

Abstract
Tyrosine hydroxylase (TyrH) catalyzes the hydroxylation of tyrosine to form 3,4-dihydroxyphenylalanine in the biosynthesis of the catecholamine neurotransmitters. The activity of the enzyme is regulated by phosphorylation of serine residues in a regulatory domain and by binding of catecholamines to the active site. Available structures of TyrH lack the regulatory domain, limiting the understanding of the effect of regulation on structure. We report the use of NMR spectroscopy to analyze the solution structure of the isolated regulatory domain of rat TyrH. The protein is composed of a largely unstructured N-terminal region (residues 1-71) and a well-folded C-terminal portion (residues 72-159). The structure of a truncated version of the regulatory domain containing residues 65-159 has been determined and establishes that it is an ACT domain. The isolated domain is a homodimer in solution, with the structure of each monomer very similar to that of the core of the regulatory domain of phenylalanine hydroxylase. Two TyrH regulatory domain monomers form an ACT domain dimer composed of a sheet of eight strands with four α-helices on one side of the sheet. Backbone dynamic analyses were carried out to characterize the conformational flexibility of TyrH65-159. The results provide molecular details critical for understanding the regulatory mechanism of TyrH.
Keywords: Tyrosine hydroxylase, regulation, ACT domain, solution structure, NMR spectroscopy
Introduction
Tyrosine hydroxylase (TyrH)a is the first and rate-limiting enzyme in catecholamine biosynthesis, catalyzing the conversion of tyrosine into 3,4-dihydroxyphenylalanine, the precursor for the catecholamines dopamine, norepinephrine and epinephrine. These neurotransmitters play key roles in the autonomic nervous system, and altered levels of TyrH are associated with hypertension and neurological disorders.1; 2; 3 TyrH is a member of the family of pterin-dependent aromatic amino acid hydroxylases together with phenylalanine hydroxylase (PheH) and tryptophan hydroxylase (TrpH).4 These proteins all catalyze the hydroxylation of the aromatic side chain of their respective amino acid substrates, using oxygen and tetrahydrobiopterin as the other substrates.
TyrH is a homotetramer; each monomer contains an N-terminal regulatory domain (~160–200 residues depending on the species and isoform), a catalytic domain (~300 residues), and a C-terminal tetramerization domain (~45 residues).5; 6 PheH and TrpH have similar structures.4 The catalytic domains of the three enzymes display sequence identities of nearly 50%, but the regulatory domains exhibit much lower sequence identities, and the regulatory domain of TyrH is significantly longer than those of PheH and TrpH. Three-dimensional structures of the catalytic domains of all three hydroxylases are available7; 8; 9 and confirm the very similar structures and active sites, indicating that these enzymes share a common catalytic mechanism.10 At present there is no structure of the regulatory domain of TyrH or TrpH; only the structure of the regulatory domain of PheH is available,8 and the N-terminal 18 residues, including the phosphorylation site Ser16, are not seen in this structure.
The three enzymes are regulated differently, consistent with the divergent structures of their regulatory domains. PheH is activated by phenylalanine, by phosphorylation of Ser16, and by inhibition by tetrahydrobiopterin,11; 12 while the regulation of TrpH is not yet understood. TyrH is regulated by phosphorylation of several residues in the regulatory domain, including Ser19, Ser31, and Ser40, and inhibited by catecholamines.13; 14 Phosphorylation of Ser40 of TyrH by cAMP-dependent protein kinase activates the enzyme by relieving it from high-affinity catecholamine inhibition;15 phosphorylation of the other residues does not significantly affect the enzyme activity but rather alters interactions with other proteins.14; 16; 17 The opposing effects of catecholamines and Ser40 phosphorylation on the activity of TyrH have been attributed to the enzyme having two conformations: a closed conformation in which the N-terminus of the regulatory domain lies over the enzyme active site, preventing substrates from binding, and an open conformation in which the N-terminus has moved away from the active site.18; 19; 20; 21 Phosphorylation of Ser40 stabilizes the active open form, while catecholamine binding stabilizes the closed form. However, the molecular basis for this process is unknown in the absence of a structure of the regulatory domain of TyrH.
In this report, we describe the use of high-field NMR methods to determine the solution structure of the regulatory domain of TyrH. The results provide insights into the regulatory properties of TyrH and the evolution of the family of aromatic amino acid hydroxylase.
Results
NMR assignments of RDTyrH
Knowledge of the structure of the regulatory domain of TyrH is clearly critical for understanding the molecular basis for regulation of the enzyme. As a starting point to determining the solution structure of the isolated regulatory domain (RDTyrH, residues 1-159), a 2D 1H-15N heteronuclear single-quantum coherence (HSQC) spectrum was obtained; the result is shown in red in Figure 1a. The spectrum is well dispersed and shows nearly 140 discrete signals corresponding to backbone amides of the expected 145 resonances. However, the clustering of signals with high intensity in the random coil region suggests that a portion of the isolated RDTyrH is disordered. The backbone resonances of the N-terminal 70 residues of RDTyrH could be assigned using sensitivity-enhanced TROSY-based HNCACB, HN(CO)CACB, HNCA and HN(CO)CA triple-resonance experiments,22; 23 with the exception of the amide resonances of Met1, Pro11, Lys12, Gln39, Ser40, Leu41, and Pro68. The weaker resonances for the remaining 89 residues were more difficult to assign. The assignments of a better-behaved N-terminally truncated mutant protein (RDTyrH65-159, discussed below) allowed us to assign many of the resonances of RDTyrH. Still, no resonances could be assigned for 17 of the 159 residues. Only the Cα assignments could be made for Asn80, Arg90, Lys93, Pro94, Glu105, Thr106, Leu114, Pro127, His128, Leu129 and Pro138. The backbone amide signals of 11 nonproline residues (Lys12, Val29, Ile42, Arg49, Ala71, Phe74, Ser95, Lys102, Ala109, Leu146 and Arg150) could not be assigned, and most Cβ chemical shift of the last 89 residues were not assigned.
Figure 1.
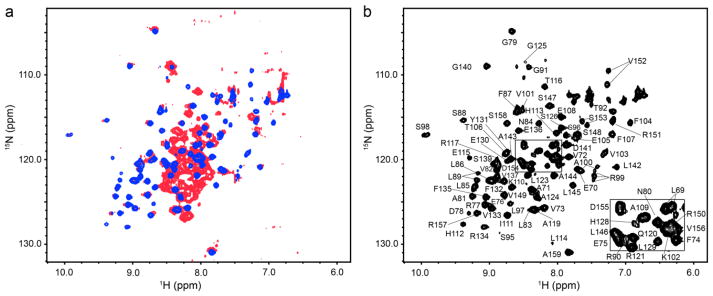
NMR spectra of the isolated regulatory domain of tyrosine hydroxylase. (a) Overlay of the 2D 1H-15N HSQC spectra of RDTyrH (red) and RDTyrH65–159 (blue). (b) 2D 1H-15N HSQC spectrum of 0.8 mM RDTyrH65–159 showing the assignments of the individual residues. Conditions: 50 mM sodium phosphate, 1 μM leupeptin, 1 μM pepstatin A and 5% D2O (pH 7.0), at 300 K at a magnetic field strength of 14.1 T (600 MHz 1H).
The available backbone chemical shifts were analyzed using the program PECAN,24 which provides the secondary structure probabilities on a residue-by-residue basis (Figure S1a). This indicated that RDTyrH is composed of a largely unstructured N-terminal region (residues 1-72) and a more ordered and well-folded C-terminal portion (residues 73-159). The chemical shifts predict that there is a helix (residues 46-56) in the N-terminal flexible region. We carried out preliminary characterization of a mutant of RDTyrH lacking the N-terminal 30 residues. This protein was proteolyzed around residue Pro64 after a couple of days at room temperature, precluding comprehensive structural analyses. This cleavage did not alter the remaining resonances (residues 65-159) in the 2D 1H-15N HSQC spectra. The T2 values for residues 46-56 in this deletion mutant of RDTyrH were much higher T2 (average of 158 ms) than the values for the other secondary structure elements (average of 62 ms), but lower than those of the more flexible N-terminal region (average of 192 ms), suggesting that residues 46-56 may have some secondary structure but still retain significant flexibility in solution.
NMR assignments of RDTyrH65-159
Since the secondary structure assignments and the intensities of the resonances suggested that the C-terminal ~90 residues are well-structured, a series of proteins lacking the N-terminal 60-70 residues were examined as NMR samples. The protein lacking the N-terminal 64 residues of RDTyrH (RDTyrH65-159) was the best behaved in terms of solubility and the quality of the HSQC NMR spectrum (results not shown). This truncation eliminated the high intensity signals in the random coil region of the spectrum but left the more dispersed lower intensity signals unperturbed (Figure 1a, blue). Thus, the N-terminal 64 residues are flexible and are not necessary for proper folding of the remaining C-terminal portion.
The backbone resonances of RDTyrH65-159 were assigned for a uniformly 13C and 15N labeled sample using sensitivity-enhanced 3D triple resonance experiments including HNCA,25 HN(CO)CA,26 HNCACB27 and CBCA(CO)NH (Figure 1b).28 Nearly all (88/93) the backbone resonances of TyrH65-159 could be assigned; the exceptions were Pro65, Gly66, Asn67, Lys93 and Pro94. Leu89, Arg99, Ala100, Val103, Val152 and Ser153, exhibited two NH peaks in the 2D 1H -15N HSQC spectrum, suggesting that they have two conformations in solution. The backbone chemical shifts of RDTyrH65-159 were analyzed using the program PECAN.24 The results are consistent with the presence of four β strands and two α-helices and are essentially identical to those for RDTyrH (Figure S1b). More than 90% of the chemical shift assignments for side chain atoms were then obtained with 3D HBHA(CO)NH,28 CC(CO)NH, (H)CCH-COSY,29 and (H)CCH-TOCSY30 experiments. 3D 15N- and 13C-edited NOESY-HSQC spectra (mixing time 120 ms) were collected to confirm the chemical shift assignments and generate distance restraints for structure calculations.31; 32
Quaternary structure of RDTyrH65-159
Equilibrium ultracentrifugation was carried out to determine the quaternary structure of RDTyrH. The results showed that RDTyrH forms a stable dimer at concentrations as low as 5 μM (Figure S2). Analyses of the quaternary structure of RDTyrH65-159 by both size exclusion chromatography and sedimentation velocity analytical centrifugation showed that RDTyrH65-159 is a dimer, so that removal of the N-terminal residues did not alter the oligomerization state.
The dimer interface of RDTyrH65-159 was identified by a differential-labeling NMR technique that relies on the 3D 15N-NOESY spectrum of a mixed dimer formed by one completely deuterated, 15N-labeled monomer and one protonated unlabeled monomer.33; 34 In such a mixed dimer, cross peaks between 15N-labeled amide protons and aliphatic protons should be due to intermonomer contacts. To ensure that the cross peaks were not due to residual aliphatic protons, a 3D 15N-NOESY spectrum of the completely deuterated 15N-labeled protein prior to mixing with the unlabeled protein was recorded as a control spectrum. Thus, the cross peaks in the 15N-NOESY spectrum from the mixed dimer that show a significant increase in intensity compared to the corresponding cross peaks from the control spectrum can be identified as intermonomer NOEs. Using such an approach, 8 cross peaks from 6 backbone amide protons were initially identified as intermonomer NOEs (Figure S3). Additional NOE assignments obtained during the course of iterative refinement yielded a total of 28 unique intermonomer distance constraints for the final structure.
Overall structure
The three-dimensional structure of the RDTyrH65-159 dimer was calculated with the program ARIA (Ambiguous Restraints for Iterative Assignment),35 using the inter-proton NOE-derived distance restraints in combination with chemical shift derived dihedral angle,36 3JHNHα coupling,37 and residual dipolar couplings (RDC) restraints.38 The superimpositions of the 10 lowest energy structures, and the ribbon diagram of one representative structure are shown in Figure 2. The structural statistics of RDTyrH65-159 are summarized in Table 1. The whole structure is well defined with an overall backbone root mean square deviation (rmsd) of 0.71 ± 0.10 Å and an rmsd for the secondary structural elements of 0.34 ± 0.08 Å. A PROCHECK_NMR39 analysis showed that more than 90% of the residues are within the most favored and additional allowed regions of the Ramachandran plot, and only 1.5% of the residues are in the disallowed regions.
Figure 2.

Structure of the regulatory domain of tyrosine hydroxylase. (a) Superposition of the backbones of the 10 lowest-energy structures of RDTyrH65–159 in two orientations with an 180° rotation along the Y-axis. β-Strands are in green and helices in red. (b) Ribbon diagram of a representative low-energy structure in two orientations with an 180° rotation along the Y-axis indicating the location of secondary structure elements.
Table 1.
Structural statistics for RDTH65–159a
| Restraints | Experimental |
|---|---|
| Total restraints | 3070 |
| Sequential restraints (| i − j | = 1) | 830 |
| Short range (2 ≤ | i − j | ≤ 5) | 414 |
| Long range (| i − j | ≥ 5) | 644 |
| Inter-subunit | 28 |
| Dihedral restraints | |
| Φ | 138 |
| Ψ | 138 |
| RDC restraints | |
| 1DNH | 112 |
| Coupling restraints | |
| 3JHNHα | 86 |
| Deviation among ensemble | |
| Bonds (Å) | 0.0054 ± 0.0004 |
| Angles (degrees) | 0.76 ± 0.04 |
| Improper (degrees) | 0.80 ± 0.12 |
| Dihedral restraints (degrees) | 1.08 ± 0.63 |
| RDC 1DNH (Hz) | 0.52 ± 0.09 |
| JHNα restraints (Hz) | 0.93 ± 0.12 |
| Ramachandran plotb | |
| Most favored (%) | 68.5 |
| Additionally allowed (%) | 25.9 |
| Generously allowed (%) | 4.1 |
| Disallowed (%) | 1.5 |
| Overall precision (Å) | |
| Secondary structure | |
| Backbonec | 0.34 ± 0.08 |
| Heavy atomsc | 0.71 ± 0.10 |
| All residues | |
| Backbonec | 3.2 ± 1.7 |
| Heavy atomsc | 3.4 ± 0.6 |
Structural statistics were calculated for the ensemble of the 10 lowest-energy structures.
Calculated using the program PROCHECK.39
Backbone atoms include NH, Cα, and CO; heavy includes all non-hydrogen atoms.
RDTyrH65-159 is a homodimer, with each monomer consisting of a four stranded antiparallel β-sheet (β1: residues 75-77, β2: residues 80-84, β3: residues 110-116, β4: residues 131-137) and two α-helices (α1: residues 97-106, α2: residues 139-152) connected by four loops. The two α-helices are parallel to and on one side of the sheet, an arrangement that is very similar to ferredoxin-like structures. The N-terminal region (residues 65-72) is defined by a limited number of restraints and is poorly ordered, while the C-terminal portion is defined by a high number of restraints and is well-ordered. There are two highly flexible loop regions, L2 (residues 85-96) and L4 (residues 119-130). The two monomers interact through strand β3 and helix α1, forming a sheet of eight strands with four α-helices on one side of the sheet. The surface area buried in the dimer interface is about 770 Å2 for each monomer; this amounts to ~12% of the monomer surface area. In addition to a number of hydrophobic interactions, there are four hydrogen bonds across the interface, two between the carbonyl oxygen of Leu114 and the amide nitrogen of Thr116 and two between the amide of Leu114 and the carbonyl oxygen of Thr116.
Dynamic properties of RDTyrH65-159
To obtain insight into the dynamic properties of RDTyrH65-159, the backbone 15N longitudinal relaxation time (T1), transverse relaxation time (T2), and heteronuclear 1H-15N NOE values were obtained at a 1H frequency of 600 MHz. Seventy-six residues yielded observable 1H-15N correlations that could be analyzed. The unanalyzed residues included six prolines, three of the unassigned residues, and 10 residues that either overlapped or were too weak to be analyzed. The experimentally determined T1, T2, and 1H-15N NOE values versus the amino acid sequence are shown in Figure S4. Overall, the entire domain is rigid, except for the N-terminal region and loops L2 and L4. The short T2 values for most of the residues in the secondary structure elements (~ 70 ms) are consistent with RDTyrH65-159 being a dimer.
It is well established that motional anisotropy can influence the measured relaxation parameters. Based on the five lowest energy structures, the ratio of the principle components of the inertia tensors for RDTyrH65-159 is 1:1.51:1.83, suggesting that the protein tumbles anisotropically. Consequently, the rotational diffusion parameters were determined using both isotropic and axially symmetric anisotropic diffusion models.40 When the model with axial symmetry was used, the χ2 value for the agreement between the experimentally measured and modeled T1/T2 values was 1.98. This was significantly lower than the χ2 value obtained for a fit to an isotropic model (2.67) or to an axially symmetric model with randomized N-H bond vector orientations (2.51), indicating that the protein tumbles anisotropically. Thus, the RDTyrH65-159 dimer is best described by an axially symmetric diffusion tensor, with a global correlation time τave of 11.90 ± 0.02 ns and a diffusional anisotropy (D||/D⊥) of 1.23 ± 0.01.
The relaxation data of RDTyrH65-159 were analyzed using the program ModelFree441 with the parameters describing an axially symmetric diffusion tensor. Five models with increasing complexity were used iteratively to reproduce the experimental data until the confidence level reached 95%. The resulting profiles are shown in Figure 3. For the 76 residues used in the analysis, the dynamics of seven residues can be described adequately using the simplest model including only the generalized order parameter S2, with an average value of 0.87 ± 0.03. Twenty-four residues also have significant but small τe values, reflecting internal motions on the ps time scale; the average S2 value for these is 0.81 ± 0.04. Eighteen residues close to the dimer interface have significant Rex values, reflecting conformational exchange on the μs-ms time scale, in addition to an average S2 value of 0.78 ± 0.05 and ps τe values. Finally, 27 residues near the termini and loops are best described by a model that includes Sf2, reflecting an S2 value on fast time scales, in addition to an average S2 value of 0.61 ± 0.04 and τe values on the ns time scale. Overall, the protein adopts a fairly rigid structure in that most residues exhibit S2 values greater than 0.75. Residues at the termini and in loops have much smaller S2 values and exhibit internal motions (τe) on the ps-ns time scale, indicating that they are highly flexible.
Figure 3.

Model-free parameters for RDTyrH65–159 backbone amides derived by the fitting of the 15N T1, 15N T2, and 1H-15N NOE data. Lipari-Szabo S2, S2f, τe, and Rex parameters are shown from top to bottom, respectively. Missing S2, S2f, τe, and Rex data points indicate that this parameter was not included in the motional model for that residue. A representation of the RDTyrH65–159 secondary structure is shown along the top.
Effect of Phosphorylation
TyrH is phosphorylated at Ser40 by protein kinase A. The isotopically labeled RDTyrH was stoichiometrically phosphorylated to determine the effect of phosphorylation on the structure. Most of the NH peaks overlap in the spectra of the phosphorylated and unphosphorylated enzymes, but additional peaks are present in the HSQC spectrum of phosphorylated RDTyrH (Figure S5). The backbone assignments of phosphorylated RDTyrH could be made using the same methods as for RDTyrH; these included Ser40 and the adjacent Gln39 and Leu41. All the assigned residues retain the same chemical shifts as in RDTyrH except for Gly36, Arg37, Gln39, Ser40, Leu41, Ile42, and Glu43. This suggests that after phosphorylation the core structure of RDTyrH remains the same as that in unphosphorylated RDTyrH, and a local structural change takes place around Ser40.
Discussion
The solution NMR data presented here establish that the isolated regulatory domain of TyrH can be described as a well-packed C-terminal core made up of residues 71-159 plus a flexible N-terminal tail. Limited proteolysis,20 N-terminal truncation mutants,42; 43 fluorescence spectrocopy,18 and hydrogen/deuterium exchange19 have been used to study the structure of the N-terminus of TyrH in the context of the intact protein. In all cases the results are consistent with the first 71 residues being dynamic and relatively unstructured. The PECAN prediction of the secondary structure of the first 70 residues of RDTyrH suggests that there is a helix containing residues 46-56 and possibly a β strand in residues 10-13. This is not consistent with a previous prediction from computational analyses that the first 60 residues contain two helices (residues 16-29 and residues 41-60) connected by a β turn.44 The previous mass spectrometric analyses and the NMR T2 values of these residues suggests that any helix formed by residues 46-56 is very dynamic. Residues 40-49 are heavily conserved across multiple species of TyrH from fish to human, while residues 50-59 form an unusual poly-alanine tract of variable length in different species. This sequence conservation suggests that this region of the protein has a functional role. The ~70 residue N-terminal flexible portion of RDTyrH is significantly longer than the corresponding region of the N-terminus of PheH (~31 residues).
Both the complete RDTyrH construct and the shorter RDTyrH65-459 construct end with residue 159. Defining the exact delineation between protein domains has some degree of uncertainty, but residues 118-123 of rat PheH and residues 164-169 of rat TyrH can be assigned to the N-terminal portion of the respective catalytic domains. These residues occupy identical positions at the N-termini in the structures of the catalytic domains of both, and all three mammalian aromatic amino acid hydroxylases show high sequence identities from these residues to the C-termini (Figures 4 and S6). While the protein used to obtain the crystal structure of the catalytic domain began with residue 156, residues 156-160 were not seen in the available structures. Thus, the regulatory domain of TyrH can be assigned to the first 159 residues.
Figure 4.

Comparison of the structures of the regulatory domains of phenylalanine hydroxylase and tyrosine hydroxylase. (a) Overlay of the ribbon diagrams of RDTyrH65–159 (green) and RDPheH (medium purple, residues 19–117, PDB code 2PHM). (b) Topology diagrams of RDTyrH65–159 (top) and RDPheH (bottom); the structural elements are labeled in green and magenta, respectively. (c) Structure-based sequence alignment of the regulatory domains of the three amino acid hydroxylases. The secondary structural elements of RDTyrH65–159 and RDPheH are labeled green and magenta, respectively. To generate the alignment, a structure-based alignment of RDTyrH65–159 and RDPheH was first performed with Chimera.84 The sequence of the regulatory domain of TrpH was then added to the alignment using Clustal X.86
The fold of the RDTyrH65-159 monomer is similar to that of the regulatory domain of rat PheH (RDPheH) determined by X-ray crystallography of the combined regulatory and catalytic domains of a dimeric form of that enzyme.8 The overall arrangement of the two helices and the beta sheet is conserved (Figure 4a), with the major differences between the two structures at the termini. Figure 4b compares the secondary structure topologies of the regulatory domains of TyrH and PheH, and Figure 4c shows a structure-based alignment. Residues 82-159 of TyrH align well with residues 32-109 of PheH, although the two sequences are only 25% identical over this span. Both structures contain four strands and two helices, but the locations of the strands in the sequence differ, in that strand β1 of TyrH has a similar location in the β sheet to strand β4 of PheH. The structures of the C-termini of the two regulatory domains are conserved through helix α2. PheH has four more residues from there to the start of the catalytic domain; this region includes strand β4, so that it is likely that the lack of this strand in RDTyrH is not due simply to the absence of the remainder of the protein. In addition, strand β1 of TyrH is located in the N-terminal portion of the sequence that does not correspond clearly to residues in PheH (Figure 4c). The formation of β1 in RDTyrH65-159 is confirmed by dN–N(i,i+1) and dα–n(i,i+1) NOE connectivities and a positive consensus chemical shift index.
The internal mobility parameters obtained from the model-free analysis correlate well with the three-dimensional structure of RDTyrH65-159 (Figure 5). The S2 values indicate that overall the enzyme shows restricted motion on the ps-ns time scale, except at the termini and loops (Figure 5a). Most residues in the secondary structural elements of the enzyme have high S2 values and very small value (~10 ps) τe values. On the other hand, residues near the termini and in loops L2 and L4 have lower S2 values and a certain degree of flexibility on ps-ns time scale (Figure 5b). There is significant conformational exchange of residues in strand β3 and helix α1 (Figure 5c), consistent with a monomer-dimer equilibrium for RDTyrH65-159. Five residues located on strands β2 and β4 also show significant Rex values, indicating that the β sheet is influenced by this equilibrium.
Figure 5.
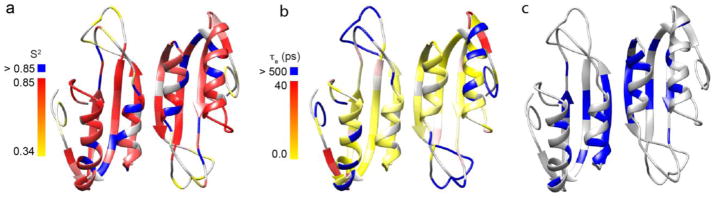
The dynamic properties of the backbone of the RDTyrH65–159 dimer: a) the generalized order parameter S2 values with colors ranging from yellow to red and blue corresponding to S2 values from 0.4 to 0.85, and >0.85; b) the internal motions on the ps to ns time scales with colors ranging from yellow to red and magenta corresponding to Te values from 0 to 40 ps and >500 ps; c) residues with conformational changes (Rex) on the microsecond to millisecond time scales colored in blue. Residues for which the respective parameter was not included in the model are colored in grey.
Available crystal structures of PheH do not predict a dimeric structure for RDTyrH. However, independent expression of residues 1-117 of rat PheH yields a polypeptide that forms a dimer,45 suggesting that the dimeric structure is intrinsic to the structure of the regulatory domains of both proteins. The regulatory domains of both PheH and TrpH have previously been identified as belonging to the ACT structural family, and TyrH has been assumed to do so also.46; 47; 48 The present NMR-based structure of RDTyrH65-159 clearly shows that the regulatory domain of TyrH contains an ACT domain, consistent with that prediction. The protein family data base (Pfam)49 lists over 20,000 proteins as containing an ACT domain (Pfam accession number PF01842), and the structures of more than 160 of them have been determined to date. ACT domains are most commonly found in bacteria, although there are examples in plants and fungi; PheH appears to be the only previous example of an ACT domain from a multi-cellular animal. 49 ACT domain monomers fold with a ferredoxin-like βαββαβ topology.46; 47; 50; 51 These generally associate in two different ways to form dimers or higher order oligomers, a side-by-side arrangement involving α1 and β246; 52; 53 (corresponding to α1 and β3 in RDTyrH65-159) and a face to face arrangement of the β sheets.54; 55 The RDTyrH dimer structure is very similar to that of a side-by-side ACT domain dimer, and the dynamic properties of the protein are consistent with a side-by-side arrangement.53 Figure 6 compares the structure of the RDTyrH65-159 dimer with three side by side ACT domain dimers,52; 53; 56; 57 further confirming that it belongs to the ACT domain family.
Figure 6.
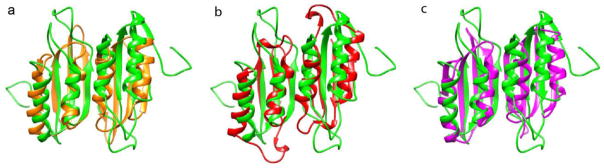
Structural comparisons between the RDTyrH65–159 dimer (green) and three ACT domain dimers: (a) phosphoglycerate dehydrogenase (orange, PDB code 1PSD), (b) aspartate kinase from Arabidopsis (red, PDB code 2CDQ), and (c) E. coli IlvH, the regulatory subunit of acetohydroxyacid synthase (magenta, PDB code 2F1F).
In many enzymes ACT domains bind amino acids as part of allosteric regulation.46 PheH is an allosteric enzyme, and there is evidence for a regulatory site for phenylalanine in the regulatory domain of that enzyme.45 In contrast, TyrH is not an allosteric enzyme, and addition of tyrosine to RDTyrH has no effect on its HSQC spectrum (Tao Huang and Paul F. Fitzpatrick, unpublished results). TyrH appears to have lost this function of its ACT domain during evolution while retaining the structure. This is not the only example of TyrH retaining residual properties of PheH during evolution. Mutagenesis of a single Asp residue in a flexible loop in TyrH can effectively abolish all activity as a TyrH while increasing its residual activity as a PheH.58
Phosphorylation of RDTyrH at Ser40 is a key regulatory mechanism for the enzyme. The effects of phosphorylation at this residue on the NMR spectrum of RDTyrH shows that there is only a very local structural change near the phosphorylation site and no change to the core structure of RDTyrH. Previous analyses of the effects of phosphorylation on the structure of TyrH suggested that the changes in the intact protein are limited to the flexible N-terminal residues.19 These results imply that a large structural rearrangement of the N-terminal domain is not responsible for the effects of phosphorylation. This is consistent with previous studies of the effects of phosphorylation of Ser40 on the dynamic properties of the enzyme,18; 19 which were consistent with phosphorylation only changing the dynamics of residues in the N-terminal 70 residues.
Knowledge of the arrangement of the regulatory and catalytic domains in the intact TyrH is critical for understanding the regulation of the enzyme. There are presently two precedents for the arrangement. The most obvious is the crystal structure of the PheH dimer containing both the regulatory and catalytic domains of that enzyme.59; 60 Figures 7a and b show the tetramer and monomer of a model for intact TyrH generated by using this structure to align the crystal structure of the tetrameric TyrH catalytic and tetramerization domains7 with the NMR-based structure of monomers of the TyrH regulatory domain. In this model, the four regulatory domains do not interact with one another, and the interface of each regulatory domain with its respective catalytic domain involves the β sheet, in contrast to the interface in the regulatory domain dimer. The N-terminal 64 residues of TyrH are not shown in this model, and the structure of the subsequent 11 residues, 64-74, is not well-defined in the NMR-based structure. In PheH the N-terminal residues 19-27 extends across the surface of the catalytic domain and the active site opening. Hydrogen-deuterium exchange analyses of TyrH show that residues at the edge of the active site opening are protected by the regulatory domain,19 supporting a model in which the flexible N-terminus of the regulatory domain in that enzyme similarly extends across the active site.
Figure 7.

Structural models of the combined regulatory, catalytic, and tetramerization domains of TyrH. a, b: Models of the tetramer (a) and monomer (b) derived from the structure of PheH (PDB code 2PHM) by replacing the regulatory domain of PheH with the RDTyrH65–159 monomer (magenta) and the catalytic domain of PheH with the combined catalytic and tetramerization domains of TyrH (PDB code 1TOH, green), respectively. c, d: Models of the tetramer (c) and monomer (d) derived from the model of Jaffe et al.57 by replacing the dimer of the regulatory domain of PheH with the RDTyrH65–159 dimer (red) and the catalytic and tetramerization domains of PheH with the catalytic and tetramerization domains of TyrH (green), respectively. The missing residues connecting the two domains were added with Modeller in Chimera.
The position of loop L4 in RDTyrH is not well-defined in the NMR-based structure (Figure 2). In the model for the intact enzyme shown in Figure 7a, this loop clashes with residues 475-484 of the C-terminal helix in most of the low-energy RDTyrH structures. In PheH, the homologous loop also clashes with a C-terminal helix if a structural model for the intact tetramer is generated by combining the structure of dimer containing the regulatory and catalytic domains but lacking the C-terminal helix8 with the structure of the tetrameric catalytic domain containing the helix but lacking the regulatory domain.61 In PheH this interaction provides a potential mechanism for communication of allosteric effects between subunits. In contrast, TyrH is not an allosteric enzyme, so the benefit of such an interaction is not clear if this structural model is correct.
Recently, Jaffe et al.57 proposed an alternative quaternary structure for PheH, suggesting that the published crystal structure of PheH is of a low activity conformation of the enzyme. This would be consistent with the observation that PheH is effectively inactive until activated by phenylalanine12 or removal of the regulatory domain.62; 63; 64 In this model, formation of the active enzyme involves a 90% rotation of the relative positions of the catalytic and regulatory domains, resulting in the regulatory domains forming side by side ACT domain dimers. This model is supported by the finding that the isolated RDPheH is a dimer in solution, with addition of phenylalanine stabilizing the dimer,45 and by the evidence from hydrogen-deuterium exchange analyses that activation of PheH by phenylalanine results in a large change in the interface between the two domains.65 Figure 7c shows a model for the intact TyrH tetramer derived by superimposing the structures of the isolated catalytic and regulatory domains of TyrH on the model of Jaffe et al. for the active form of the PheH tetramer, and Figure 7d shows the structure of the TyrH monomer in this model. Each regulatory domain is rotated nearly 90 degrees relative to its position in the crystal-based structure, allowing the formation of two regulatory domain dimers. In this structure the extended β sheet of the ACT domain dimer lies over the edge of the tetramerization domain, and loop L4 is far away from the tetramerization helix and exposed in solution, in contrast to its location in the model structure based on the PheH crystal structure (Figure 7b).
The dimeric structure of RDTyrH65-159 described here is clearly more consistent with the structural model in Figure 7c than that in Figure 7a. The regulatory properties of TyrH are substantially different from those of PheH, despite the overall similarity in structure. TyrH is not allosteric, while PheH requires allosteric activation by phenylalanine.12; 14 The regulatory domain of TyrH forms a stable dimer that is unaffected by tyrosine, while phenylalanine stabilizes the dimer of the regulatory domain of PheH.45 H/D exchange analyses have shown that treatment of PheH with phenylalanine causes extensive structural changes in the enzyme, while treatment of TyrH with tyrosine does not.19; 66 A model consistent with these observations is that the quaternary structure of PheH is an equilibrium involving two tetramers, an inactive species with a structure similar to that seen in crystals of protein containing the catalytic and regulatory domains and an active species in which phenylalanine is bound to a dimeric regulatory domain.57 In contrast, TyrH would have a quaternary structure similar to the active form of PheH, with a dimeric regulatory domain. In PheH activation by phenylalanine has been proposed to open up the active site as the change in conformation moves the N-terminus away from the active site.8 In the case of TyrH, phosphorylation of Ser40 results in increased exposure of the active site to solvent, while feedback inhibition by catecholamines results in increased interaction between the N-terminus and the active site.19 Thus, if the quaternary structure of TyrH indeed is better modeled as shown in Figure 7c, the enzyme still retains an interaction between the N-terminus and the active site analogous to that in the unactivated form of PheH.
TyrH and PheH diverged from a common evolutionary ancestor that had already acquired an ACT domain.67; 68 It is not clear whether that ancestral precursor was allosteric or if allostery was acquired by PheH after it diverged from TyrH, in that the allosteric properties of PheH from species distant in evolution have not been determined to a significant extent. However, the ability of ATC domains from various sources to bind amino acids suggests that the common ancestor shared this property.
Conclusions
The present report describes for the first time the three-dimensional structure of the regulatory domain of TyrH, and establishes that it contains an ACT domain. The dimeric structure of the isolated domain contrasts with the monomeric structure of the regulatory domain of PheH seen in the crystal structure of the combined regulatory and catalytic domains of that enzyme., suggesting that the latter may not reflect an active structure.
Materials and Methods
Protein expression and purification
The expression and purification of RDTyrH from rat were as described previously;6 the purification was modified in that a gel filtration step (Superdex 200 10/300 GL) was added after the heparin column. The expression vector for the N-terminal 64-residue deletion mutant RDTyrH65-159 was produced from pETNTERM, the plasmid used to express RDTyrH, using the QuikChange Mutagenesis protocol (Agilent Technologies). The oligonucleotides used as primers were 5′-CTTTAAGAAGGAGATATACATATGCCTGGGAACCCACTGG-3′ and 5′-CTTTAAGAAGGAGATATACATATGCCTGGGAACCCACTGG-3′. RDTyrH65-159 was expressed in E. coli BL21(DE3) and purified using the same protocol as for RDTyrH. The purity of all enzyme preparations was greater than 95% based on polyacrylamide gel electrophoresis in the presence of sodium dodecyl sulfate.
To prepare 15N-labeled or 15N/13C-labeled protein for NMR studies, cells were grown in M9 minimal medium with ampicillin (100 mg/l) and 15NH4CI in the absence or presence of 13C6-glucose for preparations of 15N-labeled or 15N/13C-labeled samples, respectively.69 For completely deuterated 15N-labeled NMR samples, the cells were grown in LB medium made up in 99.99% D2O. After overnight growth at 37 °C, the cells were added in a 1:100 ratio by volume to 5 ml M9 minimal medium in 99.99% D2O with 15NH4CI and D-glucose-1,2,3,4,5,6,6-d7 as the sole nitrogen and carbon sources, respectively. After the cells reached an absorbance of 1.2 at 600 nm (~10 h at 37 °C), they were added to 500 ml M9 minimal medium in 99.99% D2O, containing 15NH4CI and D-glucose-1,2,3,4,5,6,6-d7. All media contained 200 mg/ml ampicillin. After the cells had grown at 37 °C to an absorbance of 0.6 at 600 nm (~8 h), the temperature was decreased to 28 °C and IPTG was added to 100 mg/l. The cells were harvested by centrifugation 18 h later. The yield of purified protein was ~16 mg/l, very close to that obtained in H2O (20 mg/l).
Protein kinase A was purified from beef heart using the protocol of Flockhart and Corbin.70 Phosphorylation of RDTyrH by protein kinase A and purification of the phosphorylated protein were performed as previously described.15
NMR samples
NMR samples were prepared in 50 mM phosphate, 1 μM leupeptin, 1 μM pepstatin A, 5% D2O, at pH 7.0, and were between 0.8 and 1.0 mM. Isotopic heterodimers were prepared by mixing equal amounts of unlabeled and 15N, 2H-labeled RDTyrH65-159 (8 mg each) in ~2 ml 6 M urea for several minutes and then dialyzing this against 1 liter NMR buffer overnight; the sample was then concentrated to 1 mM using AmiconUltra Centrifugal Filters (10,000 MW cutoff). All steps were performed at 4 °C.
Resonance assignments
NMR experiments were carried out at 300 K on Bruker Avance 600- and 700-MHz spectrometers using cryogenically-cooled probes equipped with 13C and 15N decoupling and pulsed field gradient coils. All NMR spectra were processed using NMRPipe71 and analyzed using NMRView.72 Two-dimensional 1H-15N TROSY HSQC and sensitivity-enhanced TROSY-based triple-resonance experiments, including 3D HNCACB,22; 23 HN(CO)CACB,22; 23 HNCA 22; 23 and HN(CO)CA22; 23 were collected to obtain the backbone chemical shift assignments of RDTyrH and phosphorylated RDTyrH. Two-dimensional 1H-15NHSQC and three-dimensional HNCA,25 HNCACB,27 CBCA(CO)NH,28 HNCO,73 HN(CA)CO,74 HBHA-(CO)NH,28 (H)CC(CO)NH,29 and (H)CCH-TOCSY30 experiments were performed to obtain the backbone and side-chain chemical shift assignments of RDTyrH65-159. Three-dimensional 15N- and 13C-edited NOESY-HSQC spectra (mixing time 120 ms) were collected to confirm the chemical shift assignments and generate distance restraints for structure calculations.31;32
Structure calculations
Intramolecular NOE distance restraints were identified through analysis of three-dimensional 15N and 13C-edited NOESY experiments. For the intermolecular NOE distance restraints, two NMR samples were used for 15N-edited NOESY experiments with the same parameters. One was completely deuterated 15N-labeled RDTyrH65-159 and the other the isotopic heterodimers formed from equal amounts of unlabeled and 15N, 2H-labeled RDTyrH65-159. Two sets of the 15N-NOESY experiments, with mixing times of 120 ms and 200 ms, were collected. RDCs were measured using a 15N-labeled sample containing 8 mg ml−1 Pf1 phage (Asla Biotech, Riga, Latvia) by recording gradient-enhanced 2-D 1H-15N IPAP-HSQC experiments.37 Backbone ϕ and ψ restraints were obtained by analysis of the backbone chemical shifts using TALOS.36 The 3JHNα coupling constants were determined from a 3D HNHA spectrum.75
The structure calculations were performed with CNS 1.276 using ARIA 2.335. A single ARIA run consisted of eight iterations and a final water refinement step, all using the standard ARIA/CNS simulated annealing protocol. Forty starting structures were generated based on a linear template molecule with randomly associated velocities for all atoms in each of the first seven iterations, and the 7 lowest energy structures were selected to recalibrate the NOE distance restraints for the next iteration. In the eighth iteration, 100 structures were calculated. The final 10 lowest energy structures were further refined in explicit water.
Backbone 15N relaxation parameters
The backbone 15N relaxation parameters of the spin-lattice relaxation time T1, the spin-spin relaxation time T2, and the steady-state heteronuclear 1H-15N NOE relaxation were determined at 300 K on a 600-MHz spectrometer using a 15N-labeled 0.8 mM NMR sample.77 Two separate data sets were collected. The time delays used for T1 experiments were 16, 160, 320, 480, 640, 800, 1040, 1280 and 1920 ms, and those for T2 experiments were 24, 48, 72, 96, 120, 144, 168, 192, 216 and 240 ms. The T1 and T2 relaxation data were obtained by fitting the individual peak volumes to a two parameter exponential.78 1H-15N NOE values were calculated from peak intensity ratios obtained from spectra with and without 1H saturation prior to the 15N excitation pulse, and errors were derived from the background noise of the spectra.79 The duplicate data sets were merged into a single data set by averaging each value and the corresponding error.
Modeling of the rotation diffusion tensor
The relaxation data for 55 residues were used to determine the rotation diffusion tensor of RDTyrH65-159; 21 residues were excluded as undergoing exchange or high amplitude internal motion on the ns-ps time-scale.80 Amide bond vector orientations were calculated from the 5 lowest energy NMR structures. The parameters describing the diffusion tensor, θ, ϕ, τc and anisotropy were obtained by minimizing the quantity χ2 using the computational strategy described by Tjandra et al.40 The statistical significance of the fit was assessed by repeating the fitting procedure with randomized relaxation data, which removes the correlation between the orientation of the amide bond and the measured relaxation data.40 The modeled diffusion tensor was calculated using the inertial tensors calculated from the coordinates of the 5 lowest energy structures. The axial ratio of the diffusion tensor was then approximated as described by Copie et al.81
Analysis of dynamics
Model-free analysis82; 83 was performed using an axially symmetric diffusion tensor with the program ModelFree4. Five models with increasing complexity (Model1, S2; Model2, S2, τe; Model3, S2, Rex; Model4, S2, τe, Rex; Model5, Sf2, Ss2, τe) were used iteratively to reproduce the experimental data. S2 is the generalized order parameter, Sf2 and Ss2 are the order parameters for fast and slower motions where S2 = Sf2 × Ss2, τe is the effective internal correlation time, and Rex is the slow (μs-ms) motion resulting from chemical exchange. Model selection for each residue was performed using the F test strategy described by Mandel et al.41
Modeling the structure of intact TyrH
Models of the complete tetrameric TyrH structure were based on the pdb file 2TOH, which describes the structure of a tetramer of a truncated form of TyrH that contains only the catalytic and tetramerization domains, lacking residues 1–159. The model based on the structure of the PheH dimer containing both catalytic and regulatory domains (PDB code 2PHM) was obtained by replacing the respective catalytic and regulatory domains of that structure with the tetrameric catalytic domain from pdb file 2TOH and the solution structure of the RDTyrH65-159– monomer. The model based on Jaffe et al.57 was obtained by replacing the dimeric regulatory domain of PheH with that of the RDTyrH65-159 dimer, and the tetrameric catalytic domain with that of TyrH. In both models, the N-terminal 64 residues are missing. Three missing residues linking the domains (Arg160, Glu161, Asp162) were added to each model using Chimera.84 Residues 158–169 were then refined using Modeller,85 and the relative zDOPE scores were used to select among the five structures generated for each model.
Accession codes
The atomic coordinates for RDTyrH65-159 have been deposited in the Protein Data Bank with accession code 2MDA, and the chemical shift assignments for RDTyrH, RDTyrH phosphorylated at Ser40, and RDTyrH65-159 have been deposited in the Biological Magnetic Resonance Bank with accession numbers 19840, 19841 and 19842.
Supplementary data (Figures S1–S6) for this article can be found online.
Supplementary Material
Highlights.
The structure of the regulatory domain of tyrosine hydroxylase was determined by NMR.
The domain consists of an ATC domain of ~90 residues and a flexible N-terminal tail.
The domain forms a side-by-side ATC dimer.
Acknowledgments
The authors thank Dr. Borries Demeler for assisting with analytical ultracentrifugation analyses and Dr. Dmitri Ivanov for helpful discussions. This work was supported by grants from the NIH (GM047291 and GM098140) and The Welch Foundation (AQ-1245).
Footnotes
Abbreviations used: TyrH, tyrosine hydroxylase; PheH, phenylalanine hydroxylase; TrpH, tryptophan hydroxylase; RDTyrH, the regulatory domain of tyrosine hydroxylase, residues 1-159; RDTyrH65-159, the regulatory domain of tyrosine hydroxylase lacking the first 64 residues; HSQC, heteronuclear single quantum coherence; TROSY, transverse relaxation optimized spectroscopy; rmsd, root mean square deviation; RDC, residual dipolar couplings.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Rao F, Zhang K, Zhang L, Rana B, Wessel J, Fung M, Rodriguez-Flores J, Taupenot L, Ziegler M, O’Connor D. Human tyrosine hydroxylase natural allelic variation: Influence on autonomic function and hypertension. Cell Mol Neurobiol. 2010;30:1391–1394. doi: 10.1007/s10571-010-9535-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hoffmann GF, Assmann B, Brautigam C, Dionisi-Vici C, Haussler M, de Klerk JB, Naumann M, Steenbergen-Spanjers GC, Strassburg HM, Wevers RA. Tyrosine hydroxylase deficiency causes progressive encephalopathy and dopa-nonresponsive dystonia. Ann Neurol. 2003;54(Suppl 6):S56–S65. doi: 10.1002/ana.10632. [DOI] [PubMed] [Google Scholar]
- 3.Ludecke B, Dworniczak B, Bartholome K. A point mutation in the tyrosine hydroxylase gene associated with Segawa’s syndrome. Hum Genet. 1995;95:123–125. doi: 10.1007/BF00225091. [DOI] [PubMed] [Google Scholar]
- 4.Fitzpatrick PF. Tetrahydropterin-dependent amino acid hydroxylases. Ann Rev Biochem. 1999;68:355–381. doi: 10.1146/annurev.biochem.68.1.355. [DOI] [PubMed] [Google Scholar]
- 5.Lohse DL, Fitzpatrick PF. Identification of the intersubunit binding region in rat tyrosine hydroxylase. Biochem Biophys Res Commun. 1993;197:1543–1548. doi: 10.1006/bbrc.1993.2653. [DOI] [PubMed] [Google Scholar]
- 6.Daubner SC, Lohse DL, Fitzpatrick PF. Expression and characterization of catalytic and regulatory domains of rat tyrosine hydroxylase. Protein Sci. 1993;2:1452–1460. doi: 10.1002/pro.5560020909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Goodwill KE, Sabatier C, Marks C, Raag R, Fitzpatrick PF, Stevens RC. Crystal structure of tyrosine hydroxylase at 2.3 Å and its implications for inherited neurodegenerative diseases. Nat Struct Biol. 1997;4:578–585. doi: 10.1038/nsb0797-578. [DOI] [PubMed] [Google Scholar]
- 8.Kobe B, Jennings IG, House CM, Michell BJ, Goodwill KE, Santarsiero BD, Stevens RC, Cotton RGH, Kemp BE. Structural basis of autoregulation of phenylalanine hydroxylase. Nat Struct Biol. 1999;6:442–448. doi: 10.1038/8247. [DOI] [PubMed] [Google Scholar]
- 9.Wang L, Erlandsen H, Haavik J, Knappskog PM, Stevens RC. Three-dimensional structure of human tryptophan hydroxylase and its implications for the biosynthesis of the neurotransmitters serotonin and melatonin. Biochemistry. 2002;41:12569–12574. doi: 10.1021/bi026561f. [DOI] [PubMed] [Google Scholar]
- 10.Fitzpatrick PF. Mechanism of aromatic amino acid hydroxylation. Biochemistry. 2003;42:14083–14091. doi: 10.1021/bi035656u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xia T, Gray DW, Shiman R. Regulation of rat liver phenylalanine hydroxylase. III Control of catalysis by (6R)-tetrahydrobiopterin and phenylalanine. J Biol Chem. 1994;269:24657–24665. [PubMed] [Google Scholar]
- 12.Fitzpatrick PF. Allosteric regulation of phenylalanine hydroxylase. Arch Biochem Biophys. 2012;519:194–201. doi: 10.1016/j.abb.2011.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Haycock JW. Phosphorylation of tyrosine hydroxylase in situ at serine 8, 19, 31, and 40. J Biol Chem. 1990;265:11682–11691. [PubMed] [Google Scholar]
- 14.Daubner SC, Le T, Wang S. Tyrosine hydroxylase and regulation of dopamine synthesis. Arch Biochem Biophys. 2011;508:1–12. doi: 10.1016/j.abb.2010.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ramsey AJ, Fitzpatrick PF. Effects of phosphorylation of serine 40 of tyrosine hydroxylase on binding of catecholamines: Evidence for a novel regulatory mechanism. Biochemistry. 1998;37:8980–8986. doi: 10.1021/bi980582l. [DOI] [PubMed] [Google Scholar]
- 16.Bevilaqua LRM, Graham ME, Dunkley PR, von Nagy-Felsobuki EI, Dickson PW. Phosphorylation of Ser19 alters the conformation of tyrosine hydroxylase to increase the rate of phosphorylation of Ser40. J Biol Chem. 2001;276:40411–40416. doi: 10.1074/jbc.M105280200. [DOI] [PubMed] [Google Scholar]
- 17.Toska K, Kleppe R, Armstrong CG, Morrice NA, Cohen P, Haavik J. Regulation of tyrosine hydroxylase by stress-activated protein kinases. J Neurochem. 2002;83:775–783. doi: 10.1046/j.1471-4159.2002.01172.x. [DOI] [PubMed] [Google Scholar]
- 18.Wang S, Lasagna M, Daubner SC, Reinhart GD, Fitzpatrick PF. Fluorescence spectroscopy as a probe of the effect of phosphorylation at Ser40 of tyrosine hydroxylase on the conformation of its regulatory domain. Biochemistry. 2011;50:2364–2370. doi: 10.1021/bi101844p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang S, Sura GR, Dangott LJ, Fitzpatrick PF. Identification by hydrogen/deuterium exchange of structural changes in tyrosine hydroxylase associated with regulation. Biochemistry. 2009;48:4972–4979. doi: 10.1021/bi9004254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McCulloch RI, Fitzpatrick PF. Limited proteolysis of tyrosine hydroxylase identifies residues 33-50 as conformationally sensitive to phosphorylation state and dopamine binding. Arch Biochem Biophys. 1999;367:143–145. doi: 10.1006/abbi.1999.1259. [DOI] [PubMed] [Google Scholar]
- 21.Ramsey AJ, Fitzpatrick PF. Effects of phosphorylation on binding of catecholamines to tyrosine hydroxylase: specificity and thermodynamics. Biochemistry. 2000;39:773–778. doi: 10.1021/bi991901r. [DOI] [PubMed] [Google Scholar]
- 22.Loria JP, Rance M, Palmer AG. Transverse-relaxation-optimized (TROSY) gradient-enhanced triple-resonance NMR spectroscopy. J Magn Reson. 1999;141:180–184. doi: 10.1006/jmre.1999.1891. [DOI] [PubMed] [Google Scholar]
- 23.Salzmann M, Wider G, Pervushin K, Senn H, Wuthrich K. TROSY-type triple-resonance experiments for sequential NMR assignments of large proteins. J Am Chem Soc. 1999;121:844–848. [Google Scholar]
- 24.Eghbalnia HR, Wang L, Bahrami A, Assadi A, Markley JL. Protein energetic conformational analysis from NMR chemical shifts (PECAN) and its use in determining secondary structural elements. J Biomol NMR. 2005;32:71–81. doi: 10.1007/s10858-005-5705-1. [DOI] [PubMed] [Google Scholar]
- 25.Yamazaki T, Lee W, Revington M, Mattiello DL, Dahlquist FW, Arrowsmith CH, Kay LE. An HNCA pulse scheme for the backbone assignment of 15N,13C,2H-labeled proteins: Application to a 37-kDa Trp repressor-DNA complex. J Am Chem Soc. 1994;116:6464–6465. [Google Scholar]
- 26.Bax A, Ikura M. An efficient 3D NMR technique for correlating the proton and 15N backbone amide resonances with the α-carbon of the preceding residue in uniformly 15N/13C enriched proteins. J Biomol NMR. 1991;1:99–104. doi: 10.1007/BF01874573. [DOI] [PubMed] [Google Scholar]
- 27.Wittekind M, Mueller L. HNCACB, a high-sensitivity 3D NMR experiment to correlate amide-proton and nitrogen resonances with the alpha-and beta-Carbon resonances in proteins. J Magn Reson, Ser B. 1993;101:201–205. [Google Scholar]
- 28.Grzesiek S, Bax A. Amino acid type determination in the sequential assignment procedure of uniformly 13C/15N-enriched proteins. J Biomol NMR. 1993;3:185–204. doi: 10.1007/BF00178261. [DOI] [PubMed] [Google Scholar]
- 29.Grzesiek S, Anglister J, Bax A. Correlation of backbone amide and aliphatic side-chain resonances in C-13/N-15-enriched proteins by isotropic mixing of C-13 magnetization. J Magn Reson, Ser B. 1993;101:114–119. [Google Scholar]
- 30.Kay LE, Xu GY, Singer AU, Muhandiram DR, Formankay JD. A Gradient-Enhanced HCCH TOCSY Experiment for Recording Side-Chain H-1 and C-13 Correlations in H2O Samples of Proteins. J Magn Reson, Ser B. 1993;101:333–337. [Google Scholar]
- 31.Muhandiram DR, Farrow NA, Xu GY, Smallcombe SH, Kay LE. A gradient 13C NOESY-HSQC experiment for recording NOESY spectra of 13C-labeled proteins dissolved in H2O. J Magn Reson, Ser B. 1993;102:317–321. [Google Scholar]
- 32.Zwahlen C, Legault P, Vincent SJF, Greenblatt J, Konrat R, Kay LE. Methods for measurement of intermolecular NOEs by multinuclear NMR spectroscopy: Application to a bacteriophage λ N-peptide/boxB RNA complex. J Am Chem Soc. 1997;119:6711–6721. [Google Scholar]
- 33.Walters KJ, Matsuo H, Wagner G. A simple method to distinguish intermonomer nuclear Overhauser effects in homodimeric proteins with C2 symmetry. J Am Chem Soc. 1997;119:5958–5959. [Google Scholar]
- 34.Ivanov D, Stone JR, Maki JL, Collins T, Wagner G. Mammalian SCAN domain dimer is a domain-swapped homolog of the HIV capsid C-terminal domain. Mol Cell. 2005;17:137–43. doi: 10.1016/j.molcel.2004.12.015. [DOI] [PubMed] [Google Scholar]
- 35.Linge JP, O’Donoghue SI, Nilges M. Automated assignment of ambiguous nuclear Overhauser effects with ARIA. Methods Enzymol. 2001;339:71–90. doi: 10.1016/s0076-6879(01)39310-2. [DOI] [PubMed] [Google Scholar]
- 36.Cornilescu G, Delaglio F, Bax A. Protein backbone angle restraints from searching a database for chemical shift and sequence homology. J Biomol NMR. 1999;13:289–302. doi: 10.1023/a:1008392405740. [DOI] [PubMed] [Google Scholar]
- 37.Ottiger M, Delaglio F, Bax A. Measurement of J and dipolar couplings from simplified two-dimensional NMR spectra. J Magn Reson. 1998;131:373–378. doi: 10.1006/jmre.1998.1361. [DOI] [PubMed] [Google Scholar]
- 38.Bax A, Kontaxis G, Tjandra N. Dipolar couplings in macromolecular structure determination. Methods Enzymol. 2001;339:127–174. doi: 10.1016/s0076-6879(01)39313-8. [DOI] [PubMed] [Google Scholar]
- 39.Laskowski RA, Rullmannn JA, MacArthur MW, Kaptein R, Thornton JM. AQUA and PROCHECK-NMR: Programs for checking the quality of protein structures solved by NMR. J Biomol NMR. 1996;8:477–86. doi: 10.1007/BF00228148. [DOI] [PubMed] [Google Scholar]
- 40.Tjandra N, Feller SE, Pastor RW, Bax A. Rotational diffusion anisotropy of human ubiquitin from N-15 NMR relaxation. J Am Chem Soc. 1995;117:12562–12566. [Google Scholar]
- 41.Mandel AM, Akke M, Palmer AG. Backbone Dynamics of Escherichia coli Ribonuclease Hi - Correlations with Structure and Function in an Active Enzyme. J Mol Biol. 1995;246:144–163. doi: 10.1006/jmbi.1994.0073. [DOI] [PubMed] [Google Scholar]
- 42.Ota A, Yoshida S, Nagatsu T. Deletion Mutagenesis of Human Tyrosine Hydroxylase Type 1 Regulatory Domain. Biochem Biophys Res Commun. 1995;213:1099–1106. doi: 10.1006/bbrc.1995.2240. [DOI] [PubMed] [Google Scholar]
- 43.Daubner SC, Piper MM. Deletion mutants of tyrosine hydroxylase identify a region critical for heparin binding. Protein Sci. 1995;4:538–541. doi: 10.1002/pro.5560040320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Alieva I, Mustafayeva N, Gojayev N. Conformation analysis of the N-terminal sequence Met1-Val60 of the tyrosine hydroxylase. J Mol Struct. 2006;785:76–84. [Google Scholar]
- 45.Li J, Ilangovan U, Daubner SC, Hinck AP, Fitzpatrick PF. Direct evidence for a phenylalanine site in the regulatory domain of phenylalanine hydroxylase. Arch Biochem Biophys. 2011;505:250–255. doi: 10.1016/j.abb.2010.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Grant GA. The ACT domain: a small molecule binding domain and its role as a common regulatory element. J Biol Chem. 2006;281:33825–33829. doi: 10.1074/jbc.R600024200. [DOI] [PubMed] [Google Scholar]
- 47.Chipman DM, Shaanan B. The ACT domain family. Curr Opin Struct Biol. 2001;11:694–700. doi: 10.1016/s0959-440x(01)00272-x. [DOI] [PubMed] [Google Scholar]
- 48.Aravind L, Koonin EV. Gleaning non-trivial structural, functional and evolutionary information about proteins by iterative database searches. J Mol Biol. 1999;287:1023–1040. doi: 10.1006/jmbi.1999.2653. [DOI] [PubMed] [Google Scholar]
- 49.Finn RD, Mistry J, Tate J, Coggill P, Heger A, Pollington JE, Gavin OL, Gunasekaran P, Ceric G, Forslund K, Holm L, Sonnhammer ELL, Eddy SR, Bateman A. The Pfam protein families database. Nucleic Acids Res. 2010;38:D211–D222. doi: 10.1093/nar/gkp985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sträter N, Schnappauf G, Braus G, Lipscomb WN. Mechanisms of catalysis and allosteric regulation of yeast chorismate mutase from crystal structures. Structure. 1997;5:1437–1452. doi: 10.1016/s0969-2126(97)00294-3. [DOI] [PubMed] [Google Scholar]
- 51.Al-Rabiee R, Zhang Y, Grant GA. The mechanism of velocity modulated allosteric regulation in D-3-phosphoglycerate dehydrogenase: Site-directed mutagenesis of effector binding site residues. J Biol Chem. 1996;271:23235–23238. doi: 10.1074/jbc.271.38.23235. [DOI] [PubMed] [Google Scholar]
- 52.Schuller DJ, Grant GA, Banaszak LJ. The allosteric ligand site in the Vmax-type cooperative enzyme phosphoglycerate dehydrogenase. Nat Struct Biol. 1995;2:69–76. doi: 10.1038/nsb0195-69. [DOI] [PubMed] [Google Scholar]
- 53.Mas-Droux C, Curien G, Robert-Genthon M, Laurencin M, Ferrer JL, Dumas R. A novel organization of ACT domains in allosteric enzymes revealed by the crystal structure of Arabidopsis aspartate kinase. The Plant Cell. 2006;18:1681–1692. doi: 10.1105/tpc.105.040451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Leonard PM, Smits SHJ, Sedelnikova SE, Brinkman AB, de Vos WM, van der Oost J, Rice DW, Rafferty JB. Crystal structure of the Lrp-like transcriptional regulator from the archaeon Pyrococcus furiosus. EMBO J. 2001;20:990–997. doi: 10.1093/emboj/20.5.990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Devedjiev Y, Surendranath Y, Derewenda U, Gabrys A, Cooper DR, Zhang R-g, Lezondra L, Joachimiak A, Derewenda ZS. The structure and ligand binding properties of the B. subtilis YkoF gene product, a member of a novel family of thiamin/HMP-binding proteins. J Mol Biol. 2004;343:395–406. doi: 10.1016/j.jmb.2004.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kaplun A, Vyazmensky M, Zherdev Y, Belenky I, Slutzker A, Mendel S, Barak Z, Chipman DM, Shaanan B. Structure of the regulatory subunit of acetohydroxyacid synthase isozyme III from Escherichia coli. J Mol Biol. 2006;357:951–63. doi: 10.1016/j.jmb.2005.12.077. [DOI] [PubMed] [Google Scholar]
- 57.Jaffe EK, Stith L, Lawrence SH, Andrake M, Dunbrack RL., Jr A new model for allosteric regulation of phenylalanine hydroxylase: Implications for disease and therapeutics. Arch Biochem Biophys. 2013;530:73–82. doi: 10.1016/j.abb.2012.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Daubner SC, Avila A, Bailey JO, Barrera D, Bermudez JY, Giles DH, Khan CA, Shaheen N, Thompson JW, Vasquez J, Oxley SP, Fitzpatrick PF. Mutagenesis of a specificity-determining residue in tyrosine hydroxylase establishes that the enzyme is a robust phenylalanine hydroxylase but a fragile tyrosine hydroxylase. Biochemistry. 2013;52:1446–1455. doi: 10.1021/bi400031n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Solstad T, Stokka AJ, Andersen OA, Flatmark T. Studies on the regulatory properties of the pterin cofactor and dopamine bound at the active site of human phenylalanine hydroxylase. Eur J Biochem. 2003;270:981–990. doi: 10.1046/j.1432-1033.2003.03471.x. [DOI] [PubMed] [Google Scholar]
- 60.Erlandsen H, Patch MG, Gamez A, Straub M, Stevens RC. Structural Studies on Phenylalanine Hydroxylase and Implications Toward Understanding and Treating Phenylketonuria. Pediatrics. 2003;112:1557–1565. [PubMed] [Google Scholar]
- 61.Fusetti F, Erlandsen H, Flatmark T, Stevens RC. Structure of tetrametic human phenylalanine hydroxylase and its implications for phenylketonuria. J Biol Chem. 1998;273:16962–16967. doi: 10.1074/jbc.273.27.16962. [DOI] [PubMed] [Google Scholar]
- 62.Fisher DB, Kaufman S. The stimulation of rat liver phenylalanine hydroxylase by lysolecithin and α-chymotrypsin. J Biol Chem. 1973;248:4345–4353. [PubMed] [Google Scholar]
- 63.Knappskog PM, Flatmark T, Aarden JM, Haavik J, Martinez A. Structure/function relationships in human phenylalanine hydroxylase. Effect of terminal deletions on the oligomerization, activation and cooperativity of substrate binding to the enzyme. Eur J Biochem. 1996;242:813–821. doi: 10.1111/j.1432-1033.1996.0813r.x. [DOI] [PubMed] [Google Scholar]
- 64.Daubner SC, Hillas PJ, Fitzpatrick PF. Expression and characterization of the catalytic domain of human phenylalanine hydroxylase. Arch Biochem Biophys. 1997;348:295–302. doi: 10.1006/abbi.1997.0435. [DOI] [PubMed] [Google Scholar]
- 65.Li J, Dangott LJ, Fitzpatrick PF. Regulation of phenylalanine hydroxylase: Conformational changes upon phenylalanine binding detected by hydrogen/deuterium exchange and mass spectrometry. Biochemistry. 2010;49:3327–3335. doi: 10.1021/bi1001294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wang S. Ph D. Texas A&M University; 2010. Identification of Structural Changes Related to the Regulation of Tyrosine Hydroxylase. [Google Scholar]
- 67.Cao J, Shi F, Liu X, Huang G, Zhou M. Phylogenetic analysis and evolution of aromatic amino acid hydroxylase. FEBS Lett. 2010;584:4775–82. doi: 10.1016/j.febslet.2010.11.005. [DOI] [PubMed] [Google Scholar]
- 68.Siltberg-Liberles J, Steen IH, Svebak RM, Martinez A. The phylogeny of the aromatic amino acid hydroxylases revisited by characterizing phenylalanine hydroxylase from Dictyostelium discoideum. Gene. 2008;427:86–92. doi: 10.1016/j.gene.2008.09.005. [DOI] [PubMed] [Google Scholar]
- 69.Marley J, Lu M, Bracken C. A method for efficient isotopic labeling of recombinant proteins. J Biomol NMR. 2001;20:71–5. doi: 10.1023/a:1011254402785. [DOI] [PubMed] [Google Scholar]
- 70.Flockhart DA, Corbin JD. Preparation of the catalytic subunit of cAMP-dependent protein kinase. In: Maranos PJ, Campbell IC, Cohen RM, editors. Brain Receptor Methodologies, Part A. Academic Press; New York: 1984. pp. 209–215. [Google Scholar]
- 71.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:277–93. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 72.Johnson BA. Using NMRView to visualize and analyze the NMR spectra of macromolecules. Methods Mol Biol. 2004;278:313–52. doi: 10.1385/1-59259-809-9:313. [DOI] [PubMed] [Google Scholar]
- 73.Kay LE, Ikura M, Tschudin R, Bax A. Three-dimensional triple-resonance NMR spectroscopy of isotopically enriched proteins. J Magn Reson. 1990;89:496–514. doi: 10.1016/j.jmr.2011.09.004. [DOI] [PubMed] [Google Scholar]
- 74.Clubb RT, Thanabal V, Wagner G. A constant-time three-dimensional triple-resonance pulse scheme to correlate intraresidue 1HN, 15N, and 13C′ chemical shifts in 15N-13C-labelled proteins. J Magn Reson. 1992;97:213–217. [Google Scholar]
- 75.Vuister GW, Bax A. Measurement of four-bond HN-H alpha J-couplings in staphylococcal nuclease. J Biomol NMR. 1994;4:193–200. doi: 10.1007/BF00175247. [DOI] [PubMed] [Google Scholar]
- 76.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr. 1998;54:905–21. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 77.Kay LE, Nicholson LK, Delaglio F, Bax A, Torchia DA. Pulse sequences for removal of the effects of cross-correlation between dipolar and chemical-shift anisotropy relaxation mechanism on the measurement of heteronuclear T1 and T2 values in proteins. J Magn Reson. 1992;97:359–375. [Google Scholar]
- 78.Fushman D, Cahill S, Cowburn D. The main-chain dynamics of the dynamin pleckstrin homology (PH) domain in solution: Analysis of N-15 relaxation with monomer/dimer equilibration. J Mol Biol. 1997;266:173–194. doi: 10.1006/jmbi.1996.0771. [DOI] [PubMed] [Google Scholar]
- 79.Freedberg DI, Ishima R, Jacob J, Wang YX, Kustanovich I, Louis JM, Torchia DA. Rapid structural fluctuations of the free HIV protease flaps in solution: relationship to crystal structures and comparison with predictions of dynamics calculations. Protein Sci. 2002;11:221–32. doi: 10.1110/ps.33202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Barbato G, Ikura M, Kay LE, Pastor RW, Bax A. Backbone dynamics of calmodulin studied by N-15 relaxation using inverse detected two-dimensional NMR-spectroscopy - the central helix is flexible. Biochemistry. 1992;31:5269–5278. doi: 10.1021/bi00138a005. [DOI] [PubMed] [Google Scholar]
- 81.Copie V, Tomita Y, Akiyama SK, Aota S, Yamada KM, Venable RM, Pastor RW, Krueger S, Torchia DA. Solution structure and dynamics of linked cell attachment modules of mouse fibronectin containing the RGD and synergy regions: Comparison with the human fibronectin crystal structure. J Mol Biol. 1998;277:663–682. doi: 10.1006/jmbi.1998.1616. [DOI] [PubMed] [Google Scholar]
- 82.Lipari G, Szabo A. Model-free approach to the interpretation of nuclear magnetic resonance relaxation in macromolecules. 1 Theory and range of validity. J Am Chem Soc. 1982;104:4546–4559. [Google Scholar]
- 83.Clore GM, Szabo A, Bax A, Kay LE, Driscoll PC, Gronenborn AM. Deviations from the simple two-parameter model-free approach to the interpretation of nitrogen-15 nuclear magnetic relaxation of proteins. J Am Chem Soc. 1990;112:4989–4991. [Google Scholar]
- 84.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera-A visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 85.Fiser A, Do RKG, Šali A. Modeling of loops in protein structures. Protein Sci. 2000;9:1753–1773. doi: 10.1110/ps.9.9.1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.