFOH production. The production of these chlorinated lipids has previously been shown in other systems to be a result of plasmalogen oxidation by HOCl leading to α-ClFALD accumulation which is subsequently metabolized through the fatty acid-fatty alcohol cycle.13, 14 It should also be appreciated that the present studies demonstrate that intracellular levels of α-ClFA reach ~20 μM during monocyte activation, Furthermore since activation and apoptosis of monocytes are key events in the formation and rupture of atherosclerotic plaques,6 the role of this lipid in monocyte apoptosis was examined. Multiple lines of evidence showed that this concentration of α-ClFA elicits monocyte/macrophage apoptosis including increased TUNEL staining, Annexin V-PI staining, and caspase 3 activity in primary human monocytes as well as increased levels of these apoptotic measures and PARP cleavage in both THP-1 monocytes and RAW 264.7 macrophages. Interestingly, a previous study indicated hypochlorite-modified LDL caused cell death in Jurkat T-cell lines.19 Such modified LDL is likely to contain α-ClFALD, which can be further oxidized to α-ClFA intracellularly. While there are many chlorinated moieties in hypochlorite-modified LDL including tyrosine and cysteine residues, it is quite possible that α-ClFALD residues in the modified LDL may elicit at least in part the injury observed in cells treated with hypochlorite-treated LDL.
ER stress activation is frequently observed in atherosclerotic lesions, liver tissue, and adipose tissue of hyperlipidemic mice and humans.21 Lipid accumulation has been suggested to activate ER stress. Extracellular exposure to high levels of saturated fatty acids induces ER stress and apoptosis in liver cells.22 Stearic acid accumulation in macrophages results in ER stress as well as apoptosis independent of toll-like receptor 4/2 activation.23 The present study is the first to report that chlorinated lipids can also induce ER stress in monocytes/macrophages, which leads to increased CHOP expression. In fact, 2-ClHA seems to be more potent in activating ER stress than HA since HA did not initiate ER stress, or apoptosis at the equimolar concentrations. The role of ER stress in ClHA-elicited apoptosis was strongly supported by the demonstration that reducing CHOP expression with its specific siRNA significantly decreases 2-ClHA-induced apoptosis in THP-1 monocytes.
The finding that 2-ClHA upregulates CHOP expression indicates ER stress may mediate 2-ClHA’s effects. Two of the three major ER stress pathways are inositol requiring enzyme-1 (IRE1) and RNA-dependent protein kinase-like ER kinase (PERK),24 and these two pathways were involved in 2-ClHA elicited ER stress. IRE1 triggers the splicing of a specific mRNA transcript for XBP1, and activation of PERK phosphorylates eIF2α, which lead to the transcription of CHOP and GRP78.24 The possible mechanism of CHOP-induced apoptosis involves interaction with members of the BCL-2 family of proteins or a calcium signaling pathway.25 Previous studies have shown ER stress signaling can contribute to apoptotic cell death in several cell types related to the cardiovascular system. In vitro studies suggested increased CHOP expression in apoptotic smooth muscle cells treated with 7-ketocholesterol, homocysteine, or glucosamine.26–28 Also, activation of ER stress has been identified in endothelial cells both in vitro and in swine.29, 30
Our studies also suggest that ROS are also involved in increasing ER stress and apoptosis. In the present study, we demonstrated that 2-ClHA can induce ROS production using Amplex Red to monitor extracellular H2O2 production. In addition, the antioxidants, NAC and GSH, decreased 2-ClHA induced ROS production as well as apoptosis. Furthermore, NAC and GSH attenuated CHOP expression, indicating a mitigation of ER stress. Thus, ROS generation may mediate 2-ClHA-triggered ER stress. It should be noted that the interplay between ROS and ER stress has been reported in many studies although the detailed mechanism is still elusive.31 Attenuation of ER stress decreases ROS generation and increases GSH level in α-tocopheryl succinate-treated human gastric carcinoma cells.32 Some other studies have reported that oxidative stress induces ER stress in cultured human hepatoma cells and hepatocytes.31
In summary, the results of this study demonstrate that α-ClFA accumulates in activated monocytes and contributes to apoptosis by triggering ROS production and ER stress. Silencing of CHOP expression attenuates the apoptosis-inducing effect of 2-ClHA, while attenuation of ROS by antioxidants can alleviate ER stress and subsequent cell apoptosis. Thus, our study suggests that α-ClFA is a bioactive lipid that links inflammation and apoptosis in monocytes/macrophages. This is an important first step, albeit in isolated cell systems, to show the biological role of α-ClFA, which may be of considerable importance in a modulatory role in some inflammatory diseases such as atherosclerosis. Demonstrating the role of both exogenously applied, as well as endogenously produced, α-ClFA in in vivo systems of inflammation including atherosclerosis need to be examined in future studies.
Supplementary Material
SIGNIFICANCE.
Increasing evidence has suggested that chlorinated lipids produced from the myeloperoxidase-HOCl system may play important roles in inflammation and cardiovascular diseases. This study demonstrates: 1) for the first time, micromolar levels of α-ClFA are produced in activated human monocytes; 2) α-ClFA initiates ROS production and subsequent ER stress leading to monocyte death; and 3) α-ClFA initiated apoptosis is reduced by either antioxidant treatment or down-regulation of CHOP. Thus, the production of α-ClFA is a novel mechanism that potentially links inflammation and cell death, and, in particular, this pathway may be involved in the inflammation, apoptosis and ER stress that occurs in atherosclerotic lesions.
Acknowledgments
SOURCES OF FUNDING
This research was supported by NIH grants HL074214 and HL111906 (DAF).
ABBREVIATIONS
- 2-ClHDA
2-chlorohexadecanal
- 2-ClODA
2-chlorooctadecanal
- 2-ClHA
2-chlorohexadecanoic acid
- 2-ClOA
2-chlorooctadecanoic acid
- 2-ClHOH
2-chlorohexadecanol
- 2-Cl-[d4]HA
2-chloro-[d4-7,7,8,8]-hexadecanoic acid
- 2-Cl-[d4]HOH
2-chloro-[d4-7,7,8,8]-hexadecanol
- α-ClFALD
α-chlorofatty aldehyde
- α-ClFA
α-chlorofatty acid
- α-ClFOH
α-chlorofatty alcohol
- CHOP
CCAAT/enhancer-binding protein homologous protein
- ER
endoplasmic reticulum
- eNOS
endothelial nitric oxide synthase
- FBS
fetal bovine serum
- GSH
glutathione
- HA
hexadecanoic acid
- H2O2
hydrogen peroxide
- HOCl
hypochlorous acid
- MS
mass spectrometry
- MPO
myeloperoxidase
- NAC
N-acetyl cysteine
- PMA
phorbol myristate acetate
- PARP
poly (ADP-ribose) polymerase
- PI
propidium iodide
- ROS
reactive oxygen species
- TUNEL
terminal deoxynucleotidyl transferase dUTP nick end labeling
Footnotes
DISCLOSURES
None.
References
- 1.Daugherty A, Dunn JL, Rateri DL, Heinecke JW. Myeloperoxidase, a catalyst for lipoprotein oxidation, is expressed in human atherosclerotic lesions. J Clin Inv. 1994;94:437–444. doi: 10.1172/JCI117342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ford DA. Lipid oxidation by hypochlorous acid: Chlorinated lipids in atherosclerosis and myocardial ischemia. Clin Lipidol. 2010;5:835–852. doi: 10.2217/clp.10.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sugiyama S, Okada Y, Sukhova GK, Virmani R, Heinecke JW, Libby P. Macrophage myeloperoxidase regulation by granulocyte macrophage colony-stimulating factor in human atherosclerosis and implications in acute coronary syndromes. Am J Pathol. 2001;158:879–891. doi: 10.1016/S0002-9440(10)64036-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang R, Brennan ML, Shen Z, MacPherson JC, Schmitt D, Molenda CE, Hazen SL. Myeloperoxidase functions as a major enzymatic catalyst for initiation of lipid peroxidation at sites of inflammation. J Biol Chem. 2002;277:46116–46122. doi: 10.1074/jbc.M209124200. [DOI] [PubMed] [Google Scholar]
- 5.Swirski FK, Nahrendorf M, Etzrodt M, Wildgruber M, Cortez-Retamozo V, Panizzi P, Figueiredo JL, Kohler RH, Chudnovskiy A, Waterman P, Aikawa E, Mempel TR, Libby P, Weissleder R, Pittet MJ. Identification of splenic reservoir monocytes and their deployment to inflammatory sites. Science. 2009;325:612–616. doi: 10.1126/science.1175202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Van Vre EA, Ait-Oufella H, Tedgui A, Mallat Z. Apoptotic cell death and efferocytosis in atherosclerosis. Arterioscler Thromb Vasc Biol. 2012;32:887–893. doi: 10.1161/ATVBAHA.111.224873. [DOI] [PubMed] [Google Scholar]
- 7.Greig FH, Kennedy S, Spickett CM. Physiological effects of oxidized phospholipids and their cellular signaling mechanisms in inflammation. Free Radic Biol Med. 2012;52:266–280. doi: 10.1016/j.freeradbiomed.2011.10.481. [DOI] [PubMed] [Google Scholar]
- 8.Mitra S, Deshmukh A, Sachdeva R, Lu J, Mehta JL. Oxidized low-density lipoprotein and atherosclerosis implications in antioxidant therapy. Am J Med Sci. 2011;342:135–142. doi: 10.1097/MAJ.0b013e318224a147. [DOI] [PubMed] [Google Scholar]
- 9.Thukkani AK, Albert CJ, Wildsmith KR, Messner MC, Martinson BD, Hsu FF, Ford DA. Myeloperoxidase-derived reactive chlorinating species from human monocytes target plasmalogens in low density lipoprotein. J Biol Chem. 2003;278:36365–36372. doi: 10.1074/jbc.M305449200. [DOI] [PubMed] [Google Scholar]
- 10.Thukkani AK, Hsu FF, Crowley JR, Wysolmerski RB, Albert CJ, Ford DA. Reactive chlorinating species produced during neutrophil activation target tissue plasmalogens: Production of the chemoattractant, 2-chlorohexadecanal. J Biol Chem. 2002;277:3842–3849. doi: 10.1074/jbc.M109489200. [DOI] [PubMed] [Google Scholar]
- 11.Thukkani AK, Martinson BD, Albert CJ, Vogler GA, Ford DA. Neutrophil-mediated accumulation of 2- ClHDA during myocardial infarction: 2-ClHDA-mediated myocardial injury. Am J Physiol-Heart Circ Physiol. 2005;288:H2955–2964. doi: 10.1152/ajpheart.00834.2004. [DOI] [PubMed] [Google Scholar]
- 12.Thukkani AK, McHowat J, Hsu FF, Brennan ML, Hazen SL, Ford DA. Identification of alpha-chloro fatty aldehydes and unsaturated lysophosphatidylcholine molecular species in human atherosclerotic lesions. Circulation. 2003;108:3128–3133. doi: 10.1161/01.CIR.0000104564.01539.6A. [DOI] [PubMed] [Google Scholar]
- 13.Anbukumar DS, Shornick LP, Albert CJ, Steward MM, Zoeller RA, Neumann WL, Ford DA. Chlorinated lipid species in activated human neutrophils: Lipid metabolites of 2-chlorohexadecanal. J Lipid Res. 2010;51:1085–1092. doi: 10.1194/jlr.M003673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wildsmith KR, Albert CJ, Anbukumar DS, Ford DA. Metabolism of myeloperoxidase-derived 2-chlorohexadecanal. J Biol Chem. 2006;281:16849–16860. doi: 10.1074/jbc.M602505200. [DOI] [PubMed] [Google Scholar]
- 15.Brahmbhatt VV, Albert CJ, Anbukumar DS, Cunningham BA, Neumann WL, Ford DA. {omega}-oxidation of {alpha}-chlorinated fatty acids: Identification of {alpha}-chlorinated dicarboxylic acids. J Biol Chem. 2010;285:41255–41269. doi: 10.1074/jbc.M110.147157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Marsche G, Heller R, Fauler G, Kovacevic A, Nuszkowski A, Graier W, Sattler W, Malle E. 2-chlorohexadecanal derived from hypochlorite-modified high-density lipoprotein-associated plasmalogen is a natural inhibitor of endothelial nitric oxide biosynthesis. Arterioscler Thromb Vasc Biol. 2004;24:2302–2306. doi: 10.1161/01.ATV.0000148703.43429.25. [DOI] [PubMed] [Google Scholar]
- 17.Messner MC, Albert CJ, Ford DA. 2-chlorohexadecanal and 2-chlorohexadecanoic acid induce COX-2 expression in human coronary artery endothelial cells. Lipids. 2008;43:581–588. doi: 10.1007/s11745-008-3189-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ullen A, Fauler G, Bernhart E, Nusshold C, Reicher H, Leis HJ, Malle E, Sattler W. Phloretin ameliorates 2-chlorohexadecanal-mediated brain microvascular endothelial cell dysfunction in vitro. Free Radic Biol Med. 2012;53:1770–1781. doi: 10.1016/j.freeradbiomed.2012.08.575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Resch U, Semlitsch M, Hammer A, Susani-Etzerodt H, Walczak H, Sattler W, Malle E. Hypochlorite-modified low-density lipoprotein induces the apoptotic machinery in jurkat t-cell lines. Biochem Biophys Res Commun. 2011;410:895–900. doi: 10.1016/j.bbrc.2011.06.089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vance JE. Lipoproteins secreted by cultured rat hepatocytes contain the antioxidant 1-alk-1-enyl-2-acylglycerophosphoethanolamine. Biochim Biophys Acta. 1990;1045:128–134. doi: 10.1016/0005-2760(90)90141-j. [DOI] [PubMed] [Google Scholar]
- 21.Myoishi M, Hao H, Minamino T, Watanabe K, Nishihira K, Hatakeyama K, Asada Y, Okada K, Ishibashi-Ueda H, Gabbiani G, Bochaton-Piallat ML, Mochizuki N, Kitakaze M. Increased endoplasmic reticulum stress in atherosclerotic plaques associated with acute coronary syndrome. Circulation. 2007;116:1226–1233. doi: 10.1161/CIRCULATIONAHA.106.682054. [DOI] [PubMed] [Google Scholar]
- 22.Wei Y, Wang D, Topczewski F, Pagliassotti MJ. Saturated fatty acids induce endoplasmic reticulum stress and apoptosis independently of ceramide in liver cells. Am J Physiol - Endocrin Metab. 2006;291:E275–281. doi: 10.1152/ajpendo.00644.2005. [DOI] [PubMed] [Google Scholar]
- 23.Anderson EK, Hill AA, Hasty AH. Stearic acid accumulation in macrophages induces toll-like receptor 4/2-independent inflammation leading to endoplasmic reticulum stress-mediated apoptosis. Arterioscler Thromb Vasc Biol. 2012;32:1687–1695. doi: 10.1161/ATVBAHA.112.250142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Scull CM, Tabas I. Mechanisms of er stress-induced apoptosis in atherosclerosis. Arterioscler Thromb Vasc Biol. 2011;31:2792–2797. doi: 10.1161/ATVBAHA.111.224881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Thorp E, Li Y, Bao L, Yao PM, Kuriakose G, Rong J, Fisher EA, Tabas I. Brief report: Increased apoptosis in advanced atherosclerotic lesions of apoe-/- mice lacking macrophage bcl-2. Arterioscler Thromb Vasc Biol. 2009;29:169–172. doi: 10.1161/ATVBAHA.108.176495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dickhout JG, Sood SK, Austin RC. Role of endoplasmic reticulum calcium disequilibria in the mechanism of homocysteine-induced er stress. Antioxid Redox Signal. 2007;9:1863–1873. doi: 10.1089/ars.2007.1780. [DOI] [PubMed] [Google Scholar]
- 27.He C, Zhu H, Zhang W, Okon I, Wang Q, Li H, Le YZ, Xie Z. 7-ketocholesterol induces autophagy in vascular smooth muscle cells through nox4 and atg4b. Am J Pathol. 2013;183:626–637. doi: 10.1016/j.ajpath.2013.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Larroque-Cardoso P, Swiader A, Ingueneau C, Negre-Salvayre A, Elbaz M, Reyland ME, Salvayre R, Vindis C. Role of protein kinase c delta in er stress and apoptosis induced by oxidized ldl in human vascular smooth muscle cells. Cell Death Dis. 2013;4:e520. doi: 10.1038/cddis.2013.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Haas MJ, Raheja P, Jaimungal S, Sheikh-Ali M, Mooradian AD. Estrogen-dependent inhibition of dextrose-induced endoplasmic reticulum stress and superoxide generation in endothelial cells. Free Radic Biol Med. 2012;52:2161–2167. doi: 10.1016/j.freeradbiomed.2012.04.009. [DOI] [PubMed] [Google Scholar]
- 30.Civelek M, Manduchi E, Riley RJ, Stoeckert CJ, Jr, Davies PF. Chronic endoplasmic reticulum stress activates unfolded protein response in arterial endothelium in regions of susceptibility to atherosclerosis. Circ Res. 2009;105:453–461. doi: 10.1161/CIRCRESAHA.109.203711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hanada S, Harada M, Kumemura H, Bishr Omary M, Koga H, Kawaguchi T, Taniguchi E, Yoshida T, Hisamoto T, Yanagimoto C, Maeyama M, Ueno T, Sata M. Oxidative stress induces the endoplasmic reticulum stress and facilitates inclusion formation in cultured cells. J Hepatol. 2007;47:93–102. doi: 10.1016/j.jhep.2007.01.039. [DOI] [PubMed] [Google Scholar]
- 32.Huang X, Li L, Zhang L, Zhang Z, Wang X, Zhang X, Hou L, Wu K. Crosstalk between endoplasmic reticulum stress and oxidative stress in apoptosis induced by alpha-tocopheryl succinate in human gastric carcinoma cells. Br J Nutr. 2012:1–9. doi: 10.1017/S0007114512001882. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.