

Abstract
Following acute infection, herpes simplex virus (HSV) establishes latency in sensory neurons, from which it can reactivate and cause recurrent disease. Available antiviral therapies do not affect latent viral genomes; therefore, they do not prevent reactivation following therapy cessation. One possible curative approach involves the introduction of DNA double strand breaks in latent HSV genomes by rare-cutting endonucleases, leading to mutagenesis of essential viral genes. We tested this approach in an in vitro HSV latency model using the engineered homing endonuclease (HE) HSV1m5, which recognizes a sequence in the HSV-1 gene UL19, encoding the virion protein VP5. Coexpression of the 3′-exonuclease Trex2 with HEs increased HE-mediated mutagenesis frequencies up to sixfold. Following HSV1m5/Trex2 delivery with adeno-associated viral (AAV) vectors, the target site was mutated in latent HSV genomes with no detectable cell toxicity. Importantly, HSV production by latently infected cells after reactivation was decreased after HSV1m5/Trex2 exposure. Exposure to histone deacetylase inhibitors prior to HSV1m5/Trex2 treatment increased mutagenesis frequencies of latent HSV genomes another two- to fivefold, suggesting that chromatin modification may be a useful adjunct to gene-targeting approaches. These results support the continuing development of HEs and other nucleases (ZFNs, TALENs, CRISPRs) for cure of chronic viral infections.
Keywords: antiviral, endonuclease, mutagenesis
Introduction
Herpes simplex virus type-1 and -2 (HSV-1 and HSV-2) are neurotropic alphaherpesviruses belonging to the Herpesviridae family along with varicella-zoster virus. HSV-1 has a seroprevalence exceeding 50% in the USA, can cause both oral and genital ulceration, and is the most common etiology of infectious blindness (keratitis) and viral encephalitis.1 HSV-2 has a US seropositivity of 16%,2 and is the leading cause of genital ulcers worldwide. Both HSV-1 and HSV-2 cause severe and potentially fatal infections in newborns and immunocompromised hosts.3,4 In addition, HSV-2 is a key risk factor for HIV-1 acquisition and transmission.5,6
After primary infection at mucosal surfaces, HSV-1 and HSV-2 establish lifelong latency in sensory neurons of the trigeminal ganglia and/or dorsal root ganglia, from which they can later reactivate, causing recurrent lesions. While antiviral therapy with acyclovir or related drugs diminishes the severity of primary or recurrent disease, and can reduce the frequency of asymptomatic HSV shedding and ulceration, it does not eliminate latent infection7 or reduce the HSV-associated risk of HIV transmission.8 Additionally, despite much effort, an effective vaccine for HSV remains elusive.9 Therefore, there is a need for new therapeutic approaches that target the virus in its latent state and hence would cure HSV infection.
Homing endonucleases (HEs, also referred to as meganucleases) are highly specific rare-cutting endonucleases that recognize, bind, and cleave large DNA sequences (usually >14 bp), thus introducing DNA double strand breaks (DSB). In their natural hosts, HEs are present within mobile genetic elements and promote self-propagation by targeted recombination, DNA DSB repair and gene conversion of their cognate target recognition sites.10 Due to their intrinsic properties, HEs are being developed as tools for targeted gene modifications.10 Targeted mutagenesis by HE is achieved by the induction of DNA DSB at specific target sequences, which stimulates the error-prone nonhomologous end joining cellular repair mechanism. We have previously suggested that rare-cutting endonucleases might be used in a new therapeutic approach to latent viral infections, in which targeted mutagenesis of essential viral genes would disable viral genomes and render the virus incapable of replication or reactivation from latency.11 HEs in particular offer certain advantages, including their high specificity of DNA cleavage due to the length of their target site (on average 20 bp), and their small size, which facilitates vectorization and delivery.11 We have validated this approach in a culture model of HIV latency,12 and Grosse et al.13 recently showed that expression of HSV-specific HEs could inhibit active HSV-1 replication at low and moderate multiplicities of infection in cultured cells.
In this report, we used an in vitro model of HSV latency and an HSV1-specific HE to study targeted mutagenesis as an approach to disable latent virus. Our results demonstrate that mutations can be introduced in the latent HSV genome by HE expressed in combination with the 3′-5′ exonuclease Trex2. Furthermore, latently infected cells subjected to HE-mediated mutagenesis show decreased HSV production after reactivation. Finally, the mutational frequency is significantly increased by treatment of HSV-infected cells with inhibitors of chromatin modification known to have an impact on epigenetic control of the HSV genome. Taken together, these data demonstrate the potential to use HE-mediated mutagenesis as a therapeutic approach to cure HSV-infected individuals.
Results
In vitro model for HSV latency/reactivation
During latency, HSV genomes are maintained as circular episomal DNA. Only latency-associated transcripts are expressed, in most but not all latently infected cells.14 In order to test whether targeted mutagenesis could be achieved in latent episomal HSV genomes, we modified a previously described in vitro model of HSV latency and reactivation.15 Latent HSV infection was established in primary human fibroblasts (HF) in the presence of interferon-α (IFN-α) and acyclovir (Figure 1a) using the green flourescent protein (GFP)-expressing HSV-1 FΔUs5 virus (Figure 1b) at a multiplicity of infection (MOI) of 2.5. After an initial burst of viral replication detected at 1 day postinfection (dpi) by GFP expression, the presence of virions in culture supernatant, and robust immediate early (IE) and late (L) gene expression, the virus established a latent infection. Latency in this model was characterized by a lack of GFP expression, no substantial IE and L gene expression, no production of progeny virions, and low levels of latency-associated transcripts expression (Figure 1c,d and Supplementary Figure S1). HSV-1 could be reactivated from latently infected fibroblasts by infection with human cytomegalovirus (HCMV). This resulted in the accumulation of GFP, robust IE, and L gene expression and production of progeny virus in the culture supernatant (Figure 1c,d and Supplementary Figure S1). These data suggested that this in vitro model of HSV latency is a reasonable surrogate for latent HSV during in vivo infection.
Figure 1.

In vitro model of herpes simplex virus (HSV) latency. (a) Timeline of the establishment of HSV latency in human fibroblasts (HF) and human cytomegalovirus (HCMV) reactivation of latent HSV. ACV, acyclovir; IFN, interferon. (b) Schematic representation of the HSV-1 FΔUs5 genome. The long terminal and internal repeats (TRL and IRL), and the internal and terminal short repeats (TRS and IRS) bordering the unique long (UL) and unique short (US) regions are shown. A green flourescent protein (GFP) expression cassette is inserted in the Us5 gene.42 The location of the target sequence recognized by the HSV-specific HE (HSV1m5) in the UL19 gene is indicated. (c) GFP expression as assessed by fluorescence microscopy at 1, 8, 11, and 13 days postinfection with FΔUs5. At day 11, cells were infected with HCMV AD169 (multiplicity of infection (MOI) = 1) to reactivate latent HSV (×10 magnification). *Indicates sample analyzed after HSV reactivation. Presence of progeny virus in the cell culture supernatant is indicated on the right (PFU: Plaque forming unit; +++ indicates ≥ 100 PFU/ml; ++ indicates ≥ 10 PFU/ml; - indicates no virus detected). (d) Reverse transcription polymerase chain reaction (RT-PCR) products for the immediate early gene UL54 encoding ICP27, late gene UL27 encoding glycoprotein B (gB) and latency-associated transcript (LAT) were generated from total RNA extracted at 1, 8, 11, 13 days postinfection with FΔUs5 and resolved on a 2% agarose gel. Reverse transcriptase (RT) was either added (+) or omitted (−). bp, base pair; mw, molecular weight size marker. *Indicates sample analyzed after HSV reactivation.
Optimization of targeted mutagenesis efficiency and enzyme delivery
We used an HSV1-specific nuclease, HSV1m5, which was engineered from I-CreI.16 This enzyme has a single target recognition site of 24 bp in the HSV-1 genome located at the 3′ end of the UL19 gene, which encodes the major capsid component VP5, an essential viral protein (Figure 1b).13 In addition to using an HSV1-specific nuclease, we added the 3′-5′ exonuclease Trex2, which cleaves 3′ overhangs generated by HE-induced DNA DSB.17,18 Previous work has shown that combining HE with Trex2 increases the frequency of targeted mutagenesis in transformed and primary cells.18,19 We tested the utility of Trex2 in enhancing HE-mediated mutagenesis by comparing HE-mediated sequence editing efficiencies in the absence or presence of Trex2 in reporter TERT-immortalized primary HF. In the absence of Trex2, the frequency of mutation was low (5%), even after 12 days of exposure to HE. However, the mutagenesis frequency was increased up to sixfold when Trex2 was delivered concomitantly with the HE (Supplementary Figure S2 and Supplementary Table S1). We therefore chose to use HSV1-specific HE in combination with Trex2 in subsequent experiments.
To deliver our DNA modifying enzymes, we investigated the use of adeno-associated virus. Self-complementary adeno-associated virus (scAAV) constructs were generated for the delivery and expression of HSV1m5, Trex2, GFP, and the control HE NV1, which targets a sequence present in the human genome but not the HSV viral genome (Figure 2a). AAV serotype-2 was found to be the most efficient serotype for the transduction of primary human fibroblasts (Supplementary Figure S3). The AAV delivery vectors could reproducibly transduce at least 40% of fibroblasts latently infected with HSV, a substantially higher proportion than we observed with lentivirus vectors (data not shown), and thus we chose AAV for enzyme delivery in subsequent experiments.
Figure 2.

Lack of homing endonuclease (HE)-induced toxicity. (a) Schematic representation of scAAV vectors constructs for transgene delivery. CMV, cytomegalovirus early promoter; ITR, inverted terminal repeat. Human fibroblasts (HF) transduced for 4 days with scAAV2 delivery vectors expressing the indicated protein (GFP, NV1, or HSV1m5 in the presence or absence of Trex2, or Trex2 alone) at a multiplicity of infection (MOI) of 1 × 102, 1 × 103, or 1 × 104 vgs/cells per vector or untransduced (no AAV) were subjected to flow cytometry analysis following staining with either (b) live/dead stain or (c) anti-activated caspase-3 antibody. Untransduced control cells were treated with 0.5 µmol/l staurosporine (STS) 24 hours prior to analysis to provide a positive control for death and apoptosis. The insert graph shows the levels of transduction in control cells transduced with either scAAV2-GFP only or scAAV2-GFP and scAAV2-Trex2 at the indicated MOI. GFP fluorescence in control cells was measured by flow cytometry.
Lack of toxicity in HE-exposed primary human fibroblasts
A search for potential off-target sites in the human genome revealed no human sequences with two or fewer mismatches to the HSV1m5 target site, only 66 sequences with three to five mismatches, and 570 sequences with six mismatches (Supplementary Figure S4). Therefore, we predicted that expression of HSV1m5 in human cells would have no overt cellular toxicity or cell death due to induction of DNA DSB in the cellular genome. To evaluate whether exposure of cells to HSV1m5, Trex2, or the combination of HSV1m5 plus Trex2 would result in toxicity, we measured cell death by two different assays in uninfected primary HF exposed to transgene delivery by AAV. As expected, exposure to increasing AAV delivery vector doses led to increased transgene expression (Figure 2b,c). However, there was no increase in the level of dead (Figure 2b) or apoptotic (Figure 2c) cells after exposure to HE, Trex2, or the combination of HE plus Trex2 compared to cells expressing GFP alone. Thus, our cultured cells tolerate expression of these enzymes well, allowing the use of this model to evaluate their utility against latent HSV genomes.
Gene disruption of latent viral genomes following exposure to HSV1-specific HE and Trex2
To test whether our targeted mutagenesis approach could disable HSV genomes in latent reservoirs, cells with latent HSV infection were exposed to the HSV1-specific HE, HSV1m5, in combination with Trex2 by AAV transduction (Figure 3a). Control latently-infected cells were transduced with either scAAV2-GFP alone or in combination with scAAV2-Trex2 to show that transgene expression was sustained for the duration of the experiment and detected in more than 40% of the cells (Figure 3b).
Figure 3.
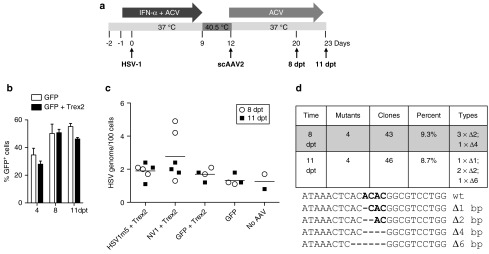
Targeted mutagenesis of latent herpes simplex virus (HSV) genomes. (a) Timeline of HSV latency establishment in human fibroblasts (HF) and exposure to scAAV2 delivery vectors. HF were plated (105/well), pretreated with ACV (30 µmol/l) and IFN-α (200 U/ml), infected with HSV-1 FΔUs5 at a MOI of 2.5 and maintained in the presence of drugs for 9 days with daily replenishment. The drugs were removed and the incubation temperature increased to 40.5 °C to maintain HSV latency. HSV-infected HFs were then transduced with scAAV2 expressing Trex2 and either HSV-1m2, NV1, or GFP at a MOI of 104 genomes/cell each and maintained in culture medium with ACV for either 8 or 11 additional days. ACV, acyclovir; IFN, interferon. (b) Assessment of transduction levels with scAAV vectors by flow cytometry for GFP at 4, 8, and 11 days posttransduction in duplicate dishes of HSV-latently infected HF exposed to scAAV2-GFP alone or in combination with scAAV2-Trex2. Mean + SD. (c) Quantitation of HSV genomes in latently-infected HF exposed to scAAV2 vectors by ddPCR in triplicate dishes at 8 (open circle) or 11 (filled circle) days posttransduction. The bars represent the mean of the combined six dishes for each condition. (d) At 8 and 11 days posttransduction, the presence of mutations at the target site of HSV1m5 in HSV genomes from HSV1m5-/Trex2-treated cells was analyzed by polymerase chain reaction (PCR) amplicon sequencing. The HSV region containing the target site was PCR amplified from total genomic DNA obtained from triplicate dishes of HSV-latently infected HF treated with HSV1m5 and Trex2. The PCR amplicons were pooled, cloned and sequenced from individual bacterial colonies. The percent and type of mutations found are presented. Wt, wild-type sequence of the HSV1m5 target site. Bold indicates the four nucleotides constituting the 3′ overhangs generated by the DNA DSB; - indicates a deleted nucleotide.
One potential outcome after exposure to HSV1-specific HE and the introduction of DNA DSB would be the degradation of the linearized episomal HSV genomes. Therefore, we first determined whether the exposure of HSV-latently infected cells to HSV1m5 and Trex2 led to a decrease in the number of HSV genomes. Quantification of HSV DNA by droplet digital polymerase chain reaction (ddPCR) in total cellular DNA extracted at 8 and 11 dpt showed no significant difference between any of the treatment groups or times of analysis (Figure 3c, P = 0.2 for both treatment and time). These data suggested that exposure to HSV1m5 and Trex2 did not result in a detectable degradation of latent HSV genomes.
A second potential outcome after induction of DNA DSB in episomal HSV genomes would be repair via the nonhomologous end joining pathway, which would be expected to lead to insertions and/or deletions at the HSV1m5 target site. Since no detectable loss of HSV genomes in HSV1m5/Trex2 exposed cells was observed by 8 or 11 dpt, we performed PCR amplicon sequencing of the region containing the HSV1m5 target sequence. Latent HSV genomes from cells treated with HSV1m5 and Trex2 revealed deletions of 1 to 6 bp in 9.3 and 8.7% of the PCR amplicons obtained from cells transduced for 8 and 11 days, respectively (Figure 3d). Thus, we conclude that expression of HSV1m5 plus Trex2 in cells carrying latent episomal HSV genomes results in targeted mutagenesis of viral sequences, but no detectable reduction in the quantity of HSV genomes.
Latently infected cells exposed to HE show decreased virus production after reactivation
Next, we determined whether exposure of latently infected cells to HSV1m5 and Trex2 had any effect on HSV reactivation. Primary HF latently infected with HSV were subjected to targeted mutagenesis and then reactivated 8 days later by infection with HCMV (Figure 4a). Following reactivation from latency, cells treated with HSV1m5/Trex2 showed an absence of virion in culture supernatant (Figure 4b; P = 0.015) and a trend toward having lower numbers of intracellular HSV genomes (Figure 4c; P = 0.14) compared to control cells that were either exposed to NV1/Trex2 or left unexposed (no enzyme). There was no statistical difference between NV1/Trex2-treated cells and the cells left unexposed (Figure 4c; P = 0.3). Both the absence of virion in culture supernatant (P < 0.001) and lower numbers of intracellular HSV genomes (P = 0.014) after HSV1m5/Trex2 treatment were confirmed in a second independent experiment (data not shown). PCR amplicon sequencing of the target site in the HSV genomes from HSV1m5-/Trex2-treated cells showed that 21% of the sequences contained deletions ranging from 1 to 13 bp (Figure 4d).
Figure 4.

Impact of homing endonuclease (HE)-mediated mutagenesis on herpes simplex virus (HSV) reactivation. (a) Timeline of HSV latency establishment in human fibroblasts (HF), exposure to scAAV2 delivery vectors and reactivation following CMV infection. Triplicate dishes of latently-infected HF transduced at a multiplicity of infection (MOI) of 104 genomes/cell/vector with scAAV2 delivery vectors expressing the indicated transgene were reactivated at day 20 (8 dpt) by infection with HCMV AD169 at a MOI of 1 for 2 days in the absence of ACV. At 2 days postinfection with human cytomegalovirus (HCMV) (day 22; 10 dpt). ACV, acyclovir; IFN, interferon. (b) The presence of virion progeny in culture supernatants was assessed by plaque assay titration and (c) the number of intracellular HSV genomes was measured by ddPCR. Bars indicate the mean from the triplicate samples. (d) The HSV region containing the target site was PCR amplified from total genomic DNA obtained from triplicate dishes of reactivated HSV infected HF treated with HSV1m5 and Trex2. The PCR amplicons were pooled, cloned, and sequenced from individual bacterial colonies. The nature and number of the mutations found are indicated on the right side of each sequence. Δ: deletion; wt, wild-type sequence of the HSV1m5 target site. Bold indicates the four nucleotides constituting the 3′ overhang generated by the DNA DSB; - indicates a deleted nucleotide.
Impact of epigenetic modification of latent HSV genomes on HE-mediated targeted mutagenesis
Several recent reports have shown that the efficiency of HE-mediated genome editing can be influenced by epigenetic modifications that dictate chromatin structure.20,21 During HSV latency, epigenetic modifications have been implicated in repression of viral gene expression. The latent HSV genome is associated with histones bearing modifications characteristic of transcriptionally inactive DNA or heterochromatin, which can confer a compact structure to the DNA.22,23
To investigate the effect of epigenetic modifications of the latent viral genome on HE-mediated mutagenesis, we used the replication-deficient HSV-1 mutant virus d106, which is deleted for four genes encoding the immediate early protein ICP4, 22, 27, and 47 but retains ICP0 and expresses GFP under the CMV promoter from the locus of the ICP27 gene (UL 54). GFP expression is used as a surrogate marker for HSV gene expression.24 Repressive chromatin marks on the HSV d106 genome are detected after 24 hours following infection of primary human fibroblasts such as MRC5 cells, and the extent of the modifications can be modulated using the histone deacetylase inhibitor (HDACi) trichostatin-A (TSA), which decreases viral genome repression. In contrast, treatment with IFN-α has been shown to increase repression of the viral genome.25
To determine the impact of TSA or IFN-α on AAV transduction and transgene expression, we analyzed control cells infected with d106 in the absence or presence of either TSA or IFN-α and subsequently transduced with both Trex2- and mCherry-expressing AAVs (Figure 5a,b). Exposure of the cells to either TSA or IFN-α during HSV infection had no effect on the efficiency of the subsequent AAV transduction, since a similar percentage of cells expressed mCherry fluorescence in these controls (Figure 5c). In contrast, drug treatment resulted in the expected increase (TSA) or decrease (IFN-α) of GFP expression from the HSV genomes compared to untreated cells (Figure 5c).
Figure 5.

Impact of epigenetic modification of the herpes simplex virus (HSV) genome on homing endonuclease (HE)-mediated mutagenesis. (a) Schematic of experimental timeline. MRC5 cells left untreated or drug-treated with IFN-α (200 U/ml) or trichostatin-A (TSA; 100 nmol/l), infected with recombinant HSV-1 d106 at a multiplicity of infection (MOI) of 0.5 were transduced with scAAV2-HSV1m5 and scAAV2-Trex2 or scAAV2-mCherry and scAAV2-Trex2 at a MOI of 5 × 104 genomes/cell/vector for 2 days in the absence of drug. (b) Schematic representation of the scAAV delivery vectors used. CMV, cytomegalovirus early promoter; ITR, inverted terminal repeat. (c) Fluorescence microscopy analysis of GFP (HSV d106 gene expression; left column) and mCherry (AAV transduction; right column) in control d106-infected MRC5 at a MOI of 5 × 104 genomes/cell/vector for 2 days. Percent of cells expressing GFP and mCherry fluorescence was also quantified by flow cytometry (percent of positive cells indicated in the upper left corner of each panel).
To evaluate the impact of TSA or IFN-α treatment on HE-mediated targeted mutagenesis of HSV, we first used the Surveyor nuclease assay (Figure 6a). Exposure to either HSV1m5 or Trex2 alone did not lead to detectable target site mutagenesis, regardless of treatment with IFN-α or TSA. In contrast, mutagenesis was readily detected when HSV1m5 and Trex2 were coexpressed, in agreement with our previous results. The degree of mutagenesis as determined by the Surveyor assay was increased approximately twofold by the inclusion of TSA, which decreases viral genome repression. In contrast, IFN-α, which increases viral genome repression, completely eliminated detectable mutagenic events. These results were supported by PCR amplicon sequencing, with a twofold increase in mutation frequency observed when HSV1m5-/Trex2-exposed cells were treated with TSA compared to untreated cells (16.5% compared to 8%, respectively), while no mutagenesis was detected for HSV1m5-/Trex2-exposed cells treated with IFN-α (Figure 6b).
Figure 6.

Mutational events in herpes simplex virus (HSV)-infected human fibroblasts (HF) treated with trichostatin-A (TSA) or interferon (IFN)-α. (a) Polymerase chain reaction (PCR) amplicons of the HSV region containing the HSV1m5 target site were treated with Surveyor nuclease, and digested products were separated on a 3% agarose gel and quantified using ImageJ. mw: molecular weight size ladder. The percent of sequence modification detected by this assay is indicated. (b) HSV1m5 target sequence analysis. The HSV region containing the HSV1m5 target site was PCR amplified from total genomic DNA obtained at 2 days post-AAV transduction. PCR amplicons were cloned and sequenced from individual bacterial colonies. The frequency and nature of the mutations found are presented. N/A, not applicable, Δ: deletion; wt, wild-type target sequence. Bold indicates the four nucleotides constituting the 3′ overhang generated by the DNA DSB.
An increase in the frequency of HE-targeted mutagenesis efficiency was also seen in a similar experiment using a panel of different HDACi to treat d106-infected cells prior to exposure with HSV1m5/Trex2 (Figure 7a). None of these drugs had a noticeable adverse effect on the subsequent levels of AAV transduction (Figure 7b). However, depending on the drug used a two- to fivefold increase in the percentage of mutations was observed at the HSV1m5 target site in HDACi-treated cells when compared to control (no drug) cells following HSV1m5/Trex2 exposure (Figure 7c). These data suggest that efficiency of targeted mutagenesis of HSV genomes can be increased if HSV-infected cells are treated with HDACi, and that TSA is the most efficient of the HDACi tested in our system.
Figure 7.
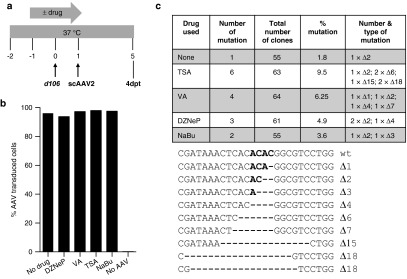
Targeted mutagenesis in herpes simplex virus (HSV)-infected cells treated with histone deacetylase inhibitor (HDACi). (a) Schematic of experimental timeline. MRC5 cells were infected with recombinant HSV-1 d106 at a multiplicity of infection (MOI) of 0.5 in the absence or presence of drug, transduced with scAAV2 at a MOI of 5 × 104 genomes/cell/vector in the absence of drug for 4 days prior to analysis for mutational events. (b) Flow cytometry for green flourescent protein (GFP) fluorescence in control uninfected MRC5 cells cultured in the absence (no drug) or presence of the indicated HDACi for 2 days, then either left untransduced (no AAV) or transduced with scAAV2-GFP and scAAV2-Trex2 at a MOI of 5 × 104 genomes/cell/vector for 4 days in the absence of drug. (c) HSV1m5 target sequence analysis. The HSV region containing the HSV1m5 target site was PCR amplified using total genomic DNA obtained at 4 days post-AAV transduction from d106-infected MRC5 exposed to HSV1m5 and Trex2 in the absence (none) or presence of the indicated HDACi. PCR amplicons were cloned and sequenced from individual bacterial colonies. The percent and nature of mutations in the HSV1m5 target site are shown. wt, wild-type sequence of the HSV1m5 target site. Bold indicates the four nucleotides constituting the 3′ overhang generated by the DNA DSB. Δ: deletion. HDACi used: trichostatin A (TSA 100 nmol/l), valproic acid (VA 1 mmol/l), 3-deazaneplanocin (DZNep 5 µmol/l), sodium butyrate (NaBu 5 mmol/l).
Discussion
The goal of this study was to demonstrate targeted mutagenesis of latent HSV genomes using HSV-specific rare-cutting endonucleases. Previously, Grosse et al.13 showed that expression of HSV-specific HEs in cultured cells could inhibit subsequent HSV-1 infection at low and moderate multiplicities of infection, but the impact of HSV-specific HE against established latent infection was not studied. The series of experiments presented in this report provide proof-of-concept that HSV-specific HE can mediate mutagenesis of latent HSV to disable the virus and limit subsequent reactivation.
The studies presented here benefited from an in vitro model of HSV latency and reactivation that was easy to establish and from which sample size was not a limiting factor. However, it was established in a non-neuronal cell type by repression of viral replication using antiviral drug treatment, and virus reactivation required induction. Establishment of virus latency and reactivation in this model therefore likely do not fully reflect in vivo infection. Despite these limitations, however, the model reproduced the main characteristic described for latent HSV in infected neurons, specifically the persistence of HSV genomes without detectable gene expression other than latency-associated transcripts, with subsequent reactivation and production of infectious virus (Figure 1). HSV1m5 was chosen from four HSV-specific HEs in a proprietary collection of engineered endonucleases for its higher cleavage activity, measured in an in vitro assay of targeted mutagenesis, compared to the other HSV-specific HEs (data not shown). This was supported by the observations made by Grosse et al.13 for HSV1m2, a variant form of HSV1m5. HSV1m5 targets VP5, a late viral gene encoding a structural protein. Targeting a late protein may have a lower impact than targeting an immediate early gene such as UL54 (encoding ICP27) or α4 (ICP4), which would exert antiviral effects at an earlier stage of the replication cycle. However, if only a fraction of viral genomes can be mutated, targeting a structural protein may be advantageous. Transdominant-negative mutant forms of the protein could be generated, preventing the virus from completing its replication cycle and/or forming infectious virus in reactivated cells when a mixture of wild-type and mutant proteins are being expressed.
The use of rare-cutting endonucleases for targeted mutagenesis relies on the error-prone nature of the nonhomologous end joining repair pathway. In this study, we showed that exposure to HSV-specific HE resulted in the introduction of mutations within the target site in the latent HSV genome. Specifically, the exposure of cells latently infected with HSV to the HE HSV1m5, which targets a unique site in the UL19 gene, in combination with the 3′ to 5′ exonuclease Trex2, resulted in deletions ranging from 1 to 13 bp at the targeted site (Figure 3 and Figure 4). Trex2, a 3′ to 5′ exonuclease, was coexpressed with HSV1m5, since several groups have now shown that the efficiency of HE-targeted mutagenesis can be greatly increased in the presence of Trex2 (Supplementary Figure S2 and Supplementary Table S1),18,19 which cleaves the 3′-overhangs resulting from HE cleavage of DNA. Removal of the 3′-overhangs before the cellular DNA repair machinery can accurately religate the DNA ends prevents precise repair and favors mutagenesis.
An alternative outcome after the introduction of a DNA DSB within an episomal DNA target (such as latent HSV) would be elimination/degradation of the linearized episome rather than repair and mutagenesis. However, in all the experiments performed for this study, no detectable loss of viral genomes was observed following exposure to the HE (Figure 3 and data not shown). In vivo, HSV establishes latency in neurons, which are terminally differentiated non-dividing cells. The experiments presented here were also performed in nondividing cells, and thus we predict that exposure of HSV-latent genomes to HSV-specific HE in neurons may preferentially result in mutagenesis rather than the loss of viral genomes.
An important adjunct to mutagenesis of latent HSV genomes was the observation that exposure to HE plus Trex2 greatly reduced viral reactivation from latently infected cells. HE/Trex2-exposed cells that were subsequently treated to induce viral reactivation had reduced levels of HSV genomes compared to controls, and produced no detectable progeny virus. In these cells 21% of the HSV genomes harbored a mutation at the site targeted by the HSV1m5 nuclease. This could account at least in part for the observed impact on viral replication following reactivation; however, there are still a substantial number of viral genomes with wild type sequence. As previously suggested by Grosse et al.,13 the impact of HE on virus replication could be the consequence of three nonexclusive mechanisms. First, mutations in an essential gene could prevent protein function and thus efficient viral replication. Second, the introduction of DNA DSB in the viral genome could lead to DNA degradation. Finally, cleavage of the viral genome could interfere with viral DNA replication, which for HSV occurs from the circular genome by a rolling circle mechanism.14 The complete absence of progeny virus production in our experiments may suggest that this latter effect is especially effective against HSV, perhaps because the newly synthesized viral genomes are particularly susceptible to HE attack.
A significant concern in the use of rare-cutting endonucleases as a therapeutic tool is the possibility of toxicity. The HSV-specific HE used in this study was selected among a collection of engineered endonucleases derived from the I-CreI nuclease16 for its ability to cleave a DNA sequence in the HSV-1 genome. As this enzyme was engineered to specifically recognize a chosen HSV-1 sequence, no extensive off-target cleavage analysis was performed. Only a small number of off-target sites with three to six mismatches were found in the human genome (Supplementary Figure S4). Nevertheless, this enzyme might be more likely to show toxicity due to off-target activity than would more refined enzymes. In any event, our assays did not reveal overt toxicity, although we cannot completely eliminate the potential for off-target activity by this enzyme, especially after prolonged expression. Furthermore, none of the transduced cells presented any phenotypic features characteristic of cell transformation in the time frame of our study (data not shown). Some vector-associated toxicity was observed at the higher AAV doses, which appeared to be related not to the enzyme activity but rather to the unpurified nature of the AAV stocks used in this study (see Materials and Methods). Future uses of nucleases for antiviral therapy will require an absence of toxicity due to off-target cleavage, and substantial recent effort has gone toward HE re-engineering strategies to generate efficacious and safe enzymes for use during in vivo therapy.16,26,27,28,29
Another potential concern regarding toxicity relates to the overexpression of Trex2. DNA DSB occur naturally in most cells, especially in mitotic cells, and some have questioned whether Trex2 might have deleterious effects on repair of these breaks. In this study, we detected no overt toxicity in cells in which Trex2 was overexpressed in the absence or presence of HE (Figure 2). Other groups have also evaluated the potential toxicity of Trex2, and reported no effects on the cell cycle or growth kinetics, and no increase in mutagenesis at off-target sites.17,18 Indeed, it was found that Trex2 expression decreased distal end-joining, suggesting that it may also reduce the susceptibility for deleterious translocations, and thus actually increase the safety of targeted gene modification.17,18
A critical issue for the use of HE as an antiviral agent will be to ensure effective enzyme delivery to the target cell. For this study, we used scAAV vectors because they allowed us to reproducibly transduce primary fibroblasts that have been maintained in culture for at least a week and therefore are no longer actively dividing. In our model system, the direct detection of HSV1m5 was not practical because the limited packaging capacity of our scAAV vectors precluded coexpression of HSV1m5 together with fluorescent protein. Therefore, fluorescent proteins in control cells were used as surrogate markers for evaluating the levels of transduction and transgene expression. In a separate study, we are currently modeling the relationship between HE efficacy and nuclease delivery and expression in the context of latent HSV infection as previously done for hepatitis B virus infection.30
AAV may also be well suited as an HE delivery vector for in vivo studies. AAV is only weakly immunogenic, and there are many different serotypes that can be used to transduce multiple cell types. AAV can infect neurons, can reach sensory neuronal cell bodies by anterograde transport along the axon, and has been successfully used for gene delivery to neurons of the PNS.31,32 Several recent reports have shown that AAV vectors can infect neurons in the dorsal root ganglia and brain, with sustained long-term transgene expression.32,33 We used two separate AAV vectors for delivering the HE and Trex2 in this work, which may not be optimal for in vivo studies. Recently, we have generated new delivery AAV vectors with shorter promoter sequences, which allows the expression of both enzymes from a single scAAV vector. In this study, we showed that HE-mediated mutagenesis of viral sequences could be achieved after delivery of the HSV-specific HE with AAV vectors, supporting the continued development of this approach. Alternatively, defective HSV vectors could be used in the event that AAV vectors do not transduce the same subpopulation of neurons latently infected by HSV. These HSV recombinant vectors have been widely used in the development of gene transfer therapeutics for the treatment of several diseases, including brain tumors and chronic pain (reviewed in ref. 34,35).
For almost any application using HEs or other rare-cutting endonucleases, it would be desirable to have the greatest efficiency of DNA cleavage. After exposure to HSV1m5/Trex2, the frequency of mutation at the target site in the HSV genome remained low (9%) despite sustained expression of the HE in 50% of the cells over an 8- to 11-day period (Figure 3). In mice, viral burden as defined by HSV copy number and the number of infected cells appears to be linked to the rate of recurrence.36,37 In immunocompetent individuals, HSV latency is established in only a small percentage of sensory neurons, ranging from 2.0 to 10.5% for HSV-1, and the number of HSV genome copies in each latently infected neuron is modest (2–50 copies/cell for HSV-1).38,39 There are ~10,000–20,000 neuronal cell bodies per ganglia, and only a few ganglia are at risk for infection. Thus, the total number of latently infected cells to target is low, and these cells are anatomically localized. Therefore, even incomplete elimination of latent genomes within infected neurons might reduce disease severity and viral shedding, or even lead to the functional cure of HSV infection.
During latency, the HSV genome is chromatinized, and the associated histones harbor modifications characteristic of transcriptionally inactive DNA or heterochromatin. These modifications confer a closed conformation to the DNA that can impede access by transcription factors and/or activators and may promote binding of transcriptional repressors leading to repression of gene expression.23 This closed DNA conformation may also limit access of the HE to its target sequence and reduce the efficiency of HSV-specific HE-mediated mutagenesis. When HSV is reactivated from latency, the heterochromatin marks on the viral genome are replaced with marks characteristics of transcriptionally active DNA. In vitro, HDACi such as TSA can reactivate latent HSV.40 In our study, when HSV-infected cells were subjected to TSA or other HDACi treatments prior to exposure to the HSV-specific HE, the frequency of targeted mutagenesis was increased by at least twofold. These data suggest that while targeted mutagenesis of latent HSV DNA can be achieved in our in vitro model of HSV latency, it may be limited due to the state of the latent viral genome, which makes the target sequence relatively inaccessible to the HE. Accessibility to the HE appears to be improved by treatment with HDACi. In fact, our experimental system using fibroblasts probably represents a particularly rigorous test for HE efficacy, since in neurons the viral genome is not as repressed as in latently infected primary fibroblasts.25 Furthermore, recent studies from Wald and colleagues demonstrated that HSV-1 or HSV-2 seropositive healthy individuals have high frequency short subclinical HSV reactivation, suggesting that HSV may reactivate more frequently than previously reported.41 Therefore, virus genomes may be more accessible to endonuclease in vivo than during our in vitro experiments.
Chronic viral infections such as HSV have long been considered incurable. Any hope of cure for these infections must by definition target the long-lived, latent form of the virus. Our work here demonstrates that the latent form of HSV can be effectively targeted by HEs introduced into cells using AAV, and that this results in mutagenesis of latent HSV episomes and elimination of viral reactivation. These results support the continued development of targeted endonucleases as a potential curative therapy. In vivo studies to determine the efficacy and tolerability of this approach are therefore critically needed.
Materials and methods
Cells. HEK 293T, HEp-2, and Vero cell lines (American Type Culture Collection), primary human foreskin fibroblasts (HF), human embryonic lung fibroblasts MRC5 (provided by Dr A. Cent) and, TERT-immortalized primary human fibroblasts (TERT-HF, obtained from Dr D. Galloway Fred Hutchinson Cancer Research Center, Seattle, WA) were propagated in Dulbecco's modified Eagle medium supplemented with 10% fetal bovine serum. F06, a Vero-derived cell line expressing ICP4 and ICP27 was obtained from Dr J.A. Blaho and propagated as previously described.24 T30BS and reporter TERT-HF bulk cell lines were established after transduction of HEp-2 and TERT-HF cells, respectively, with the lentiviral reporter vector pRRL.SFFV.I-AniI-RS-GFP, followed by fluorescence-activated cell sorting of the GFP-positive cell population.12
Herpesviruses. FΔUs5 was derived from HSV-1 strain F with the ORF coding for GFP driven by the CMV promoter inserted in the Us5 gene encoding glycoprotein J (gJ)42 and was propagated on Vero cells. The IE mutant d106 was kindly provided by Dr J.A. Blaho and originally obtained from Dr N. DeLuca, and was propagated on F06 cells as previously described.24 This recombinant HSV-1 expresses only the immediate early gene ICP0, is defective for the four other IE genes and has a transgene construct consisting of the GFP gene driven under the HCMV promoter in the UL54 locus. HCMV strain AD169 was provided by Dr A. Geballe (Fred Hutchinson Cancer Research Center), and was propagated and titered on primary human fibroblasts.
Lentiviral vectors. pRRL.SFFV.I-AniI-RS-GFP, a lentiviral reporter vector, was previously described in ref. 12. The lentiviral vectors pCVL.SFFV.(Y2 or E148D) I-AniI.T2A.Trex2.T2A.mCherry were generated by PCR amplification of (Y2 or E148D)I-AniI.T2A.Trex2 from pCVL.SFFV. (Y2 or E148D) I-AniI.T2A.Trex2.IRES.mTagBFP18 using primers pXhoI-atgHANLSF CCGCTCGAGGCCGCCACCATGGGATATC AND pBglII-Trex2L CGCGAAGATCCTGGCTTCGAGGCTTGGAC; followed by cloning into the XhoI and BamHI sites of pCVL.SFFV.MCS2.T2A.mCherry. VSV-G-pseudotyped lentiviral vectors were produced and titered as described previously.12
scAAV vectors. pscAAV-CMV-HSV1m5, pscAAV-CMV-NV1, and pscAAV-CMV-Trex2 were generated by PCR amplification of HSV1m5 or NV1 using primers KpnI-HA-nls GAGATCGGTACCGCCGCCACCATGGGATATCCATACGATGT and XhoI-HSV1m5 GAGATCCTCGAGTCAAGGAGAGGACTTTTTCTTCTCAGAGA and Trex2 using primers KpnI-Trex2 GAGATCGGTACCGCCGCCACCATGTCTGAGCCACCTCGGGCTGAGACCTTTGTA and XhoI-Trex2 GAGATCCTCGAGTCAGGCTTCGAGGCTTGGACCATCAGGTGGCAC. Each PCR amplicon was cloned into pscAAV-CMV-pA as a XhoI-NotI fragment. To make the plasmid pscAAV-CMV-pA, the CMV-pA cassette from the plasmid pShuttle-CMV43 was PCR amplified using the primers AscI-CMVF GAGATCGGCGCGCCCCTGCAGGTACTGTAATAGTAATCAATTACGGGGTCAT and NaeI-SV40R GAGATCGGCCGGCCGCGTTAAGATACATTGATGAGTTTGGACAA, digested with AscI/NaeI and then cloned into the AscI/SnaBI sites of the plasmid pscAAV-GFP.44 scAAV viruses were generated by transient transfection of HEK 293T cells using PEI according to the method of Choi et al.45 Briefly, HEK 293T cells were transfected with a scAAV vector, a plasmid that expresses the AAV capsid protein (pRepCap) from AAV serotype-2 and a helper plasmid that expresses adenovirus helper proteins (pHelper). At 72 hours posttransfection, crude AAV lysates were made by collecting and resuspending the cells in AAV lysis buffer (50 mmol/l Tris, 150 mmol/l NaCl, pH 8.5) before freeze-thawing four times. AAV stocks were filtered through a 0.45µm filter and stored at −80 °C. Low-level cellular toxicity was occasionally observed with these crude AAV stocks when used at high MOI.
Cell transduction with lentiviral vectors. For HEp-2- and TERT-HF-derived cell lines, 2 × 105 cells/well were plated in 12-well plates and either left to adhere for 12–16 hours before being transduced or immediately transduced with the indicated lentivirus construct in 1 ml culture medium containing 4–8 µg/ml polybrene. The next day, the transduction medium was replaced with fresh culture medium and cells were incubated for the indicated time. If the incubation period was longer than 5 days, cells were passaged and diluted into new 12-well plates.
In vitro HSV latency. HSV-latently infected primary HFs were established as described by Wigdahl et al.15 Briefly, 5 × 105 cells/dish were plated in 60 mm dishes, and pretreated for 24 hours with 200 U/ml IFN-α (PBL Interferon Source; PBL, Piscataway, NJ) and 30 µg/ml acyclovir (Sigma, St Louis, MO), before infection at a MOI of 2.5 with FΔUs5 virus in the presence of IFN-α and acyclovir. Culture medium was replenished daily with drugs for 8–10 days until establishment of latency. Then, the cells were incubated at 40.5 °C in culture medium without drugs to maintain latent infection. Establishment of latency was followed by microscopy analysis for the loss of GFP fluorescence and plaque assay for the absence of progeny virus in culture supernatant. Viral latency was indicated by the absence of viral cytopathic morphologies, loss of GFP expression and lack of progeny virus in the culture supernatants as assessed by plaque assay titration on Vero cells. Prior to transduction with HE-expressing viral vectors, the supernatants from HSV-latently infected cells were transferred to Vero cell monolayers to confirm that HSV was quiescent with no viral reactivation and production of infectious virus.
Plaque assay titration. HSV stocks or culture supernatants either undiluted or serially diluted were applied to monolayers of Vero cells grown in six-well plate. After 3 days incubation at 37 °C, plaques were counted.
HSV reactivation. Triplicate dishes of HSV-infected HF either left untransduced or transduced with indicated scAAV2 delivery vectors were infected with HCMV AD169 at a MOI of 1–2 for 2 days at 37 °C in culture medium without acyclovir. HSV reactivation was assessed by one or more of the following methods: flow cytometry analysis for GFP fluorescence, HSV genome quantification using ddPCR, and plaque assay titration. In this latter method, viral plaques observed are the result of infectious HSV replication and not due to HCMV, which only replicates in human fibroblasts.
DNA analysis. Genomic DNA (gDNA) was extracted using the DNeasy Tissue & Blood kit (Qiagen, Valencia, CA) after collection of the cells by trypsinization. Fifty nanogram of gDNA and Platinum Pfx DNA polymerase (Invitrogen, Carlsbad, CA) were used to PCR amplify the region containing the I-AniI target site with primers against SFFV 5′ GGAAGCTTGCCAAACAGGATATCTGCGGTGAGC-3′ and TurboGFP 5′-GACTAGTCGGGTAGGTGCCGAAGTGGTAGAAGC-3′, and PCR program: 94 °C 2 minutes, 35 cycles (94 °C 15 seconds, 60 °C 15 seconds, 68 °C 30 seconds), and then 68 °C 10 minutes. The region of the HSV genome containing the target site for HSV1m5 was PCR amplified using UL19 B primers: forward 5′-GGCCGGCGGAAGTAGTTGAC-3′ and reverse 5′-CACCGACATGGGCAACCTTC-3′, with thermocycling conditions of: 94 °C 5 minutes, 35 cycles (94 °C 30 seconds, 60 °C 30 seconds, 70 °C 30 seconds), then 70 °C 5 minutes.
PCR amplicon sequencing. PCR amplicons containing the target region obtained as described above were cleaned using the QIAquick PCR purification kit (Invitrogen) and cloned into the PCR Blunt-TOPO vector using the Zero Blunt TOPO PCR cloning kit (Invitrogen). For Y2 I-AniI target sequence analysis, plasmid DNA was isolated using the FastPlasmid mini kit (5 Prime) and sequenced as described previously.12 For HSV1m5 target sequence analysis, bacterial colony sequencing was performed by Genewiz (Seattle, WA). In the experiments described in Figure 3e and Figure 4d, PCR amplicons were obtained from triplicate dishes, purified and combined prior to TOPO blunt cloning.
Quantification of cellular HSV DNA by ddPCR. A duplex digital PCR was performed to measure the levels of cellular HSV DNA on the QX100 droplet digital PCR system (Bio-Rad Laboratories, Hercules, CA) at the University of Washington, Department of Laboratory Medicine, Molecular Virology Laboratory. DNA samples were extracted from cells as described above. Copies of HSV DNA were quantified using the following gB primers/probe set: HSV-qPCR-F CCGTCAGCACCTTCATCGA; HSV-qPCR-R CGCTGGACCTCCGTGTAGTC and probe HSVgbProbe 6FAM-CCACGAGATCAAGGACAGCGGCC-BHQ1. Cell numbers were determined using the following beta-globin primer/probe set: BETAU 5′-TGAAGGCTCATGGCAAGAAA-3′, BETAL 5′-GCTCACTCAGTGTGGCAAAGG-3′, and probe BetaPb 5′-Hex-TCCAGGTGAGCCAGGCCATCACTA-BHQ1-3′. Beta-globin is a reference gene for cell count that exists at two copies/cell.
The ddPCR reaction mixture consisted of 12.5 µl of a 2X ddPCR Mastermix (Bio-Rad), 1.25 µl of each 20X primer-probe mix, and 10 µl of template DNA in a final volume of 25 µl. 20 µl of each reaction mixture was loaded onto a disposable plastic cartridge (Bio-Rad) with 70 µl of droplet generation oil (Bio-Rad) and placed in the droplet generator (Bio-Rad). The droplets generated from each sample were transferred to a 96-well PCR plate, and PCR amplification was performed on a 2720 Thermal Cycler (Applied Biosystems, Carlsbad, CA) with the following conditions: 95 °C for 10 minutes, 40 cycles of 94 °C for 30 seconds, and 60 °C for 1 minute, followed by 98 °C for 10 minutes and ending at 4 °C. After amplification, the plate was loaded onto the droplet reader (Bio-Rad), and the droplets from each well of the plate were automatically read at a rate of 32 wells/hour. Data were analyzed with QuantaSoft analysis software (Biorad, Hercules, CA) and quantitation of target molecules presented as copies/µl of PCR reaction. For quantification of both cellular and supernatant HSV levels, values were standardized to cellular β-globin levels.
Surveyor nuclease analysis. The surveyor nuclease assay was performed using the Surveyor Mutation Detection kit (Transgenomic, Omaha, NE) according to the manufacturer's instructions. PCR amplification of the region containing the HSV1m5 target site in HSV genomes was done using the UL19B forward and reverse primers as described above. Digested PCR products were resolved in a 3% agarose gel, and quantified using ImageJ software (National Institute of Health, Bethesda, MD) to determine the levels of gene disruption. The level of gene disruption was calculated using the formula: 100 X (1-(1-fraction cleaved)1/2) where fraction cleaved = the density of cleavage product/(density of cleavage product + density of uncleaved product).
Reverse transcription polymerase chain reaction (RT-PCR) detection of HSV transcripts. RNA was extracted using QIAshredder and RNeasy kit (Qiagen) per the manufacturer's recommendations. RT-PCR was performed on 1 µg total RNA using the Quantitec RT kit (Qiagen) after treatment with DNase I to remove potential viral DNA contamination. The primers and conditions for RT-PCR have been described previously.46 No reverse transcriptase (-RT) controls were also performed (Figure 1d).
Toxicity assays. After collection by trypsinization, cells were stained either for viability/cytotoxicity using the live/dead stain (Life technologies, Grand Island, NY) as per manufacturer recommendations or apoptosis using FITC anti-activated caspase-3 antibody (BD Biosciences San Jose, CA) as previously described47 and then analyzed by flow cytometry on a BD LSRII flow cytometer.
Flow cytometry. Expression of fluorescent proteins (GFP and mCherry) was analyzed using a BD LSRII flow cytometer after collection of the cells by trypsinization. To analyze GFP fluorescence in TERT-HF, and T30BS reporter cell lines, cells were treated with 1 µmol/l MG132 (Calbiochem, La Jolla, CA) for 6 hours, collected by trypsinization and immediately analyzed. Cell sorting for GFP fluorescence to establish the TERT-HF and T30BS reporter cell lines was performed using a BD FACSVantage. All data were analyzed using FlowJo software (TreeStar, Ashland, OR).
Microscopy analysis. Images were acquired using a microscope (TE2000; Nikon, Melville, NY) equipped with a CCD camera (CoolSNAP ES; Photometrics) and analyzed using MetaVue (Universal Imaging, Downingtown, PA). Images were taken with a ×10 objective.
Inhibitors. Cells were treated with either TSA (100 nmol/l), sodium butyrate (5 mmol/l), valproic acid (1 mmol/l; Sigma), or 3-deazaneplanocin A (5 µmol/l; Cayman chemical, Ann Arbor, MI).
Statistical analysis. Multiple sample comparisons (Figure 3d) were done with two-way analysis of variance (Prism V5.0d). Multiple sample comparisons (Figure 4b,c) were made with two-way nonparametric analysis of variance using permutation tests and the Bray-Curtis dissimilarity measure on ranked data (PERMANOVA6).48
SUPPLEMENTARY MATERIAL Figure S1. GFP fluorescence in HSV-infected HF. Figure S2. HE-mediated mutagenesis in the presence or absence of Trex2 in primary and transformed human reporter cells. Figure S3. Identification of the optimal AAV serotype for efficient transduction of primary human fibroblasts. Figure S4. HSV1m5 off-target sites in the human genome. Table S1. Targeted mutagenesis in primary and transformed cells.
Acknowledgments
Portions of this work were supported by National Institutes of Health (NIH) grant U19-AI 096111. Additional funding was from the US NIH (RL1-HL092553-05 PL1-HL092557-05) in support of the Northwest Genome Engineering Consortium. A.M.S. receives salary and equity compensation as Chief Science Officer for Cellectis Therapeutics. All the other authors declared no conflict of interest.
Supplementary material
References
- Schiffer JT, Corey L. New concepts in understanding genital herpes. Curr Infect Dis Rep. 2009;11:457–464. doi: 10.1007/s11908-009-0066-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu F, Sternberg MR, Kottiri BJ, McQuillan GM, Lee FK, Nahmias AJ, et al. Trends in herpes simplex virus type 1 and type 2 seroprevalence in the United States. JAMA. 2006;296:964–973. doi: 10.1001/jama.296.8.964. [DOI] [PubMed] [Google Scholar]
- Berrington WR, Jerome KR, Cook L, Wald A, Corey L, Casper C. Clinical correlates of herpes simplex virus viremia among hospitalized adults. Clin Infect Dis. 2009;49:1295–1301. doi: 10.1086/606053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown EL, Gardella C, Malm G, Prober CG, Forsgren M, Krantz EM, et al. Effect of maternal herpes simplex virus (HSV) serostatus and HSV type on risk of neonatal herpes. Acta Obstet Gynecol Scand. 2007;86:523–529. doi: 10.1080/00016340601151949. [DOI] [PubMed] [Google Scholar]
- Serwadda D, Gray RH, Sewankambo NK, Wabwire-Mangen F, Chen MZ, Quinn TC, et al. Human immunodeficiency virus acquisition associated with genital ulcer disease and herpes simplex virus type 2 infection: a nested case-control study in Rakai, Uganda. J Infect Dis. 2003;188:1492–1497. doi: 10.1086/379333. [DOI] [PubMed] [Google Scholar]
- Wald A, Link K. Risk of human immunodeficiency virus infection in herpes simplex virus type 2-seropositive persons: a meta-analysis. J Infect Dis. 2002;185:45–52. doi: 10.1086/338231. [DOI] [PubMed] [Google Scholar]
- Corey L, Wald A, Patel R, Sacks SL, Tyring SK, Warren T, et al. Once-daily valacyclovir to reduce the risk of transmission of genital herpes. N Engl J Med. 2004;350:11–20. doi: 10.1056/NEJMoa035144. [DOI] [PubMed] [Google Scholar]
- Celum C, Wald A, Lingappa JR, Magaret AS, Wang RS, Mugo N, et al. Acyclovir and transmission of HIV-1 from persons infected with HIV-1 and HSV-2. N Engl J Med. 2010;362:427–439. doi: 10.1056/NEJMoa0904849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen J. Immunology. Painful failure of promising genital herpes vaccine. Science. 2010;330:304. doi: 10.1126/science.330.6002.304. [DOI] [PubMed] [Google Scholar]
- Stoddard BL. Homing endonucleases: from microbial genetic invaders to reagents for targeted DNA modification. Structure. 2011;19:7–15. doi: 10.1016/j.str.2010.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiffer JT, Aubert M, Weber ND, Mintzer E, Stone D, Jerome KR. Targeted DNA mutagenesis for the cure of chronic viral infections. J Virol. 2012;86:8920–8936. doi: 10.1128/JVI.00052-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aubert M, Ryu BY, Banks L, Rawlings DJ, Scharenberg AM, Jerome KR. Successful targeting and disruption of an integrated reporter lentivirus using the engineered homing endonuclease Y2 I-AniI. PLoS One. 2011;6:e16825. doi: 10.1371/journal.pone.0016825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosse S, Huot N, Mahiet C, Arnould S, Barradeau S, Clerre DL, et al. Meganuclease-mediated inhibition of HSV1 infection in cultured cells. Mol Ther. 2011;19:694–702. doi: 10.1038/mt.2010.302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roizman B, Knipe DM, Whitley RJ.2007Herpes simplex viruses. Knipe DM, Howley PM.eds.). Fields Virology5th edn, vol. 2Wolters Kluwer Health/Lippincott Williams & Wilkins; pp. 2502–2599. [Google Scholar]
- Wigdahl BL, Scheck AC, De Clercq E, Rapp F. High efficiency latency and activation of herpes simplex virus in human cells. Science. 1982;217:1145–1146. doi: 10.1126/science.6180477. [DOI] [PubMed] [Google Scholar]
- Arnould S, Chames P, Perez C, Lacroix E, Duclert A, Epinat JC, et al. Engineering of large numbers of highly specific homing endonucleases that induce recombination on novel DNA targets. J Mol Biol. 2006;355:443–458. doi: 10.1016/j.jmb.2005.10.065. [DOI] [PubMed] [Google Scholar]
- Bennardo N, Gunn A, Cheng A, Hasty P, Stark JM. Limiting the persistence of a chromosome break diminishes its mutagenic potential. PLoS Genet. 2009;5:e1000683. doi: 10.1371/journal.pgen.1000683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Certo MT, Gwiazda KS, Kuhar R, Sather B, Curinga G, Mandt T, et al. Coupling endonucleases with DNA end-processing enzymes to drive gene disruption. Nat Methods. 2012;9:973–975. doi: 10.1038/nmeth.2177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delacôte F, Perez C, Guyot V, Duhamel M, Rochon C, Ollivier N, et al. High frequency targeted mutagenesis using engineered endonucleases and DNA-end processing enzymes. PLoS One. 2013;8:e53217. doi: 10.1371/journal.pone.0053217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valton J, Daboussi F, Leduc S, Molina R, Redondo P, Macmaster R, et al. 5'-Cytosine-phosphoguanine (CpG) methylation impacts the activity of natural and engineered meganucleases. J Biol Chem. 2012;287:30139–30150. doi: 10.1074/jbc.M112.379966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Rensburg R, Beyer I, Yao XY, Wang H, Denisenko O, Li ZY, et al. Chromatin structure of two genomic sites for targeted transgene integration in induced pluripotent stem cells and hematopoietic stem cells. Gene Ther. 2013;20:201–214. doi: 10.1038/gt.2012.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshmane SL, Fraser NW. During latency, herpes simplex virus type 1 DNA is associated with nucleosomes in a chromatin structure. J Virol. 1989;63:943–947. doi: 10.1128/jvi.63.2.943-947.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knipe DM, Cliffe A. Chromatin control of herpes simplex virus lytic and latent infection. Nat Rev Microbiol. 2008;6:211–221. doi: 10.1038/nrmicro1794. [DOI] [PubMed] [Google Scholar]
- Samaniego LA, Neiderhiser L, DeLuca NA. Persistence and expression of the herpes simplex virus genome in the absence of immediate-early proteins. J Virol. 1998;72:3307–3320. doi: 10.1128/jvi.72.4.3307-3320.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferenczy MW, DeLuca NA. Epigenetic modulation of gene expression from quiescent herpes simplex virus genomes. J Virol. 2009;83:8514–8524. doi: 10.1128/JVI.00785-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang L, Gao H, Zhu X, Wang X, Zhou M, Jiang R. Construction of “small-intelligent” focused mutagenesis libraries using well-designed combinatorial degenerate primers. Biotechniques. 2012;52:149–158. doi: 10.2144/000113820. [DOI] [PubMed] [Google Scholar]
- Rosen LE, Morrison HA, Masri S, Brown MJ, Springstubb B, Sussman D, et al. Homing endonuclease I-CreI derivatives with novel DNA target specificities. Nucleic Acids Res. 2006;34:4791–4800. doi: 10.1093/nar/gkl645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith J, Grizot S, Arnould S, Duclert A, Epinat JC, Chames P, et al. A combinatorial approach to create artificial homing endonucleases cleaving chosen sequences. Nucleic Acids Res. 2006;34:e149. doi: 10.1093/nar/gkl720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thyme SB, Jarjour J, Takeuchi R, Havranek JJ, Ashworth J, Scharenberg AM, et al. Exploitation of binding energy for catalysis and design. Nature. 2009;461:1300–1304. doi: 10.1038/nature08508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiffer JT, Swan DA, Stone D, Jerome KR. Predictors of hepatitis B cure using gene therapy to deliver DNA cleavage enzymes: a mathematical modeling approach. PLoS Comput Biol. 2013;9:e1003131. doi: 10.1371/journal.pcbi.1003131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glatzel M, Flechsig E, Navarro B, Klein MA, Paterna JC, Büeler H, et al. Adenoviral and adeno-associated viral transfer of genes to the peripheral nervous system. Proc Natl Acad Sci USA. 2000;97:442–447. doi: 10.1073/pnas.97.1.442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming J, Ginn SL, Weinberger RP, Trahair TN, Smythe JA, Alexander IE. Adeno-associated virus and lentivirus vectors mediate efficient and sustained transduction of cultured mouse and human dorsal root ganglia sensory neurons. Hum Gene Ther. 2001;12:77–86. doi: 10.1089/104303401450997. [DOI] [PubMed] [Google Scholar]
- Xu Y, Gu Y, Wu P, Li GW, Huang LY. Efficiencies of transgene expression in nociceptive neurons through different routes of delivery of adeno-associated viral vectors. Hum Gene Ther. 2003;14:897–906. doi: 10.1089/104303403765701187. [DOI] [PubMed] [Google Scholar]
- Manservigi R, Argnani R, Marconi P. HSV recombinant vectors for gene therapy. Open Virol J. 2010;4:123–156. doi: 10.2174/1874357901004010123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goins WF, Cohen JB, Glorioso JC. Gene therapy for the treatment of chronic peripheral nervous system pain. Neurobiol Dis. 2012;48:255–270. doi: 10.1016/j.nbd.2012.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawtell NM. Comprehensive quantification of herpes simplex virus latency at the single-cell level. J Virol. 1997;71:5423–5431. doi: 10.1128/jvi.71.7.5423-5431.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawtell NM, Poon DK, Tansky CS, Thompson RL. The latent herpes simplex virus type 1 genome copy number in individual neurons is virus strain specific and correlates with reactivation. J Virol. 1998;72:5343–5350. doi: 10.1128/jvi.72.7.5343-5350.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K, Lau TY, Morales M, Mont EK, Straus SE. Laser-capture microdissection: refining estimates of the quantity and distribution of latent herpes simplex virus 1 and varicella-zoster virus DNA in human trigeminal Ganglia at the single-cell level. J Virol. 2005;79:14079–14087. doi: 10.1128/JVI.79.22.14079-14087.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K, Mahalingam G, Hoover SE, Mont EK, Holland SM, Cohen JI, et al. Diverse herpes simplex virus type 1 thymidine kinase mutants in individual human neurons and Ganglia. J Virol. 2007;81:6817–6826. doi: 10.1128/JVI.00166-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terry-Allison T, Smith CA, DeLuca NA. Relaxed repression of herpes simplex virus type 1 genomes in murine trigeminal neurons. J Virol. 2007;81:12394–12405. doi: 10.1128/JVI.01068-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mark KE, Wald A, Magaret AS, Selke S, Olin L, Huang ML, et al. Rapidly cleared episodes of herpes simplex virus reactivation in immunocompetent adults. J Infect Dis. 2008;198:1141–1149. doi: 10.1086/591913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aubert M, Chen Z, Lang R, Dang CH, Fowler C, Sloan DD, et al. The antiapoptotic herpes simplex virus glycoprotein J localizes to multiple cellular organelles and induces reactive oxygen species formation. J Virol. 2008;82:617–629. doi: 10.1128/JVI.01341-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He TC, Zhou S, da Costa LT, Yu J, Kinzler KW, Vogelstein B. A simplified system for generating recombinant adenoviruses. Proc Natl Acad Sci U S A. 1998;95:2509–2514. doi: 10.1073/pnas.95.5.2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kota J, Chivukula RR, O'Donnell KA, Wentzel EA, Montgomery CL, Hwang HW, et al. Therapeutic microRNA delivery suppresses tumorigenesis in a murine liver cancer model. Cell. 2009;137:1005–1017. doi: 10.1016/j.cell.2009.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi VW, Asokan A, Haberman RA, Samulski RJ. Production of recombinant adeno-associated viral vectors for in vitro and in vivo use. Curr Protoc Mol Biol. 2007;Chapter 16:Unit 16.25. doi: 10.1002/0471142727.mb1625s78. [DOI] [PubMed] [Google Scholar]
- Halford WP, Gebhardt BM, Carr DJ. Mechanisms of herpes simplex virus type 1 reactivation. J Virol. 1996;70:5051–5060. doi: 10.1128/jvi.70.8.5051-5060.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox R, Aubert M. Flow cytometric detection of activated caspase-3. Methods Mol Biol. 2008;414:47–56. doi: 10.1007/978-1-59745-339-4_5. [DOI] [PubMed] [Google Scholar]
- Anderson MJ. A new method for non-parametric multivariate analysis of variance. Austral Ecol. 2001;26:32–46. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.